

SUMMARY
Nuclear actin (N-actin) is known to participate in the regulation of gene expression. We showed previously that N-actin levels mediate the growth and quiescence of mouse epithelial cells in response to laminin-111 (LN1), a component of the mammary basement membrane (BM). We know that BM is defective in malignant cells, and we show here that it is the LN1/N-actin pathway that is aberrant in human breast cancer cells, leading to continuous growth. Photobleaching assays revealed that N-actin exit in nonmalignant cells begins as early as 30 min after LN1 treatment. LN1 attenuates the PI3K pathway leading to upregulation of exportin-6 (XPO6) activity and shuttles actin out of the nucleus. Silencing XPO6 prevents quiescence. Malignant cells are impervious to LN1 signaling. These results shed light on the crucial role of LN1 in quiescence and differentiation and how defects in the LN1/PI3K/XPO6/N-actin axis explain the loss of tissue homeostasis and growth control that contributes to malignant progression.
Graphical Abstract
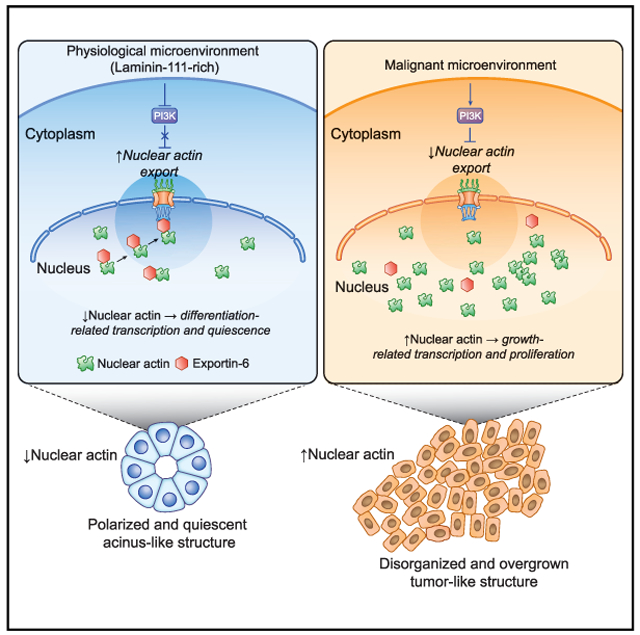
In Brief
Fiore et al. show that laminin-111 (LN1) induces a drastic decrease of nuclear actin in human mammary epithelial cells in a process mediated by XPO6 and required for acquisition of cellular quiescence. The LN1/XPO6/N-actin pathway is abnormal in malignant cells that are unresponsive to LN1 and proliferate uncontrollably.
INTRODUCTION
In vivo, the majority of cells are quiescent (O’Farrell, 2011). Unlike microorganisms, which become quiescent mainly because of nutrient deprivation (Daignan-Fornier and Sagot, 2011; O’Farrell, 2011), cells in higher organisms stop proliferating as organisms mature even when growth factors are plentiful. The pathway by which tissues in vivo stop proliferating is poorly understood. We have provided evidence that the cellular microenvironment, in particular the extracellular matrix (ECM) in the form of laminin-111 (LN1), an essential component of the basement membrane (BM) of many tissues, regulates cell death and quiescence (Boudreau et al., 1995; Petersen et al., 1992; Spencer et al., 2011). Disruption of this regulation is a fundamental step in cancer progression (Bissell and Hines, 2011; Kenny and Bissell, 2003). Signaling from LN1/BM (Streuli and Bissell, 1990) reprograms gene expression (Streuli and Bissell, 1990), leading to growth arrest and functional differentiation (Barcellos-Hoff et al., 1989; Spencer et al., 2011; Streuli et al., 1991, 1995). Malignant cells fail to respond to signals from the BM (Petersen et al., 1992), and active degradation of LN1 by matrix metalloproteinases disrupts the architecture of breast acini, reinitiating cell proliferation (Beliveau et al., 2010) and leading to eventual formation of mammary tumors (Bissell et al., 2005). However, the molecular mechanisms by which LN1 performs these functions are only partially understood.
We showed previously the surprising involvement of reduction in the N-actin level as an essential process for normal mouse mammary epithelial cells to become quiescent (Spencer et al., 2011). Preventing N-actin export from the nucleus, through expression of actin fused to a nuclear localization signal (NLS), blocked quiescence. These data, and also reports that place N-actin as an essential regulator of gene transcription and other nuclear processes (Belin and Mullins, 2013; Virtanen and Vartiainen, 2017), led us to hypothesize that the pathway regulating N-actin levels in normal epithelial cells is disrupted in malignant cells and that the inability of the malignant cells to stop growth relates to its inability to respond to quiescence-inducing extra-cellular cues, including cell and tissue polarity.
Here we used nonmalignant (“normal”) and malignant human breast epithelial cells from the syngeneic HMT-3522 progression series, a robust model of breast cancer progression, (Briand et al., 1987; Rizki et al., 2008; Weaver et al., 1997). We tested the cells in both 2D monolayers in the presence of LN1 or on top of 3D LN1-rich ECM gels (Barcellos-Hoff et al., 1989; Petersen et al., 1992) and found that, in both cases, nonmalignant cells contained less N-actin compared with their malignant counterparts. Reduction of N-actin in nonmalignant cells began as early as 30 min after LN1 treatment and was mediated by rapid upregulation of XPO6, a nuclear export receptor that actively transports N-actin out of the nucleus (Dopie et al.,2012). Post-transcriptional silencing of XPO6 overcame cell growth arrest induced by LN1 in nonmalignant cells. N-actin levels remained unchanged in tumor cells, indicating that the signaling pathway responsible for LN1-mediated control over N-actin levels is defective in malignant breast cells. Although much remains to be discovered about this fundamental pathway, in particular about how LN1 and loss of actin reorganize chromatin and the epigenome, this work lays the foundation for fundamental pathways that may be critical in therapeutic responses in cancer and other related diseases and could provide new druggable targets. Our work reveals that the level of N-actin is an intermediary in translating LN1 cues that influence transcription, proliferation, and quiescence and that perturbation of signaling from the ECM to N-actin is pivotal for disruption of tissue homeostasis and uncontrolled growth observed in cancer.
RESULTS
Nonmalignant Human Breast Epithelial Cells Become Quiescent upon Addition of LN1-Rich ECM or LN1, whereas Malignant Cells Continue to Proliferate
To address why malignant cells continue to grow even in the presence of LN1, we used S1 (normal) and T4-2 (malignant) cells from the syngeneic HMT-3522 progression series (Briand et al., 1987; Petersen et al., 1992; Rizki et al., 2008; Weaver et al., 1997). We found that, analogous to what we showed for mouse cells (Spencer et al., 2011), human S1 cells decreased proliferation, global transcription, and total RNA and mRNA levels upon addition of LN1-rich ECM (LrECM; we used commercial Matrigel) to monolayer cultures and assessed growth by 5-ethynyl-2’-deoxyuridine (EdU) incorporation assay (Figure 1A; Figure S1A), transcription by quantification of immunofluorescence (IF) of the phosphorylated form of RNA polymerase II (Pol II-Ser2-P; Figure 1B; Figure S1B), quantification of 5-ethynyl-uridine (EU) incorporation (Figure 1C; Figure S1C), and determination of total RNA and mRNA to DNA ratios (Figure S1D). Addition of LrECM to S1 cells grown as monolayers resulted in a dramatic reduction of N-actin levels (Figures 1D and 1E; Figure S1E) but only a slight reduction of cytoplasmic actin (Figure 1E). Malignant cells exposed to LrECM were unresponsive; proliferation (Figure 1A; Figure S1A), N-actin and cytoplasmic actin levels (Figures 1D and 1E), RNA synthesis (Figure 1C; Figure S1C), total RNA, mRNA, and Pol II-Ser2-P staining (Figure 1B; Figure S1B) remained similar to the untreated cultures. We also found that quiescent and tissue-like acini formed by S1 cells in the growth-suppressive 3D microenvironment of LrECM presented much lower N-actin levels than overgrown tumor-like structures formed by T4-2 cells (Figure 1F). Matrigel is LN1-rich; however, it also has significant levels of other ECM proteins as well as some minute levels of growth factors and other molecules. We therefore tested whether pure LN1 or type IV collagen (another BM molecule also abundant in LrECM) would affect N-actin levels in normal cells. Only LN1 treatment reduced N-actin (Figure S1F), confirming the specificity of this crucial BM constituent in inducing cellular quiescence in nonmalignant cells as well as tissues via depletion of N-actin (Spencer et al., 2011). Accordingly, from here on, we will refer to all LN1-rich supplements as LN1.
Figure 1. Nonmalignant and Malignant Cells Respond Differently to LrECM in Terms of Quiescence, Changes in Levels of N-Actin, and RNA Synthesis.

(A–E) S1 (nonmalignant) and T4-2 (malignant) HMT3522 cells were grown in the absence (control) or presence of 5% LrECM for 24 hr. Note that, because S1 cells are smaller than T4-2 cells, we increased the magnification of S1 cell micrographies to show similar number of cells per image (see also Figure S1A for phase-contrast images).
(A) Top: representative images of EdU incorporation (red) reflecting cell proliferation. Bottom: quantification of EdU-positive cells (>500 cells/condition from three independent experiments).
(B) Top: representative images of cells stained for active RNA polymerase II (Pol II-Ser2-P, green). Bottom: quantification of Pol II-Ser2-P staining (>250 cells/ condition from three independent experiments).
(C) Top: representative images of EU incorporation (gray) reflecting RNA synthesis. Bottom: quantification of EU incorporation (>65 cells/condition from two independent experiments).
(D) Top: representative images of S1 and T4-2 cells stained for N-actin (green). Bottom: quantification of N-actin staining (>250 cells/condition from three independent experiments).
(B–D) The nuclei are highlighted by white outlines (see also Figures S1B–S1E).
(E) Western blots of actin in the nuclear and cytoplasmic fractions. Lamin-B and tubulin were used to assess the purity of the cellular fractionation and as loading controls of nuclear and cytoplasmic fractions, respectively (three independent experiments).
(F) S1 and T4-2 cells were cultured in 3D-LrECM for 5 days. N-actin is more abundant in disorganized and overgrown structures formed by T4-2 cells, whereas polarized and quiescent acinus-like structures formed by S1 cells display low levels of N-actin. Left: representative images of breast epithelial structures in 3D LrECM stained for N-actin (green) and the basal polarity marker α6-integrin (red). In close-up images, the nuclei are highlighted by white outlines. Right: quantification of N-actin staining (50 cells/condition from three independent experiments).
Data are expressed as mean ± SEM. **p < 0.01, ***p < 0.0001. Scale bars, 10 μm.
LN1 Induces Rapid Reduction of N-Actin in Nonmalignant Cells by Increasing Nuclear Export
To gain insights into the underlying mechanism by which N-actin content is regulated, we monitored the kinetics of the N-actin levels in S1 nonmalignant cells in response to LN1. IF staining of N-actin and western blotting of the nuclear and cytoplasmic fractions from S1 cells revealed that the decline of N-actin could be detected as early as 30 min after LN1 treatment, whereas cytoplasmic actin levels started to reduce weakly (a drop of 30%) only by 4 hr after treatment. The drop of N-actin was progressive; N-actin reached ~20% of the amount measured in untreated S1 cells by 24 hr (Figures 2A and 2B; Figures S1G-S1I).
Figure 2. LN1-Induced Depletion of N-Actin in S1 Nonmalignant Cells Is Due to Increased Nuclear Export.

(A) Representative images of N-actin staining of S1 cell nuclei showing changes of content after LN1 treatment (three independent experiments) (see also Figures S1H and S1I). The nuclei are highlighted by white outlines.
(B) Western blot of the nuclear and cytoplasmic fractions of S1 cells showing rapid reduction of N-actin by 30 min after LN1 addition. Lamin-B and tubulin were used to assess the purity of the cellular fractionation and as loading controls of nuclear and cytoplasmic fractions, respectively (three independent experiments).
(C) Schematic of the fluorescence loss in photobleaching (FLIP) assay as described in Experimental Procedures.
(D) Representative images of S1 cells expressing mYPET-tagged actin analyzed by the FLIP assay.
(E) Quantification of the N-actin export (8–12 cells assayed/time point; FLIP measurements were done in two independent experiments) (see also Figure S2). The nuclei are highlighted by red outlines. Note that the N-actin export starts to increase as early as 15–30 min after LN1 addition.
Data are expressed as mean ± SEM. *p < 0.05, **p < 0.01. Scale bars, 5 μm.
Actin monomers are transported in and out of the nucleus through the nuclear pore complex via active transport (Dopie et al., 2012). We used established live-cell imaging approaches based on photobleaching to measure the dynamics of both N-actin import and export (Dopie et al., 2012; Skarp and Vartiainen, 2013) in nonmalignant cells expressing mYPET-tagged β-actin. Import of N-actin was measured by fluorescence recovery after photobleaching (FRAP). By photobleaching the entire cell nucleus, we could record the regaining of the nuclear fluorescence. Surprisingly, LN1 treatment did not change the nuclear import of actin (Figures S2A and S2B) but N-actin export, using fluorescence loss in photobleaching (FLIP), where the entire cytoplasm is photobleached intermittently and the nuclear fluorescence decay is measured (Figures 2C–2E). The decay rate of fluorescence in the nucleus then reflects the export rate of N-actin. FLIP revealed that N-actin export increased significantly in S1 cells as early as 30 min after the start of LN1 treatment, which coincided with the bulk kinetics of N-actin we documented above (Figures 2A–2E). Our attempts to document the kinetics of N-actin movement failed in malignant T4-2 cells because of their rapid mobility. However, because the total N-actin levels in malignant cells were unchanged after addition of LN1, we conclude that the balance of import and export in T4-2 cells almost certainly remains unchanged.
The Nuclear Exporter XPO6 Mediates the LN1-Induced Reduction of N-Actin in Nonmalignant Cells
The N-actin exporter XPO6 actively shuttles N-actin out of the nucleus through nuclear pore complexes (Dopie et al., 2012). To determine the mechanism by which LN1 enhances the export rate of N-actin to drive quiescence, we silenced XPO6 in S1 cells with short hairpin RNA (shRNA; S1-XPO6-shRNA-1 and S1-XPO6-shRNA-2; Figure S3A). This significantly suppressed the rate of N-actin export and prevented N-actin levels from declining in response to LN1 (Figures S3B and S3C; Figures 3A–3C), indicating the specific involvement of XPO6 in movement of N-actin from the nucleus. Interestingly, despite the fact that export of N-actin would start as early as 30 min after LN1 addition in S1 cells, the XPO6 protein level remained essentially unchanged for 2 hr after LN1 treatment (Figure 4A, left). However, co-immunoprecipitation (coIP) of myc-tagged actin in the nuclear fraction of S1 cells revealed that the XPO6 to N-actin ratio increased significantly as early as 30 min after LN1 addition (Figure 4A, right). This indicates that XPO6 must be present in excess of free N-actin under growth conditions and that the early export of N-actin engaged in nuclear processes important for transcription and proliferation is accommodated by the existing level of XPO6. The reciprocal assay, co-immunoprecipitation of endogenous XPO6, produced complementary results, further substantiating the results obtained with myc-tagged actin (Figure S3D). Concomitantly, XPO6 became concentrated to the nuclear lamina in S1 cells, as evidenced by co-staining with lamin B (Figure 4B). The nuclear lamina vicinity is where nuclear importers and exporters concentrate preferentially when actively transporting their cargos (Kutay et al., 1998). These data are consistent with the kinetics of N-actin reduction and export rate increase we observed for nonmalignant cells (Figures 2A–2E). In sharp contrast, in malignant cells treated with LN1, the level of XPO6 and the XPO6 to N-actin ratio, if anything, decreased (Figure 4C), and XPO6 localization did not change either (Figure 4D).
Figure 3. XPO6 Mediates LN1-Induced Depletion of N-Actin in Nonmalignant Cells.

(A–D) Silencing of XPO6 with shRNA prevents the increase of N-actin export in S1 cells exposed to LN1 (see also Figures S3A–S3C).
(A) Left: representative images of S1 cells expressing a control shRNA (S1-ct-shRNA) or two different shRNAs for XPO6 (S1-XPO6-shRNA-1 or -2) treated for 24 hr with LN1 and stained for N-actin (green; the nuclei are highlighted by white outlines). Right: quantification of N-actin staining (>150 cells/condition from two independent experiments).
(B) Schematic of the FLIP assay.
(C) Representative images of S1 cells expressing mYPET-tagged actin analyzed by the FLIP assay. The nuclei are highlighted by red outlines.
(D) Quantification of the export rate of N-actin (4–6 cells assayed/time point; FLIP measurements were done in two independent experiments).
Data are expressed as mean ± SEM. ***p < 0.0001; n.s., no statistical significance. Scale bars, 10 μm in (A) and 5 μm in (C).
Figure 4. LN1 Changes the Ratio of XPO6 to N-Actin in Opposite Directions in Nonmalignant and Malignant Cells.

(A) Left: western blot of total S1 cell lysate showing the changes in XPO6 content after addition of LN1 (three independent experiments). Tubulin was used as a loading control. Right: co-immunoprecipitation of myc-tagged actin reveals actin interaction with XPO6 in the nuclear fraction of S1 cells after LN1 treatment (two independent experiments). In nonmalignant cells, LN1 increases both XPO6 expression and the XPO6 to N-actin ratio (normalization of XPO6 levels to myc-tagged actin) in the co-immunoprecipitation pellet (see also Figure S3D).
(B) Left: representative images of cells stained for XPO6 (green) and lamin-B (red) showing an increase in localization of XPO6 at the nuclear lamina (NL) region after LN1 treatment. Arrowheads denote the localization of the nuclear lamina based on lamin-B staining (above). Right: quantification of the fluorescence intensity of XPO6 detected at the nuclear lamina region (10 cells assayed/time point from three independent experiments).
(C) LN1 reduces both XPO6 expression and its interaction with N-actin in malignant cells. Left: western blot of total T4-2-cell lysate showing the decrease of XPO6 after LN1 treatment. Tubulin was used as a loading control. Right: co-immunoprecipitation of myc-tagged actin revealing actin interaction with XPO6 in the nuclear fraction of T4-2 cells after LN1 treatment.
(D) The distribution of XPO6 remains unchanged in T4-2 cells after LN1 treatment. Left: Representative images of IF showing the localization of the XPO6 in T4-2 cells. Right: quantification of XPO6 at the nuclear lamina region of T4-2 cells (10 cells assayed/time point from three independent experiments).
Data are expressed as mean ± SEM. ***p < 0.0001; n.s., no statistical significance. Scale bars, 5 μm.
XPO6 Silencing Overcomes Epithelial Cell Quiescence
For decades, the disparity in growth regulation observed in culture dishes in normal and malignant cells or in vivo in malignant tumors has been considered the most defining feature of malignancy. The differences in the LN1/N-actin axis we uncovered between nonmalignant and malignant cells led us to speculate that the finding may be related to the inability of malignant cells to respond to LN1 when tissue organization is perturbed, as is the case in tumors. Thus, disrupting N-actin export would allow N-actin and growth to remain unchanged in nonmalignant cells. This was indeed the case. shRNA targeting XPO6 in normal cells prevented the drop in EdU-positive cells (Figure 5A; Figure S4A) in the levels of Pol II-Ser2-P and EU incorporation (Figures S4B and S4C) induced by LN1, where LN1-treated and untreated cells continued proliferating at the same rate. In contrast, the proliferation rate of S1 cells expressing control shRNA (ct-shRNA) was decreased appreciably by 4 hr of LN1 treatment (Figure 5A).
Figure 5. XPO6 Silencing Prevents Epithelial Cell Quiescence.

(A–C) S1 cells silenced for XPO6 (XPO6-shRNA) do not reduce the level of N-actin in the presence of LN1 and overcome cellular quiescence.
(A) Left: representative images of EdU incorporation (red) reflecting proliferation. Right: quantification of EdU-positive cells (>250 cells/condition from three independent experiments) (see also Figure S4).
(B and C) Silencing of XPO6 prevents tissue quiescence of nonmalignant cells cultured in 3D-LrECM.
(B) Representative images of S1 3D structures stained for the cell cycle marker Ki67 (green) and the basal polarization marker α6-integrin (red).
(C) Left: quantification of the area of 3D structures (>400 structures/condition from three independent experiments). Right: quantification of Ki67-positive cells (10 structures assessed/condition from two independent experiments) (see also Figure S5).
(D) Left: S1 cells in 3D LrECM that have been silenced for XPO6 present disrupted apical polarity, as shown by ZO-1 staining (green), but intact basal polarity, as represented by the discrete and continuous localization of α6-integrin (red) to the basal surfaces. Arrows point to ZO-1 at the apical region of the structures, and arrowheads indicate basal deposition of ZO-1. Right: schematic of the results shown on the left (see also Figure S5C).
(E and F) Forcing the level of N-actin by expressing an NLS-actin construct in S1 cells results in overgrown 3D structures.
(E) Representative images of S1 3D structures expressing heterogeneous levels of YFP-NLS-actin (green). The 3D structures are highlighted by white outlines.
(F) Left: scatterplot showing YFP-NLS-actin fluorescence intensity as a function of the area of the 3D structures (R2, squared Pearson’s correlation coefficient; *p < 0.05; 91 structures from two independent experiments). Right: comparison of the area of YNAlow and YNAhigh 3D structures (see also Figure S6).
Data are expressed as mean ± SEM. **p < 0.01, ***p < 0.0001. Scale bars, 10 μm in (A) and (B), 20 μm in (C and D), and 30 μm in (E).
When cultured on top of a 3D LrECM gel, S1 cells displayed an orchestrated program of tissue morphogenesis culminating in quiescent acinus-like structures, as shown previously (Petersen et al., 1992), whereas XPO6 knockdown resulted in larger, proliferative epithelial structures containing high levels of N-actin (Figures 5B and 5C; Figures S5A and S5B) and partially disrupted apical polarity, as assessed by staining for the apical marker ZO-1 (Figure 5D; Figure S5C). Importantly, cells continued to preserve basal polarity, as judged by staining for the basal marker a6-integrin (Figures 5B and 5D; Figures S5B and S5C). These structures had a filled lumen and resembled what we had observed previously when we induced high levels of ERBb2 in MCF10A cells cultivated in 3D LrECM (Muthuswamy et al., 2001). This proliferative and apically disrupted phenotype is consistent with the morphological characteristics of ductal carcinoma in situ (DCIS) and pre-invasive carcinoma (Carraro et al., 2014).
We asked whether forcing the level of N-actin in nonmalignant cells (other than silencing XPO6) would result in similar 3D phenotypes seen in S1-XPO6-shRNA cells. S1 cells were transfected with an expression plasmid containing actin fused to an NLS and yellow fluorescent protein (YFP) (YFP-NLS-actin) or a control construct lacking actin (YFP-NLS). To generate a heterogeneous population that contained cells expressing a wide range of YFP-NLS-actin or YFP-NLS levels (from negligible to high levels), the cells were not submitted to any type of selection after transfection (e.g., antibiotic selection or sorting) (Figure S6A). Pearson’s correlation analysis revealed that the expression level of YFP-NLS-actin, as measured by fluorescence microscopy, positively correlated with the size of epithelial structures formed on the top of 3D LrECM gels (Figures 5E and 5F, left). We expanded this analysis by comparing the size of epithelial 3D structures expressing low (25% lowest fluorescence intensity structures, YNAlow) and high (25% highest fluorescence intensity structures, YNAhigh) levels of YFP-NLS-actin and found that YNAhigh structures were, on average, 2.4 times larger than YNAlow structures (Figure 5F, right). The fluorescence of S1 cells transfected with the control construct did not change with size (Figure S6B).
Attenuation of PI3K Activity Is Necessary for N-Actin Export in Monolayer Cultures
The rapid effect of LN1 on N-actin export raised the question of which major kinase signaling cascades could be involved in the regulation of these events. We and others have shown that the phosphatidylinositol 3-kinase (PI3K) and mitogen-activated protein kinase (MAPK) pathways relay extracellular cues from growth factors and the ECM to mediate changes in mammary cells during acinar morphogenesis (Akhtar et al., 2009; Beliveau et al., 2010; Liu et al., 2004; Utermark et al., 2012; Wang et al., 1998, 2002). These pathways are perturbed in breast tumors and in many other cancer types (Bader et al., 2005; Dhillon et al., 2007; Liu et al., 2009). Downmodulation of MAPK or inhibition of PI3K permits cells in 3D LrECM to reprogram the gene expression circuitry promoting epithelial polarization and quiescence of mammary structures (Becker-Weimann et al., 2013; Beliveau et al., 2010). We found that PI3K signaling activation, defined as the ratio of the phosphorylated form to total PI3K protein, was attenuated by LN1 within 30 min (ratio = 0.4) in nonmalignant S1 cells (Figure 6A). In malignant T4-2 cells, LN1 enhanced activation of PI3K within the same time frame (ratio = 1.6) (Figure 6B). AKT, a crucial kinase downstream of PI3K, displayed decreased activation (activation ratio = 0.7) in nonmalignant cells 2 hr after LN1 treatment. In malignant cells, the change in AKT activation was negligible. Although MAPK was also down- and up-modulated by LN1 in S1 and T4-2 cells, respectively, it exhibited a slower activation time course than PI3K (Figures 6A and 6B). In nonmalignant S1 cells, inhibition of MAPK activation was measurable only 2 hr after LN1 addition (ratio = 1.1 at 30 min and 0.5 at 2 hr) (Figure 6A). Analogous to the PI3K pathway, MAPK was activated further after LN1 addition in malignant cells (Figure 6B). To further examine the mechanism, we inhibited either the PI3K or MAPK pathway with specific chemical inhibitors and assessed the kinetics of N-actin export between 1–2 hr after addition of the inhibitors (Figure 6C). Within this short experimental time frame, only PI3K inhibition enhanced N-actin export and, consequently, reduced N-actin concentration in monolayers of S1 cells (Figure 6C). Using the same experimental setup, we inhibited PI3K signaling in S1-XP06-shRNA cells and found no significant changes in N-actin export and N-actin levels (Figure S7A). These results indicate that attenuation of PI3K signaling most probably is the primary regulatory mechanism necessary for the decrease of N-actin levels and subsequent quiescence in cells cultured on monolayers.
Figure 6. Reduction of PI3K Signaling Is Required for N-Actin Export.

(A and B) Kinetics of the activation ratio of PI3K, AKT, and MAPK (measured as the normalized ratio of phosphorylated form to total protein) in S1 and T4-2 cells after LN1 treatment (three independent experiments).
(A) The activation ratio of PI3K is reduced 30 min after LN1 addition in S1 cells. The MAPK activation ratio is inhibited only by 2 hr after treatment, and both the total levels and phosphorylated form are reduced. The AKT activation ratio reduces in S1 cells by 2 hr after LN1 addition.
(B) Both the PI3K and MAPK activation ratios increase in T4-2 cells after LN1 addition. Tubulin was used as a loading control.
(C) Growing S1 cells expressing mYPET-tagged actin were treated with either PI3K (LY294002) or MAPK phosphorylation (PD98059) inhibitors or only the vehicle (DMSO) and submitted to a FLIP assay 1–2 hr after addition of the drugs. Inhibition of the PI3K pathway, but not MAPK, enhances N-actin export, resulting in lower levels of N-actin. Left: quantification of the N-actin export rate (4–6 cells/condition, FLIP measurements were done in two independent experiments). Right: quantification of N-actin fluorescence (10 cells/ condition from two independent experiments; see also Figure S7A).
Data are expressed as mean ± SEM. *p < 0.05, **p < 0.01.
XPO6 Silencing Prevents “Phenotypic Reversion” of Malignant Cells Induced by PI3K Inhibition
We have shown repeatedly that attenuation of any of several signaling pathways, including PI3K, that are often deregulated in malignant cells leads to phenotypic reversion in 3D LrECM gels, resulting in the formation of quiescent structures with proper basal polarity and much reduced malignancy in vivo (Liu et al., 2004; Wang et al., 1998, 2002; Weaver et al., 1997). The integration of signaling pathways in 3D, the general correlation between quiescence and epithelial polarity (Bissell and Hines, 2011), and our data revealing that reduction of PI3K signaling is necessary for N-actin export in monolayer cultures led us to test whether XPO6 knockdown could prevent phenotypic reversion of malignant cells induced by chemical inhibition. T4-2 cells phenotypically reverted with the PI3K inhibitor LY294002 displayed much lower levels of N-actin than control malignant T4-2 cells (Figure S7B). T4-2-XPO6-shRNA cells regained basal polarity, as observed with a continuous α6-integrin-containing layer, but failed to become quiescent (Figures S7C and S7D; Figures 7A and 7B); this was similar to the phenotype of S1-XPO6-shRNA cells grown in 3D LrECM (Figure 5B; Figures S5B and S5C). These findings agree with reports showing that expression of a number of oncogenes in nonmalignant cells, such HMT3522-S1 and MCF10A cells, in 3D LrECM cultures inhibits quiescence but fails to disrupt epithelial basal polarity (Debnath et al., 2002; Leung and Brugge, 2012; Liu et al., 2004; Muthuswamy et al., 2001; Spancake et al., 1999). An additional conclusion is that acquisition of basal polarity alone is not sufficient to induce quiescence in 3D cultures and that further steps, such acquisition of apical polarity and organized tissue architecture, are required.
Figure 7. XPO6 Silencing Prevents Phenotypic Reversion Induced by PI3K Inhibition.

(A and B) Malignant T4-2 cells silenced for XPO6 do not become quiescent when treated with a reverting agent in 3D-LN1-rich gels.
(A) Left: representative images of T4-2-3D structures stained for Ki67 (green), α6-integrin (red), and DNA (DAPI, blue). Right: schematic depicting the results on the left.
(B) Top: quantification of the area of the 3D structures (2,000 structures/condition in three independent experiments). Bottom: quantification of Ki67-positive cells under different conditions (6–15 structures assessed/condition from two independent experiments) (see also Figures S7B–S7E).
Data are expressed as mean ± SEM. **p < 0.01, ***p < 0.0001. Scale bar, 10 μm.
DISCUSSION
Quiescence is a fundamental attribute in organ and tissue specificity as well as functional differentiation, and its loss is one of the most important manifestations of cancer progression. We reported more than two decades ago that cultivation of normal mouse and human mammary epithelial cells in LrECM leads to quiescence and formation of mammary acini (Barcellos-Hoff et al., 1989; Petersen et al., 1992; Streuli et al., 1991, 1995). We established also that, in human primary breast cells obtained from reduction mammoplasty, myoepithelial cells play a critical role in maintaining the polarity and quiescence of breast tissue by producing and depositing LN1 (Gudjonsson et al., 2002). However, the mechanism by which cultivation in 3D gels leads to growth inhibition was not investigated until our finding that, in normal mouse epithelial cells, LN1 addition led to decreased N-actin, a step that appeared to be essential for quiescence (Spencer et al., 2011).
Here we show that LN1/XPO6/N-actin is a critical regulatory pathway that controls the proliferation and quiescence of breast epithelia. The existence of N-actin was disputed quite strongly for more than 50 years, but, in the last 10 years, it has been shown repeatedly to be involved in many fundamental nuclear processes (Belin and Mullins, 2013; Virtanen and Vartiainen, 2017). N-actin is a critical factor for the activity of different chromatin remodeling complexes, RNA synthesis, and pre-mRNA processing machineries as well as DNA repair (Belin and Mullins, 2013; Virtanen and Vartiainen, 2017). We found that LN1 treatment drastically reduced RNA synthesis and proliferation (Figure 1), but blocking the nuclear exit of N-actin by either expressing an NLS-actin construct (Spencer et al., 2011; Figure 5) or silencing XPO6 overcame the growth-suppressive signals from LN1 in both 2D and 3D cultures (Figures 5 and 7). Previous work has demonstrated a clear dependence on de novo synthesis of protein coding and noncoding RNAs for cell proliferation in normal and abnormal conditions. Total RNA synthesis is required for BALB/c 3T3 cells to proceed from G1 to S phase in the cell cycle (Olson et al., 1993), and aberrant RNA Pol I and III activities correlate with uncontrolled proliferation (White, 2008). Disruption of RNA Pol III function decreases tRNA levels and induces cell cycle arrest in proliferative tissues of the developing zebrafish (Yee et al., 2007). Enhanced PI3K/AKT signaling increases transcription of ribosomal genes (Nguyen le and Mitchell, 2013), but the tumor suppressors phosphatase and tensin homolog (PTEN), P53, and retinoblastoma protein (pRb) inhibit Pol I and Pol III machineries (White, 2008). We had previously found that LN1 decreased N-actin and destabilized RNA Pol II and III interactions with nuclear substructures, resulting in lower rates of RNA synthesis and growth of mouse epithelial cells (Spencer et al., 2011). Furthermore, impairment of N-actin import resulted in lower transcriptional rates in NIH 3T3 cells (Dopie et al., 2012). Given the importance of N-actin to the activity of all three RNA polymerases (Percipalle, 2013) and that RNA synthesis is crucial for cell cycle progression, the level of N-actin that, according to our data, is regulated by LN1 might be a key intermediary in the microenvironmental control of proliferation.
We provide here a mechanism through which LN1 acts to control N-actin levels. It is the export of N-actin by XPO6 that is pivotal for cells to become quiescent when in a growth-suppressive LN1-containing environment (Figures 2, 3, and 4); an important corollary is that this pathway is aberrant in breast cancer cells (Figures 1, 4, 5, 6, and 7). Significantly, by searching a public clinical database, we also found that low XPO6 expression correlates with poor survival of breast cancer patients (Figure S7E). Failure in this signaling cascade prevents formation of the proper mammary architecture in 3D cultures, leading to morphological changes similar to those observed in early stages of breast cancer progression (Carraro et al., 2014; Figures 5 and 7). These findings show the power of using 3D assays to understand the relevance of what is being studied in culture to what happens in vivo. These data support the notion that cues from the microenvironment are crucial for restraining cell invasion, disruption of tissue architecture, and loss of BM integrity to malignancy (Bissell and Hines, 2011).
We did not detect changes in N-actin import (Figure S2) but observed a dramatic increase in N-actin export (Figure 2). Of note, a study with nonconfluent NIH/3T3 cells found that single or double knockdown of IPO9 and CFL1, proteins implicated in the nuclear import of actin, produced a drastic reduction in transcription, whereas silencing of XP06, despite resulting in increased N-actin levels, did not change transcription (Dopie et al., 2012). There are a number of explanations for these apparently conflicting data. NIH/3T3 cells are essentially fibroblasts that are different from mammary epithelial cells in many different biological aspects, including morphology, migration, and cell proliferation. However, when nonmalignant epithelial cells are cultured in a quiescence-inducing environment, such as in the presence of LN1, reestablishing the level of N-actin either by expressing an NLS-actin construct (Spencer et al., 2011) or an shRNA against XPO6 (Figure S4) restores transcription to the level of growing cells.
We observed that, in monolayers without LN1, inhibition of PI3K, but not MAPK, resulted in enhanced N-actin export and reduced N-actin concentration (Figure 6C; Figure S7). We and others have obtained data indicating that the PI3K signaling pathway establishes a link between LN1 and its receptors in mammary architecture and function (Akhtar et al., 2009; Chaudhuri et al., 2014; Xu et al., 2010). On the other hand, we have previously provided evidence that, in 3D cultures and in vivo, inhibition of either the MAPK or PI3K pathway induces growth arrest and phenotypic reversion of malignant cells resembling normal mammary acini (Beliveau et al., 2010; Wang et al., 1998, 2002). This is because, to maintain the 3D architecture of tissues, regulatory signaling pathways must integrate reciprocally (Bissell et al., 2005). Thus, blocking MAPK leads to attenuation of PI3K and vice versa. In 3D, the MAPK pathway would also contribute to epithelial quiescence via regulation of N-actin export, albeit secondarily. The molecular details of how attenuation of PI3K signaling triggers N-actin export require further investigation. We speculate that the kinase cascade triggered LN1 signaling through its β1 integrin receptor (Streuli et al., 1991) and that PI3K leads to reciprocal conformation changes in nuclear and chromatin structure, the details of which would be forthcoming. Additionally, it is known that PI3K signaling influences the availability of actin monomers (Janmey and Stossel, 1987; Lassing and Lindberg, 1985), which could regulate the pool of nuclear transport-competent actin. It is important to also highlight the nuclear myosins that can partner with N-actin, assembling nuclear actomyosin complexes and performing functions in nuclear processes such as regulation of Pol-I, and events that require movement, such as rearrangement and positioning of the nuclear matrix, chromatin, and gene loci (de Lanerolle and Serebryannyy, 2011). There is evidence that nuclear myosin 1c (NM1) mediates Pol I activation and cell cycle progression by facilitating P300/CBP-associated factor (PCAF)-mediated histone H3 lysine 9 (H3K9) acetylation (Sarshad et al., 2013). A more recent work reported that, at early G1 phase, glycogen synthase kinase (GSK) 3b, an enzyme downstream of PI3K, phosphorylates NM1 (Sarshad et al., 2014). This modification prevents NM1 polyubiquitination by the E3 ligase UBR5 and proteasomal degradation, allowing for NM1 association with the chromatin on the rDNA. Experiments with GSK3β−/− mouse embryonic fibroblasts indicated that, at G1, rRNA synthesis is suppressed because of decreased H3K9 acetylation (Sarshad et al., 2014). We have shown previously that PI3K inhibition in malignant cells in both 2D and 3D cultures indeed resulted in decreased GSK3β activation (Liu et al., 2004). It remains to be elucidated, however, whether GSK3β is also involved in the mechanism that regulates N-actin triggered by LN1 signaling. Nevertheless, the results presented here suggest a mechanism in which LN1 cues attenuate PI3K, initiating a cascade of events that culminates in export of N-actin by XPO6. Maintenance of N-actin levels is essential for keeping several nuclear machineries and processes functioning to sustain growth.
Studies using a combination of microscopy and fluorescent N-actin probes support the existence of polymeric N-actin in somatic cells (Baarlink et al., 2013; Belin et al., 2013; McDonald et al., 2006). Serum stimulation in fibroblasts induced formation of a short-lived (<60 s) polymeric actin network in the nucleus (Baarlink et al., 2013). During spreading of fibroblasts, however, N-actin filaments appear to be thicker, shorter, and more persistent; the network lasts 2–3 hr (Plessner et al., 2015). This transient N-actin network assembly, in both serum stimulation and cellular spreading, is crucial to induce the nuclear retention and activity of the co-transcriptional activator myocardin-related transcription factor (MRTF-A) (Baarlink et al., 2013; Plessner et al., 2015). Moreover, it has been shown that, during spreading of fibroblasts, the linker of the nucleoskeleton and cytoskeleton (LINC) complex connects signals from the ECM protein fibro-nectin to the nuclear envelope, influencing polymerization of N-actin (Plessner and Grosse, 2015). Contrasting with work showing that N-actin filaments activate specific transcription machineries, polymerizing actin into stable N-actin filaments inhibits the global transcriptional activity of RNA polymerase II (Serebryannyy et al., 2016). In the cytoplasm, actin partners with a large number of molecules and forms force-generating and contractile filaments that integrate, transduce, and effect mechanical and chemical signals in many crucial cellular processes, such as regulation of cell motility, morphology, endo-cytosis, phagocytosis, and other traits (Fletcher and Mullins, 2010). Actin is a central player in the molecular plasticity and cytoplasmic and nuclear interactions (Huet et al., 2012; Jorgens et al., 2017) bridging the dynamic and reciprocal exchange of information between the nucleus and the cell microenvironment that ultimately defines and regulates tissue structure and function. Although there is compelling evidence for different forms of polymeric N-actin (Grosse and Vartiainen, 2013), the exact function of actin filaments in the nucleus is still unknown. More work is needed to determine whether the proportion of distinct N-actin forms in malignant cells is different from that of normal cells or whether LN1 promotes changes in the relative amounts of these forms. Because N-actin is transported only as monomers, and the availability of actin monomers is a limiting factor in the nuclear shuttling (Dopie et al., 2012), possibly, at earlier time points after LN1 addition, the monomeric pool of N-actin is the initial target for XPO6, and then, at later time points, N-actin molecules that are released as monomers from nuclear complexes (such as transcriptional machineries) and/or from filaments that might undergo depolymerization after LN1 treatment are exported. We are currently designing experiments to determine whether the proportion of the different N-actin forms changes along with the decrease in N-actin after LN1 treatment.
The kinetics assessment we performed revealed that, in nonmalignant cells exposed to LN1, the interaction of N-actin and XPO6 and the concentration of XPO6 at the nuclear lamina began to rise before the increase in the protein level of XPO6 (Figure 4). It is thus possible that events triggered by LN1 cues, possibly attenuation of PI3K, might lead to conformational changes of XPO6. There is evidence that N-actin is targeted by post-translational modifications such as SUMOylation (Hofmann et al., 2009), but it remains to be shown whether LN1 signaling induces covalent modifications in N-actin that modulate its nuclear roles and export. The first process we measured in the dynamics of N-actin after LN1 treatment in normal cells was the nuclear exit of N-actin by 30 min (Figures 2 and 3). The slight decrease we detected in cytoplasmic actin did not occur until 4 hr later (Figure 2), concomitant with a sharp decrease in cell proliferation, which was reversed by silencing XPO6 (Figure 5). We did not detect cytoplasmic accumulation of actin as a consequence of the increased nuclear export, most likely because the actin concentration in the cytoplasm is much higher than in the nucleus and exported N-actin molecules would not contribute significantly to the cytoplasmic pool of actin. Also, we speculate that the reduction in cytoplasmic actin that happens long after N-actin drops might be due to a decrease in β-actin gene expression or increased mRNA or protein degradation. Previous studies have shown that LN1 reduces cytoplasmic actin stress fibers, induces organization of a cortical actin (Roskelley et al., 1994), and decreases cellular rigidity by inhibiting actin polymerization and myosin II activity (Alcaraz et al., 2008). These finely regulated tensile and structural changes are required during morphogenesis and functional differentiation of mammary gland epithelial cells (Akhtar et al., 2009; Alcaraz et al., 2008) and are perturbed in malignant transformation (Paszek et al., 2005). Although there are now compelling data for the existence of a nuclear tunnel containing cytoskeleton fibers in breast epithelial cells (Jorgens et al., 2017), implicating direct mechanical connections between the ECM and nucleus, it remains to be seen whether the cytoplasmic filamentous actin mechanically connects signals from the cytoplasm to N-actin. Nevertheless, actin is actively imported and exported through the nuclear pore complexes so that the nuclear and cytoplasmic actin pools are connected by a dynamic and continual stream (Dopie et al., 2012; Figure 3). What we show here is that this process is tuned by LN1 in normal breast epithelial cells, effecting cellular quiescence, one of the most essential aspects of homeostasis.
Given the multiple tasks of actin in the nucleus, especially in chromatin remodeling and in many steps of RNA biogenesis (Belin and Mullins, 2013; Virtanen and Vartiainen, 2017), it appears that it acts as a lever for the ECM to regulate proliferation and quiescence in cells in culture and, most probably, also in vivo (Spencer et al., 2011). The parallel phenotypes between cells silenced forXPO6 in 3D cultures and DCIS is compelling (Figures 5 and 7) and suggests that the LN1/XPO6/N-actin signaling pathway may provide new targets for the early stages of breast cancer progression.
EXPERIMENTAL PROCEDURES
Cell Culture and ECM Treatments
S1 and the T4-2 cells were grown in 2D or 3D LrECM in a serum-free defined medium as described previously (Briand et al., 1987; Petersen et al., 1992). Treatments with different extracellular matrices were performed as published before (Spencer et al., 2011) with some modifications. Detailed procedures are described in the Supplemental Experimental Procedures.
Antibodies, Plasmids, Lentivirus Production, and Infection
All antibodies, plasmid specifications, and procedures for lentivirus production and infection are described in the Supplemental Experimental Procedures.
Immunofluorescence
Immunofluorescence for 2D and 3D cell cultures was performed as described previously (Lee et al., 2007; Spencer et al., 2011). Detailed procedures are described in the Supplemental Experimental Procedures.
EdU and EU Incorporation Assays
For EdU and EU assays, we used the Click-iT EdU and Click-iT RNA kits (Life Technologies) following the manufacturer’s instructions. For more details, see the Supplemental Experimental Procedures.
Purification and Quantification of Nucleic Acids
Nucleic acids were isolated using TRIzol reagent (Life Technologies) as instructed by the manufacturer with minor changes. The mRNA was isolated using the Dynabeads kit (Life Technologies). For detailed information, see the Supplemental Experimental Procedures.
Image Acquisition and Analysis
Microscope settings, photobleaching assays, quantification of fluorescence staining, and measurements of the the size of 3D structures are described in detail in the Supplemental Experimental Procedures.
Preparation of Cellular Extracts, Co-immunoprecipitation, and Western Blotting
Detailed procedures are described in the Supplemental Experimental Procedures.
Kaplan-Meier Survival Analysis
The online resource SurvExpress (Aguirre-Gamboa et al., 2013) was used to compare the survival profiles of breast cancer patients with respect to the expression of XPO6. For detailed information, see the Supplemental Experimental Procedures.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 7.0. Data are presented as mean ± SEM. The number of samples analyzed and times that the experiments were performed are indicated in the figure legends. Two-tailed unpaired t test was used for comparisons of two conditions. For comparisons of three or more conditions, ANOVA followed by Dunnett test was used. Significance was defined as p < 0.05.
Supplementary Material
Highlights.
N-actin levels control transcription and proliferation of normal mammary cells
Laminin-111 (LN1) induces a rapid decrease of nuclear actin mediated by XPO6
LN1 attenuates PI3K, leading to upregulation of XPO6 activity
The LN1/N-actin pathway is aberrant in malignant cells, causing uncontrolled growth
ACKNOWLEDGMENTS
We are grateful to Ana L. Correa, Cyrus M. Ghajar, and Deborah Schechtman for critical reading of the manuscript and Ramray Bhat, Richard Schwarz, and Carla Columbano de Oliveira for suggestions. We thank Cyrus M. Ghajar, Doug Brownfield, and Kandice Tanner for initial assistance with microscopy. We also thank Antonio Manucci for help with plasmid purification. We thank all members of the Bissell laboratory for their constructive discussions and help. This work was supported by funds (to M.J.B.) from the NCI (awards R37CA064786 and U54CA143836 – Bay Area Physical Sciences Oncology Center), the Department of Defense Breast Cancer Research Program (award W81XWH0810736), and a grant from the Breast Cancer Research Foundation. A.B.C. was supported by funds from the Bissell laboratory and by INFABIC. INFABIC is funded jointly by the Brazilian National Research Council (CNPq; award 465699/2014-6) and the Sao Paulo State Research Funding Agency (FAPESP; award 2014/50938-8). The Bruni-Cardoso laboratory is supported by FAPESP Young Investigator Grant (award 2014/10492-0). A.P.Z.P.F. is a recipient of FAPESP postdoctoral fellowship (award 2014/25832-1).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures and seven figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2017.05.050.
REFERENCES
- Aguirre-Gamboa R, Gomez-Rueda H, Martínez-Ledesma E, Martínez-Tor-teya A, Chacolla-Huaringa R, Rodriguez-Barrientos A, Tamez-Perña JG, and Treviño V (2013). SurvExpress: an online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS ONE 8, e74250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhtar N, Marlow R, Lambert E, Schatzmann F, Lowe ET, Cheung J, Katz E, Li W, Wu C, Dedhar S, et al. (2009). Molecular dissection of integrin signalling proteins in the control of mammary epithelial development and differentiation. Development 136, 1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcaraz J, Xu R, Mori H, Nelson CM, Mroue R, Spencer VA, Brownfield D, Radisky DC, Bustamante C, and Bissell MJ (2008). Laminin and biomimetic extracellular elasticity enhance functional differentiation in mammary epithelia. EMBO J 27, 2829–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baarlink C, Wang H, and Grosse R (2013). Nuclear actin network assembly by formins regulates the SRF coactivator MAL. Science 340, 864–867. [DOI] [PubMed] [Google Scholar]
- Bader AG, Kang S, Zhao L, and Vogt PK (2005). Oncogenic PI3K deregulates transcription and translation. Nat. Rev. Cancer 5, 921–929. [DOI] [PubMed] [Google Scholar]
- Barcellos-Hoff MH, Aggeler J, Ram TG, and Bissell MJ (1989). Functional differentiation and alveolar morphogenesis of primary mammary cultures on reconstituted basement membrane. Development 105, 223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker-Weimann S, Xiong G, Furuta S, Han J, Kuhn I, Akavia UD, Pe’er D, Bissell MJ, and Xu R (2013). NFkB disrupts tissue polarity in 3D by preventing integration of microenvironmental signals. Oncotarget 4, 2010–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belin BJ, and Mullins RD (2013). What we talk about when we talk about nuclear actin. Nucleus 4, 291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belin BJ, Cimini BA, Blackburn EH, and Mullins RD (2013). Visualization of actin filaments and monomers in somatic cell nuclei. Mol. Biol. Cell 24, 982–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beliveau A, Mott JD, Lo A, Chen EI, Koller AA, Yaswen P, Muschler J, and Bissell MJ (2010). Raf-induced MMP9 disrupts tissue architecture of human breast cells in three-dimensional culture and is necessary for tumor growth in vivo. Genes Dev. 24, 2800–2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissell MJ, and Hines WC (2011). Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med 17, 320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissell MJ, Kenny PA, and Radisky DC (2005). Microenvironmental regulators of tissue structure and function also regulate tumor induction and progression: the role of extracellular matrix and its degrading enzymes. Cold Spring Harb. Symp. Quant. Biol 70, 343–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau N, Sympson CJ, Werb Z, and Bissell MJ (1995). Suppression of ICE and apoptosis in mammary epithelial cells by extracellular matrix. Science 267, 891–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briand P, Petersen OW, and Van Deurs B (1987). A new diploid nontumorigenic human breast epithelial cell line isolated and propagated in chemically defined medium. In Vitro Cell. Dev. Biol 23, 181–188. [DOI] [PubMed] [Google Scholar]
- Carraro DM, Elias EV, and Andrade VP (2014). Ductal carcinoma in situ of the breast: morphological and molecular features implicated in progression. Biosci. Rep 34, e00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri O, Koshy ST, Branco da Cunha C, Shin JW, Verbeke CS, Allison KH, and Mooney DJ (2014). Extracellular matrix stiffness and composition jointly regulate the induction of malignant phenotypes in mammary epithelium. Nat. Mater 13, 970–978. [DOI] [PubMed] [Google Scholar]
- Daignan-Fornier B, and Sagot I (2011). Proliferation/Quiescence: When to start? Where to stop? What to stock? Cell Div 6, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lanerolle P, and Serebryannyy L (2011). Nuclear actin and myosins: life without filaments. Nat. Cell Biol 13, 1282–1288. [DOI] [PubMed] [Google Scholar]
- Debnath J, Mills KR, Collins NL, Reginato MJ, Muthuswamy SK, and Brugge JS (2002). The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell 111, 29–40. [DOI] [PubMed] [Google Scholar]
- Dhillon AS, Hagan S, Rath O, and Kolch W (2007). MAP kinase signalling pathways in cancer. Oncogene 26, 3279–3290. [DOI] [PubMed] [Google Scholar]
- Dopie J, Skarp KP, Rajakylä EK, Tanhuanpää K, and Vartiainen MK (2012). Active maintenance of nuclear actin by importin 9 supports transcription. Proc. Natl. Acad. Sci. USA 109, E544–E552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher DA, and Mullins RD (2010). Cell mechanics and the cytoskeleton. Nature 463, 485–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse R, and Vartiainen MK (2013). To be or not to be assembled: progressing into nuclear actin filaments. Nat. Rev. Mol. Cell Biol 14, 693–697. [DOI] [PubMed] [Google Scholar]
- Gudjonsson T, Ronnov-Jessen L, Villadsen R, Rank F, Bissell MJ, and Petersen OW (2002). Normal and tumor-derived myoepithelial cells differ in their ability to interact with luminal breast epithelial cells for polarity and basement membrane deposition. J. Cell Sci 115, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann WA, Arduini A, Nicol SM, Camacho CJ, Lessard JL, Fuller-Pace FV, and de Lanerolle P (2009). SUMOylation of nuclear actin. J. Cell Biol 186, 193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huet G, Skarp KP, and Vartiainen MK (2012). Nuclear actin levels as an important transcriptional switch. Transcription 3, 226–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janmey PA, and Stossel TP (1987). Modulation of gelsolin function by phosphatidylinositol 4,5-bisphosphate. Nature 325, 362–364. [DOI] [PubMed] [Google Scholar]
- Jorgens DM, Inman JL, Wojcik M, Robertson C, Palsdottir H, Tsai WT, Huang H, Bruni-Cardoso A, Lopez CS, Bissell MJ, et al. (2017). Deep nuclear invaginations are linked to cytoskeletal filaments - integrated bioimaging of epithelial cells in 3D culture. J. Cell Sci 130, 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny PA, and Bissell MJ (2003). Tumor reversion: correction of malignant behavior by microenvironmental cues. Int. J. Cancer 107, 688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutay U, Lipowsky G, Izaurralde E, Bischoff FR, Schwarzmaier P, Hartmann E, and Görlich D (1998). Identification of a tRNA-specific nuclear export receptor. Mol. Cell 1, 359–369. [DOI] [PubMed] [Google Scholar]
- Lassing I, and Lindberg U (1985). Specific interaction between phosphatidylinositol 4,5-bisphosphate and profilactin. Nature 314, 472–474. [DOI] [PubMed] [Google Scholar]
- Lee GY, Kenny PA, Lee EH, and Bissell MJ (2007). Three-dimensional culture models of normal and malignant breast epithelial cells. Nat. Methods 4, 359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung CT, and Brugge JS (2012). Outgrowth of single oncogene-expressing cells from suppressive epithelial environments. Nature 482, 410–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Radisky DC, Wang F, and Bissell MJ (2004). Polarity and proliferation are controlled by distinct signaling pathways downstream of PI3-kinase in breast epithelial tumor cells. J. Cell Biol 164, 603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Cheng H, Roberts TM, and Zhao JJ (2009). Targeting the phos-phoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov 8, 627–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald D, Carrero G, Andrin C, de Vries G, and Hendzel MJ (2006). Nucleoplasmic beta-actin exists in a dynamic equilibrium between low-mobility polymeric species and rapidly diffusing populations. J. Cell Biol 172, 541–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthuswamy SK, Li D, Lelievre S, Bissell MJ, and Brugge JS (2001). ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat. Cell Biol 3, 785–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen le XT, and Mitchell BS (2013). Akt activation enhances ribosomal RNA synthesis through casein kinase II and TIF-IA. Proc. Natl. Acad. Sci. U.S.A 110, 20681–0686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Farrell PH (2011). Quiescence: early evolutionary origins and universality do not imply uniformity. Philos. Trans. R. Soc. Lond. B Biol. Sci 366, 3498–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson JE, Winston JT, Whitlock JA, and Pledger WJ (1993). Cell cycle-dependent gene expression in V point-arrested BALB/c-3T3 cells. J. Cell. Physiol 154, 333–342. [DOI] [PubMed] [Google Scholar]
- Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, et al. (2005). Tensional homeostasis and the malignant phenotype. Cancer Cell 8, 241–254. [DOI] [PubMed] [Google Scholar]
- Percipalle P (2013). Co-transcriptional nuclear actin dynamics. Nucleus 4, 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen OW, Rønnov-Jessen L, Howlett AR, and Bissell MJ (1992). Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. Proc. Natl. Acad. Sci. USA 89, 9064–9068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plessner M, and Grosse R (2015). Extracellular signaling cues for nuclear actin polymerization. Eur. J. Cell Biol 94, 359–362. [DOI] [PubMed] [Google Scholar]
- Plessner M, Melak M, Chinchilla P, Baarlink C, and Grosse R (2015). Nuclear F-actin formation and reorganization upon cell spreading. J. Biol. Chem 290, 11209–11216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizki A, Weaver VM, Lee SY, Rozenberg GI, Chin K, Myers CA, Bascom JL, Mott JD, Semeiks JR, Grate LR, et al. (2008). A human breast cell model of preinvasive to invasive transition. Cancer Res 68, 1378–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskelley CD, Desprez PY, and Bissell MJ (1994). Extracellular matrix-dependent tissue-specific gene expression in mammary epithelial cells requires both physical and biochemical signal transduction. Proc. Natl. Acad. Sci. USA 91, 12378–12382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarshad A, Sadeghifar F, Louvet E, Mori R, Böhm S, Al-Muzzaini B, Vintermist A, Fomproix N, Östlund AK, and Percipalle P (2013). Nuclear myosin 1c facilitates the chromatin modifications required to activate rRNA gene transcription and cell cycle progression. PLoS Genet 9, e1003397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarshad AA, Corcoran M, Al-Muzzaini B, Borgonovo-Brandter L, Von Euler A, Lamont D, Visa N, and Percipalle P (2014). Glycogen synthase kinase (GSK) 3β phosphorylates and protects nuclear myosin 1c from protea-some-mediated degradation to activate rDNA transcription in early G1 cells. PLoS Genet 10,e1004390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serebryannyy LA, Parilla M, Annibale P, Cruz CM, Laster K, Gratton E, Kudryashov D, Kosak ST, Gottardi CJ, and de Lanerolle P (2016). Persistent nuclear actin filaments inhibit transcription by RNA polymerase II. J. Cell Sci 129, 3412–3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarp KP, and Vartiainen MK (2013). Actin as a model for the study of nucleocytoplasmic shuttling and nuclear dynamics. Methods Mol. Biol. 1042, 245–255. [DOI] [PubMed] [Google Scholar]
- Spancake KM, Anderson CB, Weaver VM, Matsunami N, Bissell MJ, and White RL (1999). E7-transduced human breast epithelial cells show partial differentiation in three-dimensional culture. Cancer Res 59, 6042–6045. [PubMed] [Google Scholar]
- Spencer VA, Costes S, Inman JL, Xu R, Chen J, Hendzel MJ, and Bissell MJ (2011). Depletion of nuclear actin is a key mediator of quiescence in epithelial cells. J. Cell Sci 124, 123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streuli CH, and Bissell MJ (1990). Expression of extracellular matrix components is regulated by substratum. J. Cell Biol 110, 1405–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streuli CH, Bailey N, and Bissell MJ (1991). Control of mammary epithelial differentiation: basement membrane induces tissue-specific gene expression in the absence of cell-cell interaction and morphological polarity. J. Cell Biol 115, 1383–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streuli CH, Schmidhauser C, Bailey N, Yurchenco P, Skubitz AP, Roskelley C, and Bissell MJ (1995). Laminin mediates tissue-specific gene expression in mammary epithelia. J. Cell Biol 129, 591–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utermark T, Rao T, Cheng H, Wang Q, Lee SH, Wang ZC, Iglehart JD, Roberts TM, Muller WJ, and Zhao JJ (2012). The p110α and p110β isoforms of PI3K play divergent roles in mammary gland development and tumorigenesis. Genes Dev 26, 1573–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virtanen JA, and Vartiainen MK (2017). Diverse functions for different forms of nuclear actin. Curr. Opin. Cell Biol 46, 33–38. [DOI] [PubMed] [Google Scholar]
- Wang F, Weaver VM, Petersen OW, Larabell CA, Dedhar S, Briand P, Lupu R, and Bissell MJ (1998). Reciprocal interactions between beta1-integrin and epidermal growth factor receptor in three-dimensional basement membrane breast cultures: a different perspective in epithelial biology. Proc. Natl. Acad. Sci. USA 95, 14821–14826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Hansen RK, Radisky D, Yoneda T, Barcellos-Hoff MH, Petersen OW, Turley EA, and Bissell MJ (2002). Phenotypic reversion or death of cancer cells by altering signaling pathways in three-dimensional contexts. J. Natl. Cancer Inst 94, 1494–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver VM, Petersen OW, Wang F, Larabell CA, Briand P, Damsky C, and Bissell MJ (1997). Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J. Cell Biol 137, 231–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RJ (2008). RNA polymerases I and III, non-coding RNAs and cancer. Trends Genet. 24, 622–629. [DOI] [PubMed] [Google Scholar]
- Xu R, Spencer VA, Groesser DL, and Bissell MJ (2010). Laminin regulates PI3K basal localization and activation to sustain STAT5 activation. Cell Cycle 9, 4315–4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee NS, Gong W, Huang Y, Lorent K, Dolan AC, Maraia RJ, and Pack M (2007). Mutation of RNA Pol III subunit rpc2/polr3b Leads to Deficiency of Subunit Rpc11 and disrupts zebrafish digestive development. PLoS Biol 5, e312. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.