

Abstract
In eukaryotic cells, many introns are constitutively, rather than alternatively, spliced and therefore do not contribute to isoform diversification. It has remained unclear what functional roles such constitutive splicing provides. To explore this issue, we asked how splicing affects the efficiency with which individual pre-messenger RNA transcripts are productively processed across different gene expression levels. We developed a quantitative single-molecule fluorescence in situ hybridization-based method to quantify splicing efficiency at transcription active sites in single cells. We found that both natural and synthetic genes in mouse and human cells exhibited an unexpected ‘economy of scale’ behavior in which splicing efficiency increased with transcription rate. Correlations between splicing efficiency and spatial proximity to nuclear speckles could explain this counter-intuitive behavior. Functionally, economy of scale splicing represents a non-linear filter that amplifies the expression of genes when they are more strongly transcribed. These results indicate that constitutive splicing plays an active functional role in modulating gene expression.
Splicing is ubiquitous in eukaryotes1. During splicing, nascent pre-messenger RNAs are processed to remove introns and to include or exclude exons. A key function of this splicing process is the ability to increase gene-product diversity by generating multiple distinct isoforms from a single gene2–6. These isoform differences result from inclusion or exclusion of particular alternatively spliced exons and(or) introns. However, the majority of individual introns (and exons) are constitutively spliced7. These splice events do not directly contribute to isoform diversification.
It has been suggested that constitutive splicing provides several possible functions. First, it may function to increase expression through a phenomenon of ‘intron-mediated enhancement’8. Second, constitutively spliced introns appear to encode a large proportion of intronic microRNAs9 and other non-coding RNAs10,11. Third, constitutive splicing could reflect a loss of alternative splicing during evolution or facilitate the evolutionary acquisition of new alternative splicing behaviors12,13. Thus, splicing can play roles beyond isoform diversification per se.
The ubiquitousness of constitutive splicing and its tight coupling with transcription14–16 together suggest a different potential function as a signal-processing filter that modulates the amount of mature mRNA depending on the rate of transcription or other parameters. As an enzymatic process17,18, the efficiency of splicing cannot be 100%. In general, only a fraction of transcribed RNA is productively spliced to enable subsequent translation or other functions. Unspliced RNA is predominantly retained in the nucleus and degraded, through a quality-control mechanism19,20. By quantifying splicing efficiency, defined as the ratio of spliced RNA to total transcribed RNA (Fig. 1a), we can investigate how splicing efficiency depends on the gene expression level and, therefore, what type of filter, if any, splicing provides.
Fig. 1 |. Splicing efficiency requires single-cell measurement.
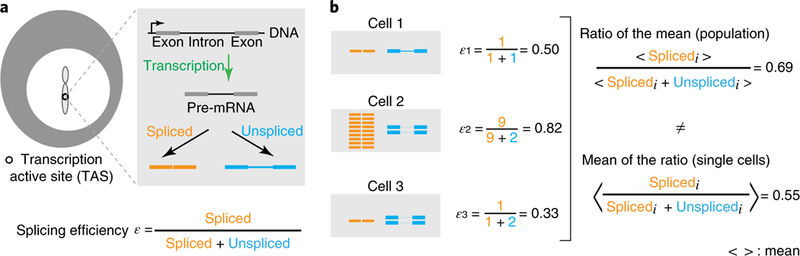
a, Splicing occurs co-transcriptionally at the TAS (white circle on chromosome). Each pre-mRNA molecule (gray) can be spliced to remove introns (orange) or left unspliced (blue). Splicing efficiency is the ratio between the number of spliced transcripts and the total number of transcripts at the TAS (equation, bottom). b, Averaging over heterogeneous cells can distort splice efficiency. We consider three hypothetical cells with different numbers of spliced (orange) and unspliced (blue) transcripts. The mean of the splicing efficiency measured in each cell individually does not equal the ratio of the mean number of spliced transcripts and the mean number of total transcripts at the population level. Thus, splicing efficiency should ideally be measured in individual cells.
In principle, splicing efficiency could behave in three qualitatively distinct ways. The simplest possibility is that splicing efficiency is determined by sequence features and concentrations of splicing machinery, and therefore is constant for a given gene in a given cell state. However, as an enzymatic process, splicing could, in principle, decline in efficiency at high substrate (pre-mRNA) concentrations. This ‘diminishing returns’ behavior would tend to disproportionately suppress processing at higher expression levels relative to lower levels. The final, formal, possibility, which is not expected under conventional models of splicing, is that splicing efficiency could increase with transcription level, in an ‘economy of scale’ fashion, and thereby disproportionately enhance the expression of more highly expressed genes. Accurate measurements of splicing efficiency across transcription levels are needed to discriminate among these or more complex behaviors.
Several approaches have been used to measure splicing efficiency21–25. Genome-wide, shot-gun, high-throughput sequencing provides a way to compute splicing efficiency by comparing the number of reads at unspliced regions to the total number of reads in constitutive exon regions of the same gene. However, much splicing occurs co-transcriptionally26 and different RNA species have different lifetimes once released from the transcription active site (TAS). Because they do not discriminate RNAs at the TAS from RNAs at other sites, these methods can distort quantification of splice ratios. To circumvent this problem, more recent studies have performed nascent-RNA-sequencing27,28 to measure co-transcriptional splicing efficiency at the TAS. These approaches have been powerful and informative. However, of necessity the average over individual cells.
Averaging over a heterogeneous cell population can, by itself, distort splicing efficiency. The key issue is that splicing efficiency is an inherently ratiometric quantity. To determine the mean splicing efficiency across a cell population, one would ideally calculate the ratio of spliced to total transcripts in each cell individually and then average this quantity over the cell population. However, because the mean of a ratio is not, in general, equal to the ratio of a mean, splicing efficiency measured from single cells does not match the population average (Fig. 1b). More specifically, population-average measurements systematically underweight contributions from lower expressing cells relative to higher expression cells. For a ‘bursty’ process such as gene expression29–32, this effect can be strong.
Several previous studies have sought to analyze splicing efficiency in single cells30,33. Pioneering studies engineered binding sites for the MS2 and PP7 RNA-binding proteins to fluorescently label individual transcripts in live cells34,35. This approach enabled simultaneous analysis of splicing and transcriptional kinetics in individual cells. However, it cannot be used on endogenous (unmodified) transcripts and insertion of binding sites could potentially perturb the splicing dynamics.
Here, we report a method for quantitative single-cell measurement of splicing efficiency based on single-molecule fluorescence in situ hybridization (smFISH). The method measures splicing efficiency at transcription active sites in individual cells. In contrast to smFISH methods based on counting the number of distinct molecules, appearing as fluorescent dots in images36–38, here we quantify dot intensity at the TAS. For accurate quantitation, we developed methods for unbiased intensity comparisons between channels and adapted a method from astrophysics for estimating stellar luminosities in crowded starfields39.
Contrary to the classic enzyme–substrate Michaelis–Menten model, splicing efficiency increased, rather than decreased, with increasing levels of gene expression, in an ‘economy of scale’ fashion. Increased transcription also correlated with spatial proximity to speckles, suggesting a mechanism for economy of scale on the basis of spatial clustering. A mathematical model based on this observation shows how economy of scale splicing could emerge if enzyme availability increases with substrate (pre-mRNA) concentration. Together, these results enable quantitative analysis of splicing in single cells and reveal a role for splicing as a gene expression filter.
Results
Intron and exon probe smFISH sets identify distinct RNA species.
We set out to measure splicing efficiency by quantifying the relative levels of different isoforms at the same TAS, across a range of expression levels. As a model system, we used the RG6 minigene, whose splicing behavior was previously characterized using fluorescent proteins40. To enable regulation of transcription, we site-specifically integrated the minigene under the control of a doxycycline (dox)-inducible promoter into HEK293 cells. To measure the splicing efficiency at the TAS, we designed three smFISH probe sets. The intron probe set targeted the spliceable constitutive intron and thus measured the number of unspliced transcripts. The other two probe sets, denoted Exonl and Exon2, targeted constitutive exons, measuring the number of total transcripts (Fig. 2a). The use of two redundant exon probe sets facilitates subsequent analysis (see below). We cultured cells under standard conditions and then fixed and imaged the cells using all three smFISH probe sets (Methods).
Fig. 2 |. Experimental method for quantifying splicing efficiency at the TAS in individual cells.
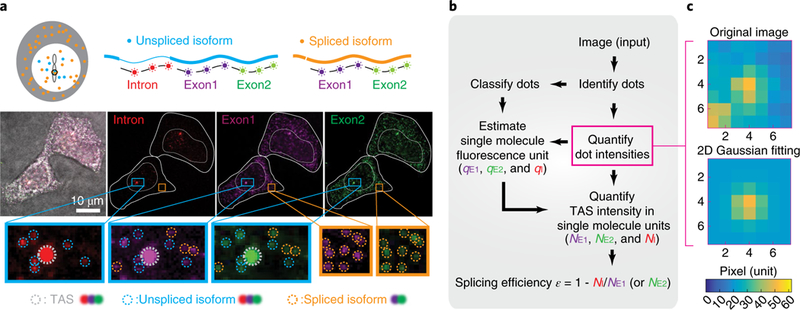
a, We designed three smFISH probe sets targeting one spliceable intron (red) and two constitutive exons (purple, green). Sequences of Intron, Exon1 and Exon2 probe sets are listed in Supplementary Table 1. Note that exon probe-set numbering does not match exon numbering. In this image of two cells, staining by each probe set is shown in the corresponding color channel, superimposed on a brightfield image (grayscale). Identified smFISH dots are circled in the zoomed insets, with TAS in gray, unspliced isoform (dots co-localized in Intron, Exon1 and Exon2 channels) in blue and spliced isoform (dots co-localized in Exon1 and Exon2 channels) in orange. b, Workflow for quantifying NE1, NE2 and NI, the number of transcripts observed within the TAS in the exon 1, exon 2 and intron channels using corresponding single-molecule fluorescence units, qE1, qE2 and qI, respectively (see also Supplementary Fig. 1a). c, Iterative fitting of individual dot intensities. Using a method from stellar photometry of crowded star fields, we iteratively remove fluorescence from adjacent dots to produce a single-dot image (bottom) from an initial image with multiple dots (top) (see Supplementary Note 1 and Supplementary Fig. 1c for more detail). Source data are available online.
We observed several types of smFISH dots that could be classified by their intensity and probe-binding patterns. The first type, representing the TAS, consisted of one or two bright dots per cell that appeared in all three channels in the nucleus (Fig. 2a). The second type consisted of many scattered dots of lower intensity that appeared in both the nucleus and cytoplasm, corresponding to single transcripts (Fig. 2a). Using co-localization of different probe sets enabled further classification of dots. All RNAs were labeled by both Exonl and Exon2 probe sets, both in the cytoplasm and in the nucleus. In contrast, unspliced isoforms were labeled in all three channels, but appeared only in the nucleus (Fig. 2a). Although we targeted a constitutively spliced intron, we also observed released unspliced molecules, consistent with imperfect splicing efficiencies (Fig. 2a, ‘unspliced isoform’). These transcripts could result from a failure to complete splicing before transcriptional termination or from competition among transcripts for limited levels of splicing machinery41. Together, these probe sets allowed identification of multiple distinct molecular species both at and away from the TAS.
Determining single-transcript intensity units.
Because individual transcript molecules cannot be spatially resolved at the TAS, we developed an intensity-based transcript-counting procedure to quantify the number of transcripts of each species from fluorescence intensities of each probe set (Fig. 2b, Supplementary Fig. 1a and Supplementary Notes 1 and 2). Briefly, we first used the Poisson-distributed individual transcript intensities to obtain the single-molecule fluorescence intensity calibration units in each color channel, denoted qE1, qE2 and qI for the two exon and one intron channels, respectively. Then, we used this calibration to quantify the number of copies of exonic (NE1, NE2) and intronic (NI) targets at the TAS, in molecular units.
In this procedure, the values of qn (n = E1, E2 or I) are estimated from the distribution of intensities of single-molecule non-TAS smFISH dots in each channel. However, dot identification can be inconsistent between channels due to differences in signal-to-back-ground levels, differences in binding efficiencies between probe sets and differences in the fluorescence properties of each fluorophore, among other issues. These factors produce systematic differences in sensitivity between channels that distort qn quantification.
To address this issue, we developed an unbiased dot-identification procedure. We first identified candidate dots using a low (permissive) threshold that captures all foreground smFISH dots as well as some background signal (false-positive dots) in each channel and quantified the integrated intensity of each dot. To correct for fluorescence ‘contamination’ from neighboring dots, we adapted an algorithm from stellar photometry of crowded starfields39, which works by iteratively removing fluorescence from neighboring objects (Fig. 2c, Supplementary Fig. 1c and Supplementary Note 1).
We also performed this analysis on negative-control images lacking true smFISH dots, to obtain the distribution of background-dot intensities. Subtracting the background histogram from the total (foreground+background) histogram generated a corrected foreground-dot intensity distribution (Fig. 3a and and Supplementary Note 1). TAS intensity measurements do not require accurate counting of scattered smFISH dots, as in conventional FISH36. This approach thus effectively sacrifices precision in individual dot identification to obtain a less-biased distribution of smFISH dot intensity in each fluorescent channel. Finally, to obtain the single-transcript intensity unit qn, we fit the resulting distributions with a continuous analog of the Poisson distribution (Fig. 3b and Supplementary Note 2).
Fig. 3 |. Quantification of single-transcript intensity units in multiple channels.

a, The single-dot transcript intensity distribution (top) can be obtained by subtracting a background distribution obtained without FISH probes (middle, histogram of n = 1,953) from the foreground plus background image obtained with FISH probes (bottom, histogram of n = 1,054). b, Applying this procedure to each channel and fitting the resulting distributions to a Poisson distribution provides the three single-transcript intensity units (indicated). c, The measured single-transcript intensity units in multiple channels are verified via dot-co-localization (Supplementary Fig. 1d). Different methods generate similar single-molecule fluorescence units. Color is labeled as in Fig. 2a. d, The properties of background dots (gray, n = 5,000) and identified true FISH dots (pink, n = 10,000). Note overlap of these properties. See more dot-fitting properties in Supplementary Note 2. e, The measured fluorescent intensity for staining of SDHA gene is proportional to the number of smFISH probes included (indicated numbers). A negative control (brown) uses a fixed number of probes and displays a relatively fixed fluorescence intensity. This indicates that intensity scales linearly with the number of probes. The data are for n = 3,000 and n = 200dots of SDHA and control staining, respectively. The measure of center is the mean. Error bars represent standard deviation. Source data for panels d and e are available online.
As an additional consistency check, we used an independent method to identify foreground dots by their co-localization across multiple fluorescence channels. This approach produced similar values for qn (Fig. 3c, Supplementary Fig. 1d and Supplementary Note 2). It also revealed that foreground and background dots exhibit overlapping distributions of key properties such as intensity, peak height and peak width (Fig. 3d and Supplementary Note 2), further supporting the need for histogram subtraction, as above. Taken together, these results provide a simple and general method for accurately estimating the fluorescence intensity at the TAS using precisely calibrated intensity units for single transcripts.
Fluorescence intensity increases linearly with number of probes.
Because intensity quantification is critical for estimating the number of molecules at the TAS, we next asked how TAS fluorescence intensity depends on the abundance of bound probes. We designed 42 smFISH probes targeting the housekeeping gene SDHA and mixed them into groups of 18, 24, 30, 36 and 42 probes. We then measured the fluorescence intensity of stained HEK293 cells with each group. Fluorescence dot intensities increased linearly with the number of included probes (Fig. 3e). By contrast, a second set of 27 probes targeting HES1, which were labeled in a different fluorescent channel and included in all experiments as a fixed control, were constant across each condition, as expected. These results indicate that fluorescence intensity provides a linear readout of probe density at the TAS. This linearity enables one to quantify the number of transcripts (NE1, NE2 and N1) at the TAS in molecular units by dividing TAS fluorescence intensity by the single-transcript fluorescence units (qE1, qE2 and qI respectively).
Splicing efficiency exhibits an ‘economy of scale’ behavior.
Having established the method, we next used it to determine how splicing efficiency changes with transcription level in individual cells. Quantifying splicing efficiency requires comparing the number of spliced transcripts to the total number of transcripts (spliced + unspliced) at the TAS. Here, the total transcript number was represented by NE1 or NE, and the number of spliced transcripts was obtained by subtracting the number of pre-spliced transcripts, NI, from NE1 or NE2. With these quantities, the splicing efficiency can be computed as , where i = 1 or 2 denotes either of the two exon probe sets, and εi should, in the absence of noise, be independent of i.
To cover a broad range of expression levels, we induced the Sonic Hedgehog (SHH) target gene Gli1 in 3T3 cells with varying concentrations of recombinant SHH and analyzed cells after 48 hours of induction42. In parallel, we analyzed the synthetic spliceable RG6 minigene described above, induced for 3 hours with a range of doxycycline concentrations. After induction, we fixed cells and performed smFISH hybridization and imaging (Methods).
The Gli1 and CMV promoters expressed at up to ~20 transcripts per TAS. Altogether, we analyzed ~3,000 (Gli1, Fig. 4a) and ~1,000 (RG6, Supplementary Fig. 2a) active sites in single cells, and computed the geometric mean for each condition (Supplementary Note 3) as well as the splicing efficiency in each cell (Fig. 4b and Supplementary Fig. 2b). For both genes, transcription level and splicing efficiency were heterogeneous, even at a single induction level29,43. This variability probably reflects both transcriptional bursting and other sources of biological variation43, as well as measurement errors from stochastic binding of probes and other sources (see Supplementary Note 3 for more details).
Fig. 4 |. Splicing efficiency increases with transcript level, exhibiting ‘economy of scale’ behavior, for the two tested genes, Gli1 induced by Shh in 3T3 cells and the Tet-off minigene RG6 induced by dox in HEK293 cells.

a, Raw data of the number of transcripts measured by our methods. Each dot (gray) in the plot is one measurement from a single TAS. The number of TAS measured is 3,010 for Gli1 (and 1,289 for RG6 in Supplementary Fig. 2). Geometric means are shown in orange or pink or blue. Error bars represent standard error of the mean. b, On the basis of a, we find splicing efficiency (1–N1/NE1 or 1–N1/NE2) increases with transcription level (orange and pink), while control (1–NE1/NE2 or 1–NE2/NE1) measurements remained constant (blue). Solid lines are guides to the eye to highlight the ‘economy of scale’ behavior. Geometric means are shown in orange or pink or blue. Error bars represent standard error of the mean. The ‘economy of scale’ mathematical model is discussed in the main text. c, Overlapped curves from b. d, Overlapped curves of the synthetic gene RG6. e, Overlapped curves from biological repeats for Gli1 and RG6. All of them showed the repeatability of ‘economy of scale’ observation. Source data are available online.
While individual cells were variable, the mean splicing efficiency systematically increased with gene expression level. This ‘economy of scale’ behavior occurred for both genes. It was robust to experimental conditions, such as the strength and duration of induction. It was also robust to data analysis parameters (see Supplementary Note 4). Additionally, splicing efficiency increased with transcription level in a similar pattern for both genes (Fig. 4c–e), reaching 80% of its maximum value at ~3.5 transcripts per TAS.
We ruled out potential artifacts that could appear to generate this ‘economy of scale’ behavior. For example, because imaging occurs at a fixed point in time, images can in general capture incomplete transcription events. If incomplete transcription were expression level-dependent, it would alter the two exon probe ratio, NE1/NE2, from its ideal value of 1. However, this ratio showed no systematic dependence on transcription level (Fig. 4 and Supplementary Fig. 2). Additionally, to rule out potential misclassification of individual transcripts as the TAS, we used single-molecule DNA–FISH to independently identify the TAS (Supplementary Fig. 3). Finally, analysis of two independent transcription-level measurements (NE1 and NE2) enabled us to compute splicing efficiency and transcription rate using distinct exon readouts, avoiding a potentially spurious correlation between transcription level and splicing efficiency due to the appearance of NE1 in the expression for splicing efficiency, (1 – NI/NE1). (Note that without the second exon probe, hardness-ratios correction methods44,45 from astrophysics could also help to address this issue (Supplementary Fig. 4 and Supplementary Note 3)). Together, these results support the validity of the economy of scale observation.
A mathematical model of ‘economy of scale’ splicing.
How can an enzymatic process such as splicing produce ‘economy of scale’ behavior? In the classic Michaelis-Menten model (Fig. 5a and Supplementary Note 4) reaction efficiency declines monotonically with increasing substrate concentration (Fig. 5b, black curve), producing the opposite ‘diminishing returns’ behavior.
Fig. 5 |. Phenomenological model for ‘economy of scale’ behavior.
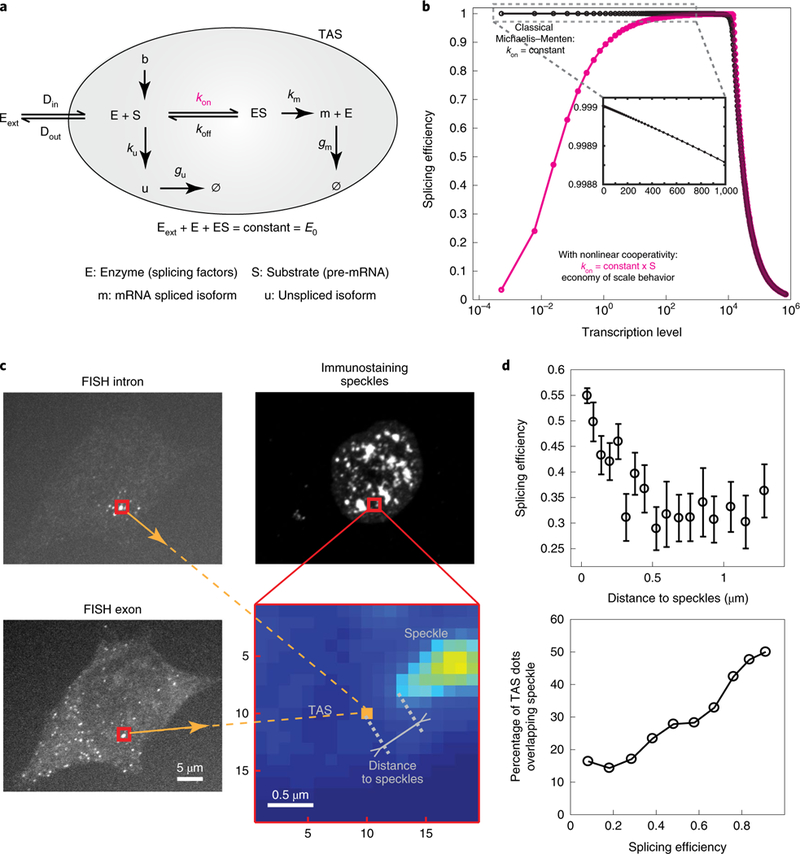
a, In a classical Michaelis-Menten model, splicing factors, collectively denoted E, bind to pre-mRNA, denoted S, at a rate kon to form an enzyme-substrate complex, ES, which can unbind, at rate koff, or transform into a mature mRNA, m, at rate km, releasing the enzyme. The rate ku denotes the production rate of unspliced RNA, denoted u, while gu and gm denote the rate at which unspliced RNA or mRNA, respectively, are degraded or released from the TAS (shaded gray area). E0 denotes the total concentration of enzyme. Din and Dout denote the diffusion rate of splicing factors, respectively, in and out of the TAS. The splicing factors outside of the TAS are denoted as Eext. b, In the classical Michaelis-Menten model, splicing efficiency declines monotonically with transcription rate, in a ‘diminishing returns’ manner (black). A variant of the model in which kon is proportional to S, rather than constant, generates ‘economy of scale’ behavior (pink). Note that at higher transcription rates, splicing efficiency declines due to enzyme saturation. These ‘diminishing returns’ and ‘economy of scale’ behaviors were parameter independent (Supplementary Fig. 6b,c and Supplementary Note 4). c, We measured the distance between TAS and its nearest speckle via smFISH and immunostaining in the same cell. FISH probes were designed as in Fig. 2a. We used SC35 antibody to immunostain the speckles. The zoomed-in area is marked in red. smFISH defined the TAS location (orange) and immunostaining intensity showed the presence of speckles (Supplementary Fig. 5). d, Splicing efficiency increases with proximity to speckles (top figure). Error bars represent standard error of the mean. TAS with higher splicing efficiency has greater probability of overlap with speckles (bottom figure). See raw data in Supplementary Fig. 5b. Source data for panel d are available online.
One limitation of the simple Michaelis–Menten model is that it does not account for the inhomogeneous concentration of splicing machinery in dynamic interchromatin granule clusters, called nuclear speckles46–48. Previous work has shown that more highly transcribed genes are closer to speckles47,49. To analyze the relationship between spatial organization and splicing efficiency, we analyzed splicing efficiency of RG6 as described above and also performed immunostaining to detect the splicing factor SC35 in the same cell (Fig. 5c and Methods). We then quantified the distance from each TAS to its nearest speckle (Supplementary Fig. 5a and Supplementary Note 4) and plotted the results as a function of splicing efficiency. This analysis revealed a correlation between splicing efficiency, transcription level and the proximity of the TAS to the splicing speckle (Fig. 5d and Supplementary Fig. 5b).
Together, these results suggest the hypothesis that stronger expression could increase the proximity of a gene to a speckle, which in turn could increase the availability of splicing machinery. To incorporate these effects into a modified version of the model, we allowed the rate of pre-mRNA binding to splicing machinery, kon, to increase with the concentration of pre-mRNA (Fig. 5). This simple modification generated economy of scale behavior at lower expression levels (Fig. 5b, pink curve, and scheme given in Supplementary Fig. 6a), switching to diminishing returns at higher expression levels as the splicing machinery eventually saturates (see additional mechanisms in the Supplementary Note 4). These results show that a positive correlation between expression level and speckle accessibility could qualitatively explain ‘economy of scale’ splicing behavior.
Discussion
Here, we introduced a quantitative imaging-based method to measure splicing efficiency in single cells and used it to characterize the dependence of splicing efficiency on transcription level. The method enables accurate intensity quantification of smFISH data to allow direct comparison of intensities of multiple channels at the same site. Although we focused on quantifying the splicing efficiency of constitutively spliced introns, the pipeline presented here can be extended to alternative splicing by incorporation of additional fluorescence channels.
The observed ‘economy of scale’ behavior is opposite to the ‘diminishing returns’ behavior one would expect from a standard enzymatic process. Mechanistically, the economy of scale behavior could reflect a disproportionate allocation of the shared splicing machinery ‘resource’ to more highly expressed genes. In fact, previous work showed that different genes can effectively compete for splicing machinery inside the nucleus41, with more weakly expressed genes receiving less access to splicing factors47,49, possibly serving to optimize the total amount of splicing that can be achieved by a fixed abundance of splicing components50. Such competition is consistent with the observed spatial correlation of highly transcribed genes with splicing speckles (Fig. 5d and Supplementary Fig. 5) and can produce economy of scale behavior in a simple phenomenological model (Fig. 5a and Supplementary Fig. 6). The observed spatial correlation could also provide insight into the long-standing question of why transgenes with introns are more robustly transcribed8. However, the causal relationships between spatial co-localization, transcription rate and splicing efficiency are not yet clear. In the future, the ability to track spatio-temporally the effects of a sudden expression perturbation on all three features could help to clarify these issues. Apart from the non-uniform distribution of splicing resources, other mechanistic factors, including polymerase speed14, epigenetic modifications51 and isoform residence times52,53, could also contribute to ‘economy of scale’ splicing (see more detailed discussion in Supplementary Note 4).
Functionally, economy of scale acts as a non-linear filter within the overall gene regulation process, enhancing more strongly expressed genes (Fig. 6a). In principle, this filter could impact many cellular regulation processes. For instance, it could help prevent pervasive low-level transcription54 from inappropriately propagating to protein synthesis. It could also amplify, suppress or reshape the mRNA distribution, depending on the underlying distribution of transcription levels (Fig. 6b,c). Thus, it could play an active role in controlling variation across a population of cells55,56. It will be interesting to see how perturbations in the strength of competing splice acceptor or donor sites57–59, the length of introns60 and other parameters61 control the shape of this splicing filter.
Fig. 6 |. Splicing can act as a gene expression ‘filter’.
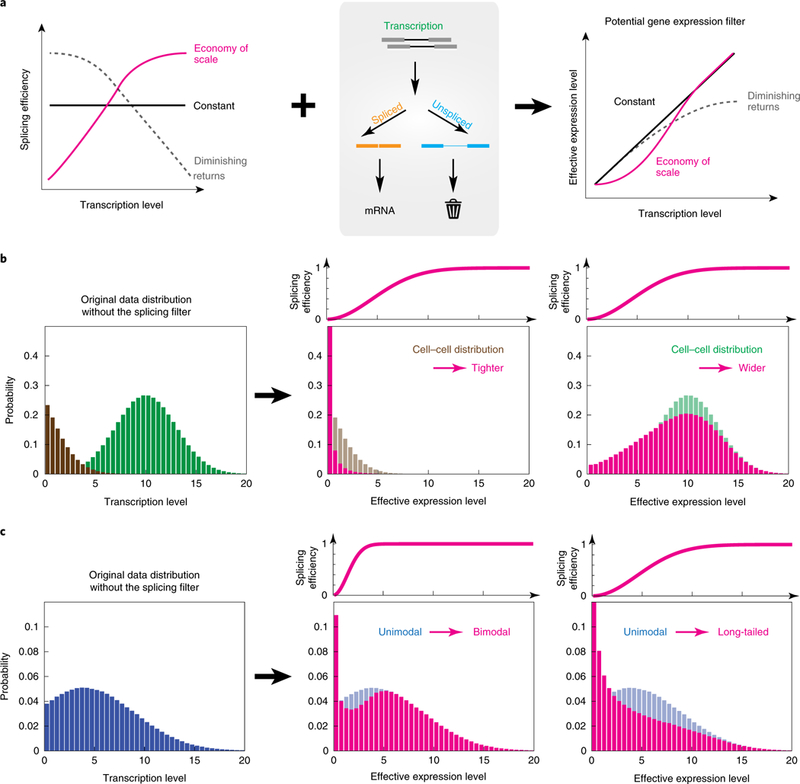
a, Different possible relationships between splicing efficiency and transcription level can generate different types of gene expression filter. ‘Economy of scale’ (pink) and ‘diminishing returns’ (gray) are high-pass and low-pass filters, respectively. b, The ‘economy of scale’ filter can narrow or broaden the distribution of mRNA levels depending on the underlying distribution of mRNA expression levels. The filter (pink) preserves higher transcription levels (green) but suppresses lower transcription levels (brown). c, Depending on its parameters, the ‘economy of scale’ filter can change the shape of an expression distribution from unimodal (blue) to bimodal (pink, center plot) or long-tailed (pink, right-hand plot).
The present method has several limitations. First, it focuses on co-transcriptional splicing and does not capture potential regulation through post-transcriptional splicing processes35,62. Second, because the method relies on direct binding of probes, occlusion of probe-binding sites by bound proteins or secondary structure of the target RNA could, in principle, affect quantitation. Third, the post-splicing residence times for different products at the TAS are unknown. Their relative values predominantly affect the absolute magnitude of the measured efficiency. However, the dependence of splicing efficiency on transcription rate could be distorted if the residence times for different molecules depend in different ways on transcription rate (see more discussion in Supplementary Note 4). To ensure that the economy of scale phenomenon is robust to measurement method, we also quantified the ratio between spliced and unspliced isoforms of RG6 across various transcription levels by quantitative PCR (qPCR) (Supplementary Fig. 9b). This independent measurement, which includes isoforms outside the TAS, produced a similar ‘economy of scale’ behavior.
Because it operates quantitatively in single cells with subcellular resolution, the method described here should provide insight into kinetic features of the splicing mechanism. For example, by simultaneously imaging the spatial locations of splicing regulatory factors such as long non-coding RNA alongside their target genes, it could enable one to determine how these factors affect splicing efficiency51,63–66. Using additional fluorescent channels, it could also allow analysis of correlations in splicing between neighboring genes and enable comparison of splicing efficiency between alleles of a gene within a single cell. Further development of the method could address additional questions. For example, recent work has shown how comparison of nascent transcript levels with total transcript levels can provide information on dynamic changes in expression (RNA ‘velocities’) from single time-point snapshots67. These approaches could be combined with the analysis shown here to provide dynamic information on the relationship between splicing and transcriptional bursting. Finally, by combining these approaches with sequential hybridization and barcoding techniques63,64,68, this method could enable genome-wide analysis of splicing efficiency in a single cell.
Supplementary Material
Acknowledgements
We thank T. Cooper for DNA constructs of minigene RG6, P. Li for providing Gli1 inducible protocol, Z. Singer and Y. Antebi for technical assistance, M. Guttman, C. Su, H. Klumpe, M. Budde and L. Bintu for critical feedbacks on the manuscript. We also thank M. Guttman, G. Seelig, D. Black, D. Baltimore, M. Moore, J.G. Ojalvo and N. Wingreen for discussion and feedback on the project. The work was funded by a Fellowship from the Schlumberger Foundation, by the Gordon and Betty Moore Foundation through Grant GBMF2809 to the Caltech Programmable Molecular Technology Initiative and the Institute for Collaborative Biotechnologies through grant W911NF-09–0001 from the U.S. Army Research Office. The content of the information does not necessarily reflect the position or the policy of the Government, and no official endorsement should be inferred. M.B.E. is a Howard Hughes Medical Institute Investigator.
Footnotes
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information is available for this paper at https://doi.org/10.1038/s41594-019-0226-x.
Reprints and permissions information is available at www.nature.com/reprints.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, statements of code and data availability and associated accession codes are available at https://doi.org/10.1038/s41594-019-0226-x.
References
- 1.Wang ET et al. Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Papasaikas P & Valcarcel J Evolution. Splicing in 4D. Science 338, 1547–1548 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Black DL Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 72, 291–336 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Barbosa-Morais NL et al. The evolutionary landscape of alternative splicing in vertebrate species. Science 338, 1587–1593 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Kalsotra A & Cooper TA Functional consequences of developmentally regulated alternative splicing. Nat. Rev. Genet. 12, 715–729 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nilsen TW & Graveley BR Expansion of the eukaryotic proteome by alternative splicing. Nature 463, 457–463 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen F-C, Wang S-S, Chen C-J, Li W-H & Chuang T-J Alternatively and constitutively spliced exons are subject to different evolutionary forces. Mol. Biol. Evol. 23, 675–682 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Brinster RL, Allen JM, Behringer RR, Gelinas RE & Palmiter RD Introns increase transcriptional efficiency in transgenic mice. Proc. Natl Acad. Sci. USA 85, 836–840 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Westholm JO & Lai EC Mirtrons: microRNA biogenesis via splicing. Biochimie 93, 1897–1904 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Filipowicz W & Pogacic V Biogenesis of small nucleolar ribonucleoproteins. Curr. Opin. Cell Biol. 14, 319–327 (2002). [DOI] [PubMed] [Google Scholar]
- 11.Rearick D et al. Critical association of ncRNA with introns. Nucleic Acids Res. 39, 2357–2366 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gilbert W Why genes in pieces? Nature 271, 501 (1978). [DOI] [PubMed] [Google Scholar]
- 13.Lev-Maor G et al. The ‘alternative’ choice of constitutive exons throughout evolution. PLoS Genet. 3, e203 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bentley DL Coupling mRNA processing with transcription in time and space. Nat. Rev. Genet. 15, 163–175 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Das R et al. SR proteins function in coupling RNAP II transcription to pre-mRNA splicing. Mol. Cell 26, 867–881 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Rosonina E & Blencowe BJ Gene expression: the close coupling of transcription and splicing. Curr. Biol. 12, R319–R321 (2002). [DOI] [PubMed] [Google Scholar]
- 17.Green MR Biochemical mechanisms of constitutive and regulated pre-mRNA splicing. Annu. Rev. Cell Biol. 7, 559–599 (1991). [DOI] [PubMed] [Google Scholar]
- 18.Reed R Mechanisms of fidelity in pre-mRNA splicing. Curr. Opin. Cell Biol. 12, 340–345 (2000). [DOI] [PubMed] [Google Scholar]
- 19.Houseley J & Tollervey D The many pathways of RNA degradation. Cell 136, 763–776 (2009). [DOI] [PubMed] [Google Scholar]
- 20.Boutz PL, Bhutkar A & Sharp PA Detained introns are a novel, widespread class of post-transcriptionally spliced introns. Genes Dev. 29, 63–80 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khodor YL, Menet JS, Tolan M & Rosbash M Cotranscriptional splicing efficiency differs dramatically between Drosophila and mouse. RNA 18, 2174–2186 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khodor YL et al. Nascent-seq indicates widespread cotranscriptional pre-mRNA splicing in Drosophila. Genes Dev. 25, 2502–2512 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carrillo Oesterreich F, Preibisch S & Neugebauer KM Global analysis of nascent RNA reveals transcriptional pausing in terminal exons. Mol. Cell 40, 571–581 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Lipp JJ, Marvin MC, Shokat KM & Guthrie C SR protein kinases promote splicing of nonconsensus introns. Nat. Struct. Mol. Biol. 22, 611–617 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Katz Y, Wang ET, Airoldi EM & Burge CB Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods 7, 1009–1015 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neugebauer KM On the importance of being co-transcriptional. J. Cell Sci. 115, 3865–3871 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Churchman LS & Weissman JS Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature 469, 368–373 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herzel L, Straube K & Neugebauer KM Long-read sequencing of nascent RNA reveals coupling among RNA processing events. Genome Res. 28, 1008–1019 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raj A & van Oudenaarden A Nature, nurture, or chance: stochastic gene expression and its consequences. Cell 135, 216–226 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Waks Z, Klein AM & Silver PA Cell-to-cell variability of alternative RNA splicing. Mol. Syst. Biol. 7, 506 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Larson DR et al. Direct observation of frequency modulated transcription in single cells using light activation. eLife 2, e00750 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raj A, Peskin CS, Tranchina D, Vargas DY & Tyagi S Stochastic mRNA synthesis in mammalian cells. PLoS Biol. 4, e309 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vargas DY et al. Single-molecule imaging of transcriptionally coupled and uncoupled splicing. Cell 147, 1054–1065 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin RM, Rino J, Carvalho C, Kirchhausen T & Carmo-Fonseca M Live-cell visualization of pre-mRNA splicing with single-molecule sensitivity. Cell Rep. 4, 1144–1155 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coulon A et al. Kinetic competition during the transcription cycle results in stochastic RNA processing. eLife 3, e03939 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A & Tyagi S Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods 5, 877–879 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zenklusen D, Larson DR & Singer RH Single-RNA counting reveals alternative modes of gene expression in yeast. Nat. Struct. Mol. Biol. 15, 1263–1271 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Femino AM, Fay FS, Fogarty K & Singer RH Visualization of single RNA transcripts in situ. Science 280, 585–590 (1998). [DOI] [PubMed] [Google Scholar]
- 39.Stetson PB DAOPHOT: a computer program for crowded-field stellar photometry. Publ. Astron. Soc. Pac. 99, 191–222 (1987). [Google Scholar]
- 40.Orengo JP, Bundman D & Cooper TA A bichromatic fluorescent reporter for cell-based screens of alternative splicing. Nucleic Acids Res. 34, e148 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Munding EM, Shiue L, Katzman S, Donohue JP & Ares M Jr. Competition between pre-mRNAs for the splicing machinery drives global regulation of splicing. Mol. Cell 51, 338–348 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li P et al. Morphogen gradient reconstitution reveals Hedgehog pathway design principles. Science 360, 543–548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Elowitz MB, Levine AJ, Siggia ED & Swain PS Stochastic gene expression in a single cell. Science 297, 1183–1186 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Park T et al. Bayesian estimation of hardness ratios: modeling and computations. Astrophys. J. 652, 610–628 (2006). [Google Scholar]
- 45.Coath CD, Steele RCJ & Fred Lunnon W Statistical bias in isotope ratios. J. Anal. Spectrom. 28, 52–58 (2013). [Google Scholar]
- 46.Lamond AI & Spector DL Nuclear speckles: a model for nuclear organelles. Nat. Rev. Mol. Cell Biol. 4, 605–612 (2003). [DOI] [PubMed] [Google Scholar]
- 47.Misteli T, Caceres JF & Spector DL The dynamics of a pre-mRNA splicing factor in living cells. Nature 387, 523–527 (1997). [DOI] [PubMed] [Google Scholar]
- 48.Phair RD & Misteli T High mobility of proteins in the mammalian cell nucleus. Nature 404, 604–609 (2000). [DOI] [PubMed] [Google Scholar]
- 49.Quinodoz SA et al. Higher-order inter-chromosomal hubs shape 3D genome organization in the nucleus. Cell 174, 744–757.e24 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wahl MC, Will CL & Luhrmann R The spliceosome: design principles of a dynamic RNP machine. Cell 136, 701–718 (2009). [DOI] [PubMed] [Google Scholar]
- 51.Luco RF, Allo M, Schor IE, Kornblihtt AR & Misteli T Epigenetics in alternative pre-mRNA splicing. Cell 144, 16–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Colgan DF & Manley JL Mechanism and regulation of mRNA polyadenylation. Genes Dev. 11, 2755–2766 (1997). [DOI] [PubMed] [Google Scholar]
- 53.Shatkin AJ & Manley JL The ends of the affair: capping and polyadenylation. Nat. Struct. Biol. 7, 838–842 (2000). [DOI] [PubMed] [Google Scholar]
- 54.Jensen TH, Jacquier A & Libri D Dealing with pervasive transcription. Mol. Cell 52, 473–484 (2013). [DOI] [PubMed] [Google Scholar]
- 55.Eldar A & Elowitz MB Functional roles for noise in genetic circuits. Nature 467, 167–173 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Losick R & Desplan C Stochasticity and cell fate. Science 320, 65–68 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Beyer AL & Osheim YN Splice site selection, rate of splicing, and alternative splicing on nascent transcripts. Genes Dev. 2, 754–765 (1988). [DOI] [PubMed] [Google Scholar]
- 58.Rosenberg AB, Patwardhan RP, Shendure J & Seelig G Learning the sequence determinants of alternative splicing from millions of random sequences. Cell 163, 698–711 (2015). [DOI] [PubMed] [Google Scholar]
- 59.Wong MS, Kinney JB & Krainer AR Quantitative activity profile and context dependence of all human 5’ splice sites. Mol. Cell 71, 1012–1026.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wieringa B, Hofer E & Weissmann C A minimal intron length but no specific internal sequence is required for splicing the large rabbit beta-globin intron. Cell 37, 915–925 (1984). [DOI] [PubMed] [Google Scholar]
- 61.Graveley BR, Hertel KJ & Maniatis T A systematic analysis of the factors that determine the strength of pre-mRNA splicing enhancers. EMBO J. 17, 6747–6756 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Girard C et al. Post-transcriptional spliceosomes are retained in nuclear speckles until splicing completion. Nat. Commun. 3, 994 (2012). [DOI] [PubMed] [Google Scholar]
- 63.Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M & Cai L Single-cell in situ RNA profiling by sequential hybridization. Nat. Methods 11, 360–361 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shah S, Lubeck E, Zhou W & Cai L In situ transcription profiling of single cells reveals spatial organization of cells in the mouse hippocampus. Neuron 92, 342–357 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zahler AM, Lane WS, Stolk JA & Roth MB SR proteins: a conserved family of pre-mRNA splicing factors. Genes Dev. 6, 837–847 (1992). [DOI] [PubMed] [Google Scholar]
- 66.Engreitz JM et al. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 539, 452–455 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.La Manno G et al. RNA velocity of single cells. Nature 560, 494–498 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen KH, Boettiger AN, Moffitt JR, Wang S & Zhuang X RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.