

Abstract
Histone lysine specific demethylase 1 (LSD1) has been recognized as an important modulator in post-translational process in epigenetics. Dysregulation of LSD1 has been implicated in the development of various cancers. Herein, we report the discovery of the hit compound 8a (IC50 = 3.93 μmol/L) and further medicinal chemistry efforts, leading to the generation of compound 15u (IC50 = 49 nmol/L, and Ki = 16 nmol/L), which inhibited LSD1 reversibly and competitively with H3K4me2, and was selective to LSD1 over MAO-A/B. Docking studies were performed to rationalize the potency of compound 15u. Compound 15u also showed strong antiproliferative activity against four leukemia cell lines (OCL-AML3, K562, THP-1 and U937) as well as the lymphoma cell line Raji with the IC50 values of 1.79, 1.30, 0.45, 1.22 and 1.40 μmol/L, respectively. In THP-1 cell line, 15u significantly inhibited colony formation and caused remarkable morphological changes. Compound 15u induced expression of CD86 and CD11b in THP-1 cells, confirming its cellular activity and ability of inducing differentiation. The findings further indicate that targeting LSD1 is a promising strategy for AML treatment, the triazole-fused pyrimidine derivatives are new scaffolds for the development of LSD1/KDM1A inhibitors.
Abbreviations: AML, acute myeloid leukemia; ATRA, all-trans retinoic acid; BTK, Bruton׳s tyrosine kinase; CDK, cyclin-dependent kinase; CuAAC, copper-catalyzed azide-alkyne cycloadditions; DABCO, triethylenediamine; DCM, dichloromethane; DNMTs, DNA methyltransferases; DIPEA, N,N-diisopropylethylamine; EA, ethyl acetate; EtOH, ethanol; GSCs, glioma stem cells; FAD, flavin adenine dinucleotide; LSD1, histone lysine specific demethylase 1; MAO, monoamine oxidase; MeOH, methanol; PAINS, pan-assay interference compound; Rt, room temperature; SAR, structure—activity relationship; TCP, tranylcypromine; TEA, triethylamine; THF, terahydrofuran; TLC, thin layer chromatography.
KEY WORDS: Epigenetic regulation, Histone demethylase, LSD1, Pyrimidine-triazole, Mercapto heterocycles, Antiproliferative ability, AML treatment, Structure–activity relationships (SARs)
Graphical abstract
A series of triazole-pyrimidine derivatives were designed and synthesized as LSD1 inhibitors based on the hit compound 8a from our in-house compound library. Among them, compound 15u was identified as the most potent, selective and reversible LSD1 inhibitor, and also demonstrated excellent cellular inhibitory activities against AML cell lines.

1. Introduction
Histone modifications such as methylation, phosphorylation, acetylation, ubiquitylation, and sumoylation are thought to regulate transcription, chromatin structure and other nuclear processes1., 2.. Among them, the post-translational histone methylation is an important chromatin modification that is known to affect many biological processes3. To date, two classes of lysine demethylases (KDMs), namely KDM1s and JmjC KDMs, have been identified to be able to remove methyl groups of N-methyl-lysine residues through the flavin adenine dinucleotide (FAD) or Fe(II)/a-ketoglutarate (α-KG)-dependent oxidative mechanisms, respectively4., 5.. Although evidence of reversible methylation of calf thymus histones was documented by Kim and co-workers in 19736, the histone methylation had generally been recognized as an irreversible modification until the lysine specific histone demethylase 1 (LSD1 or KDM1A) was first identified by Shi and co-workers in 20047.
LSD1 catalyzes the demethylation of mono- and di-methylated K4 or K9 on histone H3 (H3K4me1/2 & H3K9me1/2) under diverse biological settings via the FAD-dependent enzymatic oxidation8., 9.. LSD1 could also remove methyl groups of non-histone substrates such as p53, E2F transcription factor, DNA methyltransferases (DNMTs) and further modulate their downstream cellular functions10., 11., 12., 13., 14.. In vivo, LSD1 played a pivotal role during the process of early embryonic development and differentiation of embryonic stem cell10., 15., 16., 17.. By modulating the expression of target genes, LSD1 is closely associated with tumorigenesis18, pluripotent stem cells19, and neurodegenerative disorders20., 21.. Moreover, LSD1 has been observed to be overexpressed in various malignant tumors22., 23., 24., 25., 26., 27., 28., 29. and is closely associated with differentiation, proliferation, migration, invasion and poor prognosis30., 31.. Knockdown of LSD1 using shRNA reduced glioma stem cells (GSCs) stemness and induced the differentiation. Pharmacological inhibition of LSD1 using NCL-1 and NCD-38 significantly reduced the cell viability, neurosphere formation and induced apoptosis of GSCs32. LSD1 has also been reported to able to promote S-phase entry and tumorigenesis via chromatin co-occupation with E2F1 and selective H3K9 demethylation33. These findings unveil the biological importance of LSD1 and the therapeutic potentials of LSD1 inhibitors.
To date, TCP-based LSD1 inhibitors ORY-1001/RG-6016, GSK2879552 (Clinicaltrials.gov identifier: NCT02177812) and INCB059872 (Clinicaltrials.gov identifier: NCT02712905) alone or in combination with other therapeutic agents such as all-trans retinoic acid (ATRA), cytarabine or azacitidine, etc., have advanced into clinical trials for the treatment of acute myeloid leukemia and small-cell lung cancer, etc. (Fig. 1)34., 35., 36.. The success of TCP-based drug candidates makes TCP an attractive scaffold for the development of new LSD1 inhibitors37. Apart from TCP-based inhibitors, varieties of other different classes of LSD1 inhibitors have also been identified. However, these LSD1 inhibitors (e.g., TCP, SP-2509, and GSK-690) have showed poor specificity, off-target effects, etc. For example, polyamine derived LSD1 inhibitors generally showed a low micromolar range and poor selectivity38., 39.. The highly potent hydrzone derivatives suffered from the off-target issues due to its slow response to CD86, an important biomarker of LSD1 activity40. In addition, pyridine-derived compound GSK-690 was reported to inhibit the human ether-a-go-go-related gene (hERG) cardiac ion channel, albeit with good anti-LSD1 potency in biochemical and cellular level41., 42..
Figure 1.

TCP-based LSD1 inhibitors.
Following our previous work on the identification of reversible LSD1 inhibitors43., 44., 45., 46., 47., here we describe the identification of triazole-fused pyrimidine-based reversible LSD1 inhibitors through the biochemical screening of our in-house structurally diverse molecular library (ca. 500 compounds) and subsequent extensive medicinal chemistry efforts, leading to the identification of highly potent and selective LSD1 inhibitors (Fig. 2). Our data indicate that the triazole-fused pyrimidine is a new scaffold for the development of highly potent and selective LSD1 inhibitors.
Figure 2.

Identification of hit compound 8a from our chemical library and further optimizations leading to discovery of compound 15u.
2. Results and discussion
2.1. Synthetic routes
The synthetic routes of the designed compounds were presented in Scheme 1, Scheme 2, Scheme 3, Scheme 4. The key intermediate derivatives 7a–ab were prepared following our previously reported procedures, as depicted in Scheme 1 48. Briefly, treatment of 2-mercaptopyrimidine-4,6-diol (1) with alkyl bromide in MeOH gave compound 2a–e, which then reacted with fuming nitric acid, affording compounds 3a–e. Chlorination of 3a–e using POCl3 yielded 4a–e, which was then subjected to Fe-mediated hydrogenation, generating compounds 5a–g. Compounds 5a–g reacted with different amines in the presence of triethylamine (TEA) in EtOH to form compounds 6a–ab, which were then treated with NaNO2, generating the intermediates 7a–ab, in which the new triazole ring was formed efficiently.
Scheme 1.

Synthesis of Intermediates 7a–ab. Reagents and conditions: (a) alkyl bromide, TEA, MeOH, reflux, 2 h; (b) fuming nitric acid, AcOH, 25–45 °C, 1 h; (c) POCl3, DMA, reflux, 2 h; (d) Fe, AcOH, MeOH, reflux; (e) appropriate amines, TEA, EtOH, reflux, 48 h; (f) NaNO2, AcOH, H2O, 10 °C, 1 h.
Scheme 2.

Synthesis of compounds 8a–l, 9a–b and 10. Reagents and conditions: (a) mercapto heterocyclic analogs, TEA, MeCN, reflux, 2 h; (b) TEA,DCM, rt, overnight.
Scheme 3.

Synthesis of compounds 15a–ak. Reagents and conditions: (a) TEA or DABCO, CS2, THF, rt, overnight; (b) BTC, CHCl3, rt, overnight; (c) NaN3, H2O, reflux, 5 h; (d) TEA, MeCN, reflux, 2 h.
Scheme 4.

Synthesis of compounds 17, 19, 22a–b and 23. Reagents and conditions: (a) PhSCN, Cs2CO3, MeCN, rt, overnight; (b) 5-mercapto-1-methyltetrazole, K2CO3, i-PrOH, reflux, 5 h; (c) CH(OEt)3, aq HCl, rt, 8 h; (d) 5-mercapto-1-methyltetrazole, DIEA, DMF, 100 °C, 3 h; (e) 5-mercapto-1-phenyltetrazole, DIEA, DMF, 100 °C, 2 h; (f) DIEA, i-PrOH/DMF (v/v, 1:1 ), 90 °C, 3 h; (g) BnN3, CuSO4·5H2O, sodium ascorbate, THF/H2O (1:1), rt, 3 h.
As shown in Scheme 2A, compounds 8a–l were efficiently synthesized from compounds 7a–b in the presence of TEA through the nucleophilic substitution reactions of different mercapto heterocyclic analogs. Compounds 8k and 8l were then chosen for further modifications by reacting with 4-toluenesulfonyl chloride (TsCl), methanesulfonyl chloride (MsCl), benzoyl chloride (BzCl), respectively, affording compounds 9a–b and 10 (Scheme 2B).
In addition, replacement of heterocycles in compounds 8a–k with the tetrazole ring led to formation of compounds 15a–ak. As shown in Scheme 3, the mercapto tetrazole derivatives 14a–i were synthesized from amines 11a–i. Aromatic amine reacted with carbon disulfide in the presence of TEA or triethylenediamine (DABCO) to produce dithiocarbamic acid salts 12a–i49, followed by addition of triphosgene (BTC) in chloroform to form isothiocyanates 13a–i. Isothiocyanates 13a–i reacted with sodium azide in water to give the mercaptotetrazole analogs 14a–i, which then reacted with intermediates 7c–7ab in the presence of TEA in MeCN to yield compounds 15a–ak.
Furthermore, bioisosteric replacement and scaffold hopping have widely been recognized as two useful strategies in drug design, which have led to the identification of numerous lead compounds50., 51.. In this work, the triazole-fused pyrimidine scaffold was also replaced with the triazolo[5,4-d]pyrimidine, purine and pyrimidine scaffold, respectively, forming the corresponding compounds 17, 19, 22a–b and 23. As shown in Scheme 4, treatment of compound 4b with phenyl isothiocyanate in the presence of Cs2CO3 in MeCN gave compound 16, which then reacted with 5-mercapto-1-methyltetrazole to afford compound 17 (Scheme 4A). Compound 6j was chosen as the starting material for constructing the purine scaffold 18 by reacting with triethoxy methane (Scheme 4B). The nucleophilic substitution reaction of 5-mercapto-1-methyltetrazole with compound 18 yielded compound 19. Compounds 20a–b reacted with 5-mercapto-1-phenyltetrazole in the presence of DIEA in DMF to generate compounds 21a–b, followed by the substitution reaction with 2-aminoethanol to yield 22a–b (Scheme 4C). The terminal alkyne group of compound 22a was then employed to synthesize compound 23 bearing an additional triazole moiety via the copper-catalyzed azide-alkyne cycloadditions (CuAAC) (Scheme 4D). Conceivably, more analogs of compound 23 could be obtained from different alkynes through the CuAAC reactions and could be used to construct compound collections.
2.2. LSD1 inhibitory activity and structure—activity relationship studies (SARs)
All the compounds synthesized in this study were examined for their in vitro inhibitory effect toward LSD1, and GSK2879552 was chosen as a positive control46., 47.. The results were summarized in Table 1, Table 2, Table 3, Table 4. Besides, to avoid interference of false positive compounds, PAINS screening of the synthesized compounds was carried out by employing the online program ("PAINS-Remover", http://www.cbligand.org/PAINS/)52, and all the tested compounds passed the filter.
Table 1.
Inhibitory effect of compounds 8a–l, 9a–b, 10 and 11 on recombinant LSD1.
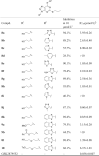 |
aData are represented as the mean of the inhibition rate.
bData are represented as mean±SD. All experiments were independently carried out at least three times.
– Not applicable.
Table 2.
Inhibitory effect of compounds 15a–s, 17 and 19 on recombinant LSD1.
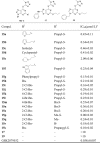 |
aData are represented as mean±SD. All experiments were independently carried out at least three times.
–, not applicable.
Table 3.
Inhibitory effect of compounds 15t–aj on recombinant LSD1.
 |
aData are represented as mean±SD. All experiments were independently carried out at least three times.
–, not applicable.
Table 4.
Inhibitory effect of compounds 15ak, 22b and 23 on recombinant LSD1.
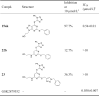 |
aData are represented as the mean of the inhibition rate.
bData are represented as mean±SD. All experiments were independently carried out at least three times.
The hit compound 8a identified from our in-house library inhibited LSD1 moderately with an IC50 value of 3.93 μmol/L. This interesting result promoted us to perform further structural elaborations. Replacement of the thiadiazole ring in 8a with other aromatic rings led to the formation of compounds 8b–k (Table 1). Clearly, compounds 8d and 8i bearing the phenyl and imidazole ring, respectively were found to be inactive toward LSD1 (IC50 > 10 μmol/L). The remaining compounds showed acceptable inhibitory effect toward LSD1 with the IC50 values less than 9.0 μmol/L. Particularly, compounds 8j–k possessing the tetrazole and benzothiazole, respectively, exhibited potent inhibition toward LSD1 with the IC50 values of 0.60 and 0.85 μmol/L, respectively. Further derivatizations based on the hydroxy group of compound 8k were carried out, yielding compounds 9a–b, 8l and 10. Except for compound 9b, compounds 9a, 8l and 10 also exerted moderate inhibitory effect, albeit with relatively lower activity than compound 8k, suggesting the importance of the scaffold for the inhibitory activity.
The first round structural modifications as shown in Table 1 led to the identification of the tetrazole containing compound 8j, which inactivated LSD1 potently (IC50 = 0.60 μmol/L). Further structural optimizations centering on variations of R1 and R2 groups attached to the triazole—pyrimidine core were carried out, leading to the discovery of compounds 15a–s. Interestingly, this series of compounds showed potent inhibition toward recombinant LSD1. Compound 15p showed the best potency with an IC50 value of 80 nmol/L, about 7.5-fold more potent than 8j. For compounds 15a–l bearing the same propylthio group (R1), only compound 15e bearing a hydrophilic morpholine group (R2) was found to have significantly decreased inhibitory effect toward LSD1 with an IC50 value of 2.09 μmol/L, the remaining compounds bearing a hydrophobic R2 group exhibited comparable and potent inhibitory activity against LSD1. These findings reveal the essential structural elements for the activity toward LSD1. Replacement of the R1 group with benzylthio, methylthio, methyl, hydrogen atom (H) and propargylthio groups led to a new series of compounds 15m–s, of which compounds 15p and 15s inhibited LSD1 with the IC50 value of 80 and 100 nmol/L, respectively. Interestingly, bioisosteric replacement of the triazole—pyrimidine in compounds 15a–s with the thiazolo[5,4-d]pyrimidine and purine rings led to significantly decreased inhibitory activity. Compounds 17 and 19 were found to be inactive against LSD1 (IC50 > 10 μmol/L).
Based on above findings, further modifications were mainly focused on variations of R3 group attached to the tetrazole ring. As shown in Table 3, this series of compounds generally exhibited excellent inhibition toward LSD1. Compound 15t inactivated LSD1 with an IC50 value of 0.15 μmol/L. While 2-chloro-benzyl (R2)-substituted compound 15u exhibited about 3-fold increase in potency (IC50 = 49 nmol/L), comparable to that of compound 15x bearing 4-isopropy benzyl group (IC50 = 55 nmol/L). Compounds 15v and 15w bearing the 3-Cl benzyl or 4-Cl benzyl group, respectively also displayed acceptable potency at nanomolar levels (IC50 = 74 and 93 nmol/L, respectively), but was slightly less potent than 15t. 4-Methoxyl-benzyl-substituted compound 15y showed slightly decreased activity against LSD1 with an IC50 value of 140 nmol/L, comparable to that of compound 15t. In contrast, compounds 15z and 15aa with a larger benzylthio group (R1) exhibited remarkably decreased inhibitory activity (IC50 = 300 and 410 nmol/L, respectively). Compared with compound 15u, compound 15ac exerted decreased inhibitory activity with the steric hindrance of R1 group increased correspondingly. When the R3 substitution was substituted with phenyl ring, naphthyl or pyridyl group, the corresponding compounds 15ad–ai showed decreased activity. Particularly, when the R3 group was the hydrophilic N,N-dimethylaminoethyl group, compound 15aj was found to be inactive against LSD1.
Finally, the scaffold hopping strategy was employed to investigate the effects of the core scaffold variations on the LSD1 inhibition, and compounds 15ak, 22b and 33 were produced and compared. As shown in Table 4, the removal of the triazole ring of compound 15ak led to the generation of compound 22b, which was found to be inactive toward LSD1 (IC50 > 10 μmol/L), highlighting the importance of the triazole-fused pyrimidine scaffold for the activity. Additionally, compound 23 bearing a triazole ring at the side chain also showed weak inhibition toward LSD1 (36.3% inhibitory rate at 10 μmol/L).
2.3. Molecular docking studies of LSD1 inhibitors
We carried out a molecular docking study to predict the possible binding mode of the studied compounds with LSD1 using MOE 2015.10. The crystal structure of LSD1 in complex with an H3K4 peptide (PDB code: 2V1D) was used as receptor protein, and was prepared by adding hydrogen atoms, removing water molecules and the peptide, while FAD was kept as a component of the receptor. Docking results showed that the 20 binding structures with the lowest-energies of the most potent compound 15u were similar (Fig. 3A). The tetrazole ring formed hydrogen bond interactions with the side chain of Gln358 and Asn535. The distance of these two hydrogen bonds was 2.7 and 2.5 Å respectively. The phenyl group attached to the tetrazole ring was surrounding by Ile356, Leu677 and Trp695, and had hydrophobic interactions with these residues (Fig. 3B). The triazole ring formed a hydrogen bond as well as electrostatic interaction with the positively charged side chain of His564, and the distance was 2.7 Å. The pyrimidine ring was predicted to form a hydrogen bond with the side chain of Asn535 while the distance was 2.4 Å. The 2-Cl phenyl group would be located at the hydrophobic pocket that consists of the flavin ring of FAD, Val333, Phe538, Trp695 and Tyr761, and had π–π stacking with flavin ring, Phe538, Trp695 and Tyr761. In addition, the propargyl group had hydrophobic interactions with Phe382, Leu536 and Trp552 (Fig. 3C). The docking results predicted the binding models of compound 15u in the active site of LSD1, and could well explain the activity discrepancy of our synthesized compounds. The hydrogen bond and strong electrostatic interaction with the positively charged side chain of His564 could explain the importance of triazole ring. Replacement of the triazole ring with thiazole or imidazole may lead to the decrease of hydrogen interaction and electrostatic interaction, which may be responsible for the loss of the anti-LSD1 activity of compounds 17, 19, 22b and 23. Besides, the substituent connecting with the triazole ring was predicted to locate at a hydrophobic pocket and had π–π stacking with some surrounding residues, which could account for the increased activity of compound 15u compared to 8j with hydroxyethyl side chain. Interestingly, the replacement of the phenyl ring with the hydrophilic N,N-dimethylaminoethyl group caused the complete loss of the activity (compound 15u vs. compound 15aj), suggesting the essential structural element for the anti-LSD1 activity. In addition, a newly released crystal structure of LSD1-CoREST in complex with a small molecular inhibitor (4-[5-(piperidin-4-ylmethoxy)-2-(p-tolyl)pyridin-3-yl]benzonitrile, PDB code: 5YJB) was also employed for binding mode prediction (Fig. 3D), presenting a quite similar docking result with that (Fig. 3B) generated from protein receptor (PDB 2V1D). The 3D-QSAR model was also established based on the data of this series (Supporting Information Tables S1, S2 and Fig. S1), which may provide directions for designing new potent and selective LSD1 inhibitors.
Figure 3.

The proposed binding mode of compound 15u with LSD1 (PDB code: 2V1D). (A) 20 lowest-energy docking structures of compound 15u, with the most stable conformation shown as a cyan stick model; (B) Close-up views of the hydrogen bonds and hydrophobic interactions between compound 15u and LSD1; (C) Close-up views of the hydrogen bond interactions between compound 15u and LSD1; (D) The binding mode of compound 15u with LSD1 (PDB code: 5YJB). Compound 15u is shown in cyan stick representation, while the residues and FAD of LSD1 are shown in white and yellow stick representation, respectively.
2.4. Selectivity and reversibility of compound 15u
As LSD1 belongs to the monoamine oxidases (MAOs) family, the most potent LSD1 inhibitor compound 15u (IC50 = 49 nmol/L, Ki = 16 nmol/L, Fig. 4A) was then examined for its enzymatic selectivity against MAO-A and MAO-B. As showed in Fig. 4B, compound 15u inhibited LSD1 with an inhibitory rate of 95.8% at 1 μmol/L, while it inhibited MAO-A/B with the inhibitory rates below 10% at 1 μmol/L and around 40% of inhibitory rates at 10 μmol/L, indicating that compound 15u may be highly selective to LSD1 over MAO-A/B. Furthermore, the reversibility of compound 15u against LSD1 was also investigated using the dilution assay. As shown in Fig. 4C, 80-fold dilution of the LSD1/compound 15u complex resulted in more than 80% recovery of LSD1 activity, while the positive control GSK2879552 failed to recover the activity of LSD1. These results indicated that compound 15u may interact with LSD1 in a reversible manner. Additionally, with classic Lineweaver—Burk plots, compound 15u was characterized as a substrate (H3K4me2) competitive inhibitor over LSD1 (Fig. 4D), as compared to the noncompetitive and uncompetitive fitting (Supporting Information Fig. S2).
Figure 4.

(A) Inhibition of compound 15u against LSD1; (B) Inhibition of compound 15u against LSD1 and MAO-A/B. Data are expressed as the mean of at least three determinations±SD; (C) Compound 15u reversibly inhibited LSD1. Data are expressed as the means±SDs of three independent experiments; (D) Lineweaver—Burk plot of competitive inhibition for compound 15u with indicated concentrations.
2.5. Kinase inhibition
Pyrimidine-containing bicyclic N-heterocycles have been reported to be inhibitors of a number of kinases, such as Bruton׳s tyrosine kinase (BTK) or cyclin-dependent kinases (CDKs)53., 54., 55., herein compound 15u was also evaluated against some selected kinases such as BTK and CDK1/2/4/6/7/9, and staurosporine was used as reference compound (Supporting Information Table S3). As shown in Fig. 5, compound 15u was found to be devoid of the inhibitory activity with <15% of the inhibitory rate at 10 μmol/L toward the tested kinases. The data indicated that compound 15u was highly selective to LSD1 over the tested kinases.
Figure 5.

Inhibition of compound 15u (10 μmol/L) against BTK and CDKs. Data are expressed as the mean of at least three determinations ±SD (n=3).
2.6. Antiproliferative ability of compounds 15s, 15u and 15y
Compound 15u was then tested for its antiproliferative ability of several LSD1-overexpressed cancer cell lines. As depicted in Table 5, treatment of compound 15u for 6 days exhibited strong antiproliferative activity against the four leukemia cell lines (OCL-AML3, K562, THP-1 and U937) as well as the lymphoma cell line Raji with the IC50 values of 1.79, 1.30, 0.45, 1.22 and 1.40 μmol/L, respectively. Clearly, THP-1 cell line was found to be more sensitive to compound 15u as it harbors a t(9;11) translocation, the cytogenetic hallmark of MLL-AF9, while LSD1 acts at genomic loci bound by MLL-AF9 to sustain expression of the associated oncogenic program, thus preventing differentiation as reported56. Another two compounds 15s and 15y (LSD1 IC50=100 and 140 nmol/L, respectively) also displayed strong antiproliferative activity against OCL-AML3, K562, THP-1, U937 and Raji at low micromolar levels and certain selectivity to THP-1 cells. In contrast, the reference compound GSK2879552 was found to be inactive toward THP-1 cells with an IC50 of more than 50 μmol/L. The activity discrepancy may be due to that our compounds may also bind to other cellular targets. Further mechanistic studies are underway in our lab and will be reported in due course.
Table 5.
Antiproliferative activity of compounds 15s, 15u and 15y.
| Cell line | Cell type | IC50 (μmol/L)a |
||
|---|---|---|---|---|
| 15s | 15u | 15y | ||
| OCI-AML3 | Leukemia | 2.50±0.54 | 1.79±0.14 | 4.72±1.11 |
| K562 | Leukemia | 7.02±1.01 | 1.30±0.36 | 7.24±0.93 |
| THP-1 | Leukemia | 1.14±0.25 | 0.45±0.11 | 2.52±0.82 |
| U937 | Leukemia | 2.31±0.33 | 1.22±0.31 | 3.23±0.28 |
| Raji | Lymphoma | 3.02±0.95 | 1.40±0.49 | 3.32±0.75 |
Cell lines were treated with compounds for 6 days. Data are expressed as the mean±SD of three independent experiments (n = 3).
We next evaluated how LSD1 inhibitors altered the colony-forming capacity of THP-1 cells with soft agar assay, coupled with high content analysis. As indicated in Fig. 6A, treatment of compound 15u for 8 days significantly inhibited colony formation of THP-1 cells. As B7-2 (CD86), one of type I transmembrane proteins that were originally identified as ligands for CD28/CTLA-4, is a surrogate cellular biomarker of LSD1 activity57, THP-1 cells were treated with compound 15u to evaluate its effect on protein expression of CD86. As indicated in Fig. 6B, increased CD86 expression was observed, suggesting that compound 15u may inactivate LSD1 in the cellular level. Moreover, considering that LSD1 inhibition could induce differentiation of human monocytic cells58., 59., the THP-1 monocytic cells line were treated with compound 15u at three different concentrations to evaluate its effect on CD11b, a well-known myelo-monocytic differentiation marker modulated by KDM1A60. As shown in Fig. 6C, compound 15u induced expression increase of CD11b in THP-1 cells concentration dependently, which suggests the differentiation induction of the THP-1 cells. Also, treatment of THP-1 cells with compound 15u caused remarkable morphological changes of granulocytic differentiation61, such as segmented nuclei and condensed chromatin (Fig. 6D).
Figure 6.

(A) Colony formation for THP-1 cells treated with indicated concentrations of compound 15u for 8 days; (B) CD86 expression of THP-1 cells analyzed by flow cytometry after treatment with compound 15u for 8 days; (C) CD11b expression of THP-1 cells analyzed by flow cytometry after treatment with compound 15u for 8 days; (D) Cell morphology of THP-1 cells analyzed by Wright—Giemsa staining after treatment with compound 15u.
3. Conclusions
In conclusion, we have reported the discovery of a series of triazole-fused pyrimidine derivatives as highly potent LSD1 inhibitors. Among them, the most potent compound 15u (IC50=49 nmol/L) inhibited LSD1 reversibly and competitively with H3K4me2 substrate, and showed selectivity to LSD1 over MAO-A/B. Molecular docking simulations and 3D-QSAR studies using the CoMFA model were performed to predict the binding models and to explain the SARs observed. The MTT assay indicated that compound 15u exerted strong antiproliferative activity against four leukemia cell lines (OCL-AML3, K562, THP-1 and U937) and the lymphoma cell line Raji. Additionally, compound 15u significantly inhibited colony formation and caused remarkable morphological changes of THP-1 cells in a concentration-dependent manner. Compound 15u also induced expression of CD86 and CD11b in THP-1 cells, confirming its cellular activity and ability of inducing differentiation. Taken together, compound 15u is a newly developed heterocyclic inhibitor of LSD1, which makes the triazole-fused pyrimidine an attractive scaffold for the discovery of potent inhibitors of LSD1.
4. Experimental
4.1. Chemistry
Reagents and solvents were purchased from commercial sources and were used without further purification. All reaction were monitored by thin-layer chromatography (TLC) and visualized with UV light. Melting points were determined on an X-5 micromelting apparatus (Haohai Inc., Nanjing, China) and are uncorrected. 1H NMR and 13C NMR spectra were recorded on a Bruker 400 and 100 MHz spectrometer (Ettlingen, Germany), respectively. High resolution mass spectra (HR-MS) were recorded on a Waters Micromass Q-T of Micromass spectrometer (Milford, MA, USA) by electrospray ionization (ESI). Final products were of >95% purity as analyzed by HPLC analysis (Phenomenex column, C18, 5.0 μm, 150 mm × 4.6 mm) on Dionex UltiMate 3000 UHPLC instrument from ThermoFisher (Waltham, MA, USA). The signal was monitored at 254 nm with a UV dector. A flow rate of 1.0 mL/min was used with a mobile phase of methanol in H2O (80:20, v/v).
4.1.1. Preparation of compound 5e
To a suspension of 2-mercaptobarbituricacid 1 (7 g, 48 mmol, 1 eq.) in methanol (40 mL) was added triethylamine (7 mL, 51 mmol, 1.05 eq.) at rt, and a clear solution was formed. Then cyclopropylmethyl bromide (4.7 mL, 48 mmol, 1 eq.) was added dropwise over 20 min and was kept stirring under reflux for 2 h. After cooling to rt, the formed precipitate was filtered and washed with methanol to afford compound 2e (4.2 g, yield 44%), which was very smelly and used for the next step without further purification. The obtained compound 2e was added cautiously in portions to a precooled solution of fuming nitric acid (3.6 mL) in acetic acid (11 mL) in an ice bath, and the resulting slurry was stirred for 1 h below 35 °C. After the completion of the reaction, the slurry was poured to crushed ice and stirred for 20 min. The precipitate was obtained by filtration and washed with water, affording compound 3e as smelly pink solid (3.9 g, yield 76%) which was directly used for the next step. Then compound 3e was dissolved in POCl3 (10 mL) at rt, following the addition of N,N-dimethylphenylamine (3.5 mL). The reaction mixture was refluxed for 3 h and monitored by TLC (PE/EA = 5:1). After cooled to rt, the mixture was hydrolyzed by pouring on the crushed ice, and extracted with ethyl acetate. The organic phase was washed with water and saturated sodium bicarbonate. The solvent was evaporated under reduced pressure to afford crude product 4e, which was used for the next step without further purification. To a mixture of acetic acid/ethanol (v/v = 1:5, 35 mL) was added compound 4e, and followed by the addition of iron powder (2.7 g, 48 mmol) portionwise. The formed slurry was heated to 50 °C for 3 h, and monitored by TLC (PE/EA = 6:1). The reaction mixture was diluted with ethyl acetate, and filtrated over celite pad. The filtrate was washed with water and saturated sodium dicarbonate. The residue was purified by flash column chromatography (PE/EA = 6:1) to give compound 5e as brown semi-solid, yield, 76%. 1H NMR (400 MHz, CDCl3) δ 4.21 (br, 2H), 3.05 (d, J = 7.2 Hz, 2H), 1.12–1.19 (m, 1H), 0.58–0.61 (m, 2H), 0.31–0.35 (m, 2H).
4.1.2. Preparation of compound 8a
The mixture of intermediate 7a (1 eq.), 5-methyl-1,3,4-thiadiazole-2-thiol (1 eq.) and triethylamine (1.2 eq.) in 20 mL of acetonitrile was refluxed for 2 h and monitored by TLC. After completion of the reaction, the mixture was concentrated under reduced pressure. The residue was dissolved in ethyl acetate and washed with water successively. The crude product was purified by flash column chromatography on silica gel to afford compound 8a as white solid, m.p. 111–112 °C, yield 65%. 1H NMR (400 MHz, CDCl3) δ 4.76–4.79 (t, J = 4.8 Hz, 2H), 4.22 (m, 2H), 3.09–3.12 (t, J = 7.4 Hz, 2H), 2.87 (s, 3H), 1.70–1.80 (m, 2H), 1.04–1.08 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 170.5, 169.1, 157.9, 155.0, 149.2, 130.9, 60.2, 50.2, 33.4, 22.1, 15.7, 13.5. HR-MS (ESI): Calcd. C12H15N7OS3, [M+H]+: 370.0578, Found: 370.0567. Compounds 8b–l and 15a–ak were prepared through similar procedures as used for the synthesis of compound 8a (see Supporting Information Section 6.2).
4.1.3. General procedure for the synthesis of compounds 9a–9b and 10
To a solution of compound 8k (1 eq.) and triethylamine (1.1 eq.) in dry DCM was added methanesulfonyl chloride (1 eq.) in an ice bath, then the reaction mixture was stirred at rt overnight. Upon completion, the reaction mixture was diluted with ethyl acetate and washed with water. The organic phase was dried over Na2SO4, and the solvent was removed under vacuum. The crude residue was purified by flash column chromatography (PE/EA = 3:1) to afford compound 9a as white solid, yield 86%. Compounds 9b and 10 were also synthesized following the same methods. m.p. 153–154 °C, 1H NMR (400 MHz, CDCl3) δ 8.12–8.14 (m, 1H), 7.97–7.99 (m, 1H), 7.55–7.59 (m, 1H), 7.49–7.53 (m, 1H), 4.95–4.97 (t, J = 5.2 Hz, 2H), 4.79–4.81 (t, J = 5.2 Hz, 2H), 3.00 (s, 3H), 3.00–2.96 (m, 2H), 1.55–1.62 (m, 2H), 0.85–0.89 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 171.3, 160.3, 154.9, 152.2, 149.2, 137.3, 131.1, 126.6, 126.1, 123.5, 121.3, 65.2, 46.1, 37.9, 33.5, 22.2, 13.3. HR-MS (ESI): Calcd. C17H18N6O3S4, [M+Na]+: 505.0221, Found: 505.0220.
4.1.3.1. 2-(7-(Benzo[d]thiazol-2-ylthio)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)ethyl benzoate (9b)
Pale yellow solid, m.p. 97–98 °C, yield 79%. 1H NMR (400 MHz, CDCl3) δ 8.11–8.13 (m, 1H), 7.96–7.98 (m, 1H), 7.91–7.94 (m, 2H), 7.54–7.58 (m, 2H), 7.48–7.52 (m, 1H), 7.40–7.44 (m, 2H), 5.02–5.05 (t, J = 5.2 Hz, 2H), 4.83–4.85 (t, J = 5.2 Hz, 2H), 2.87–2.90 (t, J = 7.2 Hz, 2H), 1.50–1.59 (m, 2H), 0.83–0.86 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 171.0, 166.0, 160.0, 155.3, 152.1, 149.3, 137.2, 133.4, 131.0, 129.8, 129.2, 128.5, 126.6, 125.9, 123.4, 121.3, 62.3, 46.1, 33.32, 22.1, 13.3. HR-MS (ESI): Calcd. C23H20N6O2S3, [M+Na]+: 531.0708, Found: 531.0709.
4.1.3.2. 2-(2-(7-(Benzo[d]thiazol-2-ylthio)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyri-midin-3-yl)et-hoxy)ethyl-4-methylbenzenesulfonate (10)
White solid, m.p. 97–99 °C, yield 82%. 1H NMR (400 MHz, CDCl3) δ 8.12–8.14 (m, 1H), 7.97–7.99 (m, 1H), 7.76 (m, 2H), 7.55–7.59 (m, 1H), 7.48–7.53 (m, 1H), 7.35–7.37 (m, 2H), 4.72–4.75 (t, J = 5.6 Hz, 2H), 4.08—4.10 (t, J = 4.6 Hz, 2H), 4.02–4.05 (t, J = 5.6 Hz, 2H), 3.67–3.69 (t, J = 4.6 Hz, 2H), 2.99—3.02 (t, J = 7.2 Hz, 2H), 2.46 (s, 3H), 1.59–1.64 (m, 2H), 0.87–0.91 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 170.7, 159.9, 155.4, 152.2, 149.2, 144.9, 137.3, 131.0, 129.9, 127.9, 126.5, 125.9, 123.4, 121.3, 68.9, 68.8, 68.5, 68.4, 46.5, 33.5, 22.2, 13.3. HR-MS (ESI): Calcd. C25H26N6O4S4, [M+H]+: 603.0977, Found: 603.0975.
4.1.4. Preparation of compounds 14a—i
To a mixture of appropriate amines 11 (1 eq.), TEA or DABCO (3 eq.) in THF was added CS2 (3 eq.) dropwise at rt. The precipitate was formed and the reaction was kept overnight. The dithiocarbamic acid salts 12 were readily obtained after filtration, which then reacted with triphosgene (0.5 eq) in chlorofrom at rt to form isothiocyanates 13 after a short flash column chromatography (PE as eluent). The obtained 13 was reacted with NaN3 (3 eq.) in water under refluxing over 5 h, and then cooled down to rt. The reaction mixture was treated with aq HCl until the pH reached to 3, the formed precipitate was filtrated to give the mercapto tetrazole 14, which was used for the next step without further purification.
4.1.5. Preparation of compound 17
To a solution of compound 4b (450 mg, 1.9 mmol, 1.1 eq.) and phenyl isothiocyanate (230 mg, 1.7 mmol, 1 eq.) in 20 mL of acetonitrile was added Cs2CO3 (1.2 g, 3.7 mmol, 2.2 eq.) portionwise at rt, and the resultant slurry was stirred overnight. After completion of the reaction, the mixture was diluted with ethyl acetate and washed with water. The organic phase was concentrated under vacuum. The residue was purified by flash column chromatography (PE/EA = 6:1) to afford 300 mg of intermediate 16 as light yellow solid, yield: 52%. A mixture of 5-mercapto-1-methyltetrazole (50 mg, 0.43 mmol, 1 eq.), compound 16 (146 mg, 0.43 mmol, 1 eq.) and K2CO3 (60 mg, 0.43 mmol, 1 eq.) in 20 mL of isopropanol was refluxed and monitored by TLC (PE/EA = 5:1). After 5 h, the reaction was done, and then the slurry was filtrated over celite pad. The filtrate was concentrated under vacuum, and the residue was purified by flash column chromatography to give compound 17, white solid, yield: 68% (120 mg), m.p. 240–241 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.09 (s, 1H), 7.72–7.74 (m, 2H), 7.42–7.45 (m, 2H), 7.11–7.15 (m, 1H), 4.09 (s, 3H), 3.35 (s, 2H), 2.61–2.65 (t, J = 7.2 Hz, 2H), 1.34–1.41 (m, 2H), 0.84–0.88 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 162.4, 161.8, 159.7, 149.6, 146.6, 139.4, 137.5, 129.2, 123.4, 118.4, 34.6, 32.3, 22.0, 13.0. HR-MS (ESI): Calcd. C16H16N8S3, [M+Na]+: 439.0558, Found: 439.0554.
4.1.6. Preparation of compound 19
A mixture of compound 6j (100 mg) and aq HCl (0.2 mL) in 5 mL of triethylorthoformate was kept overnight at rt. The resulting solid was filtrated and washed with ether, affording 60 mg of intermediate 18 (yield: 58%). Then mixture of 18 (60 mg, 0.19 mmol, 1 eq.), 5-mercapto-1-methyltetrazole (24 mg, 0.21 mmol, 1.1 eq.) and N,N-diisopropylethylamine (50 mg, 0.39 mmol, 2 eq.) in 15 mL of DMF was heated to 100 °C for 3 h. Then the reaction mixture was cooled to rt, and diluted with ethyl acetate and washed with water for three times successively. The organic phase was concentrated under vacuum, and the residue was purified by flash column chromatography (PE/EA = 4:1) to afford 54 mg of compound 19 as off-white solid, yield: 72%, m.p. 118–120 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.59 (s, 1H), 7.30–7.36 (m, 5H), 5.41 (s, 2H), 4.06 (s, 3H), 2.75–2.78 (t, J = 7.2 Hz, 2H), 1.42–1.48 (m, 2H), 0.86–0.90 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 164.1, 153.8, 150.8, 146.2, 145.4, 136.0, 128.7, 128.0, 127.9, 127.7, 46.7, 34.6, 32.4, 22.1, 13.1. HR-MS (ESI): Calcd. C17H18N8S2, [M+Na]+: 421.0994, Found: 421.0995.
4.1.7. Preparation of compounds 22a–b
A solution of compound 20 (1 eq.), mercaptophenyltetrazole (1 eq.) and DIEA (1.2 eq.) in DMF was heated at 100 °C for 2 h. The reaction mixture was then cooled to rt, diluted with ethyl acetate (EA), and washed with water. The organic phase was dried over NaSO4, and the solvent was removed under vacuum, and the residue was purified by flash column chromatography (PE/EA = 4:1) to afford intermediate 21. A mixture of 21 (1 eq.), ethanolamine (1 eq.) and DIEA (1.1 eq.) in a mixed solvent of isopropanol/DMF (v/v = 1:1) was heated at 90 °C for 3 h, and then cooled to rt. The reaction mixture was concentrated under vacuum, and then poured into water and extracted with EA. The combined organic phases were dried over Na2SO4, filtrated, concentrated and purified by flash column chromatography (PE/EA = 1:1) to give the desired product 22.
4.1.7.1. 2-((6-((1-Phenyl-1H-tetrazol-5-yl)thio)-2-(prop-2-yn-1-ylthio)pyrimidin-4-yl)-amino)ethan-1-ol (22a)
Waxy solid, yield 72%. 1H NMR (400 MHz, CDCl3) δ 7.54–7.60 (m, 5H), 6.32 (s, 1H), 5.44 (br, 1H), 3.81 (t, J = 5.0 Hz, 2H), 3.70–3.75 (m, 1H), 3.61 (d, J = 2.6 Hz, 2H), 3.55 (br, 1H), 2.08 (t, J = 2.6 Hz, 1H).
4.1.7.2. 2-((2-(Benzylthio)-6-((1-phenyl-1H-tetrazol-5-yl)thio)pyrimidin-4-yl)amino)ethan-1-ol (22b)
Light yellow oil, yield 65%. 1H NMR (400 MHz, CDCl3) δ 7.50–7.53 (m, 5H), 7.19–7.28 (m, 5H), 6.24 (s, 1H), 5.66–5.69 (t, J = 5.7 Hz, 1H), 4.11 (s, 2H), 3.71–3.74 (t, J = 5.0 Hz, 2H), 3.47 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 171.0, 161.8, 148.2, 137.3, 133.4, 130.7, 129.7, 128.8, 128.5, 127.1, 124.8, 61.5, 43.5, 35.0. HR-MS (ESI): Calcd. C20H19N7OS2, [M+Na]+: 460.0900, Found: 460.0991.
4.1.8. Preparation of compound 23
A mixture of 22a (100 mg, 0.26 mmol, 1 eq.), benzyl azido (40 mg, 0.30 mmol, 1.15 eq.), sodium ascorbate (5 mg, 0.1 eq.) and CuSO4 (5 mg, 0.1 eq.) in 8 mL of mixed solvent of THF/water (1:1) was stirred at rt for 3 h. The reaction mixture was diluted with EA, washed with water for three times. The organic phase was concentrated under vacuum, and the residue was purified by flash column chromatography (PE/EA = 2:3) to give compound 23 as pale yellow solid, yield 57%. m.p. 51–53 °C, 1H NMR (400 MHz, DMSO-d6) δ 7.94 (s, 1H), 7.66–7.68 (m, 3H), 7.62 (m, 3H), 7.33–7.37 (m, 3H), 7.27–7.29 (m, 2H), 6.04 (s, 1H), 5.54 (s, 2H), 4.73–4.76 (t, J = 5.2 Hz, 1H), 4.15 (s, 2H), 3.42–3.45 (m, 2H), 3.30–3.31 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 169.2, 161.4, 159.8, 147.9, 143.9, 136.0, 133.2, 130.8, 129.6, 128.7, 128.1, 127.8, 125.2, 123.2, 97.9, 59.3, 52.7, 42.7, 24.7. HR-MS (ESI): Calcd. C23H22N10OS2, [M+Na]+: 541.1317, Found: 541.1318.
4.2. Biology assay
4.2.1. LSD1 enzymatic assay
Inhibitory effects of the target compounds against LSD1 were evaluated according to the reported methods62. Full length LSD1 cDNA encoding LSD1 was obtained by RT-PCR and cloned into pET-28b (pET-28b-LSD1). Then the plasmid pET-28b-LSD1 was transfected into BL21 (DE). The recombinant protein was induced with 0.25 mmol/L IPTG at 20 °C and purified following affinity chromatography, ion exchange chromatography and gel filtration. Then the compounds were incubated with the 5 nmol/L recombinant LSD1 and 25 μmol/L H3K4me2 peptide in the present of FAD (50 nmol/L), Amplex Red (20 nmol/L) and horseradish peroxidase (5.5 U/mL) for 30 min. After that, the fluorescence was measured at excitation wavelength 530 nm and emission wavelength 590 nm as reported in order to evaluate the inhibition rate of the candidate compounds. In the control experiment, assay with specific concentration of H2O2 coupled with Amplex Red and HRP was performed to exclude the false positive result.
For the competitive analysis of the candidate compound, the demethylase activity of LSD1 was assessed in the presence of different concentrations of the compound (0, 0.125, 0.25 and 0.5 μmol/L) at a fixed concentration of FAD (2.5 μmol/L) and peptide concentrations from 0 to 60 μmol/L. Assays were performed triplicate, and kinetics values were obtained using Lineweaver–Burk plots63 made by GraphPad 6.0 (GraphPad Software Inc., San Diego, CA, USA).
4.2.2. Inhibitory evaluation of compound 15u against MAO-A/B
The MAO-A/B were purchased from Active Motif (Cat#31502, Cat#31503). Biochemical kits were purchased from Promega (MAO-Glo Assay, V1402). Inhibition assay was carried out according to the manufacturer׳s protocol. The tested compound solution was transferred into a 384-well plate in duplicate, then incubated with 10 μL of recombinant MAO-A or MAO-B solutions at rt for 15 min (the final concentration was 15 and 20 nmol/L respectively), followed by adding 10 μL of luciferin derivative substrate (the final concentration is 10 μmol/L) to initiate the reaction. After incubation for 60 min at rt, the reporter luciferase detection reagent (20 μL) was added and incubated with each reaction for an additional 20 min. Relative light units (RLU) were detected using plate reader.
4.2.3. BTK enzyme assay
BTK was purchased from Carna Biosciences. The kinases (5 nmol/L) were assayed with 5 μL tested compounds, 3 μmol/L peptide2 (5-FAM-EAIYAAPFAKKK), 90 μmol/L ATP, and reaction buffer (50 mmol/L HEPES, pH 7.5, 0.0015% Brij-35, 10 mmol/L MgCl2, 2 mmol/L DTT). After incubation at 28 oC for 60 min, the reactions were stopped by adding 25 μL stop buffer (100 mmol/L HEPES, pH 7.5, 0.015% Brij-35, 50 mmol/L EDTA). The reaction mixture was analyzed on Caliper, and the readout values were converted to inhibition values.
4.2.4. CDKs enzyme assay
CDK1/cyclinB, CDK2/CycA2, CDK7/cyclinH/MAT1 and CDK9/cyclinT1 kinase were purchased from Millipore, and CDK4/CycD3, CDK6/cycD3 were purchased from Carna. The kinases were assayed with 3 μmol/L peptide8 (5-FAM-IPTSPITTTYFFFKKK-COOH for CDK4 and CDK6), peptide18 (5-FAM-QSPKKG-CONH2 for CDK1 and CDK2), peptide CTD3 (5-FAM-ACSYSPTSPSYSPTSPSYSPTSPSKK for CDK7 and CDK9) in the 20/30/280/800/77/10 μmol/L ATP (for CDK1/CDK2/CDK4/CDK6/CDK7CDK9), and of the test compounds in a final volume of 5 μL, and the reaction buffer (50 mmol/L HEPES, pH 7.5, 0.0015% Brij-35, 10 mmol/L MgCl2, 2 mmol/L DTT). After incubation at 28 °C for 60 min, the reactions were stopped by adding 25 μL stop buffer (100 mmol/L HEPES, pH 7.5, 0.015% Brij-35, 50 mmol/L EDTA). The reaction mixture was analyzed on Caliper, and the readout values were converted to inhibition values.
4.2.5. Reversibility analysis
The dilution assay was done as published64. Briefly, an amount of 2.5 μg of recombinant LSD1 was incubated with 312.5 μmol/L compound 15u , 600 μmol/L GSK2879552, or DMSO. At 1 h later, 1.25 μL aliquots were removed from all samples and diluted into HRP-assay solution containing substrate and coupling reagents to a final volume of 100 μL. This represents an 80-fold dilution of the inhibitor concentration, which is expected to yield the same inhibition rate for an irreversible inhibitor or significant difference for a reversible inhibitor.
4.2.6. Morphological analysis
The morphological analysis of THP-1 cells differentiation, cytospins were prepared by centrifugation in 150 μL PBS at a speed of 300 rpm for 5 min. The cytospun slides were stained at rt with hematoxylin and eosin (Solarbio), and cellular morphology was examined using a microscope with a camera (Leica, Wetzlar, Germany).
4.2.7. Cell viability assay
The MTS/PMS method was used to evaluate the inhibition of cancer cell proliferation. Cells were treated with various concentrations of the test compounds. After the incubation of 6 d, MTS assay was performed using CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega, USA) according to the manufacturer׳s instructions. At least three independent experiments were performed for statistics. All data were represented as mean±SD, n=3.
4.2.8. Colony formation assay
THP-1 cells (500 cells/well) were seeded in 24-well plate, 1:1 mixture of RPMI 1640 containing 10% FBS (BI) and Methylcellulose-based medium as the culture medium. Cells were treated with compound 15u at indicated concentrations for 8 days. Then cells were stained with DAPI (Sigma) according to the manufacturer׳s instructions and washed with PBS. The colony formation was analyzed using High Content analyzer (ThermoFisher, Waltham, MA, USA).
4.2.9. Flow-cytometry
Over 8 days of incubation with indicated concentrations of compound 15u or 0.1% DMSO, 1 × 106 THP-1 cells were treated in the dark with PE-conjugated CD11b antibody (abcam, ab28101) or FITC-conjugated CD86 antibody (abcam, ab77131) at 4 °C for 30 min. The expression of CD11b and CD86 was analyzed by FACSCanto™II flow cytometer (BD Biosciences, San Jose, CA, USA) and the flow cytometry data was analyzed using Flowjo.
4.3. Molecular modeling simulations and docking simulations
The structures of the studied ligands were built up by MOE 2015.10 and minimized using Amber10:EHT force field. The crystal structure of LSD1 in complex with an H3K4 peptide (PDB code: 2V1D) was prepared by QuickPrep module using the default parameters. The H3K4 peptide binding pocket was set as the docking site in this study. Induced fit docking simulations were carried out to predict the possible interactions for the receptor–ligand system using the Dock module of MOE 2015.10. During the docking simulations, all the side chains of residues surrounding the defined binding site were regarded as rotatable bonds, and the ligand and its single bonds were allowed to move freely within the potential binding pocket. After the docking simulations, the 20 best-scored ligand–protein complexes of each ligand were kept for further analyses. The result of our induced fit docking simulations showed that the best-scored conformations of our ligands were similar. Therefore, only the LSD1–ligand complex of the most active compound 15u was selected to analyze the interaction mode.
Acknowledgments
This work was supported by the National Key Research Program of Proteins (Nos. 2016YFA0501800 and 2017YFD0501401, China); the National Natural Science Foundation of China (Nos. 81703326, 81773562, 81430085 and 21403200, China); the Open Fund of State Key Laboratory of Pharmaceutical Biotechnology, Nan-jing University, China (No. KF-GN-201902, China); Outstanding Young Talent Research Fund of Zhengzhou University (No. 1521331002, China); Scientific Program of Henan Province (Nos. 182102310123 and 161100310100, China), China Postdoctoral Science Foundation (No. 2018M630840, China), Key Research Program of Higher Education of Henan Province (Nos. 15A350018 and 18B350009, China), and the Starting Grant of Zhengzhou University (No. 32210533, China).
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
Supporting data associated with this article can be found in the online version at doi:10.1016/j.apsb.2019.01.001.
Contributor Information
Bin Yu, Email: yubin@zzu.edu.cn.
Yichao Zheng, Email: yichaozheng@zzu.edu.cn.
Hongmin Liu, Email: liuhm@zzu.edu.cn.
Appendix A. Supplementary material
Supplementary material.
.
References
- 1.Kooistra S.M., Helin K. Molecular mechanisms and potential functions of histone demethylases. Nat Rev Mol Cell Biol. 2012;13:297–311. doi: 10.1038/nrm3327. [DOI] [PubMed] [Google Scholar]
- 2.Niu Y., Wan A., Lin Z., Lu X., Wan G. N6-methyladenosine modification: a novel pharmacological target for anti-cancer drug development. Acta Pharm Sin B. 2018;8:833–843. doi: 10.1016/j.apsb.2018.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greer E.L., Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13:343–357. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lohse B., Kristensen J.L., Kristensen L.H., Agger K., Helin K., Gajhede M. Inhibitors of histone demethylases. Bioorg Med Chem. 2011;19:3625–3636. doi: 10.1016/j.bmc.2011.01.046. [DOI] [PubMed] [Google Scholar]
- 5.McAllister T.E., England K.S., Hopkinson R.J., Brennan P.E., Kawamura A., Schofield C.J. Recent progress in histone demethylase inhibitors. J Med Chem. 2016;59:1308–1329. doi: 10.1021/acs.jmedchem.5b01758. [DOI] [PubMed] [Google Scholar]
- 6.Paik W.K., Kim S. Enzymatic demethylation of calf thymus histones. Biochem Biophys Res Commun. 1973;51:781–788. doi: 10.1016/0006-291x(73)91383-1. [DOI] [PubMed] [Google Scholar]
- 7.Shi Y., Lan F., Matson C., Mulligan P., Whetstine J.R., Cole P.A. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 8.Metzger E., Wissmann M., Yin N., Müller J.M., Schneider R., Peters A.H. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437:436–439. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- 9.Garcia-Bassets I., Kwon Y.S., Telese F., Prefontaine G.G., Hutt K.R., Cheng C.S. Histone methylation-dependent mechanisms impose ligand dependency for gene activation by nuclear receptors. Cell. 2007;128:505–518. doi: 10.1016/j.cell.2006.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang J., Hevi S., Kurash J.K., Lei H., Gay F., Bajko J. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat Genet. 2009;41:125–129. doi: 10.1038/ng.268. [DOI] [PubMed] [Google Scholar]
- 11.Ooi S.K., Qiu C., Bernstein E., Li K., Jia D., Yang Z. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–717. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ciccone D.N., Su H., Hevi S., Gay F., Lei H., Bajko J. KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature. 2009;461:415–418. doi: 10.1038/nature08315. [DOI] [PubMed] [Google Scholar]
- 13.Huang J., Sengupta R., Espejo A.B., Lee M.G., Dorsey J.A., Richter M. p53 is regulated by the lysine demethylase LSD1. Nature. 2007;449:105–108. doi: 10.1038/nature06092. [DOI] [PubMed] [Google Scholar]
- 14.Kontaki H., Talianidis I. Lysine methylation regulates E2F1-induced cell death. Mol Cell. 2010;39:152–160. doi: 10.1016/j.molcel.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 15.Rudolph T., Yonezawa M., Lein S., Heidrich K., Kubicek S., Schäfer C. Heterochromatin formation in Drosophila is initiated through active removal of H3K4 methylation by the LSD1 homolog SU(VAR)3-3. Mol Cell. 2007;26:103–115. doi: 10.1016/j.molcel.2007.02.025. [DOI] [PubMed] [Google Scholar]
- 16.McGraw S., Vigneault C., Sirard M.A. Temporal expression of factors involved in chromatin remodeling and in gene regulation during early bovine in vitro embryo development. Reproduction. 2007;133:597–608. doi: 10.1530/REP-06-0251. [DOI] [PubMed] [Google Scholar]
- 17.Di Stefano L., Ji J.Y., Moon N.S., Herr A., Dyson N. Mutation of Drosophila Lsd1 disrupts H3-K4 methylation, resulting in tissue-specific defects during development. Curr Biol. 2007;17:808–812. doi: 10.1016/j.cub.2007.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang G.G., Allis C.D., Chi P. Chromatin remodeling and cancer, part I: covalent histone modifications. Trends Mol Med. 2007;13:363–372. doi: 10.1016/j.molmed.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 19.Di Stefano B., Collombet S., Jakobsen J.S., Wierer M., Sardina J.L., Lackner A. C/EBPα creates elite cells for iPSC reprogramming by upregulating Klf4 and increasing the levels of Lsd1 and Brd4. Nat Cell Biol. 2016;18:371–381. doi: 10.1038/ncb3326. [DOI] [PubMed] [Google Scholar]
- 20.Maes T., Mascaró C., Ortega A., Lunardi S., Ciceri F., Somervaille T.C. KDM1 histone lysine demethylases as targets for treatments of oncological and neurodegenerative disease. Epigenomics. 2015;7:609–626. doi: 10.2217/epi.15.9. [DOI] [PubMed] [Google Scholar]
- 21.Habibi E., Masoudi-Nejad A., Abdolmaleky H.M., Haggarty S.J. Emerging roles of epigenetic mechanisms in Parkinson׳s disease. Funct Integr Genom. 2011;11:523–537. doi: 10.1007/s10142-011-0246-z. [DOI] [PubMed] [Google Scholar]
- 22.Kahl P., Gullotti L., Heukamp L.C., Wolf S., Friedrichs N., Vorreuther R. Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res. 2006;66:11341–11347. doi: 10.1158/0008-5472.CAN-06-1570. [DOI] [PubMed] [Google Scholar]
- 23.Gao L., Alumkal J. Epigenetic regulation of androgen receptor signaling in prostate cancer. Epigenetics. 2010;5:100–104. doi: 10.4161/epi.5.2.10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Magerl C., Ellinger J., Braunschweig T., Kremmer E., Koch L.K., Höller T. H3K4 dimethylation in hepatocellular carcinoma is rare compared with other hepatobiliary and gastrointestinal carcinomas and correlates with expression of the methylase Ash2 and the demethylase LSD1. Hum Pathol. 2010;41:181–189. doi: 10.1016/j.humpath.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y., Zhang H., Chen Y., Sun Y., Yang F., Yu W. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell. 2009;138:660–672. doi: 10.1016/j.cell.2009.05.050. [DOI] [PubMed] [Google Scholar]
- 26.Lv T., Yuan D., Miao X., Lv Y., Zhan P., Shen X. Over-expression of LSD1 promotes proliferation, migration and invasion in non-small cell lung cancer. PLoS One. 2012;7:e35065. doi: 10.1371/journal.pone.0035065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schulte J.H., Lim S., Schramm A., Friedrichs N., Koster J., Versteeg R. Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapy. Cancer Res. 2009;69:2065–2071. doi: 10.1158/0008-5472.CAN-08-1735. [DOI] [PubMed] [Google Scholar]
- 28.Lokken A.A., Zeleznik-Le N.J. Breaking the LSD1/KDM1A addiction: therapeutic targeting of the epigenetic modifier in AML. Cancer Cell. 2012;21:451–453. doi: 10.1016/j.ccr.2012.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yokoyama A., Takezawa S., Schüle R., Kitagawa H., Kato S. Transrepressive function of TLX requires the histone demethylase LSD1. Mol Cell Biol. 2008;28:3995–4003. doi: 10.1128/MCB.02030-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ding J., Zhang Z.M., Xia Y., Liao G.Q., Pan Y., Liu S. LSD1-mediated epigenetic modification contributes to proliferation and metastasis of colon cancer. Br J Cancer. 2013;109:994–1003. doi: 10.1038/bjc.2013.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Højfeldt J.W., Agger K., Helin K. Histone lysine demethylases as targets for anticancer therapy. Nat Rev Drug Discov. 2013;12:917–930. doi: 10.1038/nrd4154. [DOI] [PubMed] [Google Scholar]
- 32.Sareddy G.R., Viswanadhapalli S., Surapaneni P., Suzuki T., Brenner A., Vadlamudi R.K. Novel KDM1A inhibitors induce differentiation and apoptosis of glioma stem cells via unfolded protein response pathway. Oncogene. 2017;36:2423–2434. doi: 10.1038/onc.2016.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He Y., Zhao Y., Wang L., Bohrer L.R., Pan Y., Wang L. LSD1 promotes S-phase entry and tumorigenesis via chromatin co-occupation with E2F1 and selective H3K9 demethylation. Oncogene. 2018;37:534–543. doi: 10.1038/onc.2017.353. [DOI] [PubMed] [Google Scholar]
- 34.Somervaille T., Salamero O., Montesinos P., Willekens C., Simon J.A., Pigneux A. Safety, phamacokinetics (PK), pharmacodynamics (PD) and preliminary activity in acute leukemia of Ory-1001, a first-in-Class inhibitor of lysine-specific histone demethylase 1A (LSD1/KDM1A): initial results from a first-in-human phase 1 study. Blood. 2016;128:4060. [Google Scholar]
- 35.Zheng Y.C., Yu B., Chen Z.S., Liu Y., Liu H.M. TCPs: privileged scaffolds for identifying potent LSD1 inhibitors for cancer therapy. Epigenomics. 2016;8:651–666. doi: 10.2217/epi-2015-0002. [DOI] [PubMed] [Google Scholar]
- 36.Lee S.H., Liu X.M., Diamond M., Dostalik V., Favata M., He C. The evaluation of INCB059872, an FAD-directed inhibitor of LSD1, in preclinical models of human small cell lung cancer. Cancer Res. 2016;76:4704. [Google Scholar]
- 37.Fu X., Zhang P., Yu B. Advances toward LSD1 inhibitors for cancer therapy. Future Med Chem. 2017;9:1227–1242. doi: 10.4155/fmc-2017-0068. [DOI] [PubMed] [Google Scholar]
- 38.Mould D.P., McGonagle A.E., Wiseman D.H., Williams E.L., Jordan A.M. Reversible inhibitors of LSD1 as therapeutic agents in acute myeloid leukemia: clinical significance and progress to date. Med Res Rev. 2015;35:586–618. doi: 10.1002/med.21334. [DOI] [PubMed] [Google Scholar]
- 39.Sorna V., Theisen E.R., Stephens B., Warner S.L., Bearss D.J., Vankayalapati H. High-throughput virtual screening identifies novel N′-(1-phenylethylidene)-benzohydrazides as potent, specific, and reversible LSD1 inhibitors. J Med Chem. 2013;56:9496–9508. doi: 10.1021/jm400870h. [DOI] [PubMed] [Google Scholar]
- 40.Hitchin J.R., Blagg J., Burke R., Burns S., Cockerill M.J., Fairweather E.E. Development and evaluation of selective, reversible LSD1 inhibitors derived from fragments. MedChemComm. 2013;4:1513–1522. [Google Scholar]
- 41.Mould D.P., Alli C., Bremberg U., Cartic S., Jordan A.M., Geitmann M. Development of (4-cyanophenyl)glycine derivatives as reversible inhibitors of lysine specific demethylase 1. J Med Chem. 2017;60:7984–7999. doi: 10.1021/acs.jmedchem.7b00462. [DOI] [PubMed] [Google Scholar]
- 42.Johnson NW. The identification of GSK2879552, a mechanism based irreversible inhibitor of the histone lysine demethylase LSD1. In: XXIV EFMC international symposium on medicinal chemistry. Manchester, UK. Cambridge: Royal society of chemsitry; Aug 28—Sep 1 2016.
- 43.Zheng Y.C., Duan Y.C., Ma J.L., Xu R.M., Zi X., Lv W.L. Triazole-dithiocarbamate based selective lysine specific demethylase 1 (LSD1) inactivators inhibit gastric cancer cell growth, invasion, and migration. J Med Chem. 2013;56:8543–8560. doi: 10.1021/jm401002r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma L.Y., Zheng Y.C., Wang S.Q., Wang B., Wang Z.R., Pang L.P. Design, synthesis, and structure—activity relationship of novel LSD1 inhibitors based on pyrimidine-thiourea hybrids as potent, orally active antitumor agents. J Med Chem. 2015;58:1705–1716. doi: 10.1021/acs.jmedchem.5b00037. [DOI] [PubMed] [Google Scholar]
- 45.Ye X.W., Zheng Y.C., Duan Y.C., Wang M.M., Yu B., Ren J.L. Synthesis and biological evaluation of coumarin-1,2,3-triazole-dithiocarbamate hybrids as potent LSD1 inhibitors. MedChemComm. 2014;5:650–654. [Google Scholar]
- 46.Li Z.H., Liu X.Q., Geng P.F., Suo F.Z., Ma J.L., Yu B. Discovery of [1,2,3]triazolo[4,5-d]pyrimidine derivatives as novel LSD1 inhibitors. ACS Med Chem Lett. 2017;8:384–389. doi: 10.1021/acsmedchemlett.6b00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang S., Zhao L.J., Zheng Y.C., Shen D.D., Miao E.F., Qiao X.P. Design, synthesis and biological evaluation of [1,2,4]triazolo[1,5-a]pyrimidines as potent lysine specific demethylase 1 (LSD1/KDM1A) inhibitors. Eur J Med Chem. 2017;125:940–951. doi: 10.1016/j.ejmech.2016.10.021. [DOI] [PubMed] [Google Scholar]
- 48.Li Z.H., Yang D.X., Geng P.F., Zhang J., Wei H.M., Hu B. Design, synthesis and biological evaluation of [1,2,3]triazolo[4,5-d]pyrimidine derivatives possessing a hydrazone moiety as antiproliferative agents. Eur J Med Chem. 2016;124:967–980. doi: 10.1016/j.ejmech.2016.10.022. [DOI] [PubMed] [Google Scholar]
- 49.Wong R., Dolman S.J. Isothiocyanates from tosyl chloride mediated decomposition of in situ generated dithiocarbamic acid salts. J Org Chem. 2007;72:3969–3971. doi: 10.1021/jo070246n. [DOI] [PubMed] [Google Scholar]
- 50.Lima L.M., Barreiro E.J. Bioisosterism: a useful strategy for molecular modification and drug design. Curr Med Chem. 2005;12:23–49. doi: 10.2174/0929867053363540. [DOI] [PubMed] [Google Scholar]
- 51.Zhao H. Scaffold selection and scaffold hopping in lead generation: a medicinal chemistry perspective. Drug Discov Today. 2007;12:149–155. doi: 10.1016/j.drudis.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 52.Baell J.B., Holloway G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 53.Vymeˇtalová L., Kryštof V. Potential clinical uses of CDK inhibitors: lessons from synthetic lethality screens. Med Res Rev. 2015;35:1156–1174. doi: 10.1002/med.21354. [DOI] [PubMed] [Google Scholar]
- 54.Shi Q., Tebben A., Dyckman A.J., Li H., Liu C., Lin J. Purine derivatives as potent Bruton׳s tyrosine kinase (BTK) inhibitors for autoimmune diseases. Bioorg Med Chem Lett. 2014;24:2206–2211. doi: 10.1016/j.bmcl.2014.02.075. [DOI] [PubMed] [Google Scholar]
- 55.Cicenas J., Valius M. The CDK inhibitors in cancer research and therapy. J Cancer Res Clin Oncol. 2011;137:1409–1418. doi: 10.1007/s00432-011-1039-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Harris W.J., Huang X., Lynch J.T., Spencer G.J., Hitchin J.R., Li Y. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell. 2012;21:473–487. doi: 10.1016/j.ccr.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 57.Fang J., Ying H., Mao T., Fang Y., Lu Y., Wang H. Upregulation of CD11b and CD86 through LSD1 inhibition promotes myeloid differentiation and suppresses cell proliferation in human monocytic leukemia cells. Oncotarget. 2017;8:85085–85101. doi: 10.18632/oncotarget.18564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Feng Z., Yao Y., Zhou C., Chen F., Wu F., Wei L. Pharmacological inhibition of LSD1 for the treatment of MLL-rearranged leukemia. J Hematol Oncol. 2016;9:24. doi: 10.1186/s13045-016-0252-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McGrath J.P., Williamson K.E., Balasubramanian S., Odate S., Arora S., Hatton C. Pharmacological inhibition of the histone lysine demethylase KDM1A suppresses the growth of multiple acute myeloid leukemia subtypes. Cancer Res. 2016;76:1975–1988. doi: 10.1158/0008-5472.CAN-15-2333. [DOI] [PubMed] [Google Scholar]
- 60.Schenk T., Chen W.C., Göllner S., Howell L., Jin L., Hebestreit K. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med. 2012;18:605–611. doi: 10.1038/nm.2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fiskus W., Sharma S., Shah B., Portier B.P., Devaraj S.G., Liu K. Highly effective combination of LSD1 (KDM1A) antagonist and pan-histone deacetylase inhibitor against human AML cells. Leukemia. 2014;28:2155–2164. doi: 10.1038/leu.2014.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Culhane J.C., Wang D.Q., Yen P.M., Cole P.A. Comparative analysis of small molecules and histone substrate analogues as LSD1 lysine demethylase inhibitors. J Am Chem Soc. 2010;132:3164–3176. doi: 10.1021/ja909996p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lineweaver H., Burk D. The determination of enzyme dissociation constants. J Am Chem Soc. 1934;56:658–666. [Google Scholar]
- 64.Willmann D., Lim S., Wetzel S., Metzger E., Jandausch A., Wilk W. Impairment of prostate cancer cell growth by a selective and reversible lysine-specific demethylase 1 inhibitor. Int J Cancer. 2012;131:2704–2709. doi: 10.1002/ijc.27555. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material.