

SUMMARY
Alternatively activated (M2) macrophages promote wound healing but weaken antimicrobial defenses. The mechanisms that enforce macrophage divergence and dictate the phenotypic and metabolic characteristics of M2 macrophages remain elusive. We show that alternative activation with interleukin (IL)-4 induces expression of metallothionein 3 (MT3) that regulates macrophage polarization and function. MT3 was requisite for metabolic reprograming in IL-4-stimulated macrophages or M(IL-4) macrophages to promote mitochondrial respiration and suppress glycolysis. MT3 fostered an M(IL-4) phenotype, suppressed hypoxia inducible factor (HIF)1α activation, and thwarted the emergence of a proinflammatory M1 program in macrophages. MT3 deficiency augmented macrophage plasticity, resulting in enhanced interferon γ (IFNγ) responsiveness and a dampened M(IL-4) phenotype. Thus, MT3 programs the phenotype and metabolic fate of M(IL-4) macrophages.
Graphical Abstract
In Brief
Metabolic reprograming in interleukin (IL)-4-stimulated macrophages (M(IL-4) macrophages) have a distinct polarization and metabolic phenotype. Chowdhury et al. show that metallothionein 3 (MT3) is required for M(IL-4) polarization and oxidative metabolism. MT3 suppresses glycolysis and the emergence of a proinflammatory M1 program. MT3 is a gatekeeper that subverts interferon (IFNγ) responses and intracellular defenses in M(IL-4) macrophages.
INTRODUCTION
Macrophage polarization involves intricate programing pathways that generate phenotypically and functionally distinct subsets. Alternative activation differentiates macrophages to an M2 phenotype, with characteristics distinct from classically activated (M1) macrophages (Mosser and Edwards, 2008). M2 macrophages are anti-inflammatory, dampen defenses against intracellular pathogens, and preferentially utilize oxidative phosphorylation for energy, as opposed to glycolytic M1 macrophages (Galván-Peña and O’Neill, 2014; Mosser and Edwards, 2008). The mechanisms that define M2 macrophage characteristics and augment permissiveness to intracellular pathogens are not thoroughly understood and continue to be uncovered.
M2 macrophages constitute a heterogeneous population classified into at least three subsets induced by (1) interleukin-4 (IL-4), IL-13, or IL-33 signaling; (2) immune complex, Toll-like receptor (TLR) or IL-1 receptor signaling; and (3) IL-10, transforming growth factor-β (TGF-β) or glucocorticoids (Murray et al., 2014). IL-4- and IL-13-mediated activation of signal transducer and activator of transcription (STAT)6 shapes the M(IL-4) macrophage phenotype and upregulates the markers arginase-1 (Arg1), chitinase 3 like-3 (Chi3l3), resistin-like molecule alpha (Retnla) that encodes the adipokine RELMα, mannose receptor CD206, programmed death-ligand 2 (PD-L2), and transferrin receptor (TFRC) (Gordon, 2003; Weiss et al., 1997). The proposed mechanisms for permissiveness to intracellular pathogens include dampening of nitric oxide (NO) by Arg-1 and elevated TFRC that facilitates iron import (Gordon, 2003; Nairz et al., 2010). M1 macrophages, on the other hand, are induced by proinflammatory signals such as IFNγ, express mediators including NO synthase 2 (NOS2), and clear intracellular pathogens. While M1 macrophages resist transitioning to an M(IL-4) phenotype, the latter exhibit plasticity under the influence of M1 signals such as lipopolysaccharide (LPS) + IFNγ (Van den Bossche et al., 2016).
M1 and M(IL-4) macrophages diverge in metabolic preferences to meet differential energy demands for their functions (Galván-Peña and O’Neill, 2014). To conduct pro-fibrotic and tissue repair processes, M(IL-4) macrophages derive a prolonged energy supply from fatty acid breakdown to generate acetyl-coenzyme A (CoA) that feeds into the tricarboxylic acid (TCA) cycle, although this is dispensable for polarization (Van den Bossche and van der Windt, 2018; Divakaruni et al., 2018). However, M(IL-4) macrophages also utilize glucose to fuel the TCA cycle through pyruvate (Huang et al., 2016). Electrons generated in this cycle enter the mitochondrial respiratory chain to drive oxidative phosphorylation for ATP generation. Peroxisome proliferator-activated receptors PPARγ, PPARγ, coactivator-1β (PGC-1β), carbohydrate kinase-like protein CLARKL, and peroxisomal Acyl-CoA Oxidase 1 are some of the key regulators of oxidative metabolism (Vats et al., 2006; Chawla, 2010; Haschemi et al., 2012; Huang et al., 2012). Conversely, M1 macrophage activation is associated with aerobic glycolysis. These macrophages primarily derive energy by converting glucose to lactate. Metabolic reprogramming is intricately linked to macrophage antimicrobial effector functions. For example, elevated glycolysis in M1 macrophages rapidly supplies energy to mount proinflammatory responses to restrict the growth of Mycobacterium tuberculosis (Mtb), whereas M(IL-4) macrophages are permissive to the growth of Mtb. β-oxidation promotes the production of type-I IFNs that support Mtb growth inside macrophages (Huang et al., 2018). Despite the critical nature of the M1-M2 balance in inflammation, the molecular cues that direct pro- or anti-inflammatory macrophage fates and inhibit transition from one to another macrophage fate remain under-defined.
Metallothioneins (MTs) are divalent cation binding proteins whose expression is enhanced by stress or inflammatory stimuli. Proinflammatory activation of macrophages by granulocyte macrophage-colony stimulating factor (GM-CSF) induces MT1 and MT2, which curtails growth of the intracellular fungal pathogen, Histoplasma capsulatum by zinc (Zn) sequestration (Subramanian Vignesh et al., 2013). MT1 and MT2 are ubiquitously expressed and upregulated by excess Zn to limit metal intoxication. MT3 expression, however, is primarily restricted to the brain and a few other tissues (Palmiter et al., 1992; Faraonio et al., 2000). We showed that MT3 is expressed in the innate immune compartment, in macrophages, where it plays a crucial role. In response to IL-4 or IL-13, macrophages upregulate MT3 expression. MT3 elevates the labile Zn pool that is exploited for Zn acquisition and survival by H. capsulatum residing within macrophages (Subramanian Vignesh et al., 2016).
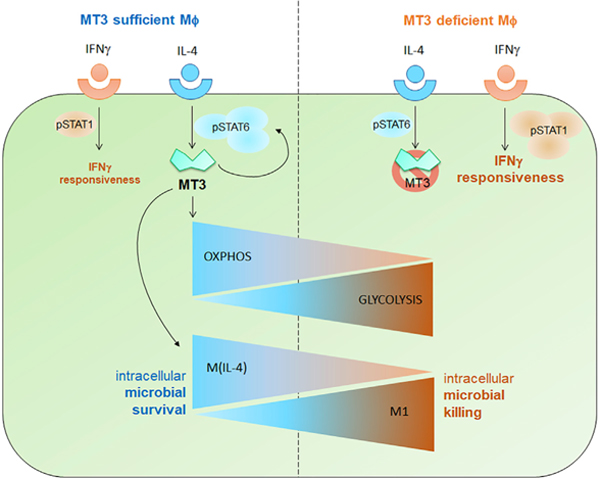
In this study, we report a previously unknown function of MT3 in macrophage biology. We show that MT3 regulates the phenotypic and metabolic signatures of M(IL-4) macrophage polarization. IL-4-driven MT3 was required for optimal expression of the M(IL-4) phenotype and suppression of macrophage plasticity to an M1 phenotype. We identified MT3 as a metabolic switch that suppressed glycolysis and directed the energy utilization preference of M(IL-4) macrophages to oxidative phosphorylation. Conversely, a lack of MT3 resulted in the transition of M(IL-4) macrophages to an M1 phenotype, lactate accumulation, and increased hypoxia inducible factor (HIF)1α activation. The lack of MT3 in an M(IL-4)-polarizing environment enhanced pro-inflammatory responsiveness to IFNγ and antibacterial defense against Escherichia coli. Our data unravel a crucial function of MT3 in shaping macrophage polarization and metabolism and open fresh avenues for exploring the function of MT3 in inflammatory pathologies driven by macrophages.
RESULTS
MT3 Regulates Alternatively Activated Macrophage Phenotypes
MT3 is induced by IL-4 and IL-13, but not by IFNγ, GM-CSF, IL-10, or IL-33 (Subramanian Vignesh et al., 2016). The specificity of this response urged us to determine if MT3 acted as a programing signal that directed macrophage polarization. Alternative activation with IL-4 strongly induced Mt3 in macrophages in agreement with our previous finding (Subramanian Vignesh et al., 2016) (Figure 1A). Induction of Mt3 expression occurred after 6 h and peaked at 24 h post IL-4 stimulation (Figure 1B). This correlated with elevated MT3 protein signal in M(IL-4) macrophages compared to unstimulated macrophages by immunostaining (Figure 1C). Next, we treated macrophages with scramble or Mt3 small interfering RNA (siRNA) for 24 h, followed by IL-4 stimulation for another 24 h. Mt3 was silenced in IL-4-stimulated macrophages by 91% compared to the scramble siRNA control (Figure 1D). Additional analysis revealed that Mt3 silencing modulated expression of the M(IL-4) markers Retnla and Tfrc, while Arg1 and Chi3l3 expressions were unaltered (Figure 1D). We further queried if expression of the M(IL-4) surface markers PD-L2 and CD206 were affected. IL-4 stimulation elevated the mean fluorescence intensity (MFI) and the proportion of PD-L2 and CD206 on macrophages, but these were reduced when Mt3 was silenced (Figure 1E). Thus, MT3 plays an important role in the optimal expression of the M(IL-4) phenotype.
Figure 1. MT3 Programs the Alternatively Activated Macrophage Phenotype.
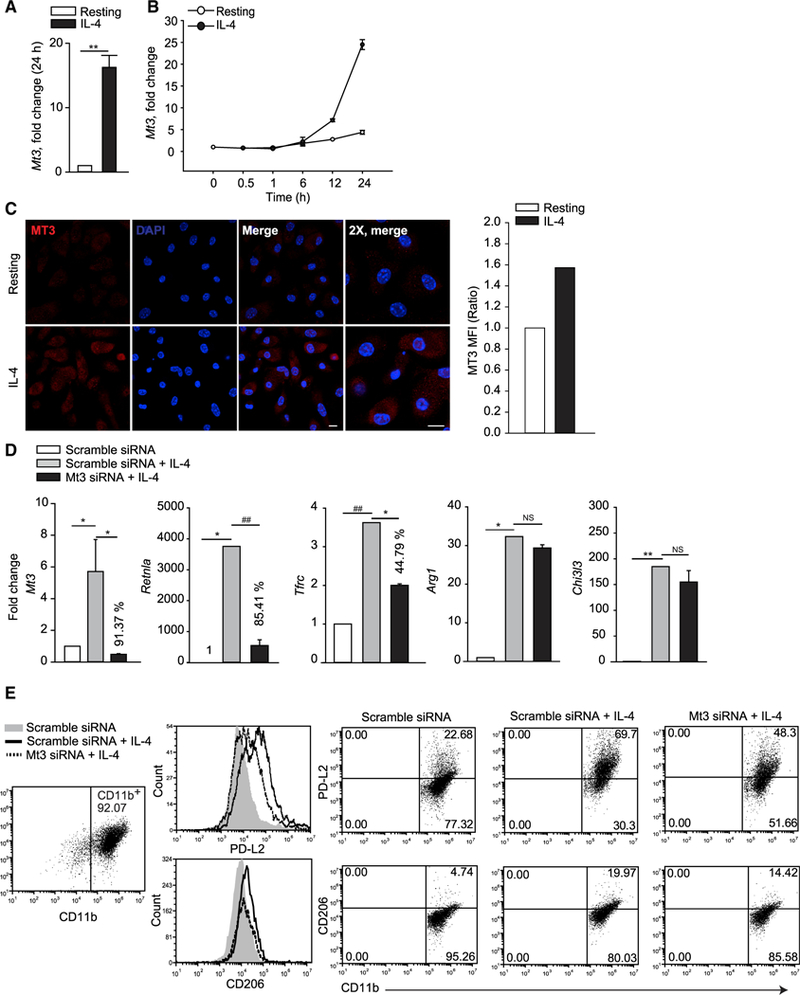
(A and B) Mt3 expression in macrophages (Mϕ) treated with IL-4 for 24 h (A) or indicated time points (B), three independent experiments, Mann-Whitney rank sum test (U statistic, 0).
(C) Immunostaining of MT3 in resting and IL-4-stimulated macrophages; cells were treated with IL-4 for 24 h, followed by change of media and re-stimulation with IL-4 for 9 h; 10 µm for scale bar; bar graph represents MFI of MT3 in IL-4-treated macrophages compared to resting cells; two independent experiments.
(D) Mt3, Retnla, Tfrc, Arg1, and Chi3l3 expression in IL-4-treated (24 h) Mt3-silenced macrophages compared to scramble siRNA treated control; percent values are decrease in gene expression compared to IL-4-treated scramble siRNA group; three independent experiments; one-way ANOVA (Holm-Sidak method).
(E) Histogram and dot plots of PD-L2 and CD206 gated on CD11b+ scramble siRNA or Mt3 siRNA treated macrophages 24 h post IL-4 stimulation; two independent experiments; data are mean ± SEM.
MT3 Programs the Metabolic Switch in Alternative Activation
We investigated how MT3 shaped the fundamental attributes of M(IL-4) macrophages. Since metabolic programing segregates with distinct macrophage polarization states, we queried if MT3 controlled the metabolic preferences of M(IL-4) macrophages. MT3 deficiency enhanced lysosomal acidification and reduced extracellular pH (5 proton µ-equivalents/106 cells) in IL-4-treated macrophages, suggesting that MT3 suppresses lysosomal and extracellular acidification in the M(IL-4) macrophages (Figures 2A and 2B). We determined if the decrease in extracellular pH was caused by lysosomal acidification. Treatment of MT3-deficient macrophages with chloroquine, a lysosomotropic agent, did not increase extracellular pH (Figure S1A).
Figure 2. MT3 Functions as a Metabolic Switch in M(IL-4) Polarization.
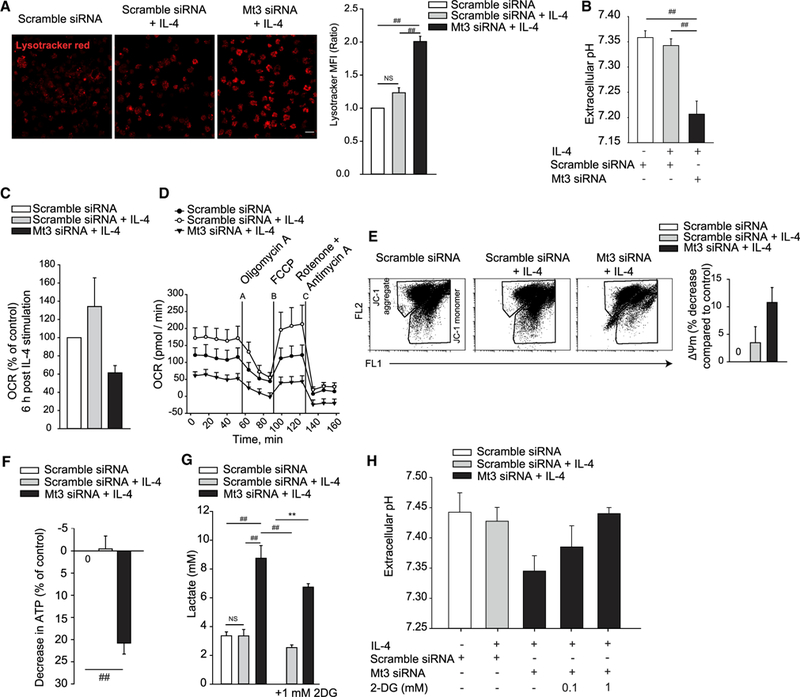
(A) Confocal images of lysotracker red stained macrophages (Mϕ) 24 h post IL-4 stimulation; 20 µm for scale bar; bar graph represents MFI of lysotracker staining compared to scramble siRNA treated control macrophages; three independent experiments; one-way ANOVA (Holm-Sidak method).
(B) Extracellular pH in culture supernatants of Mt3-silenced macrophages 24 h post IL-4 stimulation; three to seven independent experiments; one-way ANOVA (Holm-Sidak method).
(C) OCR-measured 6 h post IL-4 stimulation; percent change compared to scramble siRNA treated control; three independent experiments.
(D) OCR-measured 24 h post IL-4 stimulation for 55 min, followed by OCR in response to sequential mitochondrial stress using the ATP synthesis inhibitor oligomycin A, oxidative phosphorylation uncoupler carbonyl cyanide 4 (trifluoromethoxy) phenylhydrazone (FCCP), and the electron transport chain inhibitors rotenone and antimycin A.
(E) Flow cytometry of mitochondrial membrane potential shown as JC-1 aggregates (FL2) and JC-1 monomers (FL1) 24 h post IL-4 stimulation; bar graph; percent decrease in membrane potential compared to unstimulated control; three independent experiments for the Mt3-silenced group and six independent experiments for the scramble siRNA and scramble siRNA + IL-4 groups.
(F) ATP analysis in macrophage 24 h post IL-4 stimulation; percent decrease compared to unstimulated control; three independent experiments; ANOVA on ranks (Tukey test).
(G) Lactate in culture supernatants 24 h post IL-4 stimulation in untreated cells and cells treated with the glycolysis inhibitor 2-DG; four independent experiments with 2-DG and six independent experiments in the absence of 2-DG; one-way ANOVA (Holm-Sidak method).
(H) Extracellular pH in scramble siRNA or Mt3 siRNA treated macrophages exposed to 2-DG during the culture period and stimulated with IL-4 for 24 h; four independent experiments; data are mean ± SEM.
See also Figure S1.
The change in extracellular acidification suggested the emergence of an altered metabolic program in MT3-deficient macrophages exposed to IL-4. We hypothesized that MT3 altered cellular metabolism in M(IL-4) macrophages. MT3 deficiency reduced the mitochondrial oxygen consumption rate (OCR) in IL-4-treated macrophages (Figure 2C). M(IL-4) macrophages exert increased mitochondrial spare respiratory capacity, a measure of cellular ability to respond to elevated stress (Huang et al., 2014). Silencing Mt3 dampened mitochondrial respiration and the OCR response to mitochondrial stress (Figure 2D). We queried if mitochondrial function was intact in M(IL-4) macrophages by analyzing the mitochondrial membrane potential and ATP generation. MT3-deficient macrophages exhibited reduced mitochondrial membrane potential assessed by decreased JC-1 dye aggregate to monomer ratio and declined ATP production (Figures 2E and 2F). We examined if a diminished oxidative phosphorylation state shifted the metabolic balance of M(IL-4) macrophages. Silencing Mt3 enforced a glycolytic phenotype in macrophages measured by elevated extracellular lactate, indicating that M(IL-4) polarization suppresses aerobic glycolysis through MT3 induction (Figure 2G). Inhibition of glycolysis using the hexokinase inhibitor 2-deoxyglucose (2-DG) curtailed lactate accumulation and extracellular acidosis (Figures 2G and 2H). The monocarboxylic acid transporters, MCT1 and MCT4, encoded by the Slc16a1 and Slc16a3 genes, respectively, transport lactate across plasma membranes (Halestrap and Wilson, 2012). IL-4-induced Slc16a1 expression in both the control and Mt3 silenced macrophages, but Slc16a3 expression was specifically elevated in MT3-deficient macrophages compared to the control macrophages (Figure S1B). These data reveal that Mt3 programs an energy utilization switch that dictates the metabolic preferences in M(IL-4) macrophages.
MT3 Thwarts HIF1α Activation to Promote the HIF2α Transcriptional Program
We asked whether MT3 was positioned as a checkpoint in preventing macrophage transition to M1 macrophages. HIF1α and HIF2α exert distinct roles in macrophage polarization. HIF1α is a transcriptional regulator associated with M1 activation and HIF2α regulates M(IL-4) macrophage function (Galván-Peña and O’Neill, 2014). IL-4 suppressed expression of the downstream target of HIF1α, vascular endothelial growth factor (Vegfa) (Forsythe et al., 1996), in an MT3-dependent manner (Figure 3A). This finding prompted us to determine if MT3 prevented HIF1α activation. IL-4-exposed MT3-deficient macrophages exhibited augmented HIF1α nuclear localization (Figure 3B) and elevated the expression of Nos2, a downstream target of HIF1α (Figure 3C). To determine if MT3 suppressed the M1 phenotype by regulating HIF1α, we silenced both Hif1a and Mt3 in wild-type (WT) macrophages (Figure S2A) or silenced Mt3 in macrophages derived from Lyz2Cre Hif1afl/fl mice that lack HIF1α in myeloid cells (Figure S2B). HIF1α deficiency in Mt3-silenced macrophages decreased Nos2 and Vegfa expression, indicating that MT3 thwarts HIF1α activation in M(IL-4) macrophages (Figures 3C and S2C).
Figure 3. MT3 Suppresses HIF1α Activation and M1 Phenotype in M(IL-4) Macrophages.
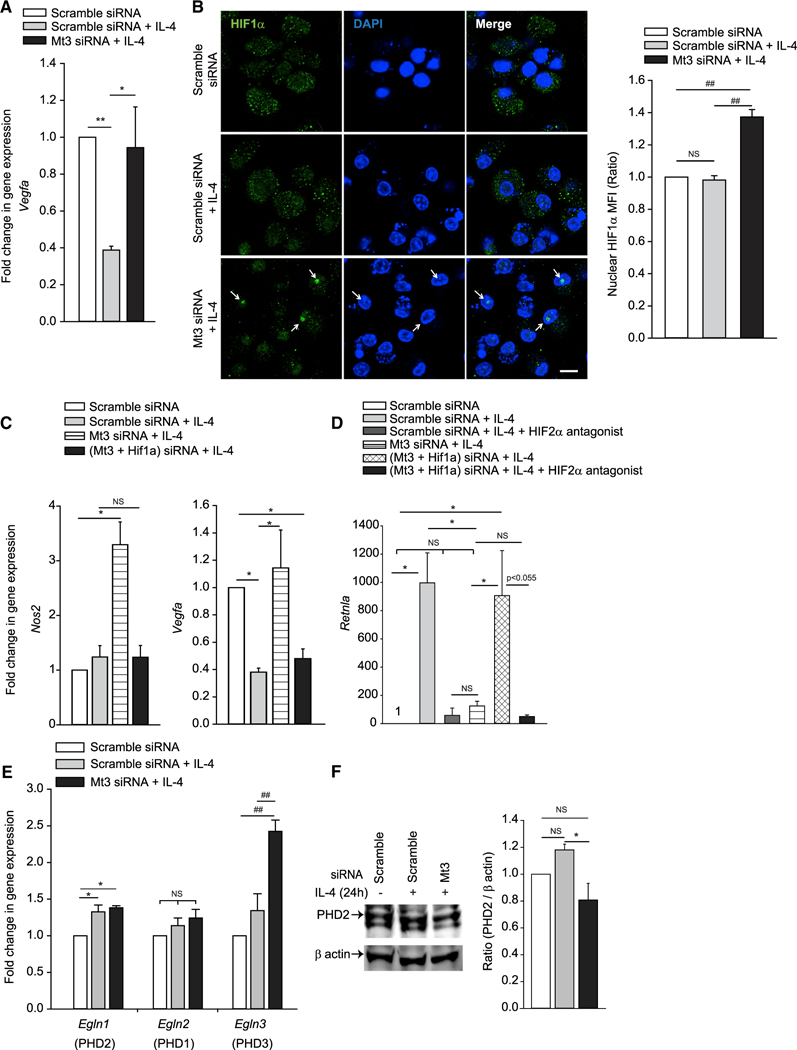
(A) Vegfa expression in Mt3-silenced or scramble siRNA treated macrophages (Mϕ) exposed to IL-4 for 24 h compared to unexposed macrophages; six independent experiments; one-way ANOVA (Holm-Sidak method).
(B) Nuclear localization of HIF1α 24 h post IL-4 stimulation; 10 µm for scale bar; arrows indicate nuclear HIF1α; bar graph represents MFI of nuclear HIF1α staining assessed from 107 to 113 nuclei in 24 to 29 different fields per group from four independent experiments; one-way ANOVA (Holm-Sidak method).
(C and D) Gene expression of (C) Nos2 and Vegfa in Mt3- and Hif1a-silenced macrophages 24 h post IL-4 stimulation; three independent experiments for Vegfa and five independent experiments for Nos2; ANOVA on ranks (Tukey and Dunn’s methods, respectively); and (D) Retnla in Mt3- and Hif1a-silenced macrophages 24 h post IL-4 stimulation and treated with 10-μM HIF2α antagonist; three independent experiments; one-way ANOVA (Holm-Sidak method).
(E) Egln gene expression in IL-4-stimulated macrophages; four independent experiments for Egln1 and 2, and five independent experiments for Egln3; one-way ANOVA (Holm-Sidak method).
(F) Western blot of PHD2 and β-actin 24 h post IL-4 stimulation; densitometry normalized to scramble siRNA treated macrophages; three independent experiments; one-way ANOVA (Holm-Sidak method); data are mean ± SEM.
See also Figure S2.
We probed whether MT3 promoted the HIF2α transcriptional program by suppressing HIF1α. Silencing Hif1a in MT3-deficient macrophages rescued the expression of Retnla, a HIF2α-dependent gene (Hickey et al., 2010). Antagonizing HIF2α in these macrophages abrogated Retnla expression, demonstrating that MT3 regulates the HIF1α-HIF2α balance in M(IL-4) macrophages (Figure 3D). Prolyl hydroxylases (PHD)2 and PHD3 preferentially degrade HIF1α and HIF2α, respectively (Appelhoff et al., 2004). Thus, to gain insight into how MT3 controlled HIF, we investigated if MT3 regulated the PHD2-PHD3 balance in M(IL-4) macrophages. MT3 silencing significantly elevated Egln3 (PHD3) expression (p < 0.001) that degrades HIF2α, while causing a marginal decrement in PHD2 protein that targets HIF1α (Figures 3E and 3F). These data indicate that MT3 governs the HIF1α-HIF2α balance to program divergent macrophage subsets.
Pyruvate Dehydrogenase Kinase Regulates Suppression of Glycolysis by MT3
HIF1α is an important transcriptional regulator of enzymes in the glycolytic pathway (Galván-Peña and O’Neill, 2014). We examined if HIF1α was responsible for the glycolytic phenotype of Mt3-silenced macrophages. We treated WT and Lyz2Cre Hif1afl/fl macrophages with scramble or Mt3 siRNA and exposed them to IL-4. Despite the lack of HIF1α, Mt3-silenced macrophages exposed to IL-4 elevated glycolysis, indicating that the metabolic shift was HIF1α independent (Figure 4A). We queried the molecular cues involved in the control of metabolic commitment by MT3. The activity of intracellular lactate dehydrogenase (LDH), which catalyzes the conversion of pyruvate to lactate, was comparable between scramble siRNA and Mt3 siRNA treated macrophages (Figure S3A). We further posited that MT3 regulated a stage that bridged pyruvate produced by glucose break-down to the TCA cycle. The enzymes pyruvate kinase M2 (PKM2) and pyruvate dehydrogenase (PDH) control metabolic transition from glycolysis to TCA (Tan et al., 2015b; Anastasiou et al., 2012). PKM2 catalyzes the conversion of phosphoenolpyruvate to pyruvate. PKM2 activation inhibits cellular dependence on glycolysis for energy provision and escalates oxidative phosphorylation (Anastasiou et al., 2012). PDH, whose activity is inhibited by pyruvate dehydrogenase kinase (PDK), bridges glycolysis and TCA by shunting pyruvate to acetyl-CoA that enters the TCA cycle (Tan et al., 2015b). Thus, we queried the role of these enzymes in the control of macrophage metabolism by MT3. We treated macrophages with the PKM2 activator, DASA-10, or the PDK inhibitor, dichloroacetate (DCA), and measured extracellular pH and lactate. PKM2 activation failed to reduce glycolysis, but PDK inhibition completely abrogated the glycolytic phenotype and extracellular acidification in MT3-deficient macrophages (Figures 4B, 4C, and S3B). These data demonstrate that MT3 suppresses glycolysis by controlling PDK function.
Figure 4. MT3-Deficient Macrophages Stabilize HIF1α through Lactate and PDK.
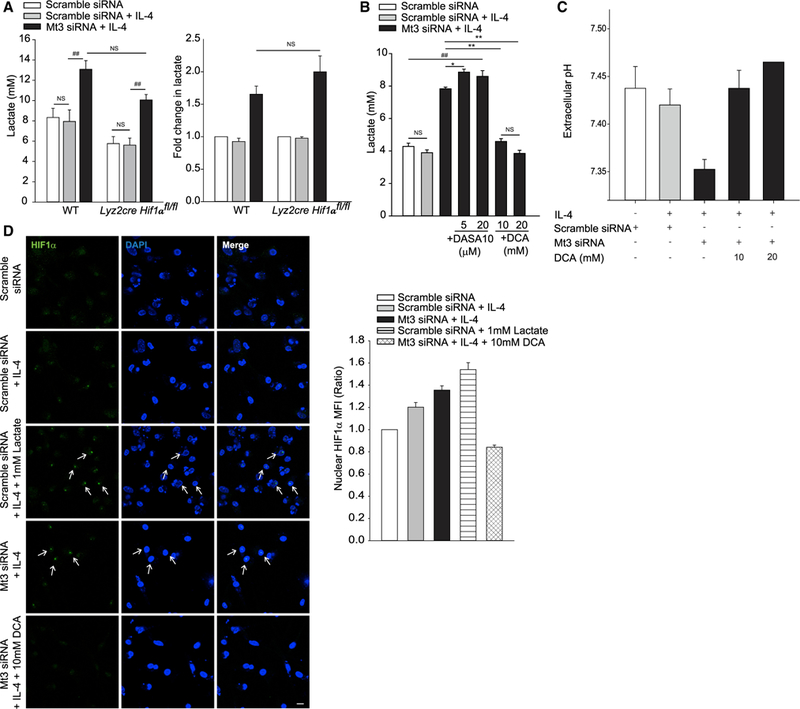
(A) (Left) Lactate in culture supernatants of WT and myeloid HIF1α deficient (Lyz2cre Hif1αfl/fl) macrophages (Mϕ) 24 h post IL-4 stimulation; one-way ANOVA (Holm-Sidak method). (Right) Fold change in lactate in WT and Lyz2cre Hif1αfl/fl macrophages compared to scramble siRNA treated macrophages of each group; four independent experiments.
(B) Lactate in culture supernatants of untreated, DASA-10, or DCA-treated macrophages stimulated with IL-4 for 24 h; four independent experiments; one-way ANOVA (Holm-Sidak method).
(C) Extracellular pH in supernatants of untreated and DCA-treated macrophages 24 h post IL-4 stimulation; two independent experiments with 20-mM DCA and four independent experiments for all other groups.
(D) Nuclear localization of HIF1α in cells treated with L-lactate or DCA throughout the culture period and stimulated with IL-4 for 24 h; 10 µm for scale bar; arrows indicate nuclear HIF1α; one representative of two experiments; bar graph represents MFI of nuclear HIF1α staining assessed from 49 to 71 nuclei in 8 to 10 different fields per group, data are mean ± SEM.
See also Figure S3.
Lactate stabilizes HIF1α by inhibiting PHD2 activity (De Saedeleer et al., 2012). We asked whether heightened HIF1α activation in MT3-deficient macrophages was a downstream effect of lactate accumulation caused by PDK. Lactate triggered HIF1α nuclear localization in M(IL-4)-polarized, scramble siRNA treated macrophages, similar to that observed in Mt3 siRNA treated macrophages. Conversely, PDK inhibition with DCA suppressed the nuclear accumulation of HIF1α in Mt3-silenced macrophages (Figure 4D). Thus, MT3 controls the metabolic switch in M(IL-4) macrophages by preventing emergence of an M1 glycolytic program and HIF1α activation occurs downstream of glycolysis in Mt3-silenced macrophages. Taken together, MT3 controls the lysosomal, mitochondrial, and metabolic facets of alternative activation to promote the M(IL-4) phenotype.
Genetic Deletion of MT3 Promotes a HIF1α-Dependent Transcriptional Signature in Macrophages
We generated C57BL/6 mice genetically deficient in exon 3 of the Mt3 gene using CRISPR/Cas9 (Figure 5A). Cell numbers in the thymus, spleen, and bone marrow of Mt3−/− mice were comparable to age-matched WT control mice (Figure S4A). WT and Mt3−/− mice also had similar proportions and numbers of thymic T cells, splenic T and B cells, dendritic cells (DCs), macrophages, and neutrophils (Figures S4B and S4C). GM-CSF differentiation yielded similar numbers of WT and Mt3−/− macrophages from the bone marrow (Figure S4D). IL-4 treatment of Mt3−/− macrophages failed to increase the intracellular labile Zn pool compared to WT macrophages, confirming that exon 3 deletion caused loss of MT3 function in Mt3−/− macrosssphages, in agreement with our previous findings (Subramanian Vignesh et al., 2016) (Figures S4E and S4F).
Figure 5. Mt3−/− Macrophages Exhibit a Glycolytic Phenotype, Increased HIF1α Activation, and a HIF1α-Dependent Transcriptional Signature.
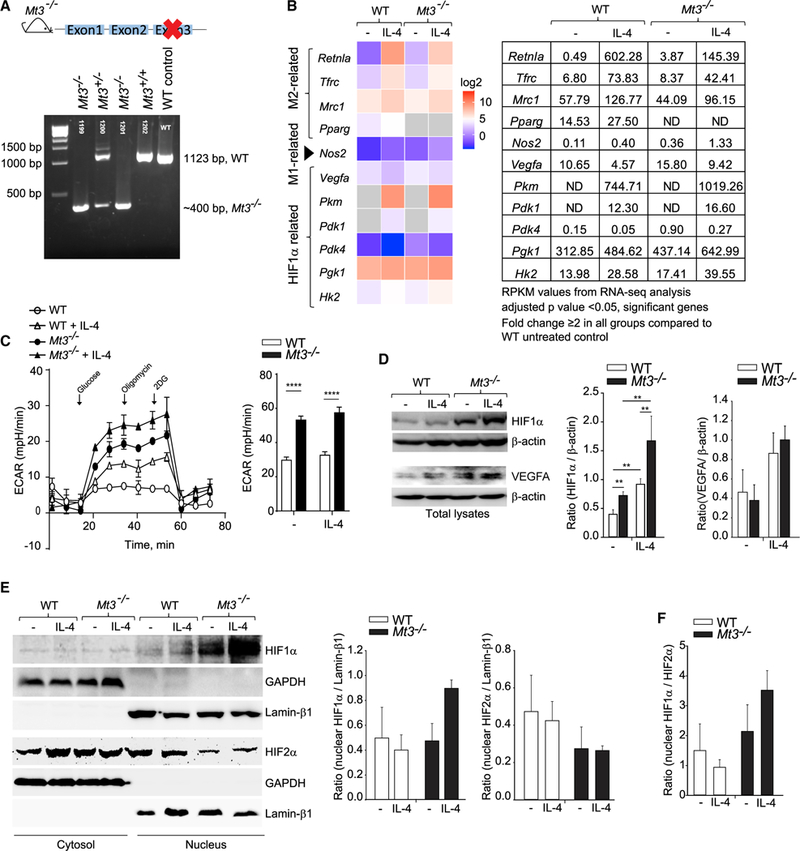
(A) Schematic of Mt3−/− mice on the C57BL/6 background obtained using CRISPR/Cas9; confirmation of deletion of exon 3 of the Mt3 gene by agarose gel electrophoresis of PCR products obtained by amplifying chromosomal DNA.
(B) Heatmap of differentially expressed M(IL-4)-, M1-, and HIF1α-related genes obtained by RNA-seq analysis of untreated and IL-4-treated (24 h) WT and Mt3−/− macrophages (Mϕ); data are log2 transformed and compared to untreated WT macrophages; fold change ≥ 2 in all other groups compared to WT macrophages; adjusted p < 0.05 (Benjamini-Hochberg) significant; n = 3 per group; table on right shows RPKM values (reads per kilobase of transcript per million mapped reads) of M1/M(IL-4)/HIF1α-related genes; ND, not detected.
(C) ECAR measured 24 h post IL-4 stimulation for 15 min, followed by ECAR in response to sequential treatment with glucose, oligomycin, and 2-DG; bar graph (right) shows glycolytic capacity of macrophages; three independent experiments; two-way ANOVA (Holm-Sidak’s multiple comparisons test).
(D) Western blots of HIF1α, VEGFA, and β-actin in total cell lysates of WT and Mt3−/− macrophages 24 h post IL-4 stimulation; three experiments for VEGFA and six experiments for HIF1α; one-way ANOVA on ranks (Student’s Newman-Keuls method).
(E) HIF1α and HIF2α in cytosolic and nuclear fractions of WT and Mt3−/− macrophages 24 h post IL-4 stimulation; GAPDH and lamin β1 are cytosolic and nuclear loading controls, respectively.
(F) Ratio of nuclear HIF1α/HIF2α; three independent experiments.
Bar graphs in (E) and (F) represent densitometric analysis of bands; data are mean ± SEM. See also Figures S4 and S5.
We queried if genetic MT3 deficiency promoted M(IL-4) macrophage plasticity and altered the metabolic phenotype of these macrophages. We performed RNA sequencing (RNA-seq) on GM-CSF-differentiated bone-marrow-derived WT and Mt3−/− macrophages left untreated or treated with IL-4 for 24 h. Similar to Mt3-silenced macrophages, IL-4- exposed Mt3−/− macrophages exhibited a subdued M(IL-4) transcriptional signature compared to IL-4-treated WT macrophages. We found reduced expression of the M(IL-4) markers Retnla, Tfrc, Mrc1 (gene encoding CD206), and Pparg (gene encoding PPARγ) in Mt3−/− macrophages in response to IL-4, while Nos2 expression was elevated (Figure 5B). Additionally, a HIF1α-dependent transcriptional signature was seen in Mt3−/− macrophages with elevated Vegfa, Pkm, Pdk1, Pdk4, phosphoglucokinase 1 (Pgk1), and hexokinase 2 (Hk2) expression (Figure 5B). Moreover, Mt3−/− macrophages left untreated or treated with IL-4 had an increased extracellular acidification rate (ECAR) compared to WT macrophages, indicating a shift toward glycolysis in these cells (Figure 5C). Concordant with our findings with Mt3-silenced macrophages, we found elevated HIF1α and VEGFA in both resting and IL-4-treated Mt3−/− macrophages compared to WT controls by western blot (Figure 5D). Cytosolic and nuclear fractionation of macrophages lysates revealed that the lack of MT3 promoted nuclear assimilation of HIF1α, but suppressed that of HIF2α in the presence of IL-4 (Figures 5E and 5F). Thus, M(IL-4) macrophages employ MT3 to guard against HIF1α stabilization. The Akt-mechanistic target of rapamycin complex 1 (mTOR) pathway is implicated in HIF1α stabilization (Majumder et al., 2004; Cheng et al., 2014). We queried whether the lack of MT3 impacted Akt or mTOR activation. Mt3−/− macrophages exhibited no changes in pAkt, pmTOR, and the downstream target p70S6K compared to WT macrophages (Figure S4G).
Macrophage-colony stimulating factor (M-CSF) is commonly used to prepare bone-marrow-derived macrophages. Thus, we assessed the role of MT3 in M(IL-4) macrophages derived by M-CSF differentiation. Similar to our findings with GM-CSF differentiation, macrophages lacking MT3 exhibited elevated glycolysis at baseline and with IL-4 treatment. Mt3−/− macrophages had increased basal OCR. However, while WT macrophages responded to IL-4 with a robust increase in OCR, Mt3−/− macrophages failed to exhibit this response (Figures S5A and S5B). We found a trend toward decreased M(IL-4) polarization marker Arg1 and an elevation in Nos2 expression in the absence of MT3, but this effect was modest (Figure S5C). Thus, while MT3 controls metabolism in M-CSF-derived M(IL-4) macrophages, its role in macrophage polarization may be influenced by the macrophage source. We examined if MT3 exerted its function on other macrophage sources such as tissue-derived macrophages. Alveolar macrophages from Mt3−/− mice exhibited a trend toward increased glycolysis and modestly decreased mitochondrial respiration (Figure S5D).
MT3 Suppresses IFNγ Responsiveness in Macrophages
We posited that if MT3 suppressed M(IL-4) macrophage plasticity, IL-4-conditioned Mt3−/− macrophages would exhibit pronounced responsiveness to IFNγ. WT and Mt3−/− macrophages were exposed to IL-4 for 24 h, followed by IFNγ for 24 h. IL-4- polarized WT macrophages induced Nos2 in response to IFNγ, but this response was higher in Mt3−/− macrophages. We also detected higher NO in culture supernatants of IFNγ-exposed Mt3−/− macrophages (Figures 6A and 6B). The activation of pSTAT1 by IFNγ was elevated, while IL-4-driven pSTAT6 activation was dampened in Mt3−/− macrophages compared to WT macrophages (Figures 6C and 6D). The decrement in pSTAT6 did not result from changes in the IL-4 receptor (IL-4Rα) in Mt3−/− macrophages (Figure S6A). To assess if Mt3−/− macrophages exhibited functional attributes of M1 polarization, we examined bacterial clearance by these cells. WT and Mt3−/− macrophages were treated with IL-4, followed by IFNγ and infected with E. coli strain K12. Mt3−/− macrophages treated with IL-4 or IFNγ alone or IL-4 + IFNγ exhibited enhanced bacterial clearance compared to WT macrophages treated with these cytokines (Figure 6E). This observation did not result from changes in bacterial uptake as WT and Mt3−/− macrophages exhibited comparable phagocytosis of E. coli (Figure S6B).
Figure 6. Genetic Deficiency of MT3 Increases IFNγ Responsiveness and Improves Microbial Clearance.
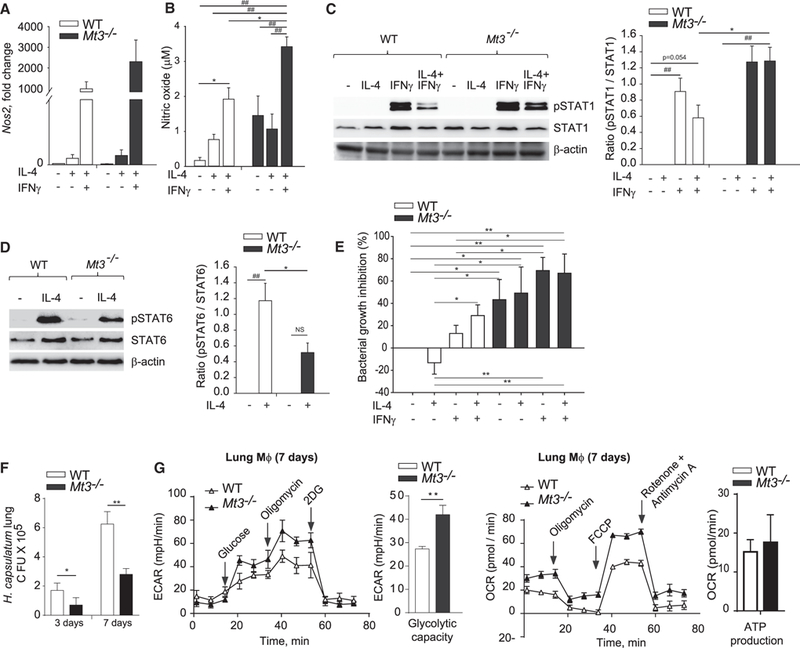
(A) Nos2 gene expression in macrophages (Mϕ) left untreated or treated with 10 ng/ml IL-4 for 24 h, followed by change of media and stimulation with 5 ng/ml IFNγ for 24 h; three independent experiments.
(B) NO in macrophages culture supernatants treated as above; three independent experiments; one-way ANOVA (Holm-Sidak method).
(C) Western blot of pSTAT1, total STAT1, and β-actin in WT and Mt3−/− macrophages left untreated or treated with IL-4 for 24 h, followed by IFNγ for 15 min; bar graphs represent densitometric analysis normalized to STAT1; one-way ANOVA (Holm-Sidak method); three independent experiments.
(D) pSTAT6, total STAT6, and β-actin in WT and Mt3−/− macrophages treated with IL-4 for 15 min; bar graphs represent densitometric analysis normalized to STAT6; one-way ANOVA (Holm-Sidak method); three independent experiments.
(F) E. coli growth inhibition in macrophages treated as indicated in experimental procedures; one-way ANOVA (Fisher least significant difference [LSD] method); three independent experiments.
(F) Colony-forming units (CFUs) in lungs of H. capsulatum infected mice on days 3 and 7 post infection; two-way ANOVA (Holm-Sidak’s method); n = 4 mice/group.
(G) ECAR and OCR analysis of macrophages obtained from H. capsulatum infected mice 7 days post infection; two-way ANOVA (Holm-Sidak’s method); n = 4 mice/group.
See also Figure S6.
We previously showed that MT3 promotes macrophages permissiveness to the intracellular fungal pathogen, H. capsulatum (Subramanian Vignesh et al., 2016). To extend the significance of our findings in vivo, we asked if fungal clearance would be enhanced in Mt3−/− mice as early as day 3 post infection, a time point at which the contribution of T cells is expected to be minimal. Lungs of Mt3−/− mice exhibited markedly enhanced clearance of H. capsulatum on days 3 and 7 post infection compared to WT mice (Figure 6F). Next, we examined the metabolic profile of macrophages from lungs of WT and Mt3−/− mice. On day 7, we did not detect changes in mitochondrial respiration, but macrophages from Mt3−/− mice manifested elevated glycolytic capacity compared to WT controls, indicating that MT3 dampens glycolysis in macrophages during fungal infection (Figure 6G). Collectively, these data indicate that MT3 thwarts proinflammatory macrophage activation and impairs antibacterial and antifungal defenses. Thus, MT3 programs the phenotypic and metabolic circuitry of M(IL-4) polarization and inhibits macrophage transition to a proinflammatory M1 phenotype.
DISCUSSION
Much is known about alternatively activated macrophage function, but the mechanisms that drive their plasticity remain incompletely understood. This study delved into understanding the central biology of M(IL-4) macrophages, and the results document MT3 as a critical regulatory module that defines the metabolic and phenotypic properties of alternative activation.
MT1 and MT2 are ubiquitously expressed in body tissues and are induced in immune cells in response to stress stimuli (Coyle et al., 2002). MT3, however, has long been considered to be restricted to the brain and has neuronal growth inhibitory functions (Palmiter et al., 1992). Other studies have identified MT3 in the testis, tongue, and cancer cells of the prostate, breast, and skin tissues (Hozumi et al., 2008; Kmiecik et al., 2015; Garrett et al., 1999; Slusser et al., 2015). Herein, we demonstrated that MT3 plays a fundamental role in shaping macrophage polarization. In M(IL-4) macrophages, MT3 regulates key manifestations that guide macrophage subset differentiation, phenotype, and metabolism.
Classical activation programs macrophages to express NOS2, while M(IL-4) programing leads to elevated ARG1, CD206, PD-L2, and RELMα (Gordon, 2003; Huber et al., 2010). Multiple facets of alternative activation depended on MT3. The M(IL-4) polarization program required MT3 for regulating RELMα, PD-L2, TFRC, and CD206, with the strongest impact on RELMα. M(IL-4)-driven RELMα expression is dependent on lysosomal lipolysis and β oxidation, indicating that breakdown of triacylglycerols into free fatty acids and their oxidation to acetyl-CoA regulates this gene (Huang et al., 2014). Mt3−/− astrocytes exhibit increased lysosomal acidification (Lee et al., 2010), and we found this role of MT3 applied to macrophages. These macrophages exhibited reduced dependence on oxidative phosphorylation as an energy source, a hallmark of the M(IL-4) metabolic phenotype. Notably, Arg1 and Chi3l3 were not affected, indicating that MT3 dictated specific attributes of the M(IL-4) phenotype, leaving others unaffected. For example, macrophages lacking MT3 retained intact Arg1 but elevated Nos2, perhaps representing a competitive mechanism for arginine utilization. Considering that MT3 is a Zn binding protein, the aspects of MT3 function attributable to Zn regulation remain to be determined. Recent studies have elucidated complex roles for increased intracellular free Zn, exogenous Zn supplementation, and Zn deficiency in vivo in negatively regulating M(IL-4) polarization (Kido et al., 2019; Aratake et al., 2018; Dierichs et al., 2018). Thus, it is plausible that changes in intracellular free Zn signals by MT3 influences macrophage polarization. Nonetheless, our data indicate that by interfering with MT3 regulation, macrophages yield to plasticity in a manner that disrupts defined aspects of the M(IL-4) program.
The failure to induce MT3 in response to type-2 cytokines resulted in a glycolytic transition of cellular metabolism with a characteristic extracellular acidosis phenotype. The lactate transporters MCT1 and MCT4 exhibit bidirectionality in shuttling monocarboxylic acids across plasma membranes. However, MCT1 has largely been associated with importation, while MCT4 functions as an exporter (Halestrap and Wilson, 2012). Likewise, M(IL-4) polarization enhanced Slc16a1 (MCT1) expression that imports lactate. The upregulation of Slc16a3 (MCT4), however, occurred exclusively in MT3-deficient macrophages in response to IL-4, indicating an intracellular lactate ‘‘overload’’ that must be shuttled into the extracellular space. MCT4, a downstream target of HIF1α, regulates proinflammatory macrophage responses to TLR stimulation and promotes glycolysis in these macrophages (Tan et al., 2015a; Ullah et al., 2006). Rather than merely resulting as a metabolic end product, lactate is a potent immunological signal that influences redox status, hypoxic responses, T cell migration and transcriptional programing through autocrine and paracrine effects (Haas et al., 2015; Lee et al., 2015; Yang et al., 2014b). From this perspective, by inducing MT3, M(IL-4) macrophages may enforce a check on extracellular lactate accumulation to favorably skew the immune response toward that of diminished inflammation and increased repair.
Macrophage metabolic preferences diverge with the polarization state. Elevated glycolytic rates supply the energy demands of proinflammatory activation, enabling M1 macrophages to kill extracellular bacteria and intracellular pathogens, while M(IL-4) macrophages preferentially utilize mitochondrial respiration as the energy source (Galván-Peña and O’Neill, 2014). This metabolic choice supports their long-term repair functions, collagen biosynthesis, longevity, and proliferation. We established that MT3 was crucial in the metabolic decision-making process, placing it at the center of the M(IL-4) functional signature. A decrement in MT3 shifted the energy preference to glucose metabolism, accompanied by a decline in O2 consumption. The emergence of MT3 as an essential component of metabolic rewiring has profound implications on understanding the functional attributes of M(IL-4) macrophages. For example, these cells populate the adipose tissue, where their metabolic phenotype drives the maintenance of lean adipose (Lumeng et al., 2007). A decline in the ratio of M2:M1 is implicated in obesity-mediated inflammation and the development of insulin resistance (Suganami and Ogawa, 2010). Our data open the ground for investigation in this area, whereby metabolic ‘‘switching’’ to oxidative phosphorylation potentiated by MT3 may impact key biological roles of macrophages in the inflamed tissue.
The shift in bioenergetic preference toward glycolysis reflects upon MT3 as a suppressor of the Warburg effect, characterized by heightened reliance on energy derivation from aerobic glycolysis even when oxygen is sufficient (Kelly and O’Neill, 2015). It is important to note that glucose is required for M(IL-4) polarization, but the pyruvate derived from it fuels the TCA cycle (Huang et al., 2016). The transition to glycolysis (lactate generation) under MT3 deficiency may appear energy inefficient, but caters to rapid energy production and the buildup of glycolytic intermediates for efficiently handling pathogen encounter and deploying antimicrobial defenses that are characteristic of M1 activation. Consistent with this view, despite M(IL-4) polarizing cues, both GM-CSF- and M-CSF-differentiated macrophages lacking MT3 transitioned to an M1 metabolic program. We noted an overall increase in the glycolytic capacity of GM-CSF-differentiated macrophages derived from both WT and Mt3−/− mice compared to M-CSF-differentiated macrophages. This observation may explain why M-CSF-differentiated macrophages induce glycolysis in response to IL-4, as reported (Huang et al., 2016), while GM-CSF-differentiated macrophages do not show a similar increase.
The HIF1α-HIF2α balance, which is tightly regulated by hypoxia and inflammatory stimuli, is recognized as a new dimension in skewing macrophage polarity (Galván-Peña and O’Neill, 2014). Our findings reveal an MT3-HIF regulatory circuit, whereby MT3 suppressed HIF1α activation to promote HIF2α, ultimately thwarting the emergence of a proinflammatory M1 phenotype. The decrease in M(IL-4) markers and corresponding rise in Nos2 expression resulting from MT3 deficiency demonstrate that commitment to M(IL-4) polarization requires constant MT3-driven inhibition of macrophage plasticity toward an M1 phenotype.
MT3 governed activation of the HIF regulatory circuit through control of the metabolite, lactate. The present data and previous reports confirm that lactate stabilizes HIF1α (De Saedeleer et al., 2012). Several glycolytic enzymes are direct HIF1α targets, marking this transcription factor as an important driver of glucose metabolism. A conundrum in the field, however, has been that though HIF1α may help perpetuate glycolysis in M1, the metabolic switch during M1 polarization occurs independent of HIF1α (Rodríguez-Prados et al., 2010). We report that this switch in macrophage metabolic reprogramming to glycolysis is suppressed by MT3. A change in the phosphofructokinase (PFK)2 isoform from L-PFK2 to u-PFK2 drives the shift to glycolysis in M1 (Rodríguez-Prados et al., 2010). Whether MT3 inhibits isoform switching in macrophages is unknown, but this protein has been reported to interact with several enzymes in the glycolytic pathway, implying functional control of metabolic regulation by MT3 (El Ghazi et al., 2010). Our data suggest that MT3 restrains the glycolytic shift by negatively regulating PDK. This mechanism promotes shunting of pyruvate into other metabolic processes, including conversion to acetyl-CoA by PDH (DeBerardinis et al., 2008). Thus, the decision to transition to aerobic glycolysis potentially necessitates two signals: (1) isoform switching to u-PFK2 and (2) reversing an MT3-imposed negative regulation on PDK (Rodríguez-Prados et al., 2010). Accordingly, PDK1 guides macrophage differentiation to an M1 phenotype (Tan et al., 2015b). The macrophage phenotypic and metabolic states are linked to their antimicrobial effector function (O’Neill and Pearce, 2016). M1 macrophages combat extracellular bacteria and intracellular fungi, while M(IL-4) macrophages poorly defend internalized pathogens. The inability of Mt3−/− macrophages to fully express an M(IL-4) phenotype, while manifesting increased IFNγ responsiveness and bacterial, as well as fungal clearance, indicates that MT3 skews macrophage polarization and obviates M(IL-4) macrophage plasticity.
Collectively, here we identified MT3 as a central regulator of M(IL-4) polarization. These macrophages turn on the ‘‘MT3 metabolic switch’’ that crucially regulates energy fate decisions. MT3 leverages a dual impact on macrophage metabolism and phenotype, making it a central checkpoint in the M(IL-4) polarization program. The finding that MT3 has a fundamental role in the innate compartment has implications for uncovering immune responses in both normal and disease processes controlled by M2 macrophages. Our observations may be extended to understanding MT3 functions in type-2 immunity-associated conditions including asthma, parasitic infections, wound healing, and metabolic disease. Finally, MT3 is at the forefront of the mechanism by which IL-4 propels the balance toward M2, directing against macrophage transition to an M1 phenotype. Thus, our findings illuminate an MT3 regulatory nexus that forms a critical component of the phenotypic, metabolic, and functional characteristics of macrophage activation.
STAR ★ METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and request for reagents may be directed to and will be fulfilled by the lead contact, Kavitha Subramanian Vignesh (Kavitha.Subramanian@uc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial Strain
E. coli (K12) was used to determine antibacterial defenses of macrophages in vitro. This strain was kindly provided by Dr. Jason Gardner at University of Cincinnati.
Fungal Strain
The strain G217B of H. capsulatum was used to analyze immune responses of wild-type and Mt3−/− mice in vivo.
Mice
C57BL/6 WT mice were from Jackson Laboratory. Lyz2cre Hif1αfl/fl mice that exhibit myeloid HIF1α deficiency were kindly provided by Dr. Tim Eubank at Ohio State University. Mt3−/− (deletion of Exon 3) mice were generated on the C57BL/6 background by the Transgenic Animal and Genome editing core facility at CCHMC. Animals were maintained by Department of Laboratory Animal Medicine, University of Cincinnati, accredited by American Association for Accreditation of Laboratory Animal Care (Frederick, MD) and experiments were in accordance with Animal Welfare Act guidelines of the National Institutes of Health. For in vivo experiments, C57BL/6 WT and Mt3−/− females that were aged 12 weeks were used.
METHODS DETAILS
Macrophage Culture
Bone marrow derived Mϕ were prepared by GM-CSF or M-CSF (10ng/ml) differentiation in RPMI complete media containing 10% fetal bovine serum (HyClone Laboratories, Utah), gentamycin sulfate (10 μg/L) and 2-mercaptoethanol as previously described (Subramanian Vignesh et al., 2013). GM-CSF cultures were fed on days 0 and 3 and M-CSF cultures were fed on days 0, 2 and 4 with media and cytokine. After 6 days of differentiation, Mϕ were harvested by trypsinization and washing 2 times with HBSS. Using this method, GM-CSF differentiation yielded 95%–96% CD11b+F480+ cells (macrophage markers) by flow cytometry. Mϕ cultures were stimulated with 10 ng/ml IL-4 for 24h. After 24h, media was replaced and cells were re-stimulated with cytokine (5 ng/ml IFNγ) where indicated. For silencing, cells were treated with siRNA overnight prior to cytokine stimulation.
Gene Expression
RNA was isolated from 2.5 X 105 to 1 X 106 Mϕ after elimination of genomic DNA using RNeasy Plus Mini kit (QIAGEN) or QUICK-RNA MINIPREP KIT (Denville Scientific). cDNA was prepared with Reverse Transcription Systems Kit (Promega, WI). Real time gene expression analysis was performed using Taqman primer/probe sets (Applied Biosystems, CA) in an ABI Prism 7500. Expression of target genes was compared to hypoxanthine guanine phosphoribosyl transferase (Hprt) as an internal control and normalized to unstimulated Mϕ. For time course analysis of Mt3 expression, 5 X 105 Mϕ were plated per well on a 24 well plate and stimulated with 10 ng/ml IL-4 for 30 min, 1h, 6h, 12h and 24h. For analyzing IFNγ responsiveness, Mϕ were stimulated with 10ng/ml IL-4 for 24h, after which media was changed and cells were stimulated with 5 ng/ml IFNγ for another 24h. Cells were harvested for RNA analysis at the indicated time points.
Gene Silencing and HIF2α Antagonism
Genes were silenced in Mϕ using TransIT TKO (Mirus Bio LLC) transfection reagent as per manufacturer’s instructions. Briefly, cells were treated with siRNA containing transfection complexes overnight, prior to 10 ng/ml IL-4 stimulation. Degree of silencing was assessed by gene expression 48h post transfection. The siRNA concentrations were, scramble (100 nM), Mt3 (100 nM), and Hif1a (50 nM). In some experiments, 50 nM Mt3 siRNA was used when combined silencing was performed. The amount of scramble siRNA used was equal to the total amount of target siRNA used in each experiment. Scramble, and Hif1a siRNA were purchased from Dharmacon (GE Healthcare), Mt3 siRNA was from Santacruz Biotechnology. In some experiments, Mϕ were treated with Mt3 + Hif1a siRNA and cultured with 10 μM HIF2α antagonist (Sigma) or DMSO control. Next day, Mϕ were stimulated with IL-4 and again treated with the antagonist or DMSO control. After 24h, cells were processed for gene expression analysis.
Western Blotting
PHD2 (Novus Biologicals), VEGFA (Proteintech), HIF1α, HIF2α, Lamin β1 (Abcam), GAPDH, β-actin (Santa Cruz), mTOR, pmTOR, Akt, pAkt and pS70S6K (Cell Signaling technologies) were assessed 24h post IL-4 stimulation (10 ng/ml) in scramble siRNA and Mt3 siRNA treated Mϕ and/or WT and Mt3−/− Mϕ. For total STAT6 and pSTAT6 (BD Biosciences) analysis, WT and Mt3−/− Mϕ were stimulated with IL-4 for 15 min; for total STAT1 and pSTAT1 (Abcam) analysis, Mϕ were stimulated with 10 ng/ml IL-4 for 24h, followed by change of media and 5 ng/ml IFNγ for 15 min. To determine the translocation of HIF, total cytosolic and nuclear proteins were isolated using NE-PER® Nuclear and Cytoplasmic Extraction Reagents according to manufacturer’s instructions (Thermo Scientific). Cell lysates were prepared using Denaturing Cell Extraction Buffer (Invitrogen). Total cell lysates as well as cytosolic and nuclear proteins were run on 4%–20% Precise Protein Gels (Pierce) and transferred on to nitrocellulose membranes. Western blots were probed with corresponding host specific HRP conjugated secondary antibodies and developed using SuperSignal West Femto Chemiluminescent Substrate (Thermofisher).
Flow Cytometry
For flow cytometric analysis of Mϕ phenotype in vitro, cells were gated on CD11b and analyzed for CD206 and PD-L2 (BioLegend) surface expression 24 h post 10 ng/ml IL-4 stimulation and run on Accuri C6 (BD Biosciences). For analysis of baseline IL-4Rα expression, freshly isolated Mϕ were stained with an IL-4Rα PE antibody (BD Biosciences) for 30 min. Cells were washed twice with HBSS and analyzed on an Accuri C6 flow cytometer. Data were analyzed using FCS Express 6 software (De Novo).
Mitochondrial membrane potential was analyzed by probing Mϕ with 50 nM JC-1 dye (Molecular Probes) for 30 min at 37°C. Cells were washed twice in HBSS + 1% FBS or bovine serum albumin (BSA) and analyzed on an Accuri C6 flow cytometer. Membrane depolarization was measured by excitation using a 488 nm laser and emission intensities at 585 nm and 530 nm were obtained. Compensation was performed using carbonyl cyanide m-chlorophenyl hydrazone (CCCP) treated Mϕ controls and the ratio of FL2 to FL1 MFI was measured. Data were analyzed using FCS Express 6 software (De Novo).
Metabolic Assays
Extracellular Acidification and Lactate Measurements
The pH of culture supernatants was measured using a pH microprobe in 500 μl – 1ml media. For assaying glycolysis, extracellular lactate was analyzed in culture supernatants 24h post IL-4 (10 ng/ml) stimulation using Glycolysis cell-based assay kit (Cayman Chemical) as per manufacturer’s instructions. In some experiments, cells were incubated with the indicated concentrations of chloroquine diphosphate salt (Sigma), 2-deoxyglucose (2-DG) (Sigma), DASA-10 (Calbiochem) or sodium dichloroacetate (DCA) (Acros Organics) during silencing and IL-4 stimulation.
Intracellular LDH Activity
Intracellular LDH activity was analyzed using CytoTox 96® NonRadioactive Cytotoxicity Assay kit (Promega). Briefly, Mϕ were treated with siRNA for 24h followed by stimulation with 10ng/ml IL-4 for 6h and 24h. Cell lysates from 106 Mϕ were prepared at these time points and intracellular LDH activity was measured as per manufacturer’s instructions using a BioTek Synergy microplate reader.
Mitochondrial Function Assays
Analysis of mitochondrial function by Seahorse technology was performed as reported before with slight modifications (Gandhi et al., 2015; Moreno-Fernandez et al., 2018). Specifically, to assess baseline OCR, 105 Mϕ were plated in XF24 cell culture microplates (Seahorse Biosciences) and transfected with scramble or Mt3 siRNA for 5h in RPMI media as described above. For OCR measurements, RPMI media was replaced with XF assay medium (Seahorse Biosciences) containing 11 mM glucose and 1 mM sodium pyruvate. Baseline OCR was measured on an XFe24 analyzer (Seahorse Biosciences) for 6h. In some experiments, siRNA-treated cells were stimulated with 10 ng/ml IL-4 for 6h, followed by OCR measurement for 50 min. For mitochondrial stress analysis, Mϕ were treated with siRNA overnight, followed by stimulation with IL-4 for 24h. Next day, media was replaced with XF assay medium and cells were re-stimulated with IL-4. OCR measurements were obtained for 55 min followed by sequential injections of oligomycin A (1 μM), carbonyl cyanide 4 (trifluoromethoxy) phenylhydrazone (FCCP, 1.5 μM) and rotenone (200 nM) + antimycin A (4 μM) to measure OCR in response to mitochondrial stress.
For ECAR analysis, an XF96 analyzer (Seahorse Biosciences) was used. GM-CSF differentiated Mϕ were plated in XF96 cell culture microplates at a density of 4 X 104 per well in RPMI media. Cells were stimulated for 24h with 10 ng/ml IL-4. Before data acquisition, media was changed to glucose-free XF base media (Seahorse Biosciences) supplemented with 1 mM glutamine. The glycolysis stress test was conducted by sequential addition of glucose (10 μM), oligomycin (10 μM) and 2-deoxy-D-glucose (50 mM).
For ATP analysis, 105 Mϕ were plated on a 96 well black assay plate (Costar) and treated with siRNA as described above. ATP production was assessed using CellTiter-Glo Luminescent Cell Viability Assay kit (Promega) as per manufacturer’s instructions.
Confocal Microscopy
For confocal imaging, 5 X 105 Mϕ were plated on Millicell 4-well glass EZ slides (Millipore) and treated as described above. Lysosomal imaging was performed 24h post IL-4 (10 ng/ml) stimulation. Mϕ were exposed to 25 nM Lysotracker Red DND-99 (Life Technologies) for 30 min prior to imaging. Cells were washed twice with Hanks Balanced Salt Solution (HBSS) containing 1% FBS and imaged immediately.
For MT3 analysis, Mϕ were stimulated with IL-4 for 24h, next day, media was changed and cells were restimulated with IL-4 for 9h, followed by fixation, permeabilization and staining with MT3 antibody (Novus Biologicals). For HIF1α analysis, Mϕ were treated with siRNA for 24h, followed by IL-4 stimulation for 24h. Where indicated, cells were treated with 1 mM L-lactate or 10 mM DCA on both days of culture period. Cells were probed with HIF1α antibody (Novus Biologicals) and Alexa Fluor 647 was used as the secondary antibody (Life Technologies). Cells were mounted using VectaShield with DAPI (Vector Labs) and images were acquired with a 63X oil immersion/1.4 NA objective and 1 – 1.5 mm optical thickness on a Zeiss LSM710 confocal connected to Zeiss Axio-observer.Z1 inverted microscope and visualized using ZEN 2011 software. MFI of MT3 and nuclear localization signal of HIF1α were analyzed using ImageJ software.
Bacterial Phagocytosis and Growth Assay
E. coli (K12) were grown in Luria-Bertani broth at 37°C overnight. This culture was inoculated at a 1:100 dilution into fresh Luria-Bertani broth, grown to log phase at 37°C and monitored turbidometrically at 600 nm. Log phase culture (OD600 = 0.5–0.7) was pelleted, washed and resuspended with 1X PBS at 4°C. The number of bacteria were determined using Agilent Technologies software (https://www.chem.agilent.com/store/biocalculators/calcODBacterial.jsp).
To determine phagocytic ability, 5X105 Mϕ were plated in each well of 24 well plates. After 24h, Mϕ were infected with 1.25X107
E. coli K12 strain for 3.5 h in antibiotic and 2-mercaptoethanol free RPMI media. After infection, Mϕ were washed with antibiotic free HBSS 3 times, lysed with water and bacteria were spread on LB-agar plates. The plates were incubated overnight at 37°C and CFUs were counted.
To analyze antimicrobial defenses, 5X105 Mϕ were left untreated, or treated with 10 ng/ml IL-4 for 24h. Next day, media was changed and indicated groups were pre-treated with 5ng/ml IFNγ for 5 h. Mϕ were infected with 1.25X107 E. coli K12 for 3.5 h. Cells were then washed with antibiotics containing HBSS 3 times to kill extracellular bacteria and incubated in complete media for 24 h. Mϕ were then washed with antibiotic free HBSS 3 times, lysed and colonies were enumerated as above. Bacterial growth was represented as percent inhibition compared to WT untreated control Mϕ.
In Vivo Infection and Fungal Burden
H. capsulatum (G217B) was grown in Ham’s F-12 solution for 3 days at 37°C with agitation. The culture was pelleted, washed and resuspended with 1X PBS at 4°C. Cells were counted using a hemocytometer. Mice were infected using a dose of 2 X 106 H. capsulatum yeasts intranasally (i.n) per mouse. Mice were euthanized after 3 and 7 days using CO2. Lungs were dissected, collected and homogenized using a glass organ grinder in cold HBSS. For fungal burden analysis, for each mouse, 2 blood agar plates were smeared with the lung homogenate and incubated at 37°C for 7 days. Colonies were counted and the average of the two plates was used to calculate CFUs.
For metabolic analysis in vivo, mice were euthanized, lungs were dissected and collected in PBS. The lungs were homogenized using gentleMACS Dissociator and gentleMACS C tubes according to the manufacturer’s recommendation. The homogenate was filtered through a 40 mm cell strainer, centrifuged and the supernatant was discarded. Cell pellets were resuspended in 5 mL of RBC lysis buffer on ice for 5 min, followed by washing with HBSS and centrifugation. The cell pellets were then resuspended in 5 mL of HBSS and lympholyte (5:1) separation was performed to remove dead cells. After that, Mϕ were enriched and counted using hemocytometer and seeded in Seahorse 96 well plates and incubated at 37°C. After 24 h, glycolysis stress test (ECAR) and mitochondrial tests were performed. Glycolysis stress test was performed by measuring ECAR for 15 min, followed by ECAR in response to sequential treatment with glucose, oligomycin and 2DG. Mitochondrial stress test was done by measuring OCR for 15 min, followed by OCR in response to sequential treatment with oligomycin, FCCP and Rotenone/Antimycin. These tests were performed in accordance to manufacturer’s recommendations. Alveolar Mϕ were analyzed using the same protocol.
RNA-Seq
Directional RNA-seq was performed by the Genomics, Epigenomics and Sequencing Core (GESC) at the University of Cincinnati.
Target RNA Enrichment-Isolation of PolyA RNA
RNA concentration was measured by Nanodrop (Thermo Scientific, Wilmington, DE). RNA integrity was determined by Bioanalyzer (Agilent, Santa Clara, CA). NEBNext Poly(A) mRNA Magnetic Isolation Module (New England BioLabs, Ipswich, MA) was used for polyA RNA purification with 1 µg total input RNA. The Core used Apollo 324 system (WaferGen, Fremont, CA) and ran PrepX PolyA script for automated ployA RNA isolation.
RNA-Seq Library Preparation
Library was prepared by using NEBNext Ultra Directional RNA Library Prep kit (New England BioLabs, Ipswich, MA). During second cDNA synthesis, dUTP was incorporated to maintain strand specificity. The isolated polyA RNA or rRNA/globin depleted RNA was Mg2+/heat fragmented (~200 bp), reverse transcribed to 1st strand cDNA, followed by 2nd strand cDNA synthesis labeled with dUTP. The purified cDNA was end repaired and dA tailed, and then ligated to adaptor with a stem-loop structure. The dUTP-labeled 2nd strand cDNA was removed by USER enzyme to maintain strand specificity. After indexing via PCR (11 cycles) enrichment, the amplified libraries together with library preparation negative control were cleaned up by AMPure XP beads for QC analysis. 1 μl library was analyzed by Bioanalyzer (Agilent, Santa Clara, CA) using DNA high sensitivity chip to assess quality and purity. To quantify library concentration for clustering, the library was 1:104 diluted in dilution buffer (10 mM Tris-HCl, pH 8.0 with 0.05% Tween 20), and qPCR measured by NEBNext Library Quant Kit (New England BioLabs) using QuantStudio 5 Real-Time PCR Systems (Thermo Fisher, Waltham, MA).
Cluster Generation and HiSeq Sequencing
To study differential gene expression, individually indexed and compatible libraries were proportionally pooled (~25 million reads per sample in general) for clustering in cBot system (Illumina, San Diego, CA). Libraries at the final concentration of 15 pM were clustered onto a single read (SR) flow cell using Illumina TruSeq SR Cluster kit v3, and sequenced to 51 bp using TruSeq SBS kit on Illumina HiSeq system.
RNA-Seq Analysis
Sequence reads were aligned to the reference genome using the TopHat2 aligner(Kim et al., 2013), and reads aligning to each known transcript were counted using Bioconductor packages for next-generation sequencing data analysis(Huber et al., 2015). The differential expression analysis between different sample types was performed using the negative binomial statistical model of read counts as implemented in the edgeR Bioconductor package(Anders et al., 2013). Significance of differential expression was set to fold change ≥ 2 and adjusted p value < 0.05. P values were adjusted for multiple hypotheses testing using Benjamini-Hochberg procedure. Each group is based on 3 biological replicates.
CRISPR/Cas9 Generation of Mt3−/− Mice
sgRNAs and donor oligo design and generation of animals were as described(Yuan and Hu, 2017). sgRNAs were selected according to off-target scores from the CRISPR design web tool (http://genome-engineering.org). Selected sgRNA target sequences were cloned(Ran et al., 2013) into a modified pX458 vector (Addgene #48138) containing an optimized sgRNA scaffold and a Cas9–2A-GFP (Chen et al., 2013). Editing activity was validated by the T7E1 assay in mouse mK4 cells(Valerius et al., 2002), compared with Tet2 sgRNA known to work efficiently in mouse embryos(Wang et al., 2013). Validated sgRNA was transcribed in vitro using the MEGA-shorscript T7 kit (ThermoFisher), purified using the MEGAclear Kit (ThermoFisher), and stored at −80°C. To prepare the injection mix, we incubated sgRNA and Cas9 protein (ThermoFisher) at 37°C for 5 min to form ribonucleoproteins. The final concentrations were 50 ng/ml for each sgRNA and 100 μg/ml Cas9 protein. The mutant mice were generated by injecting the mix into the cytoplasm of fertilized eggs derived from C57BL/6 genetic mice, using a piezo-driven microinjection technique(Yang et al., 2014a). Injected eggs were transferred into the oviductal ampulla of pseudopregnant CD-1 females on the same day. Pups were born and genotyped by PCR and Sanger sequencing. Primers used for PCR were 5ʹTTGGGGTGAGGTGTAGAGGT3′ (forward) and 5ʹGCCAAGATAAAGTCCGGGGT3′ (reverse). Animals were housed in a controlled environment with a 12-h light/12-h dark cycle, with free access to water and a standard chow diet. All animal procedures were performed in accordance with the Institutional Animal Care and Use Committee-approved protocol of Cincinnati Children’s Hospital and Medical Center.
SEC-ICP-MS-MS Analysis and Normalization of Data
SEC-ICP-MS-MS analysis of WT and Mt3−/− Mϕ cultured in 68Zn containing media was performed as described previously (Subramanian Vignesh et al., 2016). Mϕ were plated in 68Zn media, and either left untreated or treated with IL-4 for 24h. After this time, Mϕ cultures were washed twice in HBSS and cells were lysed with 0.1% SDS on ice. Cell lysates were incubated on ice for 20 min and centrifuged using 0.22 mm filter tubes. Filtrates were frozen at 80°C until further analysis by SEC-IC-MS-MS. To normalize the response of ICP-MS-MS signal from SEC separations on different days, 50 μl of 0.5 mg ml−1 carbonic anhydrase was injected into the liquid chromatography system, and area of Zn signals from samples was normalized to area of the carbonic anhydrase peak from each day. The absorbance of carbonic anhydrase at 280 nm was followed to ensure protein integrity.
The instrumentation consisted of an Agilent 1100 HPLC equipped with a degasser, a binary pump, a thermostated auto sampler, a column oven compartment and a diode array detector. For the Mϕ lysates, a TSK gel 3000SW gel filtration column (TSK Tokyo Japan) 7 × 300 mm, 10 mm particle size was used. The HPLC system was coupled to the ICP-MS-MS nebulizer via a short polyether ether ketone capillary of 0.17 mm internal diameter. An Agilent 8800 ICP-MS-MS system equipped with a micromist quartz nebulizer, a chilled double pass Scott spray chamber and a standard 2 mm insert quartz torch with shield torch was used for all experiments. The ICP-MS-MS was operated by the Agilent Mass Hunter integrated chromatographic software in the helium collision mode as reported previously (Subramanian Vignesh et al., 2016).
ICP-MS-MS and SEC-ICP-MS-MS Quality Control to Avoid External Zn Contamination
All metal analysis experiments were carried out using trace metal grade reagents with acid washed plastic vials. Reagent blanks were used to correct the background signal. The analysis was performed through a metal free encased auto sampler. The concentration of Zn in the blanks was always below 100 parts per trillion (ppt), the blank estimate concentration on the calibration curves was always below 50 ppt, while the detection limits were below 30 ppt.
For chromatographic analysis, the mobile phase was cleaned using a Chelex 100 resin, using the batch method. In brief, 3 g of Chelex-100 was added to a liter of mobile phase, stirred for 30 min and passed through a 0.45 mm membrane. This decreased the Zn concentration below 200 ppt (measured as total). By this method, the base line ICP-MS-MS 66Zn signal was below 1000 counts per second, which represents sub-ppb levels. The SEC column was cleaned using 10 volumes of 0.2 M NaCl and equilibrated with the mobile phase, followed by injection of 50 μl of 2% HNO3 three times to remove any accumulated Zn in the column. With this procedure, Zn distribution in the samples never deviated more than 10% compared to the theoretical natural Zn isotope distribution in the control Mϕ samples. Four blanks and four carbonic anhydrase standards were injected after the cleaning procedure for monitoring Zn signal by ICP-MS-MS to ensure optimal column performance. Typically, the column was cleaned every 30–40 samples.
QUANTIFICATION AND STATISTICAL ANALYSIS
p values were calculated in Sigma plot or GraphPad Prism by one-way ANOVA for multiple comparisons using the indicated ad hoc methods with at least 3 or more replicates and where two groups were compared, non-paired Student’s t test was used; in vivo data was analyzed using two-way ANOVA, *p < 0.05, **p < 0.01, ##p < 0.001, **** p < 0.0001; NS, not significant, ND, not detected.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-Metallothionein-3 | Novus Biologicals | Cat#NBP1–89772; RRID:AB_11032938 |
| anti-EGLN1/PHD2 | Novus Biologicals | Cat#NB100–2219; RRID:AB_2096717 |
| anti-VEGFA | Proteintech | Cat#19003–1-AP; RRID:AB_2212657 |
| anti-HIF1α (EPR16897) | Abcam | Cat#Ab179483; RRID:AB_2732807 |
| anti-HIF1α | Novus Biologicals | Cat#NB100–449; RRID:AB_10001045 |
| anti-HIF2α | Abcam | Cat#ab199; RRID:AB_302739 |
| anti-Lamin B1 | Abcam | Cat#ab16048; RRID:AB_10107828 |
| anti-GAPDH (FL-335) | Santacruz Biotechnology | Cat#sc-25778; RRID:AB_10167668 |
| anti-β-Actin | Cell Signaling Technology | Cat#4967s; RRID:AB_330288 |
| anti-STAT1 | Abcam | Cat#ab99415; RRID:AB_10677307 |
| anti-pSTAT1 (pY701) [M135] | Abcam | Cat#ab29045; RRID:AB_778096 |
| anti-STAT6 (Clone 23) | BD Biosciences | Cat#611290; RRID:AB_398816 |
| anti-pSTAT6 (pY641) [Clone J71–773.58.11] | BD Biosciences | Cat#558241; RRID:AB_647299 |
| Anti-Akt (pan) (C67E7) | Cell Signaling Technology | Cat#4691; RRID:AB_915783 |
| Anti-Phospho-Akt (Ser473) (D9E) XP | Cell Signaling Technology | Cat#4060; RRID:AB_2315049 |
| Anti-mTOR | Cell Signaling Technology | Cat#2972; RRID:AB_330978 |
| Anti-Phospho-mTOR (Ser2448) | Cell Signaling Technology | Cat#2971; RRID:AB_330970 |
| Anti-Phospho-p70 S6 Kinase (Thr389) | Cell Signaling Technology | Cat#9205; RRID:AB_330944 |
| Goat anti-mouse IgG (H+L), HRP conjugate | Proteintech | Cat#SA00001–1; RRID:AB_2722565 |
| Goat anti-rabbit IgG (H+L), HRP conjugate | Proteintech | Cat#SA00001–2; RRID:AB_2722564 |
| Goat anti-Rat IgM (Heavy chain), Alexa Fluor 647 | Invitrogen | Cat#A21245; RRID:AB_2535813 |
| Bacterial and Fungal Strains | ||
| Histoplasma capsulatum (G217B) | George S. Deepe, Jr (deepegs@ucmail.uc.edu) | N/A |
| Escherichia coli (K12) | Dr. Jason Gardner (gardnejr@ucmail.uc.edu) | N/A |
| Primers | ||
| Mt3 | Applied Biosystems | Mm00496661_g1 |
| Slc16a1 | Applied Biosystems | Mm01306379_m1 |
| Slc16a3 | Applied Biosystems | Mm00446102_m1 |
| Vegfa | Applied Biosystems | Mm00437306_m1 |
| Hif1a | Applied Biosystems | Mm00468869_m1 |
| Arg1 | Applied Biosystems | Mm00475988_m1 |
| Nos2 | Applied Biosystems | Mm00440502_m1 |
| Chil3 | Applied Biosystems | Mm00657889_mH |
| Retnla | Applied Biosystems | Mm00445109_m1 |
| Tfrc | Applied Biosystems | Mm01344477_g1 |
| Hprt | Applied Biosystems | Mm00446968_m1 |
| Chemicals and reagents | ||
| Dextrose | Fisher Scientific | Cat#BP350–1 |
| HEPES | Sigma | Cat#H3375 |
| Cystine | Fisher Scientific | Cat#BP377–100 |
| Agar | BD Bacto™ | Cat#214010 |
| Agar | BD BBL™ Mycosel™ | Cat#211462 |
| Sheep Blood | Colorado Serum Company | Cat#31125 |
| GM-CSF | Biolegend | Cat#576306 |
| M-CSF | PEPROTECH | Cat#315–02 |
| IL-4 | PEPROTECH | Cat#214–14 |
| IFNγ | PEPROTECH | Cat#315–05 |
| LPS E. coli 055:B5 | Sigma-Aldrich | Cat#L2880 |
| TransIT TKO | Mirus Bio LLC | Cat#MIR2156 |
| Precise Protein Gels | Invitrogen | Cat#XP04205BOX |
| SuperSignal™ West Femto Maximum Sensitivity | Thermo Scientific | Cat#34096 |
| Substrate | ||
| Nitrocellulose membranes | BIO-RAD | Cat#162–0112 |
| Protease &Phosphatase Inhibitor Cocktail | Thermo Scientific | Cat#78442 |
| Denaturing Cell Extraction Buffers | Invitrogen | Cat#FNN0091 |
| Glycine | Fisher Scientific | Cat#BP381 |
| Tris-base | Fisher Scientific | Cat#BP152 |
| SDS | Fisher Scientific | Cat#BP166 |
| Tween 20 | Acros Organics | Cat#23336–0010 |
| Triton X-100 | Fisher Scientific | Cat#BP151 |
| Millicell 4-well glass EZ slides | Millipore | Cat#PEZGS0496 |
| XF24 cell culture microplates | Seahorse Biosciences | Cat#101085–004 |
| XF assay medium | Seahorse Biosciences | Cat# 102416–100 |
| Glucose-free XF base media | Seahorse Biosciences | Cat# 103335–100 |
| Bovine serum albumin | Sigma | Cat#A7030 |
| p-Aminobenzenesulfonamide | Sigma | Cat#S9251 |
| N-(1-Naphthyl)ethylenediamine dihydrochloride | Sigma | Cat#33461 |
| Sodium Nitrite | Fisher Scientific | Cat#S347 |
| Phosphoric Acid | Fisher Scientific | Cat#A260–500 |
| Chloroquine diphosphate salt | Sigma | Cat#C6628 |
| 2-DG | Sigma | Cat#D6134 |
| DASA-10 | Calbiochem | Cat#550602 |
| Sodium dichloroacetate (DCA) | Acros Organics from Fisher Scientific | AC338280100 |
| pX458 vector | Addgene | Cat#48138 |
| GeneArt Platinum Cas9 Nuclease | ThermoFisher | Cat# B25641 |
| Alt-R SpCas9 Nuclease 3NLS | ThermoFisher | Cat# 1074181 |
| sgRNA | CCHMC transgenic core | N/A |
| TSK gel 3000SW gel filtration column | TOSOH BIOSCIENCE | Cat#05789 |
| Lysotracker Red DND-99 | Invitrogen | Cat#L7528 |
| Critical Commercial Assays | ||
| RNeasy Mini Kit | QIAGEN | Cat#74104 |
| QUICK-RNA MINIPREP KIT | Denville Scientific | Cat#R1055 |
| Reverse Transcription System | Promega | Cat#A3500 |
| Probe Lo-Rox 2X qPCR Mix | RADIANT | Cat#QP9020 |
| NE-PER® Nuclear and Cytoplasmic Extraction Reagents | Thermo Scientific | Cat#78835 |
| MitoProbe JC-1 Assay Kit | Invitrogen | Cat#M34152 |
| Glycolysis cell-based assay kit | Cayman Chemical | Cat#600450 |
| CytoTox 96® NonRadioactive Cytotoxicity Assay | Promega | Cat#G1782 |
| kit Promega | ||
| CellTiter-Glo Luminescent Cell Viability Assay Kit | Promega | Cat#G7570 |
| NEBNext Poly(A) mRNA Magnetic Isolation Module | New England BioLabs | Cat#E7490L |
| NEBNext Ultra Directional RNA Library Prep Kit | New England BioLabs | Cat# E7420L |
| NEBNext Library Quant Kit | New England BioLabs | Cat# E7630L |
| MEGAshorscript T7 Kit | Thermo Fisher | Cat#AM1354 |
| MEGAclear Kit | Thermo Fisher | Cat# AM1908 |
| Deposited Data | ||
| RNA-seq data | NCBI SRA database | NCBI SRA: PRJNA533616 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6 Mice | Jackson Laboratory | 00064 |
| C57BL/6 (Mt3–/– deletion of Exon 3) | Transgenic Animal and Genome editing core facility at CCHMC | N/A |
| C57BL/6 (Lyz2cre Hif1αfl/fl) | Dr. Tim Eubank at (tdeubank@hsc.wvu.edu) | N/A |
| Software and Algorithms | ||
| SigmaPlot 14.0 | SYSTAT | N/A |
| Adobe® Photoshop® | Adobe | N/A |
| Adobe Illustrator® | Adobe | |
| Wave 2.0 | Agilent Technologies | N/A |
| Origin 2018b | OriginLab corporation CA | N/A |
| Bacterial culture OD to CFU assessment | Agilent Technologies | N/A |
| FCS Express 6 | De Novo | N/A |
| GraphPad Prism | GraphPad | N/A |
HIGHLIGHTS.
MT3 is required for optimal M(IL-4) polarization and metabolism
MT3 programs the metabolic switch toward increased oxidative phosphorylation
MT3 suppresses HIF1α stabilization and glycolysis
IL-4 subverts IFNγ responses and antimicrobial defenses by an MT3-dependent mechanism
ACKNOWLEDGMENTS
Support for this work came from American Heart Association 19CDA34770022, AHA 15POST25700182, and CEG NIEHS P30ES006096 grants to K.S.V.; NIH grants AI106269 and AI133797 to G.S.D.; and in part by NIH grant R01DK099222 to S.D. We thank Agilent Technologies for LCMS and 8800x ICP-MS-MS instrument support; the Transgenic Animal and Genome editing core at CCHMC for Mt3−/− mice; the Genomics, Epigenomics and Sequencing core for RNA-seq; the Microscopy Laboratory at UC; Dr. Priyanka Mishra, Dr. Maria Fields, and T. Stankiewicz for technical assistance; and Dr. A. Matthis for pH-microprobe.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.05.093.
DECLARATION OF INTERESTS
The authors declare no competing interests.
DATA AND CODE AVAILABILITY
All scripts used in this manuscript are available upon request. The accession number for the RNA-seq data reported in this paper is NCBI SRA: PRJNA533616.
REFERENCES
- Anastasiou D, Yu Y, Israelsen WJ, Jiang JK, Boxer MB, Hong BS, Tempel W, Dimov S, Shen M, Jha A, et al. (2012). Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat. Chem. Biol 8, 839–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, McCarthy DJ, Chen Y, Okoniewski M, Smyth GK, Huber W, and Robinson MD (2013). Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat. Protoc 8, 1765–1786. [DOI] [PubMed] [Google Scholar]
- Appelhoff RJ, Tian YM, Raval RR, Turley H, Harris AL, Pugh CW, Ratcliffe PJ, and Gleadle JM (2004). Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J. Biol. Chem 279, 38458–38465. [DOI] [PubMed] [Google Scholar]
- Aratake T, Higashi Y, Ueba Y, Hamada T, Shimizu T, Shimizu S, Yawata T, Ueba T, and Saito M (2018). The inhibitory role of intracellular free zinc in the regulation of Arg-1 expression in interleukin-4-induced activation of M2 microglia. Metallomics 10, 1501–1509. [DOI] [PubMed] [Google Scholar]
- Chawla A (2010). Control of macrophage activation and function by PPARs. Circ. Res 106, 1559–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li GW, Park J, Blackburn EH, Weissman JS, Qi LS, and Huang B (2013). Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/ Cas system. Cell 155, 1479–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, Giamarellos-Bourboulis EJ, Martens JH, Rao NA, Aghajanirefah A, et al. (2014). mTOR-and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 345, 1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle P, Philcox JC, Carey LC, and Rofe AM (2002). Metallothionein: the multipurpose protein. Cell. Mol. Life Sci 59, 627–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, and Thompson CB (2008). The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7, 11–20. [DOI] [PubMed] [Google Scholar]
- De Saedeleer CJ, Copetti T, Porporato PE, Verrax J, Feron O, and Sonveaux P (2012). Lactate activates HIF-1 in oxidative but not in Warburg-phenotype human tumor cells. PLoS ONE 7, e46571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierichs L, Kloubert V, and Rink L (2018). Cellular zinc homeostasis modulates polarization of THP-1-derived macrophages. Eur. J. Nutr 57, 2161–2169. [DOI] [PubMed] [Google Scholar]
- Divakaruni AS, Hsieh WY, Minarrieta L, Duong TN, Kim KKO, Desousa BR, Andreyev AY, Bowman CE, Caradonna K, Dranka, et al. (2018). Etomoxir Inhibits Macrophage Polarization by Disrupting CoA Homeostasis. Cell Metab 28, 490–503e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Ghazi I, Martin BL, and Armitage IM (2010). New proteins found interacting with brain metallothionein-3 are linked to secretion. Int. J. Alzheimers Dis 2011, 208634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraonio R, Moffatt P, Larochelle O, Schipper HM, S-Arnaud R, and Séguin C (2000). Characterization of cis-acting elements in the promoter of the mouse metallothionein-3 gene. Activation of gene expression during neuronal differentiation of P19 embryonal carcinoma cells. Eur. J. Biochem 267, 1743–1753. [DOI] [PubMed] [Google Scholar]
- Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, and Semenza GL (1996). Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol 16, 4604–4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galván-Peña S, and O’Neill LA (2014). Metabolic reprograming in macrophage polarization. Front. Immunol 5, 420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi CR, Chaillet JR, Nalesnik MA, Kumar S, Dangi A, Demetris AJ, Ferrell R, Wu T, Divanovic S, Stankeiwicz T, et al. (2015). Liver-specific deletion of augmenter of liver regeneration accelerates development of steatohepatitis and hepatocellular carcinoma in mice. Gastroenterology 148, 379–391e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett SH, Sens MA, Shukla D, Nestor S, Somji S, Todd JH, and Sens DA (1999). Metallothionein isoform 3 expression in the human prostate and cancer-derived cell lines. Prostate 41, 196–202. [DOI] [PubMed] [Google Scholar]
- Gordon S (2003). Alternative activation of macrophages. Nat. Rev. Immunol 3, 23–35. [DOI] [PubMed] [Google Scholar]
- Haas R, Smith J, Rocher-Ros V, Nadkarni S, Montero-Melendez T, D’Acquisto F, Bland EJ, Bombardieri M, Pitzalis C, Perretti M, et al. (2015). Lactate Regulates Metabolic and Pro-inflammatory Circuits in Control of T Cell Migration and Effector Functions. PLoS Biol 13, e1002202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP, and Wilson MC (2012). The monocarboxylate transporter family–role and regulation. IUBMB Life 64, 109–119. [DOI] [PubMed] [Google Scholar]
- Haschemi A, Kosma P, Gille L, Evans CR, Burant CF, Starkl P, Knapp B, Haas R, Schmid JA, Jandl C, et al. (2012). The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab 15, 813–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey MM, Richardson T, Wang T, Mosqueira M, Arguiri E, Yu H, Yu QC, Solomides CC, Morrisey EE, Khurana TS, et al. (2010). The von Hippel-Lindau Chuvash mutation promotes pulmonary hypertension and fibrosis in mice. J. Clin. Invest 120, 827–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hozumi I, Suzuki JS, Kanazawa H, Hara A, Saio M, Inuzuka T, Miyairi S, Naganuma A, and Tohyama C (2008). Metallothionein-3 is expressed in the brain and various peripheral organs of the rat. Neurosci. Lett 438, 54–58. [DOI] [PubMed] [Google Scholar]
- Huang J, Jia Y, Fu T, Viswakarma N, Bai L, Rao MS, Zhu Y, Borensztajn J, and Reddy JK (2012). Sustained activation of PPARa by endogenous ligands increases hepatic fatty acid oxidation and prevents obesity in ob/ob mice. FASEB J 26, 628–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Nazarova EV, Tan S, Liu Y, and Russell DG (2018). Growth of Mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. J. Exp. Med 215, 1135–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SC, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, Beatty W, Love-Gregory L, Lam WY, O’Neill CM, et al. (2014). Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat. Immunol 15, 846–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SC, Smith AM, Everts B, Colonna M, Pearce EL, Schilling JD, and Pearce EJ (2016). Metabolic Reprogramming Mediated by the mTORC2-IRF4 Signaling Axis Is Essential for Macrophage Alternative Activation. Immunity 45, 817–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber S, Hoffmann R, Muskens F, and Voehringer D (2010). Alternatively activated macrophages inhibit T-cell proliferation by Stat6-dependent expression of PD-L2. Blood 116, 3311–3320. [DOI] [PubMed] [Google Scholar]
- Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, et al. (2015). Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 12, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly B, and O’Neill LA (2015). Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res 25, 771–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kido T, Ishiwata K, Suka M, and Yanagisawa H (2019). Inflammatory response under zinc deficiency is exacerbated by dysfunction of the T helper type 2 lymphocyte-M2 macrophage pathway. Immunology 156, 356–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kmiecik AM, Pula B, Suchanski J, Olbromski M, Gomulkiewicz A, Owczarek T, Kruczak A, Ambicka A, Rys J, Ugorski M, et al. (2015). Metallothionein-3 Increases Triple-Negative Breast Cancer Cell Invasiveness via Induction of Metalloproteinase Expression. PLoS ONE 10, e0124865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Park MH, Kim HJ, and Koh JY (2010). Metallothionein-3 regulates lysosomal function in cultured astrocytes under both normal and oxidative conditions. Glia 58, 1186–1196. [DOI] [PubMed] [Google Scholar]
- Lee DC, Sohn HA, Park ZY, Oh S, Kang YK, Lee KM, Kang M, Jang YJ, Yang SJ, Hong YK, et al. (2015). A lactate-induced response to hypoxia. Cell 161, 595–609. [DOI] [PubMed] [Google Scholar]
- Lumeng CN, Bodzin JL, and Saltiel AR (2007). Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Invest 117, 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder PK, Febbo PG, Bikoff R, Berger R, Xue Q, McMahon LM, Manola J, Brugarolas J, McDonnell TJ, Golub TR, et al. (2004). mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat. Med 10, 594–601. [DOI] [PubMed] [Google Scholar]
- Moreno-Fernandez ME, Giles DA, Stankiewicz TE, Sheridan R, Karns R, Cappelletti M, Lampe K, Mukherjee R, Sina C, Sallese A, et al. (2018). Peroxisomal beta-oxidation regulates whole body metabolism, inflammatory vigor, and pathogenesis of nonalcoholic fatty liver disease. JCI Insight 3, 93626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser DM, and Edwards JP (2008). Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol 8, 958–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, et al. (2014). Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nairz M, Schroll A, Sonnweber T, and Weiss G (2010). The struggle for iron - a metal at the host-pathogen interface. Cell. Microbiol 12, 1691–1702. [DOI] [PubMed] [Google Scholar]
- O’Neill LA, and Pearce EJ (2016). Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med 213, 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmiter RD, Findley SD, Whitmore TE, and Durnam DM (1992). MT-III, a brain-specific member of the metallothionein gene family. Proc. Natl. Acad. Sci. USA 89, 6333–6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Prados JC, Través PG, Cuenca J, Rico D, Aragonés J, Martín-Sanz P, Cascante M, and Bosca L (2010). Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J. Immunol 185, 605–614. [DOI] [PubMed] [Google Scholar]
- Slusser A, Zheng Y, Zhou XD, Somji S, Sens DA, Sens MA, and Garrett SH (2015). Metallothionein isoform 3 expression in human skin, related cancers and human skin derived cell cultures. Toxicol. Lett 232, 141–148. [DOI] [PubMed] [Google Scholar]
- Subramanian Vignesh K, Landero Figueroa JA, Porollo A, Caruso JA, and Deepe GS Jr. (2013). Granulocyte macrophage-colony stimulating factor induced Zn sequestration enhances macrophage superoxide and limits intracellular pathogen survival. Immunity 39, 697–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian Vignesh K, Landero Figueroa JA, Porollo A, Divanovic S, Caruso JA, and Deepe GS Jr. (2016). Interleukin-4 Induces Metallothionein 3- and SLC30A4-Dependent Incerased in Intracellular Zn2+ that Promotes Pathogen Persistence in Macrophages. Cell Rep 16, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suganami T, and Ogawa Y (2010). Adipose tissue macrophages: their role in adipose tissue remodeling. J. Leukoc. Biol 88, 33–39. [DOI] [PubMed] [Google Scholar]
- Tan Z, Xie N, Banerjee S, Cui H, Fu M, Thannickal VJ, and Liu G (2015a). The monocarboxylate transporter 4 is required for glycolytic reprogramming and inflammatory response in macrophages. J. Biol. Chem 290, 46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Z, Xie N, Cui H, Moellering DR, Abraham E, Thannickal VJ, and Liu G (2015b). Pyruvate dehydrogenase kinase 1 participates in macrophage polarization via regulating glucose metabolism. J. Immunol 194, 6082–6089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullah MS, Davies AJ, and Halestrap AP (2006). The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J. Biol. Chem 281, 9030–9037. [DOI] [PubMed] [Google Scholar]
- Valerius MT, Patterson LT, Witte DP, and Potter SS (2002). Microarray analysis of novel cell lines representing two stages of metanephric mesenchyme differentiation. Mech. Dev 112, 219–232. [DOI] [PubMed] [Google Scholar]
- Van den Bossche J, and van der Windt GJW (2018). Fatty Acid Oxidation in Macrophages and T Cells: Time for Reassessment? Cell Metab 28, 538–540. [DOI] [PubMed] [Google Scholar]
- Van den Bossche J, Baardman J, Otto NA, van der Velden S, Neele AE, van den Berg SM, Luque-Martin R, Chen HJ, Boshuizen MC, Ahmed M, et al. (2016). Mitochondrial Dysfunction Prevents Repolarization of Inflammatory Macrophages. Cell Rep 17, 684–696. [DOI] [PubMed] [Google Scholar]
- Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, Wagner RA, Greaves DR, Murray PJ, and Chawla A (2006). Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab 4, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, and Jaenisch R (2013). One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss G, Bogdan C, and Hentze MW (1997). Pathways for the regulation of macrophage iron metabolism by the anti-inflammatory cytokines IL-4 and IL-13. J. Immunol 158, 420–425. [PubMed] [Google Scholar]
- Yang H, Wang H, and Jaenisch R (2014a). Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nat. Protoc 9, 1956–1968. [DOI] [PubMed] [Google Scholar]
- Yang J, Ruchti E, Petit JM, Jourdain P, Grenningloh G, Allaman I, and Magistretti PJ (2014b). Lactate promotes plasticity gene expression by potentiating NMDA signaling in neurons. Proc. Natl. Acad. Sci. USA 111, 12228–12233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan CL, and Hu YC (2017). A Transgenic Core Facility’s Experience in Genome Editing Revolution. Adv. Exp. Med. Biol 1016, 75–90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.