

Abstract
Norepinephrine (NE) is released in excess into the extracellular space during oxygen–glucose deprivation (OGD) in brain, increasing neuronal metabolism and aggravating glutamate excitoxicity. We used isolated rat optic nerve and spinal cord dorsal columns to determine whether the noradrenergic system influences axonal damage in white matter. Tissue was studied electrophysiologically by recording the compound action potential (CAP) before and after exposure to 60 min of OGD at 36°C. Depleting catecholamine stores with reserpine was protective and improved CAP recovery after 1 h of reperfusion from 17% (control) to 35%. Adding NE during OGD decreased CAP recovery to 8%, and adding NE to reserpine during OGD eliminated the protective effect of the latter. Selective inhibitors of Na+-dependent norepinephrine transport desipramine and nisoxetine improved recovery to 58% and 44%, respectively. α2 adrenergic receptor agonists UK14,304 and medetomidine improved CAP recovery to 41% and 46% after 1 h of OGD. Curiously, α2 antagonists alone were also highly protective (e.g., atipamezole: 86% CAP recovery), at concentrations that did not affect baseline excitability. The protective effect of α2 receptor modulation was corroborated by imaging fluorescent Ca2+ and Na+ indicators within axons during OGD. Both agonists and antagonists significantly reduced axonal Ca2+ and Na+ accumulation in injured axons. These data suggest that the noradrenergic system plays an active role in the pathophysiology of axonal ischemia and that α2 receptor modulation may be useful against white matter injury.
Keywords: axon, ischemia, sodium, calcium, norepinephrine, confocal microscopy
Introduction
Norepinephrine (NE) is released in excess into the extracellular space during oxygen–glucose deprivation (OGD) (Globus et al., 1989; Bhardwaj et al., 1990; Perego et al., 1992) in the brain, increasing neuronal metabolism and aggravating glutamate excitotoxicity (Bickler and Hansen, 1996; Talke and Bickler, 1996). However, no data are available regarding the effect of ischemia on the release of NE and its influence on white matter. The primary regulators of NE release are α2-adrenergic autoreceptors (α2ARs). These are catecholamine receptors, which are sensitive to the neuron's own transmitter and function as release-inhibiting autoreceptors on noradrenergic neurons. Activation of these receptors by NE inhibits further release of NE during nerve stimulation, while blocking them enhances the stimulation-evoked release of the neurotransmitter (Langer, 1974; Dixon et al., 1979); therefore, they have been classified as autoinhibitory receptors.
In models of cerebral ischemia and excitotoxicity, both α2AR agonists and antagonists have been shown to be neuroprotective in in vivo and in vitro studies (Martel et al., 1998; Puurunen et al., 2001; Ma et al., 2005). The mechanisms of α2AR-mediated protection remain elusive, although different possible biochemical and physiological cascades at nerve terminals were suggested, such as inhibition of the intracellular Ca2+ rise, activation of outward rectifying K+ channels, modulation of the release of other transmitters at different nerve terminals, and enhancement of glutamate scavenging by astrocytes (Ma et al., 2005).
While in gray matter areas, α2AR were found in the perikaryon and in association with the neuropil (Talley et al., 1996; Milner et al., 1998), in mature white matter α2ARs were suggested to be present on glia and/or microvessels, with no evidence of adrenergic receptors on axons of descending or ascending white matter tracts (Venugopalan et al., 2006). The role of α2ARs in mature white matter during metabolic inhibition has not been investigated, although several reports indicate potent neuroprotective effects of α2AR modulation in perinatal models of gray and white matter injury (Laudenbach et al., 2002; Paris et al., 2006). There is also evidence for β adrenoceptor-mediated modulation of excitability of premyelinated optic nerve axons; however, this effect is lost as the tract matures and axons become fully myelinated (Honmou and Young, 1995).
Multiple reports about the neuroprotective effect of α2AR agonists in a variety of models of cerebral metabolic stress prompted us to investigate whether manipulating α2AR in adult white matter might offer neuroprotection during OGD. Using electrophysiology and confocal microscopy we demonstrate NE-dependent axonal Ca2+ and Na+ changes, modulated by α2AR in injured rat white matter in vitro. We also demonstrate prominent α2AR immunoreactivity associated with myelinated optic and dorsal column axons suggesting a direct effect of these receptors on central fibers. The robust protection afforded by α2AR modulators, together with favorable clinical tolerability observed in other studies (Ma et al., 2005), suggests that these agents may represent an attractive class of molecules for the development of protective strategies for human white matter disorders.
Materials and Methods
Electrophysiology.
For electrophysiological recordings, both optic nerves and spinal cord dorsal columns from adult Long–Evans male rats were used. For the optic nerve recordings, rats were anesthetized with 80% CO2/20% O2, decapitated and nerves were dissected out. For dorsal columns experiments, rats were anesthetized with pentobarbital, perfused with 0 Ca2+/0.1 mm EGTA CSF, thoracic spinal cord was removed, placed in cold oxygenated zero-Ca2+/0.1 mm EGTA CSF and dorsal column slices were dissected free. The tissue was then placed in an oxygenated chamber at 36°C for recording of propagated compound action potentials (CAPs) using suction electrodes. The tissue was aerated with a 95% O2/5% CO2 gas mixture, and perfused with artificial CSF (aCSF: 126 NaCl, 3.0 KCl, 2 Mg2SO4, 26 NaHCO3, 1.25 NaH2PO4, 2.0 CaCl2, 10 dextrose (in mm), pH 7.4). Under control conditions, in vitro white matter CAP amplitudes and waveshapes remain very stable for >3 h at 37°C (Stys et al., 1991; Li et al., 1999; Malek et al., 2003). OGD was induced by switching to a 0 glucose CSF (glucose replaced by equimolar sucrose) and 95% N2/5% CO2 mixture for 1 h, followed by 1 h of reperfusion/reoxygenation. Ratios of CAP area after reperfusion to pre-OGD/predrug control areas were used to quantitate the degree of functional recovery after OGD.
Confocal microscopy.
Optic nerves were dissected out of the brain and placed in an interface perfusion chamber in Ca2+-free aCSF at 36°C. One end of each nerve was inserted into a suction pipette filled with loading buffer (aCSF with NaCl replaced by 126 mm of N-methyl-d-glucamine, with CaCl2 omitted, roughly mimicking intra-axonal ion concentrations) and fluorescent dyes: either Fluo-4 dextran (Ca2+ indicator) or CoroNa Green (Na+ indicator), and the ion-insensitive Alexa Fluor 594 dextran for visualization of axonal profiles. After application of the suction pipette to the end of the nerve, perfusion was switched to a normal Ca2+-replete CSF and nerves were loaded for 1.5 h, removed from the loading pipette and rinsed in normal CSF for a further 1.5 h. Nerves were placed in a custom-built perfusion chamber, and mounted on an upright Nikon C1 confocal laser-scanning microscope. Imaging was performed at 36°C with a 60× water-immersion objective. Fluorescence changes were normalized to average basal levels and reported as a ratio of signal collected from ion-sensitive to ion-insensitive fluorophores plotted against time. Chemical “ischemia” was induced by using the mitochondrial inhibitor NaN3 (2 mm) and zero-glucose (replaced with 10 mm sucrose) in the perfusate. All drugs were applied 30 min before the onset of OGD/chemical ischemia and continued throughout OGD.
Immunohistochemistry.
Rat optic nerves (RONs) and spinal cord were dissected out, fixed in 2–4% paraformaldehyde for 1–3 h and then cryoprotected in 20% sucrose 0.1 m phosphate buffer at 4°C overnight. Tissue was then cut at 25–35 μm, mounted on a slide and air-dried for 2 h or overnight. For norepinephrine transporter (NET) staining, an antigen retrieval procedure was required: slides were boiled in a sodium citrate solution for 20 min. After cooling, the slides were incubated in ice-cold acetone for 30 min. The slides were then rinsed 2–3 times for 10 min in 0.05 m Tris buffer of with 1.5% NaCl or PBS and 1% Triton X-100 (TBS-T or PBS-T), followed by 10% normal goat serum (NGS) in TBS-T for blocking for 1 h at room temperature and incubated overnight in primary antiserum diluted in TBS-T or PBS-T with 2% NGS at the following dilutions: 1:1000 for α2AR (Sigma), 1:1000 for neurofilament 160 clone (Sigma), 1:1500 for Na+/K+ ATPase α-3 subunit (Affinity BioReagents), 1:200 for glial fibrillary acidic protein (GFAP; Boehringer Mannheim Biochemica), 1:200 for norepinephrine transporter (Santa Cruz Biotechnology). After two or three 10 min rinses in TBS-T, samples were incubated in anti-mouse Texas red at 1:100 or anti-mouse Alexa 568 at 1:200 in combination with anti-rabbit Alexa-488 at 1:500 (Invitrogen). Tissue was then rinsed 2–3 times in TBS-T or PBS-T. Some sections were then incubated in anti-neurofilament antibody directly conjugated with Alexa Fluor 660 dye (Invitrogen) for 3 h at 1:75 dilution for triple labeling. Slides were coverslipped with Prolong Antifade Reagent (Invitrogen), and images were collected on a confocal microscope with a 60× oil-immersion objective.
Statistics.
All data are expressed as means ± SD. Statistical differences were calculated by ANOVA with Tukey's HSD (Honestly Significant Difference) test for multiple comparisons. Reported n values represent numbers of individually analyzed axons (imaging) or numbers of nerves (electrophysiology).
Results
The effect of extracellular NE content on CAP-area recovery
To investigate whether there is any catecholamine effect on the ischemic pathogenesis in RONs, we depleted endogenous catecholamines using reserpine (Fig. 1). Reserpine is an irreversible inhibitor of the vesicular monoamine transporter and decreases tissue NE content by depleting its vesicular storage (Schuldiner et al., 1995). Reserpine (1 μm) pretreatment improved CAP recovery after 1 h OGD plus 1 h of reperfusion from 17 ± 7% (control) to 35 ± 9% (p = 8.8 × 10−8; n = 12). Adding NE (500 μm) during OGD decreased CAP recovery to 8.2 ± 3% (p = 0.039; n = 12), whereas adding NE to reserpine (n = 12) during OGD eliminated the protective effect of the latter (p = 4.2 × 10−6 reserpine plus NE vs reserpine), with CAPs recovering to 16 ± 7% (p = 0.99 vs control OGD). NE alone caused an insignificant 9.9% increase in mean CAP magnitude in control nerves before OGD (p = 0.125, Wilcoxon two-tail test), in agreement with previous studies showing no effect of this agent on mature optic nerve excitability (Honmou and Young, 1995).
Figure 1.

CAP-area recovery recorded from optic nerves injured by 1 h of OGD/1 h of reperfusion (control), in the presence of reserpine (1 μm), NE (500 μm), reserpine plus NE, desipramine (10 μm), nisoxetine (5 μm), demonstrating the dependence of injury on the availability of norepinephrine in the extracellular space. Numbers within the bars represent the number of nerves used for each treatment. Error bars indicate SD. *p = 0.04, **p < 10−7 versus control.
Desipramine and nisoxetine are selective NET inhibitors. The uptake of NE into cells is performed by NET, a member of a large family of transporters that concentrate NE by cotransport with Na+ and Cl− (Mandela and Ordway, 2006). During metabolic inhibition, a decrease of ATP levels followed by inhibition of Na+/K+ ATPase activity leads to intracellular Na+ accumulation and reversal of the NET, resulting in release of transmitter (Vizi, 2000). Given that extracellular NE appears deleterious (results above), and if the reverse operation of the Na+-dependent transporter leads to NE release during metabolic inhibition, then blocking the transporter during OGD should decrease release of endogenous NE, reduce extracellular NE concentration and improve outcome. The protective effects of desipramine (10 μm) and nisoxetine (5 μm) (58 ± 11%, n = 6 and 44 ± 14%, n = 12, respectively; p = 2 × 10−11 for each group vs drug-free control OGD) are consistent with such a mechanism of NET-mediated NE release. Together, these experiments suggest that lower concentrations of NE in the extracellular space are associated with better functional recovery, and implicate the noradrenergic system in the ischemic response of white matter.
Effects of α2AR agonists and antagonists on CAP recovery
The α2AR agonists UK14,304 (5′-bromo-6-[2-imidazolin-2-yl-amino]-quinoxaline; 0.1 μm) and medetomidine (10 μm) improved CAP recovery after OGD to 41 ± 17% and 46 ± 19% versus 17% in control (p = 1.1 × 10−6; n = 24 and p = 5.6 × 10−10; n = 30) (Fig. 2). Interestingly, preapplication of α2 antagonists [RX 821002 (1–10 μm), BRL 4406 (10 μm), RS 79948 (0.1 μm) or atipamezole (50 μm)] with the agonists did not decrease the effect of the agonists. On the contrary, it slightly increased their protective effect and in the case of the most specific antagonist, atipamezole (Virtanen et al., 1989), allowed CAP area to recover to 104 ± 15% of control when combined with the agonist medetomidine. Application of α2AR antagonists alone (BRL 4406, atipamezole) was also highly protective, particularly with the latter agent (86 ± 15%, p = 6.6 × 10−14; n = 12). To test whether this protective effect extends to other white matter tracts, atipamezole was tested on dorsal columns of spinal cord. The recordings were performed under the same conditions as for optic nerve and also revealed a protective effect (control: 52 ± 10% vs 76 ± 23% with atipamezole, p = 0.043; data not shown). This suggests that α2AR-dependent injury mechanisms triggered by OGD may be common to many white matter tracts.
Figure 2.

Optic nerve CAP-area recovery in the presence of α2AR agonists: UK14,304 (0.1 μm) and medetomidine (10 μm); agonists and antagonists: RX 821002 (1–10 μm), BRL 4406 (10 μm), RS 79948 (0.1 μm) and atipamezole (50 μm); antagonists alone: atipamezole and BRL 4406, illustrating the strong dependence of OGD-induced injury on α2AR modulation. Numbers within the bars represent the number of nerves used for each treatment. Error bars indicate SD. *p < 0.009 versus control.
Agonist-dependent PKC-mediated desensitization of α2AR
The similar protective effects of either α2AR agonists and antagonists was curious and unexpected. One explanation may involve receptor desensitization in response to persistent activation by agonists, so that with either treatment, the net effect was a reduction of α2AR activity. One pathway by which agonist activation results in a desensitization of α2AR signaling involves receptor phosphorylation by protein kinase C (PKC) (Liang et al., 1998). This phosphorylation may represent a mechanism by which crosstalk between different subtypes of adrenergic receptors, or between adrenergic receptors and other G-protein-coupled receptors, can occur (Liang et al., 1998, 2002). We investigated the possibility that in white matter injured by OGD, persistent stimulation of α2AR leads to an ultimate decrease of its function through PKC activation. As shown in Figure 3, PKC activation with phorbol-12-myristate-13-acetate modestly increased recovery to 32 ± 10%, which did not reach statistical significance (p = 0.09; n = 12). More importantly, however, addition of this PKC activator to the α2AR agonist medetomidine did not produce additive protective effects, suggesting a convergent pathway.
Figure 3.

PKC activation with 0.1 μm PMA showed a modest improvement in CAP recovery after OGD, which was not additive with the α2AR agonist medetomodine, suggesting a common pathway. Numbers within the bars represent the number of nerves used for each treatment. Error bars indicate SD. *p < 0.002 versus control.
Modulation of axonal Ca2+ by the α2AR-signaling pathway
The central event in the pathophysiology of white matter injury during ischemia is the excess influx of Ca2+ ions in the cytosol (Stys, 2004), which leads to irreversible cellular injury. It is therefore logical to hypothesize that the substantial protective effect of α2AR manipulation would affect axonal Ca2+ accumulation. To visualize axonal Ca2+ change during in vitro OGD in axons, RONs were loaded with the low affinity Ca2+ indicator Fluo-4 dextran (Kd ≈ 2.6 μm) and Alexa Fluor 594 dextran for visualization of axonal profiles. Images in the left panels of Figure 4 were taken during perfusion in normal aCSF (NCSF) and demonstrate the low basal level of Fluo-4 fluorescence in healthy resting axons (Fig. 4B,F,J); images in the right panels (OGD) were taken after 30 min of OGD exposure (zero-glucose + 2 mm NaN3) and show an increase in green Ca2+-sensitive fluorescence (Fig. 4D). Preapplication of medetomidine or atipamezole reduced ischemia-induced fluorescence increase of Fluo-4 in axons, indicating that axonal Ca2+ rise during OGD is modulated by the α2AR-signaling pathway.
Figure 4.
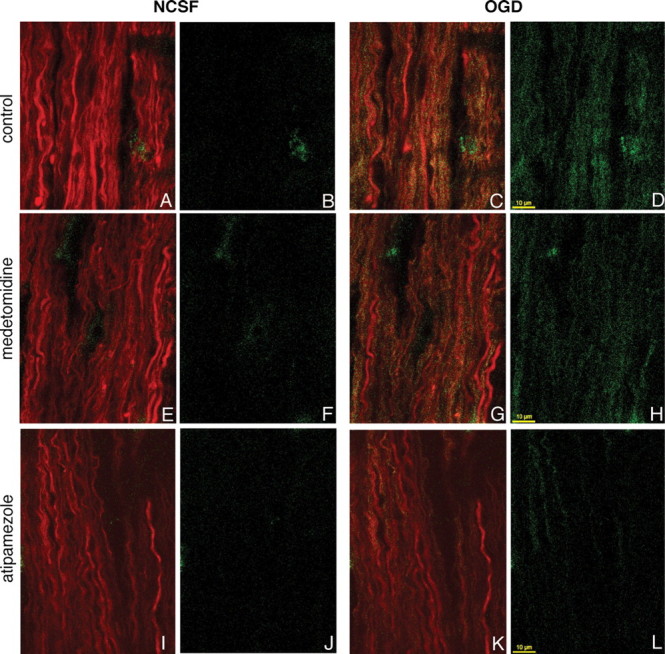
Confocal images of live rat optic nerve axons coloaded with Alexa Fluor 594 dextran (red) and Ca indicator Fluo-4 dextran (green) in vitro during perfusion in normal CSF (left panels without OGD: A, E, I, both channels; B, F, J, Ca2+-sensitive fluorescence) and after 30 min of exposure to OGD (right panels: C, G, K, both channels; D, H, L, Ca-sensitive fluorescence). Pretreatment with medetomidine (G, H) or atipamezole (K, L) reduced OGD-induced axonal Ca2+ accumulation.
To assess the extent and time course of axonal Ca2+ changes during in vitro OGD more quantitatively, images of nerves loaded with fluorescent indicators were taken at 2-min intervals during perfusion in normal aCSF and then during OGD. The fluorescence changes were normalized to the average basal fluorescence before the application of OGD, and the change in fluorescence (F/F0) was plotted against time. As shown in Figure 5A, there was a substantial rise in Ca2+-dependent fluorescence during OGD, increasing ≈7-fold over control levels after 30 min. Nisoxetine, medetomidine, atipamezole and desipramine all significantly reduced Fluo-4 fluorescence rise by ≈23, 44, 69, and 76%, respectively (p < 10−8 for all treatments) (Fig. 5B), indicating that the protective effects of noradrenergic modulators listed above are due in large part to reductions of ischemic Ca2+ overload in axons.
Figure 5.
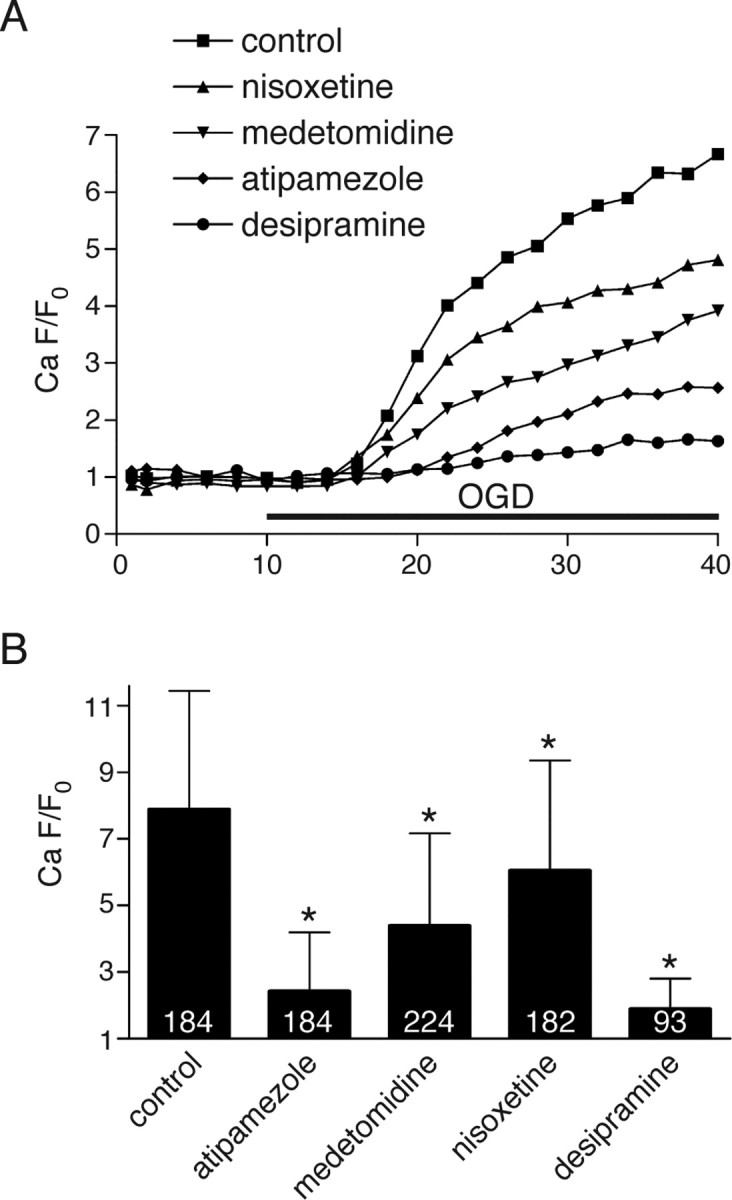
A, Time course of normalized axonal green (Ca2+-dependent)/red (Ca2+-independent) fluorescence ratio during ischemia showing ≈7-fold Ca2+-sensitive fluorescence increase over baseline after 30 min of OGD. Modulators of α2 receptors or of NE release significantly reduced OGD-induced axonal Ca2+ rise. B, Quantitative summary of Ca2+ responses in individual axons after 30 min of OGD. Numbers within the bars represent the number of individual axons analyzed for each treatment. Error bars indicate SD. *p < 10−8 versus control.
Na+ imaging
Because axonal Ca2+ overload is coupled to accumulation of axoplasmic Na+ (Stys and LoPachin, 1998; Nikolaeva et al., 2005), we then examined the question whether the robust decrease in axonal Ca2+ accumulation by noradrenergic modulators was secondarily due to reduction of ischemic Na+ influx. Axons were loaded with the Na+-sensitive dye CoroNa Green along with dextran-conjugated Alexa Fluor 594. Under normoxic control conditions, unlike the dextran-conjugated Alexa Fluor 594 whose emission dropped by only ≈5%, CoroNa Green emission decreased by ≈65% over 45 min likely because of its much lower molecular weight (it is not available as a dextran conjugate) and therefore more rapid efflux from axons (Fig. 6A). Therefore, CoroNa Green fluorescence data were always compared with time-matched normoxic controls, whose fluorescence decay was consistent enough to allow such comparisons (Nikolaeva et al., 2005); of note, percentage changes <100% do not necessarily imply a decrease in axonal Na+. As expected, OGD induced a significant axonal Na+ increase after 30 min compared with time-matched normoxic controls (CoroNa Green fluorescence at t = 30 min of OGD vs t = 0 min: 94 ± 14%, n = 46 axons, compared with 56 ± 22%, n = 86, during normoxia, P ≈ 0) (Fig. 6B). Blocking voltage-gated Na+ channels with tetrodotoxin (TTX) significantly reduced (78 ± 13%, p = 5 × 10−6 n = 108 vs drug-free OGD) but did not completely block the ischemic Na+ accumulation, with a substantial axonal Na+ increase remaining compared with control axons (P ≈ 0 vs normoxic time-matched controls), suggesting additional important routes of axonal Na+ accumulation. One additional Na+ influx pathway recently identified involves AMPA receptors (Ouardouz et al., 2006); we, therefore, measured axonal Na+ changes in the presence of both TTX and SYM2206, a potent noncompetitive AMPA receptor antagonist. The combination further reduced axonal Na+ loading compared with TTX alone (56 ± 11%; n = 75 vs 78 ± 13%, p = 8 × 10−12), which was identical to normoxic control fluorescence levels (56 ± 21%; n = 86, p = 0.99). Atipamezole alone was also highly effective at reducing ischemic axonal Na+ accumulation (63 ± 24%, n = 116) to levels that were not significantly different from normoxic time-matched controls (p = 0.18). Together, these results confirm that the two major routes of ischemic axonal Na+ influx (TTX-sensitive Na+ channels and AMPA receptors) in optic nerve are similar to those recently shown in dorsal column axons (Ouardouz et al., 2006). Importantly, the present results with atipamezole further suggest that both Na+ influx pathways are modulated by the noradrenergic system.
Figure 6.
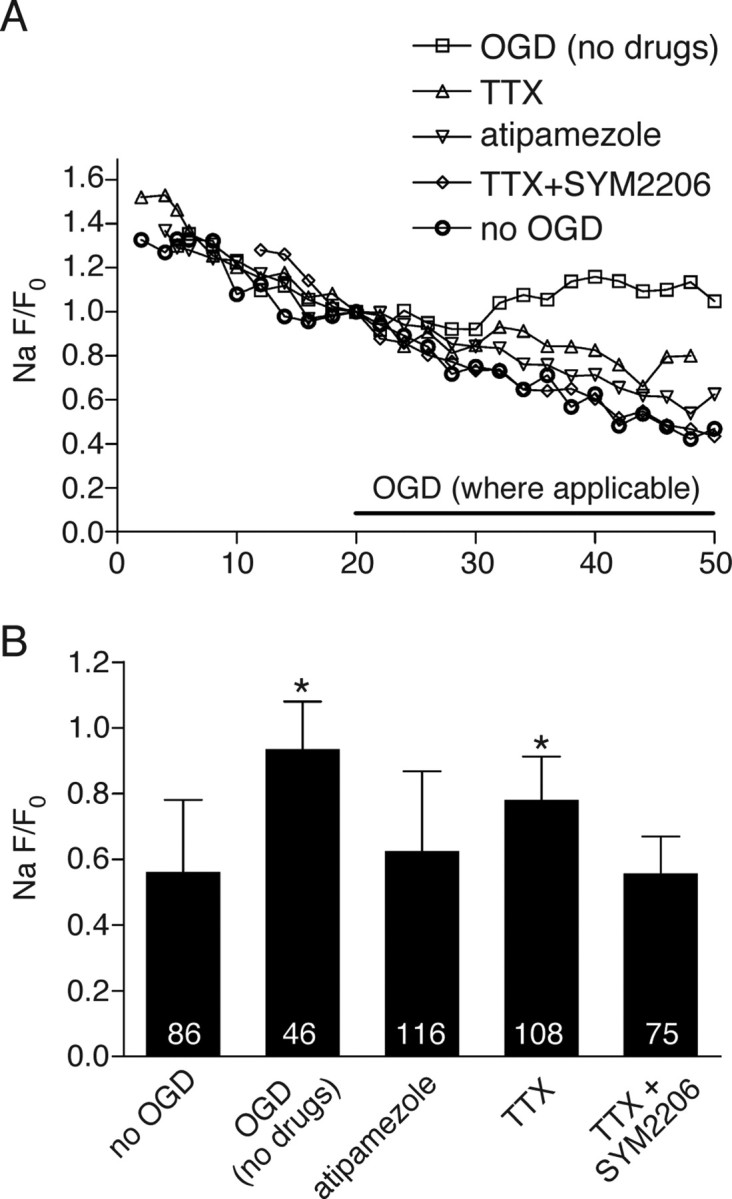
A, Representative plots of normalized axonal Na+-dependent green (Corona Green)/red (Alexa Fluor 594 dextran) fluorescence ratios versus time. The decrease in axoplasmic fluorescence due to dye leakage in NCSF was reversed in 0 glucose/NaN3 indicating a net axonal Na+ accumulation. Blocking Na+ channels with TTX (1 μm) reduced Na+ accumulation, whereas adding the AMPA receptor blocker SYM 2206 (30 μm) in addition to TTX completely abolished detectable Na+ entry. Atipamezole prevented Na+ entry almost as effectively as TTX and SYM 2206 together. B, Quantitative summary of Na+ responses in individual axons. Numbers within the bars represent the number of individual axons analyzed for each treatment. Error bars indicate SD. *p < 10−5 versus “no OGD.” There were no differences between any of the no OGD, “atipamezole” and “TTX+SYM2206” groups using multiple comparisons (p > 0.17).
α2a receptors are expressed on axons and astrocytes
Given the important role of α2ARs in the genesis of ischemic white matter damage, we proceeded to examine the subcellular distribution of these receptors immunochemically. Double labeling in RONs and dorsal columns was performed using antibodies against α2a, the most abundant α2 receptor subtype in the CNS (Zeng and Lynch, 1991) in combination with antibodies against neurofilament and GFAP. Figure 7 shows representative sections demonstrating a predominantly punctate α2a labeling pattern. Negative controls with primary antibody omitted exhibited no labeling. Positive controls were performed on sections of locus ceruleus, known to have the highest levels of α2AR expression. Clear staining observed in this area (data not shown) reinforces the specificity of our labeling approach. The distribution of α2a label in RON was heterogeneous: the highest receptor densities forming clusters of punctate labeling were seen between axonal bundles, in the space corresponding to glial localization (Fig. 7A). The overall distribution within the bundles of axons, identified with anti-neurofilament antibody, was much lower, with α2a puncta often localizing to the outer margins of neurofilament-labeled axon cylinders (Fig. 7B, arrowheads). Double labeling with GFAP revealed that some of the dense staining between axons had astroglial identity (Fig. 7C), where labeling was dense and concentrated mostly within the cell body, with less signal in the processes (Fig. 7D).
Figure 7.
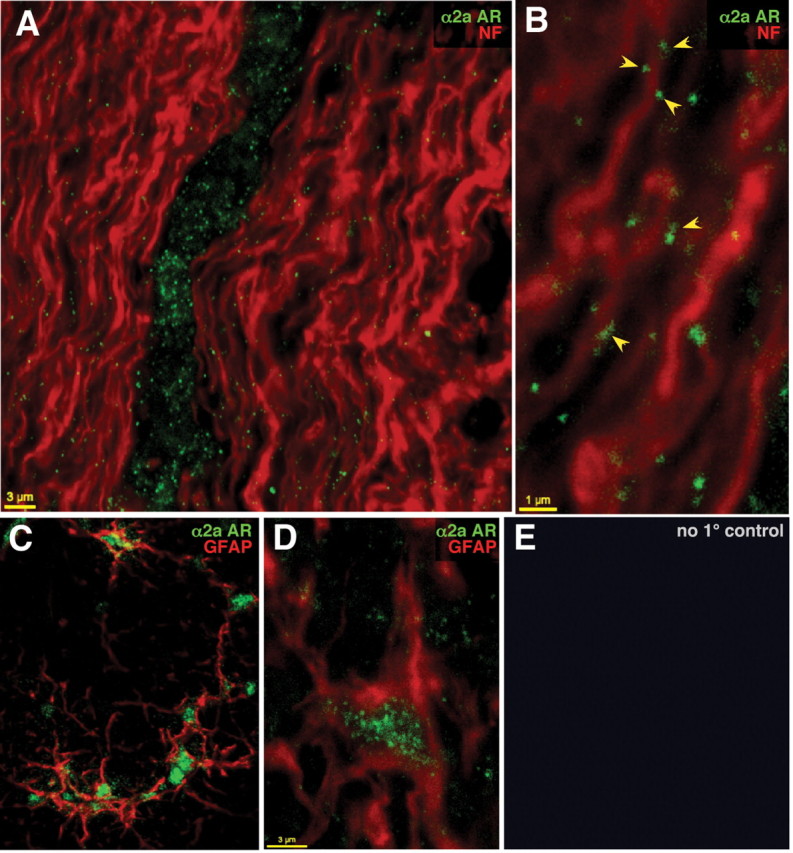
Optic nerve sections double labeled with α2a AR (green) and neurofilament (red) antibodies (A, B), andα2a AR (green) and GFAP (red) antibodies (C, D), showing punctate α2a AR in association with axonal cylinders and prominent astrocyte labeling. E, Controls with primary antibodies omitted showed no detectable signal.
To more precisely determine the spatial relationships between α2AR and axons in an attempt to confirm localization of α2ARs on the axolemma (necessary to support the hypothesis of functional axonal receptors), triple labeling for α2a AR, the α3 subunit of the Na+/K+ ATPase [known to be homogeneously distributed on the internodal axolemma of most central myelinated axons (Young et al., 2008) thus providing a reliable marker for axon membranes] and neurofilament was performed in dorsal columns (Fig. 8A) and optic nerve (Fig. 8B). On transverse sections, punctate areas of α2a labeling were often observed at the periphery of end-on axon cylinders, overlapping ring-like Na+/K+ ATPase staining representing axolemma (Fig. 8A,B, arrowheads). On Z projections, the α2a-positive clusters were frequently seen to extend in a columnar manner for a few micrometers along the length of the axon. Discrete α2a-positive regions were also seen outside of the Na+/K+ ATPase rings, representing α2ARs on glial structures. Given pharmacological evidence for the involvement of the norepinephrine transporters in the release of NE during OGD, immunostaining was performed to examine the distribution of this protein. Figure 8, C and D, shows ring-like labeling surrounding most axonal profiles. However, this signal mainly appeared outside the axolemma, colocalizing reliably with a marker for inner myelin loops (myelin-associated glycoprotein) (Sternberger et al., 1979), suggesting that this transporter is preferentially expressed in the myelin sheath. Glial processes also exhibited label (Fig. 8C1,3, arrowheads).
Figure 8.
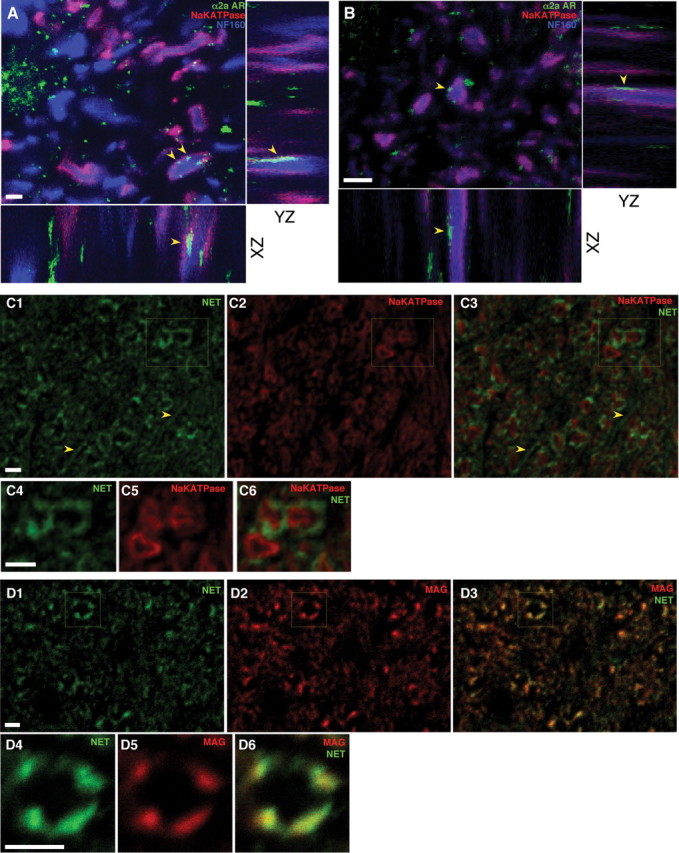
Dorsal columns (A) and optic nerve (B) sections, triple labeled with α2a adrenergic receptor (green), α3 subunit of the Na+/K+ ATPase (red) and neurofilament 160 (blue) antibodies, with XZ and YZ projection views of a confocal Z series acquired at 0.1 μm steps. Together with the Na+ and Ca2+ imaging (Figs. 5, 6), punctate α2a labeling overlapping Na+/K+ ATPase-decorated axolemma (arrowheads) provides evidence for functional axonally targeted receptors. C, Transverse sections of optic nerve labeled with NET (green) and the axolemmal marker as in A and B. C4–6, Higher-power view of the boxed area in C1–3. D, Transverse optic nerve sections stained with NET (green) and myelin-associated glycoprotein (MAG; red), which identifies the inner surface of the myelin sheath. D4–6, Higher-power view of the boxed area in D1–3. NET is mainly expressed outside the axolemma, strongly colocalizing with inner myelin loops, indicating significant localization to the sheath. Linear profiles (arrowheads in C1, 3) likely represent additional NET expression on glial processes. Controls with primary antibody omitted revealed little detectable signal (data not shown). Scale bars, 2 μm.
Discussion
The goal of the present study was to investigate the role of the noradrenergic system in the ischemic response of central white matter. We found that release of NE during oxygen glucose deprivation promotes functional damage to axons. Moreover, given our results with α2 receptor ligands affording protection against axonal injury, we conclude that OGD-mediated white matter damage is mediated at least partly by α2 receptors. The underlying mechanism of this protection involves a reduction in axonal Na+ and Ca2 +loading. The effect could be directly axonal via α2 G-protein modulation of Na+ channels and AMPA receptors and/or through α2 receptors located on astrocytes. Previous studies indicate that adrenergic receptors exert potent modulatory actions on both TTX-sensitive Na+ channels [in particular on the persistent inward current mediated by these channels (Harvey et al., 2006)] and on AMPA receptors (Pralong and Magistretti, 1995), the two major putative Na+ entry pathways in damaged myelinated axons (Stys et al., 1993; Ouardouz et al., 2006) (Fig. 6). The significant reduction in axonal Ca2+ accumulation could be secondary to a reduced Na+ influx [which will in turn restrain multiple pathways responsible for axonal Ca2+ accumulation (Stys, 2004)] and/or also due to direct modulation of Ca2+ entry mechanisms [for example, voltage-gated Ca2+ channels (Lipscombe et al., 1989)]. Overall, our results are consistent with the notion that in damaged CNS axons, the adrenergic system exerts a potent modulatory effect, likely on several pathways mediating ionic dysregulation; understanding the precise mechanisms, and the potential interplay between the many varieties of adrenergic receptors, will require further study.
Reducing NE release is protective
During cerebral ischemia, significant release of NE approaching a 40-fold rise over baseline levels has been documented (Globus et al., 1989; Gustafson et al., 1991; Sumiya et al., 2001). This high extracellular NE concentration is intensely neurotoxic (Stein and Cracco, 1982; Globus et al., 1989) and may play a key role in ischemic neuronal damage (Ma et al., 2005). The mechanism of this release under ischemic conditions involves Na+-dependent reversal of the monoamine uptake carrier (Vizi, 2000; Gerevich et al., 2001; Sumiya et al., 2001). This also seems to be the case in the current study: as in the reports cited above, it is quite likely that released extracellular NE was partially responsible for the functional axonal damage in our white matter models, because lowering tissue NE content with reserpine before OGD, or decreasing carrier-mediated NE release with desipramine or nisoxetine, both significantly reduced ischemia-induced axonal Ca2+ accumulation and improved functional recovery. Note that reserpine may have other actions, such as antioxidant effects (Chakrabarti et al., 1986), contributing to our observations; however, given that reserpine actions and those of transporter inhibitors were similar, the dominant effect is likely interference with NE release.
Agonist/antagonist effect on α2 AR-signaling pathway
Electrophysiological recording demonstrated that in our model α2 ligands protected against functional axonal injury. Moreover, many reports demonstrated the protective effect of α2AR agonists against neuronal death. For example, dexmedetomidine effectively decreased neuronal damage in a gerbil model of global cerebral ischemia (Kuhmonen et al., 1997) and in a rabbit focal model of ischemia (Maier et al., 1993). Clonidine, another α2AR agonist, improved neuronal survival from incomplete cerebral ischemia in rats (Hoffman et al., 1991). Interestingly, other studies using models of cerebral ischemia (Gustafson et al., 1989, 1990; Puurunen et al., 2001) have found α2 antagonists such as yohimbine, atipamezole and idazoxan to also be neuroprotective.
Somewhat unexpectedly, in our models of in vitro white matter injury, both agonists and antagonists were protective. We can suggest two possible explanations. One is agonist-dependent α2AR desensitization, leading to reduction of receptor signaling. α2 receptors can couple to several different G proteins, whose α and βγ subunits subsequently engage different effectors. The receptor's G-protein-coupling regions were demonstrated to be phosphorylation sites for PKC (Liang et al., 1998). This kinase phosphorylates Ser360 within the third intracellular loop of the α2a subunit (Liang et al., 2002), leading to rapid desensitization of receptor function. Because α2AR can activate PKC [via Gi-associated Gβγ-mediated activation of phospholipase C (Dorn et al., 1997)], phosphorylation by PKC may also play a role in agonist-dependent desensitization. In our experiments, PKC activation with phorbol 12-myristate-13-acetate (PMA) increased axonal recovery and the effect was not additive when PMA was combined with α2AR agonists, suggesting a common pathway. These results are in line with other studies demonstrating agonist-induced desensitization of α2ARs (Kurose and Lefkowitz, 1994; Jewell-Motz and Liggett, 1996) and may suggest one possible explanation for the protective effect of agonists in our experiments. Another possibility is related to current evidence showing that compounds that bind to G-protein-coupled receptors can either stimulate (agonists), or reduce (inverse agonists) the receptors' basal activity, depending on the affinity of ligands for different conformational states (Kenakin, 2001). The Kenakin model suggests that if a given ligand activates the system that is quiescent (no constitutive activity) by changing a receptor into an active state, it produces excitation (agonists). However, if the system exhibits significant constitutive activity, then binding of the ligand and changing a receptor into an active state, but of less efficacy, would reduce the activity, with the net effect of the “agonist” being to reduce receptor activity (inverse agonism). Examples where α2-adrenoceptor agonists act as inverse agonists are well documented, e.g., clonidine, which is often considered to be a “classic” α2-adrenoceptor agonist tool for probing the pharmacology of this receptor, produced the same effect as several selective antagonists on the release of acetylcholine in the rat prefrontal cortex (Tellez et al., 1997). Similarly, RX 801074, a partial agonist at α2-adrenoreceptors, produced a competitive antagonist effect in the CNS (Chapleo et al., 1989), as did the weak partial α2-adrenoceptor agonist levomedetomidine in human erythroleukemia cells (Jansson et al., 1998).
Action of α2 ligands on axonal Ca2+ and Na+ content
Various mechanisms of α2AR-mediated neuroprotection at nerve terminals were suggested, although the exact modes of action are not known. α2ARs belong to the G-protein-coupled receptor super family: they bind to G-proteins that inhibit adenylyl cyclase, activate K+ channels, and inhibit voltage-gated Ca2+ channels (Saunders and Limbird, 1999), leading to reduced intracellular Ca2+ accumulation. This in turn exhibits a powerful neuroprotective effect and also reduces further neurotransmitter release from nerve terminals. Lower levels of NE and glutamate release would in turn further restrain injurious events after ischemia (Bickler and Hansen, 1996; Ma et al., 2005). We found that both agonists and antagonists significantly reduced rises in axonal Ca2+. Ca2+ accumulation in ischemic axons is in part Na+-dependent (Nikolaeva et al., 2005), originating from intracellular and extracellular sources. We therefore hypothesized that α2 ligands could mediate their protective effects by restraining axonal Na+ accumulation. Indeed, blocking α2Rs with the antagonist atipamezole drastically reduced ischemic Na+ rise, suggesting that the α2-signaling pathway is coupled to a major source of Na+ entry voltage-gated Na+ channels. The persistent Na+ channel is one route of Na+ influx during ischemia, and this persistent current was shown to be upregulated by monoamine receptors in rat spinal motoneurons (Harvey et al., 2006). Our findings are consistent with the notion that noradrenergic receptors in white matter axons can modulate persistent Na+ current during ischemia, thus leading to increased Na+ entry and secondarily to a greater Ca2+ accumulation and more functional damage. Interestingly, direct measurements of axonal [Na+] revealed that blocking Na+ channels with TTX was less effective at reducing Na+ influx than α2R modulation (Fig. 6), suggesting that α2Rs also modulate other important Na+ influx pathways. Indeed, blocking AMPA receptors in addition to Na+ channels with SYM2206 together with TTX abolished ischemic Na+ rise completely, similar to the effects of α2R modulators, suggesting that α2Rs also influence AMPA receptors, directly or indirectly. This property places α2Rs in an ideal position to restrain deleterious ion fluxes in ischemic axons and may represent a very attractive point for therapeutic intervention.
Conclusion
This study shows that OGD-induced release of NE plays an important role in the pathophysiology of central white matter. The NE release was likely due to a carrier-mediated mechanism given the beneficial role of NET inhibitors; indeed, α2AR modulation itself, resulting in a profound reduction in axonal Na+ accumulation, may have in turn secondarily reduced NE release. A reduction in Na+ overload could be a key mechanism underlying the protective effect of α2 receptor ligands because it leads to a decrease in Ca2+ influx and Ca2+-related injurious events; a key consequence of reduced axonal Na+ entry would be a concomitant decrease in exchanger-mediated Ca2+ entry and in carrier-mediated release of glutamate (Li et al., 1999) and possibly other substances. The sources of axonal Na+ overload (Na+ channels and AMPA receptors) could be affected directly by α2AR located on the axonal membrane. The results of this study provide new insights into the involvement of the noradrenergic system in axonal survival in central white matter and suggest exploration of noradrenergic-based therapeutic strategies.
Footnotes
This work was supported in part by Canadian Institutes of Health Research and the Multiple Sclerosis Society of Canada (operating), and the Heart and Stroke Foundation of Ontario Center for Stroke Recovery (equipment). P.K.S. was supported by the Heart and Stroke Foundation of Ontario Career Investigator and the Alberta Heritage Foundation for Medical Research Scientist Awards.
References
- Bhardwaj A, Brannan T, Martinez-Tica J, Weinberger J. Ischemia in the dorsal hippocampus is associated with acute extracellular release of dopamine and norepinephrine. J Neural Transm Gen Sect. 1990;80:195–201. doi: 10.1007/BF01245121. [DOI] [PubMed] [Google Scholar]
- Bickler PE, Hansen BM. Alpha 2-adrenergic agonists reduce glutamate release and glutamate receptor-mediated calcium changes in hippocampal slices during hypoxia. Neuropharmacology. 1996;35:679–687. doi: 10.1016/0028-3908(96)84639-9. [DOI] [PubMed] [Google Scholar]
- Chakrabarti S, Kumar S, Shankar R. Reserpine inhibition of lipid peroxidation and protein phosphorylation in rat brain. Biochem Pharmacol. 1986;35:1611–1613. doi: 10.1016/0006-2952(86)90135-8. [DOI] [PubMed] [Google Scholar]
- Chapleo CB, Butler RC, England DC, Myers PL, Roach AG, Smith CF, Stillings MR, Tulloch IF. Heteroaromatic analogues of the alpha 2-adrenoreceptor partial agonist clonidine. J Med Chem. 1989;32:1627–1630. doi: 10.1021/jm00127a037. [DOI] [PubMed] [Google Scholar]
- Dixon WR, Mosimann WF, Weiner N. The role of presynatpic feedback mechanisms in regulation of norepinephrine release by nerve stimulation. J Pharmacol Exp Ther. 1979;209:196–204. [PubMed] [Google Scholar]
- Dorn GW, 2nd, Oswald KJ, McCluskey TS, Kuhel DG, Liggett SB. Alpha 2A-adrenergic receptor stimulated calcium release is transduced by Gi-associated G(beta gamma)-mediated activation of phospholipase C. Biochemistry. 1997;36:6415–6423. doi: 10.1021/bi970080s. [DOI] [PubMed] [Google Scholar]
- Gerevich Z, Tretter L, Adam-Vizi V, Baranyi M, Kiss JP, Zelles T, Vizi ES. Analysis of high intracellular [Na+]-induced release of [3H]noradrenaline in rat hippocampal slices. Neuroscience. 2001;104:761–768. doi: 10.1016/s0306-4522(01)00102-6. [DOI] [PubMed] [Google Scholar]
- Globus MY, Busto R, Dietrich WD, Martinez E, Valdés I, Ginsberg MD. Direct evidence for acute and massive norepinephrine release in the hippocampus during transient ischemia. J Cereb Blood Flow Metab. 1989;9:892–896. doi: 10.1038/jcbfm.1989.123. [DOI] [PubMed] [Google Scholar]
- Gustafson I, Miyauchi Y, Wieloch TW. Postischemic administration of idazoxan, an alpha-2 adrenergic receptor antagonist, decreases neuronal damage in the rat brain. J Cereb Blood Flow Metab. 1989;9:171–174. doi: 10.1038/jcbfm.1989.25. [DOI] [PubMed] [Google Scholar]
- Gustafson I, Westerberg E, Wieloch T. Protection against ischemia-induced neuronal damage by the alpha 2-adrenoceptor antagonist idazoxan: influence of time of administration and possible mechanisms of action. J Cereb Blood Flow Metab. 1990;10:885–894. doi: 10.1038/jcbfm.1990.145. [DOI] [PubMed] [Google Scholar]
- Gustafson I, Westerberg EJ, Wieloch T. Extracellular brain cortical levels of noradrenaline in ischemia: effects of desipramine and postischemic administration of idazoxan. Exp Brain Res. 1991;86:555–561. doi: 10.1007/BF00230528. [DOI] [PubMed] [Google Scholar]
- Harvey PJ, Li X, Li Y, Bennett DJ. Endogenous monoamine receptor activation is essential for enabling persistent sodium currents and repetitive firing in rat spinal motoneurons. J Neurophysiol. 2006;96:1171–1186. doi: 10.1152/jn.00341.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman WE, Cheng MA, Thomas C, Baughman VL, Albrecht RF. Clonidine decreases plasma catecholamines and improves outcome from incomplete ischemia in the rat. Anesth Analg. 1991;73:460–464. doi: 10.1213/00000539-199110000-00016. [DOI] [PubMed] [Google Scholar]
- Honmou O, Young W. Norepinephrine modulates excitability of neonatal rat optic nerves through calcium-mediated mechanisms. Neuroscience. 1995;65:241–251. doi: 10.1016/0306-4522(94)e0132-n. [DOI] [PubMed] [Google Scholar]
- Jansson CC, Kukkonen JP, Näsman J, Huifang G, Wurster S, Virtanen R, Savola JM, Cockcroft V, Akerman KE. Protean agonism at alpha2A-adrenoceptors. Mol Pharmacol. 1998;53:963–968. [PubMed] [Google Scholar]
- Jewell-Motz EA, Liggett SB. G protein-coupled receptor kinase specificity for phosphorylation and desensitization of alpha2-adrenergic receptor subtypes. J Biol Chem. 1996;271:18082–18087. doi: 10.1074/jbc.271.30.18082. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Inverse, protean, and ligand-selective agonism: matters of receptor conformation. FASEB J. 2001;15:598–611. doi: 10.1096/fj.00-0438rev. [DOI] [PubMed] [Google Scholar]
- Kuhmonen J, Pokorný J, Miettinen R, Haapalinna A, Jolkkonen J, Riekkinen P, Sr, Sivenius J. Neuroprotective effects of dexmedetomidine in the gerbil hippocampus after transient global ischemia. Anesthesiology. 1997;87:371–377. doi: 10.1097/00000542-199708000-00025. [DOI] [PubMed] [Google Scholar]
- Kurose H, Lefkowitz RJ. Differential desensitization and phosphorylation of three cloned and transfected alpha 2-adrenergic receptor subtypes. J Biol Chem. 1994;269:10093–10099. [PubMed] [Google Scholar]
- Langer SZ. Presynaptic regulation of catecholamine release. Biochem Pharmacol. 1974;23:1793–1800. doi: 10.1016/0006-2952(74)90187-7. [DOI] [PubMed] [Google Scholar]
- Laudenbach V, Mantz J, Lagercrantz H, Desmonts JM, Evrard P, Gressens P. Effects of alpha(2)-adrenoceptor agonists on perinatal excitotoxic brain injury: comparison of clonidine and dexmedetomidine. Anesthesiology. 2002;96:134–141. doi: 10.1097/00000542-200201000-00026. [DOI] [PubMed] [Google Scholar]
- Li S, Mealing GA, Morley P, Stys PK. Novel injury mechanism in anoxia and trauma of spinal cord white matter: glutamate release via reverse Na+-dependent glutamate transport. J Neurosci. 1999;19:RC16. doi: 10.1523/JNEUROSCI.19-14-j0002.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang M, Eason MG, Jewell-Motz EA, Williams MA, Theiss CT, Dorn GW, 2nd, Liggett SB. Phosphorylation and functional desensitization of the alpha2A-adrenergic receptor by protein kinase C. Mol Pharmacol. 1998;54:44–49. doi: 10.1124/mol.54.1.44. [DOI] [PubMed] [Google Scholar]
- Liang M, Eason MG, Theiss CT, Liggett SB. Phosphorylation of Ser360 in the third intracellular loop of the alpha2A-adrenoceptor during protein kinase C-mediated desensitization. Eur J Pharmacol. 2002;437:41–46. doi: 10.1016/s0014-2999(02)01280-3. [DOI] [PubMed] [Google Scholar]
- Lipscombe D, Kongsamut S, Tsien RW. Alpha-adrenergic inhibition of sympathetic neurotransmitter release mediated by modulation of N-type calcium-channel gating. Nature. 1989;340:639–642. doi: 10.1038/340639a0. [DOI] [PubMed] [Google Scholar]
- Ma D, Rajakumaraswamy N, Maze M. alpha2-Adrenoceptor agonists: shedding light on neuroprotection? Br Med Bull. 2005;71:77–92. doi: 10.1093/bmb/ldh036. [DOI] [PubMed] [Google Scholar]
- Maier C, Steinberg GK, Sun GH, Zhi GT, Maze M. Neuroprotection by the alpha 2-adrenoreceptor agonist dexmedetomidine in a focal model of cerebral ischemia. Anesthesiology. 1993;79:306–312. doi: 10.1097/00000542-199308000-00016. [DOI] [PubMed] [Google Scholar]
- Malek SA, Coderre E, Stys PK. Aberrant chloride transport contributes to anoxic/ischemic white matter injury. J Neurosci. 2003;23:3826–3836. doi: 10.1523/JNEUROSCI.23-09-03826.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandela P, Ordway GA. The norepinephrine transporter and its regulation. J Neurochem. 2006;97:310–333. doi: 10.1111/j.1471-4159.2006.03717.x. [DOI] [PubMed] [Google Scholar]
- Martel J, Chopin P, Colpaert F, Marien M. Neuroprotective effects of the alpha2-adrenoceptor antagonists (+)-efaroxan and (+/-)-idazoxan, against quinolinic acid-induced lesions of the rat striatum. Exp Neurol. 1998;154:595–601. doi: 10.1006/exnr.1998.6942. [DOI] [PubMed] [Google Scholar]
- Milner TA, Lee A, Aicher SA, Rosin DL. Hippocampal alpha2a-adrenergic receptors are located predominantly presynaptically but are also found postsynaptically and in selective astrocytes. J Comp Neurol. 1998;395:310–327. [PubMed] [Google Scholar]
- Nikolaeva MA, Mukherjee B, Stys PK. Na+-dependent sources of intra-axonal Ca2+ release in rat optic nerve during in vitro chemical ischemia. J Neurosci. 2005;25:9960–9967. doi: 10.1523/JNEUROSCI.2003-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouardouz M, Malek S, Coderre E, Stys PK. Complex interplay between glutamate receptors and intracellular Ca2+ stores during ischaemia in rat spinal cord white matter. J Physiol. 2006;577:191–204. doi: 10.1113/jphysiol.2006.116798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris A, Mantz J, Tonner PH, Hein L, Brede M, Gressens P. The effects of dexmedetomidine on perinatal excitotoxic brain injury are mediated by the alpha2A-adrenoceptor subtype. Anesth Analg. 2006;102:456–461. doi: 10.1213/01.ane.0000194301.79118.e9. [DOI] [PubMed] [Google Scholar]
- Perego C, Gatti S, Vetrugno GC, Marzatico F, Algeri S. Correlation between electroencephalogram isoelectric time and hippocampal norepinephrine levels, measured by microdialysis, during ischemia in rats. J Neurochem. 1992;59:1257–1262. doi: 10.1111/j.1471-4159.1992.tb08435.x. [DOI] [PubMed] [Google Scholar]
- Pralong E, Magistretti PJ. Noradrenaline increases K-conductance and reduces glutamatergic transmission in the mouse entorhinal cortex by activation of alpha 2-adrenoreceptors. Eur J Neurosci. 1995;7:2370–2378. doi: 10.1111/j.1460-9568.1995.tb01034.x. [DOI] [PubMed] [Google Scholar]
- Puurunen K, Jolkkonen J, Sirviö J, Haapalinna A, Sivenius J. An alpha(2)-adrenergic antagonist, atipamezole, facilitates behavioral recovery after focal cerebral ischemia in rats. Neuropharmacology. 2001;40:597–606. doi: 10.1016/s0028-3908(00)00182-9. [DOI] [PubMed] [Google Scholar]
- Saunders C, Limbird LE. Localization and trafficking of alpha2-adrenergic receptor subtypes in cells and tissues. Pharmacol Ther. 1999;84:193–205. doi: 10.1016/s0163-7258(99)00032-7. [DOI] [PubMed] [Google Scholar]
- Schuldiner S, Shirvan A, Linial M. Vesicular neurotransmitter transporters: from bacteria to humans. Physiol Rev. 1995;75:369–392. doi: 10.1152/physrev.1995.75.2.369. [DOI] [PubMed] [Google Scholar]
- Stein SC, Cracco RQ. Cortical injury without ischemia produced by topical monoamines. Stroke. 1982;13:74–83. doi: 10.1161/01.str.13.1.74. [DOI] [PubMed] [Google Scholar]
- Sternberger NH, Quarles RH, Itoyama Y, Webster HD. Myelin-associated glycoprotein demonstrated immunocytochemically in myelin and myelin-forming cells of developing rat. Proc Natl Acad Sci U S A. 1979;76:1510–1514. doi: 10.1073/pnas.76.3.1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stys PK. White matter injury mechanisms. Curr Mol Med. 2004;4:113–130. doi: 10.2174/1566524043479220. [DOI] [PubMed] [Google Scholar]
- Stys PK, Lopachin RM. Mechanisms of ion flux in anoxic myelinated CNS axons. Neuroscience. 1998;82:21–32. doi: 10.1016/s0306-4522(97)00230-3. [DOI] [PubMed] [Google Scholar]
- Stys PK, Ransom BR, Waxman SG. Compound action potential of nerve recorded by suction electrode: a theoretical and experimental analysis. Brain Res. 1991;546:18–32. doi: 10.1016/0006-8993(91)91154-s. [DOI] [PubMed] [Google Scholar]
- Stys PK, Sontheimer H, Ransom BR, Waxman SG. Non-inactivating, TTX-sensitive Na+ conductance in rat optic nerve axons. Proc Natl Acad Sci U S A. 1993;90:6976–6980. doi: 10.1073/pnas.90.15.6976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumiya Y, Torigoe K, Gerevich Z, Köfalvi A, Vizi ES. Excessive release of [3H] noradrenaline by veratridine and ischemia in spinal cord. Neurochem Int. 2001;39:59–63. doi: 10.1016/s0197-0186(00)00124-8. [DOI] [PubMed] [Google Scholar]
- Talke P, Bickler PE. Effects of dexmedetomidine on hypoxia-evoked glutamate release and glutamate receptor activity in hippocampal slices. Anesthesiology. 1996;85:551–557. doi: 10.1097/00000542-199609000-00014. [DOI] [PubMed] [Google Scholar]
- Talley EM, Rosin DL, Lee A, Guyenet PG, Lynch KR. Distribution of alpha 2A-adrenergic receptor-like immunoreactivity in the rat central nervous system. J Comp Neurol. 1996;372:111–134. doi: 10.1002/(SICI)1096-9861(19960812)372:1<111::AID-CNE8>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Tellez S, Colpaert F, Marien M. Acetylcholine release in the rat prefrontal cortex in vivo: modulation by alpha 2-adrenoceptor agonists and antagonists. J Neurochem. 1997;68:778–785. doi: 10.1046/j.1471-4159.1997.68020778.x. [DOI] [PubMed] [Google Scholar]
- Venugopalan VV, Ghali Z, Sénécal J, Reader TA, Descarries L. Catecholaminergic activation of G-protein coupling in rat spinal cord: further evidence for the existence of dopamine and noradrenaline receptors in spinal grey and white matter. Brain Res. 2006;1070:90–100. doi: 10.1016/j.brainres.2005.10.101. [DOI] [PubMed] [Google Scholar]
- Virtanen R, Savola JM, Saano V. Highly selective and specific antagonism of central and peripheral alpha 2-adrenoceptors by atipamezole. Arch Int Pharmacodyn Ther. 1989;297:190–204. [PubMed] [Google Scholar]
- Vizi ES. Role of high-affinity receptors and membrane transporters in nonsynaptic communication and drug action in the central nervous system. Pharmacol Rev. 2000;52:63–89. [PubMed] [Google Scholar]
- Young EA, Fowler CD, Kidd GJ, Chang A, Rudick R, Fisher E, Trapp BD. Imaging correlates of decreased axonal Na+/K+ ATPase in chronic multiple sclerosis lesions. Ann Neurol. 2008;63:428–435. doi: 10.1002/ana.21381. [DOI] [PubMed] [Google Scholar]
- Zeng DW, Lynch KR. Distribution of alpha 2-adrenergic receptor mRNAs in the rat CNS. Brain Res Mol Brain Res. 1991;10:219–225. doi: 10.1016/0169-328x(91)90064-5. [DOI] [PubMed] [Google Scholar]