

Abstract
In inflammatory diseases occurring outside the CNS, communication between the periphery and the brain via humoral and/or neural routes results in central neural changes and associated behavioral alterations. We have recently identified another immune-to-CNS communication pathway in the setting of organ-centered peripheral inflammation: namely, the entrance of immune cells into the brain. In our current study, using a mouse model of inflammatory liver injury, we have confirmed the significant infiltration of activated monocytes into the brain in mice with hepatic inflammation and have defined the mechanism that mediates this trafficking of monocytes. Specifically, we show that in the presence of hepatic inflammation, mice demonstrate elevated cerebral monocyte chemoattractant protein (MCP)-1 levels, as well as increased numbers of circulating CCR2-expressing monocytes. Cerebral recruitment of monocytes was abolished in inflamed mice that lacked MCP-1/CCL2 or CCR2. Furthermore, in mice with hepatic inflammation, microglia were activated and produced MCP-1/CCL2 before cerebral monocyte infiltration. Moreover, peripheral tumor necrosis factor (TNF)α signaling was required to stimulate microglia to produce MCP-1/CCL2. TNFα signaling via TNF receptor 1 (TNFR1) is required for these observed effects since in TNFR1 deficient mice with hepatic inflammation, microglial expression of MCP-1/CCL2 and cerebral monocyte recruitment were both markedly inhibited, whereas there was no inhibition in TNFR2 deficient mice. Our results identify the existence of a novel immune-to-CNS communication pathway occurring in the setting of peripheral organ-centered inflammation which may have specific implications for the development of alterations in cerebral neurotransmission commonly encountered in numerous inflammatory diseases occurring outside the CNS.
Keywords: cytokine, sickness behaviors, chemokine, CCR2, CCL2, inflammation
Introduction
Changes in behavior (termed sickness behaviors) that result from changes in central neural activity are often encountered during acute systemic infections and are felt to be an important adaptive mechanism for survival (Dantzer and Kelley, 2007). Such behavioral changes are also seen in inflammatory diseases originating within the CNS including multiple sclerosis (MS) (Gold and Irwin, 2006; Heesan et al., 2006), as well as in chronic organ specific or more generalized inflammatory diseases that occur outside of the CNS, such as rheumatoid arthritis (Maini et al., 2004; Mayoux-Benhamou, 2006), inflammatory bowel disease (Gralnek et al., 2000; Lichtenstein et al., 2002), psoriasis (Tyring et al., 2006) and liver disease (Kumar and Tandon, 2002; Swain, 2006). However, in such chronic diseases, these changes in behavior can be maladaptive and have a major impact on the quality of life of patients. Despite their prevalence, the etiology of such symptoms in the setting of systemic inflammatory diseases is poorly understood. However, to mediate changes within the CNS, pathways of communication must exist between the periphery and the CNS. Studies of fever and the neuroendocrine response to peripheral inflammatory insults have established that the periphery can communicate with the CNS via humoral and/or neural routes (Romanovsky et al., 2005; Hopkins, 2007).
Trafficking of immune cells into the CNS is a cardinal pathological feature of cerebral inflammatory diseases (Hemmer et al., 2004; Hult et al., 2008). Importantly, changes in neurotransmission that give rise to behavioral alterations in such diseases occur before the development of overt pathological CNS tissue damage, but after cerebral immune cell infiltration has occurred (Pollak et al., 2003). In previous studies, we observed that such an immune-to-brain communication pathway also exists in mice with peripheral organ specific inflammatory injury. Specifically, using a well characterized model of hepatic inflammation due to surgical bile flow disruption (Abe et al., 2004), we found a marked increase in the number of leukocytes that were rolling and adhering along the cerebral endothelium in inflamed mice (Kerfoot et al., 2006). This was paralleled by a striking fivefold increased recruitment of activated monocytes into the brains of these mice. Such an immune mediated communication pathway with the CNS had not previously been defined in an organ specific inflammatory disease occurring outside of the CNS. The potential significance of this pathway was further highlighted by our observation that the monocytes transmigrating into the brain produced the cytokine tumor necrosis factor (TNF)α, a potent neuromodulator with multiple effector functions that has been strongly implicated in the genesis of sickness behaviors (Simen et al., 2006; Gosselin and Rivest, 2007).
Therefore, we performed a series of experiments to determine important potential pathway(s) involved in recruiting monocytes into the brain in the setting of peripheral hepatic inflammation. Here, we show that peripheral TNFα signaling stimulates monocyte chemoattractant protein (MCP)-1/CCL2 production in microglia which drives the subsequent infiltration of CCR2-expressing monocytes into the brain. Moreover, inhibition of cerebral monocyte recruitment significantly improved sickness behavior in mice with hepatic inflammation.
Materials and Methods
Model of peripheral organ-specific inflammatory injury.
For all experiments, we used male C57BL/6 mice (6–8 weeks of age), purchased from The Jackson Laboratory: MCP-1/CCL2 knock-out (KO), CCR2 KO, Tumor necrosis factor receptor 1 (TNFR1) KO and TNFR2 KO mice (all on a C57BL/6 genetic background) were also purchased from The Jackson Laboratory. Mice were housed in a light-controlled room maintained at 22°C with a 12 h light/dark cycle and had ad libitum access to food and water. All procedures in this study were approved by the Animal Care Committee of the University of Calgary and conformed to the guidelines established by the Canadian Council on Animal Care.
The experimental model of organ specific inflammatory injury used was the well characterized model of hepatic inflammation resulting from bile duct ligation and resection as previously described (Koniaris et al., 2001; Sewnath et al., 2002; Abe et al., 2004; Kerfoot et al., 2006). Briefly, laparotomy was performed under halothane anesthesia and the bile duct was isolated, doubly ligated and resected between the ligatures [bile duct resected (BDR)]. In sham controls, the bile duct was identified and manipulated without ligation or resection (sham). This model was chosen to represent peripheral organ specific inflammatory injury as it has been well characterized at both the cellular and biochemical levels, and has also been associated with elevated circulating levels of a number of cytokines, including TNFα (Bemelmans et al., 1996; Fernández-Martinez et al., 2006). In addition, BDR rodents exhibit alterations in cerebral neurotransmission as well as the development of well defined sickness behaviors which include anorexia, lethargy and a loss of social interest (Swain et al., 1993, 1998; Swain and Maric, 1995, 1997; Swain and Le, 1998; Burak et al., 2002). Experiments were performed on day 5 and day 10 postsurgery at which time BDR mice had developed overt liver injury (but not liver failure or cirrhosis).
Biochemical analysis.
Blood was collected by cardiac puncture from halothane anesthetized sham and BDR mice into tubes containing EDTA. Blood samples were centrifuged for 20 min and plasma collected. Alanine aminotransferase (ALT) levels were quantified using a commercial ALT kit (Biotron Labs) and bilirubin levels were determined using a commercial bilirubin kit (Biotron Labs). ALT is released from damaged hepatocytes and as such reflects the degree of hepatic inflammatory injury in BDR mice. Serum bilirubin levels were measured to confirm adequate disruption of bile flow by the BDR surgery, as well as for a secondary measure of disruption of liver cell function in response to liver injury.
Peripheral blood monocyte isolation and flow cytometry analysis.
Whole blood was collected from sham and BDR mice and peripheral blood mononuclear cells were isolated using Lympholyte M (Cedarlane) according to the manufacturer's instructions. Monocytes were characterized by flow cytometry (FACSscan; Becton Dickinson) and identified by surface expression of F4/80 (FITC-labeled antibody, Serotec). Two color staining was used to identify MCP-1/CCL2-producing or CCR2-expressing monocytes. Briefly, cells were first preincubated with anti-mouse CD16/32 antibody (BD Biosciences) for 15 min at 4°C to block FCγRs and then stained with FITC-labeled F4/80 antibody for 30 min at 4°C. For intracellular staining of MCP-1/CCL2, cells were permeabilized with Cytofix/Cytoperm (BD Biosciences) and then incubated with a PE-labeled MCP-1/CCL2 monoclonal antibody for 45 min (BD Biosciences). CCR2-expressing monocytes were identified as those cells coexpressing F4/80 and surface CCR2. For CCR2 staining, after FCγRs were blocked (as above), cells were simultaneously incubated with PE-labeled F4/80 antibody (Serotec) and a primary rabbit CCR2 monoclonal IgG antibody (Epitomics) for 30 min at 4°C. A FITC-labeled goat anti-rabbit secondary antibody (Jackson ImmunoResearch Labs) was then applied for 45 min at room temperature.
Cerebral immune cell isolation and flow cytometry analysis.
Mononuclear cells were isolated from the cerebral cortices of sham and BDR mice using previously described protocols (Campanella et al., 2002; Kerfoot et al., 2006). Briefly, mice were anesthetized with halothane and perfused with ice-cold PBS. The brain was removed and the cortex separated and homogenized with a dounce homogenizer in cold PBS. Brain tissue was digested in collagenase (Collagenase A, Roche Diagnostics) for 30 min at 37°C and then passed through a 40 μm wire mesh (Sigma-Aldrich). The homogenate was then centrifuged at 200 g for 10 min at 4°C. The resulting pellet was resuspended in 4 ml of 70% isotonic Percoll (Amersham Biosciences) which was overlaid with 37% and 30% isotonic Percoll. The Percoll gradient was centrifuged at 500 g for 20 min at 4°C. Mononuclear cells were collected from the 70/37% interface and washed with PBS. To identify monocytes and microglia, isolated mononuclear cells were first preincubated with anti-mouse CD16/32 antibody for 15 min at 4°C to block FCγRs and then simultaneously stained with FITC-labeled anti-CD11b (BD Biosciences) and PerCP-labeled anti-CD45 antibodies (BD Biosciences) for 30 min at 4°C. Since CNS microglia are of monocytic origin, it is important to be able to distinguish between resident microglia and CNS infiltrating monocytes. Flow cytometry is a quantitative technique that can be used to differentiate between these respective cell types based on expression of CD45 (i.e., CNS infiltrating monocytes are Mac1/CD11b+ CD45high-expressing cells, whereas microglia are Mac 1/CD11b+ CD45low-expressing cells) (Babcock et al., 2003; Kerfoot et al., 2006).
To identify MCP-1/CCL2-expressing monocytes and microglia, mononuclear cells were simultaneously stained with CD45 and CD11b antibodies (as above), permeabilized with Cytofix/Cytoperm and then incubated with PE-labeled MCP-1/CCL2 antibody for 45 min at 4°C. CCR2-expressing monocytes and microglia were identified by simultaneous surface expression of CD45, CD11b and CCR2 (staining as above).
For peripheral TNFα blocking studies, BDR mice were injected intraperitoneally with 0.5 ml of either normal rabbit serum or anti-TNFα serum (kindly provided by Dr. C. Hogoboam, University of Michigan, Ann Arbor, MI) on days 2 and 4 postsurgery (Huffnagle et al., 1996; McCullough et al., 2000; Neff et al., 2003). MCP-1/CCL2 expression in microglia was examined (as above) on day 5 postsurgery (see Results).
For inhibition of cerebral monocyte recruitment, BDR mice were injected with anti-P selectin (RB40–34, 20 μg/mouse; BD Biosciences) and anti-α4 integrin (R1–2, 70 μg/mouse, BD Biosciences) antibodies on days 6 and 8 postsurgery as previously described (Kerfoot et al., 2006). Control BDR mice received rat IgG antibody. Cerebral mononuclear cells were isolated on day 10 post surgery and characterized by flow cytometry as outlined above.
Cerebral MCP-1/CCL2 levels.
Cerebral MCP-1/CCL2 protein levels were determined in brain tissue homogenates by ELISA. Briefly, cerebral cortical tissue was obtained from day 10 sham and BDR mice and homogenized immediately in cold PBS buffer containing protease mixture inhibitor (Sigma-Aldrich). Tissue homogenates were then centrifuged twice at 10,000 RPM for 20 min at 4°C. The resulting supernatant was filtered through a 0.45 μm filter (Sigma-Aldrich) and then stored at −80°C until ready for analysis by a commercially available ELISA (R&D Systems). Total protein levels were determined using a Bio-Rad protein colorimetric assay (Bio-Rad Laboratories).
Immunohistochemistry for assessment of microglial activation by morphology.
Mice at day 5 postsurgery (this time point was chosen since in day 5 BDR mice we found no significant increase in cerebral monocyte infiltration compared with sham controls; see Results) were anesthetized with halothane and perfused transcardially with PBS followed by 4% paraformaldehyde. Brains were removed and postfixed in the same fixative overnight. Brain tissue was then cryoprotected with increasing concentrations of sucrose solutions. For immunohistochemical staining, frozen tissue sections were cut at 12 μm using a cryostat. To detect microglia, sections were stained with an antibody directed against the ionized calcium-binding adapter molecule-1 (Iba-1) which recognizes both microglia and macrophages (Ahmed et al., 2007). Briefly, sections were washed three times in PBS and then 10% normal goat serum (Sigma-Aldrich) was applied for 30 min. After a PBS wash, the primary rabbit anti-Iba1 antibody (Wako) was diluted to 1:1000 and sections were incubated for 2 h at room temperature. Sections were then thoroughly washed with PBS and incubated with a secondary Alexa Flour 488 goat anti-rabbit IgG antibody (Invitrogen) for 1 h at room temperature. After a final wash with PBS, sections were mounted in glycerol and viewed using a confocal microscope (Fluoview, Version 1.6; Olympus America). Resting microglia were identified as “ramified” microglia with long processes while primed/activated microglia were identified as microglial cells that were more rounded, with retracted and shorter processes.
CFSE labeling and adoptive transfer of monocytes.
Peripheral blood mononuclear cells were isolated from donor day 8 BDR, and sham mice as described above and monocytes were then purified by magnetic cell sorting with CD11b microbeads (Miltenyi Biotech). Briefly, peripheral blood mononuclear cells were incubated with CD11b microbeads for 20 min at 4°C. CD11b-positive monocytes were then sorted using an autoMACS separator (Miltenyi Biotech). Purified monocytes were then incubated with carboxyfluorescein diacetate succinimidyl ester (CFSE, Invitrogen) dye for 30 min at 37°C followed by PBS wash. Approximately 60,000 CFSE-labeled monocytes were then injected per mouse in 200 μl of PBS intravenously by retro-orbital injection into respective recipient day 8 BDR or sham mice (Wong et al., 2006). At day 10 postsurgery, brains were removed and embedded in paraffin. For immunohistochemical staining, brain sections (6 μm) underwent heat-induced antigen retrieval in citrate buffer and then the same procedure as outlined for Iba1 immunohistochemistry was followed. Von Willebrand Factor (vWF) was used to identify cerebral vascular endothelium. The primary rabbit anti-vWF antibody (Abcam) was diluted to 1:5000, and sections were incubated for 2 h at room temperature. Sections were then thoroughly washed with PBS and incubated with a secondary Alexa Flour 555 goat anti-rabbit IgG antibody (Invitrogen) for 1 h at room temperature. Separate brain sections from BDR mice infused with CFSE-labeled monocytes were also stained with Ly6C, a marker that identifies endothelial cells as well as infiltrating monocytes but is found at low to undetectable levels on resident cerebral microglia (Mildner et al., 2007). For Ly6C staining, the primary rat anti-Ly6C antibody (Santa Cruz Biotechnology) was applied for 18 h at room temperature followed by incubation with a secondary Alexa Flour 594 goat anti-rat IgG antibody (Invitrogen). Sections were mounted in glycerol and viewed using a confocal microscope (Fluoview, Version 1.6; Olympus America).
Sickness behavior assessment.
For these studies, BDR mice (treated with anti-P selectin and anti-α4 integrin antibodies to inhibit cerebral monocyte recruitment, or isotype control IgG, as outlined above) were used at day 10 post surgery. Sham mice at day 10 postsurgery were also included in the study for comparison to the BDR mice groups. To confirm the inhibition of cerebral monocyte infiltration, BDR mice were killed at day 10 post surgery and cerebral mononuclear cells were isolated as described above.
Mice were housed in individual cages for 24 h before sickness behavior assessment. Behavioral observations were performed between 7:00 A.M. and 10:00 A.M. by two blinded observers. Sickness behavior was assessed by methods used extensively for the quantification of sickness behavior in mice (Swain and Le, 1998; Bluthé et al., 1999; Hayley et al., 1999; Bluthé et al., 2000; Johnston et al., 2005; O'Connor et al., 2005) and included (1) social investigation behavior (2) duration of immobility of the test mouse during the observation period, and (3) changes in food intake. Social investigation was quantitated by recording the time an adult mouse spent investigating a novel congenic juvenile mouse (∼3 weeks old) which was introduced into the home cage of the test mouse. To minimize aggressive interactions which tend to occur when isolated male mice are presented with juveniles, the juvenile mouse was confined in a small Plexiglas box (8 × 8 × 10 cm) which was placed in the corner of the cage of the test mouse as described by Bluthé et al. (1999). Two holes (1.5 cm diameter) were made (one per wall) in the Plexiglas box to allow the adult test mouse to smell the juvenile mouse. The following were recorded by the 2 observers for each adult mouse: (1) the duration of the time the adult mouse spent investigating the juvenile mouse (i.e., the total time in seconds the adult mouse introduced its nose into the holes in the Plexiglas box or touched and sniffed the Plexiglas box) (2) the total number of interactions of the adult mouse with the juvenile mouse during the observation period (i.e., total number of approaches of the test mouse toward the juvenile mouse), and (3) the total time in seconds that the adult mouse remained immobile after the introduction of the juvenile mouse into its home cage during the observation period. The observation period lasted for 5 min and the means for each behavior for the 2 observers were calculated for each test mouse. To assess food intake, food pellets were preweighed and then placed in a corner of the test mouse cage in a spill-proof container, and the food pellets which remained were weighed 12 h later (i.e., each mouse was exposed to the weighed food pellets from 7:00 P.M. to 7:00 A.M.).
Statistical analysis.
Data are expressed as mean ± SEM. For comparison between two means, a Student's unpaired t test was used. For comparison between sham mice and isotype control or anti-P selectin and anti-α4 integrin antibody-treated BDR mice groups, behavioral data were analyzed by a one-way ANOVA followed by Tukey post hoc test. A p value <0.05 was considered significant.
Results
Characterization of murine model of hepatic inflammatory injury
Day 10 BDR mice showed biochemical evidence of marked inflammatory liver cell injury as reflected by a ∼70-fold increase in plasma ALT levels (sham: 18.1 ± 3.4 IU/L vs BDR: 1262.6 ± 91.2 IU/L; n = 10/group, p < 0.0001). In addition, significant impairment of bile flow was confirmed in day 10 BDR mice as reflected by an ∼29-fold elevation in plasma total bilirubin levels (sham: 0.6 ± 0.1 mg/dl vs BDR: 17.5 ± 0.7 mg/dl, n = 4/group; p ≤ 0.05). Day 5 BDR mice showed a ∼24-fold increase in plasma ALT levels (sham: 10.3 ± 2.7 IU/L vs BDR: 244.7 ± 73.5 IU/L BDR, n = 8/group; p < 0.05) and an ∼21-fold elevation in plasma total bilirubin levels (sham: 0.4 ± 0.2 mg/dl vs BDR: 8.2 ± 0.5 mg/dl; n = 4/group, p < 0.05).
Monocytes are recruited into the brains of mice with hepatic inflammation
In our current study, we confirmed our previous finding of a significant ∼8-fold increased recruitment of monocytes into the brains of mice with hepatic inflammation (Fig. 1). Furthermore, we used immunohistochemistry and confocal microscopy to confirm that monocytes were indeed entering the brain parenchyma of day 10 BDR mice. To do this, monocytes were isolated and purified from the peripheral blood of donor day 8 BDR mice, labeled in vitro with CFSE, and then injected intravenously into recipient day 8 BDR mice. The recipient BDR mice were then killed at day 10 post surgery (as outlined above). CFSE-labeled monocytes were readily identified in brain sections from BDR mice (Figs. 2, 3, 4), but were not found in the brains of sham mice infused with CFSE-labeled monocytes isolated from the peripheral blood of donor sham mice (supplemental material, available at www.jneurosci.org). Moreover, CFSE-labeled monocytes in the brains of BDR mice were also Ly6C positive [a cell surface marker for monocytes, but not microglia (Mildner et al., 2007)] and were clearly identified mainly within the cerebral parenchyma (Fig. 4G–I). However, Ly6C-immunostained CFSE-labeled monocytes were also occasionally found to be adherent to cerebral endothelium (Fig. 4D–F,G–I,J–L) or present in close proximity to cerebral blood vessels (Fig. 4A–C). CFSE-labeled monocytes were observed in perivascular and periventricular regions, both of which serve as potential routes of leukocyte entry into the brain in CNS inflammatory diseases (Ubogu et al., 2006) (Figs. 2, 4). In addition, Figure 2, A and B, depicts CFSE-labeled monocytes in the choroid plexus and in the subfornical organ region, areas that could also serve as possible routes of leukocyte entry into the brain (Ubogu et al., 2006). We consistently found the most readily identifiable parenchymal CFSE-labeled monocytes in BDR mice to be present in specific regions of the brain, including the basal ganglia, hippocampus and motor cortex (Fig. 3). CFSE-labeled monocytes were found less frequently within the brain parenchyma in other brain regions examined. Interestingly, these three brain areas have been implicated in the regulation of behavior and therefore our current findings could have implications for behavioral changes seen in BDR mice (Dantzer, 2006; Dantzer et al., 2008). Furthermore, the CFSE-labeled adoptively transferred monocytes typically had a rounded or rod-like morphology, similar to that reported recently by Getts et al. (2008). Alternatively, adoptive transfer studies with SNARF-1-labeled BDR monocytes in recipient day 10 BDR mice also demonstrated findings similar to those made using CFSE-labeled monocytes (supplemental material, available at www.jneurosci.org).
Figure 1.

Monocytes infiltrate the brains of BDR mice. A, B, Representative flow cytometry profiles from sham (A) and BDR (B) mouse at day 10 postsurgery. Resident microglia were identified as CD45low CD11b+ cells (bottom right quadrant). Cerebral infiltrating monocytes were identified as CD45high CD11b+ cells (top right quadrant) and were clearly distinguishable from the CD45low CD11b+ cells. The inserted number shows the percentage of total brain mononuclear cells that are CD45high CD11b+. C, Solid bars represent the percentage of total brain mononuclear cells isolated from day 10 sham and BDR mice that are CD45high CD11b+. Hatched bars represent the total number of CD45high CD11b+ cells in the brains of day 10 sham and BDR mice. Error bars represent mean ± SEM of data from 9 sham and 9 BDR mice; *p < 0.05 versus sham controls.
Figure 2.
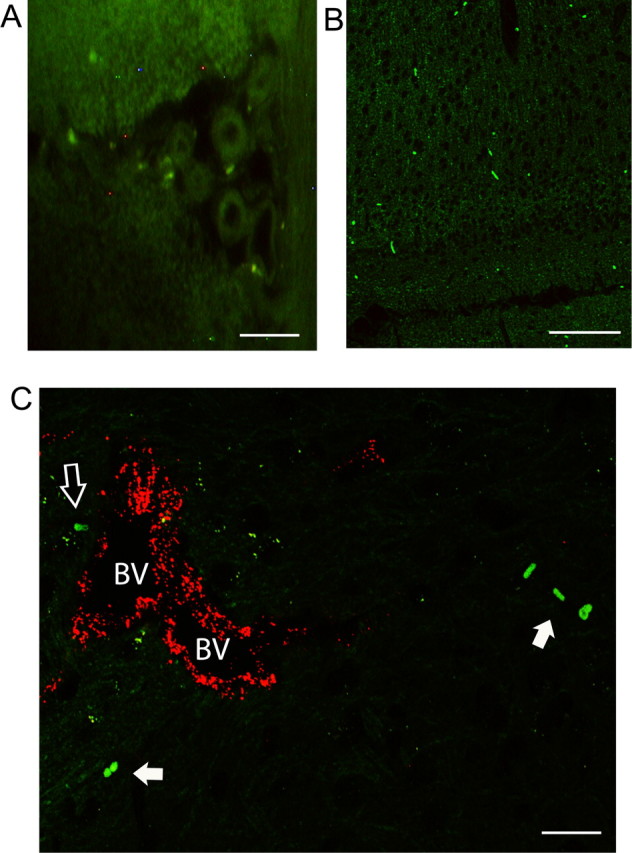
Detection of CFSE-labeled monocytes in brains of day 10 BDR mice. CFSE-labeled monocytes were observed within the parenchyma, most prominently in the periventricular and perivascular regions, in brains of day 10 recipient BDR mice. A, CFSE-labeled monocytes present in the lumen of the choroid plexus and also within the cerebral parenchymal area surrounding the choroid plexus. B, CFSE-labeled monocytes present in the cerebral parenchyma in the subfornical organ region of the brain (both the choroid plexus and the subfornical organ are potential routes used for leukocyte transmigration into brain parenchyma). C, Immunohistochemical staining for vWF (labels cerebral vascular endothelium) demonstrated that CFSE-labeled monocytes were present within the cerebral parenchyma (closed arrow) in areas clearly distinct from cerebral blood vessels, and were also observed in close proximity to cerebral blood vessels (open arrow). A, B, C, Brain sections from n = 3 day 10 BDR mice. C, A blood vessel in the periventricular region in a day 10 BDR mouse. Scale bars: A, C, 5 μm; B, 20 μm. BV, Blood vessel.
Figure 3.
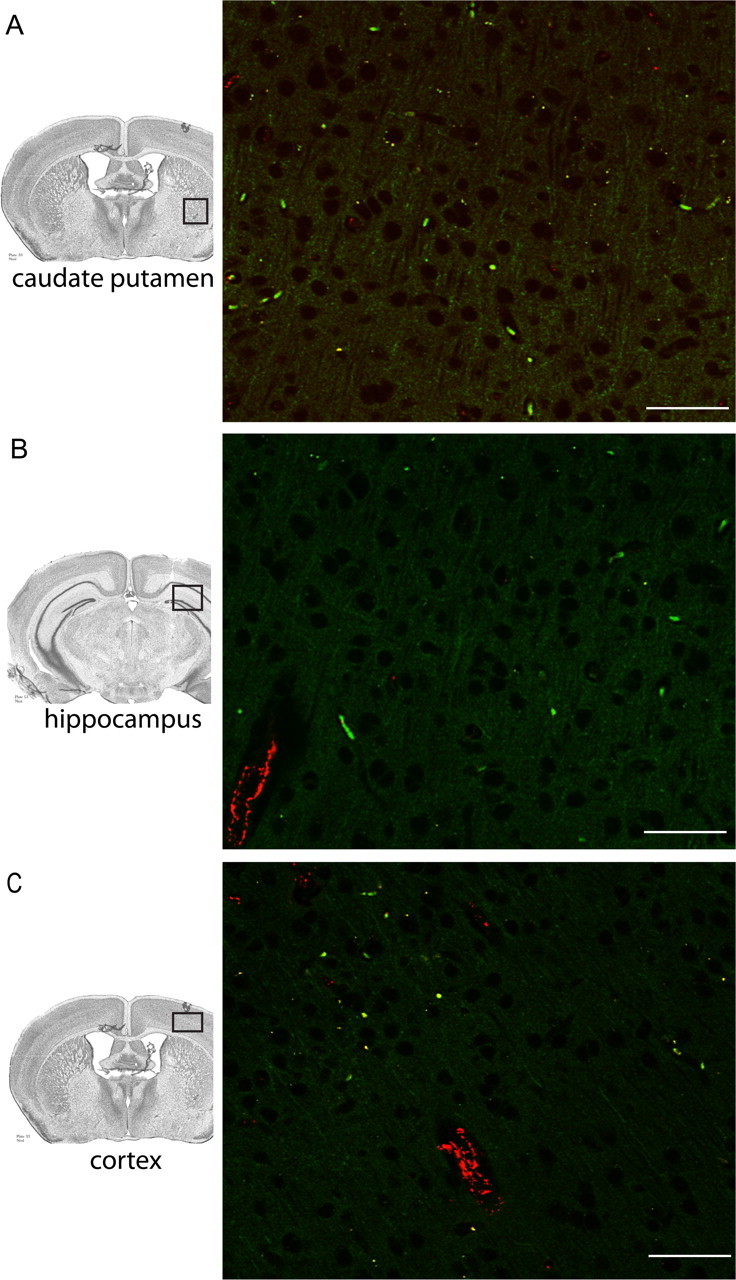
Region-specific detection of CFSE-labeled monocytes in BDR mice. Large numbers of CFSE-labeled monocytes were most readily detected in the cerebral parenchyma of BDR mice in specific regions of the brain, including the basal ganglia (A), hippocampus (B) and motor cortex (C). Immunostaining for vWF was used to identify blood vessels in all three panels. Positive VWF staining is depicted by red immunofluorescence. A, B, C, Brain sections from n = 3 day 10 BDR mice. Scale bar, 20 μm. BV, Blood vessel.
Figure 4.
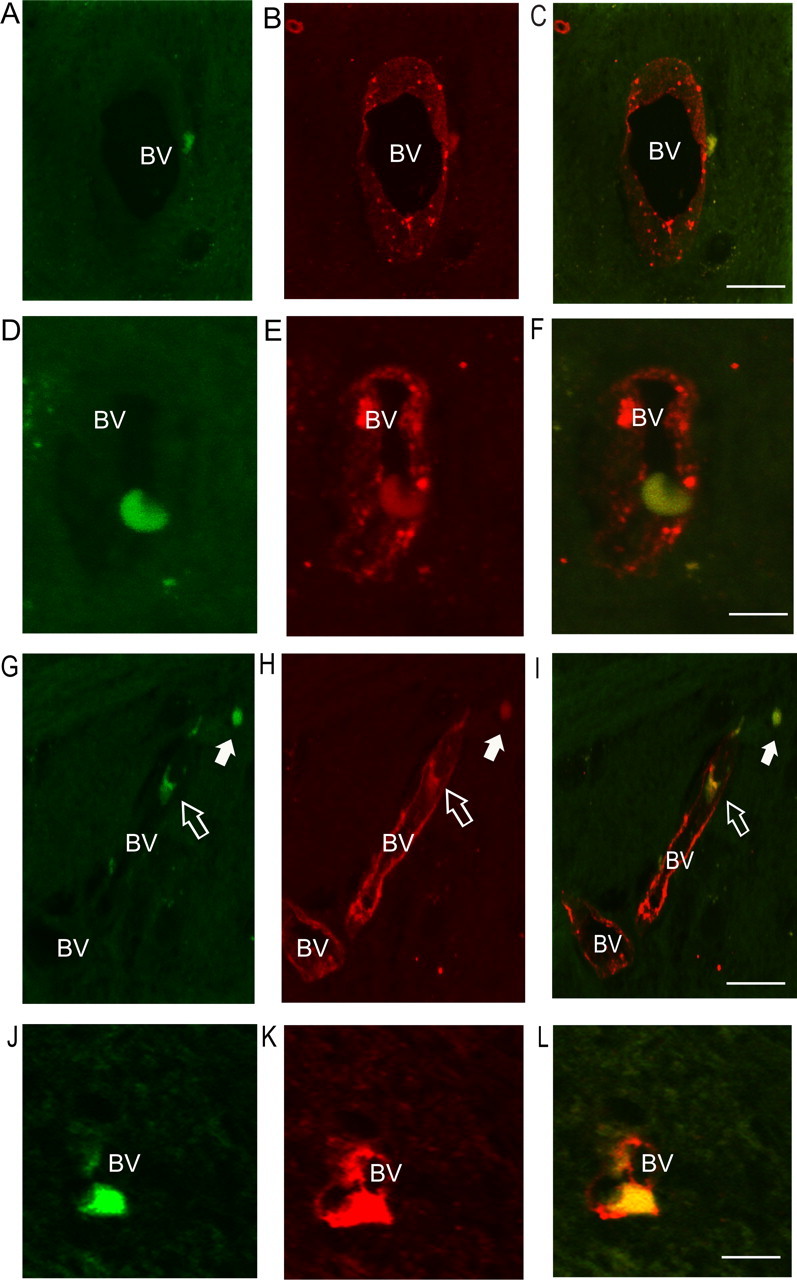
Immunohistochemical detection of Ly6C-stained endothelium and CFSE-labeled monocytes in the brains of day 10 BDR mice. A–C, A Ly6C-positive CFSE-labeled monocyte present in the perivascular region in the brain of a day 10 BDR mouse. D–F, A Ly6C-positive CFSE-labeled monocyte that appears to be adherent to the cerebral blood vessel endothelium. G–I, A Ly6C-positive CFSE-labeled monocyte that appears to be adherent to the blood vessel (open arrow) and a Ly6C-positive CFSE-labeled monocyte that is present within the cerebral parenchyma (closed arrow). J–L, A Ly6C-immunostained CFSE-labeled monocyte that appears to be adherent to the blood vessel wall or in the process of transmigrating out of the blood vessel and into the brain parenchyma. All panels are representative of blood vessels in the periventricular region in brain sections from n = 3 day 10 BDR mice. CFSE-labeled monocytes are indicated in the panels on the left (A, D, G, J). Ly6C-immunostained cerebral endothelium and monocytes are indicated by red fluorescence in the middle panels. Combined images of Ly6C-immunostained CFSE-labeled monocytes (i.e., images from respective left and middle panels were superimposed) are demonstrated in yellow (panels on the right, C, F, I, L). Scale bar, 5 μm. BV, Blood vessel.
MCP-1/CCL2, via its action on CCR2, mediates monocyte recruitment into the brains of mice with hepatic inflammation
We next wanted to determine how monocytes were being recruited into the brains of day 10 BDR mice. When we measured cerebral chemokine levels, we found cerebral MCP-1/CCL2 levels to be ∼2-fold higher in day 10 BDR (10.9 ± 2.1 ng/ml) versus sham (5.2 ± 0.6 ng/ml; n = 5–6 mice/group, p < 0.05) mice. To respond to MCP-1/CCL2, cells need to express CCR2 (the cognate receptor for MCP-1/CCL2) (Charo and Ransohoff, 2006). Monocytes are a heterogeneous population and inflammation is usually associated with an increase in an inflammatory subtype which expresses CCR2 (Geissmann et al., 2003; Gordon and Taylor, 2005; Tacke and Randolph, 2006). We found that compared with day 10 sham controls there was an ∼2.5-fold elevation in the number of peripheral-circulating monocytes that expressed surface CCR2 in day 10 BDR mice (Fig. 5A). In addition, virtually all of the monocytes that transmigrated into the brains of day 10 BDR mice expressed surface CCR2. Interestingly, in contrast to our findings for brain infiltrating monocytes, there was a decrease in the number of microglia that expressed CCR2 in day 10 BDR versus sham mice (Fig. 5B).
Figure 5.

MCP-1/CCL2 via its actions on CCR2 mediates monocyte recruitment into the brains of BDR mice. A, Solid bars represent the percentage of total peripheral blood monocytes (PBMs)-expressing CCR2 in day 10 sham and BDR mice. Hatched bars represent the total number of CCR2-expressing PBMs isolated from day 10 sham and BDR mice. Error bars represent mean ± SEM of data from 6 sham and 9 BDR mice; *p < 0.05 versus sham controls. B, Solid bars represent the percentage of total brain microglia that express CCR2 in day 10 sham and BDR mice. Hatched bars represent the total number of CCR2-expressing microglia isolated from the brains of day 10 sham and BDR mice. Error bars represent the mean ± SEM of data from 6 sham and 9 BDR mice; *p < 0.05 versus sham controls.
If MCP-1/CCL2, via its action on CCR2, mediates the recruitment of monocytes into the brains of BDR mice, then in the absence of MCP-1/CCL2 or CCR2 we would expect monocyte infiltration to be significantly reduced. Therefore, we examined monocyte migration into the brains of day 10 MCP-1/CCL2 KO BDR mice and day 10 CCR2 KO BDR mice. We found that in day 10 MCP-1/CCL2 KO BDR mice, the number of CD45high CD11b+ cells (i.e., infiltrating monocytes) isolated from the brains of these mice was significantly reduced compared with day 10 WT BDR mice, and resembled levels observed in day 10 sham controls (Fig. 6A–C). Similar observations were made using day 10 CCR2 KO BDR mice in which cerebral monocyte recruitment was once again significantly reduced compared with day 10 WT BDR mice, to levels similar to those observed in day 10 sham controls (Fig. 6D–F). Furthermore, deficiency of MCP-1/CCL2 or CCR2 did not affect the severity of inflammatory liver injury as reflected by measurements of plasma ALT levels (day 10 MCP-1/CCL2 KO BDR: 1700.1 ± 390 IU/L vs WT BDR: 1211.9 ± 96 IU/L, n = 3/group, p > 0.05; day 10 CCR2 KO BDR: 951.7 ± 85 IU/L vs WT BDR: 1154 ± 92 IU/L, n = 4/group, p > 0.05) and bilirubin levels (day 10 MCP-1/CCL2 KO BDR: 11.1 ± 2.3 mg/dl vs WT BDR 15.0 ± 0.8 mg/dl, n = 3/group, p > 0.05; day 10 CCR2 KO BDR: 17.8 ± 4.5 mg/dl vs WT BDR: 15.9 ± 1.1 mg/dl, n = 4/group, p > 0.05).
Figure 6.
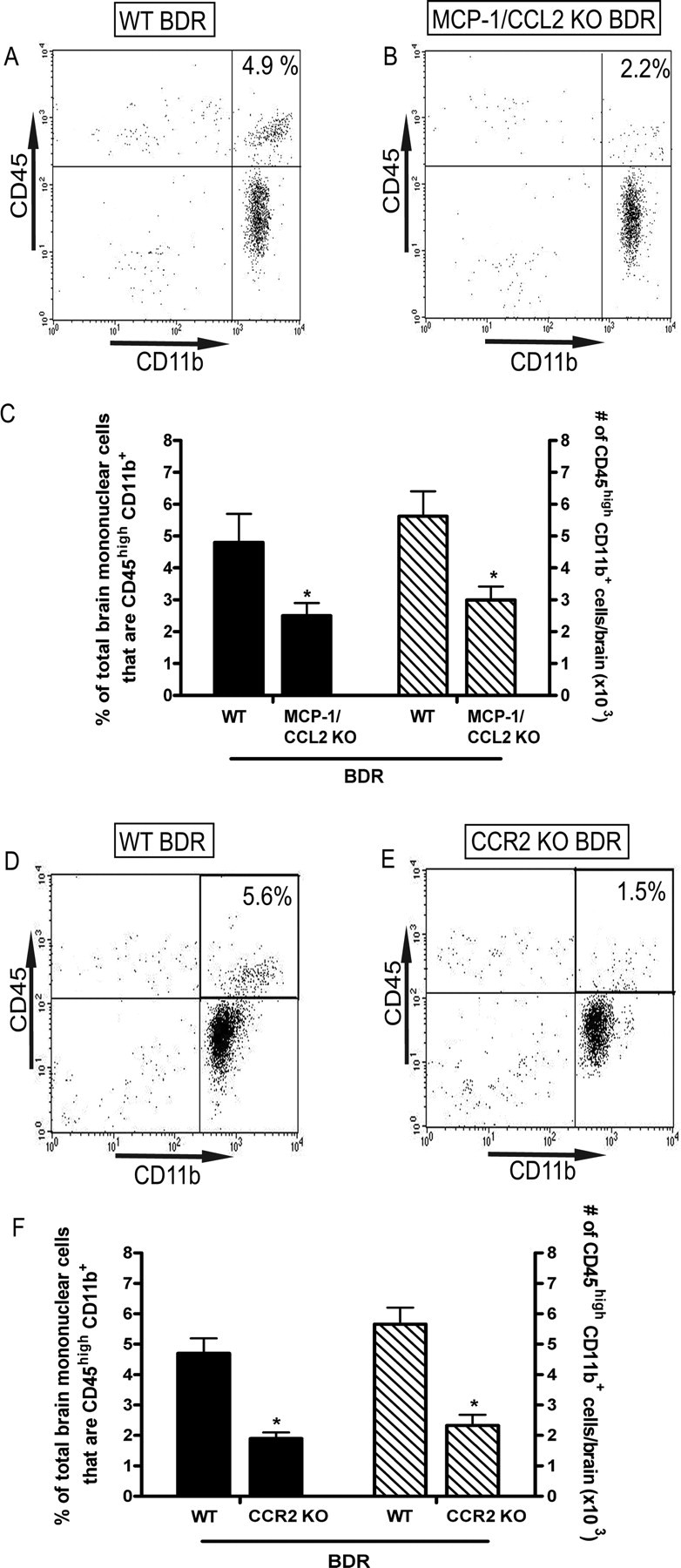
Cerebral monocyte recruitment in day 10 BDR mice is inhibited in the absence of MCP-1/CCL2 or CCR2. Representative flow cytometry profiles from day 10 WT BDR (A) versus MCP-1/CCL2 KO BDR (B) mice. The inserted number shows the percentage of total brain mononuclear cells that are CD45high CD11b+ (i.e., infiltrating monocytes). C, Solid bars represent the percentage of isolated brain mononuclear cells that are CD45high CD11b+ in day 10 WT BDR and MCP-1/CCL2 KO BDR mice. Hatched bars represent the total number of CD45high CD11b+ cells isolated from the brains of day 10 WT BDR and MCP-1/CCL2 KO BDR mice. Error bars represent the mean ± SEM of data from 4 WT BDR and 3 MCP-1/CCL2 KO BDR mice; *p < 0.05 versus WT BDR mice. Representative flow cytometry profiles from day 10 WT BDR (D) versus CCR2 KO BDR (E) mice. The inserted number shows the percentage of total brain mononuclear cells that are CD45high CD11b+. F, Solid bars represent the percentage of total brain mononuclear cells that are CD45high CD11b+ in day 10 WT BDR and CCR2 KO BDR mice. Hatched bars represent the total number of CD45high CD11b+ cells isolated from the brains of day 10 WT BDR and CCR2 KO BDR mice. Bars represent the mean ± SEM of data from 4 WT BDR and 4 CCR2 KO BDR mice; *p < 0.05 versus WT BDR mice.
Cerebral sources of MCP-1/CCL2 in day 10 BDR mice
Our data confirms a key role for MCP-1/CCL2 in mediating the trafficking of monocytes into the brains of BDR mice. We next determined whether microglia are an important cellular source of MCP-1/CCL2 production within the brains of day 10 BDR mice. In cerebral inflammatory diseases, microglia are the main intrinsic immunoeffector cells of the brain and are significant producers of a wide array of chemokines, including MCP-1/CCL2 (Kielian, 2004; Garden and Möller, 2006). Specifically, in CNS inflammatory pathologies such as MS and human immunodeficiency virus associated dementia, where cerebral infiltration of monocytes is a cardinal pathological feature, microglia are a potent cerebral source of MCP-1/CCL2 (Mahad and Ransohoff, 2003; Hult et al., 2008). We documented an ∼3-fold elevation in the number of microglia that expressed MCP-1/CCL2 in the brains of day 10 BDR compared with sham mice (Fig. 7). Moreover, we also identified a marked increase in the number of cerebral transmigrating monocytes that expressed MCP-1/CCL2 in day 10 BDR versus sham mice (Fig. 7).
Figure 7.

Cerebral sources of MCP-1/CCL2 in day 10 BDR mice. Solid bars represent the total number of microglia expressing MCP-1/CCL2 in the brains of day 10 BDR and sham mice. Hatched bars represent the total number of CD45high CD11b+ cells (i.e., infiltrating monocytes) isolated from the brains of day 10 sham and BDR mice that express MCP-1/CCL2. Error bars represent mean ± SEM of data from 7 sham and 10 BDR mice; *p < 0.05 versus sham controls.
Microglia are activated to produce MCP-1/CCL2 which mediates the subsequent recruitment of monocytes into the brain in BDR mice
Our current data indicates that MCP-1/CCL2 drives the recruitment of monocytes into the brains of BDR mice, and that microglia are a significant source of MCP-1/CCL2 produced within the brain. However, it is important to determine whether microglial production of MCP-1/CCL2 in BDR mice occurs before the infiltration of monocytes into the brain, or develops as a consequence of the entrance of monocytes into the brain. Therefore, we wanted to determine whether microglia were activated first (i.e., before monocyte recruitment) to release MCP-1/CCL2 and thereby mediate the subsequent recruitment of monocytes from peripheral blood into the brain, or whether microglial activation occurs concurrently with or after monocyte recruitment into the brains of BDR mice. To do this, we examined monocyte infiltration into the brain, as well as microglial MCP-1/CCL2 expression, in BDR mice at 5 d postsurgery. In contrast to day 10 BDR mice, we did not document a significant increase in the number of CD45high CD11b+ cells (i.e., infiltrating monocytes) isolated from the brains of day 5 BDR mice compared with sham controls (Fig. 8A–C). However, we found a significant ∼2-fold increase in the number of microglial cells that expressed MCP-1/CCL2 in day 5 BDR mice (Fig. 8D). These data strongly suggest that microglia within the brains of BDR mice are stimulated to produce MCP-1/CCL2 prior to cerebral monocyte recruitment, and therefore are consistent with MCP-1/CCL2 produced by microglia driving the subsequent infiltration of monocytes into the brains of BDR mice.
Figure 8.

Microglia are stimulated to produce MCP-1/CCL2 before monocytes are recruited into the brains of BDR mice. A, B, Representative flow cytometry profiles from day 5 sham (A) and BDR (B) mice. The inserted number shows the percentage of total brain mononuclear cells that are CD45high CD11b+ (i.e., infiltrating monocytes). C, Solid bars represent the percentage of total brain mononuclear cells that are CD45high CD11b+ in day 5 sham and BDR mice. Hatched bars represent the total number of CD45high CD11b+ cells isolated from the brains of day 5 sham and BDR mice. Error bars represent mean ± SEM of data from 5 sham and 5 BDR mice, p > 0.05 versus sham mice. D, Solid bars represent the percentage of total brain microglia that express MCP-1/CCL2 in day 5 sham and BDR mice. Hatched bars represent the total number of cerebral microglia expressing MCP-1/CCL2 in day 5 sham and BDR mice. Error bars represent mean ± SEM of data from 5 sham and 5 BDR mice; *p < 0.05 versus sham controls.
Histological evidence for microglial activation in day 5 BDR mice
We examined cerebral microglia for morphological evidence of activation in day 5 BDR versus sham mice using immunohistochemistry. Iba1 immunohistochemical staining was used to identify microglia in brain sections from day 5 sham and BDR mice. Using this technique, we observed that, in general, microglia in sham mice had a “resting ramified” morphology, with longer processes. In contrast, cerebral microglia in BDR mice were generally more rounded and appeared “primed” in appearance with retracted and thicker processes, typical of activated microglia (Ahmed et al., 2007) (Fig. 9). In day 5 BDR mice, there was diffuse microglial activation (as demonstrated by their primed appearance) apparent throughout the brain; however, there was a preponderance of activated microglia located in the periventricular regions (Fig. 9C,D) and in the areas close to the blood vessels (Fig. 9E,F) compared with sham controls. Although Iba1 cannot discriminate between microglia and macrophages, our flow cytometry data demonstrates that there is no significant increase in recruitment of monocytes into the brain in day 5 BDR mice, and therefore the Iba1-positive cells very likely identify microglia alone.
Figure 9.
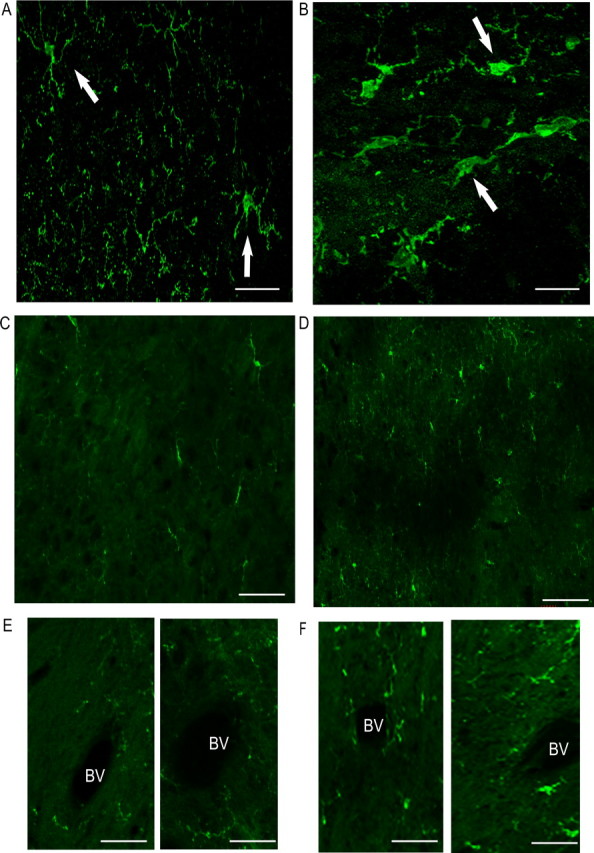
Immunohistochemical staining for Iba1-positive microglia in day 5 BDR mice. In general, microglia in sham mice (A, C, E) have a resting ramified morphology with longer processes while microglia in BDR mice (B, D, F) are more rounded with retracted and thicker processes typical of activated microglial cells. Compared with sham controls, in BDR mice there was a dominance of activated microglia in the periventricular region [sham (C); BDR (D)] and in the area close to the blood vessels [sham (E); BDR (F)]. A, B, Microglia in the periventricular regions in respective day 10 sham and BDR mice and demonstrate the typical morphology (indicated by arrows) seen for microglia in sham (A) and BDR (B) mice. C, D, The periventricular region in sham (C) and BDR (D) mice. E, F, An area close to the blood vessel in sham (E) and BDR (F) mice. All pictures are representative of brain sections from 3 sham and 3 BDR mice. Scale bars: A, B, 5 μm; C–F, 20 μm. BV, Blood vessel.
TNFα signaling via TNFR1 stimulates cerebral microglia to produce MCP-1 to subsequently mediate monocyte recruitment into the brain in BDR mice
Our findings indicate that microglia are stimulated to produce MCP-1/CCL2 before monocytes transmigrate into the brains of BDR mice. Therefore, we wanted to identify the possible pathway(s) leading to the early activation of cerebral microglia in day 5 BDR mice. TNFα is a potent inducer of MCP-1/CCL2 production (Thibeault et al., 2001; Harkness et al., 2003), and we have previously documented an elevation in the number of TNFα-expressing peripheral blood monocytes in day 10 BDR mice (Kerfoot et al., 2006). When we examined mice on day 5 postsurgery, we found that there was an ∼2-fold increase in the number of TNFα-producing peripheral-circulating monocytes in BDR mice (1.2 × 106 ± 0.7 cells/brain) versus sham controls (0.6 × 106 ± 0.2 cells/brain; n = 5/group, p < 0.05). Therefore, we next wanted to determine if peripheral TNFα, possibly signaling via cerebral endothelial cells, was important for stimulating microglia to produce MCP-1/CCL2 in day 5 BDR mice. To do this, we treated BDR mice systemically (i.e., by intraperitoneal injection on days 2 and 4 postsurgery) with either anti-TNFα serum or normal rabbit serum and then killed the mice on day 5 postsurgery. We found that the number of microglia that expressed MCP-1/CCL2 was significantly reduced in BDR mice that received anti-TNFα serum (to levels similar to those observed in sham controls) compared with BDR mice that received normal rabbit serum (Fig. 10).
Figure 10.

Peripheral TNFα signaling stimulates microglia to produce MCP-1/CCL2 in day 5 BDR mice. Solid bars represent the percentage of total cerebral microglia that express MCP-1/CCL2 in day 5 BDR mice that had received either normal rabbit serum (NRS) or anti-TNFα serum intraperitoneally on days 2 and 4 postsurgery. Hatched bars represent the total number of MCP-1/CCL2-expressing cerebral microglia in day 5 BDR mice treated with either NRS or anti-TNFα serum on days 2 and 4 postsurgery. Error bars represent mean ± SEM of data from 4 NRS-treated BDR and 3 anti-TNFα serum-treated BDR mice; *p < 0.05 versus NRS-treated BDR mice.
TNFα mediates its biological effects via two known receptors, TNFR1 and TNFR2 (Aggarwal, 2003; Sriram and O'Callaghan, 2007). To determine if TNFα signaling acts via TNFR1 or TNFR2 to stimulate microglia to produce MCP-1/CCL2 and subsequently recruit monocytes into the brain, we studied TNFR1 KO and TNFR2 KO mice at day 10 postsurgery; the time point at which we had documented a significant increase in monocyte infiltration into the brains of WT BDR mice. We found a marked reduction in the number of CD45high CD11b+ cells (i.e., infiltrating monocytes) isolated from the brains of day 10 TNFR1 KO BDR mice compared with WT BDR mice (Fig. 11A–C). In addition, we found that the number of MCP-1/CCL2-expressing cerebral microglia was also significantly reduced in day 10 TNFR1 KO BDR versus WT BDR mice (Fig. 11D). In contrast, there was no significant difference between the number of CD45high CD11b+ cells isolated from the brains of day 10 TNFR2 KO BDR compared with WT BDR mice (Fig. 11E–G), and in parallel to this observation, the number of MCP-1/CCL2-expressing microglia was also similar in day 10 TNFR2 KO BDR (7.6 × 103 ± 0.9 cells/brain) and WT BDR mice (8.5 × 103 ± 1.0 cells/brain; n = 5/group, p > 0.05).
Figure 11.
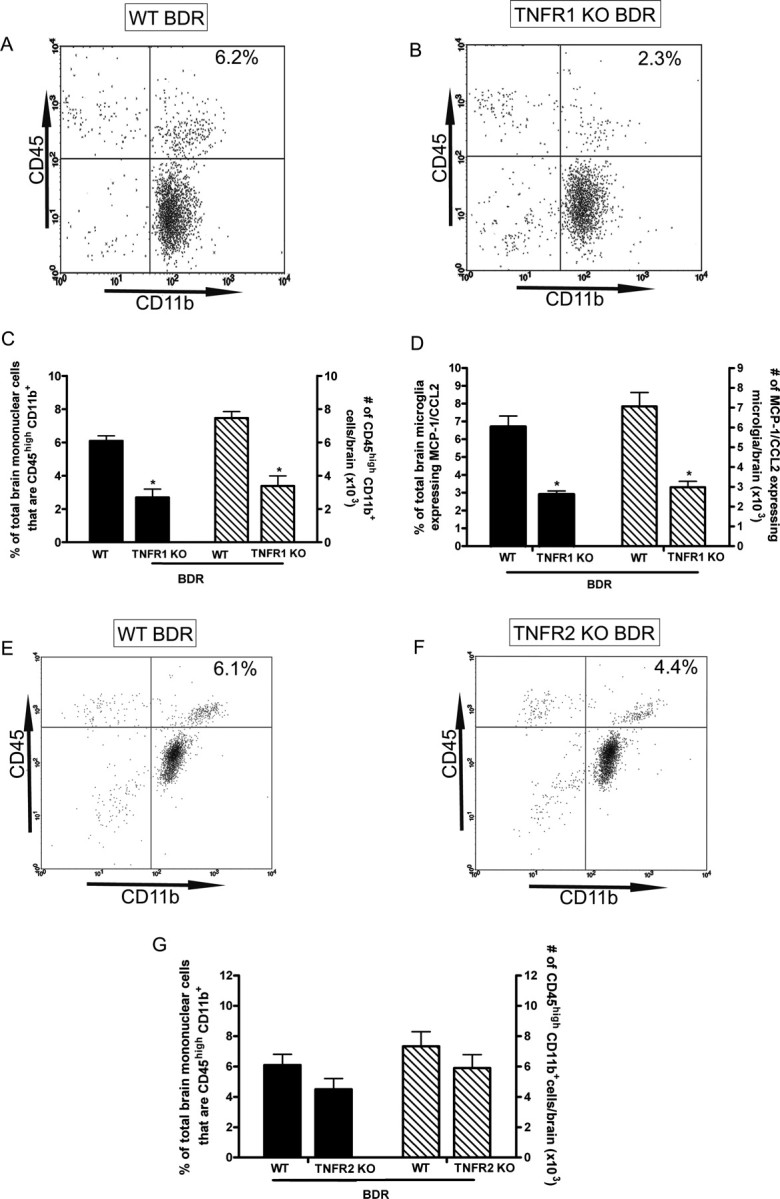
TNFα signaling via TNFR1 stimulates microglia in BDR mice to produce MCP-1/CCL2 and to subsequently recruit monocytes into the brain. A, B, Representative flow cytometry profiles from day 10 WT BDR (A) and TNFR1 KO BDR (B) mice. The inserted number shows the percentage of total brain mononuclear cells that are CD45high CD11b+ (i.e., infiltrating monocytes). C, Solid bars represent the percentage of total brain mononuclear cells that are CD45high CD11b+ in day 10 WT BDR and TNFR1 KO BDR mice. Hatched bars represent the total number of CD45high CD11b+ cells isolated from the brains of day 10 WT BDR and TNFR1 KO BDR mice. Error bars represent mean ± SEM of data from 9 WT BDR and 6 TNFR1 KO BDR mice; *p < 0.05 versus WT BDR mice. D, Solid bars represent the percentage of total cerebral microglia that express MCP-1/CCL2 in day 10 WT BDR and TNFR1 KO BDR mice. Hatched bars represent the total number of cerebral microglia that express MCP-1/CCL2 in day 10 WT BDR and TNFR1 KO BDR mice. Error bars represent mean ± SEM of data from 9 WT BDR and 6 TNFR1 KO BDR mice; *p < 0.05 versus WT BDR mice. E, F, Representative flow cytometry profiles from day 10 WT BDR (E) and TNFR2 KO BDR (F) mice. The inserted number shows the percentage of total brain mononuclear cells that are CD45high CD11b+ (i.e., infiltrating monocytes). G, Solid bars represent the percentage of total brain mononuclear cells that are CD45high CD11b+ in day 10 WT BDR and TNFR2 KO BDR mice. Hatched bars represent the total number of CD45high CD11b+ cells isolated from the brains of day 10 WT BDR and TNFR2 KO BDR mice. Error bars represent mean ± SEM of data from 5 WT BDR and 5 TNFR2 KO BDR mice; p > 0.05 versus WT BDR mice.
Inhibition of cerebral monocyte recruitment improves sickness behavior in mice with hepatic inflammation
To determine if the recruitment of monocytes into the brains of day 10 BDR mice represents a significant pathway for the induction of liver disease associated sickness behavior, we inhibited cerebral monocyte recruitment in BDR mice and examined the impact of this upon the development of sickness behavior. Our previous study demonstrated that monocyte infiltration into the brains of BDR mice could be inhibited using anti-P selectin and anti-α4 integrin antibodies (Kerfoot et al., 2006). We used the same antibody treatment protocol as previously described to inhibit cerebral monocyte infiltration (i.e., treated BDR mice systemically with anti-P selectin and anti-α4 integrin antibodies on days 6 and 8 post surgery; control BDR mice received rat IgG) and then assessed social investigative behavior, duration of immobility and food intake on day 10 postsurgery. Inhibition of cerebral monocyte recruitment caused a significant increase in the time spent by adult BDR mice in social investigative behavior and a decrease in immobility compared with BDR mice in which cerebral monocyte recruitment had not been inhibited, but all of these behaviors (except number of interactions) were still significantly impaired compared with sham mice (Fig. 12). Furthermore, no differences in food intake between the three groups were observed (grams of food consumed in 12 h: sham: 3.2 ± 0.1 g vs IgG-treated BDR mice: 2.3 ± 0.5 g vs antibody-treated BDR mice: 2.5 ± 0.4 g, n = 3–5 mice/group, p > 0.05). Therefore, inhibition of cerebral monocyte recruitment in BDR mice resulted in a significant improvement in measures of sickness behavior related to social interaction, but not in food intake, suggesting that the improvement in social interaction was a specific effect of inhibition of cerebral monocyte recruitment in BDR mice. We confirmed that anti-P selectin and anti-α4 integrin antibody treatment had prevented monocyte recruitment into the brains of BDR mice at day 10 postsurgery by sacrificing the mice after the behavioral studies had been concluded and isolating mononuclear cells from the brains of these mice as described above (number of CD45high CD11b+ cells in IgG-treated BDR: 8.4 × 103 cells/brain vs antibody-treated BDR mice: 2.4 × 103 cells/brain, n = 5–6 mice/group, p < 0.05).
Figure 12.
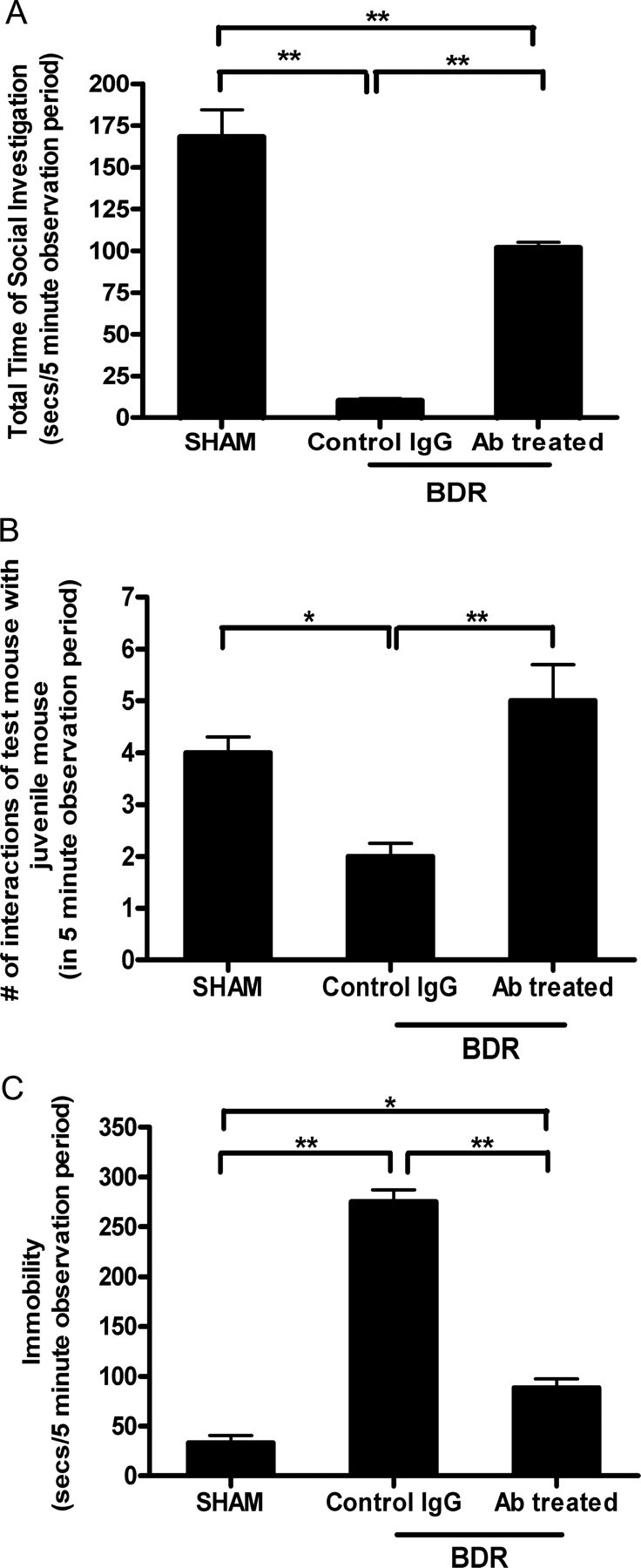
Cerebral recruitment of monocytes in BDR mice contributes significantly to the development of hepatic inflammation associated sickness behavior. Sickness behavior was assessed by quantifying (1) the duration of social exploratory behavior exhibited by an adult mouse toward a congenic juvenile mouse placed in close proximity (2), the total number of interactions of the adult test mouse with the juvenile mouse, and (3) the total time the test mouse remained immobile during the observation period. A, Total duration of social exploration in seconds during a 5 min observation period. Error bars represent mean ± SEM of data from 5 sham mice, 5 anti-P selectin and anti-α4 integrin-treated BDR mice, and 5 isotype IgG-treated control BDR mice; **p < 0.001 IgG-treated BDR versus sham controls; **p < 0.001 antibody-treated BDR versus IgG-treated BDR mice; **p < 0.001 antibody-treated BDR versus sham mice. B, Total number of interactions of the adult test mouse with a juvenile mouse during the 5 min observation period. Error bars represent mean ± SEM of data from 5 sham mice, 5 anti-P selectin and anti-α4 integrin-treated BDR mice, and 5 isotype IgG-treated control BDR mice; *p < 0.05 IgG-treated BDR versus sham mice; **p < 0.01 antibody-treated BDR versus IgG-treated BDR mice. C, Total time in seconds that the adult test mouse remained immobile during the 5 min observation period after the introduction of the juvenile mouse into the test mouse home cage. Error bars represent mean ± SEM of data from 5 sham mice, 5 anti-P selectin and anti-α4 integrin-treated BDR mice, and 5 isotype IgG-treated control BDR mice; **p < 0.001 IgG-treated BDR versus sham mice; **p < 0.001 antibody-treated BDR versus IgG-treated BDR mice; *p < 0.05 antibody-treated BDR versus sham mice. Ab, Antibody.
Discussion
In systemic inflammatory diseases, communication between the periphery and the CNS has traditionally been felt to occur via neural and/or humoral routes (Dantzer, 2006). However, using a murine model of inflammatory liver injury, we have identified the existence of a third potential communication pathway whereby immune cells, specifically monocytes, from the periphery are actively recruited into the brain (Kerfoot et al., 2006). We have confirmed this significant finding in our current study and have documented an important mechanism underlying the cerebral recruitment of monocytes.
In CNS inflammatory diseases, changes in central neurotransmission and corresponding behavioral alterations often occur before overt pathological CNS tissue damage, but after cerebral immune cell recruitment is evident (Pollak et al., 2003). During CNS pathologies, cerebral inflammation drives the migration of circulating immune cells into the brain. Therefore, it is important to understand how monocytes are recruited into the brain in the setting of peripheral inflammation, in the absence of overt CNS inflammation. BDR mice exhibit liver specific inflammatory injury; however, cerebral MCP-1/CCL2 levels were significantly elevated in day 10 BDR mice compared with controls. To respond to MCP-1/CCL2 cells need to express CCR2 on their surface (Charo and Ransohoff, 2006). In keeping with this, we found a marked elevation in the number of peripheral blood monocytes that expressed CCR2 in day 10 BDR mice, and virtually all of the monocytes that transmigrated into the brains of BDR mice expressed CCR2. In contrast, we documented reduced numbers of CCR2-expressing microglia in the brains of day 10 BDR versus sham mice. This observation may be due to elevated cerebral MCP-1/CCL2 levels in BDR mice (Bonfield et al., 2005). A central role for MCP-1/CCL2 and CCR2 in mediating cerebral monocyte recruitment in BDR mice was further demonstrated by our finding that cerebral monocyte trafficking was abolished in the absence of MCP-1/CCL2 or CCR2. Despite the redundancy in the chemokine system, others have also identified the MCP-1/CCL2-CCR2 axis as being vital in driving cerebral monocyte infiltration in models of CNS inflammatory pathologies, including experimental autoimmune encephalitis (Izikson et al., 2000; Huang et al., 2001) and human immunodeficiency virus associated dementia (Zink et al., 2001; Gonzalez et al., 2002). In addition, the absence of either of these mediators was associated with a reduction in sickness behaviors exhibited during the course of these experimental disease models; thereby highlighting the potential relevance of this immune-to-CNS communication pathway in mediating alterations in central neural activity.
Our results indicate that even in the absence of overt CNS inflammation, microglia can be activated to produce MCP-1/CCL2 which is biologically relevant for the recruitment of monocytes into the brain. Immunohistochemical staining demonstrated a dominance of activated microglia, as indicated by their primed appearance in day 5 BDR mice; predominantly located in areas around blood vessels and in the periventricular regions. These observations are potentially important since transmigration via the blood–brain barrier and the blood–CSF barrier both serve as potential routes of leukocyte entry into the brain in CNS inflammatory diseases (Ubogu et al., 2006). MCP-1/CCL2 secreted from activated microglia located in the periventricular and perivascular regions in day 5 BDR mice could potentially direct the subsequent migration of monocytes into the brain parenchyma. In keeping with this, immunohistochemical examination of brain sections from day 10 BDR mice indicated that CFSE-labeled monocytes had transmigrated into the cerebral parenchyma and were detected most prominently in both the perivascular and periventricular regions.
Understanding the signaling stimulus that leads to the early “priming” of microglia to produce mediators such as MCP-1/CCL2 is fundamental to delineating how monocytes are subsequently recruited into the brain in the setting of systemic inflammation. Activation of cerebral endothelial cells is a key step in relaying peripheral immune signals to CNS parenchymal structures (Matsumura and Kobayashi, 2004; Gosselin and Rivest, 2008). In addition, TNFα has been highlighted as an important link between peripheral inflammation and the brain. Systemic TNFα administration provokes activation of cerebral endothelial cells and brain parenchymal elements (Nadeau and Rivest, 1999). Furthermore, the absence of TNFα signaling was shown to significantly inhibit central inflammation in response to LPS induced systemic inflammation (Qin et al., 2007). In our previous study, we could not detect TNFα in the peripheral circulation of BDR mice (using a sensitive ELISA). However, we did document an increase in the number of TNFα-producing circulating monocytes in day 10 BDR versus sham mice (Kerfoot et al., 2006). In addition, we observed increased leukocyte rolling and adhesion along the cerebral endothelium in day 10 BDR mice. In our current study, in day 5 BDR mice we also documented a significant elevation in the number of TNFα-producing peripheral-circulating monocytes, suggesting that TNFα might potentially be an important link in relaying peripheral inflammation to the CNS. Hence, TNFα released from monocytes in contact with cerebral endothelium could potentially initiate a signaling stimulus into the brain parenchyma that leads to activation of microglia to produce MCP-1. In keeping with this, when we administered a TNFα neutralizing antibody systemically in day 5 BDR mice, the number of MCP-1-producing cerebral microglia was markedly reduced and were similar to numbers observed in sham controls. These findings indicate that peripheral TNFα signaling (possibly via the cerebral endothelium) is a critical early event that stimulates microglia to produce MCP-1/CCL2.
TNFα signaling via TNFR1 plays a dominant role in mediating inflammation in numerous diseases (Probert et al., 2000; Gimenez et al., 2006). To determine whether differential TNFα signaling via TNFR1 or TNFR2 affects cerebral monocyte recruitment in BDR mice, we conducted recruitment studies in day 10 BDR mice deficient in TNFR1 or TNFR2. We found that cerebral monocyte recruitment was markedly inhibited in TNFR1 deficient BDR mice, but not in TNFR2 deficient BDR mice. The reduction in monocyte recruitment in day 10 TNFR1 KO BDR mice was associated with a striking reduction in the number of microglia that produced MCP-1/CCL2. TNFα signaling via TNFR1 has also been shown to potently stimulate MCP-1/CCL2 production and mediate subsequent cerebral leukocyte recruitment in models of CNS inflammatory diseases, including experimental autoimmune encephalitis (Probert et al., 2000). These findings are consistent with peripheral TNFα, signaling via TNFR1, stimulating microglia to produce MCP-1 which subsequently mediates the recruitment of monocytes into the brains of BDR mice.
The recruitment of monocytes into the brain via the MCP-1-CCR2 axis could have potential implications for central neural changes which occur during peripheral inflammation. Monocytes that transmigrate into the brain produce MCP-1 and as we have previously shown, TNFα (Kerfoot et al., 2006). The cerebral recruitment of monocytes in BDR mice highlights an important potential communication route that enables cellular messenger molecules such as TNFα, which have been classically viewed as humoral in nature, to gain access to the CNS in systemic inflammatory diseases. TNFα production within the brain can also be augmented in the setting of increased ammonia levels (Odeh, 2007). Elevated plasma and cerebral ammonia levels have been documented in BDR rats with cirrhosis (Wright et al., 2007), suggesting that this pathway could also possibly play a role in further increasing cerebral TNFα levels in mice with hepatic inflammation and warrants further investigation.
TNFα plays a key role in the genesis of sickness behaviors (Palin et al., 2007, 2008). Importantly, patients with systemic inflammatory diseases being treated with anti-TNFα therapies demonstrate reduced clinical symptoms (which by definition are due to changes in central neurotransmission), often before overt clinical changes in the underlying inflammatory disease state have been observed (Lichtenstein et al., 2002; Satapathy et al., 2004). We have previously documented changes in a number of neurotransmitter systems, as well as associated behavioral changes, in the setting of hepatic inflammation (Swain et al., 1993; Swain and Maric, 1995, 1997; Swain and Le, 1998; Burak et al., 2001, 2002). In our current study, we have shown that inhibition of monocyte recruitment into the brains of mice with liver inflammation results in significant improvement in sickness behavior as reflected by an increase in social interaction and decreased immobility. Therefore, in mice with peripheral hepatic inflammation, the cerebral recruitment of activated monocytes which are capable of producing a number of neuromodulators (e.g., TNFα, MCP-1) (Banisadr et al., 2002; Rostène et al., 2007) potentially serves as an important route that can mediate changes in CNS neurotransmission and thereby behavior.
In conclusion, we have observed that in the setting of peripheral organ-centered inflammation there is a directed recruitment of activated monocytes into the CNS, which occurs as a result of an initial activation of cerebral microglia to produce MCP-1/CCL2. Peripheral TNFα signaling appears to be a critical component in the activation of microglia and the subsequent recruitment of monocytes into the brain in this setting. Moreover, the cerebral infiltration of monocytes plays an important role in driving hepatic inflammation associated sickness behavior. Our findings have significant implications for communication pathways between the periphery and the CNS, not only in liver disease, but also in other inflammatory diseases occurring outside of the CNS.
Footnotes
This work was supported by an operating grant from the Canadian Institutes of Health Research (CIHR) to M.G.S. C.D. is supported through the Canadian Liver Foundation and the CIHR training program. M.G.S. is an Alberta Heritage Foundation for Medical Research Senior Scholar. We thank the following individuals for their assistance with this manuscript: Dr. Maureen Ajuebor (Louisiana State University, Shreveport, LA) for assistance with developing the cerebral cell isolation techniques; Dr. Dave Stirling (University of Calgary) for assistance with the immunohistochemistry; and Laurie Kennedy and Laurie Robertson (University of Calgary Flow Cytometry Core laboratory) for their help with the flow cytometry analyses.
References
- Abe T, Arai T, Ogawa A, Hiromatsu T, Masuda A, Matsuguchi T, Nimura Y, Yoshikai Y. Kupffer cell derived interleukin 10 is responsible for impaired bacterial clearance in bile duct ligated mice. Hepatology. 2004;40:414–423. doi: 10.1002/hep.20301. [DOI] [PubMed] [Google Scholar]
- Aggarwal BB. Signaling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003;3:745–756. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- Ahmed Z, Shaw G, Sharma VP, Yang C, McGowan E, Dickson DW. Actin-binding proteins Coronin-1a and IBA-1 are effective microglial markers for immunohistochemistry. J Histochem Cytochem. 2007;55:687–700. doi: 10.1369/jhc.6A7156.2007. [DOI] [PubMed] [Google Scholar]
- Babcock AA, Kuziel WA, Rivest S, Owens T. Chemokine expression by glial cells directs leukocytes to sites of axonal injury in the CNS. J Neurosci. 2003;23:7922–7930. doi: 10.1523/JNEUROSCI.23-21-07922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banisadr G, Quéraud-Lesaux F, Boutterin MC, Pélaprat D, Zalc B, Rostène W, Haour F, Parsadaniantz SM. Distribution, cellular localization and functional role of CCR2 chemokine receptors in adult rat brain. J Neurochem. 2002;81:257–269. doi: 10.1046/j.1471-4159.2002.00809.x. [DOI] [PubMed] [Google Scholar]
- Bemelmans MH, Greve JW, Gouma DJ, Buurman WA. Increased concentrations of tumour necrosis factor (TNF) and soluble TNF receptors in biliary obstruction in mice; soluble TNF receptors as prognostic factors for mortality. Gut. 1996;38:447–453. doi: 10.1136/gut.38.3.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluthé RM, Jafarian-Tehrani M, Michaud B, Haour F, Dantzer R, Homo-Delarche F. Increased sensitivity of prediabetic nonobese diabetic mouse to the behavioral effects of IL-1. Brain Behav Immun. 1999;13:303–314. doi: 10.1006/brbi.1998.0542. [DOI] [PubMed] [Google Scholar]
- Bluthé RM, Layé S, Michaud B, Combe C, Dantzer R, Parnet P. Role of interleukin -1β and tumor necrosis factor-α in lipopolysaccharide-induced sickness behavior: a study with interluekin-1 type 1 receptor-deficient mice. Eur J Neurosci. 2000;12:4447–4456. [PubMed] [Google Scholar]
- Bonfield TL, John N, Malur A, Barna BP, Culver DA, Kavuru MS, Thomassen MJ. Elevated monocyte chemotactic proteins 1,2 and 3 in pulmonary alveolar proteinosis are associated with chemokine receptor suppression. Clin Immunol. 2005;114:79–85. doi: 10.1016/j.clim.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Burak KW, Le T, Swain MG. Increased midbrain 5-HT1A receptor number and responsiveness in cholestatic rats. Brain Res. 2001;892:376–379. doi: 10.1016/s0006-8993(00)03058-4. [DOI] [PubMed] [Google Scholar]
- Burak KW, Le T, Swain MG. Increased sensitivity to the locomotor-activating effects of corticotropin-releasing hormone in cholestatic rats. Gastroenterology. 2002;122:681–688. doi: 10.1053/gast.2002.31878. [DOI] [PubMed] [Google Scholar]
- Campanella M, Sciorati C, Tarozzo G, Beltramo M. Flow cytometric analysis of inflammatory cells in ischemic rat brain. Stroke. 2002;33:586–592. doi: 10.1161/hs0202.103399. [DOI] [PubMed] [Google Scholar]
- Charo IF, Ransohoff RM. The many roles of Chemokines and Chemokine Receptors in Inflammation. N Engl J Med. 2006;354:610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- Dantzer R. Cytokine, sickness behavior and depression. Neurol Clin. 2006;24:441–460. doi: 10.1016/j.ncl.2006.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, Kelley KW. Twenty years of research on cytokine-induced sickness behavior. Brain Behav Immun. 2007;21:153–160. doi: 10.1016/j.bbi.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Martínez E, Pérez-Alvarez V, Tsutsumi V, Shibayama M, Muriel P. Chronic bile duct obstruction induces changes in plasma and hepatic levels of cytokines and nitric oxide in the rat. Exp Toxicol Pathol. 2006;58:49–58. doi: 10.1016/j.etp.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Garden GA, Möller T. Microglia biology in health and disease. J Neuroimmune Pharmacol. 2006;1:127–137. doi: 10.1007/s11481-006-9015-5. [DOI] [PubMed] [Google Scholar]
- Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- Getts DR, Terry RL, Getts MT, Müller M, Rana S, Shrestha B, Radford J, Van Rooijen N, Campbell IL, King NJ. Ly6C+ “inflammatory monocytes” are microglial precursors recruited in a pathogenic manner in West Nile virus encephalitis. J Exp Med. 2008;205:2319–2337. doi: 10.1084/jem.20080421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez MA, Sim J, Archambault AS, Klein RS, Russell JH. A tumor necrosis factor receptor 1-dependent conversation between central nervous system-specific T cells and the central nervous system is required for inflammatory infiltration of the spinal cord. Am J Pathol. 2006;168:1200–1209. doi: 10.2353/ajpath.2006.050332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold SM, Irwin MR. Depression and immunity: inflammation and depressive symptoms in multiple sclerosis. Neurol Clin. 2006;24:507–519. doi: 10.1016/j.ncl.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Gonzalez E, Rovin BH, Sen L, Cooke G, Dhanda R, Mummidi S, Kulkarni H, Bamshad MJ, Telles V, Anderson SA, Walter EA, Stephan KT, Deucher M, Mangano A, Bologna R, Ahuja SS, Dolan MJ, Ahuja SK. From the cover: HIV-1 infection and AIDS dementia are influenced by a mutant MCP-1 allele linked to increased monocyte infiltration of tissues and MCP-1 levels. Proc Natl Acad Sci U S A. 2002;99:13795–13800. doi: 10.1073/pnas.202357499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- Gosselin D, Rivest S. Role of IL-1 and TNF in the brain: twenty years of progress on a Dr. Jekyll/Mr. Hyde duality of the innate immune system. Brain Behav Immun. 2007;21:281–289. doi: 10.1016/j.bbi.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Gosselin D, Rivest S. MyD88 signaling in brain endothelial cells is essential for the neuronal activity and glucocorticoid release during systemic inflammation. Mol Psychiatry. 2008;13:480–497. doi: 10.1038/sj.mp.4002122. [DOI] [PubMed] [Google Scholar]
- Gralnek IM, Hays RD, Kilbourne A, Naliboff B, Mayer EA. The impact of irritable bowel syndrome on health-related quality of life. Gastroenterology. 2000;119:654–660. doi: 10.1053/gast.2000.16484. [DOI] [PubMed] [Google Scholar]
- Harkness KA, Sussman JD, Davies-Jones GA, Greenwood J, Woodroofe MN. Cytokine regulation of MCP-1 expression in brain and retinal microvascular endothelial cells. J Neuroimmunol. 2003;142:1–9. doi: 10.1016/s0165-5728(03)00251-0. [DOI] [PubMed] [Google Scholar]
- Hayley S, Brebner K, Lacosta S, Merali Z, Anisman H. Sensitization to the effects of tumor necrosis factor-α: neuroendocrine, central monamine and behavioral variations. J Neurosci. 1999;19:5654–5665. doi: 10.1523/JNEUROSCI.19-13-05654.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heesen C, Nawrath L, Reich C, Bauer N, Schulz KH, Gold SM. Fatigue in multiple sclerosis: an example of cytokine mediated sickness behavior? J Neurol Neurosurg Psychiatry. 2006;77:34–39. doi: 10.1136/jnnp.2005.065805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmer B, Cepok S, Zhou D, Sommer N. Multiple sclerosis - a coordinated immune attack across the blood brain barrier. Curr Neurovasc Res. 2004;1:141–150. doi: 10.2174/1567202043480152. [DOI] [PubMed] [Google Scholar]
- Hopkins SJ. Central nervous system recognition of peripheral inflammation: a neural, hormonal collaboration. Acta Biomed. 2007;78:231–247. [PubMed] [Google Scholar]
- Huang DR, Wang J, Kivisakk P, Rollins BJ, Ransohoff RM. Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J Exp Med. 2001;193:713–726. doi: 10.1084/jem.193.6.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huffnagle GB, Toews GB, Burdick MD, Boyd MB, McAllister KS, McDonald RA, Kunkel SL, Strieter RM. Afferent phase production of TNF-α is required for the development of protective T cell immunity to cryptococcus neoformans. J Immunol. 1996;157:4529–4536. [PubMed] [Google Scholar]
- Hult B, Chana G, Masliah E, Everall I. Neurobiology of HIV. Int Rev Psychiatry. 2008;20:3–13. doi: 10.1080/09540260701862086. [DOI] [PubMed] [Google Scholar]
- Izikson L, Klein RS, Charo IF, Weiner HL, Luster AD. Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J Exp Med. 2000;192:1075–1080. doi: 10.1084/jem.192.7.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DR, O'Connor JC, Dantzer R, Freund GG. Inhibition of vagally mediated immune-to-brain signaling by vanadyl sulphate sppeds recovery from sickness. Proc Natl Acad Sci U S A. 2005;102:15184–15189. doi: 10.1073/pnas.0507191102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerfoot SM, D'Mello C, Nguyen H, Ajuebor MN, Kubes P, Le T, Swain MG. TNF-α secreting Monocytes are recruited into the brains of cholestatic mice. Hepatology. 2006;43:154–162. doi: 10.1002/hep.21003. [DOI] [PubMed] [Google Scholar]
- Kielian T. Microglia and chemokines in infectious diseases of the nervous system: views and reviews. Front Biosci. 2004;9:732–750. doi: 10.2741/1266. [DOI] [PubMed] [Google Scholar]
- Koniaris LG, Zimmers-Koniaris T, Hsiao EC, Chavin K, Sitzmann JV, Farber JM. Cytokine-responsive gene-2/IFN-inducible protein-10 expression in multiple models of liver and bile duct injury suggests a role in tissue regeneration. J Immunol. 2001;167:399–406. doi: 10.4049/jimmunol.167.1.399. [DOI] [PubMed] [Google Scholar]
- Kumar D, Tandon RK. Fatigue in cholestatic liver disease–a perplexing symptom. Postgrad Med J. 2002;78:404–407. doi: 10.1136/pmj.78.921.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenstein GR, Bala M, Han C, DeWoody K, Schaible T. Infliximab improves quality of life in patients with Crohn's disease. Inflamm Bowel Dis. 2002;8:237–243. doi: 10.1097/00054725-200207000-00001. [DOI] [PubMed] [Google Scholar]
- Mahad DJ, Ransohoff RM. The role of MCP-1 (CCL2) and CCR2 in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE) Semin Immunol. 2003;15:23–32. doi: 10.1016/s1044-5323(02)00125-2. [DOI] [PubMed] [Google Scholar]
- Maini RN, Breedveld FC, Kalden JR, Smolen JS, Furst D, Weisman MH, St Clair EW, Keenan GF, van der Heijde D, Marsters PA, Lipsky PE. Sustained improvement over two years in physical function, structural damage and signs and symptoms among patients with rheumatoid arthritis treated with infliximab and methotrexate. Arthritis Rheum. 2004;50:1051–1065. doi: 10.1002/art.20159. [DOI] [PubMed] [Google Scholar]
- Matsumura K, Kobayashi S. Signaling the brain in inflammation: the role of endothelial cells. Front Biosci. 2004;9:2819–2826. doi: 10.2741/1439. [DOI] [PubMed] [Google Scholar]
- Mayoux-Benhamou MA. Fatigue and rheumatoid arthritis. Annales de readaptation et de medecine physique. 2006;49:385–388. doi: 10.1016/j.annrmp.2006.04.011. [DOI] [PubMed] [Google Scholar]
- McCullough LK, Takahashi Y, Le T, Pittman QJ, Swain MG. Attenuated febrile response to lipopolysaccharide in rats with biliary obstruction. Am J Physiol Gastrointest Liver Physiol. 2000;279:G172–G177. doi: 10.1152/ajpgi.2000.279.1.G172. [DOI] [PubMed] [Google Scholar]
- Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, Heikenwalder M, Brück W, Priller J, Prinz M. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci. 2007;10:1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- Nadeau S, Rivest S. Effects of circulating tumor necrosis factor-α on the neuronal activity and expression of the genes encoding the tumor necrosis factor receptors (p55 and p75) in the rat brain: a view from the blood-brain barrier. Neuroscience. 1999;93:1449–1464. doi: 10.1016/s0306-4522(99)00225-0. [DOI] [PubMed] [Google Scholar]
- Neff SB, Neff TA, Kunkel SL, Hogaboam CM. Alterations in cytokine/chemokine expression during organ-to-organ communication established via acetaminophen-induced toxicity. Exp Mol Pathol. 2003;75:187–193. doi: 10.1016/s0014-4800(03)00096-0. [DOI] [PubMed] [Google Scholar]
- O'Connor JC, Satpathy A, Hartman ME, Horvath EM, Kelley KW, Dantzer R, Johnson RW, Freund GG. IL-1Β mediated innate immunity is amplified in the db/db mouse model of type 2diabetes. J Immunol. 2005;174:4991–4997. doi: 10.4049/jimmunol.174.8.4991. [DOI] [PubMed] [Google Scholar]
- Odeh M. Pathogenesis of hepatic encephalopathy: the tumor necrosis factor-α theory. Eur J Clin Invest. 2007;37:291–304. doi: 10.1111/j.1365-2362.2007.01778.x. [DOI] [PubMed] [Google Scholar]
- Palin K, Bluthé RM, McCusker RH, Moos F, Dantzer R, Kelley KW. TNFalpha-induced sickness behavior in mice with functional 55 kD TNF receptors is blocked by central IGF-I. J Neuroimmunol. 2007;187:55–60. doi: 10.1016/j.jneuroim.2007.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palin K, McCusker RH, Strle K, Moos F, Dantzer R, Kelley KW. Tumor necrosis factor-alpha-induced sickness behavior is impaired by central administration of an inhibitor of c-jun N-terminal kinase. Psychopharmacology. 2008;197:629–635. doi: 10.1007/s00213-008-1086-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak Y, Ovadia H, Orion E, Weidenfeld J, Yirmiya R. The EAE-associated behavioral syndrome: I. Temporal correlation with inflammatory mediators. J Neuroimmunol. 2003;137:94–99. doi: 10.1016/s0165-5728(03)00075-4. [DOI] [PubMed] [Google Scholar]
- Probert L, Eugster HP, Akassoglou K, Bauer J, Frei K, Lassmann H, Fontana A. TNFR1 signaling is critical for the development of demyelination and the limitation of T-cell responses during immune mediated CNS disease. Brain. 2000;123:2005–2019. doi: 10.1093/brain/123.10.2005. [DOI] [PubMed] [Google Scholar]
- Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanovsky AA, Almeida MC, Aronoff DM, Ivanov AI, Konsman JP, Steiner AA, Turek VF. Fever and hypothermia in systemic inflammation: recent discoveries and revisions. Front Biosci. 2005;10:2193–2216. doi: 10.2741/1690. [DOI] [PubMed] [Google Scholar]
- Rostène W, Kitabgi P, Parsadaniantz SM. Chemokines: a new class of neuromodulator? Nature Reviews: Neuroscience. 2007;8:895–903. doi: 10.1038/nrn2255. [DOI] [PubMed] [Google Scholar]
- Satapathy SK, Garg S, Chauhan R, Sakhuja P, Malhotra V, Sharma BC, Sarin SK. Beneficial effects of tumor necrosis factor-α inhibition by pentoxifylline on clinical biochemical and metabolic parameters of patients with nonalcoholic steatohepatitis. Am J Gastroenterol. 2004;99:1946–1952. doi: 10.1111/j.1572-0241.2004.40220.x. [DOI] [PubMed] [Google Scholar]
- Sewnath ME, Van Der Poll T, Van Noorden CJ, Ten Kate FJ, Gouma DJ. Endogenous Interferon γ Protects Against Cholestatic Liver Injury in Mice. Hepatology. 2002;36:1466–1477. doi: 10.1053/jhep.2002.37196. [DOI] [PubMed] [Google Scholar]
- Simen BB, Duman CH, Simen AA, Duman RS. TNF-α signaling in depression and anxiety: behavioral consequences of individual receptor targeting. Biol Psychiatry. 2006;59:775–785. doi: 10.1016/j.biopsych.2005.10.013. [DOI] [PubMed] [Google Scholar]
- Sriram K, O'Callaghan JP. Divergent roles for tumor necrosis factor-α in the brain. J Neuroimmune Pharmacol. 2007;2:140–153. doi: 10.1007/s11481-007-9070-6. [DOI] [PubMed] [Google Scholar]
- Swain MG. Fatigue in liver disease: pathophysiology and clinical management. Can J Gastroenterol. 2006;20:181–188. doi: 10.1155/2006/624832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain MG, Le T. Chronic cholestasis in rats induces anhedonia and a loss of social interest. Hepatology. 1998;28:6–10. doi: 10.1002/hep.510280102. [DOI] [PubMed] [Google Scholar]
- Swain MG, Maric M. Defective corticotropin-releasing hormone mediated neuroendocrine and behavioral responses in cholestatic rats: implications for cholestatic liver disease - related sickness behaviors. Hepatology. 1995;22:1560–1564. [PubMed] [Google Scholar]
- Swain MG, Maric M. Improvement in cholestasis associated fatigue with a serotonin receptor agonist using a novel rat model of fatigue assessment. Hepatology. 1997;25:291–294. doi: 10.1002/hep.510250206. [DOI] [PubMed] [Google Scholar]
- Swain MG, Patchev V, Vergalla J, Chrousos G, Jones EA. Suppression of hypothalamic pituitary-adrenal axis responsiveness to stress in a rat model of acute cholestasis. J Clin Invest. 1993;91:1903–1908. doi: 10.1172/JCI116408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain MG, Beck P, Rioux K, Le T. Augmented interleukin -1β induced depression of locomotor activity in cholestatic rats. Hepatology. 1998;28:1561–1565. doi: 10.1002/hep.510280616. [DOI] [PubMed] [Google Scholar]
- Tacke F, Randolph GJ. Migratory fate and differentiation of blood monocyte subsets. Immunobiology. 2006;211:609–618. doi: 10.1016/j.imbio.2006.05.025. [DOI] [PubMed] [Google Scholar]
- Thibeault I, Laflamme N, Rivest S. Regulation of the gene encoding the monocyte chemoattractant protein 1 (MCP-1) in the mouse and rat brain in response to circulating LPS and proinflammatory cytokines. J Comp Neurol. 2001;434:461–477. doi: 10.1002/cne.1187. [DOI] [PubMed] [Google Scholar]
- Tyring S, Gottlieb A, Papp K, Gordon K, Leonardi C, Wang A, Lalla D, Woolley M, Jahreis A, Zitnik R, Cella D, Krishnan R. Etanercept and clinical outcomes, fatigue, and depression in psoriasis: double-blind placebo-controlled randomised phase III trial. Lancet. 2006;367:29–35. doi: 10.1016/S0140-6736(05)67763-X. [DOI] [PubMed] [Google Scholar]
- Ubogu EE, Cossoy MB, Ransohoff RM. The expression and function of chemokines involved in CNS inflammation. Trends Pharmacol Sci. 2006;27:48–55. doi: 10.1016/j.tips.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Wong CW, Christen T, Roth I, Chadjichristos CE, Derouette JP, Foglia BF, Chanson M, Goodenough DA, Kwak BR. Connexin37 protects against atherosclerosis by regulating monocyte adhesion. Nat Med. 2006;12:950–954. doi: 10.1038/nm1441. [DOI] [PubMed] [Google Scholar]
- Wright G, Davies NA, Shawcross DL, Hodges SJ, Zwingmann C, Brooks HF, Mani AR, Harry D, Stadlbauer V, Zou Z, Zou Z, Williams R, Davies C, Moore KP, Jalan R. Endotoxemia produces coma and brain swelling in bile duct ligated rats. Hepatology. 2007;45:1517–1526. doi: 10.1002/hep.21599. [DOI] [PubMed] [Google Scholar]
- Zink MC, Coleman GD, Mankowski JL, Adams RJ, Tarwater PM, Fox K, Clements JE. Increased macrophage chemoattractant protein1 in cerebrospinal fluid precedes and predicts simian immunodeficiency virus encephalitis. J Infect Dis. 2001;184:1015–1021. doi: 10.1086/323478. [DOI] [PubMed] [Google Scholar]