

Abstract
The perirhinal cortex of the temporal lobe is essential for the familiarity discrimination component of recognition memory. In view of the importance of changes in calcium ion concentration for synaptic plasticity, the present study examined the effects of L-type voltage-dependent calcium channel (VDCC) antagonism on rat perirhinal-based familiarity discrimination processes and plasticity including long-term depression (LTD), long-term potentiation (LTP), and depotentiation. Single doses of three different types of L-type VDCC antagonists, verapamil, diltiazem, and nifedipine, administered systemically, or verapamil administered locally into the perirhinal cortex, impaired acquisition of long-term (24 h) but not shorter-term (20 min) recognition memory. L-type VDCC antagonism also disrupted memory retrieval after 24 h but not 20 min. Differential neuronal activation produced by viewing novel or familiar visual stimuli was measured by Fos expression. L-type VDCC antagonism by verapamil in perirhinal cortex during memory acquisition disrupted the normal pattern of differential Fos expression, so paralleling the antagonist-induced memory impairment. In slices of perirhinal cortex maintained in vitro, verapamil was without effect on baseline excitability or LTP but blocked LTD and depotentiation. The consistency of effects across the behavioral and cellular levels of analysis provides strong evidence for the involvement of perirhinal L-type VDCCs in long-term recognition memory processes.
Introduction
The perirhinal cortex (PRH) of the temporal lobe is essential for the familiarity discrimination component of recognition memory (Brown and Aggleton, 2001). It has become a valuable region for studying the neural substrates of memory (Warburton et al., 2003, 2005; Hannesson et al., 2004, 2005; Winters and Bussey, 2005a,b; Barker at al., 2006a,b, 2007; Winters et al., 2006, 2008; Barker and Warburton, 2008). Experience-dependent synaptic modifications underlying memory are believed to share mechanisms with synaptic plasticity processes such as long-term potentiation (LTP) and long-term depression (LTD) (Bliss and Collingridge, 1993; Martin et al., 2000). Such synaptic plasticity is dependent on changes in the concentration of intracellular calcium ions (Chittajallu et al., 1998; Cho et al., 2001; Franks and Sejnowski, 2002). Previous work established roles for glutamatergic, including NMDA receptors in perirhinal plasticity and recognition memory processes (Ziakopoulos et al., 1999; Cho et al., 2000; Winters and Bussey, 2005b; Barker et al., 2006a,b; Voglis and Tavernarakis, 2006; Barker and Warburton, 2008). The role of NMDA receptors was investigated because their activation results in changes in the concentration of intracellular calcium ions and consequent synaptic plasticity (Bliss and Collingridge, 1993; Chittajallu et al., 1998; Franks and Sejnowski, 2002; West et al., 2002; Voglis and Tavernarakis, 2006). L-type voltage-dependent calcium channels (VDCCs) are also major sites for calcium ion entry into neurones (Chittajallu et al., 1998; West et al., 2002; Voglis and Tavernarakis, 2006), but their role in recognition memory and perirhinal synaptic plasticity remains to be elucidated.
L-type VDCCs are involved in both LTP and LTD in the hippocampus and amygdala (Grover and Teyler, 1990; Bolshakov and Siegelbaum, 1994; Morgan and Teyler, 1999; Freir and Herron, 2003; Niikura et al., 2004; Voglis and Tavernarakis, 2006; Tchekalarova and Albrecht, 2007) and their transient inactivation impairs fear conditioning, passive avoidance learning, and spatial memory (Borroni et al., 2000; Bauer et al., 2002; Woodside et al., 2004; Lashgari et al., 2006). It was therefore important to test whether L-type VDCCs played a similar role in perirhinal plasticity and recognition memory.
To test for a role in PRH memory, the effects of L-type VDCC antagonists on familiarity discrimination acquisition and retrieval processes were determined using a novel object preference task (Ennaceur and Delacour, 1988). To test for a role in plasticity, the effects of L-type VDCC antagonism on LTP, LTD, and depotentiation were determined in slices of adult PRH. Additionally, to test an additional link between L-type VDCC activation and PRH memory processes, the effects of L-type VDCC antagonism on neuronal activation produced by viewing novel and familiar stimuli were assessed. Many PRH neurones respond strongly to novel but only weakly to familiar stimuli (Brown and Xiang, 1998; Brown and Aggleton, 2001): we imaged this differential neuronal activation using Fos expression (Sheng and Greenberg, 1990; Zhu et al., 1995, 1996; Wan et al., 1999).
As a check for specificity of action, we tested the effects on memory of an exemplar of each of the three main types of L-type VDCC antagonists—phenylalkylamines (e.g., verapamil), benzothiazepines (e.g., diltiazem), and dihydropyridines (e.g., nifedipine)—as they have different chemical structures and bind at different sites on the channels (Striessnig et al., 1990, 1991; Nakayama et al., 1991; Nakayama and Kuniyasu, 1996; Hockerman et al., 2000).
Materials and Methods
Animals and drugs
Adult male pigmented Dark Agouti rats (220–250 g; Bantin and Kingman) were maintained on a 14 h light/10 h dark cycle, with the dark phase during normal daylight. Rats with implanted cannulae were housed in pairs, and others in groups of four. All rats had 24 h access to food and water, except those used for the paired-viewing test when water was restricted to 2 h a day for a maximum of 6 consecutive days. All experiments were performed in accordance with the United Kingdom Animals Scientific Procedures Act (1986) and had the approval of the University of Bristol Ethical Review Committee.
In in vivo experiments, (±) verapamil hydrochloride (Sigma-Aldrich) was administered intraperitoneally at a solution strength of 10 mg/ml in a solution volume of 1 ml of normal saline per kilogram of rat weight (n = 12 for object recognition test), or infused locally into the PRH through bilaterally implanted cannulae at 4 μg/side in 1 μl of normal saline (n = 9 for object recognition test and n = 7 for paired-viewing test). (+)-cis-Diltiazem hydrochloride (Sigma-Aldrich) and nifedipine (Sigma-Aldrich) were administered intraperitoneally at 10 mg/ml in 1 ml/kg normal saline for the former and in 1 ml/kg polyethylene glycol (PEG) (molecular weight 400; Sigma-Aldrich) for the latter (n = 12 for object recognition test).
Cannula implantation into PRH
Cannula implantation was performed in rats deeply anesthetized with isoflurane (Merial Animal Health) and placed in a stereotaxic frame in which the skull was maintained in a flat position (the height difference between bregma and lambda was <0.1 mm). Two stainless-steel guide cannulae (26 gauge; Plastics One; via Semat) were implanted through holes in the skull, at an angle of 20° to the vertical and according to the following coordinates (relative to bregma): anteroposterior, −5.6 mm; lateral, ±4.5 mm; and ventral, −6.7 mm (relative to the skull surface) (Paxinos and Watson, 1998). The guide cannulae were anchored to the skull with two stainless-steel screws, Epoxy resin (Araldite; Bostik), and dental cement (CMW1 Radiopaque with gentamycin; DePuy International). Cannulae were protected with dummy inserts (Plastics One) except at the time of the infusion. The rats were allowed to recover for at least 20 d before the experiment began. The infusion of verapamil or saline bilaterally into the PRH through a cannula in each hemisphere was performed by inserting a 33 gauge cannula (Plastics One), which protruded 1 mm beyond the guide cannula tip. Each infusion cannula was attached to a Hamilton syringe (Hamilton Bonaduz) via PVC (polyvinylchloride) tubing (Barloworld Scientific). The syringe was advanced with an infusion pump (Harvard Bioscience) at a rate of 0.5 μl/min for 2 min, and 5 min later the injection cannulae were withdrawn.
Novel object preference task
Procedure.
The methodology of the novel object preference test has been described in detail in previous studies (Warburton et al., 2003; Barker et al., 2006a,b, 2007). In brief, this task took place in an arena (50 × 90 × 100 cm) surrounded with black curtains and with sawdust on the floor. The rat's behavior was monitored using a camera and a video recorder. The objects were made of Duplo bricks (Lego Produktion) or were junk objects, and varied in size (range, 10 × 5 × 15 to 20 × 20 × 30 cm), color, and shape, and were placed near the two corners at either end of one side of the arena (15 cm from each adjacent wall). Before the start of memory testing, each rat was habituated to the empty arena for 5 min daily for 4 d.
The novel object preference test comprised two phases, acquisition and test, separated by a delay of 20 min or 24 h. In the acquisition phase, an animal was allowed to explore two identical objects for 40 s of exploration or a maximum of 4 min spent in the arena. In the test phase, the rat was allowed to explore an identical third copy of the object explored in the acquisition phase and a novel object for 3 min from the start of the objects' exploration. In both acquisition and test phases, the time spent exploring each of the objects was recorded. Exploration was considered only when the animal's nose was directed toward the object at a distance of <1 cm. If the time of exploration was <15 s in the acquisition phase or <10 s in the test phase, the animal was discarded from the analysis of that experiment. The objects used as novel or familiar, and their position in the arena, were counterbalanced between animals in a group, and between control and drug-treated animals. The experimenter was blind as to the treatment of each animal.
Experimental design.
Animals were administered drug or vehicle intraperitoneally or locally into the PRH through the bilaterally implanted cannulae. To evaluate shorter-term and long-term memory, delays between acquisition and test phases of 20 min and 24 h were used. To assess the effects on acquisition, the drug or its vehicle was administered 1 h before the acquisition phase. To study the effects on long-term memory retrieval, the drug or its vehicle was administered 1 h before the test phase, with the 24 h delay period. To evaluate whether a drug effect was state dependent, administration was both 1 h before the acquisition phase and 1 h before the test phase, with the 24 h delay period.
Each experiment was performed in a counterbalanced crossed-over design: thus, rats treated with the drug in one experimental trial were administered the vehicle on the following trial, and vice versa. The animals were allowed to rest between experimental trials for at least 1 week to ensure the drug or vehicle had cleared from the animal's body.
Histology
Cannula locations were checked to confirm that infusions were in PRH; rats were anesthetized with Euthatal and perfusion fixed with 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4. The brain was removed and immersed in the same fixative for at least 24 h before being transferred to 30% sucrose in 0.1 m phosphate buffer for at least 2 d. The right hemisphere of the brain was marked with an incision before being sectioned (40 μm) coronally on a cryostat and stained with cresyl violet. All animals had the tips of their cannulae within the PRH from bregma −5.5 to −4.5 (Paxinos and Watson, 1998; Shi and Cassell, 1999) (Fig. 1A).
Figure 1.
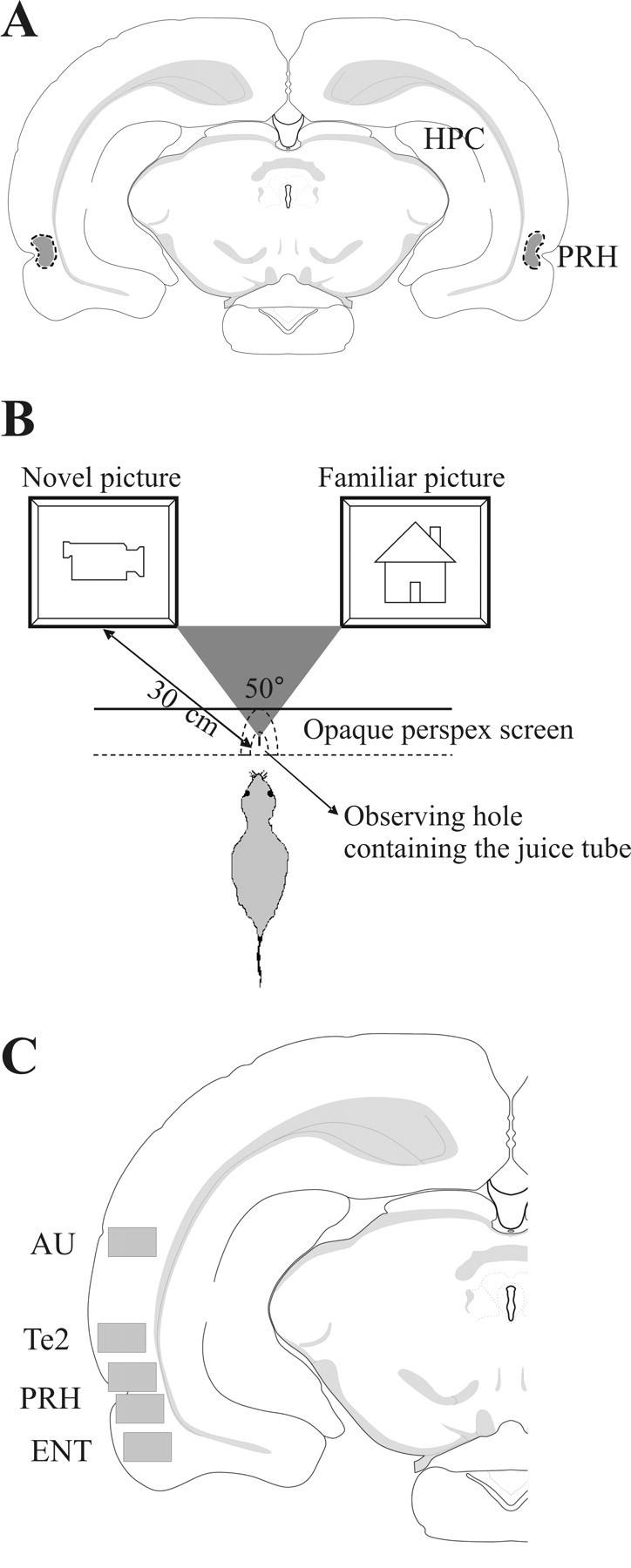
Procedures. A, The location of cannula tips, as observed in cresyl violet-stained sections, for all implanted animals used in object recognition test (n = 9) and paired-viewing test (n = 15) fell within the dotted outlined area, which was located within the PRH from bregma −5.5 to −4.5. HPC, Hippocampus. B, Paired-viewing apparatus: a rat, lured by the availability of juice, places its head in a hole in a Perspex barrier. By using a central partition, each eye can see the picture only on the computer monitor on that side. Pictures were presented simultaneously on both monitors. C, Fos-labeled cell counting was performed in rectangular areas (shown in gray) from four sections from bregma −5.5 to −6.0 in the following regions: PRH, Te2, ENT, and AU (Paxinos and Watson, 1998; Shi and Cassell, 1999).
Paired-viewing test
Procedure.
The paired-viewing procedure is designed to allow measurement of differential neuronal activation produced by novel and familiar visual stimuli. The methodology has been described in detail in previous studies (Zhu et al., 1995, 1996; Warburton et al., 2003). In brief, each rat, lured by the availability of juice, viewed two pictures presented simultaneously on video monitors positioned so that each picture was seen only within the monocular visual field of one eye (Fig. 1B). The visual information was therefore chiefly processed by the opposite cerebral hemisphere, making possible the comparison of within rat differences in activation between the two hemispheres. Across 6 d, the rat was repeatedly shown a “repeat set” of pictures together with sets of novel pictures; 30 pictures were shown to each eye in the morning and 30 in the afternoon. This period of training familiarized the rat with the whole procedure, including the occurrence of new pictures, and in nontreated rats allows the repeatedly shown pictures to become familiar. At the end of the sixth day, both eyes had had equal exposure to the pictures of the repeat set and to novel pictures. On the seventh day, 24 h after the previous viewing of the repeat set (i.e., the memory delay was 24 h), this set was seen again by one eye, while the other eye viewed an additional set of 30 novel pictures (the “novel set”). Which set of pictures was used as the novel and which as the repeat set, and which eye was exposed to novel and which to the repeat pictures were counterbalanced across rats.
Experimental design.
Drugs, verapamil (n = 7) or saline (n = 8), were infused via cannulae implanted bilaterally into the PRH. One rat from the verapamil-treated group died before completion of the experiment. Four rats in each group were the same as those used in the object recognition experiments; the remainder were naive. Saline or verapamil was infused 1 h before both morning and afternoon sessions of exposure to pictures on the first 6 d so as to block (verapamil group) or not (saline group) the acquisition of familiarity for the repeat set. No infusions were made on the seventh day, so the final critical comparison was made in the absence of verapamil (or saline), 24 h after the previous showing of the repeat set (i.e., at a memory delay of 24 h).
Fos immunohistochemistry.
Each rat was anesthetized with Euthatal (Merial Animal Health) and perfusion fixed with 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4, 1.5 h after the final sets of pictures were presented. The brain was removed and postfixed in the same fixative for at least 24 h before being immersed in 30% sucrose in 0.1 m phosphate buffer for at least 2 d. The right hemisphere of the brain was marked with an incision before being sectioned (40 μm) coronally on a cryostat. Two series of floating sections were collected. The first series was stained with cresyl violet to determine the location of the cannula tracks. For all rats, the infusion sites were within the PRH (Fig. 1A). The second series were immunohistochemically processed for Fos. The sections were washed in 0.1 m phosphate buffer saline containing 0.2% Triton X-100 (PBST), pH 7.4, immersed in 0.3% hydrogen peroxidase for 20 min, and washed again before being incubated with primary Fos antibody [anti-c-Fos (Ab-5) rabbit pAb; Calbiochem] diluted 1:5000 in PBST for 48 h at 4°C. After washes with PBST, the sections were incubated with secondary antibody (biotinylated anti-rabbit IgG; BA-1000; Vector Laboratories) diluted 1:200 in PBST with 1.5% of normal goat serum for 2 h at room temperature. After several washes, sections were processed with avidin-biotinylated horseradish peroxidase complex (Vectastain ABC kit; Vector Laboratories) in PBST for 1 h at room temperature, and the reaction was visualized using 3,3′-diaminobenzidine (SigmaFast; Sigma-Aldrich).
Fos counting.
The Fos-labeled cells were counted using an automated image analysis system using in-house software that counted round or oval objects with at least 10 μm of diameter (CellCountMainv.3.1; J. Leendertz; University of Bristol, Bristol, UK). All processing was blind as to the treatment of each animal. Cell counts were obtained in both hemispheres for rectangular areas (0.94 × 0.67 mm) from four sections from bregma −5.5 to −6.0 (sections caudal to the cannula track, which was found approximately from bregma −5.5 to −4.5). The rectangular area included all cortical layers. Stereological corrections were not used as relative changes were all that were sought. The regions in which counting was performed were the PRH, temporal visual association cortex (here termed Te2), entorhinal cortex (ENT), and auditory cortex (AU) (Paxinos and Watson, 1998; Shi and Cassell, 1999) (Fig. 1C).
In vitro electrophysiology
Animals were anesthetized with an isoflurane/oxygen mixture and decapitated, and the brain was rapidly removed. The brain was placed in ice-cold artificial CSF (aCSF) (bubbled with 95% O2/5% CO2) which comprised the following (in mm): 124 NaCl, 3 KCl, 26 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 1 MgSO4, 10 d-glucose. A midsagittal section was made, the rostral and caudal parts of the brain were removed by single scalpel cuts at ∼45° to the dorsoventral axis, and each hemisphere glued by its caudal end to a vibroslice stage (Campden Instruments). Slices (400 μm) of PRH were taken in the region 4 mm behind bregma. Slices were stored submerged in aCSF (20–25°C) for 1–6 h before transferring to the recording chamber. A single slice was placed in a submerged recording chamber (28–30°C; flow rate, ∼2 ml/min) when required.
Standard in vitro extracellular field recordings were made from the PRH (Ziakopoulos et al., 1999; Massey et al., 2004). Evoked field EPSPs (fEPSPs) were recorded from layers II/III from directly below the rhinal sulcus (area 35). Two stimulating electrodes were placed on both sides (∼0.5 mm) of the recording electrode and designated entorhinal input (area 35) and temporal input (area 36). Stimuli (constant voltage) were delivered alternately to the two stimulating electrodes (each electrode, 0.033 Hz). fEPSPs were reduced to 60–70% of maximum amplitude and a baseline of synaptic transmission established before induction of synaptic plasticity. After establishing a baseline for at least 30 min, high-frequency stimulation (HFS) (four trains, each of 100 Hz, 1 s, every 30 s) was delivered to induce LTP. Subsequently, to induce depotentiation, low-frequency stimulation (LFS) (900 stimuli, 1 Hz, 15 min) was applied 60 min after LTP induction. To induce de novo synaptic LTD, LFS (3000 stimuli, 5 Hz, 10 min) was delivered (Massey et al., 2004). fEPSPs were monitored and reanalyzed off-line using the acquisition and analysis software WinLTP (Anderson and Collingridge, 2007). The peak amplitude of evoked fEPSPs was measured and expressed relative to the preconditioning baseline. LTP, LTD, and depotentiation were measured at 60 min after conditioning. The pathway in which plasticity was induced was randomly allocated; on some occasions, it was the entorhinal, and on others, the temporal input.
To investigate the effect of VDCCs on synaptic plasticity, 20 μm verapamil was added to the perfusate and allowed to wash on for 30 min before, during, and 10 min after high- or low-frequency stimulation. To investigate the effect of VDCCs on LTD expression, 20 μm verapamil was added to the perfusate 60 min after induction of LTD.
Statistical analysis
Novel object preference test.
The discrimination ratio (DR) was calculated as the time in the test phase spent exploring the novel object minus that spent exploring the previously experienced object divided by the total exploration time. The total exploration time was also calculated for both the acquisition and test phases. If the time of exploration was <15 s in the acquisition phase or <10 s in the test phase, the animal was discarded from the analysis of that experiment. Comparisons between treatment groups were analyzed by within-subject ANOVAs. One-sample t tests were used to determine whether the DR for each group was significantly different from zero, as zero corresponds to a lack of discrimination between novel and familiar objects. All tests used a significance level of p = 0.05 and were two-tailed.
Fos-labeled cell counts.
Each count was normalized by dividing it by the mean of all the counts for that area across both hemispheres for each rat. The normalized and absolute (non-normalized) counts were analyzed by ANOVA with repeat measures, with the factor treatment (saline or verapamil) being measured between subjects, and the factors novelty/familiarity and area (PRH, Te2, AU, and ENT) being measured within subjects.
In vitro electrophysiology.
Data were analyzed from one slice per rat (n, number of rats). Data pooled across slices are expressed as the mean ± SEM. LTP and LTD were compared with baseline, and depotentiation was compared with preestablished LTP. Data comparisons were made using two-tailed t tests.
Results
Familiarity discrimination
Preferential exploration of a novel rather than a familiar object was used to determine the effects of antagonists of L-type VDCCs on recognition memory.
Experiment 1: systemic administration of verapamil
Verapamil administered 1 h before acquisition caused an impairment in familiarity discrimination when the memory delay was 24 h, but not when it was 20 min (Fig. 2A). With a 20 min delay, both saline- and verapamil-treated rats spent a significantly greater proportion of time exploring the novel rather than the familiar object (DR > 0: saline: t(7) = 2.65, p = 0.03; verapamil: t(7) = 8.04, p < 0.001). In contrast, with a 24 h delay, the verapamil-treated rats were significantly impaired compared with the saline group (F(1,11) = 10.2; p = 0.009); the verapamil group failed to discriminate the familiarity of objects, whereas the saline group did discriminate (verapamil: DR ∼ 0, t(11) < 1, p > 0.1; saline: DR > 0, t(11) = 3.91, p = 0.002).
Figure 2.

Effects of verapamil on familiarity discrimination. A, Systemic verapamil produced impairment of long-term memory (24 h delay; n = 12), but not of shorter-term memory (20 min delay; n = 8). B, Local infusion of verapamil into PRH (n = 9) caused impairment of long-term memory (24 h delay) and in retrieval memory (24 h delay), but not of shorter-term memory (20 min delay). DR, Discrimination ratio (difference in time exploring the novel and familiar objects divided by the total exploration time in the test phase). Within-subject ANOVA treatment difference: *p < 0.05, **p < 0.01. One-sample t test of DR value against zero: Op < 0.05. Error bars indicate SEM.
The above results establish that there was no impairment after a delay of 20 min (when verapamil was active during both acquisition and retrieval) but there was impairment after a delay of 24 h (when the drug was active during acquisition but not during retrieval). Test was therefore made of whether the impairment with a 24 h delay was attributable to the drug state being different during both acquisition and retrieval (i.e., the amnesia was state dependent) (Colpaert, 1990). Verapamil was administered both 1 h before acquisition and 1 h before the recognition memory test with a 24 h delay. The rats were still unable to discriminate between the familiarity of objects (DR = 0.06 ± 0.14; DR ∼ 0, t(11) < 1, p > 0.1), indicating that the memory impairment at 24 h was not drug state dependent.
Experiment 2: local infusion of verapamil into PRH
To determine whether the critical region for action of the drug could be localized, verapamil was infused bilaterally via cannulae implanted in PRH. With such infusion, familiarity discrimination was again impaired with a 24 h but not a 20 min delay. Additionally, it was found that, with a 24 h delay, verapamil impaired retrieval (Fig. 2B). With the 20 min delay, both saline- and verapamil-treated animals discriminated between novel and familiar objects (DR > 0: saline: t(8) = 2.51, p = 0.04; verapamil: t(8) = 2.42, p = 0.04). With the 24 h delay, the verapamil group differed from the saline group (F(1,8) = 9.25; p = 0.02) when infusion was before acquisition. When infusion was before test rather than before acquisition, the verapamil group were again impaired compared with controls when the memory delay was 24 h (F(1,8) = 11.0; p = 0.01). With the 24 h delay, control rats discriminated whether saline was given before acquisition or before test (DR > 0: acquisition: t(8) = 3.36, p = 0.01; retrieval: t(8) = 2.86, p = 0.02), whereas the verapamil group did not (DR ∼ 0 in each case).
Experiment 3: systemic administration of diltiazem
To determine whether the impairments seen with verapamil (a phenylalkylamine) generalized to benzothiazepine VDCC antagonists, familiarity discrimination was tested after systemic administration of diltiazem. The same pattern of impairment was found: with a 24 h memory delay, diltiazem produced impairment when active during acquisition or test, but memory was normal with a 20 min delay (Fig. 3A). With a 20 min delay, both saline- and diltiazem-treated rats preferred the novel object (DR > 0; saline: t(11) = 2.97, p = 0.01; diltiazem: t(11) = 4.88, p < 0.001). With a 24 h delay, the diltiazem group was impaired compared with the saline group whether administration was before acquisition (F(1,11) = 6.88; p = 0.02) or before test (F(1,11) = 6.06; p = 0.03). In neither case did the diltiazem group show discrimination (DR ∼ 0); however, the saline group discriminated (DR > 0; acquisition: t(11) = 2.98, p = 0.01; retrieval: t(11) = 4.04, p = 0.002).
Figure 3.
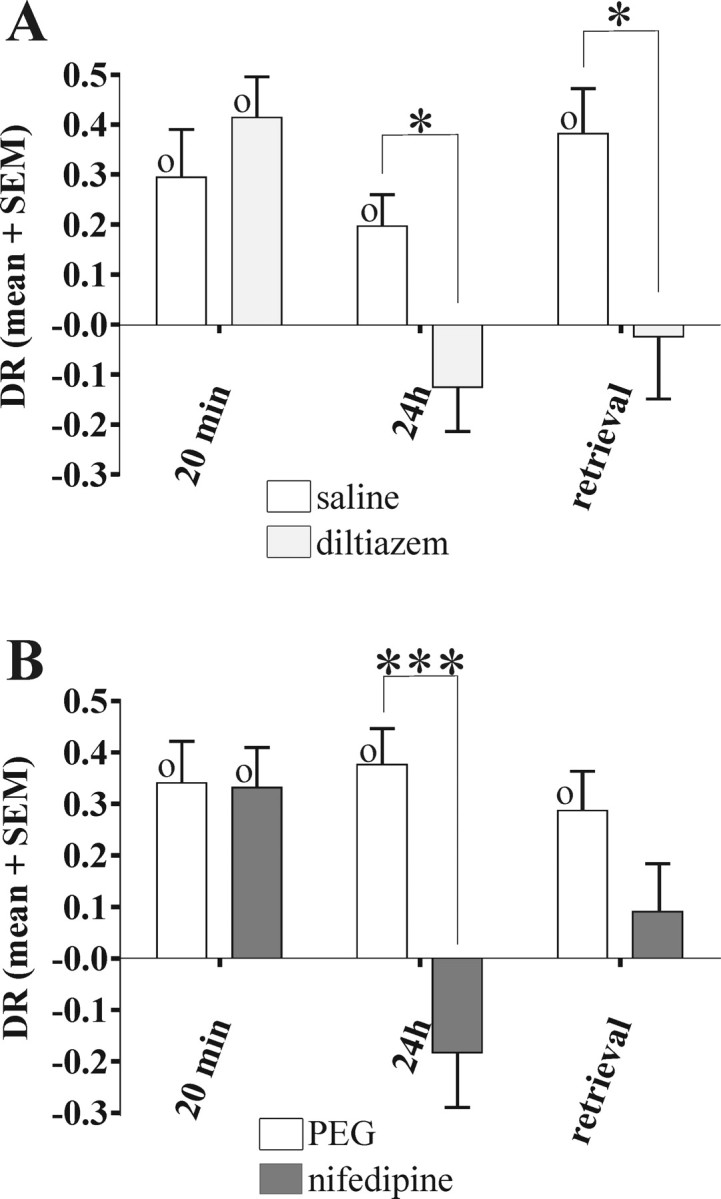
Effects of diltiazem and nifedipine in familiarity discrimination. A, Systemic diltiazem (n = 12) caused impairment of long-term memory (24 h delay) and of retrieval memory (24 h delay), but not of shorter-term memory (20 min delay). B, Systemic nifedipine caused impairment of long-term memory (24 h delay; n = 9) but not of shorter-term memory (20 min delay; n = 10). In the test of retrieval (24 h delay; n = 9), the nifedipine group did not discriminate, but did not differ significantly from the control group (which did discriminate). Within-subject ANOVA, *p < 0.05, ***p < 0.001. One-sample t test against zero, Op < 0.05. Error bars indicate SEM.
Experiment 4: systemic administration of nifedipine
To determine whether the impairments seen above generalized to dihydropyridine VDCC antagonists, recognition memory was tested after systemic administration of nifedipine. A similar pattern of impairment was again found (Fig. 3B). With the 20 min delay, both vehicle- and nifedipine-treated rats preferred the novel object (DR > 0; vehicle: t(11) = 2.97, p = 0.01; nifedipine: t(11) = 4.88, p < 0.001). In contrast, with the 24 h delay, the nifedipine group was impaired compared with controls (F(1,8) = 25.62; p < 0.001); the nifedipine group did not discriminate (DR ∼ 0), whereas the controls did (DR > 0; t(8) = 5.09, p = 0.001). When administration was before the memory test for the 24 h delay, the nifedipine-treated rats did not show significant discrimination (DR ∼ 0), unlike the controls (DR > 0; t(9) = 4.52, p = 0.001); however, the difference between the two groups did not reach significance (F(1,8) = 2.04; p = 0.2). Accordingly, the effect of nifedipine on retrieval was equivocal.
To investigate whether there was a difference in the effects of the different VDCC antagonists on retrieval, the results for the different drugs were compared. No significant difference in impairments was found: the interaction between treatment (drug or saline) and experiment (type of VDCC antagonist) was not significant (F(2,27) < 1; p > 0.1), in contrast, the treatment-related impairment across the different drugs was highly significant (F(1,27) = 15.29; p = 0.001).
Time of exploration in object recognition test
In all of these object recognition experiments, the times of exploration in both acquisition and test phases were recorded to confirm that the familiarity discrimination impairments observed could not be ascribed to changes in overall exploration (Tables 1–4). There were no significant differences between drug and vehicle groups in the time spent exploring, with the exception of one case. In the acquisition phase with a 24 h memory delay, nifedipine-treated rats explored the objects for significantly longer than vehicle-treated animals (F(1,8) = 6.92; p = 0.03). Despite this potential advantage, the nifedipine-treated rats showed impaired familiarity discrimination. Overall, there was thus no consistent evidence that the memory impairments were attributable to problems with general exploration.
Table 1.
Exploration time in acquisition and test phases (novel object preference task): systemic verapamil
| 20 min delay |
24 h delay |
|||
|---|---|---|---|---|
| Saline | Verapamil | Saline | Verapamil | |
| Acquisition | 27.1 ± 2.8 | 29.2 ± 1.5 | 36.3 ± 3.9 | 32.4 ± 3.7 |
| Test | 28.9 ± 4.2 | 29.2 ± 2.1 | 29.4 ± 2.4 | 29.7 ± 2.6 |
Data are in seconds (mean ± SEM).
Table 2.
Exploration time in acquisition and test phases (novel object preference task): verapamil via cannula
| 20 min delay |
24 h delay |
Retrieval (24 h) |
||||
|---|---|---|---|---|---|---|
| Saline | Verapamil | Saline | Verapamil | Saline | Verapamil | |
| Acquisition | 24.4 ± 2.3 | 23.3 ± 1.3 | 21.7 ± 2.0 | 19.8 ± 1.3 | 25.3 ± 2.1 | 27.1 ± 2.7 |
| Test | 31.3 ± 3.8 | 31.1 ± 4.3 | 20.9 ± 2.1 | 23.1 ± 1.8 | 24.0 ± 2.7 | 27.4 ± 3.4 |
Data are in seconds (mean ± SEM).
Table 3.
Exploration time in acquisition and test phases (novel object preference task): systemic diltiazem
| 20 min delay |
24 h delay |
Retrieval (24 h) |
||||
|---|---|---|---|---|---|---|
| Saline | Diltiazem | Saline | Diltiazem | Saline | Diltiazem | |
| Acquisition | 31.9 ± 2.5 | 38.2 ± 1.1 | 34.3 ± 1.7 | 36.8 ± 1.4 | 33.8 ± 2.0 | 37.5 ± 1.1 |
| Test | 35.2 ± 3.2 | 35.8 ± 4.9 | 38.4 ± 2.0 | 29.1 ± 1.5 | 37.1 ± 3.9 | 45.5 ± 4.4 |
Data are in seconds (mean ± SEM).
Table 4.
Exploration time in acquisition and test phases (novel object preference task): systemic nifedipine
| 20 min delay |
24 h delay |
Retrieval (24 h) |
||||
|---|---|---|---|---|---|---|
| PEG | Nifedipine | PEG | Nifedipine | PEG | Nifedipine | |
| Acquisition | 29.6 ± 2.4 | 30.3 ± 1.9 | 21.1 ± 5.1 | 31.4 ± 9.7* | 25.1 ± 2.3 | 28.6 ± 2.7 |
| Test | 26.1 ± 2.1 | 28.6 ± 3.4 | 22.4 ± 7.0 | 17.6 ± 3.1 | 18.8 ± 1.0 | 17.9 ± 1.4 |
Data are in seconds (mean ± SEM).
*PEG–nifedipine difference significant at P < 0.05.
Differential Fos expression
The different neuronal activations produced by novel and familiar pictures were imaged using immunohistochemistry for Fos. The activations were compared between hemispheres after a rat had viewed novel pictures with one eye and familiar pictures with the other eye using the paired-viewing procedure. In normal rats, the hemisphere primarily processing novel information (“novel hemisphere”) has higher Fos counts in PRH and Te2 than that primarily processing familiar information (“familiar hemisphere”) (Zhu et al., 1996; Wan et al., 1999). Verapamil was infused locally into PRH on each day so as to block acquisition while the animals were repeatedly shown pictures. No drug was given on the final day when the Fos expression induced by novel and repeatedly shown pictures was to be measured. The memory delay was ≥24 h as at test the repeatedly shown pictures had not been seen for at least 24 h.
Verapamil treatment disrupted the normal pattern of differential Fos expression in PRH and Te2, without changing Fos expression in the control areas of auditory (AU) and entorhinal (ENT) cortices (Fig. 4). For normalized Fos counts, a repeated-measures ANOVA with factors for area, novelty/familiarity, and treatment (verapamil or saline) revealed a significant three-way interaction (F(4,52) = 3.34; p = 0.02). The analysis was therefore continued by analyzing the data for each region separately. For PRH, there was a significant interaction between novelty/familiarity and treatment (F(1,13) = 51.30; p < 0.001). For the saline-treated group, Fos counts were significantly higher in the novel than the familiar hemisphere (F(1,7) = 64.99; p < 0.001). In contrast, for the verapamil-treated group, the familiar hemisphere had higher counts than the novel (F(1,6) = 6.24; p < 0.05), a reversed expression pattern previously also seen with scopolamine treatment (Warburton et al., 2003). The findings for Te2 were similar; indeed, no significant interaction involving area was found when the PRH and Te2 data were analyzed together. In Te2, there was a significant interaction between novelty and treatment (F(1,13) = 5.02; p = 0.04). For the saline group, Fos counts in Te2 were higher in the novel than the familiar hemisphere (F(1,7) = 9.44; p = 0.02), whereas there was no significant difference between the two hemispheres in the verapamil-treated rats. In control areas, AU and ENT, no significant interaction was found between novelty and treatment; and no difference was found between Fos counts in the novel and familiar hemispheres for either the saline or verapamil groups.
Figure 4.

Differential Fos expression. A, Images of Fos-labeled cells in PRH of a control rat. Paired-viewing procedure: I, hemisphere chiefly processing familiar pictures; II, hemisphere chiefly processing novel pictures. Note the higher number of Fos-positive cells on the novel side (II). Scale bar, 100 μm. B, Systemic normalized Fos counts for novel and familiar hemispheres for saline- or verapamil-treated rats for the following regions: PRH, Te2, AU, and ENT. In PRH and Te2, a significant interaction between novelty/familiarity and drug treatment was found, indicating that the differential in Fos expression between novel and familiar hemispheres of saline-treated rats was significantly different from that for verapamil-treated animals. Repeated-measures ANOVA effect of novelty/familiarity: *p < 0.05, ***p < 0.001. Error bars indicate SEM.
The Fos counts before normalization (Table 5) did not differ significantly between the saline and verapamil groups, indicating that verapamil did not have an overall effect on the Fos counts. In confirmation of the analysis on the normalized counts, a significant interaction was found between novelty and treatment for the absolute Fos counts in PRH (F(1,13) = 31.43; p < 0.001).
Table 5.
Non-normalized Fos-labeled cell counts in sample areas
| Areas | Control |
Verapamil |
||
|---|---|---|---|---|
| Novel | Familiar | Novel | Familiar | |
| PRH | 162 ± 41*** | 125 ± 28 | 133 ± 21 | 145 ± 20 |
| TE | 202 ± 58 | 163 ± 41 | 173 ± 51 | 170 ± 29 |
| AU | 291 ± 75 | 305 ± 77 | 272 ± 35 | 300 ± 45 |
| ENT | 200 ± 50 | 193 ± 42 | 190 ± 20 | 206 ± 24 |
Data are represented as mean ± SEM.
***Novel counts significantly higher than familiar, P < 0.001.
Synaptic plasticity
To evaluate the effect of blocking L-type VDCCs on synaptic plasticity in PRH slices, 20 μm verapamil was bath-applied to the perfusate before and during plasticity-inducing stimulation. At this concentration, 20 μm verapamil had no effect on baseline transmission measured by fEPSPs (Fig. 5).
Figure 5.

Effects of verapamil on synaptic plasticity in PRH slices. The presence of verapamil in the perfusate is indicated by a gray box. The traces illustrate fEPSPs taken at the time points indicated. Stimulus artifacts are blanked and replaced by arrows. Calibration: 0.2 mV, 10 ms. A, LTP (induced by 100 Hz HFS repeated 4 times, as indicated by the upward arrow) was readily induced in the presence of 20 μm verapamil (left; open circles). LTP induced in the presence of verapamil was not different from that induced in control slices (right; closed circles). B, De novo LTD (induced with 5 Hz stimulation indicated by the filled bar) was prevented in the presence of 20 μm verapamil; after washout, delivering an additional train of 5 Hz stimulation resulted in significant LTD (left; open circles). Verapamil had no significant effect on the expression of LTD, which was tested by bath applying 20 μm verapamil 60 min after induction of LTD in control slices (right; closed circles). C, After induction of LTP, depotentiation (induced by 1 Hz stimulation for 15 min as indicated by the filled bar) was also prevented by 20 μm verapamil (left; open circles). This contrasts with control experiments, in which 1 Hz LFS resulted in reversal of LTP to baseline (right; closed circles).
Delivering HFS in the presence of 20 μm verapamil produced LTP, which at 60 min after HFS was not significantly different from that induced in interleaved control slices (verapamil: +22 ± 5% from baseline, n = 8; control: +24 ± 4% from baseline, n = 7; p > 0.1) (Fig. 5A). In contrast, however, LTD normally inducible by delivering LFS (5 Hz; 10 min) was blocked by verapamil (response amplitude 60 min after LFS in the presence of verapamil, −3 ± 3% from baseline; p > 0.1; n = 6) (Fig. 5B, left). This block of LTD was reversible, as 5 Hz LFS subsequently delivered 50 min after verapamil washout resulted in significant LTD (60 min after washout of verapamil, −20 ± 5% from baseline; p = 0.01; n = 6). The level of LTD induced after verapamil washout was not significantly different (p > 0.1) from that induced in separate control experiments (response amplitude 60 min after LFS, −23 ± 5%; n = 7) (Fig. 5B, right). In addition, to test the effect of L-type VDCC blockade on LTD expression, verapamil was applied after LTD had been established (Fig. 5B, right). Verapamil did not affect LTD expression: expression was not significantly different from that measured at 60 min after LFS (response amplitude 30 min after verapamil application, −25 ± 6%; p > 0.1; n = 6).
It has previously been shown that 1 Hz LFS (900 stimuli; 15 min) applied following established LTP effectively reverses this LTP (Ziakopoulos et al., 1999; Massey et al., 2004). Such depotentiation was also blocked by verapamil: 60 min after LFS in the presence of verapamil, response amplitude was not significantly different (p > 0.1) when compared with LTP, but was significantly different (p = 0.025) when compared with baseline (LTP, +23 ± 6%; depotentiation, +19 ± 5%; n = 5) (Fig. 5C, left). In control experiments, LFS induced depotentiation (Fig. 5C, right). Sixty minutes after LFS, response amplitude was significantly (p = 0.025) different compared with LTP but not significantly different (p > 0.1) from baseline (LTP, +26 ± 8%; depotentiation, +5 ± 5%; n = 5).
Thus, verapamil blocked LTD and depotentiation but not LTP in perirhinal cortical slices.
Discussion
The results establish that L-type VDCC antagonists impair perirhinal-based recognition memory processes and plasticity. An example of each of the three main types of L-type VDCC antagonists—phenylalkylamines (verapamil), benzothiazepines (diltiazem), and dihydropyridines (nifedipine)—impaired acquisition of long-term (24 h) familiarity discrimination. This impairment was found both with systemic administration (verapamil, diltiazem, and nifedipine) and with localized infusion into PRH (verapamil). The acquisition impairment was paralleled by disruption of the normally greater neuronal activation produced by novel than familiar stimuli in PRH. Additionally, L-type VDCC antagonism impaired recognition memory retrieval processes when acquisition was normal. Our finding of involvement of L-type VDCCs in perirhinal familiarity discrimination and plasticity, adds to that in hippocampal and amygdalar-dependent memory and plasticity (Grover and Teyler, 1990; Bolshakov and Siegelbaum, 1994; Disterhoft et al., 1996; Morgan and Teyler, 1999; Borroni et al., 2000; Bauer et al., 2002; Woodside et al., 2004). However, in perirhinal cortex, our findings indicate that it is synaptic weakening rather than strengthening that is critically affected.
Thus, in parallel to the acquisition impairment, verapamil impaired LTD and depotentiation, but not LTP, in PRH slices. Although there have been reports highlighting a role for VDCCs in the induction of LTD in rat hippocampus and visual cortex (Bolshakov and Siegelbaum, 1994; McCoy and McMahon, 2007), our investigation appears to be the first reporting a role of L-type VDCCs in both LTD and depotentiation in PRH. Synaptic weakening, as is produced by LTD or depotentiation, has been argued to underlie perirhinal familiarity discrimination (Brown and Bashir, 2002; Bogacz and Brown, 2003). In contrast to the lack of effect on LTP found here in PRH, L-type VDCCs have been found necessary for LTP induction in amygdala and hippocampus (Grover and Teyler, 1990; Magee and Johnston, 1997; Kapur et al., 1998; Morgan and Teyler, 1999; Freir and Herron, 2003; Niikura et al., 2004; Voglis and Tavernarakis, 2006). It is unlikely that VDCC antagonism produces its effects through changes in synaptic transmission (Bolshakov et al., 1994; Morgan and Teyler, 1999; Freir and Herron, 2003; Niikura et al., 2004). Importantly, in support of this conclusion, verapamil produced no change in baseline transmission in perirhinal cortex in our in vitro experiments nor any change in overall perirhinal Fos counts in vivo.
The impairment of familiarity discrimination was found for a memory delay of 24 h but not 20 min. A similar pattern of impairment of long-term but not shorter-term recognition memory has been found previously for antagonism of either NMDA (Barker et al., 2006a) or metabotropic (Barker et al., 2006b) glutamate receptors in PRH. As perirhinal kainate receptor antagonism resulted in the opposite pattern of memory loss (amnesia after a 20 min but not a 24 h delay), there must be more than one underlying mechanism that supports perirhinal familiarity discrimination (Barker et al., 2006a). Thus, the current results are consistent with L-type VDCC antagonism affecting the NMDA and metabotropic glutamate receptor-dependent rather than the kainate receptor-dependent mechanism. Given that L-type VDCC antagonism does not affect kainate receptor-dependent memory, then any effects of VDCC antagonism on shorter-term (20 min) memory mechanisms would be masked by intact kainate-dependent memory. In particular, because of such masking, any impairment of the induction of LTD or depotentiation-like mechanisms would not be detectable as a memory impairment at a 20 min delay.
In the present experiments, the verapamil-induced change in Fos expression provides an additional potential link between memory and plasticity mechanisms: L-type VDCC antagonism disrupts perirhinal neuronal activation related to long-term recognition memory in addition to its effects on familiarity discrimination and perirhinal plasticity. One potential explanation for these effects is that calcium ion entry through L-type VDCCs can lead to activation of the transcription factor cAMP response element-binding protein (CREB) (Dolmetsch et al., 2001; West et al., 2002). CREB activated transcriptional processes have been linked with learning and memory, and synaptic plasticity (Bourtchuladze et al., 1994; Deisseroth et al., 1996; Silva et al., 1998; Genoux et al., 2002). More specifically, phosphorylation of CREB within PRH is essential for long-term plasticity and recognition memory mechanisms (Warburton et al., 2005). Moreover, CREB phosphorylation can lead to Fos production (Ahn et al., 1998; Silva et al., 1998), L-type VDCC antagonism in cell cultures modifies c-fos activation (Morgan and Curran, 1986), and oligonucleotide antagonism of Fos production in PRH impairs familiarity discrimination (Seoane and Brown, 2007). Hence it is possible that VDCC antagonism might act through this pathway to produce disruption of Fos expression and memory impairment. Future experiments need to explore the specificity of links between activation of VDCCs and CREB phosphorylation, as well as cooperations or occlusions between VDCCs and calcium entry via other transmitter receptor channels.
Interestingly, VDCC antagonism produced an impairment of recognition memory retrieval that has not been found with antagonism of NMDA, kainate, or metabotropic glutamate receptors (Barker et al., 2006a,b), nor with scopolamine antagonism or benzodiazepine agonism (Warburton et al., 2003; Wan et al., 2004). Retrieval deficits have been found previously with AMPA glutamate receptor antagonism (Winters and Bussey, 2005b; Winters et al., 2006), and verapamil impairs retrieval of a passive avoidance task (Lashgari et al., 2006). A retrieval deficit was not apparent with a memory delay of 20 min, although the VDCC antagonists given before acquisition were likely to have still been active. It is unlikely that state dependency masked such an effect, because the impairment remained for the 24 h memory delay when verapamil was given both before acquisition and before retrieval. Thus, as for acquisition, the retrieval effect also only affects those substrates of recognition memory necessary for long-term (24 h) familiarity discrimination and not those necessary for shorter-term (20 min) memory. The physiological substrate of this retrieval deficit remains to be determined. However, because verapamil has no effect on basal transmission and does not reverse previously established LTD (at least when applied at 60 min after LTD induction), neither of these potential effects of verapamil on synaptic function can explain the deficit in retrieval.
The memory impairments could not be ascribed to generalized behavioral effects of the drugs. Observation detected no differences in the general behavior of the animals with either systemic or local drug delivery. Moreover, the exploration times in each phase of the novel object preference task were similar for both drug-treated and control animals. For systemic administration and in vitro experiments, the chosen single doses of the antagonists were similar to the ones used by previous authors (Morgan and Teyler, 1999; Borroni et al., 2000; Freir and Herron, 2003; Niikura et al., 2004; Woodside et al., 2004), which correspond to dose levels at which the actions of the drugs are likely to be mainly on L-type VDCCs; at higher concentrations, actions have been reported on potassium channels and nicotinic receptors (Hume, 1985; Zhang et al., 1999; Houlihan et al., 2000; Madeja et al., 2000). That the same pattern of memory impairment was seen with exemplars of all three groups of antagonists, which bind to different regions of the channel (Striessnig et al., 1990, 1991; Nakayama et al., 1991; Nakayama and Kuniyasu, 1996; Hockerman et al., 2000), adds weight to the conclusion that the effects of the drugs are attributable to antagonism of L-type VDCCs. The parallel results produced by the localized infusion of verapamil establish that an action within the PRH region is sufficient to produce memory impairment. In particular, the results from local PRH infusion and the plasticity impairment found in PRH slices allow exclusion of the possibility that the memory deficits arose from an action on L-type VDCCs on the cardiovascular system. When verapamil was given locally, the normal pattern of Fos expression was changed in Te2 as well as PRH. Thus, part of the memory impairment could have arisen from drug action within Te2 as well as PRH. However, studies by others (Martin, 1991; Izquierdo et al., 2000; Attwell et al., 2001; Winters and Bussey, 2005a) suggest that the spread of infused drugs will have affected most of PRH but only part of Te2.
It is important to appreciate that the effects of L-type VDCC antagonism on memory appear to be dependent on the dose regimen: chronically repeated low doses have been reported to have, if any, beneficial effects on memory (Disterhoft et al., 1996; Batuecas et al., 1998; Meneses and Hong, 1998; Kane and Robinson, 1999; Veng et al., 2003; Lashgari et al., 2006)—although these might be indirect, because of improved cardiac performance. Thus, the amnesic effects of single acute doses in rats should not be taken as directly relevant to the chronic treatment of human cardiovascular problems.
In conclusion, exemplars of all three types of L-type VDCC antagonists produced similar impairments in both the acquisition and retrieval of long-term recognition memory. Moreover, the same pattern of impairment was found when verapamil was infused locally within PRH rather than being given systemically. The acquisition impairment was paralleled by disruption by verapamil of the normal differential neuronal activation in PRH produced by viewing novel and familiar stimuli, and the impairment of LTD and depotentiation but not LTP in PRH slices. Accordingly, familiarity discrimination and plasticity mechanisms in PRH are dependent on activation of L-type VDCCs.
Footnotes
We are grateful to the Medical Research Council for financial support and to J. Robbins and K. Narduzzo for technical assistance.
References
- Ahn S, Olive M, Aggarwal S, Krylov D, Ginty DD, Vinson C. A dominant-negative inhibitor of CREB reveals that it is a general mediator of stimulus-dependent transcription of c-fos. Mol Cell Biol. 1998;18:967–977. doi: 10.1128/mcb.18.2.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson WW, Collingridge GL. Capabilities of the Win LTP data acquisition program extending beyond basic LTP experimental functions. J Neurosci Methods. 2007;162:346–356. doi: 10.1016/j.jneumeth.2006.12.018. [DOI] [PubMed] [Google Scholar]
- Attwell PJ, Rahman S, Yeo CH. Acquisition of eyeblink conditioning is critically dependent on normal function in cerebellar cortical lobule HVI. J Neurosci. 2001;21:5715–5722. doi: 10.1523/JNEUROSCI.21-15-05715.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker GR, Warburton EC. NMDA receptor plasticity in the perirhinal and prefrontal cortices is crucial for the acquisition of long-term object-in-place associative memory. J Neurosci. 2008;28:2837–2844. doi: 10.1523/JNEUROSCI.4447-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker GR, Bashir ZI, Brown MW, Warburton EC. A temporally distinct role for group I and group II metabotropic glutamate receptors in object recognition memory. Learn Mem. 2006a;13:178–186. doi: 10.1101/lm.77806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker GR, Warburton EC, Koder T, Dolman NP, More JC, Aggleton JP, Bashir ZI, Auberson YP, Jane DE, Brown MW. The different effects on recognition memory of perirhinal kainate and NMDA glutamate receptor antagonism: implications for underlying plasticity mechanisms. J Neurosci. 2006b;26:3561–3566. doi: 10.1523/JNEUROSCI.3154-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker GR, Bird F, Alexander V, Warburton EC. Recognition memory for objects, place, and temporal order: a disconnection analysis of the role of the medial prefrontal cortex and perirhinal cortex. J Neurosci. 2007;27:2948–2957. doi: 10.1523/JNEUROSCI.5289-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batuecas A, Pereira R, Centeno C, Pulido JA, Hernández M, Bollati A, Bogónez E, Satrústegui J. Effects of chronic nimodipine on working memory of old rats in relation to defects in synaptosomal calcium homeostasis. Eur J Pharmacol. 1998;350:141–150. doi: 10.1016/s0014-2999(98)00250-7. [DOI] [PubMed] [Google Scholar]
- Bauer EP, Schafe GE, LeDoux JE. NMDA receptors and L-type voltage-gated calcium channels contribute to long-term potentiation and different components of fear memory formation in the lateral amygdala. J Neurosci. 2002;22:5239–5249. doi: 10.1523/JNEUROSCI.22-12-05239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bogacz R, Brown MW. Comparison of computational models of familiarity discrimination in the perirhinal cortex. Hippocampus. 2003;13:494–524. doi: 10.1002/hipo.10093. [DOI] [PubMed] [Google Scholar]
- Bolshakov VY, Siegelbaum SA. Postsynaptic induction and presynaptic expression of hippocampal long-term depression. Science. 1994;264:1148–1152. doi: 10.1126/science.7909958. [DOI] [PubMed] [Google Scholar]
- Borroni AM, Fichtenholtz H, Woodside BL, Teyler TJ. Role of voltage-dependent calcium channel long-term potentiation (LTP) and NMDA LTP in spatial memory. J Neurosci. 2000;20:9272–9276. doi: 10.1523/JNEUROSCI.20-24-09272.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994;79:59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- Brown MW, Aggleton JP. Recognition memory: what are the roles of the perirhinal cortex and hippocampus? Nat Rev Neurosci. 2001;2:51–61. doi: 10.1038/35049064. [DOI] [PubMed] [Google Scholar]
- Brown MW, Bashir ZI. Evidence concerning how neurons of the perirhinal cortex may effect familiarity discrimination. Philos Trans R Soc Lond B Biol Sci. 2002;357:1083–1095. doi: 10.1098/rstb.2002.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MW, Xiang JZ. Recognition memory: neuronal substrates of the judgement of prior occurrence. Prog Neurobiol. 1998;55:149–189. doi: 10.1016/s0301-0082(98)00002-1. [DOI] [PubMed] [Google Scholar]
- Chittajallu R, Alford S, Collingridge GL. Ca2+ and synaptic plasticity. Cell Calcium. 1998;24:377–385. doi: 10.1016/s0143-4160(98)90061-6. [DOI] [PubMed] [Google Scholar]
- Cho K, Kemp N, Noel J, Aggleton JP, Brown MW, Bashir ZI. A new form of long-term depression in the perirhinal cortex. Nat Neurosci. 2000;3:150–156. doi: 10.1038/72093. [DOI] [PubMed] [Google Scholar]
- Cho K, Aggleton JP, Brown MW, Bashir ZI. An experimental test of the role of postsynaptic calcium levels in determining synaptic strength using perirhinal cortex of rat. J Physiol. 2001;532:459–466. doi: 10.1111/j.1469-7793.2001.0459f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colpaert FC. Amnesic trace locked into the benzodiazepine state of memory. Psychopharmacology (Berl) 1990;102:28–36. doi: 10.1007/BF02245740. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Bito H, Tsien RW. Signaling from synapse to nucleus: postsynaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron. 1996;16:89–101. doi: 10.1016/s0896-6273(00)80026-4. [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Thompson LT, Moyer JR, Jr, Mogul DJ. Calcium-dependent afterhyperpolarization and learning in young and aging hippocampus. Life Sci. 1996;59:413–420. doi: 10.1016/0024-3205(96)00320-7. [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME. Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science. 2001;294:333–339. doi: 10.1126/science.1063395. [DOI] [PubMed] [Google Scholar]
- Ennaceur A, Delacour J. A new one-trial test for neurobiological studies of memory in rats. 1: Behavioral data. Behav Brain Res. 1988;31:47–59. doi: 10.1016/0166-4328(88)90157-x. [DOI] [PubMed] [Google Scholar]
- Franks KM, Sejnowski TJ. Complexity of calcium signaling in synaptic spines. Bioessays. 2002;24:1130–1144. doi: 10.1002/bies.10193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freir DB, Herron CE. Inhibition of L-type voltage dependent calcium channels causes impairment of long-term potentiation in the hippocampal CA1 region in vivo. Brain Res. 2003;967:27–36. doi: 10.1016/s0006-8993(02)04190-2. [DOI] [PubMed] [Google Scholar]
- Genoux D, Haditsch U, Knobloch M, Michalon A, Storm D, Mansuy IM. Protein phosphatase 1 is a molecular constraint on learning and memory. Nature. 2002;418:970–975. doi: 10.1038/nature00928. [DOI] [PubMed] [Google Scholar]
- Grover LM, Teyler TJ. Two components of long-term potentiation induced by different patterns of afferent activation. Nature. 1990;347:477–479. doi: 10.1038/347477a0. [DOI] [PubMed] [Google Scholar]
- Hannesson DK, Howland JG, Phillips AG. Interaction between perirhinal and medial prefrontal cortex is required for temporal order but not recognition memory for objects in rats. J Neurosci. 2004;24:4596–4604. doi: 10.1523/JNEUROSCI.5517-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannesson DK, Howland JG, Pollock M, Mohapel P, Wallace AE, Corcoran ME. Anterior perirhinal cortex kindling produces long-lasting effects on anxiety and object recognition memory. Eur J Neurosci. 2005;21:1081–1090. doi: 10.1111/j.1460-9568.2005.03938.x. [DOI] [PubMed] [Google Scholar]
- Hockerman GH, Dilmac N, Scheuer T, Catterall WA. Molecular determinants of diltiazem block in domains IIIS6 and IVS6 of L-type Ca2+ channels. Mol Pharmacol. 2000;58:1264–1270. doi: 10.1124/mol.58.6.1264. [DOI] [PubMed] [Google Scholar]
- Houlihan LM, Slater EY, Beadle DJ, Lukas RJ, Bermudez I. Effects of diltiazem on human nicotinic acetylcholine and GABAA receptors. Neuropharmacology. 2000;39:2533–2542. doi: 10.1016/s0028-3908(00)00116-7. [DOI] [PubMed] [Google Scholar]
- Hume JR. Comparative interactions of organic Ca++ channel antagonists with myocardial Ca++ and K+ channels. J Pharmacol Exp Ther. 1985;234:134–140. [PubMed] [Google Scholar]
- Izquierdo LA, Barros DM, Ardenghi PG, Pereira P, Rodrigues C, Choi H, Medina JH, Izquierdo I. Different hippocampal molecular requirements for short- and long-term retrieval of one-trial avoidance learning. Behav Brain Res. 2000;111:93–98. doi: 10.1016/s0166-4328(00)00137-6. [DOI] [PubMed] [Google Scholar]
- Kane KA, Robinson GB. Effect of chronic nimodipine on spatial learning and on long-term potentiation. Behav Brain Res. 1999;98:95–101. doi: 10.1016/s0166-4328(98)00057-6. [DOI] [PubMed] [Google Scholar]
- Kapur A, Yeckel MF, Gray R, Johnston D. L-Type calcium channels are required for one form of hippocampal mossy fiber LTP. J Neurophysiol. 1998;79:2181–2190. doi: 10.1152/jn.1998.79.4.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashgari R, Motamedi F, Zahedi Asl S, Shahidi S, Komaki A. Behavioral and electrophysiological studies of chronic oral administration of L-type calcium channel blocker verapamil on learning and memory in rats. Behav Brain Res. 2006;171:324–328. doi: 10.1016/j.bbr.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Madeja M, Müller V, Musshoff U, Speckmann EJ. Sensitivity of native and cloned hippocampal delayed-rectifier potassium channels to verapamil. Neuropharmacology. 2000;39:202–210. doi: 10.1016/s0028-3908(99)00110-0. [DOI] [PubMed] [Google Scholar]
- Magee JC, Johnston D. A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science. 1997;275:209–213. doi: 10.1126/science.275.5297.209. [DOI] [PubMed] [Google Scholar]
- Martin JH. Autoradiographic estimation of the extent of reversible inactivation produced by microinjection of lidocaine and muscimol in the rat. Neurosci Lett. 1991;127:160–164. doi: 10.1016/0304-3940(91)90784-q. [DOI] [PubMed] [Google Scholar]
- Martin KC, Barad M, Kandel ER. Local protein synthesis and its role in synapse-specific plasticity. Curr Opin Neurobiol. 2000;10:587–592. doi: 10.1016/s0959-4388(00)00128-8. [DOI] [PubMed] [Google Scholar]
- Massey PV, Johnson BE, Moult PR, Auberson YP, Brown MW, Molnar E, Collingridge GL, Bashir ZI. Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci. 2004;24:7821–7828. doi: 10.1523/JNEUROSCI.1697-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy PA, McMahon LL. Muscarinic receptor dependent long-term depression in rat visual cortex is PKC independent but requires ERK1/2 activation and protein synthesis. J Neurophysiol. 2007;98:1862–1870. doi: 10.1152/jn.00510.2007. [DOI] [PubMed] [Google Scholar]
- Meneses A, Hong E. Spontaneously hypertensive rats: a potential model to identify drugs for treatment of learning disorders. Hypertension. 1998;31:968–972. doi: 10.1161/01.hyp.31.4.968. [DOI] [PubMed] [Google Scholar]
- Morgan JI, Curran T. Role of ion flux in the control of c-fos expression. Nature. 1986;322:552–555. doi: 10.1038/322552a0. [DOI] [PubMed] [Google Scholar]
- Morgan SL, Teyler TJ. VDCCs and NMDARs underlie two forms of LTP in CA1 hippocampus in vivo. J Neurophysiol. 1999;82:736–740. doi: 10.1152/jn.1999.82.2.736. [DOI] [PubMed] [Google Scholar]
- Nakayama H, Kuniyasu A. Identification of binding sites for calcium channel antagonists. Jpn Heart J. 1996;37:643–650. doi: 10.1536/ihj.37.643. [DOI] [PubMed] [Google Scholar]
- Nakayama H, Taki M, Striessnig J, Glossmann H, Catterall WA, Kanaoka Y. Identification of 1,4-dihydropyridine binding regions within the alpha 1 subunit of skeletal muscle Ca2+ channels by photoaffinity labeling with diazipine. Proc Natl Acad Sci U S A. 1991;88:9203–9207. doi: 10.1073/pnas.88.20.9203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niikura Y, Abe K, Misawa M. Involvement of L-type Ca2+ channels in the induction of long-term potentiation in the basolateral amygdala-dentate gyrus pathway of anesthetized rats. Brain Res. 2004;1017:218–221. doi: 10.1016/j.brainres.2004.04.064. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. New York: Academic; 1998. The rat brain in stereotaxic coordinates. [DOI] [PubMed] [Google Scholar]
- Seoane A, Brown MW. Interfering with Fos expression impairs recognition memory in rats. Br Neurosci Assoc Abstr. 2007;19:103. [Google Scholar]
- Sheng M, Greenberg ME. The regulation and function of c-fos and other immediate early genes in the nervous system. Neuron. 1990;4:477–485. doi: 10.1016/0896-6273(90)90106-p. [DOI] [PubMed] [Google Scholar]
- Shi CJ, Cassell MD. Perirhinal cortex projections to the amygdaloid complex and hippocampal formation in the rat. J Comp Neurol. 1999;406:299–328. doi: 10.1002/(sici)1096-9861(19990412)406:3<299::aid-cne2>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Kogan JH, Frankland PW, Kida S. CREB and memory. Annu Rev Neurosci. 1998;21:127–148. doi: 10.1146/annurev.neuro.21.1.127. [DOI] [PubMed] [Google Scholar]
- Striessnig J, Glossmann H, Catterall WA. Identification of a phenylalkylamine binding region within the alpha 1 subunit of skeletal muscle Ca2+ channels. Proc Natl Acad Sci U S A. 1990;87:9108–9112. doi: 10.1073/pnas.87.23.9108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striessnig J, Murphy BJ, Catterall WA. Dihydropyridine receptor of L-type Ca2+ channels: identification of binding domains for [3H](+)-PN200-110 and [3H]azidopine within the alpha 1 subunit. Proc Natl Acad Sci U S A. 1991;88:10769–10773. doi: 10.1073/pnas.88.23.10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchekalarova J, Albrecht D. Angiotensin II suppresses long-term depression in the lateral amygdala of mice via L-type calcium channels. Neurosci Lett. 2007;415:68–72. doi: 10.1016/j.neulet.2006.12.040. [DOI] [PubMed] [Google Scholar]
- Veng LM, Mesches MH, Browning MD. Age-related working memory impairment is correlated with increases in the L-type calcium channel protein alpha1D (Cav1.3) in area CA1 of the hippocampus and both are ameliorated by chronic nimodipine treatment. Brain Res Mol Brain Res. 2003;110:193–202. doi: 10.1016/s0169-328x(02)00643-5. [DOI] [PubMed] [Google Scholar]
- Voglis G, Tavernarakis N. The role of synaptic ion channels in synaptic plasticity. EMBO Rep. 2006;7:1104–1110. doi: 10.1038/sj.embor.7400830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan H, Aggleton JP, Brown MW. Different contributions of the hippocampus and perirhinal cortex to recognition memory. J Neurosci. 1999;19:1142–1148. doi: 10.1523/JNEUROSCI.19-03-01142.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan H, Warburton EC, Zhu XO, Koder TJ, Park Y, Aggleton JP, Cho K, Bashir ZI, Brown MW. Benzodiazepine impairment of perirhinal cortical plasticity and recognition memory. Eur J Neurosci. 2004;20:2214–2224. doi: 10.1111/j.1460-9568.2004.03688.x. [DOI] [PubMed] [Google Scholar]
- Warburton EC, Koder T, Cho K, Massey PV, Duguid G, Barker GR, Aggleton JP, Bashir ZI, Brown MW. Cholinergic neurotransmission is essential for perirhinal cortical plasticity and recognition memory. Neuron. 2003;38:987–996. doi: 10.1016/s0896-6273(03)00358-1. [DOI] [PubMed] [Google Scholar]
- Warburton EC, Glover CP, Massey PV, Wan H, Johnson B, Bienemann A, Deuschle U, Kew JN, Aggleton JP, Bashir ZI, Uney J, Brown MW. cAMP responsive element-binding protein phosphorylation is necessary for perirhinal long-term potentiation and recognition memory. J Neurosci. 2005;25:6296–6303. doi: 10.1523/JNEUROSCI.0506-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AE, Griffith EC, Greenberg ME. Regulation of transcription factors by neuronal activity. Nat Rev Neurosci. 2002;3:921–931. doi: 10.1038/nrn987. [DOI] [PubMed] [Google Scholar]
- Winters BD, Bussey TJ. Transient inactivation of perirhinal cortex disrupts encoding, retrieval, and consolidation of object recognition memory. J Neurosci. 2005a;25:52–61. doi: 10.1523/JNEUROSCI.3827-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters BD, Bussey TJ. Glutamate receptors in perirhinal cortex mediate encoding, retrieval, and consolidation of object recognition memory. J Neurosci. 2005b;25:4243–4251. doi: 10.1523/JNEUROSCI.0480-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters BD, Saksida LM, Bussey TJ. Paradoxical facilitation of object recognition memory after infusion of scopolamine into perirhinal cortex: implications for cholinergic system function. J Neurosci. 2006;26:9520–9529. doi: 10.1523/JNEUROSCI.2319-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters BD, Saksida LM, Bussey TJ. Object recognition memory: neurobiological mechanisms of encoding, consolidation and retrieval. Neurosci Biobehav Rev. 2008;32:1055–1070. doi: 10.1016/j.neubiorev.2008.04.004. [DOI] [PubMed] [Google Scholar]
- Woodside BL, Borroni AM, Hammonds MD, Teyler TJ. NMDA receptors and voltage-dependent calcium channels mediate different aspects of acquisition and retention of a spatial memory task. Neurobiol Learn Mem. 2004;81:105–114. doi: 10.1016/j.nlm.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Zhang S, Zhou Z, Gong Q, Makielski JC, January CT. Mechanism of block and identification of the verapamil binding domain to HERG potassium channels. Circ Res. 1999;84:989–998. doi: 10.1161/01.res.84.9.989. [DOI] [PubMed] [Google Scholar]
- Zhu XO, Brown MW, McCabe BJ, Aggleton JP. Effects of the novelty or familiarity of visual stimuli on the expression of the immediate early gene c-fos in rat brain. Neuroscience. 1995;69:821–829. doi: 10.1016/0306-4522(95)00320-i. [DOI] [PubMed] [Google Scholar]
- Zhu XO, McCabe BJ, Aggleton JP, Brown MW. Mapping visual recognition memory through expression of the immediate early gene c-fos. Neuroreport. 1996;7:1871–1875. doi: 10.1097/00001756-199607290-00037. [DOI] [PubMed] [Google Scholar]
- Ziakopoulos Z, Tillett CW, Brown MW, Bashir ZI. Input- and layer-dependent synaptic plasticity in the rat perirhinal cortex in vitro. Neuroscience. 1999;92:459–472. doi: 10.1016/s0306-4522(98)00764-7. [DOI] [PubMed] [Google Scholar]