

Summary
The epigenetic landscape is dynamically remodeled during neurogenesis. However, it is not understood how chromatin modifications in neural stem cells instruct the formation of complex structures in the brain. We report that the histone methyltransferase PRDM16 is required in radial glia to regulate lineage-autonomous and stage-specific gene expression programs that control number and position of upper layer cortical projection neurons. PRDM16 regulates the epigenetic state of transcriptional enhancers to activate genes involved in intermediate progenitor cell production and repress genes involved in cell migration. The histone methyltransferase domain of PRDM16 is necessary in radial glia to promote cortical neuron migration through transcriptional silencing. We show that repression of the gene encoding the E3 ubiquitin ligase PDZRN3 by PRDM16 determines the position of upper layer neurons. These findings provide insights into how epigenetic control of transcriptional enhancers in radial glial determines the organization of the mammalian cerebral cortex.
Introduction
The mammalian cerebral cortex is composed of a vast diversity of neuronal cell types, which form the complex circuits necessary for high level cognition. The cerebral cortex is organized into six layers of excitatory projection neurons that originate during embryonic development from a relatively simple neuroepithelium. All cortical neurons derive from a population of highly specialized neural stem cells known as radial glia (RG). During early neurogenesis, RG self-renew and give rise to neurons destined for deep layers of the cortex. As neurogenesis proceeds, RG give rise to intermediate progenitor (IP) cells that will go through one or two rounds of cell division before producing pairs of neurons destined for upper layers of the cortex (Kwan et al., 2012; Noctor et al., 2004). Newly generated neurons reach their final position by migrating along the basal processes of RG, which span the thickness of the developing cortex (Noctor et al., 2004). Assembly of the complex circuitry of the cerebral cortex requires precisely timed transcriptional programs that define the position, connectivity and function of specific neuronal subtypes.
Chromatin modifying enzymes are transcriptional regulators that control gene expression through covalent modification of DNA or histones. Modifications of chromatin structure by histone methylation and acetylation are thought to play a key role in regulating the cell type and stage-specific context in which transcriptional complexes can act by modifying the availability of cis-regulatory elements (Nashun et al., 2015; Tuoc et al., 2013). Genome-wide epigenetic profiling experiments have shown that as cells progress toward their ultimate fates, they acquire a more restrictive chromatin state that promotes cell specific transcriptional programs (Zhu et al., 2013). However, it is not understood how chromatin modifying enzymes function in RG to establish the epigenetic landscape that determines cell type and stage specific gene expression. The importance of chromatin remodeling for brain development is underscored by recent studies showing a critical role of chromatin modifying factors in neurodevelopmental disorders (Mastrototaro et al., 2017).
The transcriptional regulator PRDM16 is a chromatin modifying enzyme that belongs to the larger PRDM (Positive Regulatory Domain) protein family, that is structurally defined by the presence of a conserved N-terminal histone methyltransferase PR domain (Hohenauer and Moore, 2012). PRDM16 has been shown to function in vitro as a histone 3 lysine 9 (H3K9) and histone 3 lysine 4 (H3K4) mono-methyltransferase (Pinheiro et al., 2012; Zhou et al., 2016). PRDM16 also regulates gene expression by forming complexes with transcriptional co-factors and other histone modifying proteins (Chi and Cohen, 2016). PRDM16 was previously shown to control embryonic and postnatal neural stem cell maintenance and differentiation in the brain (Chuikov et al., 2010; Inoue et al., 2016; Shimada et al., 2017). How PRDM16 functions to regulate transcriptional programs in the developing cerebral cortex remains largely unknown. Here, we show that PRDM16 regulates the sequential activation of stage-specific gene expression programs to establish the organization of the cortex. This function is executed by 1) transcriptional activation of genes that control the production of IP cells and the number of upper layer neurons; and 2) transcriptional repression of genes that control migration of upper layer neurons. PRDM16 controls gene expression primarily by regulating the activity of transcriptional enhancers. The histone methyltransferase domain of PRDM16 participates in transcriptional repression in RG and influences gene expression during early stages of neuronal differentiation. Together, our findings suggest that PRDM16 regulates the epigenetic state of transcriptional enhancers in RG to instruct neurogenesis and migration of late-born cortical neurons.
Results
Expression of Prdm16 in radial glia is conserved in different mammalian species
Prdm16 is expressed in the dorsal telencephalon throughout the neurogenic period (Figure S1A). At mid- and late stages of cortical neurogenesis (E13.5 and E15.5) and early post-natal development, Prdm16 was predominantly expressed in the cortical ventricular zone (VZ), overlapping with the RG-marker PAX6 (Figures 1A–1B and S1A)(Chuikov et al., 2010). In the rodent, Pax6 and Tbr2 are transiently co-expressed in RG transitioning into IP cells (Arai et al.,2011; Englund et al., 2005). PAX6+/TBR2+ cells in the VZ and subventricular zone (SVZ) expressed Prdm16 (Figure 1B). In contrast, PRDM16 was almost completely absent from PAX6−/TBR2+ IP cells in the SVZ (Figure 1B). We did not detect PRDM16 in mature post-mitotic neurons in the cortical plate (Figure 1SB), though it was detected in cortical astrocytes (Figure S1C). Hence, Prdm16 is selectively expressed in mouse RG and silenced upon differentiation into IP cells.
Figure 1. Prdm16 expression in radial glia is conserved in mammals.
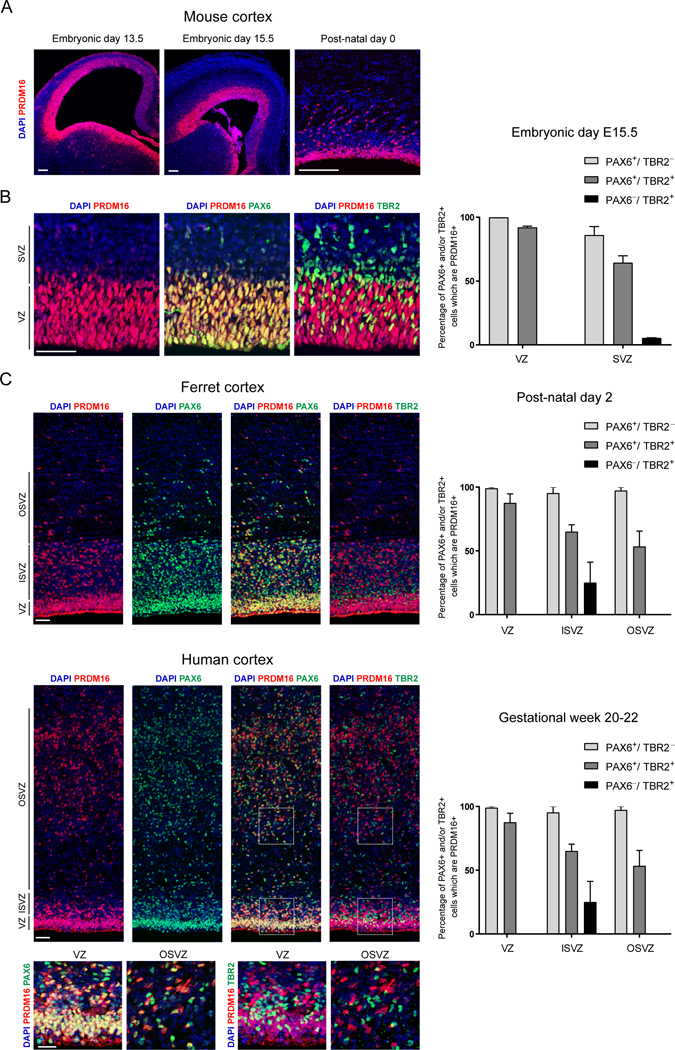
(A) Immunostaining of PRDM16 in coronal sections of the developing mouse brain
(B) PRDM16 overlaps with PAX6+/TBR2− and PAX6+/TBR2+ cells in the ventricular zone (VZ) and subventricular zone (SVZ) of mouse cortex and does not overlap with PAX6−/TBR2+ cells in the SVZ.
(C) PRDM16 is detected in PAX6+/TBR2− and PAX6+/TBR2+ cells in the VZ, inner SVZ (ISVZ) and outer SVZ (OSVZ) in ferret and human cortex. PRDM16 is present in a minority of PAX6−/TBR2+ cells.
Data represents mean ± SE for mouse (n=3), ferret (n=2) and human (n=2). Scale bars: 100 μm (A) and 50 μm (B, C).
Previous studies have suggested that Prdm16 is part of a core set of genes specifically expressed in mouse and human RG (Lui et al., 2014). Unlike the lissencephalic mouse cortex, gyrencephalic species display an expanded outer subventricular zone (OSVZ) that contains a basal population of outer RG (Lui et al., 2011). To determine if PRDM16 is expressed in both VZ and OSVZ, we examined ferret cortex at post-natal day 2 (P2) and human cortex at gestational weeks 20–22 (Figure 1C). PRDM16 is expressed in PAX6+ RG in the VZ, inner SVZ and OSVZ, while most PAX6−/TBR2+ IP cells within the ferret and human SVZ did not express PRDM16 (Figure 1C). Thus, specific PRDM16 expression in RG is conserved in gyrencephalic species.
PRDM16 activity in radial glia controls cortical neuron position
To determine the function of PRDM16 during cerebral cortex development, we crossed a mouse line carrying a conditional allele of Prdm16 (Prdm16flox/flox) with Emx1Ires-Cre mice (animals carrying both mutant alleles will be referred to as Prdm16 cKO)(Cohen et al., 2014; Gorski et al., 2002). Prdm16 cKO mice display a specific loss of PRDM16 in the developing cerebral cortex but not in the ganglionic eminences or the choroid plexus (Figure S2A). In contrast to Prdm16 null KO mice, Prdm16 cKO mice survive into adulthood and the size of the early post-natal brains is not affected (Figure S2B)(Chuikov et al., 2010).
We analyzed the laminar organization of P15 WT and cKO cortex by labeling neurons in upper layers (i.e. layers II-IV) with CUX1 and deep layers (i.e. layers V-VI) with CTIP2. There was a significant decrease in the thickness of cortical upper layers and a slight expansion of deep layers in cKO brains, whereas the overall thickness of the cortex did not change significantly in comparison to controls (Figure 2A). Quantification of CUX1+ cells through entire cortical hemisphere sections confirmed a decrease in layer II-IV projection neurons in cKO brains (Figure 2A). In contrast, the number of CTIP2+ or SATB2+ neurons was not significantly different between WT and cKO cortex (Figure 2A and S2C).
Figure 2. PRDM16 controls number and position of upper layer neurons.
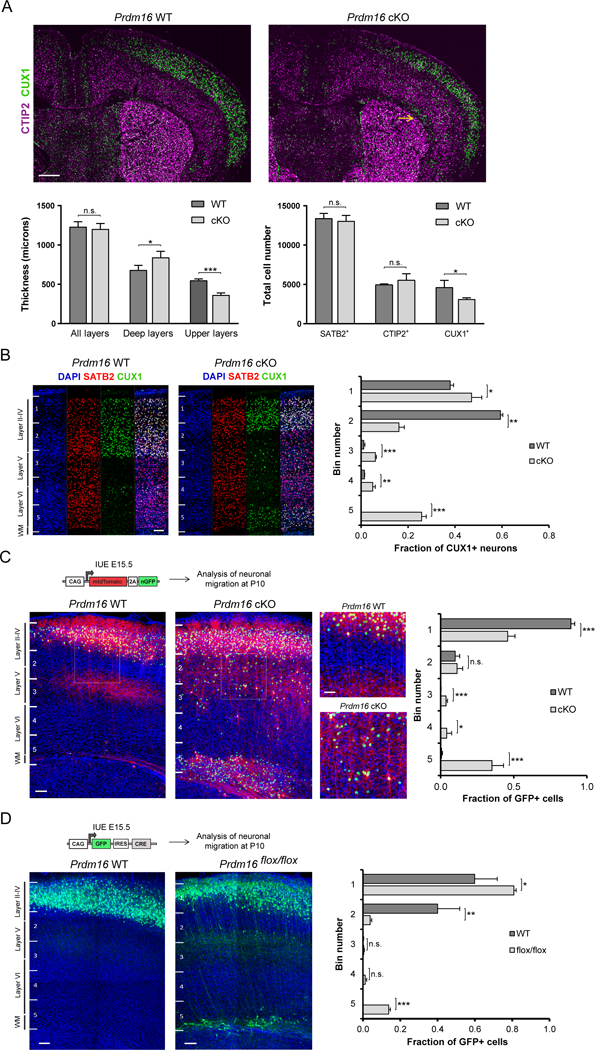
(A) Coronal sections of Prdm16 WT and cKO brains at P15. A CUX1+ heterotopia is shown in the white matter of the cKO cortex (arrow). Left plot shows quantification of deep and upper layer thickness (n=3). Right plot shows quantification of cortical neuron subtypes (n=3).
(B) Laminar distribution of CUX1+ neurons in Prdm16 WT and cKO cortex at P15. Cortical columns were divided into 5 bins. The fraction of CUX1+ neurons in each bin was quantified (n=3).
(C) E15.5 In utero electroporation (IUE) of plasmid encoding membrane tdTomato and nuclear GFP followed by analysis of migration at P10. The fraction of GFP+ nuclei in each bin was quantified (n=4 WT and n=7 cKO brains). Insets show lamination defects at higher magnification.
(D) E15.5 IUE of a plasmid encoding GFP and Cre followed by analysis of migration at P10. The fraction of GFP+ cells in each bin was quantified (n=3).
Results represent mean ± SD; statistical analysis is unpaired Student’s t test (*p < 0.05, **p < 0.01, *** p < 0.001; n.s., not significant). Scale bars: 500 μm (A) and 100 μm (B–D).
Numerous ectopic CUX1+ neurons were scattered across deep layers of the cKO cortex, as well as within distinctive neuronal heterotopias in layer VI and the white matter (WM) (Figure 2A, 2B and S2D). RORβ+ layer IV neurons were located close to the pial surface of the cKO cortex at P15, while whisker barrels in layer IV of the somatosensory cortex were small, disorganized and superficial in cKO brains, compared to controls (Figure S2C). Hence, Prdm16 cKO brains show cortical lamination defects. Quantification of S100β+ cells in WT and cKO cortex at P15 showed no difference, suggesting that PRDM16 does not affect astrocyte numbers (Figure S2E).
The lamination defects in Prdm16 cKO cortex indicate that late born upper layer neuron migration is impaired in mutant brains. To test this possibility, we electroporated control and cKO cortex at E15.5 with a plasmid encoding membrane tdTomato and nuclear GFP in order to label cortical RG and their progeny, which was analyzed at P10. As expected, control RG generated upper layer neurons that migrated into layers II/III projecting axons that branched extensively in layer V before entering the WM (Figure 2C). In contrast, mutant RG generated a large number of cortical neurons that did not reach the upper layers and either remained ectopic in deep cortical layers or formed heterotopias in the WM, resulting in disruption of the laminar organization of the cortex (Figure 2C).
Migration defects in cortical neurons could result from an inability of mutant RG to provide the structural scaffold that guides migration. However, there were no gross structural defects in the basal processes of mutant RG at E16.5 (Figure S3A), a stage when layer II-IV neurons are actively migrating to the cortical plate (Kwan et al., 2012). We next mutated Prdm16 in a subset of cortical RG by electroporating a plasmid encoding Cre and GFP into the ventricles of E15.5 Prdm16flox/flox embryos, and analyzed at P10. A group of electroporated neurons derived from mutant RG did not migrate into upper layers, remaining ectopic in the WM (Figure 2D). In addition, knock-down (KD) of Prdm16 by electroporating a short hairpin RNA (shRNA) into WT brains at E14.5 resulted in ectopic GFP+/CUX1+ neurons in deep cortical layers and heterotopias in the WM at P5 (Figure S3B and S3C). Together, our results suggest that PRDM16 has a lineage-autonomous role in directing upper layer cortical neuron migration. To rule out the possibility that low levels of Prdm16 in newborn cortical neurons might influence migration, we mutated Prdm16 specifically in newborn cortical neurons using the NexCre mouse line which is active as early as E11.5 (Figure S3D)(Goebbels et al., 2006). In utero electroporation (IUE) of a plasmid expressing nuclear GFP into NexCre Prdm16flox/flox cortex at E15.5 indicated that migration of upper layer neurons was not impaired (Figure S3E). Therefore, Prdm16 expression in newborn cortical neurons is not necessary for their migration into upper layers.
The presence of ectopic upper layer neurons in Prdm16 cKO cortex could cause disruptions in the organization of long range axonal projections connecting different brain areas. To test this, we analyzed cortical neurons electroporated with a plasmid encoding membrane tdTomato and nuclear GFP (Figure S4A). In control brains, most callosal axons projected into a single contralateral column that mirrors the ipsilateral electroporation site (Figure S4B and S4C). In contrast, callosal axons in mutant brains were more broadly dispersed along the medial-lateral axis of the contralateral hemisphere (Figure S4B and S4C) and displayed increased axonal projections into the ipsilateral motor area (Figure S4B and S4C). These observations suggest that Prdm16 might play a role in determining the pattern of axonal projections in the cortex.
PRDM16 activity in radial glia controls intermediate progenitor cell production and proliferation
The fact that Prdm16 expression is restricted to RG, together with the effect of Prdm16 loss of function on both the number and organization of upper layer neurons, led us to investigate the function of Prdm16 in cortical neurogenesis. There was no difference in the total number of dividing cells or the number of PAX6+ RG in the germinal zones (i.e. VZ plus SVZ) in WT and cKO cortex at E15.5 (Figure 3A, S5A, and S5B). In contrast, the number of mitotic and interphase TBR2+ IP cells was decreased in the cKO cortex, suggesting defects in the production of IP cells (Figure 3A).
Figure 3. PRDM16 controls intermediate progenitor generation and expansion.
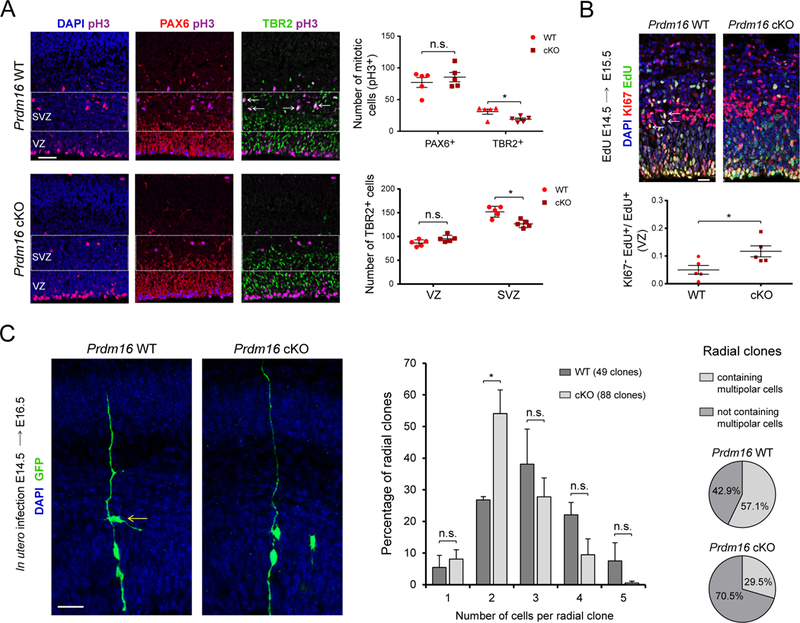
(A) E15.5 WT and Prdm16 cKO cortical sections immunostained for PAX6, TBR2 and pH3. The ventricular zone (VZ) and subventricular zone (SVZ) are indicated. Arrows show examples of TBR2+/pH3+ cells in the SVZ of WT cortex. Top plot shows quantification of PAX6+/pH3+ and TBR2+/pH3+ cells. Lower plot indicates the number of TBR2+ cells (n=5).
(B) Cell cycle exit analysis. Arrows indicate examples of KI67+/EdU+ cells. The fraction of KI67−/EdU+ cells relative to EdU+ cells was quantified (n=5).
(C) Clonal analysis of radial glia. The number of cells per clone was analyzed. Pie charts indicate the percentage of clones containing cells with multipolar morphology (arrow). Data represents mean ± SE; statistical analysis is unpaired Student’s t test (*p < 0.05, **p < 0.01, *** p < 0.001; n.s., not significant). Scale bars: 50 μm (A), 20 μm (B, C).
Reduced numbers of dividing IP cells in the mutant SVZ could result from changes in the number of progenitors re-entering the cell cycle. To test this hypothesis, we injected EdU at E14.5 to label cycling progenitors and 24 hrs later analyzed the fraction of EdU-labeled cells that co-stained with Ki67, a marker of cell proliferation. The fraction of cells exiting the cell cycle (i.e. EdU+Ki67−cells / EdU+ cells) was increased in the VZ of the cKO cortex (Figure 3B). An increase in cell cycle exit in the mutant VZ might reduce the number of progeny generated by an individual RG. In the developing cerebral cortex, radial clones consist of a single RG and its progeny, which remain in close association to the basal process of the parent cell (Noctor et al., 2001). To label individual RG and their progeny, we injected a retrovirus expressing GFP into the ventricles of E14.5 WT and cKO embryos, and analyzed the size of cortical radial clones at E16.5. To estimate the number of progeny generated by individual RG, we only took into account radial units, meaning that all one-cell clones consisted of a single RG. Quantification of the number of cells per radial clone indicated that WT RG generate a “normal distribution” of clonal sizes, with a high percentage of clones composed of three cells (Figure 3C). In contrast, mutant RG produced a larger proportion of clones composed of two cells (Figure 3C). Most IP cells in the SVZ display a multipolar morphology (Kowalczyk et al., 2009). We found that the percentage of clones containing multipolar cells was reduced in cKO radial clones compared to WT clones, further supporting a decrease in IP cell generation by mutant RG (Figure 3C). Together, our results indicate that PRDM16 decreases cell cycle exit in the VZ, thereby promoting the production of IP cells and upper layer cortical neurons.
Intrinsic control of neuronal migration by PRDM16 activity in radial glia
The migration defects observed in Prdm16 cKO cortex could be the result of changes in the timing of neurogenesis or an intrinsic migration defect in the lineage of mutant RG. To discriminate between these possibilities, we assessed the timing of deep layer and upper layer neurogenesis in WT and cKO cortex. Since the majority of deep layer neurons are generated between E12 and E14 (Molyneaux et al., 2007), we injected EdU at E14.5 to label proliferating progenitors at the final stages of deep layer neurogenesis and analyzed the percentage of EdU+/CTIP2+ cells relative to EdU+ cells at E17.5. The relative percentage of EdU+/CTIP2+ cells generated after E14.5 did not change significantly between WT and cKO cortex, indicating that the birthdate of deep layer neurons is not affected in cKO mice (Figure S5C). We next analyzed if ectopic neurons in the cKO cortex were generated during the normal phase of upper layer neurogenesis between E14 and E17 (Molyneaux et al., 2007). We injected EdU at E17.5 and P0 and analyzed the distribution of EdU+/CUX1+ cells at P2 (Figure S5D). Most ectopic CUX1+ cells in P2 cKO brains are located in the WM adjacent to the ventricle (Figure S5D). The majority of these ectopic CUX1+ neurons in the WM at P2 had not incorporated EdU, suggesting that they were generated before E17.5, and likely produced during the normal period of upper layer neurogenesis (Figure S5D). These observations suggest that a change in the birthdate of upper layer neurons is not the main cause of their impaired migration phenotype and thus support an intrinsic migration defect in the progeny of mutant RG.
PRDM16 regulates stage-specific gene expression programs for progenitor amplification and neuronal migration
Our evidence indicates that PRDM16 regulates the production and migration of late born projection neurons, possibly by controlling gene expression programs that are critical for these processes. To analyze changes of gene expression during neurogenesis, we used antibody labeling of intracellular markers in single cell suspensions, followed by cell sorting and transcriptional profiling using RNA sequencing (RNA-seq)(Figure 4A)(Hrvatin et al., 2014). Sorting gates were adjusted to purify PAX6+ cortical RG and TBR2+ IP cells, whereas sorting of prospective post-mitotic neurons was based on the absence of PAX6 and TBR2 (Figure 4B). In comparison to unsorted cell suspensions, sorted cell suspensions show a high enrichment (i.e. > 99%) of the desired cell types (Figure 4C). Quantification of gene expression levels in sorted WT cells confirmed high Pax6 expression in RG and high Tbr2 expression in IP cells (Figure 4D). Consistent with previous reports, we found medium to high levels of NeuroD1, Dcx and Tubb3 expression in sorted IP cells, indicating that a subset of TBR2+ cells are immature neurons (Figure 4D and S6A)(Arai et al., 2011; Englund et al., 2005). In contrast, PAX6−/TBR2− sorted cells showed low levels of NeuroD1 and high levels of Dcx, Tubb3 and Map2, indicating a mixture of immature and mature cortical neurons (Figure 4D and S6A). We confirmed high Prdm16 expression in sorted PAX6+ cells and low Prdm16 expression in TBR2+ IP cells and PAX6−/TBR2− neurons (Figure 4E). Together, these observations demonstrate the isolation of purified cell types for subsequent transcriptional profiling during cortical neurogenesis. We next determined the gene expression differences between E15.5 WT and cKO sorted cells. Real-time PCR amplification of Prdm16 (exon 9) indicated very low expression in sorted cKO cells (Figure S6B). We sequenced RNA libraries from four biological replicates per genotype and evaluated the data by hierarchical clustering and principal component analysis, which demonstrated that major differences between samples are driven by genotype and not by batch effects (Figure S6C). Differential gene expression analysis revealed consistent changes between WT and cKO RG across all four replicates (Figure 4F). A group of 22 differentially expressed genes was confirmed by real-time PCR analysis (Figure S6D and supplementary tables S1–S4). RNA-seq of the isolated cell types indicated that most gene expression changes occur in cKO RG and IP cells (Figure 4G). We found 417 up-regulated genes and 247 down-regulated genes in cKO RG using an adjusted p-value < 0.05 (Figure 4G and SD1). There were 241 up-regulated and 164 down-regulated genes in sorted cKO IP cells, while in mature cortical neurons only a handful of genes displayed gene expression changes (Figure 4G and SD1). In general, up-regulated genes in all cell types presented larger fold changes in gene expression than down-regulated genes (Figure 4G). Most differentially expressed genes in RG and IP cells did not overlap (Figure 4H), indicating that Prdm16 can regulate stage-specific transcriptional programs.
Figure 4. Transcriptional profiling of WT and Prdm16 cKO cells at three stages of neurogenesis.
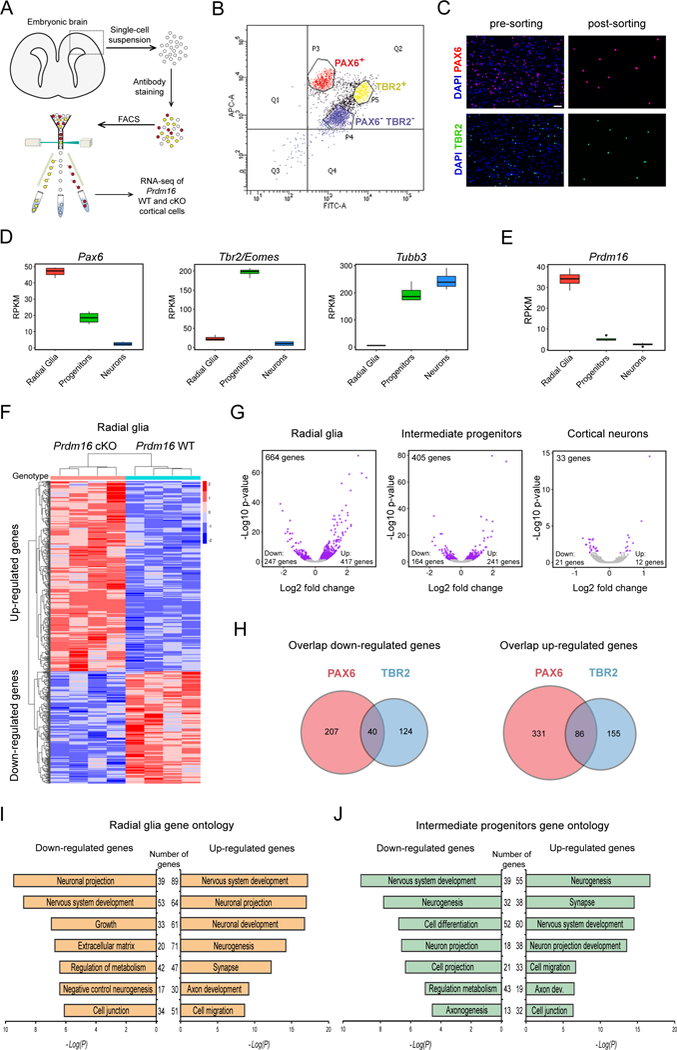
(A) Experimental design for transcriptional profiling of E15.5 cortical cells.
(B) Representative plot showing sorting gates for PAX6+ and TBR2+ cells. Double negative cells (PAX6−/TBR2−) are cortical neurons.
(C) Representative images of cell suspensions stained for DAPI, PAX6 and TBR2. Scale bar: 50 μm.
(D) Expression of Pax6, Tbr2 and Tubb3 in sorted cells from E15.5 WT cortex.
(E) Expression of Prdm16 in sorted cells from E15.5 WT cortex.
(F) Heat-map representing changes of gene expression in Prdm16 cKO radial glia (p < 0.05).
(G) Volcano plots showing fold changes of gene expression in cKO cells. Each plot indicates up-regulated and down-regulated genes (purple circles).
(H) Venn diagrams showing the overlap of down-regulated or up-regulated genes between mutant PAX6+ and TBR2+ cells. The number of genes is indicated.
(I) Gene ontology analysis of differentially expressed genes between WT and Prdm16 cKO radial glia.
(J) Gene ontology analysis of differentially expressed genes between WT and Prdm16 cKO intermediate progenitors.
All transcriptome analyses used 4 biological replicates per genotype. RPKM: Reads Per Kilobase of transcript per Million mapped reads.
Functional classification of differentially expressed genes in RG and IP cells by gene ontology revealed that up-regulated genes in cKO RG have reported roles in neuronal differentiation (i.e. neurogenesis), neuronal maturation (i.e. synapse and axon development) and cell migration (Figure 4I and SD1). In contrast, down-regulated genes in RG are involved in processes related to stem cell self-renewal and progenitor expansion (i.e. growth and negative regulation of neurogenesis) as well as maintenance of the progenitor niche (i.e. extracellular matrix and cell junction) (Figure 4I). Functional classification of differentially expressed genes in IP cells indicated up-regulation of genes involved in cell migration, whereas genes promoting neurogenesis, neuronal projection and axon development are enriched in both, up-regulated and down-regulated groups (Figure 4J). Thus, PRDM16 might regulate developmental programs that control early stages of RG progeny amplification, differentiation and migration.
Distal regulation by PRDM16 sets the gene expression programs for expansion and migration of radial glia progeny
Absence of Prdm16 in cortical RG results in the misregulation of over a thousand genes during neurogenesis. To identify the subset of genes that are transcriptional targets of PRDM16 and to understand how these genes are directly regulated, we performed chromatin immunoprecipitation followed by sequencing (ChIP-seq) at E15.5, when upper layer neurons are being generated. In two independent biological replicates, we detected 13864 and 26666 ChIP-seq peaks that represent putative PRDM16 binding sites. To identify the most reproducible binding regions, we used irreproducible discovery rate (IDR) analysis using an IDR < 0.05, which resulted in 3151 highly reproducible PRDM16 binding sites across the entire genome (Figure 5A and SD2). To validate the specificity of these binding sites, we performed PRDM16 ChIP-seq in two separate pools of E15.5 Prdm16 cKO cortices. The majority of PRDM16 binding sites in WT cortex show a significantly higher read density than equivalent genomic regions in cKO cortex (Figure S7A). Visualization of PRDM16 ChIP-seq peaks within genomic tracks demonstrated absence of binding sites in cKO samples (Figure S7A). In the embryonic cortex, most PRDM16 binding sites are located either at intergenic regions (1596 peaks) or introns (1363 peaks), while few binding sites map to transcription start sites (TSS) and proximal promoter regions (115 peaks)(Figure 5B). Analysis of DNA sequence motifs within all reproducible PRDM16 binding regions indicated high enrichment of binding motifs for transcriptional regulators known to play a key role in cortical neurogenesis, such as LHX2, SOX2/3, NEUROD1, TBR2 and MEIS1 (Figure 5C)(Guillemot and Hassan, 2017; Hsu et al., 2015; Sessa et al., 2008). Of note, our analysis did not recover a putative DNA binding motif for PRDM16, which is consistent with previous evidence suggesting that PRDM16 is likely to function as a co-factor with other proteins that directly bind to DNA (Harms et al., 2015). PRDM16 binding at genomic regions distal from most TSS precludes an easy identification of direct transcriptional targets among differentially expressed genes between WT and cKO cells. To overcome this limitation, we applied a statistical method that integrates fold changes of gene expression between WT and cKO cells with the number and proximity of PRDM16 binding sites to the TSS of differentially expressed genes (Wang et al., 2013). This method predicts whether a transcription factor functions as activator or repressor and determines the probability of direct transcriptional regulation on individual genes (Wang et al., 2013). This analysis predicted a robust function of PRDM16 as a transcriptional repressor (p = 1.85E-12) and a moderate function as a transcriptional activator (p = 7.85E-4)(Figure 5D). Statistical prediction of individual transcriptional targets in RG defined 221 repressed genes associated with 379 PRDM16 binding regions and 106 activated genes associated with 174 PRDM16 binding regions using a False Discovery Rate (FDR) < 0.05 (Figure 5E and SD3). Many cell migration genes are predicted to be directly repressed by PRDM16 activity in RG, while a number of progenitor amplification genes are predicted to be directly activated by PRDM16 in the same cells (Figure 5F). The list of transcriptional targets activated by PRDM16 includes genes that participate in IP cell generation and differentiation, such as Insm1 and Tbr2/Eomes (Figure 5F and S7B)(Arnold et al., 2008; Farkas et al., 2008; Sessa et al., 2008). Together, our results show that PRDM16 activity in RG provides a dichotomy of transcriptional repression and activation of functionally distinct groups of genes to coordinate the production and migration of late born cortical neurons. By combining the information on PRDM16 binding sites across the genome of cortical RG with the identification of 279 misregulated genes in mutant IP cells, we determined the subset of direct PRDM16 transcriptional targets for which gene expression is exclusively affected at the IP stage (Figure 5G and SD3). This analysis suggests that PRDM16 promotes transcriptional activation or silencing of cis-regulatory regions during RG differentiation into IP cells (Figure 5G). These findings suggest that PRDM16 activity in RG sets the gene expression program for subsequent stages of IP cell differentiation.
Figure 5. Distal binding by PRDM16 regulates functionally distinct groups of genes.
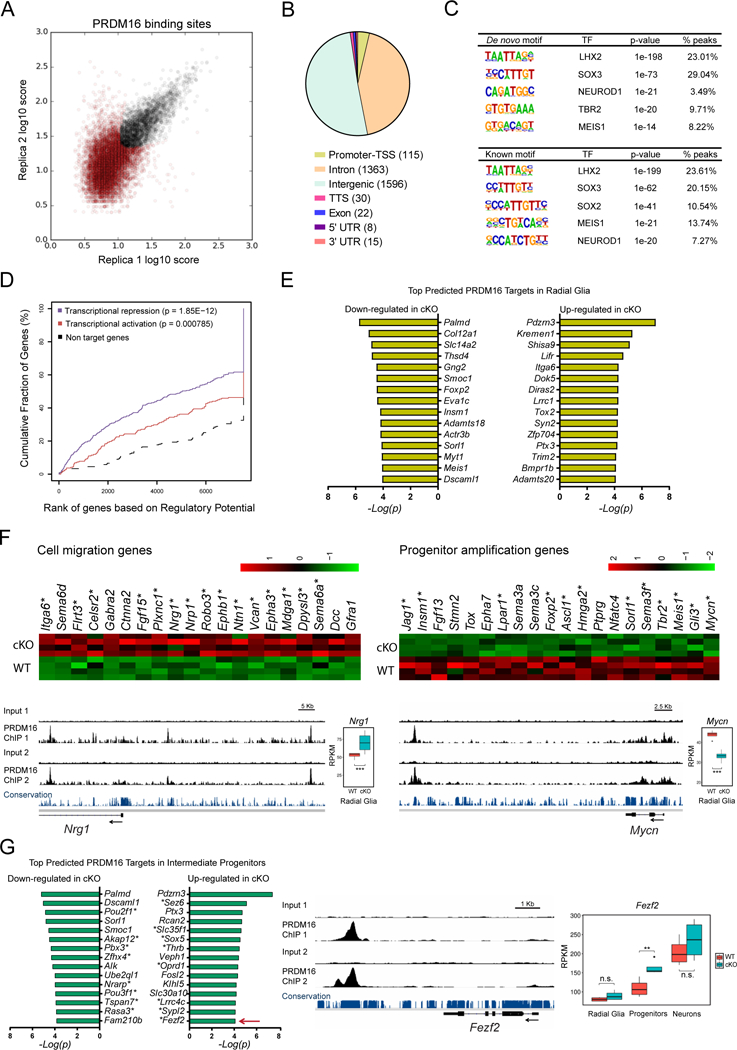
(A) Identification of genome-wide PRDM16 binding sites in the E15.5 cortex by ChIP-seq. The plot represents the irreproducible discovery rate (IDR) analysis of two independent biological replicates. A total of 3151 high-confidence PRDM16 binding sites were identified over an IDR < 0.05 (black circles).
(B) Distribution of PRDM16 binding sites across the genome. TSS, transcription start site; TTS, transcription termination site.
(C) De novo and known motif discovery within PRDM16 binding sites.
(D) Statistical integration of PRDM16 binding sites with differentially expressed genes in the cKO cortex predicts a dual function of PRDM16 as repressor (blue line) and activator (red line), using a set of non-differentially expressed genes as reference (black dashed line). The p-value for each function is indicated.
(E) Top predicted PRDM16 transcriptional targets in radial glia.
(F) Heat-maps and ChIP-seq tracks indicate examples of cell migration genes repressed by PRDM16 and progenitor amplification genes activated by PRDM16. Asterisks indicate predicted direct transcriptional targets of PRDM16. Nrg1 and Mycn are shown as examples for each group of genes.
(G) Top predicted PRDM16 transcriptional targets in intermediate progenitor cells. Asterisks indicate genes uniquely misregulated in intermediate progenitors, such as Fezf2 (arrow), which is shown as example of this group of genes.
Significant gene expression changes were identified using a FDR < 0.05 (*p < 0.05, **p < 0.01, *** p < 0.001; n.s., not significant).
Enhancer regulation by PRDM16 controls gene expression in the embryonic cortex
To understand the transcriptional mechanisms by which PRDM16 controls gene expression during cortical development, we sought to determine the histone methylation pattern within PRDM16 binding regions using the ENCODE project datasets collected from E14.5 mouse brains (Stamatoyannopoulos et al., 2012). We analyzed the overlap of PRDM16 binding regions with histone 3 lysine 4 tri-methylation (H3K4me3), an epigenetic modification enriched in promoter regions, and histone 3 lysine 4 mono-methylation (H3K4me), a chromatin mark associated with poised and active enhancers (Heintzman et al., 2007). In addition, we analyzed the overlap of PRDM16 binding with histone 3 lysine 27 tri-methylation (H3K27me3), a polycomb modification linked to transcriptional repression during neurogenesis (Hirabayashi and Gotoh, 2010). We found moderate H3K4me3 and H3K4me levels in regions bound by PRDM16, whereas there was little overlap with H3K27me3 (Figure 6A).
Figure 6. PRDM16 regulates transcriptional enhancers in the embryonic cortex.
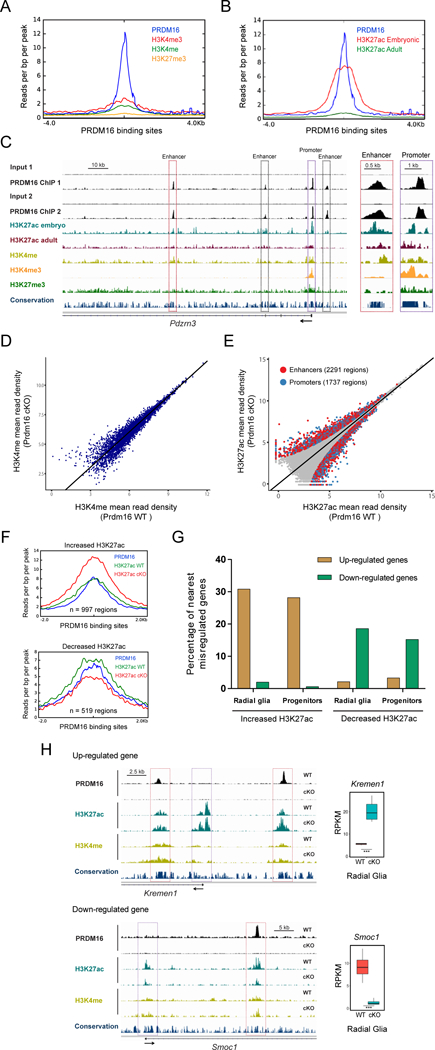
(A) H3K4me3, H3K4me, and H3K27me3 profile within a genomic window centered on PRDM16 binding sites in the embryonic cortex.
(B) H3K27ac embryonic and adult profile within a genomic window centered on PRDM16 binding sites in the embryonic cortex.
(C) Profile of histone modifications and PRDM16 binding near the transcription start site of Pdzrn3. Inset shows closer view of the Pdzrn3 promoter (purple box) and one enhancer (red box).
(D) Genome-wide comparison of H3K4me in E15.5 WT and Prdm16 cKO cortex (n=2).
(E) Genome-wide comparison of H3K27ac in E15.5 WT and Prdm16 cKO cortex (n=3). Differential H3K27ac enrichment within enhancers and promoters (FDR < 0.05) are highlighted as red and blue circles, respectively.
(F) Overlap of PRDM16 binding sites with regions showing differential H3K27ac between WT and Prdm16 cKO cortex.
(G) Percentage of nearest misregulated genes in the cKO cortex with regions of differential H3K27ac enrichment.
(H) Genome tracks showing regions with differential H3K27ac enrichment near Kremen1 and Smoc1, which are shown as examples of up-regulated and down-regulated genes, respectively. Significant changes in H3K27ac (FDR < 0.05) are indicated at enhancers (red boxes) and promoters (purple boxes).
Statistically significant gene expression changes were identified using a FDR < 0.05 (*** adjusted p-value < 0.001).
The moderate enrichment of H3K4me within PRDM16 binding sites led us to assess the overlap with histone 3 lysine 27 acetylation (H3K27ac), another chromatin modification associated with active enhancer regions (Creyghton et al., 2010; Rada-Iglesias et al., 2011). PRDM16 binding regions overlapped extensively with embryonic H3K27ac (Figure 6B). Given that PRDM16 is mostly associated with distal genomic regions relative to promoters, the enrichment of H3K27ac indicates that PRDM16 primarily binds to active enhancers. PRDM16 binding sites show much less H3K27ac enrichment in the adult cerebral cortex compared to embryonic stages (Figure 6B). We compared PRDM16 ChIP-seq peaks with embryonic and adult H3K27ac peaks in order to classify cortical enhancers as “developmental” (i.e. overlapping only with embryonic H3K27ac) or “developmental and adult” (i.e. overlapping with embryonic and adult H3K27ac), and found that around two-thirds of PRDM16-bound genomic regions are active enhancers only during cortical development (Figure S7C).
The gene encoding for the ubiquitin E3 ligase PDZRN3 is the top predicted transcriptional target of PRDM16 in both RG and IP cells (Figure 5E and 5G). We selected a genomic region near the TSS of Pdzrn3 to illustrate the pattern of histone modifications at PRDM16 binding sites. Pdzrn3 belongs to a small group of 18 out of 327 predicted transcriptional targets that show PRDM16 binding at the promoter, which is identified by high levels of H3K4me3 (Figure 6C). PRDM16 also binds at multiple enhancers within the genomic region of Pdzrn3; these regions show high levels of H3K4me and H3K27ac (Figure 6C). Hence, our analyses using the ENCODE datasets indicate that PRDM16 is primarily associated with active enhancers in the embryonic cortex. Next, we performed ChIP-seq to determine genome-wide differences in H3K4me and H3K27ac between E15.5 WT and cKO cortex. We identified 76,807 H3K4me peaks (2 replicates per genotype) and 67,723 H3K27ac peaks (3 replicates per genotype) with high concordance across all samples (Figure 6D and 6E). The overall levels of H3K4me did not change significantly between different genotypes (Figure 6D). In contrast, 4028 genomic regions exhibited differential H3K27ac enrichment between WT and cKO cortex (Figure 6E and SD2). These genomic regions correspond to 2291 enhancers and 1737 promoters and both types of regulatory regions show either gain or loss of H3K27ac in the cKO cortex (Figure 6E). Given that PRDM16 is mostly associated to enhancers, the large number of promoter regions showing changes in H3K27ac might reflect modifications in the long range enhancer-promoter interactions in the cKO cortex. PRDM16 binds to 997 regions that show increased H3K27ac and 519 regions that show reduced H3K27ac in the cKO cortex (Figure 6F). To evaluate the impact of these epigenetic changes on gene expression, we determined the percentage of misregulated genes that represent the nearest genes to PRDM16-bound regions showing H3K27ac changes in the cKO cortex. In general, up-regulated genes in mutant RG and IP cells are associated with genomic regions showing increased H3K27ac, whereas down-regulated genes are near regions showing reduced H3K27ac (Figure 6G and 6H). Therefore, our results suggest that PRDM16 represses and activates gene expression by modifying the epigenetic state of transcriptional enhancers.
The epigenetic state of PRDM16-regulated enhancers correlates with gene expression levels in the progeny of radial glia.
To further assess the impact of epigenetic changes on gene expression in the cKO cortex, we investigated whether enhancer activity is associated with misregulated gene expression in RG progeny (i.e. IP cells and neurons). We selected three of the top genes repressed by PRDM16 activity: Ptx3, Itga6 and Gabra2 (Supplementary tables S1 and S3). These genes are part of a small group of upregulated genes in the cKO cortex for which expression consistently changes in RG, IP cells and neurons (Figure S8A–S8C and SD1). We confirmed increased PTX3 levels in the VZ, SVZ and IZ of the cKO cortex by immunostaining (Figure S8A). Fluorescent in situ hybridization (FISH) of the cKO cortex indicated increased levels of Itga6 and Gabra2 in the VZ, SVZ and IZ (Figure S8B and S8C). Of note, Itga6 and Gabra2 transcript levels are increased in the cKO cortex only during early stages of neuronal differentiation in the IZ (Figure S8B and S8C). This observation correlates well with our RNA-seq results showing progressive silencing of up-regulated genes as the progeny of mutant RG differentiate into mature cortical neurons (Figure S8A–S8C). We found that Ptx3, Itga6 and Gabra2 are the nearest genes to a group of PRDM16-bound enhancers that show significantly increased H3K27ac in the mutant cortex (Figure S8A–S8C). The promoter regions of these genes also show an increase in H3K27ac, even in cases where there is complete absence of PRDM16 binding at the promoter (Figure S8A and S8B). Thus, changes in the epigenetic state of PRDM16-regulated enhancers may influence gene expression at early stages of neuronal differentiation.
Silencing of Pdzrn3 expression by PRDM16 promotes upper layer neuron migration
The function of PDZRN3 in cortical development is currently unknown, although previous research has shown that it regulates endothelial cell migration in vitro (Sewduth et al., 2014). Notably, the probability that Pdzrn3 is a direct transcriptional target of PRDM16 is two orders of magnitude higher than the second predicted target in both RG and IP cells (Figure 5E, 5G and SD3). Transcriptional profiling of cortical cells indicates that PRDM16 activity strongly represses Pdzrn3 expression in RG and IP cells (Figure 7A). PRDM16 binding sites at the promoter of Pdzrn3 and nearby enhancer regions show a significant increase of H3K27ac in the cKO cortex, suggesting that these regulatory regions are more active in the absence of PRDM16 (Figure 7B). In the E15.5 WT cortex, Pdzrn3 expression is relatively low in the VZ and SVZ and high at the apical region of the IZ, as shown by FISH (Figure 7C). In the cKO cortex, Pdzrn3 is up-regulated in the VZ, SVZ and IZ (Figure 7C). Pdzrn3 is down-regulated in fully differentiated neurons in the CP and there is no difference in Pdzrn3 expression between WT and cKO cortex in this region (Figure 7C). Thus, PRDM16 silences Pdzrn3 in RG and during subsequent stages of differentiation by directly repressing the activity of multiple regulatory regions of this gene. To investigate the role of PDZRN3 in the migration defects observed in the Prdm16 cKO cortex, we used a previously described shRNA that efficiently depletes PDZRN3 (Figure S9A)(Honda et al., 2010). We simultaneously reduced Prdm16 and Pdzrn3 expression by in utero electroporation of WT mice at E14.5, followed by analysis of cell migration at P5. The fraction of ectopic GFP+ cells in the cortical WM was not different between brains electroporated with Prdm16 shRNA alone or in combination with Pdzrn3 scrambled RNA (Figure 7D). In contrast, co-electroporation of Prdm16 shRNA and Pdzrn3 shRNA resulted in a significant reduction in the fraction of ectopic GFP+ cells in the WM (Figure 7D). In all electroporations, double KD of Prdm16 and Pdzrn3 resulted in absence of large heterotopias and only few scattered GFP+ cells were observed in the WM (Figure 7D and S9B). Quantification of GFP+ cells across 4 bins encompassing the entire cortex confirmed the reduction of the GFP+ fraction close to the WM in the double KD (Figure S9B and S9C). Thus, PRDM16 repression of Pdzrn3 expression is necessary to establish the position of upper layer neurons.
Figure 7. PRDM16 silencing of Pdzrn3 controls cortical neuron position.
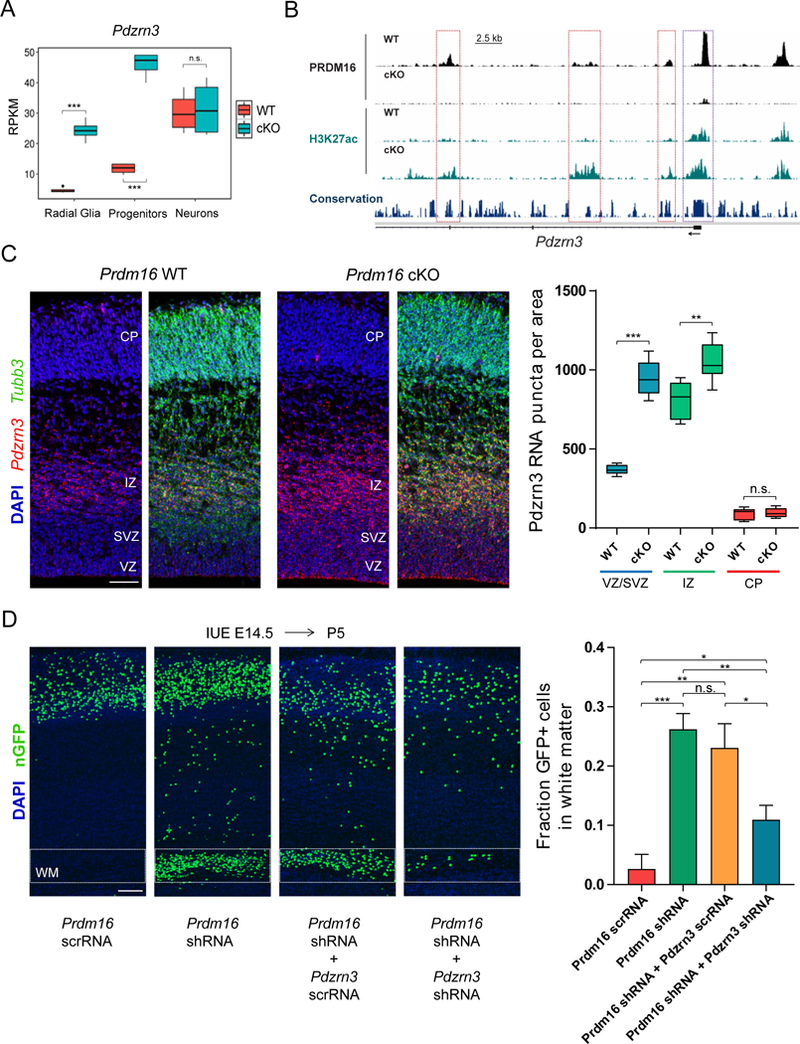
(A) Pdzrn3 expression at three stages of differentiation in WT and Prdm16 cKO cells.
(B) Genome tracks showing regions with differential H3K27ac enrichment near the transcription start site of Pdzrn3. Significant changes in H3K27ac (FDR < 0.05) are indicated at enhancers (red boxes) and promoter (purple box).
(C) Pdzrn3 expression was analyzed by FISH in E15.5 WT and Prdm16 cKO cortex. The number of Pdzrn3 RNA puncta per area (104 μm2) was quantified in the ventricular/subventricular zone (VZ/SVZ), intermediate zone (IZ) and cortical plate (CP) (n=3).
(D) Analysis of cell migration after single knockdown (KD) of Prdm16 or double KD of Prdm16 and Pdzrn3. After E14.5 IUE the fraction of GFP+ cells in the white matter (WM) was quantified at P5 (n=3, Prdm16 scrRNA; n=4, Prdm16 shRNA; n=5, Prdm16 shRNA and Pdzrn3 scrRNA; n=7, Prdm16 shRNA and Pdzrn3 shRNA).
Data represents mean ± SE; statistical analysis is unpaired Student’s t test (*p < 0.05, **p < 0.01, *** p < 0.001; n.s., not significant). Scale bars: 50 μm (C), 100 μm (D).
We tested the ability of overexpressed Pdzrn3 to disrupt migration by electroporating full-length Pdzrn3 into E14.5 WT cortex and analyzing the position of electroporated neurons at P5 (Figure S9D). We confirmed high levels of PDZRN3 in cortical neurons electroporated with Pdzrn3 (Figure S9E). Ectopic GFP+/CUX1+ neurons in layers V-VI and the WM were present in brains electroporated with Pdzrn3 compared to an empty vector (Figure S9D). Altogether, our results suggest that Pdzrn3 is a primary target of PRDM16 in the embryonic cortex and its transcriptional repression during early stages of neurogenesis promotes cortical neuron migration.
The histone methyltransferase domain of PRDM16 promotes neuronal migration through transcriptional repression
PRDM16 controls gene expression programs by regulating the epigenetic state of transcriptional enhancers. This function could be mediated by the intrinsic chromatin modifying activity of the PR domain of PRDM16 (Pinheiro et al., 2012; Zhou et al., 2016). To study the role of this domain in the cortex, we generated vectors driving expression of full-length Prdm16 (Prdm16 F.L.) or mutant Prdm16 lacking the sequence encoding the PR domain (ΔPRdm16), under the RG-specific Hes5 promoter (Figure 8A)(Mizutani et al., 2007). In utero electroporation of Prdm16 F.L. into E13.5 cKO cortex resulted in Prdm16 expression in the VZ/SVZ and absence of expression in the IZ and CP at E15.5 (Figure 8B). The majority of PRDM16+ electroporated cells in the mutant VZ and SVZ were also PAX6+ (Figure 8C). Thus, by driving Prdm16 expression under the Hes5 promoter in vivo, we could recapitulate the endogenous expression pattern of Prdm16.
Figure 8. The histone methyltransferase domain of PRDM16 promotes transcriptional silencing.
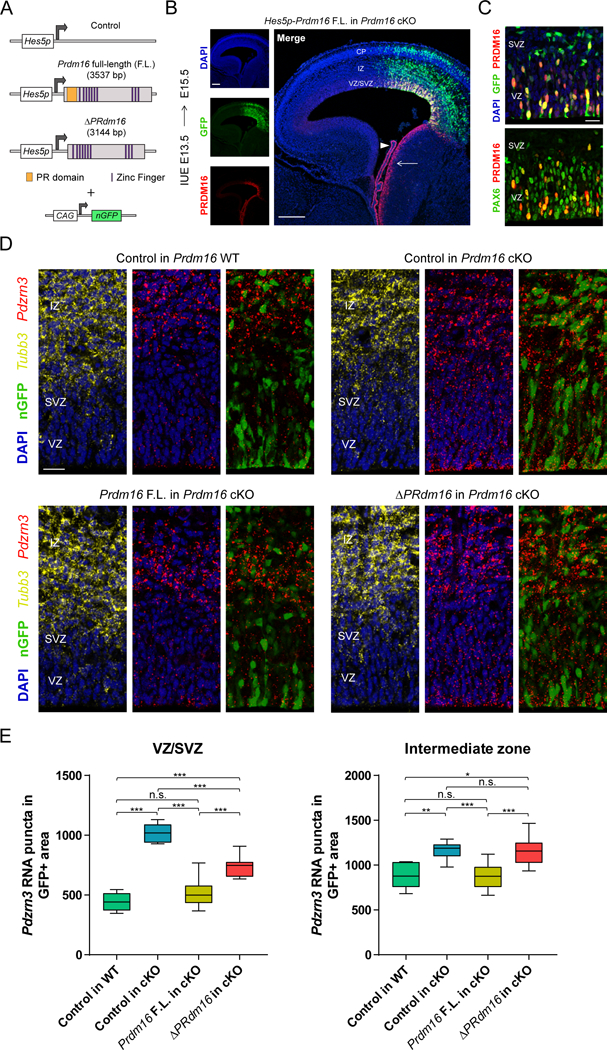
(A) Vectors driving Prdm16 full-length (F.L.) and ΔPRdm16 expression from the Hes5 proximal promoter. The experimental vectors were co-electroporated with a plasmid driving constitutive expression of nuclear GFP.
(B) IUE of Hes5p-Prdm16 F.L. into Prdm16 cKO cortex. Endogenous Prdm16 expression in the choroid plexus (arrowhead) and lateral ganglionic eminence (arrow) are indicated.
(C) IUE of Hes5p-Prdm16 F.L. into E13.5 Prdm16 cKO cortex shows that PRDM16 overlaps with PAX6 in most electroporated cells in the ventricular zone (VZ) and few cells in the subventricular zone (SVZ) at E15.5.
(D) In vivo transcriptional assay of PRDM16 activity in the embryonic cortex. The indicated vectors were electroporated into E13.5 WT or cKO cortex and Pdzrn3 expression in GFP+ cells in the VZ/SVZ and intermediate zone (IZ) was evaluated at E15.5 by fluorescent in situ hybridization. The IZ was identified by Tubb3 expression.
(E) Quantification of Pdzrn3 puncta in GFP+ cells in the cortical VZ/SVZ and IZ of electroporated brains. Results represent number of RNA puncta in 104 μm2 (n=3). Statistical analysis is unpaired Student’s t test (*p < 0.05, **p < 0.01, *** p < 0.001; n.s., not significant). Scale bars: 200 μm (B) and 20 μm (C, D).
We next tested if electroporation of Prdm16 F.L. or ΔPRdm16 rescued the neuronal migration defects in the cKO cortex. As expected, in utero electroporation of control vector into E13.5 cKO cortex resulted in relatively high numbers of ectopic GFP+ cells in the WM and IZ at E18.5 in comparison to WT cortex (Figure S9D and S9E). Electroporation of Prdm16 F.L in the cKO cortex rescued the migration defects of GFP+ cells, while electroporation of ΔPRdm16 did not (Figure S9D and S9E). Hence, the histone methyltransferase domain of PRDM16 promotes cortical neuron migration.
PRDM16 represses the expression of genes involved in migration. Thus, we tested whether the histone methyltransferase domain of PRDM16 was necessary for silencing gene expression. Since Pdzrn3 silencing has an important role in promoting neuronal migration, we used Pdzrn3 expression levels in electroporated GFP+ cells as a transcriptional read-out of PRDM16 activity in vivo. In utero electroporation of control vector at E13.5 showed high levels of Pdzrn3 expression in GFP+ cells in the VZ/SVZ of the cKO cortex at E15.5 in comparison to the WT cortex (Figure 8D and 8E). Introduction of Prdm16 F.L in the cKO cortex reduced Pdzrn3 expression down to WT levels in GFP+ cells in the VZ/SVZ, whereas ΔPRdm16 did not restore WT levels of Pdzrn3 expression (Figure 8D and 8E). Nonetheless, ΔPRdm16 did repress Pdzrn3 expression to some degree in the VZ/SVZ (Figure 8D and 8E). Given that some transcriptional targets of PRDM16 are up-regulated in immature neurons in the IZ of the cKO cortex, we evaluated the influence of the PR domain on gene expression in cortical neurons. For this assay, we quantified Pdzrn3 expression in GFP+ cells in the IZ, which was identified by Tubb3 expression (Figure 8D). Similar to our results in the VZ/SVZ, Prdm16 F.L fully restored Pdzrn3 expression to WT levels in immature cortical neurons in the IZ of the cKO cortex, whereas ΔPRdm16 does not (Figure 8D and 8E). Together, our findings suggest that epigenetic silencing of gene expression by PRDM16 activity in RG determines the position of upper layer cortical neurons.
Discussion
The complexity of the mammalian cerebral cortex is developmentally encoded by gene expression programs, which orchestrate the production neuronal subtypes and their migration to specific positions in the cortical plate. The dynamics of this process depends on intricate interactions between transcriptional complexes and changes in the permissive or repressive chromatin state of cis-regulatory elements across the genome. Our results suggest that PRDM16 modifies the genome-wide pattern of H3K27ac at distal enhancers to directly regulate gene expression in the embryonic cortex. This process is critical to regulate cortical neurogenesis and neuronal migration.
We found that a group of genes activated by PRDM16, such as Insm1 and Tbr2/Eomes, have roles in IP cell generation/differentiation (Farkas et al., 2008; Mihalas et al., 2016; Sessa et al., 2008). In addition, almost 10% of PRDM16-regulated regions contain the TBR2/EOMES binding motif. We propose that PRDM16 activates a transcriptional program that promotes indirect neurogenesis, thereby establishing the correct number of upper layer neurons.
Late-born cortical upper layer neurons cannot reach their final positions when they originate from RG lacking PRDM16 activity. Loss of Prdm16 in cortical RG leads to upregulation of cell migration genes, some of which have reported roles in repressing migration in the cortex, such as Itga6 and Gabra2 (Georges-Labouesse et al., 1998; Heck et al., 2007). Thus, PRDM16 might keep a subset of cell migration genes repressed in RG and IP cells, thereby establishing the proper timing for the onset of cortical neuron migration. We established that repression of Pdzrn3 by PRDM16 activity promotes migration of upper layer neurons. The precise function of Pdzrn3 in migrating cortical neurons is unknown. Identifying the substrates of PDZRN3 ubiquitin ligase activity should provide insight into the molecular regulation of neuron migration.
Transcriptional enhancers have been extensively mapped in the developing mouse and human cerebral cortex (de la Torre-Ubieta et al., 2018; Visel et al., 2013). However, much less is known about how genome-wide modifications in enhancer activity control specific cellular behaviors such as proliferation and migration to assemble cortical circuits. We identified over two thousand PRDM16-bound enhancers showing changes in H3K27ac levels in the developing Prdm16 cKO cortex. Up-regulated genes in the cKO cortex are preferentially associated with enhancers showing increased H3K27ac, whereas down-regulated genes are near enhancers showing decreased H3K27ac. Therefore, PRDM16 might regulate the permissive or repressive chromatin state of enhancer elements, thereby promoting or antagonizing gene expression. These findings raise the possibility that during embryonic neurogenesis, the number and position of cortical neurons is encoded in RG by the dynamic patterns of histone methylations and acetylations within enhancer regions.
Chromatin modifications influence gene expression across cell lineages during neurogenesis (Hirabayashi and Gotoh, 2010). We found that loss of PRDM16 activity in cortical RG promotes up-regulation of Ptx3, Gabra2, Itga6 and Pdzrn3 expression in IP cells in the SVZ and immature neurons in the IZ. Upregulation of these genes in immature cortical neurons is associated with increased activity of nearby enhancers in the cKO cortex. Thus, a role of PRDM16 as a chromatin-modifying enzyme involved in the inheritance of transient transcriptional states during cortical neurogenesis is an interesting possibility. Additional experiments are needed to assess whether PRDM16-bound enhancers remain misregulated once Prdm16 is no longer expressed in cortical neurons and whether this is functionally relevant for the migration defects in the cKO cortex. Regardless of the mechanisms involved in this process, our results show that transcriptional changes in RG have a permanent impact on the final laminar position of their neuronal progeny. This evidence highlights how transiently executed transcriptional programs have long lasting effects in the organization of the cerebral cortex.
Previous studies in vitro of PRDM16 function have shown that is can methylate H3K9 to promote transcriptional repression, or H3K4 to activate transcription (Pinheiro et al., 2012; Zhou et al., 2016). In this study, we tested the role of the PR domain of PRDM16 in transcriptional regulation and cortical neuron migration. Our data suggest that the activity of the PR domain is critical in RG to repress gene expression and to determine the migration of upper layer neurons. Moreover, we found that ΔPRdm16 retains some gene silencing activity, suggesting that other repressor domains of PRDM16, such as the CtBP-binding motifs, might also be important to repress gene expression in the embryonic cortex (Kajimura et al., 2008). Together, our findings suggest that the histone methyltransferase activity of PRDM16 regulates chromatin states at enhancer regions in RG, and this mechanism encodes the position of their neuronal progeny. Further experiments will be necessary to understand how the activity of the PR domain and PRDM16-interacting co-factors operate in conjunction to modify the pattern of histone methylations and chromatin accessibility within cortical enhancer elements.
PRDM16 haploinsufficiency is correlated with brain malformations and intellectual disability in humans with 1p36 deletion syndrome (Jordan et al., 2015). Human-gained enhancers are preferentially associated with genes expressed in outer RG and are important for human specific neural development (de la Torre-Ubieta et al., 2018). Defining the role of PRDM16 and other histone modifying enzymes at human-specific enhancers and their importance in neurodevelopmental disorders is an outstanding area for future research.
Methods details
All animal procedures conducted in this study were performed in accordance with the protocol approved by the Institutional Animal Care and Use Committee of Harvard Medical School. The mouse lines used in this study and their source are indicated in the supplementary resource table. Mouse housing and husbandry conditions followed the standards set by the Division of Comparative Medicine at Harvard Medical School.
Human tissue specimens and processing
Research performed on samples of human origin was conducted according to protocols approved under expedited category 5 with waiver of consent (45 CFR 46.110) by the institutional review boards of Beth Israel Deaconess Medical Center and Boston Children’s Hospital. Fetal brain tissue was received after release from clinical pathology, with a maximum post-mortem interval of four hours. Cases with known anomalies were excluded. Gestational ages were determined using fetal foot length. Tissue was transported in HBSS medium on ice to the laboratory for research processing.
In situ hybridization
Two digoxigenin-labeled RNA probes were generated to detect Prdm16 transcripts. One probe hybridizes with exon 4–8 and the other hybridizes with exon 9. Both probes gave almost identical Prdm16 expression patterns. To generate probe templates, exon 4–8 and exon 9 sequences were amplified by PCR from E14.5 mouse brain cDNA and cloned into pGEM-T easy vector (Promega). Antisense and sense probes were transcribed using T3 and T7 RNA polymerases and digoxigenin-labeled dNTPs (Roche). Probes were purified in polyacrylamide micro Bio-Spin columns (Bio-Rad), recovered in 20 μl of RNase-free water and diluted in 1 ml of hybridization buffer (see below) to generate a 10X probe solution. We performed colorimetric in situ hybridization on RNase-free cryostat sections using standard protocols. Briefly, brain sections were hybridized overnight at 65°C with probe solution in hybridization buffer (50% formamide, 5X SSC pH 5.0, 50 μg/ml yeast RNA, 1% SDS and 50 μg/ml heparin). The following day, sections were washed, treated with RNase A (20 μg/ml), incubated for 2 hrs in blocking buffer (100 mM maleic acid pH 7.5, 150 mM NaCl, 0.1% Tween 20, 20% goat serum and 2% Roche blocking reagent) and then incubated overnight at 4°C with anti-DIG antibody conjugated to alkaline phosphatase (Roche) diluted 1:2000 in blocking buffer. The following day, sections were washed and alkaline phosphatase was developed with BM purple (Roche). Sections were washed and mounted using Fluoromount-G (Southern Biotech).
Fluorescent in situ hybridization (FISH) was done by following the RNAscope protocol (Advanced Cell Diagnostics) on cryostat sections of PFA-fixed tissue according to the manufacturer’s instructions. Double FISH was done by combining RNAscope probes against mouse Pdzrn3, Itga6 or Gabra2 with a probe against Tubb3. For electroporation experiments, double FISH was followed by immunofluorescence to detect GFP on the same tissue sections.
Immunofluorescence and EdU labeling
Immunostaining of cellular proteins was done according to standard protocols. Briefly, mice pups at postnatal day 5, 8, 10 and 15 were anesthetized by intraperitoneal injection of ketamine (100 mg/Kg) - xylazine (12.5 mg/Kg) mix and transcardially perfused with PBS 1X pH 7.4 followed by 4% PFA. Brains were dissected and fixed in 4% PFA overnight at 4°C with rocking. Next day, brains were washed with PBS and sectioned into 100 μm vibratome sections or cryoprotected overnight in 30% sucrose to generate 20 μm cryostat sections. We performed antigen retrieval in all our immunostainings in order to enhance antibody signal. Vibratome and cryostat sections were incubated in antigen retrieval solution (10 mM sodium citrate, 0.05% Tween 20, pH 6.0) for 1 hr at 70°C. After incubation, sections were allowed to cool down at room temperature (RT), washed 3 times in PBS, incubated in blocking buffer (10% goat serum, 0.1% triton X-100, 0.01% sodium azide in PBS) for 1–2 hrs and then incubated with primary antibodies in blocking buffer at 4°C overnight. The next day, sections were washed 3 times with PBS and incubated with Alexa Fluor-conjugated secondary antibodies in blocking buffer for 1–2 hrs at RT. Sections were washed and nuclei stained with DAPI (4’,6-diamidino-2-phenylindole) before mounting with Fluoromount-G (Southern Biotech). DNA synthesis in neural progenitors was detected by EdU (5-ethynyl-2´-deoxyuridine) injection (approximately 5–20 μg/g of body weight) into the peritoneal cavity of pregnant mice or newborn pups. EdU incorporation was detected with the Click-iT assay (Invitrogen) using Alexa Fluor 647 azide according to the manufacturer’s instructions. Images were acquired with a Leica DM6000 FS epifluorescence microscope, a Leica SP8 confocal microscope and a Zeiss LSM 700 confocal microscope. Images were processed with Leica LAS X and Zen 2011 software or Image J.
In utero electroporation
Timed pregnant mice were anesthetized using an isoflurane vaporizer and placed on a warming pad. An abdominal incision of about 1 inch in length was made and the uterine horns were carefully exposed on top of a sterile gauze pad. Embryos were kept moist with pre-warmed PBS at 37°C during the entire procedure. Approximately, 0.5–1.0 μl of endotoxin-free DNA (1–3 μg/μl) diluted in PBS/0.025% Fast Green (SIGMA) was injected into the lateral ventricles of the forebrain using heat-pulled glass micropipettes (Drummond). Once all embryos were injected, 5 pulses of 30–40 volts (50 msec duration and 950 msec intervals) were applied with 5 mm or 7 mm platinum electrodes (BTX) connected to an ECM 830 square wave electroporator (BTX). The abdominal cavity was then sutured and stapled before administering buprenorphine (0.05–0.1 mg/kg) and ketoprofen (5–10 mg/kg). Mice were allowed to recover in a 37°C chamber for 2 hrs after surgery. Plasmids coding for Prdm16 shRNA and scrambled control were electroporated at 1 μg/μl, whereas vectors coding for Pdzrn3 shRNA and scrambled control were injected at 1.5 μg/μl. All shRNA and scrambled controls were combined with 0.5 μg/μl of pCAG-TAG (Addgene) to permanently label the nuclei of cortical neurons with GFP. Vectors coding for full-length Prdm16 or ΔPrdm16 were electroporated at 2.5 μg/μl in combination with 0.5 μg/μl of pCIG (Addgene). When needed, we used a neutral plasmid as ‘filler DNA’ in order to reach a final plasmid concentration of 3.0 μg/μl in all in utero electroporations.
Retrovirus production
Human embryonic kidney 293gp NIT-GFP retrovirus packaging cell line was grown to 90% confluency in DMEM (Gibco) supplemented with 10% Fetal Bovine Serum and transfected with pCMV-VSV-G vector using lipofectamine 2000 (Invitrogen) and Optimem (Gibco). Two days after transfection, cell supernatant was collected, filtered through 0.45 μm filter (VWR International) and centrifuged at 25000 r.p.m. for 90 min at 4°C. After centrifugation, 100 μl of cold PBS plus Ca+2 were added to the pellet and incubated at 4°C over 12 hrs. Viral particles were gently resuspended with a pipette and split into aliquots that were stored at ‒ 80°C. Viral particles with a titer of 106−107 PFU/ml were used for in utero injections into the lateral ventricles of E14.5 mouse embryos.
Fluorescence Activated Cell Sorting using intracellular antibodies
Sorting of immunostained cortical cells for transcriptional profiling was based on a previously described protocol (Hrvatin et al., 2014). Briefly, cortical tissue was dissected from E15.5 embryos and kept on ice in Hibernate-E (GIBCO) supplemented with B27 (GIBCO) during identification of WT and cKO genotypes among littermates. WT and cKO cortices were pooled by genotype and resuspended in digest solution containing 0.25% Trypsin (Invitrogen) and 0.01% DNase (SIGMA) in Hibernate-E minus Ca (BrainBits). Tissues were incubated at 37°C for 10 min in digest solution, washed twice in fresh Hibernate-E/B27 plus 0.01% DNase and mechanically dissociated into a single cell suspension. Cells were fixed in 4% PFA plus 0.1% saponin for 30 min at 4°C, then washed twice in washing buffer (0.1% saponin, 0.2% BSA in PBS) and resuspended in antibody buffer (0.1% saponin, 1% BSA in PBS) containing anti-PAX6 and anti-TBR2 antibodies and incubated for 0.5–1 hr at 4°C with rocking. Cells were washed twice, then incubated with Alexa Fluor-conjugated secondary antibodies for 0.5 hr at 4°C with rocking, washed twice and resuspended in 400 μl of recovery buffer (0.5% BSA in PBS). All steps from fixation onwards were carried out with RNase-free reagents and solutions were treated with either 1:25 RNasin (fixing/antibody/recovery buffers) or 1:100 RNasin (washing buffer). Cells were sorted with a FACS Aria IIU sorter (BD Biosciences) using FACS Diva 8.0 software. Thresholds for 488 nm and 633 nm sorting gates were set using cells stained only with secondary antibodies as reference. Approximately 170,000–210,000 cells were collected for each population (PAX6+, TBR2+ and PAX6−/TBR2−) in every biological replicate.
RNA sequencing
Sorted cells were centrifuged and resuspended in 100 μl of lysis solution of RecoverAll total nucleic acid isolation kit (Ambion) and incubated for 3 hrs at 50°C. RNA was then purified according to manufacturer’s instructions. The concentration and quality of purified RNA was determined using BioAnalyzer (Agilent). Normally, we recovered around 0.1 pg of RNA per cell and the total amount of RNA recovered was in the range of 5–40 ng for each sorted cell type. RNA integrity numbers (RIN) were in the range of 7.3 to 9.6. RNA was reverse-transcribed into cDNA and amplified by RNA-based single primer isothermal amplification (SPIA) using the Ovation RNA-seq system V2 (NuGEN). Synthesized cDNA was sonicated using a Covaris S2 ultrasonicator to reduce the fragment size range to 100–600 bp. The genotype of littermate embryos was further confirmed at this point by qPCR amplification of Prdm16 exon 9. For library preparation, 100 ng of sheared cDNA was end repaired, ligated with barcoded adaptors, amplified for 9 PCR cycles and purified using the Ovation Ultralow System V2 (NuGEN). Libraries were sequenced in an Illumina HiSeq 2500 sequencer to a sequencing depth of 28–40 million reads per sample. Gene expression differences among RNA-seq samples were confirmed for a subset of genes by real-time PCR analysis using purified cDNA of sorted cells, custom primers (supplementary key resources table) and SYBR Green master mix (Bio-Rad). Normalization of gene expression levels was done using Gapdh as reference gene. Reactions were run in a CFX96 Real-Time PCR Thermal Cycler (Bio-Rad).
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat monoclonal anti-CTIP2 | Abcam | Cat #ab18465 |
| Mouse monoclonal anti-S100β | Abcam | Cat #ab11178 |
| Rabbit polyclonal anti-TBR1 | Abcam | Cat #ab31940 |
| Rat monoclonal anti-pH3 | Abcam | Cat #ab10543 |
| Mouse monoclonal anti-NEUN | Millipore | Cat #MAB377 |
| Mouse monoclonal anti-VGLUT2 | Millipore | Cat #MAB5504 |
| Chicken polyclonal anti-TBR2 | Millipore | Cat #AB15894 |
| Mouse monoclonal anti-NESTIN | Millipore | Cat #MAB353 |
| Mouse monoclonal anti-SATB2 | SCBT | Cat #sc-81376 |
| Rabbit polyclonal anti-CUX1 | SCBT | Cat #sc-13024 |
| Mouse monoclonal anti-RORβ | R&D Systems | Cat #PP-H3925-00 |
| Chicken polyclonal anti-GFP | Aves labs | Cat #GFP-1020 |
| Rabbit polyclonal anti-pH3 | Sigma-Aldrich | Cat #H0412 |
| Mouse monoclonal anti-KI67 | BD Pharmingen | Cat #550609 |
| Rat monoclonal anti-KI67 | Affymetrix | Cat #14-5698 |
| Rabbit polyclonal anti-PAX6 | BioLegend | Cat #901301 |
| Mouse monoclonal anti-PAX6 | DSHB | Cat #AB_528427 |
| Rabbit polyclonal anti-RFP | MBL | Cat #PM005 |
| Rabbit polyclonal anti-BRN2 | GeneTex | Cat #GTX114650 |
| Rabbit polyclonal anti-PDZRN3 Novus | Biologicals | Cat #NBP2-55802 |
| Rabbit polyclonal anti-PTX3 | Barbara Bottazzi Humanitas research, Italy | N/A |
| Rabbit polyclonal anti-PRDM16 | Patrick Seale UPenn, U.S.A. | N/A |
| Rabbit polyclonal anti-H3K27ac | abcam | Cat #ab4729 |
| Rabbit polyclonal anti-H3K4me | abcam | Cat #ab8895 |
| Goat polyclonal anti-Mouse Alexa Fluor 488 | Thermo Fisher | Cat #A11001 |
| Goat polyclonal anti-Mouse Alexa Fluor 546 | Thermo Fisher | Cat #A11003 |
| Goat polyclonal anti-Mouse Alexa Fluor 647 | Thermo Fisher | Cat #A21235 |
| Goat polyclonal anti-Chicken Alexa Fluor 488 | Thermo Fisher | Cat #A11039 |
| Goat polyclonal anti-Rabbit Alexa Fluor 488 | Thermo Fisher | Cat #A11034 |
| Goat polyclonal anti-Rabbit Alexa Fluor 546 | Thermo Fisher | Cat #A11010 |
| Goat polyclonal anti-Rabbit Alexa Fluor 647 | Thermo Fisher | Cat #A21244 |
| Goat polyclonal anti-Rat Alexa Fluor 488 | Thermo Fisher | Cat #A11006 |
| Bacterial and Virus Strains | ||
| Replication-incompetent enhanced GFP-expressing retrovirus | (Palmer et al., 1999) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| EGS [ethylene glycolbis(succinimidylsuccinate)] | Thermo Scientific | Cat #21565 |
| Blocking reagent for nucleic acid hybridization | Roche | Cat #11096176001 |
| Critical Commercial Assays | ||
| Ovation Ultralow System V2 1–16 | NuGEN | Cat #0344 |
| Ovation RNA-Seq System V2 | NuGEN | Cat #7102 |
| RecoverAll Total Nucleic Acid Isolation | Ambion | Cat #AM1975 |
| Click-iT EdU Imaging | Invitrogen | Cat #C10340 |
| Deposited Data | ||
| RNA-seq data | This paper | GEO: GSE111660 |
| ChIP-seq data | This paper | GEO: GSE111657 GSE111658, GSE11 1659, GSE111661 |
| Experimental Models: Cell Lines | ||
| HEK-293 retrovirus packaging cell line | (Palmer et al., 1999) | 293gp NIT-GFP |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 | Charles River laboratories | Strain code: 027 |
| Mouse: CD-1 | Charles River laboratories | Strain code: 022 |
| Mouse: B6.129-Prdm16tm1.1Brsp/J | The Jackson laboratory | Stock #024992 |
| Mouse: Emx1tm1(cre)Krj/J | The Jackson laboratory | Stock #005628 |
| Mouse: B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J | The Jackson laboratory | Stock #007914 |
| Mouse: Neurod6tm1(cre)Kan | (Goebbels et al., 2006) | MGI: 5308766 |
| Oligonucleotides | ||
| Primers: in situ
hybridization, exon 4–8 probe. Forward: 5′-CTGGCTCAAGTACATCCGTGT-3′ Reverse: 5′-CGTGCTGTGGATATGCTTGT-3′ |
(Bjork et al., 2010a) | N/A |
| Primers: in situ
hybridization, exon 9 probe. Forward: 5′-CTGTACCCACGACCACCTCT-3′ Reverse: 5′-GACTTTGGCTCAGCCTTGAC-3′ |
This paper | N/A |
| Primers: Prdm16 shRNA, exon
5. Forward: 5′-GTTGGTGCATGTGAAAGAATTCAAGAGATTCTTTCACATGCACCAAC-3′ Reverse: 5′-GTTGGTGCATGTGAAAGAATCTCTTGAATTCTTTCACATGCACCAAC-3′ Primers: scrambled control. Forward: 5′-GCGGAGAAAGTGGATTTATTTCAAGAGAATAAATCCACTTTCTCCGC-3′ Reverse: 5′-GCGGAGAAAGTGGATTTATTCTCTTGAAATAAATCCACTTTCTCCGC-3′ |
(Bjork et al., 2010b) | N/A |
| Primers: Pdzrn3 shRNA, exon
10. Forward: 5′-GCTCAGAACAGGAGAATAACGTTCAAGAGACGTTATTCTCCTGTTCTGAGCCTTTTTG-3′ Reverse: 5′-AATTCAAAAAGGCTCAGAACAGGAGAATAACGTCTCTTGAACGTTATTCTCCTGTTCTGAGC-3′ Primers: scrambled control. Forward: 5′-GCAAGGACAGACACGGAATATTTCAAGAGAATATTCCGTGTCTGTCCTTGCCTTTTTG-3′ Reverse: 5′-AATTCAAAAAGGCAAGGACAGACACGGAATATTCTCTTGAAATATTCCGTGTCTGTCCTTGC-3′ |
(Honda et al., 2010) | N/A |
| Primers: PR domain deletion. Forward: 5′-GAAGGTGCCTACTCCTTG-3′ Reverse: 5′-TGGGATTGGAATGTCTTC-3′ |
This paper | N/A |
| Primers: Real-time PCR analysis
Prdm16, exons 9 and10. Forward: 5′-TGCTAAGCCTTCACCGTTCT-3′ Reverse: 5′-TCTTTCAGGACTCCCACAGG-3′ Gabra2, exons 5 and 6. Forward: 5′-ATGCCCACTGAAATTTGGAA-3′ Reverse: 5′-AGCAACCTGAACGGAGTCAG-3′ Diras2, single exon. Forward: 5′-CAGGAACTCCTCAACCTGGA-3′ Reverse: 5′-CCCTTGAGCTTCTCCTTCCT-3′ Pdzrn3, exons 5 and 6. Forward: 5′-GAGAAGAGCTGGAGCTGGAA-3′ Reverse: 5′-TCCCAATGTCATCTTCGTCA-3′ Sema6d, exons 13 and 14. Forward: 5′-CCTTCTCTAGCTGCGTGGTC-3′ Reverse: 5′-ACCAACCACAGTACGGGTCA-3′ Ptx3, exons 1 and 2. Forward: 5′-TGGACAACGAAATAGACAATGG-3′ Reverse: 5′-TCTCCAGCATGATGAACAGC-3′ Tmem35, exons 1 and 2. Forward: 5′-CAGCAAGGATGCCTACAGTG-3′ Reverse: 5′-GATGGAATTGATGCCCATTT-3′ Kremen1, exons 2 and 3. Forward: 5′-GCGAGCACAATTATTGCAGA-3′ Reverse: 5′-GCAGGCAGGAATTTCACAGT-3′ Itga6, exons 20 and 21. Forward: 5′-CCAAAGGTTTGGAGCAGATT-3′ Reverse: 5′-TTTCAGGAAGTTCCCGTTTC-3′ Adamts20, exons 6–9. Forward: 5′-GTCAGAATGTCCCGGATGAT-3′ Reverse: 5′-AAGCCTAACGTGTCGCATTT-3′ Lifr, exons 10–12. Forward: 5′-ACCACAGAAGCCACTCCTTC-3′ Reverse: 5′-GGTAAAGGCTTCCAGTAGACGA-3′ Epha3, exons 8 and 9. Forward: 5′-TCCTCACGGTGGTCACCTAT-3′ Reverse: 5′-TTAAGTGGCCATTCCCAAAG-3′ Lama1, exons 14 and 15. Forward: 5′-TTCTGCTAAAATGGCGCTCT-3′ Reverse: 5′-GGGACATTCACAGTGTTCCA-3′ Veph1, exons 10 and 11. Forward: 5′-TTGAAGTCGCCATGACAGAG-3′ Reverse: 5′-TTTCACTGAAGCCAAACACG-3′ Slc14a2, exons 8 and 9. Forward: 5′-CTGTGCATACATGGGAGCTG-3′ Reverse: 5′-GGTGAGGGTGGAGAGACAGA-3′ Smoc1, exons 5 and 6. Forward: 5′-TTGAGCCAGGGTAATTCAGG-3′ Reverse: 5′-ACACAGGTTGGGTCTCCATC-3′ Palmd, exons 3 and 4. Forward: 5′-CAAAATCAGCAAGACCAGCA-3′ Reverse: 5′-TTTGCAGTTCAGCCTTTTCA-3′ Eva1c, exons 5–7. Forward: 5′-TACGCATGTGTTCCCAAGAA-3′ Reverse: 5′-CCGCTTGGGTTGAATGTTAT-3′ |
This paper | N/A |
|
Dscaml1, exons 12 and
13. Forward: 5′-AATGGCGATGAAGTGGTCTC-3′ Reverse: 5′-ATCAAACCTCGGTCCTCTCC-3′ Tox1, exons 1–3. Forward: 5′-TGCCTGGACCCCTACTATTG-3′ Reverse: 5′-CTTGGTCCAGGGTAGGACTG-3′ Thsd4, exons 4 and 5. Forward: 5′-GGCCTATCGGCAGTACAAAC-3′ Reverse: 5′-CCATGAATGGCTTGTTGTTG-3′ Adamts18, exons 6 and 7. Forward: 5′-CAAAGATGGGACCATTGGAA-3′ Reverse: 5′-TGGTCTGCATGGTGATTGAT-3′ Meis1, exons 9 and 10. Forward: 5′-AAAAAGCAGTTGGCACAAGA-3′ Reverse: 5′-GGTCTATCATGGGCTGCACT-3′ |
This paper | N/A |
| Fluorescent in situ hybridization | ||
| RNAscope probe Mm Pdzrn3 | Advanced Cell Diagnostics | Cat #517061 |
| RNAscope probe Mm Itga6 | Advanced Cell Diagnostics | Cat #441701 |
| RNAscope probe Mm Gabra2 | Advanced Cell Diagnostics | Cat #435011 |
| RNAscope probe Mm Tubb3 | Advanced Cell Diagnostics | Cat #423391 |
| Recombinant DNA | ||
| pCAG-TAG | Addgene | Plasmid #26771 |
| CAG-GFP-IRES-CRE | Addgene | Plasmid #48201 |
| pCAGIG | Addgene | Plasmid #11159 |
| pCMV-VSV-G | Addgene | Plasmid #8454 |
| Hes5-Luc | Addgene | Plasmid #41724 |
| pcDNA3.1 Prdm16 | Addgene | Plasmid #15503 |
| pCAG-Prdm16-IRES-GFP | This paper | N/A |
| Hes5p-Prdm16-IRES-GFP | This paper | N/A |
| Hes5p-ΔPRdm16-IRES-GFP | This paper | N/A |
| pCAGIG Pdzrn3 | This paper | N/A |
| Software and Algorithms | ||
| ImageJ/ Fiji 1.49S | Wayne Rasband National Institutes of Health, U.S.A. | https://imagej.nih.gov/ij/ |
| Imaris 7.0 | Bitplane | http://www.bitplane.com/releasenotes/imaris700.aspx |
| Integrative Genomics Viewer (IGV) 2.3 | Broad Institute MIT/Harvard | http://software.broadinstitute.org/software/igv/ |
| MATLAB R2017b | MathWorks | https://www.mathworks.com/products/matlab.html |
| Cutadapt | (Martin, 2011) | http://code.google.com/p/cutadapt/ |
| STAR | (Dobin et al., 2013) | http://code.google.com/p/rna-star/ |
| FeatureCounts | (Liao et al., 2014) | http://subread.sourceforge.net |
| DESeq2 | (Love et al., 2014) | http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html |
| gProfiler | (Reimand et al., 2007) | http://biit.cs.ut.ee/gprofiler/ |
| REVIGO | (Supek et al., 2011) | http://revigo.irb.hr/ |
| Bowtie2 2.2.8 | (Langmead et al., 2009) | http://bowtie.cbcb.umd.edu |
| MACS2 2.1.1 | (Zhang et al., 2008) | https://pypi.python.org/pypi/MACS2 |
| IDR R package | (Li et al., 2011) | http://cran.rproject.org/web/packages/idr/index.html |
| HOMER v4.6 suite | (Heinz et al., 2010) | http://homer.ucsd.edu/homer/ |
| DeepTools2 | (Ramírez et al., 2016) | deeptools.iefreiburg.mpg.de |
| BETA v1.0.7 | (Wang et al., 2013) | http://cistrome.org/BETA/ |
Chromatin immunoprecipitation and sequencing
Approximately 20–40 million cortical cells were dual crosslinked by incubating in 1.5 mM EGS (ethylene glycol bis[succinimidyl succinate]) solution (Thermo Scientific) for 20 min at RT with rotation and then 1% PFA plus 1.5 mM EGS for an additional 10 min at RT. Crosslinking was quenched by adding glycine to a final concentration of 125 mM and rotating for 5 min at RT. Cells were then washed twice with cold PBS 1X plus EDTA-free protease inhibitor (Roche), centrifuged and stored at −80°C or freshly resuspended in lysis buffer (20 mM Tris-HCl pH 8.0, 85 mM KCl, 0.5% NP40) and incubated on ice for 30 min. Nuclei were pelleted by centrifugation at 1500 g for 5 min, resuspended in SDS buffer (0.2% SDS, 20 mM Tris-HCl pH 8.0, 1 mM EDTA) and incubated on ice for 10 min. Nuclei were then sonicated using a Covaris S2 ultrasonicator for shearing chromatin in the range of 100–500 bp fragments. After spinning chromatin at 18,000 g for 10 min, supernatant was transferred to a clean tube and one volume of 2X ChIP dilution buffer (0.1% sodium deoxycholate, 2% Triton X-100, 2 mM EDTA, 30 mM Tris-HCl pH 8.0, 300 mM NaCl) was added. At this step, a volume of supernatant containing around 0.5 million nuclei was set aside as input control and the remaining supernatant was incubated with 5 μg of anti-PRDM16 antibody overnight at 4°C with rotation. Next day, 50 μl of washed protein G beads (22.5 mg/ml; Novex) were added to the chromatin solution and incubated for 2 hrs at 4°C. After incubation, beads were washed twice with low salt wash buffer (0.1% SDS, 1% triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH 8.0, 150 mM NaCl ) followed by two washes with high salt wash buffer (0.1% SDS, 1% triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH 8.0, 500 mM NaCl ) then two washes with LiCl wash buffer (0.25 M LiCl, 0.5% NP40, 0.5% sodium deoxycholate, 1 mM EDTA, 10 mM Tris-HCl pH 8.0) and finally two washes with TE pH 8.0 (10 mM Tris-HCl, 1 mM EDTA). Beads were then resuspended in 90 μl of freshly prepared ChIP elution buffer (1% SDS, 0.1 M NaHCO3) and incubated at 65oC for 30 min with rotation. The recovered supernatant was incubated in reverse crosslinking solution (250 mM Tris-HCl pH 6.5, 62.5 mM EDTA pH 8.0, 1.25 M NaCl, 5 mg/ml of Proteinase K) at 65°C overnight. DNA was then extracted with phenol/chloroform/isoamyl alcohol, precipitated with 3 M sodium acetate pH 5.0 and resuspended in TE pH 8.0 low EDTA (10 mM Tris-HCl, 0.1 mM EDTA). Finally, samples were treated with RNase A (100 μg/ml) for 30 min at 37°C.
For library preparation, genomic DNA was purified, end repaired, ligated with barcoded adaptors, amplified for 11 PCR cycles and purified using the Ovation Ultralow System V2 (NuGEN) according to manufacturer’s instructions. Library fragments in the range of 100–800 bp were size-selected using agarose gel electrophoresis followed by DNA gel extraction (Qiagen). Recovered DNA was further cleaned and concentrated using a column (Zymo Research). Libraries were sequenced in an Illumina HiSeq 2500 sequencer to a sequencing depth of 30–40 million reads per sample.
Native chromatin immunoprecipitation and sequencing
Embryonic cortical tissue was dissected and ground on ice using 1.5 ml tubes and a plastic pestle. The tissue was resuspended in 100 μl of buffer 1 (0.3 M Sucrose, 60 mM KCl, 15 mM NaCl, 5 mM MgCl2, 0.1 mM EGTA, 15 mM Tris-HCl pH7.5, 0.5 mM DTT, 1 mM PMSF, 1X EDTA-free protease inhibitor) and lysed on ice for 7 min by adding 100 μl of buffer 2 (buffer 1 plus 0.4% Noniodet-40 substitute). Samples were centrifuged at 8500 r.p.m. for 7 min and resuspended in 100 μl of micrococcal nuclease buffer (0.3 M Sucrose, 50 mM Tris-HCl pH 7.5, 4 mM MgCl2, 1 mM CaCl2, 0.1 mM PMSF and 1X EDTA-free protease inhibitor) before adding another 100 μl of micrococcal nuclease buffer containing 1 μl of micrococcal nuclease at 2×106 U/ml (New England Biolabs). Samples were incubated for 10 min at 37°C and the reaction was stopped with 10 μl of 0.5 M EDTA. Samples were centrifuged at 10000 r.p.m. for 10 min, the supernatants (sup #1) were collected and the pellets incubated overnight in 200 μl of dialysis buffer (1 mM Tris-HCl pH 7.5, 0.2 mM EDTA, 1 mM PMSF, 1X EDTA-free protease inhibitor) with rocking at 4°C. The dialyzed samples were centrifuged and the recovered supernatants were combined with sup #1. The supernatants (400 μl total volume) were diluted to 4 ml with ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl pH 8.0, 167 mM NaCl, 1 mM PMSF, 1X EDTA-free protease inhibitor) and 40 μl of protein G Dyna Beads were added to pre-clear the chromatin for 1 hr. Up to this step, all solutions contained 50 mM Na Butyrate for subsequent immunoprecipitation of H3K27ac. Beads were removed and the chromatin samples were incubated overnight with 4 μg of anti-H3K27ac or anti-H3K4me antibody with rocking at 4°C. The following day, chromatin samples were incubated for 3 hrs with 40 μl of protein G Dyna Beads that were previously blocked overnight with 2 mg/ml BSA and 3 mg/ml tRNA. Bead-bound chromatin samples were sequentially washed with low salt buffer, high salt buffer and LiCl buffer (one time each) and then twice with TE buffer. Bead-bound chromatin was eluted with ChIP elution buffer (1% SDS, 0.1% NaHCO3) for 40 min at 65°C and treated with 0.1 μg/μl of RNAse A, followed by 0.05 μg/μl of Proteinase K. Genomic DNA was purified using the ChIP Clean and Concentrator kit (Zymo Research). Before proceeding to library preparation, immunoprecipitation of H3K27ac or H3K4me was confirmed by ChlP-qPCR using primers designed to amplify genomic regions with high H3K27ac or H3K4me enrichment, according to the ENCODE datasets. Additional sets of primers were designed to amplify genomic regions with low H3K27ac or H3K4me enrichment and used as negative controls. Library preparation and size selection were done as described above for transcription factor ChlPseq.
Prdm16 and Pdzrn3 Knock-down
Oligonucleotides coding for shRNA directed against exon 5 of Prdm16 or exon 10 of Pdzrn3 and scrambled controls (supplementary key resources table) were annealed, phosphorylated and cloned downstream of the human U6 promoter of the pBluescript/U6 plasmid. The specificity of Prdm16 shRNA was determined by in utero electroporation using an anti-PRDM16 antibody. The specificity of Pdzrn3 KD was determined by transfection of HEK293 fibroblasts with vectors expressing either Pdzrn3 shRNA or scrambled control (both at 1 μg/μl) in combination with a plasmid encoding full-length Pdzrn3 under a CAG promoter (0.2 μg/μl). HEK293 cells were transfected for 2 hrs at 37°C with Optimem solution (Gibco) containing plasmid DNA and Lipofectamine 2000 (Invitrogen). After incubation, media was removed and cells were incubated for 2 days. Total RNA was isolated from transfected cells with TRIzol reagent (Invitrogen) and reverse transcribed. The relative expression of Pdzrn3 in transfected cells was quantified by real-time PCR using custom primers.
Cloning and site-directed mutagenesis
The full-length sequence of Pdzrn3 was cloned into the EcoRI and NotI sites of pGAGIG (Addgene). To generate the Hes5p-Prdm16 F.L.-IRES-GFP and Hes5p-ΔPRdm16-IRES-GFP vectors, the proximal promoter region of Hes5 (−685 to +28 bp from TSS) was PCR-amplified from Hes5p-Luc plasmid (Addgene) and cloned into the Sal I and EcoRI sites of pCAGIG (Addgene), resulting in replacement of the entire CAG promoter by the Hes5 promoter (Hes5p-IRES-GFP control vector). The full length coding sequence of Prdm16 was obtained from the pcDNA3.1-Prdm16 vector (Addgene). To optimize mRNA translation, we changed the sequence immediately upstream of the start codon (GTAGTCATG) to a consensus Kozak sequence (GCCACCATG) using QuikChange II site-directed mutagenesis (Agilent). Kozak-corrected Prdm16 was cloned into the EcoRI site of the control vector to generate the Hes5p-Prdm16 F.L.-IRES-GFP final vector. An in-frame deletion of the nucleotide sequence coding for the entire PR domain of PRDM16 (amino acids 82–211, based on UniProt) was done with the Q5 site-directed mutagenesis kit (New England Biolabs) to generate the Hes5p-ΔPRdm16-IRES-GFP vector. The sequence of all vectors was confirmed using custom primers. Antibody staining was necessary to detect GFP expression from these vectors. For simplicity, vectors are indicated as Hes5p-Prdm16 F.L. and Hes5p-ΔPRdm16 in the main text.
RNAseq analysis
Read mapping and expression level estimation.
All samples were processed using an RNA-seq pipeline implemented in the bcbio-nextgen project (https://bcbio-nextgen.readthedocs.org/en/latest/). Raw reads were examined for quality issues using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) to ensure library generation and sequencing data were suitable for further analysis. If necessary, adapter sequences, other contaminant sequences such as polyA tails and low quality sequences were trimmed from reads using Cutadapt. Trimmed reads were aligned to the UCSC build mm10 of the mouse genome using STAR, and counts of reads aligning to known genes were generated by featureCounts. Quality of alignments was assessed by checking for evenness of coverage, rRNA content, genomic context of alignments (for example, alignments in known transcripts and introns), complexity and other quality checks. Principal components analysis (PCA) and hierarchical clustering methods validated clustering of samples from the same library run across different sequencing lanes. Read counts were aggregated for each library by taking the sum across lanes for each gene.
Differential gene expression and functional enrichment analysis.
Differential expression was performed at the gene level using the R Bioconductor package DESeq2. For each cell type, significant genes were identified using an FDR threshold of 0.05. Lists of differentially expressed genes were separated by direction of expression change and examined by gProfiler for statistical enrichment of information such as Gene Ontology (GO) terms, biological pathways and human disease annotations. Functional redundancy in over-represented GO terms identified by gProfiler was reduced with REVIGO for a visual representation of the most prominent processes.
ChlP-Seq analysis
Read mapping and peak calling.
For all ChIP-seq datasets, sequence quality was evaluated using FASTQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/), and if required, reads were trimmed with Cutadapt. High quality sequencing reads were mapped to the mouse genome UCSC build mm10 using Bowtie2 v2.2.8. Alignments were filtered to retain only reads with a unique mapping to the genome. ChIP-seq peaks were called with MACS2 v2.1.1 using default parameters and a narrow peak cutoff of q < 0.05 for PRDM16 WT and cKO samples. For histone modifications (i.e. H3K27ac and H3K4me) broad peak calling was used and a broad peak cutoff was set to q < 0.05. The reproducibility of peaks across replicates within each dataset was assessed by running samples through the IDR pipeline, in which peak consistency was validated by use of pooled pseudo-replicates. The pooled pseudo-replicates resulted in a number of peaks within a factor of 2 of the original results suggesting highly reproducible replicates. A consensus set of IDR-optimized peaks was identified for each dataset by merging peaks across replicates for regions with an IDR < 0.05. For comparison of PRDM16 binding sites with H3K27ac peaks, a less stringent consensus set of peaks without IDR filtering was used. This consensus set was comprised of peaks with a minimum of 1 bp overlap between replicates.
Peak annotation.
ChIP-seq peaks were annotated using the HOMER v4.6 suite of tools. Annotations were determined using nearest gene analysis which is based on distance of the peak to the nearest TSS and RefSeq annotations were used to determine associated genomic features.
Motif analysis.
Only the top 100 peaks, ranked by signal value (i.e. -log 10 p-value of the merged region), were used as input for motif analysis. HOMER conducts de novo motif analysis by parsing input sequences into unique 100 bp oligos from the peak center and read into a table. Each oligo is counted and the hypergeometric test is used to calculate the oligos significant enrichment across all target sequences. The q-value represents the FDR-corrected probability of that motif being over-represented amongst target sequences. Enrichment of known motifs is calculated similar to de novo analysis but screening against a database of previously determined high quality motifs.
Differential enrichment analysis. .
Comparisons of peak read density between WT and cKO samples was evaluated using the R Bioconductor package DiffBind. For each dataset, the full set of MACS2 peak calls from each sample was used as input rather than the IDR-optimized regions. Peaks were identified as differentially enriched if the DESeq2 analysis reported an FDR < 0.05. Differences in PRDM16 binding between WT and cKO samples were assessed by reducing the full set of cKO peak calls down to only those regions overlapping with the IDR-optimized WT peaks, and normalized read density values were plotted against each other to illustrate differential enrichment.
External data resources.
For comparison of PRDM16 binding sites to other known histone marks, the ENCODE database was used to obtain ChIP-seq data from mouse E14.5 whole brains (https://www.encodeproject.org/reference-epigenomes/ENCSR205YGI/). For samples in which bigWig files existed, those files were downloaded directly from ENCODE, otherwise mm10 aligned BAM files were downloaded and converted to bigWig format using deepTools. ChIP-seq tracks were visualized using the Integrative Genomics Viewer 2.3 (Broad Institute, Harvard/MIT). For the assessment of PRDM16 binding overlap with developmental and adult enhancer regions, we used the IDR-optimized PRDM16 peaks and evaluated overlaps with H3K27ac ENCODE datasets using Bedtools v.2.26.0. Genomic regions bound by PRDM16 within 1 Kb upstream of the TSS were classified as promoters and removed from the analysis. Developmental enhancers were defined as PRDM16 peaks that overlap with H3K27ac peaks in E14.5 brains. Developmental and adult enhancers were defined as PRDM16 peaks that overlap with H3K27ac peaks in both E14.5 brains and 8 weeks old adult cortex. Analysis was carried out based on a minimal reciprocal fraction of peak overlap set at 20%.
Binding and expression target analysis (BETA)
To identify significant associations between PRDM16 binding sites and differentially expressed genes from RNA-seq analysis, we used the BETA-plus sub-protocol from BETA v1.0.7 (Wang et al., 2013). Instead of assigning one-to-one mapping between binding sites and genes, BETA models the influence of a binding site on the expression of a gene. Two files are required as input: the IDR-optimized set of PRDM16 binding sites and RNA-seq analysis results containing statistics from differential expression analysis. BETA first calculates a Regulatory Potential Score for each gene, which is based on the number of sites within 200 kb of the TSS for that gene and the sum of distances between those sites and the gene’s TSS. A rank product is then computed, by taking the differential expression statistic for each gene and the RP value, which reflects its significance as a potential target gene. The final target gene lists only include genes from the RNA-seq expression file that i) meets the defined threshold of FDR < 0.05 and ii) contains binding sites within the 200 kb region.
Quantifications and statistical analysis
Cell counting was done either manually with ImageJ/Fiji cell counter (National Institute of Health, USA) or automatically with Imaris 7.0 software (Bitplane) using anatomically equivalent regions at the somatosensory cortex. Analysis of cell migration was done in Image J or Imaris by dividing the thickness of the cortex into four or five bins of equal size and the number of CUX1+ or GFP+ cells in each bin was counted and presented as the fraction of the total CUX+ or GFP+ neurons. Quantification of RG units only included those clones with a primary RG extending a basal process into the cortical plate. Axonal quantifications were done using ImageJ/Fiji software. Briefly, the straighten tool was used to define a broad ipsilateral or contralateral area of interest in pCAG-TAG electroporated brains. A square (1500 × 600 pixels) was drawn at the region of interest and background was subtracted in that area using the rolling ball radius tool set at 2 pixels. After background subtraction, mean fluorescence intensity and integrated density were measured in the squared area. A region of the image with no axonal projections was selected and the mean fluorescence intensity was determined; this value was used as background intensity. The axonal projections were quantified using the following formula: corrected fluorescence intensity = integrated density - (area of square × background intensity).
Quantification of neuronal nuclei in whole hemisphere sections and quantification of RNA puncta per unit area or overlapping GFP signal was carried out automatically using a data processing pipeline in MATLAB R2017b guided by MatBots (https://hms-idac.github.io/MatBots). To count neuronal nuclei, we used nuclei fragmentation value of 0.9 and adjusted the threshold values (range 0.06 to 0.1) according to the signal intensity of CUX1, CTIP2 and SATB2.
Quantification of RNA puncta was done using the SpotsInNucleiBot by selecting the Advanced Log function, sigma value of 2.0, and alpha values of 0.01 for Pdzrn3 puncta and 0.001 for Itga6 and Gabra2 puncta. The total area of GFP+ cells was quantified using the simple thresholding option with a fragmentation value in the range of 0.90 to 0.99, threshold value of 0.05 and sigma value of 1.0, whereas default settings were used for all other parameters. The number of RNA puncta in 104 μm2 of VZ/SVZ, IZ or CP tissue is reported for WT and cKO cortex. RNA puncta on GFP+ cells is reported as puncta number in 104 μm2 of GFP+ area. After automatic quantification of nuclei numbers, GFP+ nuclear area and RNA puncta, a subset of the images was overlapped with the output masks to verify accuracy. Data outliers were verified and discarded if the generated mask did not represent the real data Values represent mean ± SEM or mean ± SD as indicated in figure legends. For each experiment, the number of replicates (n) is also indicated in figure legends. Significance was determined using the two-tailed unpaired Student’s t test and reported as: *p < 0.05, **p < 0.01, ***p < 0.001.
Supplementary Material
Highlights:
PRDM16 activity in radial glia exerts lineage-autonomous control of cortical neuron position.
Enhancer regulation by PRDM16 represses cell migration genes.
PRDM16 controls cortical neuron position by silencing Pdzrn3 expression.
The PR domain of PRDM16 promotes migration through transcriptional silencing.
Acknowledgements
We are grateful to Bruce Spiegelman (Dana Farber Cancer Institute) and Paul Cohen (The Rockefeller University) for providing the Prdm16flox/flox mouse line. We thank Patrick Seale (University of Pennsylvania) and Barbara Bottazzi (Humanitas Research, Italy) for sharing reagents. We thank Lisa Goodrich, Michael Greenberg and Constance Cepko (Harvard Medical School) for critical reading of the manuscript. JMB was supported by the Pew Latin American fellow program, Alice and Joseph Brooks fellowship and the William Randolph Hearst Fund. MTG was supported by the Ellen and Melvin Gordon fellowship. This work was supported by grants from NIH (R01NS102228) and the Edward R. and Anne G. Lefler Center to CCH.
References
- Arai Y, Pulvers JN, Haffner C, Schilling B, Nüsslein I, Calegari F, and Huttner WB (2011). Neural stem and progenitor cells shorten S-phase on commitment to neuron production. Nat. Commun. 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold SJ, Huang GJ, Cheung AFP, Era T, Nishikawa SI, Bikoff EK, Molnár Z, Robertson EJ, and Groszer M (2008). The T-box transcription factor Eomes/Tbr2 regulates neurogenesis in the cortical subventricular zone. Genes Dev. 22, 2479–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi J, and Cohen P (2016). The multifaceted roles of PRDM16: Adipose biology and beyond. Trends Endocrinol. Metab. 27, 11–23. [DOI] [PubMed] [Google Scholar]
- Chuikov S, Levi BP, Smith ML, and Morrison SJ (2010). Prdm16 promotes stem cell maintenance in multiple tissues, partly by regulating oxidative stress. Nat. Cell Biol. 12, 999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Levy JD, Zhang Y, Frontini A, Kolodin DP, Svensson KJ, Lo JC, Zeng X, Ye L, Khandekar MJ, et al. (2014). Ablation of PRDM16 and beige adipose causes metabolic dysfunction and a subcutaneous to visceral fat switch. Cell 156, 304–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato M. a, Frampton GM, Sharp P. a, et al. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U. S. A. 107x, 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englund C, Fink A, Charmaine L, Pham D, Ray DAM, Bulfone A, Kowalczyk T, and Hevner RF (2005). Pax6, Tbr2, and Tbr1 Are Expressed Sequentially by Radial Glia, Intermediate Progenitor Cells, and Postmitotic Neurons in Developing Neocortex. J. Neurosci. 25, 247–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas LM, Haffner C, Giger T, Khaitovich P, Nowick K, Birchmeier C, Pääbo S, and Huttner WB (2008). Insulinoma-Associated 1 Has a Panneurogenic Role and Promotes the Generation and Expansion of Basal Progenitors in the Developing Mouse Neocortex. Neuron 60, 40–55. [DOI] [PubMed] [Google Scholar]
- Georges-Labouesse E, Mark M, Messaddeq N, and Gansmüller a (1998). Essential role of alpha 6 integrins in cortical and retinal lamination. Curr. Biol. 8, 983–986. [DOI] [PubMed] [Google Scholar]
- Goebbels S, Bormuth I, Bode U, Hermanson O, Schwab MH, and Nave KA (2006). Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis 44, 611–621. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JLR, and Jones KR (2002). Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J. Neurosci. 22, 6309–6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemot F, and Hassan BA (2017). Beyond proneural: emerging functions and regulations of proneural proteins. Curr. Opin. Neurobiol. 42, 93–101. [DOI] [PubMed] [Google Scholar]
- Harms MJ, Lim HW, Ho Y, Shapira SN, Ishibashi J, Rajakumari S, Steger DJ, Lazar MA, Won KJ, and Seale P (2015). PRDM16 binds MED1 and controls chromatin architecture to determine a brown fat transcriptional program. Genes Dev. 29, 298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck N, Kilb W, Reiprich P, Kubota H, Furukawa T, Fukuda A, and Luhmann HJ (2007). GABA-A receptors regulate neocortical neuronal migration in vitro and in vivo. Cereb. Cortex 17, 138–148. [DOI] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. (2007). Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 39, 311–318. [DOI] [PubMed] [Google Scholar]
- Hirabayashi Y, and Gotoh Y (2010). Epigenetic control of neural precursor cell fate during development. Nat. Rev. Neurosci. 11, 377–388. [DOI] [PubMed] [Google Scholar]
- Hohenauer T, and Moore AW (2012). The Prdm family: expanding roles in stem cells and development. Development 139, 2267–2282. [DOI] [PubMed] [Google Scholar]
- Honda T, Yamamoto H, Ishii A, and Inui M (2010). PDZRN3 negatively regulates BMP-2-induced osteoblast differentiation through inhibition of Wnt signaling. Mol. Biol. Cell 21, 3269–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrvatin S, Deng F, O’Donnell CW, Gifford DK, and Melton DA (2014). MARIS: Method for analyzing RNA following intracellular sorting. PLoS One 9, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu LC-L, Nam S, Cui Y, Chang C-P, Wang C-F, Kuo H-C, Touboul JD, and Chou S-J (2015). Lhx2 regulates the timing of β-catenin-dependent cortical neurogenesis. Proc. Natl.Acad. Sci. U. S. A. 112, 12199–12204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Iwai R, Tabata H, Konno D, Komabayashi-Suzuki M, Watanabe C, Iwanari H, Mochizuki Y, Hamakubo T, Matsuzaki F, et al. (2016). Prdm16 is critical for progression of the multipolar phase during neural differentiation of the developing neocortex. Development dev.136382. [DOI] [PubMed] [Google Scholar]
- Jordan VK, Zaveri HP, and Scott DA (2015). 1P36 Deletion Syndrome: an Update. Appl. Clin. Genet. 8, 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimura S, Seale P, Tomaru T, Erdjument-Bromage H, Cooper MP, Ruas JL, Chin S, Tempst P, Lazar MA, and Spiegelman BM (2008). Regulation of the brown and white fat gene programs through a PRDM16/CtBP transcriptional complex. Genes Dev. 22, 1397–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczyk T, Pontious A, Englund C, Daza RAM, Bedogni F, Hodge R, Attardo A,Bell C, Huttner WB, and Hevner RF (2009). Intermediate neuronal progenitors (basal progenitors) produce pyramidal-projection neurons for all layers of cerebral cortex. Cereb. Cortex 19, 2439–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan KY, Sestan N, and Anton ES (2012). Transcriptional co-regulation of neuronal migration and laminar identity in the neocortex. Development 139, 1535–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre-Ubieta L, Stein JL, Won H, Opland CK, Liang D, Lu D, and Geschwind DH (2018). The Dynamic Landscape of Open Chromatin during Human Cortical Neurogenesis. Cell 172, 289–304.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui JH, Hansen DV, and Kriegstein AR (2011). Development and evolution of the human neocortex. Cell 146, 18–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui JH, Nowakowski TJ, Pollen A. a, Javaherian A, Kriegstein A, and Oldham MC(2014). Radial glia require PDGFD–PDGFRb signalling in human but not mouse neocortex. Nature 515, 264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastrototaro G, Zaghi M, and Sessa A (2017). Epigenetic Mistakes in Neurodevelopmental Disorders. J. Mol. Neurosci. 1–13. [DOI] [PubMed] [Google Scholar]
- Mihalas AB, Elsen GE, Bedogni F, Daza RAM, Ramos-Laguna KA, Arnold SJ, and Hevner RF (2016). Intermediate Progenitor Cohorts Differentially Generate Cortical Layers and Require Tbr2 for Timely Acquisition of Neuronal Subtype Identity. Cell Rep. 16, 92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani K, Yoon K, Dang L, Tokunaga A, and Gaiano N (2007). Differential Notch signalling distinguishes neural stem cells from intermediate progenitors. Nature 449, 351–355. [DOI] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Menezes JRL, and Macklis JD (2007). Neuronal subtype specification in the cerebral cortex. Nat. Rev. Neurosci. 8, 427–437. [DOI] [PubMed] [Google Scholar]
- Nashun B, Hill PWS, and Hajkova P (2015). Reprogramming of cell fate: epigenetic memory and the erasure of memories past. EMBO J. 34, 1296–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noctor SC, Flint AC, Weissman TA, Dammerman RS, and Kriegstein AR (2001). Neurons derived from radial glial cells establish radial units in neocortex. Nature 409, 714–720. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Martinez-Cerdeno V, Ivic L, Kriegstein AR, Martínez-Cerdeño V, Ivic L, Kriegstein AR, Martinez-Cerdeno V, Ivic L, and Kriegstein AR (2004). Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci 7, 136–144. [DOI] [PubMed] [Google Scholar]
- Pinheiro I, Margueron R, Shukeir N, Eisold M, Fritzsch C, Richter FM, Mittler G, Genoud C, Goyama S, Kurokawa M, et al. (2012). Prdm3 and Prdm16 are H3K9me1 methyltransferases required for mammalian heterochromatin integrity. Cell 150, 948–960. [DOI] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann S. a, Flynn R. a, and Wysocka J (2011).A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessa A, Mao C. an, Hadjantonakis AK, Klein WH, and Broccoli V (2008). Tbr2 Directs Conversion of Radial Glia into Basal Precursors and Guides Neuronal Amplification by Indirect Neurogenesis in the Developing Neocortex. Neuron 60, 56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sewduth RN, Jaspard-Vinassa B, Peghaire C, Guillabert A, Franzl N, Larrieu-Lahargue F, Moreau C, Fruttiger M, Dufourcq P, Couffinhal T, et al. (2014). The ubiquitin ligase PDZRN3 is required for vascular morphogenesis through Wnt/planar cell polarity signalling. Nat. Commun. 5, 4832. [DOI] [PubMed] [Google Scholar]
- Shimada IS, Acar M, Burgess RJ, Zhao Z, and Morrison SJ (2017). Prdm16 is required for the maintenance of neural stem cells in the postnatal forebrain and their differentiation into ependymal cells. 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatoyannopoulos JA, Snyder M, Hardison R, Ren B, Gingeras T, Gilbert DM, Groudine M, Bender M, Kaul R, Canfield T, et al. (2012). An encyclopedia of mouse DNA elements (Mouse ENCODE). Genome Biol. 13, 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuoc TC, Boretius S, Sansom SN, Pitulescu ME, Frahm J, Livesey FJ, and Stoykova,(2013). Chromatin Regulation by BAF170 Controls Cerebral Cortical Size and Thickness. Dev. Cell 25, 256–269. [DOI] [PubMed] [Google Scholar]
- Visel A, Taher L, Girgis H, May D, Golonzhka O, Hoch RV, McKinsey GL, Pattabiraman K, Silberberg SN, Blow MJ, et al. (2013). A high-resolution enhancer atlas of the developing telencephalon. Cell 152, 895–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Sun H, Ma J, Zang C, Wang C, Wang J, Tang Q, Meyer CA, Zhang Y, and Liu XS (2013). Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat. Protoc. 8, 2502–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Wang J, Lee SY, Xiong J, Bhanu N, Guo Q, Ma P, Sun Y, Rao RC, Garcia BA, et al. (2016). PRDM16 Suppresses MLL1r Leukemia via Intrinsic Histone Methyltransferase Activity. Mol. Cell 62, 222–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Adli M, Zou JY, Verstappen G, Coyne M, Zhang X, Durham T, Miri M, Deshpande V, De Jager PL, et al. (2013). Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell 152, 642–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.