

SUMMARY
Tissue-resident lymphocytes play a key role in immune surveillance, but it remains unclear how these inherently stable cell populations respond to chronic inflammation. In the setting of celiac disease (CeD), where exposure to dietary antigen can be controlled, gluten-induced inflammation triggered a profound depletion of naturally occurring Vγ4+/Vδ1+ intraepithelial lymphocytes (IELs) with innate cytolytic properties and specificity for the butyrophilin-like (BTNL) molecules BTNL3/BTNL8. Creation of a new niche with reduced expression of BTNL8 and loss of Vγ4+/Vδ1+ IELs was accompanied by the expansion of gluten-sensitive, interferon-γ-producing Vδ1+ IELs bearing T cell receptors (TCRs) with a shared non-germ-line-encoded motif that failed to recognize BTNL3/BTNL8. Exclusion of dietary gluten restored BTNL8 expression but was insufficient to reconstitute the physiological Vγ4+/Vδ1+ subset among TCRγδ+ IELs. Collectively, these data show that chronic inflammation permanently reconfigures the tissue-resident TCRγδ+ IEL compartment in CeD.
In Brief
Chronic inflammation, driven in the context of celiac disease by persistent antigenic challenge with dietary gluten, permanently reshapes the tissue-resident innate-like TCRγδ+ intraepithelial lymphocyte compartment.
Graphical Abstract
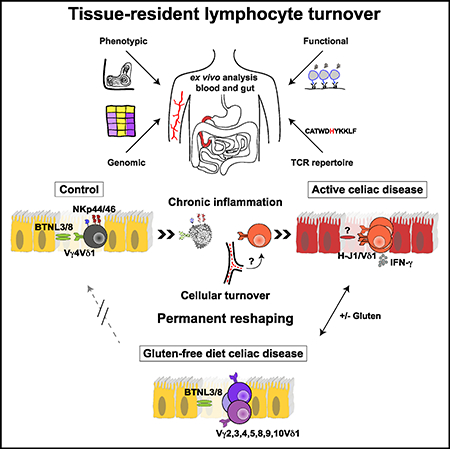
INTRODUCTION
Tissue-resident lymphocytes have been investigated extensively under steady-state conditions and during the induction of local memory populations in response to acute infections (Mueller and Mackay, 2016). In mice, the tissue-resident TCRαβ+ CD8αβ+ pool is highly stable and responds to secondary antigenic challenge via local proliferation of pre-existing memory cells (Beura et al., 2018; Park et al., 2018), which endure over time despite the accumulation of new tissue-resident populations driven by subsequent infections (Park et al., 2018). However, it remains unclear whether chronic inflammation can permanently reconfigure the tissue-resident T cell compartment.
Intraepithelial lymphocytes (IELs) expressing γδ T cell receptors (TCRs) are tissue-resident T cells that play a key role in immune surveillance via dynamic scanning of the intestinal epithelium (Hoytema van Konijnenburg et al., 2017). Murine TCRγδ+ cells seed the intestine early in life, irrespective of microbial colonization or exposure to dietary antigen (Di Marco Barros et al., 2016), and persist in situ as naturally occurring IELs (Cheroutre et al., 2011). Moreover, the peripheral and intraepithelial TCRγδ+ compartments are largely non-overlapping as a consequence of distinct migratory characteristics, especially a lack of recirculating IELs (Chennupati et al., 2010; Sugahara et al., 1999).
Celiac disease (CeD) is a gastrointestinal inflammatory disorder triggered and maintained by dietary exposure to gluten (Jabri and Sollid, 2009). Antigen exposure can therefore be controlled in vivo, providing a unique opportunity to study the dynamics of human tissue-resident T cells in the setting of chronic inflammation. Active disease is characterized histologically by villous atrophy and immunologically by expanded populations of IELs (Jabri and Sollid, 2009). Adherence to a gluten-free diet (GFD) leads to resolution of the villous abnormalities, together with a decrease in the frequencies of gluten-specific TCRαβ+ CD4+ T cells in the lamina propria and cytolytic TCRαβ+ CD8+ IELs, which are consequently implicated in the pathogenesis of CeD (Jabri and Sollid, 2009). In contrast, TCRγδ+ IEL expansions generally persist in situ, despite a lack of exposure to gluten (Kutlu et al., 1993). For this reason, tissue-resident TCRγδ+ IELs are thought to regulate disease activity (Hayday, 2000), potentially by suppressing the influx of circulating T cells and/or by maintaining tissue integrity (Hayday et al., 2001), rather than participate actively in the pathogenesis of CeD (Kutlu et al., 1993). However, these propositions remain speculative, pending a detailed functional evaluation of TCRγδ+ IELs in patients with CeD. We set out to address this knowledge gap and more fundamentally to determine the effects of chronic inflammation on human tissue-resident TCRγδ+ IELs.
RESULTS
Vδ1+ T Cells Displaying Hallmarks of Tissue-Resident Lymphocytes Are Permanently Expanded in CeD
In line with previous studies (Halstensen et al., 1989; Kutlu et al., 1993), we found higher frequencies (Figure 1A) and absolute numbers (Figure 1B) of Vδ1+ IELs in patients with CeD relative to healthy controls (Table S1), irrespective of adherence to a GFD (Figure 1C). Moreover, Vδ1+ T cells constituted a significantly higher fraction of all TCRγδ+ IELs in patients with active CeD (Figure 1D). A majority of Vδ1+ IELs in healthy controls and patients with CeD expressed markers of tissue residency, namely CD69 and CD103 (Mueller and Mackay, 2016) (Figure 1E). Vδ1+ IELs also expressed low levels of CD45RA and CCR7, indicative of an effector memory (TEM) phenotype (Sallusto et al., 1999), whereas the corresponding Vδ1+ peripheral blood lymphocytes (PBLs) comprised a mixture of naive and terminally differentiated effector (TEMRA) cells (Figure 1F). Expanded populations of Vδ1+ IELs were therefore integrated as bona fide tissue-resident lymphocytes in patients with CeD.
Figure 1. Vδ1+ IELs with Hallmarks of Tissue Residency Are Permanently Expanded in CeD.

(A) Frequency of Vδ1+ cells among CD3+ lymphocytes. Right: boxplots display first and third quartiles. ***p < 0.001. One-way ANOVA with Tukey’s test for multiple comparisons.
(B) Absolute numbers of Vδ1+ IELs from 3–5 biopsies per donor. Boxplot displays first and third quartiles ***p < 0.001. One-way ANOVA with Tukey’s test for multiple comparisons.
(C) Frequency of Vδ1+ IELs among CD3+ lymphocytes versus the duration of treatment with a GFD. Linear regression.
(D) Frequency of Vδ1+ IELs among TCRγδ+ cells. Bottom: cumulative distribution. Healthy controls: n = 99. Patients with active CeD: n = 62. Patients with GFD-treated CeD: n = 57. Kolmogorov-Smirnov test.
(E) Frequency of CD69+/CD103+ cells among Vδ1+ PBLs and IELs. Bottom: boxplot displays first and third quartiles.
(F) Fraction of cells defined as naive, central memory (TCM), effector memory (TEM), or terminal effector (TEMRA) based on expression of CD45RA and CCR7.
See also Figure S6.
Innate-like Vδ1+ IELs Are Lost in CeD
TCRαβ+ CD8αβ+ IELs in patients with CeD typically express increased levels of NKG2D and activating CD94/NKG2A− NK receptors (Jabri and Sollid, 2009). We found no evidence of a similar phenotype among Vδ1+ IELs from patients with active CeD (Figures S1A and S1B). However, a vast majority of control Vδ1+ IELs expressed the activating natural cytotoxicity receptors (NCRs) NKp46 and/or NKp44 (Figure 2A), irrespective of age (Figure 2B) and in situ expansion (Figure S1C). These observations suggested a definitive tissue-resident phenotype, reinforced by a lack of NCR expression on the surface of Vδ1+ PBLs (Figure S1D). In contrast, Vδ1+ IELs from patients with active or GFD-treated CeD rarely expressed NKp46 and almost exclusively lacked NKp44 (Figure 2A). This disease-associated loss of NCR+ Vδ1+ IELs was refractory to long-term treatment with a GFD, unlike the concomitant loss of NCR+ CD3− IELs, indicating a cell type-restricted effect distinct from the generic microenvironmental perturbations induced by CeD (Figures 2A and S1E). Moreover, Vδ1+ IELs expressing NKp46 and NKp44 were absent from the duodenum, the site of tissue destruction, but present in the colon of patients with GFD-treated CeD (Figure 2C). These results suggested that chronic inflammation precipitated an irretrievable and active site-specific loss of NK-like Vδ1+ IELs in patients with CeD.
Figure 2. Innate-like Vδ1+ IELs Are Lost in CeD.

(A) Frequency of IELs expressing NKp46 with or without NKp44. Right: boxplots display first and third quartiles. ***p < 0.001. One-way ANOVA with Tukey’s test for multiple comparisons.
(B) Expression of NKp46 or NKp46/NKp44 on control Vδ1+ IELs versus age.
(C) Expression of NKp46 and NKp44 on Vδ1+ IELs from donor-matched duodenal and right colonic biopsies.
(D) Expression of CD107a on IL-15-treated IELs after stimulation with plate-bound αTCRγδ ± αNKp46. *p < 0.05. Paired t test.
(E) Expression of CD107a on Vδ1+ IELs after stimulation with phorbol myristate acetate and ionomycin. Right: boxplot displays first and third quartiles. **p < 0.01, ***p < 0.001. One-way ANOVA with Tukey’s test for multiple comparisons.
See also Figures S1 and S6.
To determine the functional relevance of these NCRs, we measured cellular degranulation ex vivo and in response to stimulation with IL-15, which is upregulated under inflammatory conditions and is known to promote NK receptor-mediated cytolytic activity (Jabri and Abadie, 2015). Co-engagement of NKp46 and NKp44 in conjunction with TCR ligation significantly increased granule exocytosis among control Vδ1+ IELs relative to TCR ligation alone (Figure 2D), but only after pre-stimulation with IL-15 (Figures 2D and S1F). Vδ1+ IELs from healthy controls also expressed high levels of the cytolytic molecule granzyme B (Figure S1G) and degranulated at significantly higher frequencies than Vδ1+ IELs from patients with CeD (Figure 2E). These findings revealed that the healthy small intestine harbored a unique set of innate-like and potentially cytolytic Vδ1+ IELs that were displaced in the setting of CeD.
Dietary Gluten Drives the Emergence of Interferon-γ-Producing Vδ1+ IELs in CeD
To examine the functional properties of Vδ1+ IELs in more depth, we extended our analysis to cytokine production directly ex vivo. Control Vδ1+ IELs produced very little interferon (IFN)-γ or tumor necrosis factor (TNF)-α on a percent cell basis (Figure 3A). In contrast, approximately 50% of Vδ1+ IELs from patients with active CeD produced IFN-γ, but not TNF-α (Figure 3A). Significantly lower frequencies of IFN-γ+ Vδ1+ IELs were detected in patients with GFD-treated CeD (Figure 3A). Irrespective of disease status, Vδ1+ PBLs produced both IFN-γ and TNF-α, and neither TCRαβ+ CD8αβ+ nor TCRαβ+ CD4+ IELs displayed enhanced production of IFN-γ in patients with active CeD (Figure 3A). A significant increase in IFN-γ production was also detected among Vδ1+ IELs isolated from patients with GFD-treated CeD after gluten challenge relative to donor-matched Vδ1+ IELs isolated prior to gluten challenge, whereas no such chronological differences were observed in the corresponding TCRαβ+ CD8αβ+ or TCRαβ+ CD4+ IEL compartments (Figure 3B). These data suggested a gluten-dependent and cell type-restricted gain of IFN-γ-producing function among Vδ1+ IELs in patients with active CeD.
Figure 3. Dietary Gluten Drives the Emergence of IFN-γ-Producing Vδ1+ IELs in CeD.

(A) Expression of IFN-γ and TNF-α in cells stimulated ex vivo with phorbol myristate acetate and ionomycin. Bottom: boxplots display first and third quartiles. *p < 0.05, **p < 0.01, ***p < 0.001. One-way ANOVA with Tukey’s test for multiple comparisons.
(B) Expression of IFN-γ and TNF-α in cells from patients with GFD-treated CeD stimulated as in (A) before and after gluten challenge. Duration of GFD: 1.5 years, 4 years, 7 years, and 20.5 years. Right: boxplots display first and third quartiles. *p < 0.05. Paired t test.
See also Figure S6.
The Transcriptional Program of Vδ1+ IELs Is Permanently Altered in CeD
In further analyses, we performed ex vivo RNA sequencing (RNA-seq) to determine whether naturally occurring Vδ1+ IELs, represented by the prevalent NKp46+ (NCR+) subset in healthy controls, were fundamentally distinct from disease-associated Vδ1+ IELs, represented by the prevalent NKp46− (NCR−) subset in patients with CeD. A total of 645 genes exhibited differential expression between control NCR+ Vδ1+ IELs and NCR− Vδ1+ IELs from patients with active CeD (Figure S2A). Similar differences were observed in comparisons between control NCR+ Vδ1+ IELs and NCR− Vδ1+ IELs from patients with GFD-treated CeD (Figure S2B). Moreover, this overarching dichotomy was confirmed using minimum spanning tree (MST) analysis (Xu et al., 2002), which showed that control NCR+ Vδ1+ IELs formed distinct clusters relative to NCR− Vδ1+ IELs from patients with active or GFD-treated CeD (Figure 4A).
Figure 4. The Transcriptional Program of Vδ1+ IELs Is Permanently Altered in CeD.

(A) Transcriptional profiles of NCR+ Vδ1+ IELs from healthy controls, NCR− Vδ1+ IELs from patients with active CeD, and NCR− Vδ1+ IELs from patients with GFD-treated CeD compared using minimum spanning tree analysis.
(B) Differentially expressed genes from the NK module passing a false discovery rate < 10% from any two-way contrast between the Vδ1+ IEL populations in (A). Expression values were standardized (mean centered) on a per gene basis.
(C) Genes from the cytokine module passing the criteria in (B).
(D) Genes from the tissue healing module passing the criteria in (B).
(E) Genes from the transcription factor module passing the criteria in (B).
See also Figures S2 and S6.
Targeted analysis of genes encoding archetypal NK receptors and cytolytic effector molecules (Table S2; Meresse et al., 2006) confirmed the innate-like nature and cytolytic potential of control Vδ1+ IELs (Figure 4B). In particular, GZMK, FCGR3A, and TYROBP were significantly overexpressed among control NCR+ Vδ1+ IELs (Figure 4B). A similar analysis of genes encoding various cytokines, chemokines, and growth factors (Table S2) revealed that IFNG, CCL4, and IL10 were significantly overexpressed among NCR− Vδ1+ IELs from patients with CeD (Figure 4C). Transcripts encoding IL-4, IL-9, IL-13, and IL-17 were not detected. Moreover, control NCR+ Vδ1+ IELs expressed significantly more AREG, which encodes a growth factor implicated in tissue repair (Zaiss et al., 2015), relative to NCR− Vδ1+ IELs from patients with active CeD (Figure 4C). Analogous patterns were observed for other genes associated with tissue repair (Linehan et al., 2018) (Figure 4D and Table S2). Clear differences also emerged with respect to the expression of transcription factors associated with immune function (Table S2). For example, control NCR+ Vδ1+ IELs specifically expressed GATA3 and IRF8, which have been implicated in the regulation of innate immunity (Adams et al., 2018; Zhu, 2017), whereas IRF1 (Kano et al., 2008) and RUNX1 (Wang et al., 2014) were selectively overexpressed among NCR− Vδ1+ IELs from patients with active CeD (Figure 4E), consistent with the ability of these cells to produce IFN-γ (Figure 3A).
To assess the relative impact of origin versus phenotype, we also compared NCR+ Vδ1+ IELs from healthy controls with NCR+ Vδ1+ IELs from patients with active CeD. The latter were occasionally found in younger patients (Figure S2C), but lacked expression of NKp44, in contrast to control NCR+ Vδ1+ IELs. Unbiased multidimensional scaling (MDS) analysis revealed that control NCR+ Vδ1+ IELs formed a distinct subset, whereas NCR+ and NCR− Vδ1+ IELs from patients with active CeD clustered together (Figure S2D). Accordingly, differences observed between control NCR+ Vδ1+ IELs and NCR− Vδ1+ IELs from patients with active CeD (Figure S2B) were mimicked between control NCR+ Vδ1+ IELs and NCR+ Vδ1+ IELs from patients with active CeD (Figure S2E). Transcriptional differences among Vδ1+ IELs were therefore driven by disease state rather than NCR expression, irrespective of adherence to a GFD.
In line with the functional and phenotypic data, these results demonstrated that Vδ1+ IELs formed distinct subsets characterized by divergent gene expression programs in healthy controls and patients with CeD.
The Vδ1+ IEL TCR Repertoire Is Permanently Reshaped in CeD
To determine whether the switch from innate-like NCR+ Vδ1+ IELs in the healthy state to IFN-γ-producing NCR− Vδ1+ IELs in CeD was associated with cellular turnover, we used an unbiased molecular approach to characterize all expressed TRG and TRD gene rearrangements in flow-sorted Vδ1+ T cell populations isolated directly ex vivo from donor-matched PBLs and IELs (Figures S3A–S3C; Tables S3 and S4; Davey et al., 2017; Quigley et al., 2011). Control Vδ1+ IELs almost exclusively used the TRGV4 gene, unlike donor-matched Vδ1+ PBLs (Figures 5A and 5B; Table S5A). No such preference was observed in patients with active or GFD-treated CeD (Figures 5A and 5B; Table S5A). Accordingly, TRGV4 gene transcripts were significantly enriched among control Vδ1+ IELs relative to Vδ1+ IELs from patients with active or GFD-treated CeD (Figures 5A and 5B; Table S5A). These results were corroborated in a separate cohort via RNA-seq analysis, which also showed that TRGV gene use was not related to NCR expression (Figure S3D). Moreover, control Vδ1− IELs displayed a similarly extreme preference for the TRGV4 gene and expressed NCRs at frequencies equivalent to those observed among control Vδ1+ IELs (Figures 5C and S3E). In contrast, Vδ1− IELs from patients with CeD displayed no obvious preference for a particular TRGV gene and lacked expression of NCRs, akin to Vδ1+ IELs from patients with CeD (Figures 5C and S3E). This disease-associated loss of TRGV4 gene transcripts occurred independently of human leukocyte antigen (HLA) class II alleles linked with CeD (Table S3). Of note, no TRGJ gene bias was observed among groups or tissues, reflecting widespread use of the TRGJ1 gene (Figure S3F and Table S5B). Collectively, these data suggested that TRGV4 gene rearrangements facilitated the selection of naturally occurring TCRγδ+ IELs, which were supplanted in patients with CeD.
Figure 5. The Vδ1+ IEL TCR Repertoire Is Permanently Reshaped in CeD.

(A) Proportion of unique CDR3γ sequences using a particular TRGV gene among Vδ1+ PBLs and IELs. White lines demarcate individual contributions. Healthy controls: PBLs, n = 7; IELs, n = 8. Patients with active CeD: PBLs, n = 8; IELs, n = 8. Patients with GFD-treated CeD: PBLs, n = 5; IELs, n = 7. *p < 0.05, **p < 0.01, ***p < 0.001. Firth’s penalized logistic regression and beta regression. See Table S5A.
(B) Data in (A) summarized by individual.
(C) Proportion of unique CDR3γ sequences using a particular TRGV gene among Vδ1− IELs summarized by individual.
See also Figures S3 and S6.
A Molecular Signature Defines Vδ1+ IEL Expansions in Active CeD
Next, we sought evidence of antigen-driven clonal expansions within the remodeled Vδ1+ IEL TCR repertoires in patients with CeD. A subset of patients with active CeD harbored low-diversity repertoires, which were not apparent in patients with GFD-treated CeD (Figure S4A). These results suggested that gluten consumption stimulated the expansion of particular clonotypes in the Vδ1+ IEL pool, at least in some patients with active CeD, whereas gluten withdrawal allowed diversification, potentially via a loss of antigenic drive and/or de novo recruitment of non-expanded Vδ1+ IELs.
To address this possibility, we tested for amino acid (aa) preferences among unique CDR3δ sequences, with the aim of identifying Vδ1+ TCR motifs associated with active CeD. No group-specific aa enrichments were detected to indicate a disease-associated molecular signature (Figure S4B and Table S5C). Of note, TRDJ1 gene transcripts predominated across study groups (Figure S4C). At the genetic level, preferential use of TRDD3 and, to a lesser extent, TRDD2 in the forward frame was observed across the CDR3δ dataset as a whole (Figure S4D), but there was no evidence for preferential use of a specific TRDD gene among Vδ1+ IELs from patients with active CeD (Figure S4E). These data suggested that gluten-induced reshaping of the Vδ1+ IEL repertoire was not associated with a clear CDR3δ motif. In line with this interpretation, two previously described CDR3δ motifs associated with gluten challenge, CxxxxxPxLGD (PxLGD) and CxxxxxxxxYWGI (YWGI) (Han et al., 2013), were distributed at low frequencies in the corresponding Vδ1+ repertoires obtained from patients with active or GFD-treated CeD (Figure S4E). However, the fraction of Vδ1+ IEL-derived CDR3δ sequences that incorporated no TRDD gene-encoded aas was significantly higher in healthy controls relative to patients with CeD (Figure S4F and Table S5D), and the corresponding CDR3δ sequences were significantly shorter in healthy controls relative to patients with CeD (Figure S4G).
In contrast, a similar analysis of CDR3γ sequences revealed that histidine (H) was significantly enriched in the Vδ1+ IEL repertoires obtained from patients with active CeD relative to the corresponding repertoires obtained from healthy controls and patients with GFD-treated CeD (Figure 6A and Table S5E). This observation was supported by iceLogo motif analysis (Colaert et al., 2009), which revealed a significant enrichment for H adjacent to the Jγ segment among CDR3γ sequences obtained from patients with active CeD (Figure S4H). In addition, H was found adjacent to the TRGJ1-encoded Jγ segment (H-J1 motif) in four public CDR3γ sequences, collectively shared across six patients with active CeD (Figure 6B and Table S6). It was notable that these H-J1 CDR3γ sequences associated with various TRGV gene segments (Figure 6B and Table S6). This pattern of mosaicism hinted at a unique selection pressure focused on the somatically rearranged CDR3γ loop in patients with CeD. A significantly higher fraction of unique CDR3γ sequences carried the H-J1 motif in the Vδ1+ IEL repertoires obtained from patients with active CeD relative to the corresponding repertoires obtained from healthy controls and patients with GFD-treated CeD (Figure 6C). More specifically, H-J1+ CDR3γ sequences were present in seven patients with active CeD versus only two patients with GFD-treated CeD, irrespective of HLA-DQ2/DQ8 genotype (Figure S4I and Table S3). The most common Vδ1+ IEL-derived CDR3γ sequence detected in four patients with active CeD incorporated the H-J1 motif (Figures 6D and 6E). In contrast, the associated Vδ1− IEL repertoires in these patients did not exhibit H-J1+ expansions, highlighting the lineage specificity of this molecular signature (Figure 6F). The most common Vδ1+ PBL-derived CDR3γ sequence detected in three patients with active CeD also incorporated the H-J1 motif (Figures 6D and 6E). Accordingly, Vδ1+ TCR repertoires incorporating a dominant H-J1+ CDR3γ sequence were significantly enriched among patients with active CeD relative to healthy controls and patients with GFD-treated CeD, compared across PBLs and IELs (Figure 6E).
Figure 6. A Molecular Signature Defines Vδ1+ IEL Expansions in Active CeD.

(A) Proportion of unique CDR3γ sequences using a particular amino acid (aa). White lines demarcate individual contributions. Donor numbers as in Figure 5A. Double dagger (‡) denotes aas with significant differences between two groups. Firth’s penalized logistic regression and beta regression. See Table S5E.
(B) Overlapping CDR3γ sequences incorporating the H-J1 motif.
(C) Frequency of unique H-J1+ CDR3γ sequences. Boxplots display first and third quartiles. **p < 0.01. Kruskal-Wallis rank sum test with Dunn’s test for multiple comparisons.
(D) Dominant H-J1+ CDR3γ sequences among patients with active CeD.
(E) Frequency of dominant H-J1+ CDR3γ sequences. 0 denotes samples in which the dominant CDR3γ sequence lacked H-J1. Kruskal-Wallis rank sum test with Dunn’s test for multiple comparisons.
(F) Frequency of unique H-J1+ CDR3γ sequences among Vδ1+ and Vδ1− IELs from patients with active CeD.
(G) Genes from the TCR activation module passing the criteria described in Figure 4B.
(H) Expression of Nur77 in Vδ1+ IELs versus CD3. Right: boxplot displays first and third quartiles. **p < 0.01. One-way ANOVA with Tukey’s test for multiple comparisons.
See also Figures S4 and S6.
Collectively, these observations indicated that gluten-induced reshaping of the Vδ1+ IEL compartment in patients with active CeD was associated with preferential recruitment and/or expansion of clonotypes bearing the H-J1+ CDR3γ motif, which in turn suggested that the underlying chronic inflammatory process incorporated a degree of TCR-mediated specificity for a putative ligand associated with CeD.
Vδ1+ IELs Display Hallmarks of TCR-Mediated Activation in Patients with CeD
The combined observations that Vδ1+ IELs acquired the ability to produce IFN-γ in response to gluten challenge (Figure 3B) and preferentially incorporated clonotypes bearing a shared H-J1+ CDR3γ motif in patients with active but not GFD-treated CeD (Figures 6D and 6E) suggested the possibility of in vivo activation via the TCR. Analysis of a set of genes associated with T cell activation and TCR signaling (Table S2; Fabregat et al., 2018) showed that NCR− Vδ1+ IELs from patients with active CeD significantly overexpressed transcripts associated with antigenic stimulation relative to control NCR+ Vδ1+ IELs (Figure 6G). More specifically, MKI67, CTLA4, PDCD1, and a subset of genes encoding HLA class II molecules were significantly overexpressed among NCR− Vδ1+ IELs from patients with active CeD relative to NCR+ Vδ1+ IELs from healthy controls and NCR− Vδ1+ IELs from patients with GFD-treated CeD (Figure 6G). Gluten withdrawal was therefore associated with curtailed activation and a corresponding lack of TCR-driven selection among NCR− Vδ1+ IELs (Figures 6C and S4A). Direct ex vivo analysis of the transcription factor Nur77, which indicates signaling via the TCR (Ashouri and Weiss, 2017), further supported a direct link between gluten exposure and the activation status of NCR− Vδ1+ IELs (Figure 6H). Accordingly, these data provided evidence for gluten-driven immune responses mediated via the engagement of specific Vδ1+ IEL TCRs in patients with active CeD.
BTNL3/8-Reactive Vδ1+ IELs Are Permanently Lost in CeD
Recent studies have shown that butyrophilin (BTN) and butyrophilin-like (BTNL) molecules play a key role in γδ T cell biology (Di Marco Barros et al., 2016; Melandri et al., 2018; Vantourout et al., 2018). In line with the notion that human colonic Vγ4+ IELs may be selected under physiological conditions by BTNL molecules (Di Marco Barros et al., 2016), the loss of TRGV4 gene transcripts among Vδ1+ IELs from patients with CeD (Figure 5A) was associated with corresponding decreases in BTNL8 gene transcript (Figure 7A) and protein expression levels (Figure 7B). Moreover, gluten withdrawal failed to restore TRGV4 gene use among Vδ1+ IELs, despite normalization of BTNL8 expression (Figures 7A, 7B, and S5A). In addition, Vδ1+ IELs from patients with active or GFD-treated CeD were not activated in the presence of BTNL3/8+ HEK293T cells (Figures 7C and S5B), but responded efficiently to generic stimulation via cross-linking of TCRs (Figure S5C). Similar results were obtained with Vδ1− IELs from patients with CeD, which lacked expression of TRGV4, but not with control Vδ1+ and Vδ1− IELs, which displayed an extreme preference for TRGV4 (Figure 5) and responded in the presence of BTNL3/8+ HEK293T cells (Figures 7C and 7D). Importantly, BTNL3/8-reactive Vδ1+ IELs were lost from the duodenum, but not the colon, in a patient with GFD-treated CeD (Figure S5D). This finding concurred with the site-specific depletion of NCR+ Vδ1+ IELs (Figure 2C). Collectively, these results suggested that the recovery of BTNL molecules after exclusion of dietary gluten was insufficient to reconstitute the niche favored by naturally occurring Vγ4+/Vδ1+ IELs, potentially reflecting an absolute homeostatic requirement for sustained expression of BTNL3/8.
Figure 7. BTNL3/8-Reactive Vδ1+ IELs Are Lost in CeD.

(A) Expression of BTNL3 and BTNL8 relative to GAPDH in small intestinal biopsies via qPCR. Boxplots display first and third quartiles. **p < 0.01, ***p < 0.001. Kruskal-Wallis rank sum test with Dunn’s test for multiple comparisons.
(B) Immunohistochemical analysis of BTNL8 expression in duodenal sections.
(C) Downregulation of CD3 and Vδ1 on the surface of IELs pre-gated for Vδ1 expression after overnight incubation with HEK293T-BTNL8+ or HEK293T-BTNL3/8+ cells. Gating was patient specific based on the HEK293T-BTNL8+ condition. Right: boxplot displays first and third quartiles. *p < 0.05, ***p < 0.001. Kruskal-Wallis rank sum test with Dunn’s test for multiple comparisons.
(D) Downregulation of CD3 on the surface of Vδ1− IELs. Details and statistics as in (C).
(E) Proportion of unique CDR3γ sequences expressing TRGV4 gene transcripts. Circles highlighted in black for patients with active CeD denote cases where the dominant TRGV4 transcript incorporated the H-J1 motif. Boxplot displays first and third quartiles. **p < 0.01, ***p < 0.001. Kruskal-Wallis rank sum test with Dunn’s test for multiple comparisons.
(F) SKW3 cells stably expressing clonal TCRs were cultured for 2 hr with varying numbers of untransduced HEK293T cells (HEK293T-UT) or HEK293T-BTNL3/8+ cells. Activation was assessed via the induction of intracellular Nur77 from 3–5 independent experiments per TCR. Error bars display SD. *p < 0.05. Paired t test (HEK293T-UT versus HEK293T-BTNL3/8+).
(G) Frequency of Vδ1+ IELs (left), expression of NKp46 and NKp44 on Vδ1+ IELs (middle), and expression of CD3 and Vδ1 on IELs after overnight incubation with HEK293T-BTNL8+ or HEK293T-BTNL3/8+ cells (right) for two patients with potential CeD.
See also Figures S5–S7.
In some cases, patients with active CeD harbored populations of Vγ4+/Vδ1+ IELs, which tended to express TCRs that were enriched for the H-J1 motif (Figure 7E). As Vγ4+ T cells from healthy human colon have been shown to react with BTNL3/8 (Di Marco Barros et al., 2016; Melandri et al., 2018), we formally tested the ability of Vγ4+/Vδ1+ IEL H-J1+ TCRs from patients with active CeD to recognize BTNL3/8. Two Vγ4+/Vδ1+ IEL TCRs from healthy controls triggered dose-dependent responses to BTNL3/8, as expected, whereas a Vγ3+/Vδ1+ IEL TCR from a patient with active CeD failed to recognize BTNL3/8 (Figures 7F, S5E, and S5F).
Strikingly, a Vγ4+/Vδ1+ IEL H-J1+ TCR from a patient with active CeD also failed to recognize BTNL3/8 (Figures 7F, S5E, and S5F). This particular TCR was characterized by the presence of a long CDR3δ loop (22 aas), whereas control Vγ4+/Vδ1+ IEL TCRs typically incorporated shorter CDR3δ loops (Figure S4G). Moreover, the control Vγ4+/Vδ1+ IEL TCR with the shortest CDR3δ loop (12 aas, TCR 95) displayed the strongest reactivity against BTNL3/8 (Figure 7F). These observations suggested that the inability of the Vγ4+/Vδ1+ IEL H-J1+ TCR from the patient with active CeD to recognize BTN3/8 was related to the presence of a long CDR3δ loop, consistent with a recent study in which a Vγ4+/Vδ1+ IEL H-J1+ TCR with a short CDR3δ loop recognized BTNL3/8 (Melandri et al., 2018). It remains to be determined whether the HJ-1 motif can impede Vγ4+/Vδ1+ TCR interactions with BTNL3/8. Of note, all expressed TCRs transduced a functional signal in the presence of αCD3/αCD28 beads (Figures S5G and S5H). Collectively, these results provided direct evidence to support the contention that gluten-induced inflammatory remodeling of the TCRγδ+ IEL compartment in patients with CeD was associated with a loss of productive TCR-mediated interactions with BTNL3/8.
Dynamic Remodeling of the Vδ1+ IEL Compartment Precedes Tissue Damage in CeD
The Vδ1+ IEL compartment underwent dynamic changes both during the chronic inflammatory process associated with active CeD and during the resolution phase associated with strict adherence to a GFD (Figure S6). However, it was still unclear whether these alterations were causally or reactively linked with tissue damage. To address this issue, we analyzed patients with potential CeD, defined as a state of CD4+ T cell-mediated intolerance to dietary gluten without histological evidence of villous atrophy (Husby et al., 2012). This heterogeneous group encompassed patients with various levels of Vδ1+ IEL infiltration (Figure S7A), loss of BTNL8 gene transcripts (Figure S7B), loss of NCR+ Vδ1+ IELs (Figure S7C), and loss of TRGV4 gene transcripts (Figure S7D). This heterogeneity was further exemplified by two patients with potential CeD, one of which exhibited an expansion of NCR+ Vδ1+ IELs with conserved ex vivo reactivity against BTNL3/8, and the other of which harbored physiological numbers of Vδ1+ IELs that lacked NCR expression and ex vivo reactivity against BTNL3/8 (Figure 7G). In conjunction with the presence of occasional Vδ1+ IEL TCRs bearing the H-J1+ CDR3γ motif in patients with potential CeD (Figure S7E), these observations suggested that the loss of naturally occurring innate-like Vδ1+ IELs may precede tissue destruction and the emergence of adaptive features within the disease-associated Vδ1+ IEL repertoire.
DISCUSSION
Current paradigms stipulate that tissue-resident immunity is established and maintained by long-lived, anatomically compartmentalized populations of lymphocytes, which adapt via local homeostasis and proliferation to novel antigenic challenges without displacing pre-existing memory specificities (Beura et al., 2018; Mueller and Mackay, 2016; Park et al., 2018). However, the impact of chronic inflammation on the tissue-resident lymphocyte pool has not been defined in previous studies, in part due to a lack of suitable models. We addressed this knowledge gap by taking advantage of the fact that antigen exposure can be controlled in CeD, a complex T cell-mediated inflammatory disorder with an autoimmune component (Jabri and Sollid, 2009).
In healthy controls, we found that the tissue-resident TCRγδ+ IEL compartment was dominated by a unique subset of innate-like, semi-invariant Vγ4+/Vδ1+ and Vγ4+/Vδ1− IELs that recognized BTNL3/BTNL8, constitutively expressed NCRs, and persisted throughout life, exemplifying the longevity and stability required to maintain durable immunity at barrier sites under physiological conditions. Importantly, these Vγ4+/Vδ1+ IELs were ideally poised to maintain homeostasis in the local microenvironment, either by eliminating virus-infected or malignant cells in response to innate signals, such as NCR ligands and IL-15, or by promoting tissue healing via the production of growth factors, such as amphiregulin (Zaiss et al., 2015).
In patients with CeD, the tissue-resident Vδ1+ IEL compartment was profoundly altered, even after exclusion of the inciting antigen and resolution of the associated inflammation. The key changes included an irretrievable loss of innate-like Vγ4+/Vδ1+ IELs and the emergence of gluten-sensitive, IFN-γ-producing Vδ1+ IELs characterized by clonal expansions incorporating H-J1+ TCRs that lacked reactivity against BTNL3/BTNL8. These observations challenge the assumption that pre-existing tissue-resident Vδ1+ IELs expand in CeD (Hayday et al., 2001). We therefore propose a multistep model, in which naturally occurring Vγ4+/Vδ1+ IELs expand in response to inflammation, triggered by the loss of tolerance to dietary gluten, allowing initial preservation of NCR expression and BTNL3/BTNL8 reactivity, as observed in some patients with potential CeD. The ongoing disease process then reaches a tipping point, as observed in a subset of patients with potential or active CeD, where the chronic loss of BTNL8 expression eventually leads to the loss of Vγ4+/Vδ1+ IELs, which likely depend on this ligand for survival. Circulating TCRγδ+ cells are then recruited into the vacant immunological space and subsequently acquire a tissue-resident phenotype in response to local signals. In line with this proposition, Vδ1+ PBLs from several patients with active CeD expressed TCRs incorporating the H-J1+ CDR3γ motif, indicative of priming by a locally induced ligand in gut-associated lymphoid tissue (Guy-Grand et al., 2013). Moreover, gluten withdrawal precipitated a contraction of H-J1+ TCRs among Vδ1+ IELs, consistent with the in situ expression of an antigen-dependent ligand in patients with active CeD. It is important to note that NCR+ Vδ1+ IELs were occasionally found in patients with active CeD. However, these cells aligned transcriptionally with disease-associated NCR− Vδ1+ IELs rather than control NCR+ Vδ1+ IELs, lacked expression of NKp44 and the TRGV4 gene, and were only found in children with CeD. Accordingly, NCR expression is likely regulated by site-specific signals, which are retained to some extent in children with active CeD, selectively facilitating the expression of NKp46 on Vδ1+ IELs.
A previous study in mice showed that Vγ7+ IELs (the mouse homolog of human Vγ4+ IELs) could not be rescued if the expression of BTNL molecules was delayed beyond the neonatal period, during which these cells typically expand in situ (Di Marco Barros et al., 2016). In conjunction with our finding that innate-like Vγ4+/Vδ1+ IELs failed to recover in patients with GFD-treated CeD, despite restoration of the mucosal architecture and BTNL8 expression, this observation suggests that tissue-resident TCRγδ+ IELs are exquisitely sensitive to the presence of BTNL molecules in the intestinal epithelium. It therefore seems likely that the profound changes we observed in the tissue-resident compartment of patients with CeD were underpinned by a loss of BTNL molecules in the local microenvironment, although it remains to be determined how long this state of depletion needs to persist to trigger the irretrievable destruction of a fully established niche constituted by mature populations of Vγ4+/Vδ1+ IELs.
It has been proposed that TCRγδ+ IELs act to limit the infiltration of systemic T cells in CeD (Hayday et al., 2001), without playing a direct role in disease pathogenesis (Hayday, 2000; Kutlu et al., 1993). Our data challenge this assumption. In particular, we found that Vδ1+ IELs in patients with CeD expressed high levels of the chemoattractant CCL4 and adopted a Th1-like phenotype, characterized by expression of IRF1 and RUNX1 and the production of IFN-γ. Moreover, only Vδ1+ IELs showed enhanced IFN-γ production in response to gluten challenge, which is highly relevant in the context of a Th1-mediated disorder like CeD. Previous studies have shown that IFN-γ induces the upregulation of HLA-E on intestinal epithelial cells (Meresse et al., 2006) and further upregulates major histocompatibility complex (MHC) class I molecules (Früh and Yang, 1999), which in turn can contribute to the activation of TCRαβ+ CD8αβ+ IELs, the primary mediators of intestinal epithelial cell destruction in CeD (Jabri and Sollid, 2009). On the flip side of this argument, Vδ1+ IELs also expressed genes associated with the regulation of chronic inflammatory responses, such as IL-10, CTLA4, PDCD1, and ZNF683, in patients with CeD. However, the corresponding molecules can equally act as proxies of chronic inflammation, in line with a pathogenic role for IFN-γ-producing Vδ1+ IELs. For example, Th1 cells self-regulate via the production of IL-10 (O’Garra and Vieira, 2007; Saraiva et al., 2009), and gluten-specific CD4+ T cells can express both IFN-γ and IL-10 (Nilsen et al., 1995).
In conclusion, we have shown that chronic site-specific inflammation permanently reconfigures the tissue-resident TCRγδ+ IEL compartment in patients with CeD. A similar process of “immunological scarring” may contribute to the pathogenesis of other intestinal immune disorders such as ulcerative colitis, which is also characterized by decreased levels of BTNL8 in situ (Lebrero-Fernández et al., 2016). Further studies are therefore required to establish the general applicability of our findings across disease states and to determine the impact of chronic inflammatory processes on the stability of adaptive immune cell populations. In anticipation of these complementary data, we speculate that the irretrievable loss of tissue-resident subsets with unique innate-like specificities and cytolytic properties may have long-term implications for the health of patients with CeD.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Bana Jabri (bjabri@bsd.uchicago.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patients were classified into four groups for the purposes of this study. Control – symptoms of upper gastrointestinal tract disease, with no histological evidence of duodenal inflammation, no family history of CeD, and negative serology for TG2. Active – histological evidence of villous atrophy, with positive serology for TG2. GFD – established diagnosis of CeD, with no histological evidence of villous atrophy, and negative serology for TG2. Potential – no histological evidence of villous atrophy, with positive serology for TG2. Exclusion criteria included immunosuppressive medication and coincident diagnoses of Barrett’s esophagus, eosinophilic esophagitis, cancer, cirrhosis, or inflammatory bowel disease (IBD). In challenge experiments, patients with GFD-treated CeD consumed 6 g of gluten daily for six weeks. Duodenal biopsies were taken from 3–5 distinct sites, together with 2–8 mL of venous blood, under protocol 12623B approved by the Chicago Biomedicine Institutional Review Board.
METHOD DETAILS
Lymphocyte isolation
PBLs were isolated from whole blood via standard density gradient centrifugation. IELs were isolated from duodenal biopsies via mechanical disruption. Briefly, duodenal tissues were shaken at 250 rpm for 30 min at 37°C in 7 mL of RPMI 1640 medium supplemented with 1% dialyzed fetal bovine serum (Biowest), 2 mM EDTA (Corning), and 1.5 mM MgCl2 (Thermo Fisher Scientific). The procedure was repeated once with fresh medium to enhance cell recovery. Cells were harvested from the biopsy-free media via centrifugation and pooled for subsequent analyses.
Flow cytometry
The following directly conjugated antibodies were used to identify cellular markers: αVδ1 APC (REA173), αVδ1 FITC (TS8.2), αVd2 PE (B6), αVδ2 PerCP (B6), αTCRαβ BV421 (IP26), αTCRγδ PE (5A6.E9), αTCRγδ PE-Cy5 (5A6.E9), αCD3 APC (UCHT1), αCD3 APC-Cy7 (UCHT1), αCD3 PE-Cy7 (UCHT1), αCD3 V450 (UCHT1), αCD4 APC (RPA-T4), αCD4 BV786 (SK3), αCD8a BUV496 (RPA-T8), αCD8a BV510 (RPA-T8), αCD8a BV650 (RPA-T8), αCD45 BV711 (HI30), αCD45RA BV510 (HI100), αCD69 PE (FN50), αCD69 PE-CF594 (FN50), αCD103 BUV395 (Ber-ACT8), αCD107a BUV395 (H4A3), αCCR7 PE-Cy7 (G043H7), αNKp44 APC (p44–8), αNKp44 PE (p44–8), αNKp46 BV605 (9E2), αNKp46 PE (9E2), αNur77 PE (12.14), αMyc-Tag PE (9B11), and αHA-Tag Alexa Fluor 647 (6E2). For intracellular cytokine detection, cells were fixed/permeabilized using a BD Cytofix/Cytoperm Plus Fixation/Permeabilization Solution Kit (BD Biosciences) and stained with the following directly conjugated antibodies: αIFN-γ APC (4S.B3) and αTNF-α PE-Cy7 (MAb11). Dead cells were excluded from the analysis using LIVE/DEAD Fixable Aqua or LIVE/DEAD Fixable Near-IR (Thermo Fisher Scientific). All flow cytometry data were analyzed using FlowJo software (version 10.2, Tree Star).
Transcriptome sequencing
Full-length cDNA and sequencing libraries were generated using a modified version of the single-cell Smart-seq2 protocol (Picelli et al., 2014). Briefly, 50–100 NCR+ (NKp46+) or NCR− (NKp46−) Vδ1+ IELs were sorted into a lysis buffer containing oligo-dT and dNTPs. Reverse transcription was performed after hybridization of oligo-dT to poly-A RNA. The resulting cDNA was amplified over 18 thermocycles, purified using an AMPure XP Kit (Beckman Coulter), and quality controlled using a Bioanalyzer High Sensitivity DNA Kit (Agilent). A total of 0.25 ng of cDNA from each sample was tagmented, ligated with adapters (Nextera XT), amplified over 12 thermocycles, and quality controlled using a Bioanalyzer High Sensitivity DNA Kit (Agilent). Libraries were then pooled in equimolar amounts and sequenced on two lanes in duplicate using a HiSeq4000 System (Illumina).
HLA genotyping
DNA samples were genotyped using an ImmunoArray BeadChip (Illumina) with single nucleotide polymorphism (SNP) probes as described previously (Trynka et al., 2011). Data analysis was performed using PLINK v1.9. Five of the six HLA-tagging SNPs reported previously (Monsuur et al., 2008) were covered on the ImmunoArray BeadChip. Genotype call rates were higher than 95%. HLA-DQ was inferred from a combination of genotypes at these five SNPs.
TCR sequencing
Lymphocytes were stained for viability and surface expression of CD3, TCRαβ, TCRγδ, Vδ1, and Vδ2. Live Vδ1+ T cells were then sorted directly into 100 μL of RNAlater (Thermo Fisher Scientific) using a FACSAria III flow cytometer (BD Biosciences) according to the gating strategy presented in Figure S3A. Cell numbers and patient details are summarized in Table S3. All expressed TRG and TRD gene transcripts were amplified using an unbiased template-switch anchored RT-PCR (Davey et al., 2017; Quigley et al., 2011). Amplicons were subcloned, sampled, and sequenced as described previously (Price et al., 2005). Gene use was assigned using the ImMunoGeneTics (IMGT) nomenclature (Lefranc, 2003).
TCR/NKR stimulation assay
Polystyrene flat-bottom 96-well plates (Corning) were coated overnight at 4°C with one of the following antibody cocktails: mix A – 0.5 μg/mL αTCRγδ (B1, BioLegend), 1 μg/mL mouse IgG2a κ (MOPC-173, BioLegend), and 1 μg/mL mouse IgG2b κ (MPC-11, BioLegend); or mix B – 0.5 μg/mL αTCRγδ (B1, BioLegend), 1 μg/mL αNKp44 (195314, R&D Systems), and 1 μg/mL αNKp46 (253415, R&D Systems). Lymphocytes were isolated from whole biopsies either pre-treated or not pre-treated with 10 ng/mL recombinant human IL-15 (BioLegend) for 18 hr at 4°C and plated at a density of 2 × 105 cells per well (1 × 106 cells/mL) in RPMI 1640 medium supplemented with 10% human AB serum (Corning), 0.1% GolgiPlug (BD Biosciences), 0.1% GolgiStop (BD Biosciences), and 10 ng/mL recombinant human IL-15 (if pre-treated with recombinant human IL-15). Surface upregulation of CD107a was quantified via flow cytometry after incubation for 3 hr at 37°C.
Phorbol myristate acetate/ionomycin stimulation assay
Lymphocytes were suspended in RPMI 1640 medium supplemented with 10% human AB serum (Corning), 10 ng/mL phorbol myristate acetate (Sigma-Aldrich), 150 ng/mL ionomycin calcium salt (Sigma-Aldrich), 100 U/mL recombinant human IL-2 (NIH AIDS Reagent Program), 0.1% GolgiPlug (BD Biosciences), and 0.1% GolgiStop (BD Biosciences) and distributed at 2 × 105 cells per well (1 × 106 cells/mL) in polystyrene flat-bottom 96-well plates (Corning). Surface expression of CD107a and intracellular cytokine production were quantified via flow cytometry after incubation for 3 hr at 37°C.
Quantitative RT-PCR
Duodenal biopsies were collected in RNAlater RNA Stabilization Reagent (QIAGEN). After incubation for 48 hr at 4°C, excess solution was removed, and the biopsies were stored at −80°C. On the day of processing, biopsy material was thawed, suspended in 500 μL of Buffer RLT (QIAGEN) supplemented with 1% β-mercaptoethanol (Sigma-Aldrich), and homogenized in a Bullet Blender 24 (Next Advance) using a 1:1 mix of 0.5 mm and 1.0 mm zirconium oxide beads (Next Advance). RNA was then isolated using an AllPrep DNA/RNA/Protein Mini Kit (QIAGEN). cDNA was generated from 200 ng of total RNA using a GoScript Reverse Transcriptase Kit (Promega). Expression of the genes of interest was measured via quantitative RT-PCR using SYBR Advantage qPCR Premix (Clontech) on a Light Cycler 480 Instrument II (Roche). The following parameters were used for amplification: denaturation for 10 s at 95°C, annealing for 10 s at 60°C, and extension for 10 s at 72°C. Human primer sequences were as follows: GAPDH forward: 5′-ATGGGGAAGGTGAAGGTCG-3′; GAPDH reverse: 5′-GGGGTCATTGATGGCAACAATA-3′; BTNL3 forward: 5′-TCAGTTTCTACGAGCTGGTGTC-3′; BTNL3 reverse: 5′-CCAAGGCCTGGACAAACTT-3′; BTNL8 forward: 5′-GCTCTCATGCTCAGTTTGGTT-3′; and BTNL8 reverse: 5′-GTCTGGCCCAAACACCTG-3′. The BTNL3 and BTNL8 primers were described previously (Lebrero-Fernández et al., 2016).
Generation of HEK293T cell lines expressing BTNL3 and BTNL8
Synthetic DNA (Genscript) encoding full-length human BTNL8 alone or full-length human BTNL3 and BTNL8 separated by a P2A (Teschovirus A) ribosomal skip sequence was subcloned into pMIG (Holst et al., 2006) and used to make retroviral particles (Gras et al., 2010; Holst et al., 2006). Retrovirally transduced (GFP+) HEK293T cells were flow-sorted based on expression of the N-terminal myc (EQKLISEEDL(GGS)) tag to identify BTNL3 and the N-terminal HA (YPYDVPDYA(GSG)) tag to identify BTNL8.
Generation of SKW3 cell lines expressing Vδ1+ TCRs
Flow-sorted Vδ1+ IELs isolated from healthy controls and patients with GFD-treated CeD were stimulated in vitro for two weeks at a starting density of 3 cells per well with irradiated B-lymphoblastoid cell lines and heterologous PBLs in RPMI 1640 medium supplemented with 300 U/mL recombinant human IL-2 (NIH AIDS Reagent Program), 1 μg/mL phytohemagglutinin (Calbiochem), and 10% human AB serum (Atlanta Biologicals). Paired TCRγ and TCRδ sequences were obtained from clonally expanded Vδ1+ IELs. Clonal expansions containing the H-J1 motif were sufficiently large in two patients with active CeD to allow frequency-based pairing of TCRγ and TCRδ sequences obtained directly ex vivo from bulk populations of Vδ1+ IELs. Representative Vδ1+ TCRs were stably expressed via retroviral transduction of the TCRαβ-deficient T cell leukemia cell line SKW3 (German Collection of Microorganisms and Cell Cultures) (Gras et al., 2010; Holst et al., 2006).
BTNL3/8 reactivity assay ex vivo
Round-bottom 96-well plates were seeded overnight with 2 × 105 HEK293T-BTNL8+ or HEK293T-BTNL3/8+ cells per well to create a monolayer. IELs were isolated from whole biopsies pre-treated with 10 ng/mL recombinant human IL-15 (BioLegend) for 16 hr at 4°C and overlaid at 1 × 105 cells per well on top of the pre-plated HEK cell monolayer. Cells were co-cultured for 24 hr and analyzed via flow cytometry for surface downregulation of CD3 (UCHT1) and Vδ1 (REA173). As a positive control, IELs were simulated for 2 hr with 1.5 μg/mL of plate-bound purified αCD3 (UCHT1).
BTNL3/8 reactivity assay in vitro
For CD3 downregulation, round-bottom 96-well plates were seeded overnight with 2 × 105 HEK293T-UT or HEK293T-BTNL3/8+ cells per well to create a monolayer, and 1 × 105 TCRγδ-SKW3 cells were then added to each well and incubated for 24 hr at 37°C. For Nur77 induction, round-bottom 96-well plates were seeded overnight with 3.125 X 103, 6.25 × 103, 1.25 × 104, or 2.5 × 105 HEK293T-UT or HEK293T-BTNL3/8+ cells per well to create a monolayer, and 1 × 105 TCRγδ-SKW3 cells were then added to each well and incubated for 2 hr at 37°C. Flow cytometry was used to measure surface downregulation of CD3 (UCHT1) or intracellular induction of Nur77 (12.14). As a positive control, 1 × 105 TCRγδ-SKW3 cells were stimulated for 2 hr with 2.5 μL of Dynabeads Human T-Activator CD3/CD28 for T Cell Expansion and Activation (Thermo Fisher Scientific).
Immunohistochemistry
Duodenal biopsies were preserved in formalin and embedded in paraffin. Sections were cut to a thickness of 5 μm and stained with Bond RX Automatic Stainer (Leica Biosystems). Slides were then dewaxed three times with xylene, ethanol, and water, treated for 20 min with Epitope Retrieval Solution II (Leica Biosystems), incubated for 5 min with 0.5% casein to prevent non-specific binding, and stained for 1 hr with a 1:200 dilution of αBTNL8 (2187B, R&D Systems). Antigen-antibody binding was revealed using Bond Polymer Refine Detection (Leica Biosystems). Tissue sections were then blocked with peroxidase, stained with DAB, and counter-stained with hematoxylin. Images were acquired using a ScanScope XT microscope (Leica Biosystems).
QUANTIFICATION AND STATISTICAL ANALYSIS
Transcriptome analysis
A total of 34 RNA-seq libraries were generated from flow-sorted Vδ1+ IELs, including 8 from healthy controls (8 NCR+), 18 from patients with active CeD (9 NCR+ and 9 NCR−), and 8 from patients with GFD-treated CeD (3 NCR+ and 5 NCR−). Adaptor sequences and low-quality score bases (Phred score < 20) were trimmed using Trim Galore (version 0.4.4). The resulting reads were merged per sample and mapped to the human genome reference sequence (Ensembl GRCh38 release 87) using kallisto (version 0.43.0) (Bray et al., 2016). To account for differences in read counts at the tails of the distribution, samples were normalized using the weighted trimmed mean of M-values algorithm (TMM), as implemented in the R package edgeR (Robinson et al., 2010). Data were then log-transformed using the voom function in the limma package (Ritchie et al., 2015). Non-coding and lowly-expressed genes with an average (across all samples) log2(CPM) lower than 0 were excluded from downstream analyses, leaving a total of 16,066 genes. Linear models that accounted for differences in age and sex were used to identify genes with expression levels that varied between NCR+ samples from healthy controls, NCR+/NCR− samples from patients with active CeD, and NCR+/NCR− samples from patients with GFD-treated CeD, implemented using the lmFit function in the limma package (Ritchie et al., 2015).
General procedure for analysis of TCR characteristics
Sequences were first annotated for TRGV/J or TRDV/J using the IMGT database and V-QUEST tool (Brochet et al., 2008). Annotated CDR3 sequences were then imported into R for further analysis. All detected sequences were captured in the analysis, including potentially non-functional TRGV10 gene transcripts. Clonal distribution in each sample was assessed using the Shannon diversity index. Sequence characteristics (TRV, TRD, and TRJ gene use, and AA composition) were tested for association with patient groups and tissues. For each potentially distinguishing sequence characteristic (TRV, TRD, and TRJ gene use, and AA composition), enrichment was scored by overall occurrence per group and by trends in occurrence per individual. Results that weighted each unique sequence equally were found to be more robust to potentially non-representative sampling than results that weighted each unique sequence by clonal size. Unique sequences were therefore weighted equally throughout the analysis, and the most strongly enriched features were identified as those passing both group and individual tests. For overall group enrichment testing, each unique TCR sequence labeled with or without the characteristic under question was used as the response variable in Firth’s penalized logistic regression performed with the group label as the predictor (logistf R package). This method allowed nearly complete separation of each characteristic by the group label predictor. Linear hypothesis testing on the resulting group coefficients was then used to estimate the significance of each group comparison (multcomp R package). To test if the occurrence of a characteristic per individual shifted consistently between groups, characteristic proportions were explicitly modeled using beta regression (Smithson and Verkuilen, 2006). The proportion of unique TCR sequences with the characteristic under question was first calculated for each individual. These proportion values were then used as the response variable in beta regression performed with the group label as the predictor (betareg R package). Due to the common presence of 0.0 and 1.0 proportion values (completely absent or present characteristics, respectively), raw proportion values (p) were transformed using (p⋅(n − 1) + 0.5)/n, where n is the sample size, to ensure modeled values fell within the open interval (0,1) required for beta regression modeling (Ferrari and Cribari-Neto, 2004). Linear hypothesis testing on beta distribution mean coefficients per group was then used to estimate the significance of each group comparison (car R package).
Shannon diversity index
For each sample, the Shannon diversity index was calculated as H = Σn piln pi, which provides a measure of repertoire diversity. To account for variation in the number of recovered CDR3 sequences, individual datasets were subsampled 100 times to 50 CDR3 sequences. The median index was then used to represent the score for each sample (vegan R package).
Analysis of TRGV and TRGJ gene use
As described above, logistic regression was used to test for enrichment of TRGV and TRGJ gene segments in each group and each tissue. Significantly enriched gene segments were then tested for enrichment at the individual level using beta regression.
Analysis of CDR3 AAs
For each full-length CDR3 sequence, the longest substring of AAs from the V and J ends that matched the corresponding TRV and TRJ germline sequences was identified using reference data provided by the IMGT database (Lefranc, 2003). The remaining AAs were considered non-germline, irrespective of potential contributions from the TRDD segment. The absence or presence of each AA was first determined in each unique CDR3. As described above, logistic regression was then used to test for the enrichment of each AA in each group, and significantly enriched AAs were tested for enrichment at the individual level using beta regression.
Analysis of TRDD gene use
To determine the likely contribution of TRDD genes to each TCRδ sequence, an empirical statistic was generated to distinguish true germline matches from potentially spurious matches arising from random nucleotide additions. For each possible TRDD gene-derived nucleotide sequence, the longest substring match was first identified in each candidate CDR3 sequence, and 100,000 random nucleotide strings weighted by the observed nucleotide composition among all non-germline sequences were generated for each possible CDR3 length. By collecting match length statistics between a TRDD sequence and these random sequence sets, empirical null distributions of match length were obtained for each TRDD gene in both the forward and reverse frames. In each of these distributions, each match length bin contained at least 10 positive occurrences, assuring sufficient sampling coverage. These calculations allowed us to estimate an empirical p value for an observed TRDD gene sequence match, representing the probability of obtaining a match of the observed length or longer by chance. To account for testing a TRDD gene sequence against every unique CDR3 sequence, the set of empirical p values was corrected according to the Benjamini-Hochberg multiple testing procedure, which quantifies the false discovery rate (FDR). This procedure was applied independently to each of three possible TRDD genes across both the forward and reverse nucleotide sequences. Significant TRDD gene use was assigned at FDR values < 0.05. Sequences were further annotated for each TRDD gene to identify the active reading frame. As described above, logistic regression spanning all annotated unique CDR3s was used to test for enrichment of TRDD gene segments in each group. Significantly enriched gene segments were then tested for enrichment at the individual level using beta regression. Analysis of TRDD gene use, statistical testing, and sequence annotation were performed using custom code written in R.
iceLogo motifs
Non-germline-encoded CDR3 AA sequences were right-justified at the junction with the J segment and analyzed using the iceLogo java application (Maddelein et al., 2015). Unique Vδ1+ IEL CDR3 sequences from healthy controls and patients with GFD-treated CeD were pooled to serve as a fixed position background/reference set of sequences for visualization of AA enrichment among Vδ1+ IEL CDR3 sequences from patients with active CeD.
DATA AND SOFTWARE AVAILABILITY
RNA-sequencing data were submitted to the Gene Expression Omnibus under accession code GEO: GSE123649.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Vδ1 APC (TS8.2) | Miltenyi Biotec | Cat#130–100-519 |
| Vδ1 FITC (TS8.2) | Thermo Fisher Scientific | Cat#TCR2730 |
| Vδ2 PE (B6) | BioLegend | Cat#331408 |
| Vδ2 PerCP (B6) | BioLegend | Cat#331410 |
| TCRαβ BV421 (IP26) | BioLegend | Cat#306722 |
| TCRγδ PE (5A6.E9) | Thermo Fisher Scientific | Cat#MHGD04 |
| TCRγδ PE-Cy5 (5A6.E9) | Thermo Fisher Scientific | Cat#MHGD06 |
| CD3 APC (UCHT1) | BioLegend | Cat#300412 |
| CD3 APC-Cy7 (UCHT1) | BioLegend | Cat#300426 |
| CD3 PE-Cy7 (UCHT1) | BioLegend | Cat#300420 |
| CD4 BV786 (SK3) | BD Biosciences | Cat#563877 |
| CD8a BUV496 (RPA-T8) | BD Biosciences | Cat#564804 |
| CD8a BV650 (RPA-T8) | BD Biosciences | Cat#563821 |
| CD45RA BV510 (HI100) | BioLegend | Cat#304142 |
| CCR7 PE-Cy7 (G043H7) | BioLegend | Cat#353226 |
| CD45 BV711 (HI30) | BD Biosciences | Cat#564357 |
| CD103 BUV395 (Ber-ACT8) | BD Biosciences | Cat#564346 |
| CD69 PE-CF594 (FN50) | BD Biosciences | Cat#562617 |
| CD107a BUV395 (H4A3) | BD Biosciences | Cat#565113 |
| NKp44 PE (p44–8) | BD Biosciences | Cat#558563 |
| NKp44 APC (p44–8) | BD Biosciences | Cat#558564 |
| NKp46 PE (9E2) | BioLegend | Cat#331908 |
| NKp46 BV605 (9E2) | BioLegend | Cat#331926 |
| Granzyme B PE (GB12) | Invitrogen | Cat#MHGB04 |
| TNF-α PE-Cy7 (MAb11) | BioLegend | Cat#502930 |
| IFN-γ APC (4S.B3) | eBioscience | Cat#17–7319-82 |
| Nur77 PE (12.4) | eBioscience | Cat#12–5965-82 |
| Purified anti-human TCR γδ (B1) | BioLegend | Cat#331202 |
| Human NKp46/NCR1 (195314) | R&D Systems | Cat#MAB1850 |
| Human NKp44/NCR2 (253415) | R&D Systems | Cat#MAB22491 |
| LEAF Purified Mouse IgG2b, k (MPC-11) | BioLegend | Cat#400323 |
| LEAF Purified Mouse IgG2a, k (MOPC-173) | BioLegend | Cat#400223 |
| Myc-Tag PE (9B11) (αBTNL3) | Cell Signaling Technology | Cat#CST.3739S |
| HA-Tag Alexa Fluor 647 (6E2) (αBTNL8) | Cell Signaling Technology | Cat#CST.3444S |
| Human BTNL8 (2187B) | R&D Systems | Cat#MAB9359–100 |
| Dynabeads™ Human T-Activator CD3/CD28 for T Cell Expansion and Activation | Thermo Fisher Scientific | Cat#11131D |
| Biological Samples | ||
| Fetal Bovine Serum | Biowest | Cat#S01520; Lot#A11504E |
| Human AB Serum | Corning | Cat#35–060-CI; Lot#35060115 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| EDTA, 0.5M, pH8.0 | Corning | Cat#46–034-CI |
| Magnesium Chloride Hexahydrate (MgCl2) | Thermo Fisher Scientific | Cat#BP214–500 |
| Ficoll-Paque PLUS | GE Healthcare Life Sciences | Cat#17–1440-03 |
| RNAlater Stabilization Solution | Ambion | Cat#AM7020 |
| RNAlater RNA Stabilization Reagent | QIAGEN | Cat#76104 |
| 2-Mercaptoethanol (BME) | Sigma-Aldrich | Cat#M7154 |
| Phorbol Myristate Acetate | Sigma-Aldrich | Cat#P1585 |
| Ionomycin Calcium Salt from Streptomyces conglobatus | Sigma-Aldrich | Cat#10634 |
| BD GolgiPlug Protein Transport Inhibitor | BD Biosciences | Cat#555029 |
| BD GolgiStop Protein Transport Inhibitor | BD Biosciences | Cat#554724 |
| Recombinant Human IL-2 | NIH AIDS Reagent Program | Cat#136 |
| Recombinant Human IL-15 (carrier-free) | BioLegend | Cat#570304 |
| Critical Commercial Assays | ||
| S.O.C. Medium | Thermo Fisher Scientific | Cat#15544034 |
| Triton X-100 | Sigma-Aldrich | Cat#T9284 |
| Nuclease-free Water | Ambion | Cat#AM9932 |
| Recombinant RNase Inhibitor (25,000 units) | Clontech | Cat#2313B |
| 5M Betaine Solution | Sigma-Aldrich | Cat#B0300–1VL |
| 1M MgCl2 | Thermo Fisher Scientific | Cat#AM9530G |
| Ethanol 200 Proof | Decon Labs Inc | Cat#DSP-MD 43 |
| Buffer EB | QIAGEN | Cat#19086 |
| Critical Commercial Assays | ||
| LIVE/DEAD® Fixable Aqua Dead Cell Stain Kit | Thermo Fisher Scientific | Cat#L34966 |
| LIVE/DEAD® Fixable Near-IR Dead Cell Stain Kit | Thermo Fisher Scientific | Cat#L34975 |
| BD Cytofix/Cytoperm Plus Fixation/Permeabilization Solution Kit | BD Biosciences | Cat#554714 |
| AllPrep DNA/RNA/Protein Mini Kit | QIAGEN | Cat#80004 |
| GoScript Reverse Transcriptase Kit | Promega | Cat#A5001 |
| SYBR Advantage qPCR Premix | Clontech | Cat#639676 |
| μMACS mRNA Isolation Kit | Miltenyi Biotec | Cat#130–075-201 |
| SMARTer® RACE 5′/3′ Kit (contains NucleoSpin Gel and PCR Clean-Up Kit) | Clontech | Cat#634859 |
| SuperScript II Reverse Transcriptase | Thermo Fisher Scientific | Cat#18064014 |
| RNaseOUT Recombinant Ribonuclease Inhibitor | Thermo Fisher Scientific | Cat#10777019 |
| SYBR Gold Nucleic Acid Gel Stain | Thermo Fisher Scientific | Cat#S-11494 |
| TOPO TA Cloning Kit for Sequencing with One Shot® MAX Efficiency DH5α-T1R E. coli | Thermo Fisher Scientific | Cat#K459540 |
| SYBR® Safe DNA Gel Stain | Thermo Fisher Scientific | Cat#S33102 |
| TrackIt Cyan/Orange Loading Buffer | Thermo Fisher Scientific | Cat#10482–028 |
| GeneRuler 1kb DNA Ladder (ready-to-use) | Thermo Fisher Scientific | Cat#SM0313 |
| PrimeScript Reverse Transcriptase | Clontech | Cat#2680A |
| KAPA HiFi HotStart ReadyMix | Kapa Biosystems | Cat#KK2602 |
| Agilent High Sensitivity DNA Kit | Agilent | Cat#5067–4626 |
| Nextera XT DNA Sample Preparation Kit (96 samples) | Illumina | Cat#FC-131–1096 |
| Nextera XT Index Kit (96 indices, 384 samples) | Illumina | Cat#FC-131–1002 |
| Epitope Retrieval Solution 2 BOND | Leica Biosystems | Cat#AR9640 |
| Bond Polymer Refine Detection | Leica Biosystems | Cat#DS9800 |
| Deposited Data | ||
| RNA-seq from Vδ1+ IELs | GEO Database | GEO: GSE123649 |
| Experimental Models: Cell Lines | ||
| HEK293T (ATCC 293T) | American Type Culture Collection | Cat#CRL-3216 |
| SKW-3 | German Collection of Microorganisms and Cell Cultures | Cat#ACC 53 |
| Oligonucleotides | ||
|
GAPDH Primers: Forward: 5′-ATGGGGAAGGTGAAGGTCG-3′ Reverse: 5′-GGGGTCATTGATGGCAACAATA-3′ |
This paper | N/A |
|
BTNL3 Primers: Forward: 5′-TCAGTTTCTACGAGCTGGTGTC-3′ Reverse: 5′-CCAAGGCCTGGACAAACTT-3′ |
Lebrero-Fernández et al., 2016 | N/A |
|
BTNL8 Primers: Forward: 5′-GCTCTCATGCTCAGTTTGGTT-3′ Reverse: 5′-GTCTGGCCCAAACACCTG-3′ |
Lebrero-Fernández et al., 2016 | N/A |
| 10mM dNTP Mix | Thermo Fisher Scientific | Cat#18427013 |
| OligoDT-5′Biotinylated: 5′-Biot-AAGCAGTGGTATCAACGCAGAGTACT30VN-3′ |
Picelli et al., 2014 | N/A |
| 100mM dATP | Roche | Cat#11934511001 |
| 100mM dCTP | Roche | Cat#11934520001 |
| 100mM dGTP | Roche | Cat#11934538001 |
| 100mM dTTP | Roche | Cat#11934546001 |
| TSO-5′Biotinylated: 5′-Biot-AAGCAGTGGTATCAACGCAGAGTA CATrGrG+G-3′ |
Picelli et al., 2014 | N/A |
| ISPCR-5′Biotinylated: 5′-Biot-AAGCAGTGGTATCAACGCAGAGT-3′ |
Picelli et al., 2014 | N/A |
| Recombinant DNA | ||
| pMIG II (pMSCV-IRES-GFP II) | Holst et al., 2006 | N/A |
| cDNA: BTNL3 (GenBank: NM_197975.2), BTNL8 (GenBank: NM_001040462.2) | Genscript | N/A |
| cDNA: Control 94 and 95, Active 46 and 81, γδTCRs | Integrated DNA Technologies | N/A |
| cDNA: TRDC-P2A-TRGC | Integrated DNA Technologies | N/A |
| Software and Algorithms | ||
| FlowJo (version 10.2) | FlowJo | https://www.flowjo.com/solutions/flowjo/downloads |
| R (version 3.4.0) | R | https://cran.r-project.org |
| RStudio (version 1.0.143) | RStudio | https://www.rstudio.com/products/rstudio/download2/ |
| iceLogo | Colaert et al., 2009 | https://iomics.ugent.be/icelogoserver/ |
| Diva 8 | BD Biosciences | http://www.bdbiosciences.com |
| Sequencher (version 5.2.3) | Gene Codes Corporation | http://www.genecodes.com/sequencher |
| IMGT | IMGT®, the international ImMunoGeneTics information system | http://www.imgt.org |
| PLINK (version 1.9) | PLINK | https://www.cog-genomics.org/plink2 |
| edgeR | Robinson et al., 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| limma | Ritchie et al., 2015 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| Trim Galore (version 0.4.4) | Krueger n.d. | http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| kallisto (version 0.43.0) | Bray et al., 2016 | https://pachterlab.github.io/kallisto/ |
| Other | ||
| Zirconium Oxide Beads 0.5mm RNase Free | Next Advance | SKU#ZROB05-RNA |
| Zirconium Oxide Beads 1.0mm RNase Free | Next Advance | SKU#ZROB10-RNA |
| Corning 96-Well Clear Flat Bottom Polystyrene Not Treated Microplate | Corning | Cat#3370 |
| Hard-Shell 96-Well PCR Plates (thin-wall) | Bio-Rad | Cat#HSP9631 |
| Microseal ‘F’ Foil Seals | Bio-Rad | Cat#MSF1001 |
| RT-LTS-A-10uL-/F-960/10 Tips | Rainin | Cat#30389225 |
| RT-LTS-A-200uL-/F-960/10 Tips | Rainin | Cat#30389239 |
| Invitrogen Magnetic Stand-96 | Thermo Fisher Scientific | Cat#AM10027 |
Highlights.
Innate-like Vδ1+ IELs are superseded by interferon-γ-producing Vδ1+ IELs in CeD
The Vδ1+ IEL TCR repertoire is permanently reshaped in CeD
A molecular signature defines Vδ1+ IEL expansions in active CeD
Loss of BTNL8 expression coincides with permanent loss of BTNL3/8-reactive γδ+ IELs in CeD
ACKNOWLEDGMENTS
We thank the patients and their families for making this study possible; Robert Kavitt, Edwin McDonald, Atsushi Sakuraba, Joel Pekow, Neil Sengupta, and Sushila Dalal for patient care; Kathryn Lesko, Diane McKiernan, Sarbani Adhikari, and Fengshi Dong for patient recruitment; Mathieu Platteel for assistance with HLA genotyping; Steven Erickson and Delphine Guy-Grand for editorial input and thoughtful discussions; and staff at the University of Chicago Flow Cytometry Core Facility. This work was supported by the National Institutes of Health via T32 AI07090 to T.M., T32 GM007281 to D.G.S., R01 DK067180 to B.J., and the Digestive Diseases Research Core Center P30 DK42086 at the University of Chicago to B.J. J.R. is an Australian Research Council Laureate Fellow. D.A.P. is a Welcome Trust Senior Investigator (100326Z/12/Z).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and six tables and can be found with this article online at https://doi.org/10.1016/j.cell.2018.12.039.
A video abstract is available at https://doi.org/10.1016/j.cell.2018.12.039#mmc4.
REFERENCES
- Adams NM, Lau CM, Fan X, Rapp M, Geary CD, Weizman O-E, Diaz-Salazar C, and Sun JC (2018). Transcription factor IRF8 orchestrates the adaptive natural killer cell response. Immunity 48, 1172–1182.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashouri JF, and Weiss A (2017). Endogenous Nur77 is a specific indicator of antigen receptor signaling in human T and B cells. J. Immunol 198, 657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beura LK, Mitchell JS, Thompson EA, Schenkel JM, Mohammed J, Wijeyesinghe S, Fonseca R, Burbach BJ, Hickman HD, Vezys V, et al. (2018). Intravital mucosal imaging of CD8+ resident memory T cells shows tissue-autonomous recall responses that amplify secondary memory. Nat. Immunol 19, 173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Brochet X, Lefranc MP, and Giudicelli V (2008). IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res. 36, W503–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chennupati V, Worbs T, Liu X, Malinarich FH, Schmitz S, Haas JD, Malissen B, Förster R, and Prinz I (2010). Intra- and intercompartmental movement of γδ T cells: intestinal intraepithelial and peripheral γδ T cells represent exclusive nonoverlapping populations with distinct migration characteristics. J. Immunol. 185, 5160–5168. [DOI] [PubMed] [Google Scholar]
- Cheroutre H, Lambolez F, and Mucida D (2011). The light and dark sides of intestinal intraepithelial lymphocytes. Nat. Rev. Immunol 11, 445–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colaert N, Helsens K, Martens L, Vandekerckhove J, and Gevaert K (2009). Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 6, 786–787. [DOI] [PubMed] [Google Scholar]
- Davey MS, Willcox CR, Joyce SP, Ladell K, Kasatskaya SA, McLaren JE, Hunter S, Salim M, Mohammed F, Price DA, et al. (2017). Clonal selection in the human Vδ1 T cell repertoire indicates γδ TCR-dependent adaptive immune surveillance. Nat. Commun. 8, 14760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marco Barros R, Roberts NA, Dart RJ, Vantourout P, Jandke A, Nussbaumer O, Deban L, Cipolat S, Hart R, Iannitto ML, et al. (2016). Epithelia use butyrophilin-like molecules to shape organ-specific γδ T cell compartments. Cell 167, 203–218.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, Haw R, Jassal B, Korninger F, May B, et al. (2018). The Reactome Pathway Knowledgebase. Nucleic Acids Res. 46 (D1), D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari S, and Cribari-Neto F (2004). Beta regression for modeling rates and proportions. J. Appl. Stat 31, 799–815. [Google Scholar]
- Früh K, and Yang Y (1999). Antigen presentation by MHC class I and its regulation by interferon gamma. Curr. Opin. Immunol 11, 76–81. [DOI] [PubMed] [Google Scholar]
- Gras S, Chen Z, Miles JJ, Liu YC, Bell MJ, Sullivan LC, Kjer-Nielsen L, Brennan RM, Burrows JM, Neller MA, et al. (2010). Allelic polymorphism in the T cell receptor and its impact on immune responses. J. Exp. Med 207, 1555–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy-Grand D, Vassalli P, Eberl G, Pereira P, Burlen-Defranoux O, Lemaitre F, Di Santo JP, Freitas AA, Cumano A, and Bandeira A (2013). Origin, trafficking, and intraepithelial fate of gut-tropic T cells. J. Exp. Med 210, 1839–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halstensen TS, Scott H, and Brandtzaeg P (1989). Intraepithelial T cells of the TcR γ/δ+ CD8− and Vδ1/Jδ1+ phenotypes are increased in celiac disease. Scand. J. Immunol 30, 665–672. [DOI] [PubMed] [Google Scholar]
- Han A, Newell EW, Glanville J, Fernandez-Becker N, Khosla C, Chien Y-H, and Davis MM (2013). Dietary gluten triggers concomitant activation of CD4+ and CD8+ αβ T cells and γδ T cells in celiac disease. Proc. Natl. Acad. Sci. USA 110, 13073–13078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayday AC (2000). γδ cells: a right time and a right place for a conserved third way of protection. Annu. Rev. Immunol 18, 975–1026. [DOI] [PubMed] [Google Scholar]
- Hayday A, Theodoridis E, Ramsburg E, and Shires J (2001). Intraepithelial lymphocytes: exploring the Third Way in immunology. Nat. Immunol 2, 997–1003. [DOI] [PubMed] [Google Scholar]
- Holst J, Szymczak-Workman AL, Vignali KM, Burton AR, Workman CJ, and Vignali DAA (2006). Generation of T-cell receptor retrogenic mice. Nat. Protoc 1, 406–417. [DOI] [PubMed] [Google Scholar]
- Hoytema van Konijnenburg DP, Reis BS, Pedicord VA, Farache J, Victora GD, and Mucida D (2017). Intestinal epithelial and intraepithelial T cell crosstalk mediates a dynamic response to infection. Cell 171, 783–794.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husby S, Koletzko S, Korponay-Szabó IR, Mearin ML, Phillips A, Shamir R, Troncone R, Giersiepen K, Branski D, Catassi C, et al. ; ESPGHAN Working Group on Coeliac Disease Diagnosis; ESPGHAN Gastroenterology Committee; European Society for Pediatric Gastroenterology, Hepatology, and Nutrition (2012). European Society for Pediatric Gastroenterology, Hepatology, and Nutrition guidelines for the diagnosis of coeliac disease. J. Pediatr. Gastroenterol. Nutr 54, 136–160. [DOI] [PubMed] [Google Scholar]
- Jabri B, and Abadie V (2015). IL-15 functions as a danger signal to regulate tissue-resident T cells and tissue destruction. Nat. Rev. Immunol 15, 771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabri B, and Sollid LM (2009). Tissue-mediated control of immunopathology in coeliac disease. Nat. Rev. Immunol 9, 858–870. [DOI] [PubMed] [Google Scholar]
- Kano S, Sato K, Morishita Y, Vollstedt S, Kim S, Bishop K, Honda K, Kubo M, and Taniguchi T (2008). The contribution of transcription factor IRF1 to the interferon-gamma-interleukin 12 signaling axis and TH1 versus TH17 differentiation of CD4+ T cells. Nat. Immunol 9, 34–41. [DOI] [PubMed] [Google Scholar]
- Kutlu T, Brousse N, Rambaud C, Le Deist F, Schmitz J, and Cerf-Bensussan N (1993). Numbers of T cell receptor (TCR) αβ+ but not of TcR γδ+ intraepithelial lymphocytes correlate with the grade of villous atrophy in coeliac patients on a long term normal diet. Gut 34, 208–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrero-Fernández C, Wenzel UA, Akeus P, Wang Y, Strid H, Simrén M, Gustavsson B, Börjesson LG, Cardell SL, Öhman L, et al. (2016). Altered expression of butyrophilin (BTN) and BTN-like (BTNL) genes in intestinal inflammation and colon cancer. Immun. Inflamm. Dis 4, 191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefranc MP (2003). IMGT, the international ImMunoGeneTics database. Nucleic Acids Res. 31, 307–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linehan JL, Harrison OJ, Han S-J, Byrd AL, Vujkovic-Cvijin I, Villarino AV, Sen SK, Shaik J, Smelkinson M, Tamoutounour S, et al. (2018). Non-classical immunity controls microbiota impact on skin immunity and tissue repair. Cell 172, 784–796.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddelein D, Colaert N, Buchanan I, Hulstaert N, Gevaert K, and Martens L (2015). The iceLogo web server and SOAP service for determining protein consensus sequences. Nucleic Acids Res. 43 (W1), W543–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melandri D, Zlatareva I, Chaleil RAG, Dart RJ, Chancellor A, Nussbaumer O, Polyakova O, Roberts NA, Wesch D, Kabelitz D, et al. (2018). The γδTCR combines innate immunity with adaptive immunity by utilizing spatially distinct regions for agonist selection and antigen responsiveness. Nat. Immunol 19, 1352–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meresse B, Curran SA, Ciszewski C, Orbelyan G, Setty M, Bhagat G, Lee L, Tretiakova M, Semrad C, Kistner E, et al. (2006). Reprogramming of CTLs into natural killer-like cells in celiac disease. J. Exp. Med 203, 1343–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsuur AJ, de Bakker PIW, Zhernakova A, Pinto D, Verduijn W, Romanos J, Auricchio R, Lopez A, van Heel DA, Crusius JBA, and Wijmenga C (2008). Effective detection of human leukocyte antigen risk alleles in celiac disease using tag single nucleotide polymorphisms. PLoS ONE 3, e2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller SN, and Mackay LK (2016). Tissue-resident memory T cells: local specialists in immune defence. Nat. Rev. Immunol 16, 79–89. [DOI] [PubMed] [Google Scholar]
- Nilsen EM, Lundin KE, Krajci P, Scott H, Sollid LM, and Brandtzaeg P (1995). Gluten specific, HLA-DQ restricted T cells from coeliac mucosa produce cytokines with Th1 or Th0 profile dominated by interferon gamma. Gut 37, 766–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Garra A, and Vieira P (2007). TH1 cells control themselves by producing interleukin-10. Nat. Rev. Immunol 7, 425–428. [DOI] [PubMed] [Google Scholar]
- Park SL, Zaid A, Hor JL, Christo SN, Prier JE, Davies B, Alexandre YO, Gregory JL, Russell TA, Gebhardt T, et al. (2018). Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat. Immunol 19, 183–191. [DOI] [PubMed] [Google Scholar]
- Picelli S, Faridani OR, Björklund AK, Winberg G, Sagasser S, and Sandberg R (2014). Full-length RNA-seq from single cells using Smartseq2. Nat. Protoc 9, 171–181. [DOI] [PubMed] [Google Scholar]
- Price DA, Brenchley JM, Ruff LE, Betts MR, Hill BJ, Roederer M, Koup RA, Migueles SA, Gostick E, Wooldridge L, et al. (2005). Avidity for antigen shapes clonal dominance in CD8+ T cell populations specific for persistent DNA viruses. J. Exp. Med. 202, 1349–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley MF, Almeida JR, Price DA, and Douek DC (2011). Unbiased molecular analysis of T cell receptor expression using template-switch anchored RT-PCR. Curr Protoc Immunol Chapter 10, Unit10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallusto F, Lenig D, Förster R, Lipp M, and Lanzavecchia A (1999). Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401, 708–712. [DOI] [PubMed] [Google Scholar]
- Saraiva M, Christensen JR, Veldhoen M, Murphy TL, Murphy KM, and O’Garra A (2009). Interleukin-10 production by Th1 cells requires interleukin-12-induced STAT4 transcription factor and ERK MAP kinase activation by high antigen dose. Immunity 31, 209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smithson M, and Verkuilen J (2006). A better lemon squeezer? Maximum-likelihood regression with beta-distributed dependent variables. Psychol. Methods 11, 54–71. [DOI] [PubMed] [Google Scholar]
- Sugahara S, Shimizu T, Yoshida Y, Aiba T, Yamagiwa S, Asakura H, and Abo T (1999). Extrathymic derivation of gut lymphocytes in parabiotic mice. Immunology 96, 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trynka G, Hunt KA, Bockett NA, Romanos J, Mistry V, Szperl A, Bakker SF, Bardella MT, Bhaw-Rosun L, Castillejo G, et al. ; Spanish Consortium on the Genetics of Coeliac Disease (CEGEC); PreventCD Study Group; Wellcome Trust Case Control Consortium (WTCCC) (2011). Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat. Genet 43, 1193–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vantourout P, Laing A, Woodward MJ, Zlatareva I, Apolonia L, Jones AW, Snijders AP, Malim MH, and Hayday AC (2018). Heteromeric interactions regulate butyrophilin (BTN) and BTN-like molecules governing γδ T cell biology. Proc. Natl. Acad. Sci. USA 115, 1039–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Godec J, Ben-Aissa K, Cui K, Zhao K, Pucsek AB, Lee YK, Weaver CT, Yagi R, and Lazarevic V (2014). The transcription factors T-bet and Runx are required for the ontogeny of pathogenic interferon-γ-producing T helper 17 cells. Immunity 40, 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Olman V, and Xu D (2002). Clustering gene expression data using a graph-theoretic approach: an application of minimum spanning trees. Bioinformatics 18, 536–545. [DOI] [PubMed] [Google Scholar]
- Zaiss DMW, Gause WC, Osborne LC, and Artis D (2015). Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity 42, 216–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J (2017). GATA3 regulates the development and functions of innate lymphoid cell subsets at multiple stages. Front. Immunol 8, 1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-sequencing data were submitted to the Gene Expression Omnibus under accession code GEO: GSE123649.