

SUMMARY
To ensure a successful infection, herpesviruses have developed elegant strategies to counterbalance the host anti-viral responses. Sterile alpha motif and HD domain 1 (SAMHD1) was recently identified as an intrinsic restriction factor for a variety of viruses. Aside from HIV-2 and the related simian immunodeficiency virus (SIV) Vpx proteins, the direct viral countermeasures against SAMHD1 restriction remain unknown. Using Epstein-Barr virus (EBV) as a primary model, we discover that SAMHD1-mediated anti-viral restriction is antagonized by EBV BGLF4, a member of the conserved viral protein kinases encoded by all herpesviruses. Mechanistically, we find that BGLF4 phosphorylates SAMHD1 and thereby inhibits its deoxynucleotide triphosphate triphosphohydrolase (dNTPase) activity. We further demonstrate that the targeting of SAMHD1 for phosphorylation is a common feature shared by beta- and gamma-herpesviruses. Together, our findings uncover an immune evasion mechanism whereby herpesviruses exploit the phosphorylation of SAMHD1 to thwart host defenses.
Graphical Abstract
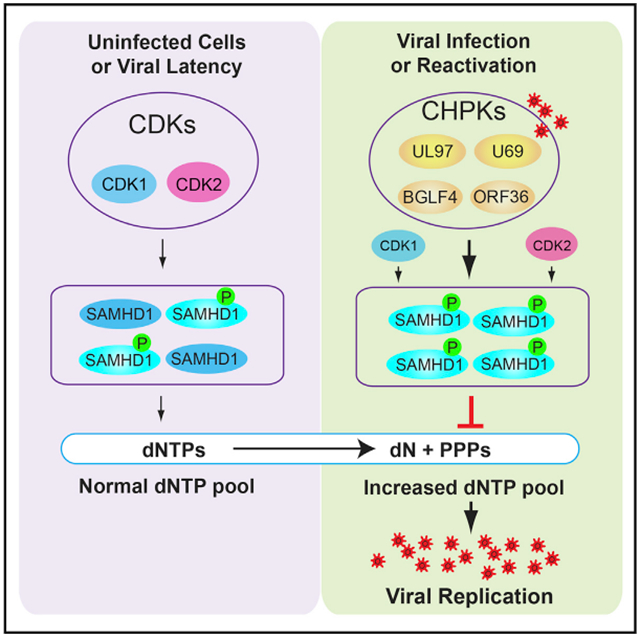
In Brief
Herpesviruses have evolved elegant strategies to dampen the host anti-viral responses. Zhang et al. discover a mechanism by which herpesviruses evade SAMHD1-mediated host defenses through phosphorylation, expanding the functional repertoire of viral protein kinases in herpesvirus biology.
INTRODUCTION
Human herpesviruses are ubiquitous pathogens that establish lifelong persistent infections and are associated with a variety of diseases ranging from cold sores to mononucleosis, birth defects, and cancers. During a long period of co-evolution with their hosts, these viruses have established multiple strategies to combat the cellular defense systems. The eight human herpesviruses discovered to date are classified into three subfamilies, alpha-, beta-, and gamma-herpesviruses. The alpha-herpesviruses consist of herpes simplex 1 and 2 (HSV-1 and HSV-2) and varicella-zoster virus (VZV); the beta-herpesviruses include human cytomegalovirus (HCMV), human herpesvirus 6 (HHV-6), and human herpesvirus 7 (HHV-7); and the gamma-herpesviruses are Epstein-Barr virus (EBV) and Kaposi sarcoma-associated herpesvirus (KSHV) (Gershburg and Pagano, 2008; Li and Hayward, 2013). Although these herpesviruses express a limited number of latency-associated genes, they each encode a large number of functionally conserved lytic genes that are critical for efficient virus replication and spread. Among the conserved lytic gene products are the orthologous serine and threonine protein kinases, namely HSV-1/2 UL13, VZV ORF47, HCMV UL97, HHV-6/7 U69, EBV BGLF4, and KSHV ORF36 (Gershburg and Pagano, 2008; Li et al., 2011). These viral protein kinases are referred to as conserved herpesvirus protein kinases (CHPKs). CHPKs have been shown to interact with and potentially phosphorylate a variety of cellular proteins to benefit viral replication (Calderwood et al., 2007; Li et al., 2015, 2011). Deletion and inhibition of CHPKs block the replication of EBV, HCMV, HSV-1, and murine gamma-herpesvirus 68 (MHV68) (El-Guindy et al., 2014; Feederle et al., 2009; Gershburg et al., 2007; Li et al., 2011; Prichard et al., 1999; Shibaki et al., 2001; Tarakanova et al., 2007; Wolf et al., 2001).
Although CHPKs from beta- and gamma-herpesviruses are structurally similar to the cellular cyclin-dependent kinases 1/2 (CDK1 and CDK2) (Kuny et al., 2010; Romaker et al., 2006), recent studies from our lab and others suggest that these CHPKs have a broader substrate recognition than the cellular ortholog CDKs (Li et al., 2011, 2015; Oberstein et al., 2015; Umaña et al., 2018; Zhu et al., 2009). The mimicry of CDK activity by CHPKs results in the phosphorylation of cell-cycle-related proteins to create a pseudo-S-phase environment suitable for efficient viral DNA replication (Chen et al., 2010; Hume et al., 2008; Iwahori et al., 2009, 2015; Kawaguchi and Kato, 2003; Kudoh et al., 2006; Kuny et al., 2010; Lee et al., 2007). CHPKs actively trigger the host DNA damage response through the TIP60-ATM-H2AX axis but suppress interferon response through inhibiting IRF3 activity (Hwang et al., 2009; Li et al., 2011; Ma et al., 2015; Tarakanova et al., 2007; Wang et al., 2009). CHPKs also phosphorylate lamin A/C, nuclear pore complex, and viral nuclear egress complex to enhance the egress of virus capsids from the nucleus (Chang et al., 2012, 2015; Hamirally et al., 2009; Lee et al., 2008; Li et al., 2015; Oberstein et al., 2015; Sharma et al., 2014, 2015; Sharma and Coen, 2014).
In addition to protein phosphorylation, some CHPKs are responsible for the phosphorylation of the nucleoside analog drugs acyclovir and ganciclovir in virus-infected cells (Gershburg et al., 2004; Meng et al., 2010; Moore et al., 2001; Sullivan et al., 1992). Given the important roles of CHPKs in herpesvirus infection, drugs targeting CHPKs are promising for treating herpesvirus-associated diseases. One protein kinase inhibitor, maribavir, targeting HCMV protein kinase UL97 has entered clinical trials (Prichard, 2009). Inhibitors that can decrease the expression of CHPKs also show promise in blocking virus replication (Sun et al., 2013).
Although significant progress has been made toward CHPK substrate identification, the functional importance of these CHPK substrates in virus replication remains to be determined. One potential CHPK target, sterile alpha motif and HD domain 1 (SAMHD1), was recently identified by our phospho-proteomic screening (Li et al., 2015). SAMHD1 is an anti-viral host restriction factor that limits the infection of HIV-1 (Laguette et al., 2011), HSV-1 (Hollenbaugh et al., 2013; Kim et al., 2013), vaccinia virus (Hollenbaugh et al., 2013), human T cell leukemia virus type 1 (Sze et al., 2013), and hepatitis B virus (HBV) (Chen et al., 2014). Phosphorylation of SAMHD1 by CDK1 and CDK2 plays a critical role in suppressing its lentiviral restriction. Initial studies suggest that the anti-viral restriction function, but not the deoxynucleotide triphosphate triphosphohydrolase (dNTPase) activity, of SAMHD1 is controlled by phosphorylation (Kim et al., 2013; Welbourn et al., 2013; White et al., 2013). However, more recent studies demonstrate that the dNTPase activity is downregulated, but not completely inactivated, by phosphorylation to finely tune the function of SAMHD1 during both viral and cellular DNA replication (Arnold et al., 2015; Badia et al., 2016; Ruiz et al., 2015; Tang et al., 2015; Wittmann et al., 2015; Yan et al., 2015). HIV-2/simian immunodeficiency virus (SIV) virion-associated Vpx accessory proteins target SAMHD1 for rapid proteasome-dependent degradation to ensure efficient transduction of myeloid cells (Hrecka et al., 2011; Schwefel et al., 2014, 2015). For all other viruses restricted by SAMHD1, the viral anti-SAMHD1 restriction strategies remain to be determined.
In this study, we identify SAMHD1 as an anti-EBV restriction factor. Our results show that depletion of SAMHD1 leads to a significant increase of viral DNA replication without affecting viral protein expression. Interestingly, we demonstrate that EBV protein kinase BGLF4 interacts with and phosphorylates SAMHD1 to inhibit its dNTPase activity. More importantly, CHPKs from all three herpesvirus subfamilies interact with SAMHD1, and those from beta- and gamma-herpesviruses phosphorylate SAMHD1 in cells. Together, our results suggest that SAMHD1 is a key regulator for all herpesviruses, and CHPKs are direct viral countermeasures against SAMHD1.
RESULTS
SAMHD1 Is Phosphorylated by the EBV Protein Kinase BGLF4
To better understand the function of EBV protein kinase BGLF4 in the viral life cycle, we recently used a Stable Isotope Labeling by Amino acids in Cell culture (SILAC)-based quantitative mass spectrometry (MS) approach to identify potential targets for EBV BGLF4 (Li et al., 2015). More than 1,000 hyperphosphorylated cellular proteins were identified in BGLF4-expressing cells. Among these proteins, we detected SAMHD1. As shown in Figure 1A, the phosphorylation of SAMHD1 on T592 is significantly increased in BGLF4-expressing cells. To further confirm these results, we monitored the phosphorylation status of SAMHD1 in Akata (EBV+) cells carrying either control or BGLF4-expressing vectors. As expected, the phosphorylation of SAMHD1 was gradually increased with BGLF4 induction by doxycycline (Figure 1B, lanes 4–6). In contrast, the phosphorylation of SAMHD1 was even decreased with the same treatment in the vector control cells (Figure 1B, lanes 1–3). Our previous study demonstrated that EBV protein kinase BGLF4 is a Small Ubiquitin-like Modifier (SUMO) binding protein. The SUMO binding-deficient mutant BGLF4 could not trigger the DNA damage response, although the BGLF4 kinase activity was not affected in vitro (Li et al., 2012). Here we also tested the function of this SUMO binding-deficient (m-SIM-N) mutant together with kinase-dead mutant (KD) in SAMHD1 phosphorylation. Interestingly, we found that the SUMO binding-deficient BGLF4 also triggered the phosphorylation of SAMHD1, whereas the expression of BGLF4-KD mutant reduced the phospho-SAMHD1 level (Figure 1B, lanes 7–9 versus lanes 10–12). These results suggested that the EBV protein kinase BGLF4 triggered the phosphorylation of SAMHD1 in a kinase activity-dependent manner and the SUMO-binding function of BGLF4 was dispensable for SAMHD1 phosphorylation. The basal SAMHD1 phosphorylation levels in the four cell lines correlated with the cellular CDK2 expression, suggesting that CDK2 might be a major kinase responsible for SAMHD1 phosphorylation (Figure 1B, lanes 1, 4, 7, and 10). In the BGLF4-KD cells, we found that the CDK1/CDK2 levels remained constant, but dephosphorylation of SAMHD1 was observed at 48 h (Figure 1B, lane 12). One possibility is that BGLF4-KD mutant may compete with CDK1/CDK2 for binding to SAMHD1, which in turn inhibits SAMHD1 phosphorylation by these cellular kinases. By using phos-tag SDS-PAGE to separate phospho- and non-phospho-proteins (Kinoshita et al., 2006), we found that nearly all SAMHD1 is converted to the phosphorylated form in BGLF4-expressing cells (Figure 1C).
Figure 1. SAMHD1 Regulation by EBV Protein Kinase BGLF4.
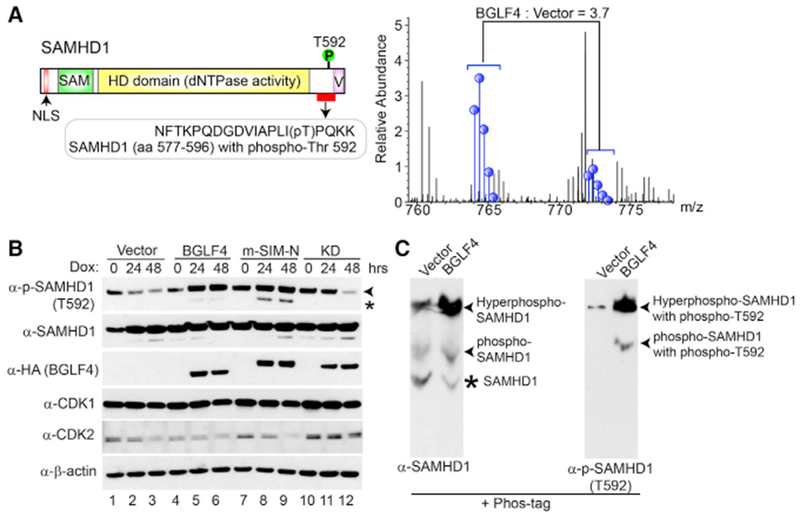
(A) SAMHD1 is phosphorylated upon BGLF4 induction. Schematic representation of SAMHD1 protein is presented in the top panel with the relative positions for each functional domain as indicated. SILAC-based quantitative proteomic analyses revealed that the SAMHD1 phosphorylation on T592 is increased upon BGLF4 induction for 48 h.
(B) Phosphorylation of SAMHD1 is induced by wild-type and SUMO binding-deficient mutant BGLF4. Western blot analysis was performed on cell lysates from Akata (EBV+) cells carrying control vector (Vector), wild-type, SUMO binding-deficient (m-SIM-N), or kinase-dead mutant (KD) BGLF4 using antibodies as indicated. The cells were either untreated (0 h) or treated with doxycycline (Dox) for 24 or 48 h to induce BGLF4 expression. Arrowhead denotes the major 72-kDa phospho-SAMHD1 band, and asterisk denotes a 69-kDa non-specific band or a phospho-SAMHD1 band derived from a SAMHD1 isoform protein.
(C) BGLF4 induces hyperphosphorylation of SAMHD1. Akata (EBV+) cells carrying control vector (Vector) or wild-type BGLF4 were treated with doxycycline for 48 h. Cell extracts were separated using Phos-tag acrylamide gel and then analyzed with western blot using antibodies against SAMHD1 (left) and phopho-SAMHD1 (T592) (right). Different forms of SAMHD1 and phospho-SAMHD1 are indicated.
HD, histidine-aspartic domain; NLS, nuclear localization signal peptide; SAM, sterile alpha motif; V, Vpx interacting domain. See also Figure S1.
To further test whether BGLF4-triggered phosphorylation of SAMHD1 is physiologically relevant during EBV lytic replication, we monitored the phosphorylation of SAMHD1 in lytically induced Akata-BX1 (EBV+) B cells. As a control, the same immunoglobulin G (IgG) cross-linking treatment was also applied to Akata-4E3 (EBV−) B cells. Consistent with the results from BGLF4-expressing cells (Figure 1B), we observed a time-dependent increase of phospho-SAMHD1 during the course of EBV lytic reactivation, which correlates nicely with the BGLF4 expression level (Figure 2A, lanes 1–4). In contrast, the phospho-SAMHD1 level gradually decreased in Akata-4E3 (EBV−) B cells following the anti-IgG treatment (Figure 2A, lanes 5–8). In addition to the major phospho-SAMHD1 band (72 kDa) (Figure 2A, arrowhead), we also noticed a 69-kDa band (Figure 2A, asterisk). This faster-migrating band was possibly derived from another isoform of SAMHD1 (Shi et al., 2014; Welbourn et al., 2012). In addition to B cells, we also monitored the phosphorylation of SAMHD1 HeLa-(EBV+) cells (Feng et al., 2007) following transfection of EBV BamHI-Z transactivator (ZTA) and BamHI-R transactivator (RTA) to induce EBV lytic replication. Interestingly, we found that the transfection of both ZTA and RTA triggers BGLF4 expression and SAMHD1 phosphorylation (Figure 2B, lane 3). Together, these results suggested that the phosphorylation of SAMHD1 is mediated by EBV protein kinase BGLF4.
Figure 2. SAMHD1 Is Phosphorylated by EBV Protein Kinase BGLF4.
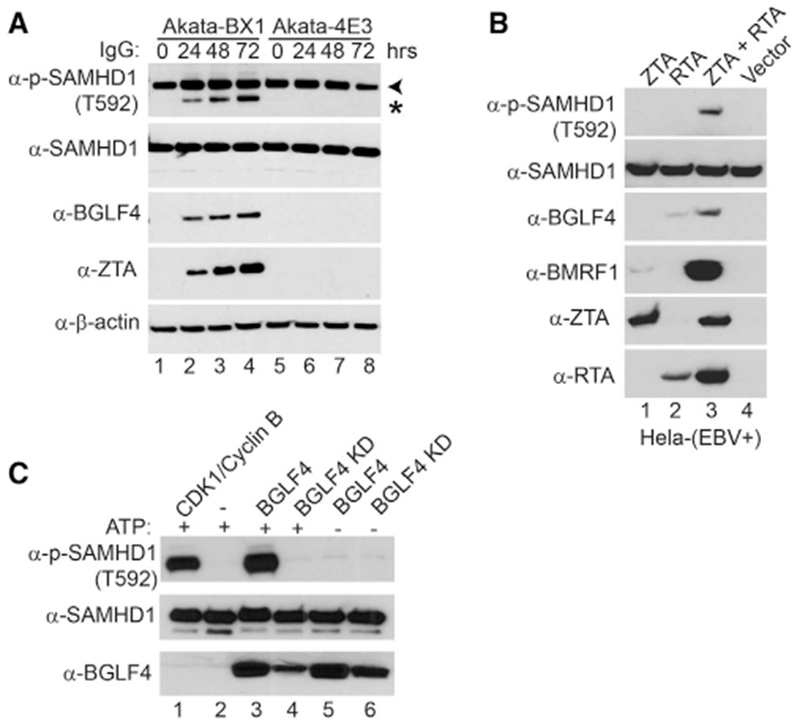
(A) SAMHD1 is phosphorylated upon lytic induction of EBV. Western blot analysis was performed on cell lysates from Akata-BX1 (EBV+) and Akata 4E3 (EBV−) cells using antibodies as indicated. The cells were either untreated (0 hr) or treated with anti-human IgG for 24, 48, or 72 h to induce lytic reactivation. Arrowhead denotes the major 72-kDa phospho-SAMHD1 band, and asterisk denotes a 69-kDa non-specific band or a phospho-SAMHD1 band derived from a SAMHD1 isoform protein.
(B) SAMHD1 is phosphorylated in EBV-replicating HeLa cells. Western blot analysis was performed on cell lysates from HeLa (EBV+) transfected with EBV ZTA, RTA, or ZTA plus RTA as indicated for 48 h to induce lytic reactivation. The phosphorylation of SAMHD1 correlated with BGLF4 expression.
(C) SAMHD1 is phosphorylated by EBV protein kinase BGLF4 in vitro. Recombinant SAMHD1 protein was mixed with purified wild-type or KD BGLF4 for 30 min at 30°C. As a positive control, SAMHD1 was incubated with CDK1/Cyclin B (lane 1), which is a known kinase for SAMHD1. As negative controls, either kinase or ATP was omitted in the reaction mixture (lanes 2, 5, and 6). The phospo-SAMHD1, SAMHD1, and BGLF4 were detected by western blot using antibodies as indicated.
See also Figure S1.
To unambiguously prove SAMHD1 as a direct substrate for EBV protein kinase BGLF4, we performed an in vitro kinase assay. We found that the wild-type EBV protein kinase BGLF4, but not the KD mutant, phosphorylates on SAMHD1 T592, which is similar to the phosphorylation induced by the kinase complex CDK1/Cyclin B (Welbourn et al., 2013) (Figure 2C, lanes 1 and 3 versus 4). In contrast, we did not detect the phosphorylation signal in reactions lacking BGLF4 or ATP (Figure 2B, lanes 2, 5, and 6). Taken together, our data suggested that EBV protein kinase BGLF4 phosphorylates SAMHD1 in vivo and in vitro.
SAMHD1 Restricts EBV Replication
Because SAMHD1 plays a critical role in restricting HIV and HSV replication, and the phosphorylation on T592 can block it anti-viral restriction function, we hypothesized that SAMHD1 is also a restriction factor for EBV. To test our hypothesis, we examined whether SAMHD1 expression affected viral protein expression. Akata-BX1 (EBV+) cells were infected with individual single-guide RNA (sgRNA) lentivirus to deplete SAMHD1 by CRISPR/Cas9-mediated genome editing. The depletion efficiency of SAMHD1 was high for both sgRNAs compared with control (Figure 3B, lanes 1–6 versus lanes 7–9). We measured accumulation of viral proteins during the course of EBV lytic reactivation. The levels of viral proteins in SAMHD1-depleted cells were similar to those seen in control cells, suggesting that SAMHD1 did not significantly affect the expression of viral proteins (Figure 3B, ZTA, BGLF4, and p18 blots). To test whether SAMHD1 plays a role in EBV replication, we measured the infectious EBV particles following lytic induction by IgG cross-linking. Interestingly, we found that the relative EBV titers were significantly higher in SAMHD1-knockout cells than those in the control cells at 48 h post-lytic induction (Figure 3B, lower panel, lanes 3 and 6 versus 9). In the EBV-replicating Akata cells, the cellular CDK1 protein level, but not CDK2, was dramatically reduced upon anti-IgG treatment (Figure 3B, CDK1 blot), suggesting that SAMHD1 phosphorylation was not mediated by CDK1 but mainly mediated by the EBV protein kinase BGLF4 and/or CDK2 (Figure 3B, phospho-SAMHD1 and BGLF4 blots, lanes 7–9).
Figure 3. SAMHD1 Depletion Facilitates EBV Lytic Replication.
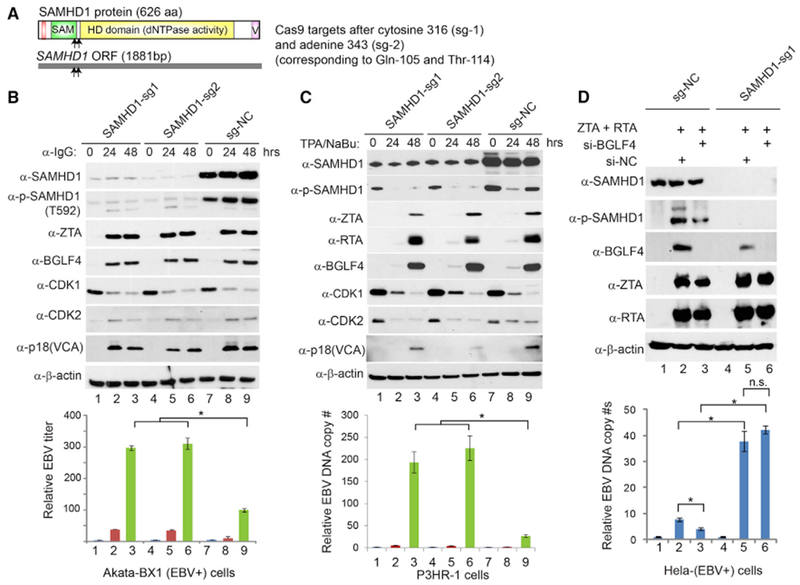
(A) Schematic representation of Cas9 target sites within the 1,881-bp SAMHD1 open read frame (ORF). The corresponding positions of amino acids are also shown.
(B) SAMHD1 depletion facilitates EBV replication in Akata cells. SAMHD1-depleted (sg-1 and sg-2) and control (sg-NC) Akata-BX1 (EBV+) B cells were treated with IgG cross-linking to induce EBV lytic reactivation. Western blot analysis was performed using antibodies as indicated. β-Actin served as a loading control. Relative EBV titer of lytically induced Akata-BX1 (EBV+) cells carrying SAMHD1-deletion or control was measured using a Raji cell infection assay.
(C) SAMHD1 depletion facilitates EBV replication in P3HR-1 cells. SAMHD1-depleted (sg-1 and sg-2) and control (sg-NC) P3HR-1 cells were treated with TPA and sodium butyrate (NaBu) to induce EBV lytic reactivation. Western blots were performed using antibodies as indicated. β-Actin served as a loading control. Relative EBV DNA copy numbers were determined by qPCR using primers to BALF5 gene normalized by β-actin.
(D) BGLF4 knockdown suppresses EBV replication in SAMHD1-expressing cells, but not in SAMHD1-depleted cells. Control (sg-NC) and SAMHD1-depleted (sg-1) HeLa (EBV+) cells were transfected with ZTA plus RTA to induce EBV lytic reactivation. The siBGLF4-expressing plasmid was co-transfected to knock down BGLF4. The transfection of si-NC served as a negative control for siBGLF4. The EBV genome copy numbers were measured by qPCR using primers specific to EBV BALF5 gene normalized by β-actin.
Representative results from three biological replicates are presented. Data are represented as mean ± SD of technical replicates (n = 3). *p < 0.05. n.s., not significant. See also Figures S1–S4.
To further demonstrate SAMHD1-mediated restriction toward EBV replication, we depleted SAMHD1 by a similar CRISPR/Cas9 method in the parental Akata (EBV+) and the P3HR-1 cells. Similarly, knocking down SAMHD1 promoted EBV DNA replication in the parental Akata (EBV+) cells (Figure S2). In P3HR-1 cells, we also observed that SAMHD1 depletion significantly enhanced EBV DNA replication with minimal effects on viral protein expression following 12-O-tetradecanoylphorbol-13-acetate or phorbol-12-myristate-13-acetate (TPA) and sodium butyrate treatment (Figure 3C, lanes 3 and 6 versus 9), reinforcing that SAMHD1 plays a key role in restricting EBV replication. Similar to SAMHD1 dephosphorylation observed in THP-1 cells differentiated by TPA treatment (Cribier et al., 2013; Kim et al., 2013; Welbourn et al., 2013), we also noticed an initial SAMHD1 de-phosphorylation upon TPA and sodium butyrate treatment of P3HR-1 cells (Figure 3C, phospho-SAMHD1 blot, lane 7 versus 8). However, SAMHD1 phosphorylation re-emerged at 48 h post-treatment when EBV protein kinase BGLF4 was highly expressed, whereas cellular CDK1 and CDK2 levels were significantly reduced (Figure 3C, phospho-SAMHD1 blot, lane 9), further suggesting BGLF4 may be responsible for the re-occurrence of SAMHD1 phosphorylation. To determine the cellular kinases responsible for basal level SAMHD1 phosphorylation in these EBV-positive Akata and P3HR-1 cells, we treated the cells with increasing amounts of CDK1 or CDK2 inhibitors for 48 h. We found that CDK2 and, to a lesser extent, CDK1 inhibition suppressed SAMHD1 phosphorylation in a dose-dependent manner (Figure S1), suggesting that both CDK1 and CDK2 contribute to SAMHD1 phosphorylation while CDK2 is the major kinase.
To further evaluate the function of SAMHD1 and its regulation by EBV BGLF4 in EBV lytic replication, we created a SAMHD1 depletion cell line using HeLa-(EBV+) cells. The advantage of using HeLa-(EBV+) cells is that EBV lytic replication can be triggered by ZTA and RTA transfection, and that BGFL4 can be easily knocked down by co-transfection of a plasmid encoding siBGLF4 (Gershburg et al., 2007) to evaluate the role of BGLF4-SAMHD1 axis in EBV replication. As expected, ZTA/RTA transfection triggered EBV lytic replication and SAMHD1 phosphorylation, whereas BGLF4 knocking down led to reduced SAMHD1 phosphorylation and EBV replication (Figure 3D, lanes 2 and 3). SAMHD1 depletion significantly enhanced EBV replication and, interestingly, BGLF4 knocking down did not affect viral replication in SAMHD1-depleted cells (Figure 3D, lanes 2 and 3), suggesting that BGLF4 promotes EBV DNA replication mainly through inhibiting SAMHD1. To corroborate these findings, we depleted SAMHD1 using a HEK293 cell line carrying BGLF4/BGLF5-knockout EBV (ΔBGLF5/BGLF5) genomes. To generate a BGLF4-knockout background, we co-transfected the control and SAMHD1-depleted cells with both ZTA and BGLF5 to induce the lytic cycle. Consistent with the results obtained above, the depletion of SAMHD1 significantly promoted EBV DNA replication even in the absence of BGLF4 (Figure S3, lane 2 versus 4). Although we did not observe significant changes on the expression of EBV late gene product p18 in SAMHD1-depleted Akata and P3HR-1 cells (Figure 3; Figure S2), we did notice a higher p18 level when SAMHD1 is ablated (Figure S3, p18 blot, lane 2 versus 4). The results obtained in HEK293 cells are consistent with most of the publications on viral DNA replication and late gene expression using similar HEK293 cells (Djavadian et al., 2016, 2018; McKenzie et al., 2016). These results suggest that the higher EBV DNA level may contribute to late gene expression in a cell-type-dependent manner, or the viral DNA level in lytically induced control B cells is high enough for late gene expression.
Phospho-Mimicking SAMHD1 Promotes whereas Phospho-Deficient SAMHD1 Blocks EBV Lytic Replication
To further prove BGLF4-mediated SAMHD1 phosphorylation is important for EBV lytic replication, we created a phospho-mimicking (T592D) and a phospho-deficient (T592A) SAMHD1. We then used the SAMHD1-depleted Akaka (EBV+) cells to reconstitute wild-type and the mutant SAMHD1. During the course of EBV lytic reactivation, the levels of viral proteins were similar among these cell lines, suggesting that SAMHD1 and its phosphorylation status did not affect the expression of viral proteins (Figure 4, upper panel). As expected, the reconstitution of wild-type SAMHD1 suppressed EBV DNA replication (Figure 4, lower panel, lane 3 versus 6). Interestingly, the reconstitution of phospho-mimicking SAMHD1 (T592D) promoted EBV DNA replication compared with cells carrying the vector control or wild-type (WT) SAMHD1 (Figure 4, lower panel, lane 9 versus 3 and 6). The higher DNA replication in phospho-mimicking (T592D) cells compared with the SAMHD1-depleted cells suggested that phosphorylation of SAMHD1 on T592 may even promote DNA repair and/or fork progression to promote viral DNA replication (Coquel et al., 2018; Daddacha et al., 2017). In contrast, the phospho-deficient SAMHD1 (T592A) blocks EBV DNA replication compared with cells carrying WT SAMHD1 or T592D mutant (Figure 4, lower panel, lane 12 versus 6 and 9). Although SAMHD1 has been implicated in cell-cycle regulation (Bonifati et al., 2016), the depletion or overexpression of SAMHD1 had minimal effect on the cell numbers at 48 h post-lytic induction (Figure S4). Together, these results indicated that SAMHD1 phosphorylation by a viral kinase plays a critical role in promoting EBV DNA replication.
Figure 4. SAMHD1 Reconstitution Suppresses EBV Lytic Replication.
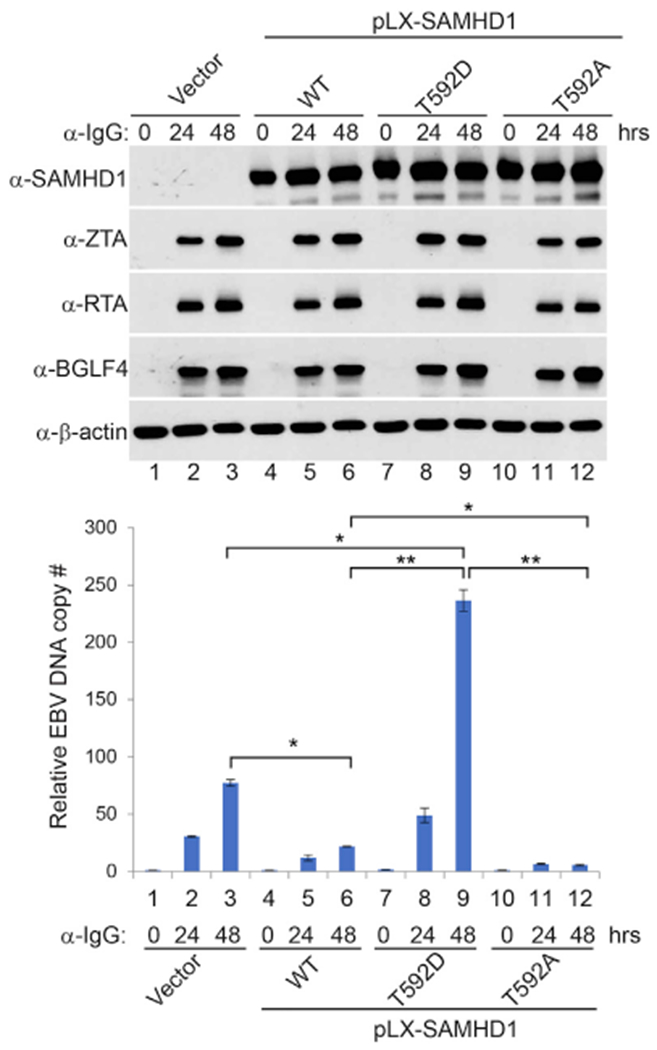
SAMHD1-depleted (sg1) Akata (EBV+) cells were used to establish SAMHD1-expressing stable cell lines using pLX-304 lentiviral constructs containing wild-type (WT), T592D, and T592A SAMHD1. Western blot analyses show SAMHD1, ZTA, RTA, and BGLF4 expression levels in different cell lines upon IgG cross-linking as indicated. Viral DNA replication was measured by qPCR using primersto BALF5. Representative results from three biological replicates are presented. The value of Vector control at 0 h (lane 1) was set as 1. Data are represented as mean ± SD of technical replicates (n = 3). *p < 0.05; **p < 0.001. See also Figure S4.
EBV BGLF4 Inhibits SAMHD1 dNTPase Activity through Phosphorylation
We hypothesized that EBV protein kinase BGLF4 can alleviate the anti-viral activity of SAMHD1 through phosphorylation. To get mechanistic insights into how BGLF4 regulates SAMHD1, we measured the dNTPase activity of SAMHD1 and phospho-SAMHD1 triggered by BGLF4. SAMHD1 was first incubated with BGLF4, BGLF4-KD, CDK1/Cyclin B, or buffer control; then SAMHD1 and phospho-SAMHD1 were activated by incubating with GTP and a mixture of all deoxyribonucleotide triphosphate (dNTP) at equal concentration. Subsequently, each individual dNTP was added to the activated SAMHD1 mixture for testing SAMHD1’s dNTPase activity. Interestingly, we found that although the tetramerization of SAMHD1 was not affected by phosphorylation (Figure S5), phospho-SAMHD1 triggered by BGLF4 and CDK1/Cyclin B shows reduced deoxycytidine triphosphate triphosphohydrolase (dCTPase) and thymidine triphosphate triphosphohydrolase (dTTPase) activity, whereas the dATPase and dGTPase activity was less affected (Figure 5). The results suggested that EBV BGLF4 promotes viral replication through selectively inhibiting SAMHD1’s dNTPase activity.
Figure 5. BGLF4 Suppresses the dCTPase and dTTPase Activity of SAMHD1 through Phosphorylation.
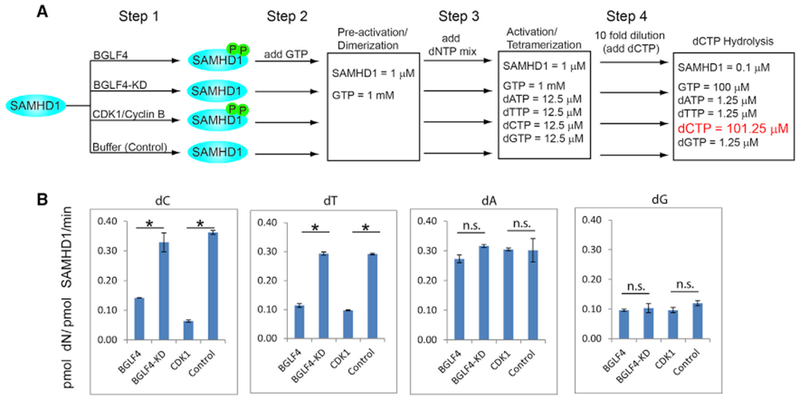
(A) Schematic representation of SAMHD1 phosphorylation, activation, and dCTP hydrolysis analysis. Step 1: recombinant SAMHD1 was phosphorylated by BGLF4 or CDK1 in vitro. Non-phosphorylated SAMHD1 was similarly processed by incubating with BGLF4-KD or buffer control. Step 2: the phosphorylated and non-phosphorylated SAMHD1 were converted to GTP-bound dimer. Step 3: SAMHD1 dimer was further activated by adding dNTP mix for 60s. Step 4: the mixture was diluted 10-fold and the substrate dCTP is added for 0–240 min. For analysis of other dNTP hydrolysis, 100 μM dTTP, dATP, or dGTP was added in step 4.
(B) The SAMHD1 dNTPase activity (dN generation rate, pmol dN/pmol-SAMHD1/min) was analyzed by HPLC usinga Synergi C18 column 150 × 4.6 mm. Dataare represented as mean ± SEM of two independent biological replicates. *p < 0.05. n.s., not significant.
See also Figure S5.
Common Targeting of SAMHD1 by the CHPKs
The eight human herpesviruses diverged around 100–200 million years ago, but they each encode a CHPK(McGeoch et al., 1995). Because the kinase domain of EBV BGLF4 shares high sequence homology with other CHPKs (Figure 6A), we reasoned that the targeting of SAMHD1 might be a common feature for all CHPKs. To test this possibility, we transfected seven CHPKs representative of the alpha-, beta-, and gamma-herpesvirus subfamilies into 293T cells and examined the phosphorylation status of endogenous SAMHD1 in individual CHPK-transfected cells. Interestingly, we found that WT CHPKs from all beta- and gamma-herpesviruses (HHV-6/7 U69, HCMV UL97, EBV BGLF4, and KSHV ORF36) induced a strong phosphorylation of SAMHD1 compared with their corresponding KD mutants and non-transfection controls [Figure 6B, p-SAMHD1 (T592) blot, lanes 3, 5, 7, 9, and 11 versus 4, 6, 8, 10, 12, and 13]. In contrast, the alpha-herpesvirus CHPKs (HSV-1 UL13 and VZV ORF47) were unable to induce SAMHD1 phosphorylation on T592 (Figure 6B, lanes 1 and 2). The total SAMHD1 protein level was not affected by all CHPKs, suggesting that the CHPK-induced increase of SAMHD1 phosphorylation was independent of its steady-state level. We did not notice a correlation between CHPKs expression level and their ability to phosphorylate SAMHD1. For example, CHPKs with high expression levels either did (HHV-6 U69, HCMV UL97, EBV BGLF4, and KSHV ORF36) or did not (VZV ORF47) trigger the phosphorylation of SAMHD1. CHPKs with low expression levels either did (HHV-7 U69) or did not (HSV-1 UL13) trigger the phosphorylation of SAMHD1 (Figure 6B). These results suggested that beta- and gamma-herpesvirus CHPKs, but not the alpha-herpesvirus CHPKs, phosphorylate the T592 residue of SAMHD1.
Figure 6. Conserved Herpesvirus Protein Kinases Target SAMHD1.
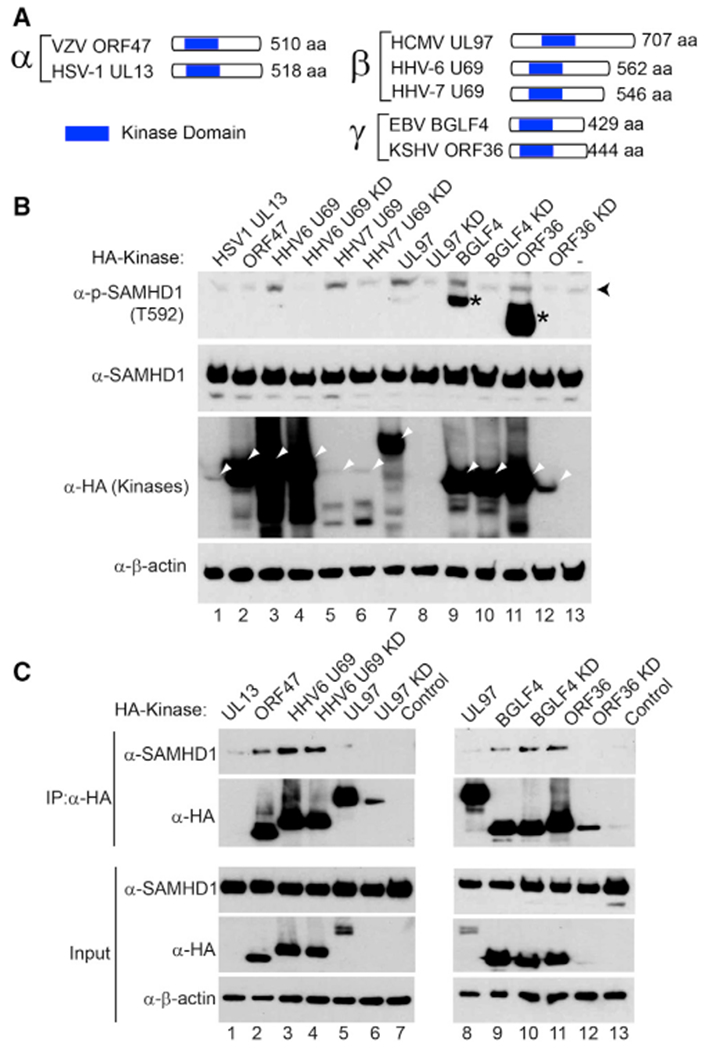
(A) Schematic representation of the conserved herpesvirus protein kinases (CHPKs) from alpha-, beta-, and gamma-herpesviruses. The relative position of the conserved kinase domain was shown.
(B) Beta- and gamma-herpesvirus CHPKs induce the phosphorylation of SAMHD1. Western blot analysis of cell extracts showing that the phosphorylation of SAMHD1 on T592 was induced by overexpression of WT HHV-6/7 U69, HCMV UL97, EBV BGLF4, and KSHV ORF36. The 293T cells were transfected with individual CHPKs and KD mutants, and the cells were harvested 48 h post-transfection. Black arrowhead denotes the major 72-kDa phospho-SAMHD1 band, and asterisks denote non-specific bands or phospho-SAMHD1 bands derived from a SAMHD1 isoform protein. The positions of CHPKs were indicated by white arrowheads. β-Actin served as a loading control.
(C) alpha-, beta-, and gamma-herpesvirus CHPKs interact with SAMHD1. Western blot analysis showing co-immunoprecipitation (coIP) of SAMHD1 with alpha-, beta-, and gamma-herpesvirus CHPKs using 293T cells transfected with SAMHD1 and CHPKs. Input, 2% whole-cell lysate used for immunoprecipitation (IP).
See also Figure S6.
To test whether CHPKs interact with SAMHD1 in cells, we transfected CHPKs into 293T cells and then performed co-immunoprecipitation experiments. We found that VZV ORF47, HHV6 UL69, EBV BGLF4, and KSHV ORF36 all interacted with SAMHD1 (Figure 6C, lanes 2, 3, 5, 8, 9, and 11). KD HHV-6 U69 and EBV BGLF4 preserved the interactions with SAMHD1 (Figure 6C, lanes 4 and 10), suggesting that the kinase activity was not required for the interaction. We detected weak or no interactions for SAMHD1 with HSV1 UL13, HCMV UL97/UL97 KD mutant, and KSHV ORF36 KD mutant, possibly because of their low expression levels compared with other viral kinases (Figure 6C, lanes 6 and 12).
To further test whether other CHPKs directly phosphorylate SAMHD1, we performed in vitro kinase assays using purified CHPKs from HCMV and HSV-1. We found that the WT HCMV protein kinase UL97, but not the KD mutant, triggered a strong phosphorylation of SAMHD1 on T592, similar to that induced by EBV BGLF4 and CDK1/Cyclin B (Figure 7A, lanes 1, 2, and 8). In contrast, we did not detect the phosphorylation signal in reactions containing WT HSV-1 UL13 in vitro (Figure 7A, lane 4). To test whether UL97-induced phosphorylation of SAMHD1 is physiologically relevant during HCMV infection, we monitored the phosphorylation of SAMHD1 in HCMV-infected cells. As shown in Figure 7B, HCMV infection also triggered the phosphorylation of SAMHD1 on T592. In addition, we also confirm the previous observation that HCMV infection led to an increase in CDK1 protein level (Gill et al., 2012). These results suggest that HCMV infection may trigger the phosphorylation of SAMHD1 by both the viral protein kinase UL97 and the cellular CDK1.
Figure 7. SAMHD1 Is Phosphorylated by HCMV Protein Kinase In Vitro and upon HCMV Infection in Cells.
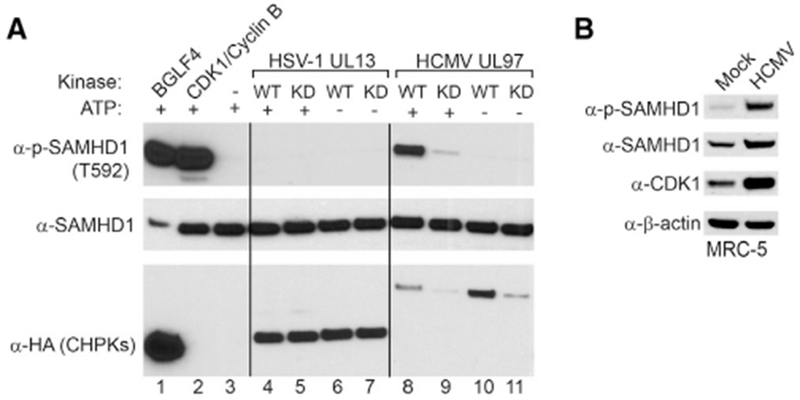
(A) SAMHD1 is phosphorylated by HCMV protein kinase UL97. Recombinant SAMHD1 protein was mixed with purified WT or KD CHPKs as indicated for 30 min at 30°C. As positive controls, SAMHD1 was incubated with BGLF4 or CDK1/Cyclin B. As negative controls, either CHPKs or ATP was omitted in the reaction mixture. The phospo-SAMHD1, SAMHD1, and CHPKs were detected by western blot using antibodies as indicated.
(B) SAMHD1 is phosphorylated upon HCMV infection. Western blot analysis was performed on cell lysates from MRC-5 cells using antibodies as indicated. The cells were either mock infected or infected by HCMV (MOI = 1) for 72 h.
See also Figure S6.
Taken together, our results suggested that antagonizing SAMHD1-mediated anti-viral activity by phosphorylation is evolutionary conserved for beta- and gamma-herpesviruses.
DISCUSSION
The alpha-, beta-, and gamma-herpesviruses have co-evolved with human over long periods of time (McGeoch et al., 1995). Despite the numerous cellular defense mechanisms to prevent viral replication, herpesviruses are able to replicate efficiently. Not only have the viruses evolved ways to dampen the immune responses, they also have elegant strategies to counteract host restriction factors.
SAMHD1, originally identified as a human homolog of mouse interferon-γ-induced protein (Li et al., 2000), is a restriction factor for a variety of viruses including HIV-1, HSV-1, vaccinia virus, and HBV (Chen et al., 2014; Hollenbaugh et al., 2013; Kim et al., 2013; Laguette et al., 2011). SAMHD1 also plays important roles in DNA repair and the degradation of nascent DNA at stalled replication forks (Coquel et al., 2018; Daddacha et al., 2017). We have shown that SAMHD1 depletion leads to a significant increase in viral replication for EBV (Figure 3; Figure S2). We have demonstrated that most CHPKs counteract the restriction of SAMHD1 by phosphorylation, indicating that beta- and gamma-herpesviruses have evolved within their conserved kinase an anti-SAMHD1 mechanism to foster viral DNA replication.
In contrast with HIV-2 and the related simian immunodeficiency viruses, which encode Vpx proteins to antagonize SAMHD1 through ubiquitin-proteasome-dependent degradation (Hrecka et al., 2011; Lenzi et al., 2015), the beta- and gamma-herpesviruses utilize CHPKs to phosphorylate SAMHD1 on T592 without affecting its protein level (Figures 1, 2, and 6). SAMHD1 phosphorylation by HCMV protein kinase UL97 was also confirmed by the Weitzman group (Kim et al., 2019). The phosphorylation of SAMHD1 on T592 plays an important role in blocking the restriction of SAMHD1 toward HIV-1 (Arnold et al., 2015; Cribier et al., 2013; Tang et al., 2015; White et al., 2013; Yan et al., 2015). In a transfection-based overexpression system, it appears that the dNTPase activity is not regulated by phospho-mimicking SAMHD1 mutants (Kim et al., 2013; White et al., 2013). However, more recent studies support that the phosphorylation of SAMHD1 on T592 could lead to the inhibition of its dNTPase activity in low dNTP level conditions (Arnold et al., 2015; Badia et al., 2016; Ruiz et al., 2015; Tang et al., 2015; Yan et al., 2015). Similar to cellular CDK1/Cyclin B (Jang et al., 2016), we show evidence that EBV protein kinase BGLF4 phosphorylates SAMHD1 and inhibits its dCTPase and dTTPase activity in vitro (Figure 5). During herpesvirus lytic reactivation, the massive viral DNA replication may require a large amount of dNTPs. Therefore, the CHPK-induced phosphorylation of SAMHD1 would ensure the dNTP supply for viral DNA replication.
Because T592 of SAMHD1 is the target site of cellular CDK1 and CDK2, our results further support the previous observation that CDK-like activity is shared by beta- and gamma-herpesvirus CHPKs (Kuny et al., 2010). Although only CHPKs from beta- and gamma-herpesviruses triggered the phosphorylation of SAMHD1 on T592, the interaction of alpha-herpesvirus CHPKs with SAMHD1 suggested that these kinases may also regulate SAMHD1 activity without affecting T592 phosphorylation level or with phosphorylation on different sites (Figure 6). Alternatively, the alpha-herpesvirus CHPKs may coordinate with a second protein kinase (US3 from HSV1/2 and ORF66 from VZV) (Erazo and Kinchington, 2010; Kato et al., 2006) within this subfamily to induce the phosphorylation of SAMHD1.
In summary, our data suggest that SAMHD1 is a restriction factor for EBV, and SAMHD1 is antagonized by CHPKs (Figure S6). Our findings about the mechanistic linkage between CHPKs and SAMHD1 have wide-ranging implications, especially in the discovery of viral countermeasures for SAMHD1. Furthermore, the broad anti-viral activity of SAMHD1 toward the infection of HIV, vaccinia virus, HBV, and herpesviruses suggests that SAMHD1 might control the replication of other viruses that require dNTPs to complete their life cycles, and these viruses may also have evolved with similar or different anti-SAMHD1 mechanisms.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Renfeng Li (rli@vcu.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines and Cultures
All Akata-derived cells and P3HR-1 cells were grown in RPMI 1640 media supplemented with 10% FBS (Cat# 26140079, Thermo Fisher Scientific) in 5% CO2 at 37°C (Li et al., 2011, 2012, 2015; Lv et al., 2018; Zhang et al., 2017). HEK293 cells carrying BGLF4/BGLF5-knockout B95.8 EBV genome, 293T cells and HeLa-(EBV+) cells were grown in DMEM media supplemented with 10% FBS in 5% CO2 at 37°C. Human MRC-5 fibroblasts were obtained from ATCC and propagated in high-glucose DMEM (GIBCO BRL) supplemented with 10% FBS, 10,000 IU/liter penicillin, and 10 mg/liter streptomycin (GIBCO-BRL).
METHOD DETAILS
Plasmids Construction
Halo-BGLF4 (WT), Halo-BGLF4 (KD), Halo-UL97 (WT), Halo-UL97 (KD), Halo-UL13 (WT) and Halo-UL13 (KD) were constructed by cloning WT and KD kinases into pHTN HaloTag CMV-neo Vector (Cat# G7721, Promega) (Li et al., 2015). The full-length SAMHD1 was cloned into a pGEX-5x-2 vector (pGEX-5x-2-SAMHD1) with an N-terminal GST tag and Factor Xa cleavage site using Gibson assembly methods (primers in Table S1) as we previous described (Zhang et al., 2017).
SAMHD1 Depletion by CRISPR/Cas9 Genome Editing
To knockout SAMHD1, two different sgRNAs targeting human SAMHD1 were designed and cloned into lentiCRISPR v2 vector (a gift from Feng Zhang; Addgene plasmid #52961)(Sanjana et al., 2014). Packaging 293T cells were transfected with SAMHD1 sgRNAs or a negative control (non-targeting sgRNA-NC) and helper vectors (pMD2.G and psPAX2; gifts from Didier Trono; Addgene plasmid #s 12259 and 12260) using Lipofectamine 2000 reagent (Cat# 11668019, Life Technologies). Medium containing lentiviral particles and 8 mg/mL polybrene (Sigma-Aldrich, St. Louis) was used to infect Akata (EBV+) cells, Akata-BX1 (EBV+) cells, HeLa-(EBV+) cells, P3HR-1 cells and HEK293 cells carrying a BGLF4/BGLF5-knockout EBV (ΔBGLF5/BGLF5). Infected cells were selected in medium containing 2 μg/mL puromycin. The target guides sequences are listed in Table S1.
Lentiviral Transduction of SAMHD1
The pLX304-SAMHD1 was purchased from DNASU Plasmid Repository. The CRISPR-resistant SAMHD1 (nucleotide G321A, silent mutation) was generated by site-directed mutagenesis using the QuikChange II site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer’s instructions. Subsequently, the pLX304-SAMHD1 T592D and T592A mutants were generated individually using the CRISPR-resistant SAMHD1 as a template. Primers sequences are listed in Table S1. To prepare lentiviruses, 293T cells were transfected with lentiviral vector pLX304 containing the gene of wild-type SAMHD1, T592D or T592A mutants and the help vectors (pMD2.G and psPAX2) using Lipofectamine 2000 reagent. The supernatants were collected 48 hr after transfection and used for infection of SAMHD1-depleted (sg1) cells. Infected cells were selected in medium containing 10 μg/mL blasticidin. Expression of SAMHD1 was examined by western blot analysis.
Cell Lysis and Immunoblotting
Cells were harvested and lysed in 2x SDS-PAGE sample buffer and boiled for 5 minutes. The samples were separated on 4%–20% TGX gels (Cat# 4561096, Biorad), transferred onto PVDF membranes, and probed with primary and horseradish peroxidase-conjugated secondary antibodies.
Phos-tag SDS-PAGE
The level of SAMHD1 phosphorylation was analyzed by applying samples to 6% (w/v) acrylamide SDS-PAGE gels supplemented with 50 μM Phos-tag and MnCl2 according to manufacturer’s instructions (Wako, Cat# AAL-107) (Kinoshita et al., 2006). This method allows simultaneous analysis of phosphoprotein isoforms and its non-phosphorylated counterpart in SDS-PAGE. The acrylamidependant Phos-tag ligand provides a phosphate affinity SDS-PAGE for mobility shift detection of phosphorylated proteins. When cell extracts were analyzed on Phos-tag acrylamide gel, cell extracts were prepared in 1x SDS-PAGE sample buffer without EDTA.
Protein Expression and Purification
Halo-tagged WT and KD CHPKs proteins were expressed and purified as previously described (Li et al., 2015; Zhang et al., 2017). Briefly, Halo-tagged WT and KD CHPKs were transfected into 293T cells. Two T175 flasks of transfected cells were harvested 48 hr post-transfection at 100% confluence and lysed with 25 mL HaloTag Protein Purification Buffer (50 mM HEPES pH7.5, 150 mM NaCl, 1mM DTT, 1mM EDTA and 0.005% NP40/IGEPAL CA-630) with Protease Inhibitor Cocktail. WT and KD Halo-CHPKs were enriched using the Halo-tag resin and CHPKs were eluted from the resin by washing 3 times with 0.5 mL HaloTag Protein Purification Buffer containing 20 μL Halo-TEV protease.
For SAMHD1 purification from E. coli, the pGEX-5x-2-SAMHD1 plasmid was transformed into competent E.coli BL21. The transformed bacterial were then cultured in LB media at 37°C to OD600 = 0.8. Protein expression was induced in the presence of 0.1 mM IPTG at 16°C overnight. The cells were harvested by centrifugation at 8000rpm for 10 min. The cell pellets were frozen and thawed two times and then re-suspended in PBS solution with 1 mM DTT and 0.1 mM PMSF. After sonication and centrifugation, the lysates were incubated overnight with prepared glutathione Sepharose 4B (GE Healthcare, 17-0756-01). The beads were then washed and SAMHD1 protein was eluted with elution buffer (50 mM Tris-HCl, pH 7.5, 150mM NaCl, 5 mM MgCl2, 0.5 mM TCEP and 10% glycerol) containing Factor Xa overnight at 4°C. The purified protein was stored at −80°C.
Immunoprecipitation Assay
293T cells were transfected with indicated plasmids using Lipofectamine 2000. The cells were harvested at 48 hr post-transfection and lysed in RIPA lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 1% NP40, 1% deoxycholate, 0.1% SDS and 1 mM EDTA) containing protease inhibitors and phosphatase cocktail I and II. The immunoprecipitation was carried out as previously described (Zhang et al., 2017).
In Vitro Kinase Assay
Each sample was incubated in 40 μL Kinase Buffer containing 0.75 (v/v) magnesium-ATP cocktail buffer (Cat# 20-113; Upstate) and 6 μL of WT and KD CHPKs or CDK1/Cyclin B (Cat# 14-450, Upstate) for 30 min at 30°C. Reaction mixtures in Kinase Buffer without ATP were included as negative controls. Finally, reaction mixtures were separated by gel electrophoresis and phospho-SAMHD1 proteins were detected by Western Blot.
Togenerate phospho-SAMHD1 for dNTPase activity assay, SAMHD1 was in vitro phosphorylated using EBV BGLF4. CDK1/Cyclin B was used as a positive control. The kinase-dead BGLF4 and buffer were included as negative controls. In a phosphorylation reaction, full-length SAMHD1 was incubated at a ratio of 50:1 (w/w) with the kinases in 10 mM Tris-HCl pH 7.5, 150 mM NaCl, 5 mM MgCl2, 0.5 mM TCEP and 2mM ATP. The reaction was initiated by the addition of SAMHD1 and incubated for 6 hours at 4°C. The phosphorylation of SAMHD1 on T592 was confirmed by Western Blot (Arnold et al., 2015).
In Vitro dNTPase Activity Assay
All SAMHD1 dNTPase activity assays were performed in a reaction buffer (100 μL per reaction) containing 50 mM Tris-HCl, pH 7.5, 150 Mm NaCl, 5 mM MgCl2 and 0.5 mM TCEP. Recombinant SAMHD1 was phosphorylated by BGLF4 or CDK1 in vitro. Non-phosphorylated SAMHD1 was similarly processed by incubating with kinase-dead BGLF4 (BGLF4-KD) or Buffer control. Then phosphorylated and non-phosphorylated SAMHD1 (1 μM) were converted to GTP-bound dimer by adding 1mM GTP for 1 min. SAMHD1 dimer was further activated by adding dNTP mixture (12.5 μM each) for 1 min. The mixture was diluted 10-fold and the substrate dCTP, dTTP, dATP or dGTP (100 μM) is added for 0 to 240 min at room temperature. The reaction samples were collected at 0, 120 and 240 min and quenched by 10-fold dilution into ice cold buffer containing 10 mM EDTA, followed by spinning through an Amicon Ultra 0.5 mL 10 kDa Alter at 16000 xg for 20 min (Jang et al., 2016; Tang et al., 2015).
Deproteinized samples were analyzed by HPLC using a Synergi C18 Column 150 × 4.6mm (Phenomenex). The column was pre-equilibrated in 20mM ammonium acetate, pH 4.5 (buffer A). Injected samples (100 μl) were eluted with a linear methanol gradient over 14 min at a flow rate of 1 mL/min. The dN yields were quantified by integration of the calibrated UV absorption peak at 260 nm.
Chemical Cross-linking Assay
Sequential activation of SAMHD1 was performed as described in in vitro dNTPase activity assay. The reaction mixture (25 μL) was subjected to cross-linking with 2.5 mM glutaraldehyde for 10 min after 30 min of enzyme catalysis reaction. The chemical cross-linking was quenched with 1 M Tris-HCl, pH 8.0. The mixtures were separated by SDS-PAGE and analyzed by western blot (Jang et al., 2016). Monomer and tetramer were detected with anti-SAMHD1 antibody.
Lytic Induction and Cell Treatment
To induce the EBV lytic cycle in Akata B cells, Akata-BX1 (EBV+) and Akata (EBV+) cells were treated with IgG (1:200, Cat# 55087, MP Biomedicals) for 24, 48 and 72 hr. Akata-4E3(EBV−) cells were treated similarly as controls. For lytic induction of EBV in P3HR-1 cells, the cells were treated by TPA (20 ng/ml; Cat# NC9325685, Fisher Scientific) and sodium butyrate (3 mM; Cat# 19-137, Millipore) for 24 and 48 hr. For lytic induction of EBV in HeLa-(EBV+) cells, the cells are transfected with EBV ZTA, RTA and ZTA plus RTA using Lipofectamine 2000 reagent (Cat# 11668019, Life Technologies) for 48 hr. In some experiments, pHTPsiRNA-PK (siBGLF4) was co-transfected with ZTA/RTA to knockdown BGLF4. For lytic induction of EBV in HEK293 (EBV+) ΔBGLF4/BGLF5 cells, the cells are transfected with EBV ZTA plus myc-BGLF5 using Lipofectamine 2000 reagent for 48 hr.
For CDKs inhibition assay, Akata and P3HR-1 cells were treated with vehicle control (DMSO) or CDK1 and CDK2 inhibitors (10, 20 and 50 μM) for 48 hr and the phospho-SAMHD1 level was analyzed by immunoblotting.
HCMV Infection
MRC-5 cells were either mock infected or infected at a multiplicity of infection (MOI) of 1 by HCMV virus TS15-rN, an epithelial-tropic variant of HCMV strain Towne (Cui et al., 2013) for 72 hr. Stocks of virus TS15-rN were prepared and infectious titers determined as described previously (Cui et al., 2012).
Virus Titration
EBV titers were determined using a Raji cell infection assay (Li et al., 2011; Meng et al., 2010). In brief, GFP-EBV recombinant virus was harvested from lytically induced Akata BX1 cells carrying sgRNAs targeting SAMHD1 or non-targeting control sgRNA. Raji cells (2 × 105 in 1 mL medium/well in 24-well plates) was infected with the GFP-virus and phorbol-12-myristate-13-acetate (TPA) (20 ng/ml) and sodium butyrate (3 mM) were added 24 h later. After a further 24 h, the GFP-positive Raji cells were scored using a fluorescence microscope. The number of green Raji cells was used to determine the concentration of infectious virus particles.
EBV DNA Detection
To measure cell associated viral DNA, total genomic DNA was extracted using the Genomic DNA Purification Kit (Cat# A1120, Promega). The relative viral genome copy numbers were determined by quantitative PCR (qPCR) using primers to BALF5 gene normalized by β-actin.
Primers sequences listed in Table S1.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses employed a two-tailed Student’s t test using Microsoft Excel software for comparison of two groups. A p value of ≤ 0.05 was considered statistically significant. Values are given as the mean ± standard error of the mean (SEM) or standard deviation (SD) of biological or technical replicate experiments detailed in figure legends (indicated by the “n” in each figure legend). Technical replicates are replicate samples processed in parallel. Biological replicates are samples prepared from different sets of experiments.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-phospho-SAMHD1-T592 | Cell Signaling Tech | Cat# 15038; Discontinued |
| Anti-SAMHD1 | Cell Signaling Tech | Cat# 12361; Discontinued |
| Anti-SAMHD1 | Bethyl | Cat# A311-354; RRID:AB_11204405; RRID:AB_11205288 |
| Anti-CDC2/CDK1 | Cell Signaling Tech | Cat# 9112; RRID:AB_2074654 |
| Anti-CDK2 | Cell Signaling Tech | Cat# 2546; RRID:AB_2276129 |
| Anti-V5 | Invitrogen/Thermo Fisher | Cat# R960-25; RRID:AB_2556564 |
| Anti-V5-HRP | Invitrogen/Thermo Fisher | Cat# R961-25; RRID:AB_2556565 |
| Anti-HA | Roche | Cat# 11-867-431-001; RRID:AB_390919 |
| Anti-HA-HRP | Cell Signaling Tech | Cat# 14031; RRID:AB_2798368 |
| Anti-Myc | Cell Signaling Tech | Cat# 2278; RRID:AB_490778 |
| Anti-HA magnetic beads | Thermo Fisher | Cat# 88836; RRID:AB_2749815 |
| Mouse anti-β-actin antibody | MP Biomedicals | Cat# 691001; RRID:AB_2336056 |
| Anti-human IgG (for IgG cross-linking) | MP Biomedicals | Cat# 55087; RRID:AB_2334305 |
| Anti-ZTA | Argene | Cat# 11–007; Discontinued |
| Anti-ZTA(BZ1) | Santa Cruz | Cat# sc-53904; RRID:AB_783257 |
| Anti-RTA | Argene | Cat# 11–008; Discontinued |
| Anti-BGLF4 | Wang et al., 2005 | Clone #s 2616 and 2224 |
| Anti-BMRF1/EAD | Millipore | Cat# MAB8186; RRID:AB_95286 |
| Anti-EBV p18 | Invitrogen/Thermo Fisher | Cat# PA1-73003; RRID:AB_1017120 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| CDK1 inhibitor (RO3306) | Santa Cruz | Cat# SC-358700 |
| CDK2 inhibitor II | Santa Cruz | Cat# SC-221409 |
| Factor Xa | NEB | Cat# P8010 |
| TPA | Fisher Scientific | Cat# NC9325685 |
| Sodium Butyrate (NaBu) | Millipore | Cat# 19137 |
| Glutaraldehyde solution | Sigma | Cat# G7776 |
| SAMHD1 human recombinant protein | Origene | Cat# TP306013 |
| Critical Commercial Assays | ||
| Halo-tag protein purification kit | VWR/Promega | Cat# PAG6790 |
| Glutathione Sepharose 4B | GE Healthcare | Cat# 17–0756-01 |
| Lipofectamine 2000 | Life Technologies | Cat# 11668019 |
| Phos-tag Acrylamide | Wako | Cat# AAL-107 |
| Genomic DNA Purification Kit | Promega | Cat# A1120 |
| Experimental Models: Cell Lines | ||
| Akata (EBV+)-tet-Vector | Hayward Lab Collection (Zhu et al., 2009) | N/A |
| Akata (EBV+)-tet-BGLF4 | Hayward Lab Collection (Zhu et al., 2009) | N/A |
| Akata (EBV+)-tet-BGLF4 (mSIM-N) | Hayward Lab Collection (Li et al., 2012) | N/A |
| Akata (EBV+)-tet-BGLF4 (KD) | Hayward Lab Collection (Li et al., 2012) | N/A |
| Akata (EBV+) | Hayward Lab Collection | N/A |
| Akata-4E3 (EBV−) | Hayward Lab Collection | N/A |
| Akata-BX1 (EBV+) | Molesworth et al., 2000 | N/A |
| HeLa-(EBV+) | Feng et al., 2007 | N/A |
| Akata (EBV+)-SAMHD1-sg1 | This study | N/A |
| Akata (EBV+)-SAMHD1-sg2 | This study | N/A |
| Akata-BX1 (EBV+)-SAMHD1-sg1 | This study | N/A |
| Akata-BX1 (EBV+)-SAMDH1-sg2 | This study | N/A |
| Akata (EBV+)-SAMHD1-sg1-pLX-SAMHD1 | This study | N/A |
| Akata (EBV+)-SAMHD1-sg1-pLX-Vector | This study | N/A |
| Akata (EBV+)-SAMHD1-sg1-pLX-SAMHD1-T592D | This study | N/A |
| Akata (EBV+)-SAMHD1-sg1-pLX-SAMHD1-T592A | This study | N/A |
| 293T cells | Hayward Lab Collection | N/A |
| P3HR-1 | ATCC | Cat# HTB-62 |
| MRC-5 | ATCC | Cat# CCL-171 |
| HEK293 (EBV+) DBGLF4/BGLF5 | Feederle et al., 2009 | N/A |
| Oligonucleotides | ||
| See Table S1 for Primer sequences used for qPCR and cloning | This study | N/A |
| Recombinant DNA | ||
| pLX304-SAMHD1 | DNASU | Cat# HsCD00442083 |
| pLX304-SAMHD1 (with PAM mutated) | This study | pKZ175 |
| pLX304-SAMHD1-T592A (with PAM mutated) | This study | pKZ176 |
| pLX304-SAMHD1-T592D (with PAM mutated) | This study | pKZ177 |
| pLX304 | Addgene | Cat# 25890 |
| lentiCRISPR v2 vector | Addgene | Cat# 52961 |
| pMD2.G | Addgene | Cat# 12259 |
| psPAX2 | Addgene | Cat# 12260 |
| HCMV UL97 | Addgene | Cat# 26687 |
| HCMV UL97 KD | Addgene | Cat# 26688 |
| HHV6 U69 | Addgene | Cat# 26689 |
| HHV6 U69 KD | Addgene | Cat# 26690 |
| EBV BGLF4 | Addgene | Cat# 26691 |
| EBV BGLF4 KD | Addgene | Cat# 26692 |
| HHV7 U69 | Addgene | Cat# 26693 |
| HHV7 U69 KD | Addgene | Cat# 26694 |
| KSHV ORF36 | Addgene | Cat# 26695 |
| KSHV ORF36 KD | Addgene | Cat# 26696 |
| HSV-1 UL13 | Addgene | Cat# 26697 |
| VZV ORF47 | Addgene | Cat# 26698 |
| SAMHD1-sg-1 | This study | pKZ119 |
| SAMHD1-sg-2 | This study | pKZ120 |
| sg-NC (Non-targeting control) | This study | pKZ5 |
| ZTA | Hayward Lab Collection | N/A |
| HA-RTA | Hayward Lab Collection | pGL196 |
| Myc-BGLF5 | Hayward Lab Collection | pGL173 |
| Halo-BGLF4 | Hayward Lab Collection | pGL771 |
| Halo-BGLF4 KD | Hayward Lab Collection | pGL772 |
| Halo-UL97 | Hayward Lab Collection | pGL798 |
| Halo-UL97 KD | Hayward Lab Collection | pGL799 |
| Halo-HSV-1 UL13 | Hayward Lab Collection | pGL801 |
| Halo-HSV-1 UL13 KD | Hayward Lab Collection | pGL803 |
| pCVM-XL4-SAMHD1 | Origene | Cat# SC114650 |
| pGEX-5x-2-SAMHD1 | This study | pKZ49 |
| pHTPsiRNA-PK (siBGLF4) | Gershburg et al., 2007 | N/A |
| pHTPsiRNA-NC (siNC) | Gershburg et al., 2007 | N/A |
Highlights.
SAMHD1 depletion facilitates EBV lytic replication
EBV protein kinase BGLF4 directly phosphorylates SAMHD1
BGLF4 phosphorylation of SAMHD1 inhibits its dCTPase and dTTPase activity
SAMHD1 is targeted by the conserved herpesvirus protein kinases
ACKNOWLEDGMENTS
We thank S. Diane Hayward (Johns Hopkins) for providing reagent and cells lines and for critically reading the manuscript. We thank Feng Zhang (MIT/Broad) for sharing the lentiCRISPR v2 plasmid (Addgene plasmid no. 52961). We thank Didier Trono (EPFL) for providing the pMD2.G and psPAX2 plasmid (Addgene plasmid no. 12259 and 12260). We thank Robert Kalejta (University of Wisconsin-Madison) for providing the human influenza hemagglutinin (HA)-tagged viral kinases (Addgene plasmid no. 26689-26698). We thank Michael McVoy and Ronzo Lee (Virginia Commonwealth University) for assistance with HCMV experiments. We thank Henri-Jacques Delecluse (German Cancer Research Centre) and Ayman El-Guindy (Yale University) for providing HEK293 cells carrying BGLF4/BGLF5-knockout B95.8 EBV genomes. We thank Yong Xiong (Yale University) for providing high-performance liquid chromatography (HPLC) column information. We thank Lindsey Hutt-Fletcher (Louisiana State University) for providing the Akata-BX1 (EBV+) cells and Mei-Ru Chen (National Taiwan University) for anti-BGLF4 antibody. We also thank Shannon Kenney (University of Wisconsin-Madison) for providing the Hela (EBV+) cell line and Edward Gershburg (Southern Illinois University) for providing small interfering RNA (siRNA)-expressing plasmids. This work was supported by NIH grant K99AI104828/R00AI104828 to R.L. The work was also supported by Institutional Research Grant IRG-14- 192-40 from the American Cancer Society. R.L. received support from the VCU Philips Institute for Oral Health Research, the VCU NCI Designated Massey Cancer Center (NIH grant P30 CA016059) (https://grants.nih.gov/grants/oer.htm), and the VCU Presidential Quest for Distinction Award. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.04.020.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Arnold LH, Groom HC, Kunzelmann S, Schwefel D, Caswell SJ, Ordonez P, Mann MC, Rueschenbaum S, Goldstone DC, Pennell S, et al. (2015). Phospho-dependent Regulation of SAMHD1 Oligomerisation Couples Catalysis and Restriction. PLoS Pathog. 11, e1005194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badia R, Angulo G, Riveira-Munoz E, Pujantell M, Puig T, Ramirez C, Torres-Torronteras J, Marti R, Pauls E, Clotet B, et al. (2016). Inhibition of herpes simplex virus type 1 by the CDK6 inhibitor PD-0332991 (palbociclib) through the control of SAMHD1. J. Antimicrob. Chemother 71, 387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifati S, Daly MB, St Gelais C, Kim SH, Hollenbaugh JA, Shepard C, Kennedy EM, Kim DH, Schinazi RF, Kim B, and Wu L (2016). SAMHD1 controls cell cycle status, apoptosis and HIV-1 infection in monocytic THP-1 cells. Virology 495, 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderwood MA, Venkatesan K, Xing L, Chase MR, Vazquez A, Holthaus AM, Ewence AE, Li N, Hirozane-Kishikawa T, Hill DE, et al. (2007). Epstein-Barr virus and virus human protein interaction maps. Proc. Natl. Acad. Sci. USA 104, 7606–7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CW, Lee CP, Huang YH, Yang PW, Wang JT, and Chen MR (2012). Epstein-Barr virus protein kinase BGLF4 targets the nucleus through interaction with nucleoporins. J. Virol 86, 8072–8085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CW, Lee CP, Su MT, Tsai CH, and Chen MR (2015). BGLF4 kinase modulates the structure and transport preference of the nuclear pore complex to facilitate nuclear import of Epstein-Barr virus lytic proteins. J. Virol 89, 1703–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PW, Lin SJ, Tsai SC, Lin JH, Chen MR, Wang JT, Lee CP, and Tsai CH (2010). Regulation of microtubule dynamics through phosphorylation on stathmin by Epstein-Barr virus kinase BGLF4. J. Biol. Chem 285, 10053–10063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Zhu M, Pan X, Zhu Y, Yan H, Jiang T, Shen Y, Dong X, Zheng N, Lu J, et al. (2014). Inhibition of Hepatitis B virus replication by SAMHD1. Biochem. Biophys. Res. Commun 450, 1462–1468. [DOI] [PubMed] [Google Scholar]
- Coquel F, Silva MJ, Técher H, Zadorozhny K, Sharma S, Nieminuszczy J, Mettling C, Dardillac E, Barthe A, Schmitz AL, et al. (2018). SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 557, 57–61. [DOI] [PubMed] [Google Scholar]
- Cribier A, Descours B, Valadão AL, Laguette N, and Benkirane M (2013). Phosphorylation of SAMHD1 by cyclin A2/CDK1 regulates its restriction activity toward HIV-1. Cell Rep. 3, 1036–1043. [DOI] [PubMed] [Google Scholar]
- Cui X, Adler SP, Davison AJ, Smith L, Habib SE, and McVoy MA (2012). Bacterial artificial chromosome clones of viruses comprising the towne cytomegalovirus vaccine. J. Biomed. Biotechnol 2012, 428498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X, Lee R, Adler SP, and McVoy MA (2013). Antibody inhibition of human cytomegalovirus spread in epithelial cell cultures. J. Virol. Methods 192, 44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daddacha W, Koyen AE, Bastien AJ, Head PE, Dhere VR, Nabeta GN, Connolly EC, Werner E, Madden MZ, Daly MB, et al. (2017). SAMHD1 Promotes DNA End Resection to Facilitate DNA Repair by Homologous Recombination. Cell Rep. 20, 1921–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djavadian R, Chiu YF, and Johannsen E (2016). An Epstein-Barr Virus-Encoded Protein Complex Requires an Origin of Lytic Replication In Cis to Mediate Late Gene Transcription. PLoS Pathog. 12, e1005718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djavadian R, Hayes M, and Johannsen E (2018). CAGE-seq analysis of Epstein-Barr virus lytic gene transcription: 3 kinetic classes from 2 mechanisms. PLoS Pathog. 14, e1007114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Guindy A, Lopez-Giraldez F, Delecluse HJ, McKenzie J, and Miller G (2014). A locus encompassing the Epstein-Barr virus bglf4 kinase regulates expression of genes encoding viral structural proteins. PLoS Pathog. 10, e1004307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erazo A, and Kinchington PR (2010). Varicella-zoster virus open reading frame 66 protein kinase and its relationship to alphaherpesvirus US3 kinases. Curr. Top. Microbiol. Immunol 342, 79–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feederle R, Mehl-Lautscham AM, Bannert H, and Delecluse HJ (2009). The Epstein-Barr virus protein kinase BGLF4 and the exonuclease BGLF5 have opposite effects on the regulation of viral protein production. J. Virol 83, 10877–10891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng WH, Kraus RJ, Dickerson SJ, Lim HJ, Jones RJ, Yu X, Mertz JE, and Kenney SC (2007). ZEB1 and c-Jun levels contribute to the establishment of highly lytic Epstein-Barr virus infection in gastric AGS cells. J. Virol 81, 10113–10122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershburg E, and Pagano JS (2008). Conserved herpesvirus protein kinases. Biochim. Biophys. Acta 1784, 203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershburg E, Marschall M, Hong K, and Pagano JS (2004). Expression and localization of the Epstein-Barr virus-encoded protein kinase. J. Virol 78, 12140–12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershburg E, Raffa S, Torrisi MR, and Pagano JS (2007). Epstein-Barr virus-encoded protein kinase (BGLF4) is involved in production of infectious virus. J. Virol 81, 5407–5412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill RB, James SH, and Prichard MN (2012). Human cytomegalovirus UL97 kinase alters the accumulation of CDK1. J. Gen. Virol 93, 1743–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamirally S, Kamil JP, Ndassa-Colday YM, Lin AJ, Jahng WJ, Baek MC, Noton S, Silva LA, Simpson-Holley M, Knipe DM, et al. (2009). Viral mimicry of Cdc2/cyclin-dependent kinase 1 mediates disruption of nuclear lamina during human cytomegalovirus nuclear egress. PLoS Pathog. 5, e1000275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbaugh JA, Gee P, Baker J, Daly MB, Amie SM, Tate J, Kasai N, Kanemura Y, Kim DH, Ward BM, et al. (2013). Host factor SAMHD1 restricts DNA viruses in non-dividing myeloid cells. PLoS Pathog. 9, e1003481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, Florens L, Washburn MP, and Skowronski J (2011). Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 474, 658–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume AJ, Finkel JS, Kamil JP, Coen DM, Culbertson MR, and Kalejta RF (2008). Phosphorylation of retinoblastoma protein by viral protein with cyclin-dependent kinase function. Science 320, 797–799. [DOI] [PubMed] [Google Scholar]
- Hwang S, Kim KS, Flano E, Wu TT, Tong LM, Park AN, Song MJ, Sanchez DJ, O’Connell RM, Cheng G, and Sun R (2009). Conserved herpesviral kinase promotes viral persistence by inhibiting the IRF-3-mediated type I interferon response. Cell Host Microbe 5, 166–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwahori S, Murata T, Kudoh A, Sato Y, Nakayama S, Isomura H, Kanda T, and Tsurumi T (2009). Phosphorylation of p27Kip1 by Epstein-Barr virus protein kinase induces its degradation through SCFSkp2 ubiquitin ligase actions during viral lytic replication. J. Biol. Chem 284, 18923–18931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwahori S, Hakki M, Chou S, and Kalejta RF (2015). Molecular Determinants for the Inactivation of the Retinoblastoma Tumor Suppressor by the Viral Cyclin-dependent Kinase UL97. J. Biol. Chem 290, 19666–19680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang S, Zhou X, and Ahn J (2016). Substrate Specificity of SAMHD1 Triphosphohydrolase Activity Is Controlled by Deoxyribonucleoside Triphosphates and Phosphorylation at Thr592. Biochemistry 55, 5635–5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato A, Yamamoto M, Ohno T, Tanaka M, Sata T, Nishiyama Y, and Kawaguchi Y (2006). Herpes simplex virus 1-encoded protein kinase UL13 phosphorylates viral Us3 protein kinase and regulates nuclear localization of viral envelopment factors UL34 and UL31. J. Virol 80, 1476–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, and Kato K (2003). Protein kinases conserved in herpesviruses potentially share a function mimicking the cellular protein kinase cdc2. Rev. Med. Virol 13, 331–340. [DOI] [PubMed] [Google Scholar]
- Kim ET, White TE, Brandariz-Núñez A, Diaz-Griffero F, and Weitzman MD (2013). SAMHD1 restricts herpes simplex virus 1 in macrophages by limiting DNA replication. J. Virol 87, 12949–12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim ET, Roche KL, Kulej K, Spruce LA, Seeholzer SH, Coen DM, Diaz-Griffero F, Murphy EA, and Weitzman MD (2019). SAMHD1 Modulates Early Steps during Human Cytomegalovirus Infection by Limiting NF-kB Activation. Cell Rep. 28, this issue, 434–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita E, Kinoshita-Kikuta E, Takiyama K, and Koike T (2006). Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell. Proteomics 5, 749–757. [DOI] [PubMed] [Google Scholar]
- Kudoh A, Daikoku T, Ishimi Y, Kawaguchi Y, Shirata N, Iwahori S, Isomura H, and Tsurumi T (2006). Phosphorylation of MCM4 at sites inactivating DNA helicase activity of the MCM4-MCM6-MCM7 complex during Epstein-Barr virus productive replication. J. Virol 80, 10064–10072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuny CV, Chinchilla K, Culbertson MR, and Kalejta RF (2010). Cyclin-dependent kinase-like function is shared by the beta- and gamma-subset of the conserved herpesvirus protein kinases. PLoS Pathog. 6, e1001092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Ségeral E, Yatim A, Emiliani S, Schwartz O, and Benkirane M (2011). SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474, 654–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CP, Chen JY, Wang JT, Kimura K, Takemoto A, Lu CC, and Chen MR (2007). Epstein-Barr virus BGLF4 kinase induces premature chromosome condensation through activation of condensin and topoisomerase II. J. Virol 81, 5166–5180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CP, Huang YH, Lin SF, Chang Y, Chang YH, Takada K, and Chen MR (2008). Epstein-Barr virus BGLF4 kinase induces disassembly of the nuclear lamina to facilitate virion production. J. Virol 82, 11913–11926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenzi GM, Domaoal RA, Kim DH, Schinazi RF, and Kim B (2015). Mechanistic and Kinetic Differences between Reverse Transcriptases of Vpx Coding and Non-coding Lentiviruses. J. Biol. Chem 290, 30078–30086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, and Hayward SD (2013). Potential of protein kinase inhibitors for treating herpesvirus-associated disease. Trends Microbiol. 21, 286–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Zhang W, and Cao X (2000). Identification of human homologue of mouse IFN-gamma induced protein from human dendritic cells. Immunol. Lett 74, 221–224. [DOI] [PubMed] [Google Scholar]
- Li R, Zhu J, Xie Z, Liao G, Liu J, Chen MR, Hu S, Woodard C, Lin J, Taverna SD, et al. (2011). Conserved herpesvirus kinases target the DNA damage response pathway and TIP60 histone acetyltransferase to promote virus replication. Cell Host Microbe 10, 390–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Wang L, Liao G, Guzzo CM, Matunis MJ, Zhu H, and Hayward SD (2012). SUMO binding by the Epstein-Barr virus protein kinase BGLF4 is crucial for BGLF4 function. J. Virol 86, 5412–5421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Liao G, Nirujogi RS, Pinto SM, Shaw PG, Huang TC, Wan J, Qian J, Gowda H, Wu X, et al. (2015). Phosphoproteomic Profiling Reveals Epstein-Barr Virus Protein Kinase Integration of DNA Damage Response and Mitotic Signaling. PLoS Pathog. 11, e1005346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv DW, Zhang K, and Li R (2018). Interferon regulatory factor 8 regulates caspase-1 expression to facilitate Epstein-Barr virus reactivation in response to B cell receptor stimulation and chemical induction. PLoS Pathog. 14, e1006868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Jacobs SR, West JA, Stopford C, Zhang Z, Davis Z, Barber GN, Glaunsinger BA, Dittmer DP, and Damania B (2015). Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc. Natl. Acad. Sci. USA 112, E4306–E4315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeoch DJ, Cook S, Dolan A, Jamieson FE, and Telford EA (1995). Molecular phylogeny and evolutionary timescale for the family of mammalian herpesviruses. J. Mol. Biol 247, 443–458. [DOI] [PubMed] [Google Scholar]
- McKenzie J, Lopez-Giraldez F, Delecluse HJ, Walsh A, and El-Guindy A (2016). The Epstein-Barr Virus Immunoevasins BCRF1 and BPLF1 Are Expressed by a Mechanism Independent of the Canonical Late Pre-initiation Complex. PLoS Pathog. 12, e1006008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Q, Hagemeier SR, Fingeroth JD, Gershburg E, Pagano JS, and Kenney SC (2010). The Epstein-Barr virus (EBV)-encoded protein kinase, EBV-PK, but not the thymidine kinase (EBV-TK), is required for ganciclovir and acyclovir inhibition of lytic viral production. J. Virol 84, 4534–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molesworth SJ, Lake CM, Borza CM, Turk SM, and Hutt-Fletcher LM (2000). Epstein-Barr virus gH is essential for penetration of B cells but also plays a role in attachment of virus to epithelial cells. J. Virol 74, 6324–6332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SM, Cannon JS, Tanhehco YC, Hamzeh FM, and Ambinder RF (2001). Induction of Epstein-Barr virus kinases to sensitize tumor cells to nucleoside analogues. Antimicrob. Agents Chemother 45, 2082–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberstein A, Perlman DH, Shenk T, and Terry LJ (2015). Human cytomegalovirus pUL97 kinase induces global changes in the infected cell phosphoproteome. Proteomics 15, 2006–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prichard MN (2009). Function of human cytomegalovirus UL97 kinase in viral infection and its inhibition by maribavir. Rev. Med. Virol 19, 215–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prichard MN, Gao N, Jairath S, Mulamba G, Krosky P, Coen DM, Parker BO, and Pari GS (1999). A recombinant human cytomegalovirus with a large deletion in UL97 has a severe replication deficiency. J. Virol 73, 5663–5670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romaker D, Schregel V, Maurer K, Auerochs S, Marzi A, Sticht H, and Marschall M (2006). Analysis of the structure-activity relationship of four herpesviral UL97 subfamily protein kinases reveals partial but not full functional conservation. J. Med. Chem 49, 7044–7053. [DOI] [PubMed] [Google Scholar]
- Ruiz A, Pauls E, Badia R, Torres-Torronteras J, Riveira-Muñoz E, Clotet B, Martí R, Ballana E, and Esté JA (2015). Cyclin D3-dependent control of the dNTP pool and HIV-1 replication in human macrophages. Cell Cycle 14, 1657–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwefel D, Groom HC, Boucherit VC, Christodoulou E, Walker PA, Stoye JP, Bishop KN, and Taylor IA (2014). Structural basis of lentiviral subversion of a cellular protein degradation pathway. Nature 505, 234–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwefel D, Boucherit VC, Christodoulou E, Walker PA, Stoye JP, Bishop KN, and Taylor IA (2015). Molecular determinants for recognition of divergent SAMHD1 proteins by the lentiviral accessory protein Vpx. Cell Host Microbe 17, 489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, and Coen DM (2014). Comparison of effects of inhibitors of viral and cellular protein kinases on human cytomegalovirus disruption of nuclear lamina and nuclear egress. J. Virol 88, 10982–10985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Kamil JP, Coughlin M, Reim NI, and Coen DM (2014). Human cytomegalovirus UL50 and UL53 recruit viral protein kinase UL97, not protein kinase C, for disruption of nuclear lamina and nuclear egress in infected cells. J. Virol 88, 249–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Bender BJ, Kamil JP, Lye MF, Pesola JM, Reim NI, Hogle JM, and Coen DM (2015). Human cytomegalovirus UL97 phosphorylates the viral nuclear egress complex. J. Virol 89, 523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lv G, Chu Z, Piao L, Liu X, Wang T, Jiang Y, and Zhang P (2014). Identification of natural splice variants of SAMHD1 in virus-infected HCC. Oncol. Rep 31, 687–692. [DOI] [PubMed] [Google Scholar]
- Shibaki T, Suzutani T, Yoshida I, Ogasawara M, and Azuma M (2001). Participation of type I interferon in the decreased virulence of the UL13 gene-deleted mutant of herpes simplex virus type 1. J. Interferon Cytokine Res 21, 279–285. [DOI] [PubMed] [Google Scholar]
- Sullivan V, Talarico CL, Stanat SC, Davis M, Coen DM, and Biron KK (1992). A protein kinase homologue controls phosphorylation of ganciclovir in human cytomegalovirus-infected cells. Nature 358, 162–164. [DOI] [PubMed] [Google Scholar]
- Sun X, Bristol JA, Iwahori S, Hagemeier SR, Meng Q, Barlow EA, Fingeroth JD, Tarakanova VL, Kalejta RF, and Kenney SC (2013). Hsp90 inhibitor 17-DMAG decreases expression of conserved herpesvirus protein kinases and reduces virus production in Epstein-Barr virus-infected cells. J. Virol 87, 10126–10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sze A, Belgnaoui SM, Olagnier D, Lin R, Hiscott J, and van Grevenynghe J (2013). Host restriction factor SAMHD1 limits human T cell leukemia virus type 1 infection of monocytes via STING-mediated apoptosis. Cell Host Microbe 14, 422–434. [DOI] [PubMed] [Google Scholar]
- Tang C, Ji X, Wu L, and Xiong Y (2015). Impaired dNTPase activity of SAMHD1 by phosphomimetic mutation of Thr-592. J. Biol. Chem 290, 26352–26359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarakanova VL, Leung-Pineda V, Hwang S, Yang CW, Matatall K, Basson M, Sun R, Piwnica-Worms H, Sleckman BP, and Virgin HW 4th. (2007). Gamma-herpesvirus kinase actively initiates a DNA damage response by inducing phosphorylation of H2AX to foster viral replication. Cell Host Microbe 1, 275–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umaña AC, Iwahori S, and Kalejta RF (2018). Direct Substrate Identification with an Analog Sensitive (AS) Viral Cyclin-Dependent Kinase (v-Cdk). ACS Chem. Biol 13, 189–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JT, Yang PW, Lee CP, Han CH, Tsai CH, and Chen MR (2005). Detection of Epstein-Barr virus BGLF4 protein kinase in virus replication compartments and virus particles. J. Gen. Virol 86, 3215–3225. [DOI] [PubMed] [Google Scholar]
- Wang JT, Doong SL, Teng SC, Lee CP, Tsai CH, and Chen MR (2009). Epstein-Barr virus BGLF4 kinase suppresses the interferon regulatory factor 3 signaling pathway. J. Virol 83, 1856–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welbourn S, Miyagi E, White TE, Diaz-Griffero F, and Strebel K (2012). Identification and characterization of naturally occurring splice variants of SAMHD1. Retrovirology 9, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welbourn S, Dutta SM, Semmes OJ, and Strebel K (2013). Restriction of virus infection but not catalytic dNTPase activity is regulated by phosphorylation of SAMHD1. J. Virol 87, 11516–11524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White TE, Brandariz-Nuñez A, Valle-Casuso JC, Amie S, Nguyen LA, Kim B, Tuzova M, and Diaz-Griffero F (2013). The retroviral restriction ability of SAMHD1, but not its deoxynucleotide triphosphohydrolase activity, is regulated by phosphorylation. Cell Host Microbe 13, 441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann S, Behrendt R, Eissmann K, Volkmann B, Thomas D, Ebert T, Cribier A, Benkirane M, Hornung V, Bouzas NF, and Gramberg T (2015). Phosphorylation of murine SAMHD1 regulates its antiretroviral activity. Retrovirology 12, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf DG, Courcelle CT, Prichard MN, and Mocarski ES (2001). Distinct and separate roles for herpesvirus-conserved UL97 kinase in cytomegalovirus DNA synthesis and encapsidation. Proc. Natl. Acad. Sci. USA 98, 1895–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Hao C, DeLucia M, Swanson S, Florens L, Washburn MP, Ahn J, and Skowronski J (2015). CyclinA2-Cyclin-dependent Kinase Regulates SAMHD1 Protein Phosphohydrolase Domain. J. Biol. Chem 290, 13279–13292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Lv DW, and Li R (2017). B Cell Receptor Activation and Chemical Induction Trigger Caspase-Mediated Cleavage of PIAS1 to Facilitate Epstein-Barr Virus Reactivation. Cell Rep 21, 3445–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Liao G, Shan L, Zhang J, Chen MR, Hayward GS, Hayward SD, Desai P, and Zhu H (2009). Protein array identification of substrates of the Epstein-Barr virus protein kinase BGLF4. J. Virol 83, 5219–5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.