

Abstract
Preconditioning is an endogenous mechanism in which a nonlethal exposure increases cellular resistance to subsequent additional severe injury. Here we show that connexin 43 (Cx43) plays a key role in protection afforded by preconditioning. Cx43 null mice were insensitive to hypoxic preconditioning, whereas wild-type littermate mice exhibited a significant reduction in infarct volume after occlusion of the middle cerebral artery. In cultures, Cx43-deficient cells responded to preconditioning only after exogenous expression of Cx43, and protection was attenuated by small interference RNA or by channel blockers. Our observations indicate that preconditioning reduced degradation of Cx43, resulting in a marked increase in the number of plasma membrane Cx43 hemichannels. Consequently, efflux of ATP through hemichannels led to accumulation of its catabolic product adenosine, a potent neuroprotective agent. Thus, adaptive modulation of Cx43 can offset environmental stress by adenosine-mediated elevation of cellular resistance.
Keywords: gap junction, astrocytes, hemichannels, stroke, adenosine, GFAP
Introduction
Preconditioning is an adaptive cellular response to noxious exposures (del Zoppo, 2006). Subthreshold injury activates endogenous protective mechanisms that increase cellular resistance to subsequent more severe injury. Essentially any injury paradigm, including hypoxia, spreading depression, inflammation, epilepsy, hypothermia and hyperthermia, and metabolic inhibition, will induce preconditioning if applied below the threshold for irreversible cell damage (Kirino, 2002; Dirnagl et al., 2003; Lo et al., 2003). The signaling pathways involved in injury tolerance have been extensively studied, and a key role of adenosine, a potent neuroprotective and cardioprotective agent, has been identified, but other pathways, including erythropoietin, inflammatory cytokines, ceramide, nitric oxide, vascular endothelial growth factor, and protein SUMOylation (for small ubiquitin-related modifier), have also been implicated (Zimmermann et al., 2001; Kariko et al., 2004; Gustavsson et al., 2007; Lee et al., 2007).
We here tested the postulate that an upregulation of the major astrocytic gap-junction protein connexin 43 (Cx43) is essential for protection afforded by preconditioning. Previous work has documented that cellular injury is linked to an upregulation of Cx43 expression in a variety of cells and tissues (Daleau et al., 2001; VanSlyke and Musil, 2005). For example, alveolar epithelial cells increased Cx43 expression after radiation (Kasper et al., 1996), smooth muscle cells exhibited increased Cx43 levels after balloon catheter injury of rat carotid artery (Yeh et al., 1997), multiple Cxs were upregulated after peripheral nerve injury (Chandross, 1998; Chang et al., 2000; Lin et al., 2002), and one of the earliest responses to kidney damage is an increase of Cx43 expression (Yaoita et al., 2002). Similarly, Cx43 is upregulated after spinal cord injury (Theriault et al., 1997; Lee et al., 2005) and in cortical astrocytes exposed to ischemia (Cotrina et al., 1998a; Lin et al., 2003). Traditionally the impact of Cx expression has been linked to gap-junction coupling, but the discovery of functional hemichannels and their potential for release of transmitters has vastly expanded our conception of the role of Cx proteins in physiological processes and pathobiology (Bennett et al., 2003; Goodenough and Paul, 2003). Hemichannels serve as a pathway for efflux of cytosolic ATP (Cotrina et al., 1998b; Stout et al., 2002; Gomes et al., 2005; Lankford et al., 2006). Once released, ATP is degraded to adenosine with a rapid time constant of 200 ms (Dunwiddie and Masino, 2001).
We asked whether the upregulation of Cx43 after preconditioning is linked to an increased number of plasma membrane hemichannels. We found that Cx43 hemichannel opening exhibited a marked increase after preconditioning, and, strikingly, Cx43 expression was required for protection afforded by preconditioning in cultures and in a mouse model of focal stroke. Furthermore, extracellular ATP and adenosine increased ∼5- to 100-fold after preconditioning of Cx-expressing cells but not when Cx-deficient cells were exposed to a similar treatment. Antagonizing Cx43 hemichannel opening with siRNA or pharmacological inhibition reduced both extracellular accumulation of purines and cellular resistance. Because stress-induced increases in Cx43 occur in a variety of injury models and adenosine broadly dampens cellular activity and preserves energy metabolism, we propose that dynamic regulation of the abundance of Cx43 hemichannels may represent a conserved protective response to cellular stress.
Materials and Methods
Cultures, preconditioning, small interfering RNA, adenosine measurements, and injury paradigms.
Astrocytes were prepared from postnatal day 0 (P0) to P1 rat pups as described previously (Cotrina et al., 1998a). C6 cells expressing Cx43, Cx43–enhanced green fluorescent protein (eGFP), or GFP under the cytomegalovirus (CMV) promoter (C6–Cx43+, C6–Cx43–eGFP, or C6–GFP, respectively) or deficient of Cx43 (C6–Cx43− cells, transfected with empty vector under the CMV promoter) were generated as described previously (Lin et al., 1998; John et al., 1999). Initial experiments indicated that cultured astrocytes and C6 cells exhibited increased injury tolerance after exposure to a variety of subthreshold insults, including exposure to ischemic conditions (Hillion et al., 2005). We chose to focus on preconditioning evoked by sublethal exposure to H2O2 and tamoxifen because of the reproducibility of the cellular responses. Essentially any stimulus capable of causing cellular injury can activate the endogenous protective pathways involved in preconditioning, exemplified by cross tolerance, e.g., one type of injury induces tolerance against another one (Dirnagl et al., 2003).
For preconditioning, astrocytes were seeded onto culture plates at 60–70% confluence and preconditioned when confluent (3–4 d) with a sublethal concentration of tamoxifen {25–30 μm in DMEM–F-12 (Invitrogen, Carlsbad, CA) containing 2.5% FBS for 45 min} or H2O2 (0.7–1.2 mm in DMEM–F-12 for 45 min). C6–Cx43+, C6–Cx43−, C6–Cx43–eGFP, and C6–GFP cells were seeded to near confluency onto poly-l-lysine-coated multiwell culture plates. After >24 h incubation in DMEM–F-12 containing 10% FBS, cells were treated for 30–60 min with tamoxifen in DMEM–F-12 (12.5 μm for C6–Cx43−, C6–Cx43–eGFP, and C6–GFP; 15–20 μm for C6–Cx43+) and returned to DMEM–F-12 containing 10% FBS. Control cultures were exposed to vehicle (H2O for H2O2 or ethanol for tamoxifen) and processed identically. After recovery for 18–24 h, the cells were reexposed to a lethal concentration of either H2O2 (1.4–1.6 mm) or tamoxifen (35–40 μm in 2.5% FBS for astrocytes; 25–30 μm in DMEM–F-12 for C6–Cx43+; 15–20 μm in DMEM–F-12 for C6–Cx43− and C6–Cx43–eGFP cells). The viability was assessed 8–24 h later by the Alamar Blue assay (BioSource, Camarillo, CA). Each data point represented the average of at least five different runs (range, 5–145). Small interference RNA (siRNA) targeting rat Cx43 (GenBank accession number X06656) was designed using a web-based program (http://www.dharmacon.com/sidesign) and manufactured by Dharmacon (Chicago, IL). The sequence ACAUCAUUGAGCUCUUCUA and its complimentary strand were chosen for the current study. Transfection mixture containing double-stranded annealed Cx43siRNA (42 pmol/24-well) and Lipofectamine 2000 (0.6 μl/24-well) was added directly to C6–Cx43 cells during seeding. Sister cultures were transfected with Stealth RNAi low GC duplex (Invitrogen) as negative controls. The effect of precondition on these siRNA-treated cultures was evaluated on 5–6 d after transfection. Gap-junction blockers (carbenoxelone, 50–100 μm; octanol, 500 μm; 18-α glycyrrhetinic acid, 50 μm; flufenamic acid, 50–100 μm; niflumic acid, 100–200 μm; all from Sigma, St. Louis, MO) were added 30–60 min before and during precondition but also in the recovery medium. Blockers used to prevent degradation via the lysosome pathway were NH4Cl (10 mm; Mallinckrodt Baker, Phillipsburg, NJ) and chloroquine (CLQ) (100 μm; Sigma). Blockers used to prevent proteasome degradation were lactacystin, epoxomicin (both at 10 μm), and MG132 (carbobenzoxy-l-leucyl-l-leucyl-l-leucinal) (40 μm; all from Boston Biochem, Cambridge, MA). They were added directly to culture medium at the above final concentrations for 4–6 h before the cultures were analyzed for their dye uptake or ATP release capacity or their sensitivity to tamoxifen. No morphological changes were apparent at the end of the treatment.
Electrophysiology, dye uptake, ATP release, adenosine measurements, immunocytochemistry, Western blot, and functional coupling assay.
Astrocytes and C6 cells were plated on 12 mm coverslips and grown in 24-well plates. The coverslips were placed in perfusion chamber, and cells visualized with a 63× water immersion objective and differential inference contrast (DIC) optics (BX51 upright microscope; Olympus Optical, Center Valley, PA). Patch electrodes with a resistance of 6–10 MΩ were pulled from KG-33 glass capillaries (inner diameter of 1.0 mm, outer diameter of 1.5 mm; Garner Glass, Claremont, CA) using a P-97 electrode puller (Sutter Instruments, Novato, CA). The pipette solution contained 130 mm Na-ATP and 20 mm tetraethylammonium (TEA) to block K+ channels. The high concentration of ATP was used, because the recordings were part of a larger study analyzing the permeability of ATP anion through Cx43 hemichannels (J. Kang, N. Kang, D. Lovatt, A. Torres, Z. Zhao, J. Lin, and M. Nedergaard, unpublished observation). Cell-attached patch recordings were performed using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA). Patches with seal resistance <5 GΩ were rejected. Signals were sampled every 50 μs with pClamp Clampex 9.0 and filtered through an eight-pole Bessel low-pass filter with a 1 kHz cutoff frequency. Cx43 hemichannel activity was analyzed 24 h after preconditioning similar to viability analysis.
Propidium iodide uptake and ATP release, both indicators of hemichannel opening, were assayed in parallel on duplicated cultures in 24-well plates after 16–24 h of post-precondition recovery similar to described previously (Cotrina et al., 1998b; Arcuino et al., 2002). A second aliquot of samples from the ATP release study was collected to correlate the release of ATP versus adenosine. Adenosine was detected using (platinum/iridium wire) biosensors (Sarissa Biomedical, Coventry, UK) (Wall et al., 2007). Calibration was performed at the beginning and the end of each experiment (Frenguelli et al., 2007). ATP and adenosine release, and uptake of propidium iodide, were in all experiments evoked by exposure to low Ca2+ as described previously (Cotrina et al., 1998b; Arcuino et al., 2002). Quantification of adenosine in vivo was attempted, but the experiments were aborted because tissue depolarization and cell swelling associated with ischemia gave rise to artificial signals.
For immunocytochemistry, astrocytes and C6 cells were grown on poly-l-lysine-coated 12 mm cover glass and fixed with 4% paraformaldehyde at specified times after treatment with blockers or post-precondition recovery. Cultures were permeabilized with 0.1% Triton X-100, blocked with 10% normal goat serum (Cotrina et al., 1998a), and immunoreacted with anti-Cx43 polyclonal antibody (Zymed, South San Francisco, CA.) Western blot of total proteins from vehicle- and tamoxifen-treated C6–Cx43+ cells using the same Cx43 antibody and anti-β-actin monoclonal antibody (Sigma) was performed as described previously (Cotrina et al., 1998a). To evaluate gap-junction coupling, cultures were loaded with calcein AM (2 μm) and subjected to the fluorescence recovery after photobleach (FRAP) analysis as described previously (Cotrina et al., 1998a; Lin et al., 1998). The cortical and hippocampal tissues from wild-type (WT) and Cx43 knock-out (KO) mice were immunostained and analyzed with Western blot as the cultured cells describe above and with additional monoclonal antibodies against GFAP (Sigma).
Connexin knock-out transgenics, slice preparation, and Lucifer yellow diffusion.
Conditional deletion of Cx43 in astrocytes was achieved by the inactivation of floxed Cx43 by Cre recombinase, the expression of which is driven by the human GFAP promoter (Theis et al., 2003). Because deletion of Cx43 is partly compensated by increased expression of Cx30, double transgenic mice lacking both Cx43 and Cx30 were generated (Wallraff et al., 2006). In all experiments, Cx30−/−Cx43fl/fl and Cx30−/−Cx43fl/fl:hGFAP–Cre (double deletion of Cx30 and Cx43) littermates were compared. Cortical slices from wild-type Cx43:Cx30 double knock-out juvenile mice (P18–P25) were prepared as described previously (Kang et al., 1998). Astrocytes were distinguished by small, round cell bodies (<10 μm) without visible processes under DIC microscope, a high resting membrane potential (∼80 mV), small input resistance (<20 MΩ), and the failure of depolarizing voltage steps to evoke action potentials. Whole-cell recordings were obtained by using a Multiclamp amplifier (Molecular Devices). The pipette solution contained 2 mg/ml Lucifer yellow, and images were collected 20 min after a stable patch was established. Lucifer yellow was excited at 850 nm by a Mai Tai laser (Spectra-Physics, San Jose, CA) attached to a scanning system (Fluoview; Olympus Optical). Emission was collected at 575–645 nm.
Hypoxic preconditioning and middle cerebral artery occlusion.
Adult C57BL/6 mice (8–10 weeks) with conditional deletion of Cx43 in astrocytes were used (Wallraff et al., 2006). In all experiments, Cx43fl/fl (control) and Cx43fl/fl:hGFAP–Cre (deletion of Cx43) littermates were compared. Because deletion of Cx43 is partly compensated by increased expression of Cx30, double transgenic mice lacking both Cx43 and Cx30 were used (Wallraff et al., 2006). C57BL/6 mice are relatively sensitive to ischemia, and the middle cerebral artery (MCA) was maximally occluded for 45 min (Connolly et al., 1996). For hypoxic preconditioning, animals were transferred to an airtight chamber for 5 h with a total gas flow of 1000 ml/min of 92% N2 and 8% O2 or compressed air (controls). A total gas flow of 1000 ml/min of 92% N2 and 8% O2 was maintained by use of a flowmeter (Dwyer, Michigan City, IN) and partial oxygen pressure was measured intermittently by means of an oxygen analyzer (OXOR; Bacharach, Pittsburgh, PA). For the control groups, compressed air was used at the same flow rate. Seventy-two hours later, the mice were anesthetized with ketamine/xylazine (100 and 10 mg/kg, respectively). The right middle cerebral artery was occluded for 45 min by a 7-0 polypropylene monofilament (Ethicon, New Brunswick, NJ) coated with silicon resin inserted through the internal carotid artery. Cortical blood flow was continuously monitored by laser Doppler flowmetry positioned 2 mm posterior and 5 mm lateral from the bregma (Perimed, Piscataway, NJ). Rectal temperature was maintained at 37 ± 0.5°C using a feedback-controlled heating system (Harvard Apparatus, Holliston, MA). DPCPX (8-cyclopentyl-1,3-dipropylxanthine) was administered in a single dose of 1 mg/kg (intraperitoneally) immediately before hypoxic preconditioning or immediately before 30 min occlusion of the MCA (Sigworth and Rea, 2003). Twenty-four hours after reperfusion, the animals were decapitated, and 1-mm-thick Vibratome sections prepared. The sections were stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC) (Sigma) and fixed with 4% paraformaldehyde. The size of the ischemic lesion was measured on digital images of the caudal face of all sections, and infarct volume was calculated using NIH ImageJ software.
Results
Preconditioning upregulates Cx43 expression in cultured astrocytes
We first asked whether cultured astrocytes exhibited increased resistance to injury after exposure to a sublethal, preconditioning stimulation. Primary astrocytic cultures were exposed to H2O2, tamoxifen, or their respective vehicles (Lin et al., 1998). Twenty-four hours later, the cultures were again exposed to H2O2 or tamoxifen but at higher concentrations to induce irreversible cell damage. Consistent with previous studies (Furuichi et al., 2005; Xiao-Qing et al., 2005; Yu et al., 2006), preexposure to sublethal injury markedly increased cellular resistance: 89.6 ± 1.9% of H2O2 preexposed astrocytes survived subsequent exposure to H2O2, whereas only 30.9 ± 14.6% survival was evident in control vehicle-treated cultures (Fig. 1A) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Similarly, tamoxifen increased the resistance of astrocytes exposed to a preconditioning stimulation to 72.6 ± 7.1% compared with 13.9 ± 5.6% survival of cultures exposure to vehicle (Fig. 1B).
Figure 1.

Preconditioning increased cellular resistance and Cx43 expression of cultured astrocytes. A, Exposure to a preconditioning or sublethal concentration of H2O2 (0.8 mm) increased resistance to subsequent exposure to a higher concentration of H2O2 (1.4 mm, 24 h later). Data represent average ± SE (n = 3–5). **p < 0.01, Tukey–Kramer test. B, Exposure to a sublethal concentration of tamoxifen (27.5 μm) increased resistance to subsequent exposure to higher concentration of tamoxifen (37.5 μm, 24 h later). Data represent average ± SE (n = 7–9). **p < 0.01, Tukey–Kramer test. C, Top, Cx43 immunolabeling (white) increased after a preconditioning exposure to tamoxifen. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). Scale bar, 11 μm. Bottom, Comparison of relative changes in Cx43 immunolabeling as a function of time after exposure to vehicle and preconditioning (sublethal concentration of tamoxifen). Mean fluorescence intensity across a field (450 × 600 μm) was measured in three to eight matched cultures. Data represent average ± SE. **p < 0.01, Tukey–Kramer test. D, The increase in Cx43 expression was not matched by an increase in gap-junction coupling analyzed by fluorescence recovery after photobleach. Data represent average ± SE (n = 4).
Astrocytes express several different types of connexins, including Cx30 and Cx43 (Theis et al., 2004). Cx43 is the predominant connexin isoform and accounts for most functional gap junctions in astrocytes (Dermietzel et al., 2000). Cx43 was present in the plasma membrane of cultured astrocytes in variable-sized plaques located primarily in regions of cell–cell contacts. Preconditioning stimulation lead to an intense increase in Cx43 immunolabeling and a spatial reorganization. The Cx43 plaques increased in both size and number. At the same time, diffuse labeling of the cytosol increased and the Golgi apparatus was in many cells outlined by high Cx43 immunoreactivity after preconditioning stimulation (Fig. 1C, inset). Quantification of the relative changes in Cx43 immunoreactivity showed that Cx43 labeling peaked at 6–8 h but remained elevated at 24 h (Fig. 1C). Gap-junction coupling was evaluated in sister cultures using the FRAP technique. This non-invasive assay showed that gap-junction coupling remained stable after preconditioning, despite the increase in Cx43 expression (Fig. 1D).
Combined, these observations indicate that preconditioning markedly increased the resistance of cultured astrocytes to subsequent additional severe insults. The gap-junction protein Cx43 exhibited a transient increase in expression after preconditioning, but functional gap-junction coupling remained essentially constant.
Preconditioning requires Cx43 expression
To establish the role of Cx43 in preconditioning, we next analyzed the effect of forced Cx43 expression in C6 cells, a glial cell line with low endogenous levels of connexins (Zhu et al., 1991; Cotrina et al., 1998b; Lin et al., 1998). Stably transfected C6 clones expressing Cx43 (C6–Cx43+ cells) were compared with Cx-deficient sister cells expressing an empty vector (C6–Cx43− cells). Similar to primary cultures of astrocytes, C6–Cx43+ cells exhibited a marked increase in cellular resistance after preconditioning, whereas similar exposure had no effect on the injury tolerance of C6–Cx43− cells (Fig. 2A). Conversely, C6–Cx43+ cells lost their injury tolerance when Cx43 expression was reduced by siRNA before preconditioning compared with control cultures treated with Stealth RNAi low GC duplex, 24.3 ± 7.6 vs 60 ± 8.1% survival, respectively (p < 0.05) (Fig. 2B). Thus, both sets of experiments suggested that Cx43 expression is a prerequisite for preconditioning. Furthermore, several Cx channel inhibitors, including carbenoxelone, octanol, flufenamic acid, and nifluphemic acid, attenuated the increase in injury resistance after preconditioning, indicating that open Cx43 channels are required for protection afforded by preconditioning (Fig. 2C). Because selective blockers of gap junctions versus hemichannels do not exist, these experiments could not discriminate between the two types of channels formed by Cx43. To address this question, C6–Cx43+ cells were next plated at a low density to prevent cell–cell contact and thereby formation of gap junctions. We found that low-density cultures of C6–Cx43+ cells increased injury resistance after preconditioning, thus indicating that gap junctions play an insignificant role in preconditioning (Fig. 2D). Of note, C6–Cx43 cells are flat epitheloid cells, whereas C6–Cx43− cells are more rounded and exhibit less cell–cell contact (Lin et al., 2003). The changes in morphology induced by transfection with Cx43 are not responsible for the increased injury resistance of C6–Cx43+ cells (Lin et al., 2003).
Figure 2.

Cx43 expression is required for preconditioning of C6 cells. A, Comparison of preconditioning stimulation of C6–Cx43+ and C6–Cx43− cells (preconditioning exposure to tamoxifen, 17.5 μm; lethal exposure, 35 μm, respectively). Data represent average ± SE (n = 3–4). **p < 0.01, Tukey–Kramer test. B, Reduced Cx43 expression in C6–Cx43+ cells by siRNA abrogated the protection afforded by precondition. Top shows Cx43 immunoreactivity (white) with DAPI (blue) in cultures treated with Stealth RNAi and Cx43 siRNA in parallel. Scale bar, 13 μm. Data represent average ± SE (n = 4–5). *p < 0.05, Tukey–Kramer test. C, Several Cx channel inhibitors, including carbenoxolone (CBX; 100 μm), octanol (OCT; 500 μm), flufenamic acid (FFA; 100 μm), and niflemic acid (NFA; 100 μm), attenuated the increased resistance after preconditioning. Data represent average ± SE (n = 6–9). **p < 0.01, Tukey–Kramer test. D, Top, A low-density culture of C6–Cx43+ that is devoid of cell–cell contact as opposed to the confluent culture (inset). Scale bars, 50 μm. Bottom, Precondition-afforded resistance does not require cell–cell contact. Data represent average ± SE (n = 4–14). **p < 0.01, Tukey–Kramer test. E, Top left, Cx43 immunolabeling (white) increased after preconditioning (tamoxifen). Nuclei were stained with DAPI (blue). Scale bar, 50 μm. Top right, Dose-dependent increase of Cx43 protein after exposure to sublethal concentrations of tamoxifen. Bottom, Comparison of relative changes in Cx43 immunolabeling (white) as a function of time after exposure to vehicle and preconditioning (20 μm tamoxifen). Scale bar, 10 μm. Data represent average ± SE (n = 4). **p < 0.01, Tukey–Kramer test.
C6–Cx43 cells expressed Cx43 under the constitutive CMV promoter. Nevertheless, Cx43 expression was clearly increased in C6–Cx43 cells after precondition (Fig. 2E). The increase in abundance of Cx43 likely results from slowing of the rate of protein degradation: Cx43 has a rapid half-life of only 1.5–5 h, and cytosolic stress, including mild hyperthermia and oxidative stress, has been shown previously to slow degradation of Cx43 (VanSlyke and Musil, 2002, 2005; Berthoud et al., 2004). In support of these reports, we found that cellular stress associated with preconditioning increased the abundance Cx43 protein in both a time- and dose-dependent manner (Fig. 2E).
These studies indicated that Cx43 expression is a prerequisite for preconditioning in cultured C6 cells and astrocytes. Furthermore, gap-junction formation did not appear to be critical, because low-density cultures responded well to preconditioning despite their lack of cell–cell contact. Combined with the potent effect of pharmacological inhibition of channel opening, the observations point to a role of hemichannels rather than gap junctions in injury resistance afforded by preconditioning.
Preconditioning is linked to a marked increase in Cx43 hemichannels
To examine the involvement of Cx43 hemichannel in preconditioning, we next asked whether hemichannel opening was modulated by preconditioning. Hemichannels are exposed to the interstitial space and gated by extracellular Ca2+ (Bennett et al., 2003). Uptake of fluorescence indicators, such as propidium iodine (molecular weight of 500 Da, 0.5 mm), in response to removal of extracellular Ca2+ is a commonly used experimental approach to evaluate hemichannel opening (Li et al., 1996; Cotrina et al., 1998b; Arcuino et al., 2002). We found that C6–Cx43+ cells exhibited a striking increase in propidium iodide uptake after preconditioning, with a more than 20-fold increase in number of propidium iodine-positive cells (Fig. 3A). The percentage of propidium iodide-positive cells increased from 0.91 ± 0.05 to 20.5 ± 1.56% after preconditioning. In contrast, preconditioning had no effect on the very low fraction of C6–Cx43− cells, which were labeled with propidium iodide, when exposed to a Ca2+-free solution (Fig. 3A). The increase in propidium uptake by preconditioned C6–Cx43+ cells was not attributable to compromised cellular integrity. Propidium iodide was barely detected in these cells before their exposure to a Ca2+-free solution. The health state of these preconditioned cells was further reflected in their ability to take up and retain the viability indicator calcein (supplemental Fig. 3, available at www.jneurosci.org as supplemental material)
Figure 3.

Preconditioning cause an increase in Cx43 hemichannels. A, Assessment of hemichannels by dye uptake. Left, Phase-contrast images of C6–Cx43+ and C6–Cx43− cells incubated for 2 min in a Ca2+-free solution containing 0.5 mm propidium iodide (white arrows) 24 h after preconditioning. Scale bar, 100 μm. Right, Preconditioning caused a significant increase in propidium iodide uptake in C6–Cx43+ cells but not in C6–Cx43− cells. Data represent average ± SE (n = 22). **p < 0.01, Tukey–Kramer test. B, ATP release was similarly increased by preconditioning (PC). Data represent average ± SE (n = 5–6). *p = 0.0115, t test. C, Cx channel blockers reduced ATP release from C6–Cx43+ cells: carbenoxolone (CBX; 100 μm), flufenamic acid (FFA; 100 μm), niflemic acid (NFA; 100 μm), and 18-α glycyrrhetinic acid (18GA; 50 μm). Data represent average ± SE (n = 5). **p < 0.01, Tukey–Kramer test. D, Recordings of Cx43 hemichannel openings in cell-attached patches from C6–Cx43 cells. Representative cell-attached patch recordings with the pipette solution containing 130 mm Na-ATP and 20 mm TEA, depict opening (o) and closing (c) of a Cx43 hemichannel in a cell-attached patch from a C6–Cx43+ cell (Cx43+ Con). The bottom trace is enlarged scale of the squared area, showing the slow gating subconductance. E, Cell-attached recordings at different membrane potentials. Voltage steps of −160 to 0 mV with each step of 20 mV were applied to the patch pipette. Membrane potential (Vm) that was the difference between a voltage step and the RMP was presented at the left of each trace (in millivolts). F, The I–V curve from recordings at different voltage steps (left) and I–V current (right) for the channel in A in response to a ramp command applied to the patch pipette from −160 to 0 mV. The membrane potential was corrected by RMP. The reversal potential (VRev) is close to 0 mV. G, A cell-attached patch from a preconditioned C6–Cx43+ cell showed opening of two Cx43 hemichannels (o1 and o2). H, The I–V current for the channels in G. The VRev is similar to the control Cx43 hemichannel in F. I, Representative cell-attached patch recordings from a C6–Cx43− cell (Cx43− control) and a preconditioned C6–Cx43− cell (Cx43− precondition) with no Cx43 hemichannel activity. J, Comparison of the numbers of Cx43 hemichannels per patch in C6–Cx43+ cells (Cx43+ control), preconditioned C6–Cx43+ cells (Cx43+ PC), C6–Cx43− cells (Cx43− control), and preconditioned C6–Cx43− cells (Cx43− PC). Number in each column indicates sample size.
The large pore diameter of Cx43 hemichannels (∼1.2 nm) allows diffusion of several cytosolic metabolites, including ATP and glutamate (Cotrina et al., 1998b; Stout et al., 2002; Ye et al., 2003). We reasoned that, if preconditioning increased the number of functional hemichannels, then we would expect that ATP release also rose after preconditioning. As expected, preconditioned C6–Cx43+ cells, but not C6–Cx43− cells, exhibited a fivefold increase in ATP release compared with vehicle-treated control cells (Fig. 3B). Of note, the preconditioning paradigms used in this study did not significantly alter cytosolic ATP concentrations or cellular metabolic state (cytosolic ATP content of preconditioned C6–Cx43+ cells was 103 ± 4% relative to vehicle-treated control). Several Cx channel inhibitors reduced ATP release (Fig. 3C), indicating that hemichannels, rather than alternative ATP release mechanisms, such as exocytosis or multiple drug resistance proteins, constitute the primary efflux pathway for ATP (Ballerini et al., 2002; Coco et al., 2003). Interestingly, the same channel inhibitors attenuated protection afforded by preconditioning (Fig. 2C), suggesting a link between increased ATP release and injury protection after preconditioning. We were unable to induce precondition by exposure to purinergic receptor agonists. Purinergic receptors are characterized by a marked and prolonged desensitization. Chronic exposure to purinergic agonists is associated with reduced responses to the agonists because the receptors are internalized (Arcuino et al., 2002).
Cell-attached patch recordings from C6–Cx43+ cells were next used to examine whether precondition was associated with an increased number of hemichannel number or changes in opening times of individual channels. To activate Cx43 hemichannels, a voltage step of −160 mV was applied to the cell-attached patch pipette (Vp), so that intracellular depolarization of +80 mV was obtained if the resting membrane potential (RMP) was −80 mV (Contreras et al., 2003). The actual membrane depolarization for each cell was justified by measuring RMP at the end of experiments. An upward current indicated an outward current across the patch membrane. A channel with a low opening probability (Table 1, Po) was observed in 31% (20 of 64) of cell-attached patches (Fig. 3D, o). Putative Cx43 hemichannels in a cell-attached patch showed a mean amplitude of 15.5 ± 0.3 pA (Table 1, A). To examine the I–V relationship and reversal potentials (VRev) for putative Cx43 hemichannels, voltage steps or a ramp command (Vp) from −160 to 0 mV were applied to cell-attached patch pipettes. Recordings at different membrane potentials showed that channel openings occurred at more depolarization range (Fig. 3E), suggesting that channel openings are voltage dependent. The I–V curve (Fig. 3F, left) and I–V current (Fig. 3F, right) indicated that channel openings reversed at near the membrane potential of 0 mV (Fig. 3F, VRev). The slope conductance of putative Cx43 hemichannels was calculated with I–V curves. The putative Cx43 hemichannel had a fast opening conductance of 165 ± 6 pS (n = 10 patches) that is approximately double of the fast gating gap-junction channel current (85 pS) (Bukauskas and Verselis, 2004). A subconductance status with slowly gating and long opening property was occasionally observed (Fig. 3D, bottom enlarged trace). The conductance of the slowly gating state was 15 ± 0.5 pS (Table 1, γsub). The conductance of Cx43 hemichannel openings was smaller than observed previously using whole-cell recordings (Endoh et al., 2001; Contreras et al., 2003) and may reflect that the opening state of Cx43 channels is sensitive to the recordings conditions. A possible explanation is that loss of cytosolic components modulate the gating properties of Cx43 hemichannels similar to other channels (Donevan and Rogawski, 1995). Precondition induced a significant increase in the number of hemichannels per patch to 73% (45 of 62 patches) (Fig. 3D,G, Cx43+, PC), suggesting that precondition enhances the channel density of Cx43 hemichannels in the plasma membrane (Fig. 3G). The channel characteristics, including conductance, VRev, and kinetics (γ, VRev, τo, τc1, and τc2) were unaltered by preconditioning (Fig. 3G–I, Table 1). Although the mean open probability of hemichannels showed a trend toward an increase in preconditioned cells compared with controls, it was not statistically significant (Table 1, Po) (p = 0.22). Because P2X7 channels have been reported to mediate ATP release (Suadicani et al., 2006), we next added the P2X7 receptor blocker Brilliant blue G (1 mm) to the pipette solution. In the presence of Brilliant blue G in the pipette, 30% (6 of 20) of cell-attached patches still showed similar hemichannel openings compared with 35% (7 of 20) of control patches. Thus, both the electrophysiological characteristics of the putative Cx43 channels as well as the lack of sensitivity to Brilliant G blue indicate that it is not a P2X7 channel. In sharp contrast to C6–Cx43+ cells, cell-attached patch recordings from control C6–Cx43− cells showed no channel openings in response to depolarization. Furthermore, preconditioning failed to promote channel opening in C6–Cx43− cells (Fig. 3J, C6–Cx43−).
Table 1.
Cx43 hemichannels from control and preconditioned C6–Cx43 cells
| A (pA) | γ (pS) | Asub (pA) | γsub (pS) | Po | τo (ms) | τc1 (ms) | τc2 (ms) | |
|---|---|---|---|---|---|---|---|---|
| Con | 15.5 ± 0.3 | 180 ± 3.0 | 1.4 ± 0.1 | 15 ± 0.5 | 0.008 ± 0.003 | 5.2 ± 0.9 | 3.4 ± 1.0 | 147 ± 30 |
| Prec | 15.9 ± 0.4 | 180 ± 2.5 | 1.4 ± 0.1 | 15 ± 0.6 | 0.016 ± 0.003 | 5.9 ± 0.8 | 2.6 ± 0.7 | 104 ± 34 |
The putative Cx43 hemichannel had a full opening conductance of 180 ± 3 pS (n = 10 patches). A subconductance status with slowly gating and long opening property was occasionally observed (Fig. 3D, bottom enlarged trace). The conductance of the slowly gating status was 15 ± 0.5 pS (γsub) smaller than the slowly gating status for whole Cx43 gap-junction channels reported previously by other groups (Valiunas et al., 1997), suggesting that the slow gating status of Cx43 channels is variable under different conditions. The fast gating conductance was 165 ± 3 pS, which is approximately double of the fast gating gap-junction channel current (85 pS) (Bukauskas and Verselis, 2004).
This set of observations indicated that the primary mechanism by which preconditioning increases hemichannel openings is by increasing the number of open plasma membrane Cx43 channels rather than changing the gating properties or opening time. Because the abundance of Cx43 protein increased after preconditioning in C6–Cx43+ cells (expression is driven by the CMV promoter) (Figs. 1, 2), these observations support the notion that the increase in channel opening was the result of a reduced rate of removal/degradation of Cx43 hemichannels from the plasma membrane.
Injury tolerance requires functional hemichannels
To gain additional insight into the mechanisms involved in injury tolerance after preconditioning, we next took advantages of the observation that Cx43 fused with enhanced green fluorescent protein (Cx43–eGFP) is inserted into the plasma membrane and forms plaques but not functional hemichannels. Cx43–eGFP hemichannels exhibited only the slow not the fast transition between closed and fully open states (Bukauskas and Verselis, 2004). C6 cells stably transfected with Cx43–eGFP under the CMV promoter, C6–Cx43–eGFP cells, exhibited low eGFP fluorescence signal in accordance with previous reports (John et al., 1999) (Fig. 4A). After preconditioning, Cx43–eGFP fluorescence increased markedly similar to Cx43 wild type (Figs. 1, 2). The enhanced fluorescence signal was evident in plaques and in the cytosol, as well as an intense increase in Cx43–eGFP over the Golgi apparatus (Fig. 4A). C6 cells expressing GFP, rather than the Cx43–eGFP fusion protein, displayed an even fluorescence signal across the cell body that remained unaffected by preconditioning. In accordance with the notion that Cx43–eGFP channels are not functional, propidium iodide uptake remained below 1% after preconditioning (Fig. 4B), despite the intense increase in Cx43–eGFP fluorescence signal. Cell-attached patch recordings provided clear evidence that C6–Cx43–eGFP did not form functional Cx43 hemichannels during control condition and after preconditioning (Fig. 4C). Similar to previous reports (Contreras et al., 2003), we never observed fast openings in C6–Cx43–eGFP cells (Fig. 4C) but occasionally observed opening of the slowly gating subconductance state. These slow openings were observed both before and after preconditioning similar to the observation in cells expressing Cx43 wild-type (Fig. 3D). Not unexpectedly, ATP release did not increase after preconditioning (Fig. 4D), and C6–Cx43–eGFP cells were insensitive to preconditioning (Fig. 4E).
Figure 4.

Cx43–eGFP fluorescence signal increase is not linked to increased hemichannel openings or increased injury tolerance. A, Sequence of images of C6–Cx43–eGFP after preconditioning stimulation. Cx43–eGFP fluorescence signal increased after preconditioning, similar to Cx43 wild-type (Figs. 1, 2). Inset, C6 cell transfected with GFP only displays an even fluorescent signal across the cell body that was unaffected by precondition. Scale bar, 15 μm. B, Uptake of propidium iodine remained below 1% despite the marked increase in Cx43–eGFP signal. Nevertheless, the relative number of cells with propidium iodide uptake increased significantly. Data represent average ± SE (n = 15–18). *p < 0.05, t test. Scale bar, 100 μm. C, Cx43–eGFP cells did not display fast gating Cx43 hemichannel subconductance either before or after preconditioning. Small arrows indicate opening and closing of the slow gating subconductance, which was observed both before and after preconditioning. Histogram compare the effect of preconditioning on number of open patches with hemichannel activity (fast gating subconductance) in C6 cells expressing Cx43 or Cx43–eGFP before and after preconditioning. Number in each column indicates sample size. D, ATP release did not increase after preconditioning of Cx43–eGFP cells. Data represent average ± SE (n = 12). E, Preconditioning failed to increase injury resistance after preconditioning of C6–Cx43–eGFP cells. Data represent average ± SE (n = 3–4).
Combined, these data add further support to the concept that an increase in Cx43 hemichannel openings is a prerequisite for injury tolerance induced by preconditioning. Although, the increase in Cx43–eGFP fluorescence closely mimicked the dynamic changes observed of Cx43 wild type, it was not linked to an increase in functional hemichannels and thereby not to an increase in ATP release and injury resistance.
Inhibitors of lysosomes replicate the effect of preconditioning
Changes in the abundance of Cx43 can be achieved through regulation of Cx43 synthesis or degradation (Laird, 2005; Salameh, 2006). Cx43 is translated in the rough endoplasmic reticulum and traffics through the Golgi compartment, before six connexins oligomerize to form a connexon. Connexon-containing vesicles are transported from the Golgi to the cell surface in which connexons move laterally to dock with connexons in neighboring cells to become a part of Cx43 gap-junction plaques (Lauf et al., 2002). Cx43 is degraded by both lysosomes and proteasome with cell-specific involvement of the two pathways (Laing et al., 1998; Musil et al., 2000). Cx43 targeted for degradation is transported from the plasma membrane in endosomes but is recycled back to the plasma membrane in cells treated with inhibitors of protein degradation (VanSlyke and Musil, 2003). As discussed above, increases in the abundance of Cx43 likely resulted from a slowing of degradation in C6–Cx43+ cells, because Cx43 expression is driven by the constitutive CMV promoter (Cotrina et al., 1998b).
We reasoned that, if a reduced rate of degradation of Cx43 was critical for injury resistance after preconditioning, then pharmacological inhibition of protein degradation should replicate the effect of preconditioning, e.g., increase the abundance of Cx43 and raise cellular injury resistance in parallel. This postulate was correct in that exposure to inhibitors of lysosomal activity, NH4Cl or CLQ (VanSlyke and Musil, 2005), increased the abundance of Cx43 and protected against injury, thereby mimicking the cellular response to preconditioning stimulation (Fig. 5A,B). Interestingly, exposure to either NH4Cl or CLQ was linked to a marked increase in plasma membrane hemichannels detected by propidium iodide uptake and a several fold increase in ATP release (Fig. 5C,D). In contrast, inhibitors of proteasomes, including MG132, lactacystin, and epoxomicin, only insignificantly increased the Cx43 immunolabeling and had no effect on cellular resistance (Fig. 5A,B). Furthermore, MG132, lactacystin, and epoxomicin failed to increase propidium iodide uptake or ATP release (Fig. 5C,D), suggesting that Cx43 is degraded primarily by lysosomes in C6–Cx43 cells, similar to some but not all cell lines studied so far (Berthoud et al., 2004).
Figure 5.

Inhibitors of protein degradation induced preconditioning. A, Cx43 immunocytochemistry (red) of C6–Cx43+ cells 6 h after exposure to control vehicle (control), tamoxifen-preconditioned (precondition), or 5 h after exposure to inhibitors of lysosomes NH4Cl (10 mm) and chloroquine (100 μm) and of proteasomes MG132 (40 μm), lactacystin (10 μm), and epoxomycin (10 μm). Preconditioning and lysosomal inhibitors increased the Cx43 expression, whereas proteasome inhibitors were ineffective. Scale bar, 8 μm. B, Comparison of tamoxifen resistance after exposure to same agents as in A. Data represent average ± SE (n = 3–7). *p < 0.05, Tukey–Kramer test. C, Propidium iodide (red) uptake after similar exposure as in A. Scale bar, 100 μm. D, ATP release after similar exposure as in A. Data represent average ± SE (n = 4–9). *p < 0.05, **p < 0.01, Tukey–Kramer test.
Thus, these experiments indicate that preconditioning slows Cx43 degradation, resulting in an increase in plasma membrane Cx43 hemichannels, which in turn increases ATP release. Because lysosomal inhibitors replicate the effect of preconditioning, our data also suggest that preconditioning protects against injury by interfering with delivery of Cx43 to the lysosomes.
Adenosine mediates injury resistance after preconditioning
Adenosine is a potent neuroprotective agent that reduced neuronal injury in the setting of ischemia, traumatic injury, epilepsy, and hypoglycemia (Dunwiddie and Masino, 2001; Lankford et al., 2006). Interestingly, preconditioning also involved adenosine, which in an A1 receptor-mediated pathway reduced ischemic injury after preconditioning (Heurteaux et al., 1995). Because preconditioning of C6–Cx43+ cells was associated with a marked increase of ATP release, we next tested whether preconditioning led to accumulation of extracellular adenosine. ATP is, after release, rapidly hydrolyzed to adenosine by extracellular ectonucleotidases (Dunwiddie et al., 1997). Adenosine was measured in samples of culture medium collected before and after preconditioning of C6–Cx43+ cells using adenosine biosensors. We found that preconditioning consistently increased adenosine concentrations from undetectable values during resting conditions to ∼2–3 μm after preconditioning (Fig. 6A). In contrast, adenosine concentrations were below detection in culture medium harvested from C6–Cx43− cells (data not shown).
Figure 6.

Adenosine mediates injury tolerance after preconditioning. A, Adenosine concentration detected by an adenosine biosensor in the medium of C6–Cx43+ and C6–Cx43− cells after exposure to vehicle or preconditioning (PC) stimulation. Data represent average ± SE (n = 5–6). **p < 0.0001, t test. B, MRS1754, an antagonist of adenosine A2b receptors, attenuated injury tolerance after preconditioning. Data represent average ± SE (n = 12). **p < 0.01, Tukey–Kramer test. C, An array of P2 receptors antagonist {suramin, reactive blue (RB), PPADS, and MRS2179} and A1–2 receptor antagonist (caffeine, DPCPX) had no impact on the protection afforded by preconditioning. Data represent average ± SE (n = 3–22). *p < 0.05, **p < 0.01, Tukey–Kramer test.
Given that C6–Cx43+ cells express both P1 and P2 purinergic receptors, ATP has the potential to activate multiple types of receptors, known to modulate the cellular injury resistance (Brismar, 1995; Abbracchio and Verderio, 2006; Franke et al., 2006; Kobayashi et al., 2006). To evaluate the relative impact of purinergic receptors on the resistance of C6–Cx43+ cells after preconditioning, we compared the effect of several P1 or P2 receptor antagonists. The analysis showed that neither reactive blue (10–30 μm), suramin (50–400 μm), PPADS (pyridoxal-phosphate-6-azophenyl-2′,4′-disulfonic acid) (50–100 μm), MRS2179 (2′-deoxy-N6-methyladenosine-3′,5′-bisphosphate) (1–100 μm), nor any of the P2 receptor antagonists, had a significant effect on injury protection after preconditioning. In contrast, an adenosine A2b antagonist, MRS1754 {N-(4-cyanophenyl)-2-{4-(2,3,6,7-tetrahydro-2,6-dioxo-1, 3-dipropyl-1H-purin-8-yl)phenoxy}-acetamide} (100 nm), attenuated the protection afforded by preconditioning. Adenosine A2b receptors have a relatively low-affinity constant of 5.1 μm, but the increases in extracellular adenosine after preconditioning were sufficiently high to activate A2b receptors. No effect of the A1 receptor antagonist DPCPX (0.1–1 μm) was noted, consistent with the low expression of A1 receptors in C6 cells and in cultured astrocytes (Jimenez et al., 1999; Rebola et al., 2005) (our unpublished observations) (Fig. 6B).
No protection after hypoxic preconditioning of Cx43-deficient mice
Astrocytes are the predominant cell type in CNS, which express Cx43 (Dermietzel et al., 2000; Simard et al., 2003). To evaluate the role of Cx43 in protection afforded by hypoxic preconditioning in intact brain, we next examined mice with deletion of Cx43 (Frisch et al., 2003; Theis et al., 2003). These mice (Cx43fl/fl:hGFAP–Cre) were generated by crossing Cx43fl/fl mice with hGFAP–Cre mice. Astrocytes also at a low level express Cx30. Because deletion of Cx43 is partly compensated by increased expression of Cx30 in astrocytes, double transgenic mice lacking both Cx43 and Cx30 were used (Wallraff et al., 2006). Littermates of these animals without Cre recombinase and with deletion of Cx30, e.g., Cx43fl/flCx30−/− mice, served as controls. As expected, Western blot or immunohistochemical analysis showed that Cx43 protein was absent in Cx43fl/fl:hGFAP–Cre (KO) but not in Cx43fl/fl mice (WT) (Fig. 7A). Functional coupling was quantified by intra-astrocytic injection of Lucifer yellow in slice preparations (P18–P25). Slices from Cx43fl/fl:hGFAP–Cre mice exhibited essentially no intercellular diffusion of Lucifer yellow, confirming the absence of functional astrocytic gap-junction coupling. In contrast, astrocytes in slices from littermate Cx43fl/fl mice were extensively coupled to neighboring cells (Fig. 7B).
Figure 7.

Cx43 expression is required for preconditioning in vivo. A, Western blot of hippocampus and cortex from a WT mouse (Cx43fl/fl) and from a Cx43 KO mouse (Cx43fl/fl:hGFAP–Cre). B, Intercellular diffusion of Lucifer yellow in astrocytes in slices from WT (red arrows indicate Lucifer yellow labeling of neighboring cells) and KO mice. Hippocampal slices were prepared from P18–P25 mice. Histogram compares diffusion of Lucifer yellow into neighboring cells in WT and KO. Only Lucifer yellow-positive cells within the field of view was included in the analysis (150 × 150 μm). Scale bar, 30 μm. Data represent average ± SE (n = 4–8). *p < 0.05, Tukey–Kramer test. C, Cx43 (white, small red arrows) and GFAP (green) immunohistochemistry from wild-type and Cx43 KO before or 24 and 72 h after preconditioning. Nuclei were stained with DAPI (blue). D, Comparison of infarct volume after MCA occlusion, preconditioning and MCA occlusion, and preconditioned mice that received one injection of the adenosine A1 receptors antagonist DPCPX (1 mg/kg, i.p.) immediately before hypoxic preconditioning. Data represent average ± SE (n = 6–8). E, Comparison of infarct volumes in mice with 30 min occlusion of the MCA. DPCPX-treated mice received an injection of 1 mg/kg intraperitoneally immediately before the occlusion. Data represent average ± SE (n = 6–8). *p < 0.05, **p < 0.01, Tukey–Kramer test.
After hypoxic preconditioning (8% O2, 5 h), Cx43 immunoreactivity increased markedly, similar to observations in vitro (Figs. 1, 2). The Cx43-positive plaques increased in both size and number in Cx43fl/fl mice exposed to preconditioning stimulation, whereas Cx43 immunolabeling was below detection in Cx43fl/fl:hGFAP–Cre mice, both before and after preconditioning. In addition, GFAP expression was increased at both 24 and 72 h in Cx43fl/fl:hGFAP–Cre and Cx43fl/fl mice without notable differences (Fig. 7C).
Focal ischemia was next induced by inserting a filament in the middle cerebral artery during continuous recording of cerebral blood flow by laser Doppler flowmetry. The filament was withdrawn after 45 min, and TTC-stained coronal sections were prepared 24 h later (Nakase et al., 2003b). Cx43fl/fl:hGFAP–Cre mice developed significantly larger infarcts than Cx43fl/fl mice in accordance with previous reports (p < 0.01, Tukey–Kramer test) (Nakase et al., 2003a,b). The larger infarct volume in Cx43fl/fl:hGFAP–Cre mice was not a consequence of a more severe reduction of cortical perfusion either during or after occlusion of the MCA, because flow reduction averaged 14.7 ± 1.3 and 14.8 ± 1.0% of control, preischemic values during MCA occlusion and rose to 71.4 ± 2.3 and 75.6 ± 2.3% in Cx43fl/fl:hGFAP–Cre mice and Cx43fl/fl mice, respectively (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Cx43fl/fl mice exposed to hypoxic preconditioning (5 h exposure to 8% O2, 72 h before MCA occlusion) exhibited a significant decrease in infarct volume (p < 0.001; n = 5–8) (Fig. 7D). Strikingly, the infarct-sparing effect of hypoxic preconditioning was abolished in Cx43fl/fl:hGFAP–Cre mice. Infarct volume in Cx43fl/fl:hGFAP–Cre mice averaged 59.5 ± 1.8 and 57.7 ± 3.3 mm3 (p > 0.05, Tukey–Kramer test) with and without hypoxic preconditioning (Fig. 7D). The failure of Cx43fl/fl:hGFAP–Cre mice to respond to preconditioning suggests that Cx43 expression is a prerequisite for hypoxic preconditioning, similar to the in vitro observations (Figs. 1–3). Because a large literature has shown previously that adenosine plays a central role in preconditioning (Nakamura et al., 2002; Liu et al., 2006b) and our in vitro analysis indicated that preconditioning was associated with a Cx43-dependent increase in adenosine (Fig. 6), we next tested the effect of the blood–brain barrier (BBB)-permeable adenosine A1 receptor antagonist DPCPX during preconditioning. We confirmed that DPCPX (1 mg/kg) administered before preconditioning eliminating protection afforded by hypoxic precondition in Cx43fl/fl mice (177% increase in infarct volume) and aggravated ischemic injury in Cx43fl/fl:hGFAP–Cre mice (25% increase in infarct volume). Thus, similar to the in vitro observations (Figs. 1–5), we found that Cx43 and adenosine play key roles in protection afforded by preconditioning.
Adenosine is also a neuroprotective agent that directly reduces ischemic injury in an adenosine A1 receptor-mediated pathway (Tsuchida et al., 1993; Dunwiddie and Masino, 2001). To evaluate whether Cx43 expression interferes with the direct neuroprotective action of adenosine, DPCPX was administered immediately before 30 min occlusion of the MCA. Interestingly, we found that DPCPX (1 mg/kg) increased the infarct volume of both Cx43fl/fl mice (56 ± 14%; p < 0.05) and Cx43fl/fl:hGFAP–Cre mice (16 ± 10%; p > 0.05; n = 8) (Fig. 7E). This observation indicates that activation of A1 receptors by endogenously released adenosine limits stroke injury in both Cx43fl/fl mice and Cx43fl/fl:hGFAP–Cre mice. However, the infarct reduction was only statistically significant in Cx43fl/fl mice (Fig. 7E). Of note, we were unable to test the effect of adenosine A2b receptor antagonist in the murine stroke model, because BBB-permeable A2b antagonists or mice with deletion of A2b receptors are not available (Yaar et al., 2005).
Thus, the in vivo experiments added additional critical support for a role of Cx43 in protection afforded by hypoxic preconditioning. Furthermore, the observations suggested that Cx43-dependent preconditioning was mediated, at least in part, by adenosine A1 receptors (Fig. 8).
Figure 8.
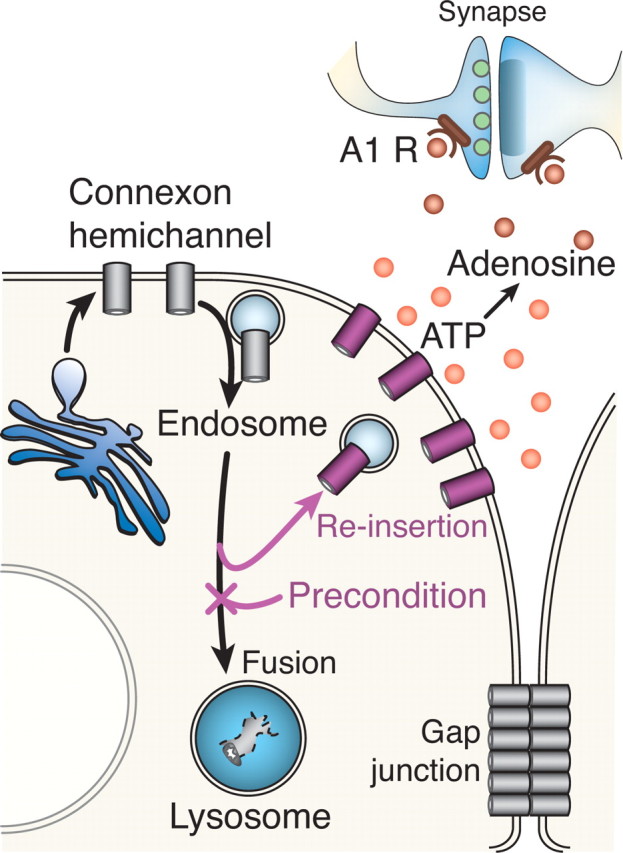
Proposed model for the role of Cx43 in preconditioning. Cx43 hemichannels are, after synthesis, transported from the Golgi to the plasma membrane. After insertion in the plasma membrane, Cx43 remains as hemichannels or hemichannels are either recruited into gap-junction plaques or endocytosed for degradation. Preconditioning reduces lysosomal degradation, resulting in reinsertion of endosomes containing Cx43 in the plasma membrane. In turn, the increased number of plasma membrane hemichannels leads to increased ATP release, resulting in accumulation of adenosine. Adenosine is a potent neuroprotective agent that reversibly inhibits excitatory transmission and thereby reduces excitotoxic neuronal injury.
Discussion
The main observation reported here is that protection afforded by preconditioning requires Cx43 expression and is linked to a marked increase in the number of functional hemichannels in the plasma membrane of glial cells. Cx43-deficient cells failed to increase injury resistance after preconditioning, and protection of Cx43-expressing cells was antagonized by siRNA against Cx43 or by pharmacologically blocking Cx43 channel opening. In parallel, an in vivo analysis showed that transgenic mice with deletion of Cx43 under the GFAP promoter were unresponsive to hypoxic preconditioning, whereas control littermates exhibited the expected reduction in infarct volume after occlusion of MCA. These observations indicate that Cx43, in addition to the pathways previously identified, is a prerequisite for preconditioning (Zimmermann et al., 2001; Kariko et al., 2004; Gustavsson et al., 2007; Lee et al., 2007). A key question is thereby how Cx43 participates in the pathways, which increases injury resistance after preconditioning. Our analysis showed that preconditioning caused a sharp increase in hemichannel opening detected by both whole-cell recordings and uptake of fluorescence indicators. Hemichannels serve as an efflux pathway for the intracellular energy metabolite ATP, which, during release, is degraded to adenosine. The notion that adenosine was an intermediary in Cx43-dependent injury protection was supported by the finding that adenosine accumulated in the culture medium after preconditioning of Cx43-expressing cells but remained below detection after similar treatment of Cx43-deficient cells. Furthermore, the adenosine A1-specific antagonist DPCPX reduced the neuroprotective effect of hypoxic preconditioning in WT (Cx43fl/fl) mice and Cx43 KO (Cx43fl/fl:hGFAP–Cre) mice but only statistically significant in WT (Cx43fl/fl) mice (Fig. 7).
During ischemia, extracellular adenosine increases to as high as 40 μm (Hagberg et al., 1987; Matsumoto et al., 1992). A number of studies suggest that A1 receptor activation plays a primary role in protection of excitatory neurons, whereas A2b receptors may aggravate injury. A1 receptors have a higher affinity for adenosine (∼70 nm) than both A2a (150 nm) and A2b receptors (5100 nm) (Dunwiddie and Masino, 2001), suggesting that a more widespread effect of A1 receptors in focal ischemia.
Experiments spanning several decades have documented that connexins are important participants in a variety of cellular processes during development, differentiation, morphogenesis, and cell death (Kumar and Gilula, 1996). In the setting of ischemia, connexins appear to play multiple roles. It is clear that gap junctions can expand injury in the process of bystander death. Gap-junction coupling is reduced but not blocked in the early stages of ischemia. As a consequence, proapoptotic agents can diffuse from dying to otherwise viable cells in the periphery of the lesion (Budd and Lipton, 1998; Cotrina et al., 1998a) and cause a significant degree of secondary injury in experimental models of stroke (Rawanduzy et al., 1997; Lin et al., 1998), head trauma (Perez Velazquez et al., 2003), and in early development (Cusato et al., 2003; de Pina-Benabou et al., 2005; Udawatte and Ripps, 2005). However, the impact of Cx expression extends beyond bystander death, because expression of Cx43, Cx36, Cx32, or Cx26 increased cellular injury resistance in a gap junction-independent pathway (Oguro et al., 2001; Lin et al., 2003; Striedinger et al., 2005). The phenotypic transformation that increases the injury threshold of Cx-expressing cells has not been established (Lin et al., 2003; Vetterlein et al., 2006) but may be similar to preconditioning involving efflux of neuroprotective agents through open hemichannels. Depending on the severity of injury, physiological parameters of the animal, or the experimental design, bystander death may outstrip adenosine-mediated neuroprotection or vice versa. For example, because infarct volume was quantified 24 h after MCA occlusion, we cannot exclude that bystander-dependent injury caused a delayed expansion of the ischemic lesions in Cx43fl/fl mice. It is also important to note that uncontrolled opening of hemichannels during prolonged ischemia is incompatible with cellular survival (Thompson et al., 2006).
A review of the literature suggests that bystander death plays a predominant role in models of permanent ischemia or traumatic brain injury (Rawanduzy et al., 1997; Frantseva et al., 2002; Farahani et al., 2005), whereas the neuroprotective effect of hemichannels dominates in models of transient or distal artery occlusion and is associated with smaller lesions (Nakase et al., 2003a,b) (Fig. 7). Exceptions exist with bystander death mediating secondary injury in smaller lesion in the developing retina and in a model of transient perinatal ischemia (Cusato et al., 2003; de Pina-Benabou et al., 2005). Because adenosine is a vasodilator (Iadecola and Nedergaard, 2007), it is also possible that Cx43 hemichannels protects against injury by adenosine-mediated vasodilation in penumbral regions.
It is of interest that studies of heart showed remarkable similarities to observations in brain (Davis et al., 1994; Garcia-Dorado et al., 2004). Bystander death is prominent in ischemic heart and was first observed in the myocardium (Garcia-Dorado et al., 1989; Ruiz-Meana et al., 1999). Minor ischemic damage in heart typically occurs in the form of contraction band necrosis (Miyazaki et al., 1987). Death of cardiomyocytes is not scattered randomly in the reperfused myocardium but rather in small groups within well delimited areas that do not follow the pattern of the local microvasculature (Garcia-Dorado et al., 2004). Gap-junction blockers applied at the time of reperfusion or reoxygenation decreased the area of necrosis (Schlack et al., 1997; Siegmund et al., 1997). Transgenic mice with reduced Cx43 expression (Cx43+/−) exhibited smaller infarcts (Kanno et al., 2003), but an analysis of Cx43−/− mice have not been possible because these mice die at birth (Liu et al., 2006a).
Another intriguing aspect of Cx43 expression in the myocardium is its involvement in ischemic preconditioning. The preconditioned heart exhibited a higher level of Cx43 and redistribution of Cx43-immunoreactive plaques (Daleau et al., 2001; Vetterlein et al., 2006). Cx43 was not confined to myocyte abutments, as in normal heart, but was dispersed diffusely along the sarcolemma after preconditioning (Daleau et al., 2001; Vetterlein et al., 2006). Heptanol administered before preconditioning abolished the protective effect in isolated heart (Li et al., 2002), and several groups found that Cx43-deficient mice (Cx43+/−) were insensitive to preconditioning (Schwanke et al., 2003; Li et al., 2004; Miura et al., 2004; Schulz and Heusch, 2004). Taking the literature in heart and CNS into account, it is clear that Cx43 gap junctions can mediate bystander death of both myocytes and astrocytes but also that Cx43 expression has cardioprotective and neuroprotective effects (Lin et al., 2003; Li et al., 2004). Furthermore, adenosine appears to play an essential role in ischemic tolerance after preconditioning of both organs (Heurteaux et al., 1995; de Jong et al., 2000). Other lines of work have compared ischemic preconditioning with hibernation, a natural model of tolerance to extreme reduction of blood flow (∼80–90%) in brain or heart (Schulz and Heusch, 1996; Stenzel-Poore et al., 2003, 2007; Lee et al., 2007), which also involves adenosine-mediated suppression of metabolic rate (Gao et al., 1995).
The expression of Cx43 is transcriptionally regulated (Teunissen and Bierhuizen, 2004), but the rate by which Cx43 is degraded can also impact the abundance of Cx43 in a short timescale (Laing et al., 1998; Berthoud et al., 2004; VanSlyke and Musil, 2005). Most plasma membrane proteins have half-lives of 20–80 h, which is contrasted by the short 1.5–5 h half-life of Cx43 (Berthoud et al., 2004). The slowing of protein degradation that occurs during cellular stress is linked to a rapid increase in the abundance of Cx43 (Berthoud et al., 2004; VanSlyke and Musil, 2005). Dynamic regulation of the rate of Cx43 degradation represents thereby a powerful pathway to offset cellular stress and improve cellular survival. In support of this concept, we observed that inhibitors of lysosomal function mimicked the effect of preconditioning by increasing Cx43 abundance, intensifying hemichannel opening, and raising cellular resistance (Fig. 5). Based on the work of others and our own, we suggest that adaptive reduction in the rate of degradation of Cxs represents a general protective mechanism. Connexins are universally expressed, and adenosine preserves energy homeostasis by dampening metabolic demands in a variety of tissues, including kidney, heart, and brain (Dunwiddie and Masino, 2001).
Our study adds a novel concept to the mechanism of preconditioning. We show that Cx43 hemichannels play an essential role in protection afforded by preconditioning by facilitating ATP release, which in turn increases the accumulation of extracellular adenosine, a potent neuroprotective agent (Fig. 8). Several of the key points in this report have been reported previously, including the stress-induced reduction of Cx43 degradation (VanSlyke and Musil, 2005), an increased abundance of Cx43 after injury (Theriault et al., 1997; Yaoita et al., 2002), the opening of Cx43 hemichannels during metabolic inhibition (Kondo et al., 2000), release of ATP/adenosine in response to hypoxia (Gribkoff and Bauman, 1992), and the neuroprotective actions of adenosine A1 receptors (Halle et al., 1997; Dunwiddie and Masino, 2001). Our data link these previous disparate observations into a coherent scheme. The result and synthesis suggest a mechanism by which hemichannels serve as a novel therapeutic target for ischemic brain injury.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke/National Institutes of Health Grant NS50315, New York State Spinal Cord Injury Research Board, and the Dana Foundation.
References
- Abbracchio MP, Verderio C. Pathophysiological roles of P2 receptors in glial cells. Novartis Found Symp. 2006;276:91–103. discussion 103–112, 275–281. [PubMed] [Google Scholar]
- Arcuino G, Lin JH, Takano T, Liu C, Jiang L, Gao Q, Kang J, Nedergaard M. Intercellular calcium signaling mediated by point-source burst release of ATP. Proc Natl Acad Sci USA. 2002;99:9840–9845. doi: 10.1073/pnas.152588599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballerini P, Di Iorio P, Ciccarelli R, Nargi E, D'Alimonte I, Traversa U, Rathbone MP, Caciagli F. Glial cells express multiple ATP binding cassette proteins which are involved in ATP release. NeuroReport. 2002;13:1789–1792. doi: 10.1097/00001756-200210070-00019. [DOI] [PubMed] [Google Scholar]
- Bennett MV, Contreras JE, Bukauskas FF, Saez JC. New roles for astrocytes: gap junction hemichannels have something to communicate. Trends Neurosci. 2003;26:610–617. doi: 10.1016/j.tins.2003.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthoud VM, Minogue PJ, Laing JG, Beyer EC. Pathways for degradation of connexins and gap junctions. Cardiovasc Res. 2004;62:256–267. doi: 10.1016/j.cardiores.2003.12.021. [DOI] [PubMed] [Google Scholar]
- Brismar T. Physiology of transformed glial cells. Glia. 1995;15:231–243. doi: 10.1002/glia.440150305. [DOI] [PubMed] [Google Scholar]
- Budd SL, Lipton SA. Calcium tsunamis: do astrocytes transmit cell death messages via gap junctions during ischemia? Nat Neurosci. 1998;1:431–432. doi: 10.1038/2147. [DOI] [PubMed] [Google Scholar]
- Bukauskas FF, Verselis VK. Gap junction channel gating. Biochim Biophys Acta. 2004;1662:42–60. doi: 10.1016/j.bbamem.2004.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandross KJ. Nerve injury and inflammatory cytokines modulate gap junctions in the peripheral nervous system. Glia. 1998;24:21–31. doi: 10.1002/(sici)1098-1136(199809)24:1<21::aid-glia3>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Chang Q, Pereda A, Pinter MJ, Balice-Gordon RJ. Nerve injury induces gap junctional coupling among axotomized adult motor neurons. J Neurosci. 2000;20:674–684. doi: 10.1523/JNEUROSCI.20-02-00674.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coco S, Calegari F, Pravettoni E, Pozzi D, Taverna E, Rosa P, Matteoli M, Verderio C. Storage and release of ATP from astrocytes in culture. J Biol Chem. 2003;278:1354–1362. doi: 10.1074/jbc.M209454200. [DOI] [PubMed] [Google Scholar]
- Connolly ES, Jr, Winfree CJ, Stern DM, Solomon RA, Pinsky DJ. Procedural and strain-related variables significantly affect outcome in a murine model of focal cerebral ischemia. Neurosurgery. 1996;38:523–531. doi: 10.1097/00006123-199603000-00021. discussion 532. [DOI] [PubMed] [Google Scholar]
- Contreras JE, Saez JC, Bukauskas FF, Bennett MV. Gating and regulation of connexin 43 (Cx43) hemichannels. Proc Natl Acad Sci USA. 2003;100:11388–11393. doi: 10.1073/pnas.1434298100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotrina ML, Kang J, Lin JH, Bueno E, Hansen TW, He L, Liu Y, Nedergaard M. Astrocytic gap junctions remain open during ischemic conditions. J Neurosci. 1998a;18:2520–2537. doi: 10.1523/JNEUROSCI.18-07-02520.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotrina ML, Lin JH, Alves-Rodrigues A, Liu S, Li J, Azmi-Ghadimi H, Kang J, Naus CC, Nedergaard M. Connexins regulate calcium signaling by controlling ATP release. Proc Natl Acad Sci USA. 1998b;95:15735–15740. doi: 10.1073/pnas.95.26.15735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusato K, Bosco A, Rozental R, Guimaraes CA, Reese BE, Linden R, Spray DC. Gap junctions mediate bystander cell death in developing retina. J Neurosci. 2003;23:6413–6422. doi: 10.1523/JNEUROSCI.23-16-06413.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daleau P, Boudriau S, Michaud M, Jolicoeur C, Kingma JG., Jr Preconditioning in the absence or presence of sustained ischemia modulates myocardial Cx43 protein levels and gap junction distribution. Can J Physiol Pharmacol. 2001;79:371–378. [PubMed] [Google Scholar]
- Davis LM, Kanter HL, Beyer EC, Saffitz JE. Distinct gap junction protein phenotypes in cardiac tissues with disparate conduction properties. J Am Coll Cardiol. 1994;24:1124–1132. doi: 10.1016/0735-1097(94)90879-6. [DOI] [PubMed] [Google Scholar]
- de Jong JW, de Jonge R, Keijzer E, Bradamante S. The role of adenosine in preconditioning. Pharmacol Ther. 2000;87:141–149. doi: 10.1016/s0163-7258(00)00044-9. [DOI] [PubMed] [Google Scholar]
- de Pina-Benabou MH, Szostak V, Kyrozis A, Rempe D, Uziel D, Urban-Maldonado M, Benabou S, Spray DC, Federoff HJ, Stanton PK, Rozental R. Blockade of gap junctions in vivo provides neuroprotection after perinatal global ischemia. Stroke. 2005;36:2232–2237. doi: 10.1161/01.STR.0000182239.75969.d8. [DOI] [PubMed] [Google Scholar]
- del Zoppo GJ. Stroke and neurovascular protection. N Engl J Med. 2006;354:553–555. doi: 10.1056/NEJMp058312. [DOI] [PubMed] [Google Scholar]
- Dermietzel R, Gao Y, Scemes E, Vieira D, Urban M, Kremer M, Bennett MV, Spray DC. Connexin43 null mice reveal that astrocytes express multiple connexins. Brain Res Brain Res Rev. 2000;32:45–56. doi: 10.1016/s0165-0173(99)00067-3. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Simon RP, Hallenbeck JM. Ischemic tolerance and endogenous neuroprotection. Trends Neurosci. 2003;26:248–254. doi: 10.1016/S0166-2236(03)00071-7. [DOI] [PubMed] [Google Scholar]
- Donevan SD, Rogawski MA. Intracellular polyamines mediate inward rectification of Ca2+-permeable alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors. Proc Natl Acad Sci USA. 1995;92:9298–9302. doi: 10.1073/pnas.92.20.9298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwiddie TV, Masino SA. The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci. 2001;24:31–55. doi: 10.1146/annurev.neuro.24.1.31. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Diao L, Proctor WR. Adenine nucleotides undergo rapid, quantitative conversion to adenosine in the extracellular space in rat hippocampus. J Neurosci. 1997;17:7673–7682. doi: 10.1523/JNEUROSCI.17-20-07673.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endoh T, Abe M, Suzuki T. Decay in prepulse facilitation of calcium channel currents by Gi/o-protein attenuation in hamster submandibular ganglion neurons, but not Gq/11. Bull Tokyo Dent Coll. 2001;42:235–241. doi: 10.2209/tdcpublication.42.235. [DOI] [PubMed] [Google Scholar]
- Farahani R, Pina-Benabou MH, Kyrozis A, Siddiq A, Barradas PC, Chiu FC, Cavalcante LA, Lai JC, Stanton PK, Rozental R. Alterations in metabolism and gap junction expression may determine the role of astrocytes as “good samaritans” or executioners. Glia. 2005;50:351–361. doi: 10.1002/glia.20213. [DOI] [PubMed] [Google Scholar]
- Franke H, Krugel U, Illes P. P2 receptors and neuronal injury. Pflügers Arch. 2006;452:622–644. doi: 10.1007/s00424-006-0071-8. [DOI] [PubMed] [Google Scholar]
- Frantseva MV, Kokarovtseva L, Naus CG, Carlen PL, MacFabe D, Perez Velazquez JL. Specific gap junctions enhance the neuronal vulnerability to brain traumatic injury. J Neurosci. 2002;22:644–653. doi: 10.1523/JNEUROSCI.22-03-00644.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenguelli BG, Wigmore G, Llaudet E, Dale N. Temporal and mechanistic dissociation of ATP and adenosine release during ischaemia in the mammalian hippocampus. J Neurochem. 2007;101:1400–1413. doi: 10.1111/j.1471-4159.2006.04425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch C, Theis M, De Souza Silva MA, Dere E, Sohl G, Teubner B, Namestkova K, Willecke K, Huston JP. Mice with astrocyte-directed inactivation of connexin43 exhibit increased exploratory behaviour, impaired motor capacities, and changes in brain acetylcholine levels. Eur J Neurosci. 2003;18:2313–2318. doi: 10.1046/j.1460-9568.2003.02971.x. [DOI] [PubMed] [Google Scholar]
- Furuichi T, Liu W, Shi H, Miyake M, Liu KJ. Generation of hydrogen peroxide during brief oxygen-glucose deprivation induces preconditioning neuronal protection in primary cultured neurons. J Neurosci Res. 2005;79:816–824. doi: 10.1002/jnr.20402. [DOI] [PubMed] [Google Scholar]
- Gao ZP, Downey HF, Fan WL, Mallet RT. Does interstitial adenosine mediate acute hibernation of guinea pig myocardium? Cardiovasc Res. 1995;29:796–804. [PubMed] [Google Scholar]
- Garcia-Dorado D, Theroux P, Desco M, Solares J, Elizaga J, Fernandez-Aviles F, Alonso J, Soriano J. Cell-to-cell interaction: a mechanism to explain wave-front progression of myocardial necrosis. Am J Physiol. 1989;256:H1266–H1273. doi: 10.1152/ajpheart.1989.256.5.H1266. [DOI] [PubMed] [Google Scholar]
- Garcia-Dorado D, Rodriguez-Sinovas A, Ruiz-Meana M. Gap junction-mediated spread of cell injury and death during myocardial ischemia-reperfusion. Cardiovasc Res. 2004;61:386–401. doi: 10.1016/j.cardiores.2003.11.039. [DOI] [PubMed] [Google Scholar]
- Gomes P, Srinivas SP, Van Driessche W, Vereecke J, Himpens B. ATP release through connexin hemichannels in corneal endothelial cells. Invest Ophthalmol Vis Sci. 2005;46:1208–1218. doi: 10.1167/iovs.04-1181. [DOI] [PubMed] [Google Scholar]
- Goodenough DA, Paul DL. Beyond the gap: functions of unpaired connexon channels. Nat Rev Mol Cell Biol. 2003;4:285–294. doi: 10.1038/nrm1072. [DOI] [PubMed] [Google Scholar]
- Gribkoff VK, Bauman LA. Endogenous adenosine contributes to hypoxic synaptic depression in hippocampus from young and aged rats. J Neurophysiol. 1992;68:620–628. doi: 10.1152/jn.1992.68.2.620. [DOI] [PubMed] [Google Scholar]
- Gustavsson M, Mallard C, Vannucci SJ, Wilson MA, Johnston MV, Hagberg H. Vascular response to hypoxic preconditioning in the immature brain. J Cereb Blood Flow Metab. 2007;27:928–938. doi: 10.1038/sj.jcbfm.9600408. [DOI] [PubMed] [Google Scholar]
- Hagberg H, Andersson P, Lacarewicz J, Jacobson I, Butcher S, Sandberg M. Extracellular adenosine, inosine, hypoxanthine, and xanthine in relation to tissue nucleotides and purines in rat striatum during transient ischemia. J Neurochem. 1987;49:227–231. doi: 10.1111/j.1471-4159.1987.tb03419.x. [DOI] [PubMed] [Google Scholar]
- Halle JN, Kasper CE, Gidday JM, Koos BJ. Enhancing adenosine A1 receptor binding reduces hypoxic-ischemic brain injury in newborn rats. Brain Res. 1997;759:309–312. doi: 10.1016/s0006-8993(97)00364-8. [DOI] [PubMed] [Google Scholar]
- Heurteaux C, Lauritzen I, Widmann C, Lazdunski M. Essential role of adenosine, adenosine A1 receptors, and ATP-sensitive K+ channels in cerebral ischemic preconditioning. Proc Natl Acad Sci USA. 1995;92:4666–4670. doi: 10.1073/pnas.92.10.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillion JA, Takahashi K, Maric D, Ruetzler C, Barker JL, Hallenbeck JM. Development of an ischemic tolerance model in a PC12 cell line. J Cereb Blood Flow Metab. 2005;25:154–162. doi: 10.1038/sj.jcbfm.9600003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10:1369–1376. doi: 10.1038/nn2003. [DOI] [PubMed] [Google Scholar]
- Jimenez AI, Castro E, Mirabet M, Franco R, Delicado EG, Miras-Portugal MT. Potentiation of ATP calcium responses by A2B receptor stimulation and other signals coupled to Gs proteins in type-1 cerebellar astrocytes. Glia. 1999;26:119–128. [PubMed] [Google Scholar]
- John SA, Kondo R, Wang SY, Goldhaber JI, Weiss JN. Connexin-43 hemichannels opened by metabolic inhibition. J Biol Chem. 1999;274:236–240. doi: 10.1074/jbc.274.1.236. [DOI] [PubMed] [Google Scholar]
- Kang J, Jiang L, Goldman S, Nedergaard M. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat Neurosci. 1998;1:683–692. doi: 10.1038/3684. [DOI] [PubMed] [Google Scholar]
- Kanno S, Kovacs A, Yamada KA, Saffitz JE. Connexin43 as a determinant of myocardial infarct size following coronary occlusion in mice. J Am Coll Cardiol. 2003;41:681–686. doi: 10.1016/s0735-1097(02)02893-0. [DOI] [PubMed] [Google Scholar]
- Kariko K, Weissman D, Welsh FA. Inhibition of toll-like receptor and cytokine signaling—a unifying theme in ischemic tolerance. J Cereb Blood Flow Metab. 2004;24:1288–1304. doi: 10.1097/01.WCB.0000145666.68576.71. [DOI] [PubMed] [Google Scholar]
- Kasper M, Traub O, Reimann T, Bjermer L, Grossmann H, Muller M, Wenzel KW. Upregulation of gap junction protein connexin43 in alveolar epithelial cells of rats with radiation-induced pulmonary fibrosis. Histochem Cell Biol. 1996;106:419–424. doi: 10.1007/BF02473301. [DOI] [PubMed] [Google Scholar]
- Kirino T. Ischemic tolerance. J Cereb Blood Flow Metab. 2002;22:1283–1296. doi: 10.1097/01.WCB.0000040942.89393.88. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Fukuoka T, Yamanaka H, Dai Y, Obata K, Tokunaga A, Noguchi K. Neurons and glial cells differentially express P2Y receptor mRNAs in the rat dorsal root ganglion and spinal cord. J Comp Neurol. 2006;498:443–454. doi: 10.1002/cne.21066. [DOI] [PubMed] [Google Scholar]
- Kondo RP, Wang SY, John SA, Weiss JN, Goldhaber JI. Metabolic inhibition activates a non-selective current through connexin hemichannels in isolated ventricular myocytes. J Mol Cell Cardiol. 2000;32:1859–1872. doi: 10.1006/jmcc.2000.1220. [DOI] [PubMed] [Google Scholar]
- Kumar NM, Gilula NB. The gap junction communication channel. Cell. 1996;84:381–388. doi: 10.1016/s0092-8674(00)81282-9. [DOI] [PubMed] [Google Scholar]
- Laing JG, Tadros PN, Green K, Saffitz JE, Beyer EC. Proteolysis of connexin43-containing gap junctions in normal and heat-stressed cardiac myocytes. Cardiovasc Res. 1998;38:711–718. doi: 10.1016/s0008-6363(98)00060-1. [DOI] [PubMed] [Google Scholar]
- Laird DW. Connexin phosphorylation as a regulatory event linked to gap junction internalization and degradation. Biochim Biophys Acta. 2005;1711:172–182. doi: 10.1016/j.bbamem.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Lankford AR, Yang JN, Rose'Meyer R, French BA, Matherne GP, Fredholm BB, Yang Z. Effect of modulating cardiac A1 adenosine receptor expression on protection with ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2006;290:H1469–H1473. doi: 10.1152/ajpheart.00181.2005. [DOI] [PubMed] [Google Scholar]
- Lauf U, Giepmans BN, Lopez P, Braconnot S, Chen SC, Falk MM. Dynamic trafficking and delivery of connexons to the plasma membrane and accretion to gap junctions in living cells. Proc Natl Acad Sci USA. 2002;99:10446–10451. doi: 10.1073/pnas.162055899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee IH, Lindqvist E, Kiehn O, Widenfalk J, Olson L. Glial and neuronal connexin expression patterns in the rat spinal cord during development and following injury. J Comp Neurol. 2005;489:1–10. doi: 10.1002/cne.20567. [DOI] [PubMed] [Google Scholar]
- Lee YJ, Miyake S, Wakita H, McMullen DC, Azuma Y, Auh S, Hallenbeck JM. Protein SUMOylation is massively increased in hibernation torpor and is critical for the cytoprotection provided by ischemic preconditioning and hypothermia in SHSY5Y cells. J Cereb Blood Flow Metab. 2007;27:950–962. doi: 10.1038/sj.jcbfm.9600395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Whittaker P, Yao M, Kloner RA, Przyklenk K. The gap junction uncoupler heptanol abrogates infarct size reduction with preconditioning in mouse hearts. Cardiovasc Pathol. 2002;11:158–165. doi: 10.1016/s1054-8807(02)00102-3. [DOI] [PubMed] [Google Scholar]
- Li H, Liu TF, Lazrak A, Peracchia C, Goldberg GS, Lampe PD, Johnson RG. Properties and regulation of gap junctional hemichannels in the plasma membranes of cultured cells. J Cell Biol. 1996;134:1019–1030. doi: 10.1083/jcb.134.4.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Heinzel FR, Boengler KD, Schulz R, Heusch G. Role of connexin 43 in ischemic preconditioning does not involve intercellular communication through gap junctions. J Mol Cell Cardiol. 2004;36:161–163. doi: 10.1016/j.yjmcc.2003.10.019. [DOI] [PubMed] [Google Scholar]
- Lin JH, Weigel H, Cotrina ML, Liu S, Bueno E, Hansen AJ, Hansen TW, Goldman S, Nedergaard M. Gap-junction-mediated propagation and amplification of cell injury. Nat Neurosci. 1998;1:494–500. doi: 10.1038/2210. [DOI] [PubMed] [Google Scholar]
- Lin JH, Yang J, Liu S, Takano T, Wang X, Gao Q, Willecke K, Nedergaard M. Connexin mediates gap junction-independent resistance to cellular injury. J Neurosci. 2003;23:430–441. doi: 10.1523/JNEUROSCI.23-02-00430.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SH, Lu CY, Muhammad R, Chou WY, Lin FC, Wu PC, Lin CR, Yang LC. Induction of connexin 37 expression in a rat model of neuropathic pain. Brain Res Mol Brain Res. 2002;99:134–140. doi: 10.1016/s0169-328x(02)00112-2. [DOI] [PubMed] [Google Scholar]
- Liu S, Liu F, Schneider AE, St Amand T, Epstein JA, Gutstein DE. Distinct cardiac malformations caused by absence of connexin 43 in the neural crest and in the non-crest neural tube. Development. 2006a;133:2063–2073. doi: 10.1242/dev.02374. [DOI] [PubMed] [Google Scholar]
- Liu Y, Xiong L, Chen S, Wang Q. Isoflurane tolerance against focal cerebral ischemia is attenuated by adenosine A1 receptor antagonists. Can J Anaesth. 2006b;53:194–201. doi: 10.1007/BF03021827. [DOI] [PubMed] [Google Scholar]
- Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4:399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Graf R, Rosner G, Shimada N, Heiss WD. Flow thresholds for extracellular purine catabolite elevation in cat focal ischemia. Brain Res. 1992;579:309–314. doi: 10.1016/0006-8993(92)90066-i. [DOI] [PubMed] [Google Scholar]
- Miura T, Ohnuma Y, Kuno A, Tanno M, Ichikawa Y, Nakamura Y, Yano T, Miki T, Sakamoto J, Shimamoto K. Protective role of gap junctions in preconditioning against myocardial infarction. Am J Physiol Heart Circ Physiol. 2004;286:H214–H221. doi: 10.1152/ajpheart.00441.2003. [DOI] [PubMed] [Google Scholar]
- Miyazaki S, Fujiwara H, Onodera T, Kihara Y, Matsuda M, Wu DJ, Nakamura Y, Kumada T, Sasayama S, Kawai C, Hamashima Y. Quantitative analysis of contraction band and coagulation necrosis after ischemia and reperfusion in the porcine heart. Circulation. 1987;75:1074–1082. doi: 10.1161/01.cir.75.5.1074. [DOI] [PubMed] [Google Scholar]
- Musil LS, Le AC, VanSlyke JK, Roberts LM. Regulation of connexin degradation as a mechanism to increase gap junction assembly and function. J Biol Chem. 2000;275:25207–25215. doi: 10.1074/jbc.275.33.25207. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Nakakimura K, Matsumoto M, Sakabe T. Rapid tolerance to focal cerebral ischemia in rats is attenuated by adenosine A1 receptor antagonist. J Cereb Blood Flow Metab. 2002;22:161–170. doi: 10.1097/00004647-200202000-00004. [DOI] [PubMed] [Google Scholar]
- Nakase T, Fushiki S, Naus CC. Astrocytic gap junctions composed of connexin 43 reduce apoptotic neuronal damage in cerebral ischemia. Stroke. 2003a;34:1987–1993. doi: 10.1161/01.STR.0000079814.72027.34. [DOI] [PubMed] [Google Scholar]
- Nakase T, Fushiki S, Sohl G, Theis M, Willecke K, Naus CC. Neuroprotective role of astrocytic gap junctions in ischemic stroke. Cell Commun Adhes. 2003b;10:413–417. doi: 10.1080/cac.10.4-6.413.417. [DOI] [PubMed] [Google Scholar]
- Oguro K, Jover T, Tanaka H, Lin Y, Kojima T, Oguro N, Grooms SY, Bennett MV, Zukin RS. Global ischemia-induced increases in the gap junctional proteins connexin 32 (Cx32) and Cx36 in hippocampus and enhanced vulnerability of Cx32 knock-out mice. J Neurosci. 2001;21:7534–7542. doi: 10.1523/JNEUROSCI.21-19-07534.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez Velazquez JL, Frantseva MV, Naus CC. Gap junctions and neuronal injury: protectants or executioners? The Neuroscientist. 2003;9:5–9. doi: 10.1177/1073858402239586. [DOI] [PubMed] [Google Scholar]
- Rawanduzy A, Hansen A, Hansen TW, Nedergaard M. Effective reduction of infarct volume by gap junction blockade in a rodent model of stroke. J Neurosurg. 1997;87:916–920. doi: 10.3171/jns.1997.87.6.0916. [DOI] [PubMed] [Google Scholar]
- Rebola N, Rodrigues RJ, Lopes LV, Richardson PJ, Oliveira CR, Cunha RA. Adenosine A1 and A2A receptors are co-expressed in pyramidal neurons and co-localized in glutamatergic nerve terminals of the rat hippocampus. Neuroscience. 2005;133:79–83. doi: 10.1016/j.neuroscience.2005.01.054. [DOI] [PubMed] [Google Scholar]
- Ruiz-Meana M, Garcia-Dorado D, Hofstaetter B, Piper HM, Soler-Soler J. Propagation of cardiomyocyte hypercontracture by passage of Na+ through gap junctions. Circ Res. 1999;85:280–287. doi: 10.1161/01.res.85.3.280. [DOI] [PubMed] [Google Scholar]
- Salameh A. Life cycle of connexins: regulation of connexin synthesis and degradation. Adv Cardiol. 2006;42:57–70. doi: 10.1159/000092562. [DOI] [PubMed] [Google Scholar]
- Schlack W, Preckel B, Barthel H, Obal D, Thamer V. Halothane reduces reperfusion injury after regional ischaemia in the rabbit heart in vivo. Br J Anaesth. 1997;79:88–96. doi: 10.1093/bja/79.1.88. [DOI] [PubMed] [Google Scholar]
- Schulz R, Heusch G. Ischemic preconditioning and myocardial hibernation: is there a common mechanism? Basic Res Cardiol. 1996;91:50–52. doi: 10.1007/BF00788864. [DOI] [PubMed] [Google Scholar]
- Schulz R, Heusch G. Connexin 43 and ischemic preconditioning. Cardiovasc Res. 2004;62:335–344. doi: 10.1016/j.cardiores.2003.12.017. [DOI] [PubMed] [Google Scholar]
- Schwanke U, Li X, Schulz R, Heusch G. No ischemic preconditioning in heterozygous connexin 43-deficient mice—a further in vivo study. Basic Res Cardiol. 2003;98:181–182. doi: 10.1007/s003950300002. [DOI] [PubMed] [Google Scholar]
- Siegmund B, Schlack W, Ladilov YV, Balser C, Piper HM. Halothane protects cardiomyocytes against reoxygenation-induced hypercontracture. Circulation. 1997;96:4372–4379. doi: 10.1161/01.cir.96.12.4372. [DOI] [PubMed] [Google Scholar]
- Sigworth LA, Rea MA. Adenosine A1 receptors regulate the response of the mouse circadian clock to light. Brain Res. 2003;960:246–251. doi: 10.1016/s0006-8993(02)03896-9. [DOI] [PubMed] [Google Scholar]
- Simard M, Arcuino G, Takano T, Liu QS, Nedergaard M. Signaling at the gliovascular interface. J Neurosci. 2003;23:9254–9262. doi: 10.1523/JNEUROSCI.23-27-09254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenzel-Poore MP, Stevens SL, Xiong Z, Lessov NS, Harrington CA, Mori M, Meller R, Rosenzweig HL, Tobar E, Shaw TE, Chu X, Simon RP. Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet. 2003;362:1028–1037. doi: 10.1016/S0140-6736(03)14412-1. [DOI] [PubMed] [Google Scholar]
- Stenzel-Poore MP, Stevens SL, King JS, Simon RP. Preconditioning reprograms the response to ischemic injury and primes the emergence of unique endogenous neuroprotective phenotypes: a speculative synthesis. Stroke. 2007;38:680–685. doi: 10.1161/01.STR.0000251444.56487.4c. [DOI] [PubMed] [Google Scholar]
- Stout CE, Costantin JL, Naus CC, Charles AC. Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J Biol Chem. 2002;277:10482–10488. doi: 10.1074/jbc.M109902200. [DOI] [PubMed] [Google Scholar]
- Striedinger K, Petrasch-Parwez E, Zoidl G, Napirei M, Meier C, Eysel UT, Dermietzel R. Loss of connexin36 increases retinal cell vulnerability to secondary cell loss. Eur J Neurosci. 2005;22:605–616. doi: 10.1111/j.1460-9568.2005.04228.x. [DOI] [PubMed] [Google Scholar]
- Suadicani SO, Brosnan CF, Scemes E. P2X7 receptors mediate ATP release and amplification of astrocytic intercellular Ca2+ signaling. J Neurosci. 2006;26:1378–1385. doi: 10.1523/JNEUROSCI.3902-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teunissen BE, Bierhuizen MF. Transcriptional control of myocardial connexins. Cardiovasc Res. 2004;62:246–255. doi: 10.1016/j.cardiores.2003.12.011. [DOI] [PubMed] [Google Scholar]
- Theis M, Jauch R, Zhuo L, Speidel D, Wallraff A, Doring B, Frisch C, Sohl G, Teubner B, Euwens C, Huston J, Steinhauser C, Messing A, Heinemann U, Willecke K. Accelerated hippocampal spreading depression and enhanced locomotory activity in mice with astrocyte-directed inactivation of connexin43. J Neurosci. 2003;23:766–776. doi: 10.1523/JNEUROSCI.23-03-00766.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theis M, Speidel D, Willecke K. Astrocyte cultures from conditional connexin43-deficient mice. Glia. 2004;46:130–141. doi: 10.1002/glia.10350. [DOI] [PubMed] [Google Scholar]
- Theriault E, Frankenstein UN, Hertzberg EL, Nagy JI. Connexin43 and astrocytic gap junctions in the rat spinal cord after acute compression injury. J Comp Neurol. 1997;382:199–214. [PubMed] [Google Scholar]
- Thompson RJ, Zhou N, MacVicar BA. Ischemia opens neuronal gap junction hemichannels. Science. 2006;312:924–927. doi: 10.1126/science.1126241. [DOI] [PubMed] [Google Scholar]
- Tsuchida A, Liu GS, Wilborn WH, Downey JM. Pretreatment with the adenosine A1 selective agonist, 2-chloro-N6-cyclopentyladenosine (CCPA), causes a sustained limitation of infarct size in rabbits. Cardiovasc Res. 1993;27:652–656. doi: 10.1093/cvr/27.4.652. [DOI] [PubMed] [Google Scholar]
- Udawatte C, Ripps H. The spread of apoptosis through gap-junctional channels in BHK cells transfected with Cx32. Apoptosis. 2005;10:1019–1029. doi: 10.1007/s10495-005-0776-8. [DOI] [PubMed] [Google Scholar]
- VanSlyke JK, Musil LS. Dislocation and degradation from the ER are regulated by cytosolic stress. J Cell Biol. 2002;157:381–394. doi: 10.1083/jcb.200111045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanSlyke JK, Musil LS. Degradation of connexins from the plasma membrane is regulated by inhibitors of protein synthesis. Cell Commun Adhes. 2003;10:329–333. doi: 10.1080/cac.10.4-6.329.333. [DOI] [PubMed] [Google Scholar]
- VanSlyke JK, Musil LS. Cytosolic stress reduces degradation of connexin43 internalized from the cell surface and enhances gap junction formation and function. Mol Biol Cell. 2005;16:5247–5257. doi: 10.1091/mbc.E05-05-0415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetterlein F, Muhlfeld C, Cetegen C, Volkmann R, Schrader C, Hellige G. Redistribution of connexin43 in regional acute ischemic myocardium: influence of ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2006;291:H813–H819. doi: 10.1152/ajpheart.01177.2005. [DOI] [PubMed] [Google Scholar]
- Wall M, Atterbury A, Dale N. Control of basal extracellular adenosine concentration in rat cerebellum. J Physiol (Lond) 2007;582:137–151. doi: 10.1113/jphysiol.2007.132050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallraff A, Kohling R, Heinemann U, Theis M, Willecke K, Steinhauser C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci. 2006;26:5438–5447. doi: 10.1523/JNEUROSCI.0037-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao-Qing T, Jun-Li Z, Yu C, Jian-Qiang F, Pei-Xi C. Hydrogen peroxide preconditioning protects PC12 cells against apoptosis induced by dopamine. Life Sci. 2005;78:61–66. doi: 10.1016/j.lfs.2005.04.048. [DOI] [PubMed] [Google Scholar]
- Yaar R, Jones MR, Chen JF, Ravid K. Animal models for the study of adenosine receptor function. J Cell Physiol. 2005;202:9–20. doi: 10.1002/jcp.20138. [DOI] [PubMed] [Google Scholar]
- Yaoita E, Yao J, Yoshida Y, Morioka T, Nameta M, Takata T, Kamiie J, Fujinaka H, Oite T, Yamamoto T. Up-regulation of connexin43 in glomerular podocytes in response to injury. Am J Pathol. 2002;161:1597–1606. doi: 10.1016/s0002-9440(10)64438-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye ZC, Wyeth MS, Baltan-Tekkok S, Ransom BR. Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J Neurosci. 2003;23:3588–3596. doi: 10.1523/JNEUROSCI.23-09-03588.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh HI, Lupu F, Dupont E, Severs NJ. Upregulation of connexin43 gap junctions between smooth muscle cells after balloon catheter injury in the rat carotid artery. Arterioscler Thromb Vasc Biol. 1997;17:3174–3184. doi: 10.1161/01.atv.17.11.3174. [DOI] [PubMed] [Google Scholar]
- Yu HM, Zhi JL, Cui Y, Tang EH, Sun SN, Feng JQ, Chen PX. Role of the JAK-STAT pathway in protection of hydrogen peroxide preconditioning against apoptosis induced by oxidative stress in PC12 cells. Apoptosis. 2006;11:931–941. doi: 10.1007/s10495-006-6578-9. [DOI] [PubMed] [Google Scholar]
- Zhu D, Caveney S, Kidder GM, Naus CC. Transfection of C6 glioma cells with connexin 43 cDNA: analysis of expression, intercellular coupling, and cell proliferation. Proc Natl Acad Sci USA. 1991;88:1883–1887. doi: 10.1073/pnas.88.5.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann C, Ginis I, Furuya K, Klimanis D, Ruetzler C, Spatz M, Hallenbeck JM. Lipopolysaccharide-induced ischemic tolerance is associated with increased levels of ceramide in brain and in plasma. Brain Res. 2001;895:59–65. doi: 10.1016/s0006-8993(01)02028-5. [DOI] [PubMed] [Google Scholar]