

Abstract
The formation of the highly organized cortical structure depends on the production and correct placement of the appropriate number and types of neurons. The Zic family of zinc-finger transcription factors plays essential roles in regulating the proliferation and differentiation of neuronal progenitors in the medial forebrain and the cerebellum. Examination of the expression of Zic genes demonstrated that Zic1, Zic2, and Zic3 were expressed by the progenitor cells in the septum and cortical hem, the sites of generation of the Cajal-Retzius (CR) cells. Immunohistochemical studies have revealed that Zic proteins were abundantly expressed in the meningeal cells and that the majority of the CR cells distributed in the medial and dorsal cortex also expressed Zic proteins in the mid-late embryonic and postnatal cortical marginal zones. During embryonic cortical development, Zic1/Zic3 double-mutant and hypomorphic Zic2 mutant mice showed a reduction in the number of CR cells in the rostral cortex, whereas the cell number remained unaffected in the caudal cortex. These mutants also showed mislocalization of the CR cells and cortical lamination defects, resembling the changes noted in type II (cobblestone) lissencephaly, throughout the brain. In the Zic1/3 mutant, reduced proliferation of the meningeal cells was observed before the thinner and disrupted organization of the pial basement membrane (BM) with reduced expression of the BM components and the meningeal cell-derived secretory factor. These defects correlated with the changes in the end feet morphology of the radial glial cells. These findings indicate that the Zic genes play critical roles in cortical development through regulating the proliferation of meningeal cells and the pial BM assembly.
Keywords: Cajal-Retzius cells, meninges, neuronal migration, type II lissencephaly, Zic, corticogenesis
Introduction
Mammalian cortical development proceeds in a highly orchestrated manner in which neurons migrate along radial glial fibers toward the surface of the brain to form a multilayered “inside-out” cortex. Cajal-Retzius (CR) cells, a subpopulation of early-born preplate neurons, play critical roles in this migration process (D'Arcangelo et al., 1995; Ogawa et al., 1995). Recent studies have revealed several focal sources of CR cells in mice: the cortical hem, the pallial–subpallial boundary (PSB), and the septum. From these foci, CR cells migrate tangentially to populate the cortical marginal zone (MZ) (Meyer et al., 2002; Shinozaki et al., 2002; Takiguchi-Hayashi et al., 2004; Bielle et al., 2005; García-Moreno et al., 2007) in which they regulate radial migration of late-born neurons via secretion of the extracellular protein reelin (Frotscher, 1998; Rice and Curran, 2001; Tissir and Goffinet, 2003).
In the radial migration process of the cortical neurons, spindle-shaped radial glial cells with their somata in the ventricular zone (VZ) extend long radial processes throughout the wall of the cortical plate (CP), being attached via their end feet to the basement membrane (BM). The radial processes serve as guiding structures for the neurons to migrate from their place of birth in the VZ or subventricular zone (SVZ) toward their final position in the CP (Rakic, 2003). The pial BM also plays an important role in cortical development, as indicated by the abnormal brain development observed after chemical ablation of the meningeal cells (Super et al., 1997) and after targeted deletion of BM constituents such as laminin and perlecan and of the receptors for the BM proteins. Disruption of the BM is known to cause type II (cobblestone) lissencephaly, characterized by cortical neurons protruding into the subarachnoid space (Graus-Porta et al., 2001; Halfter et al., 2002; Beggs et al., 2003; Niewmierzycka et al., 2005).
Some recent reports have drawn our attention to the relationship between corticogenesis and the Zic family of zinc-finger transcription factors (for review, see Aruga, 2004). It has been reported that Zic1, Zic2, and Zic3 play essential roles in medial forebrain development (Nagai et al., 2000; Inoue et al., 2007a). Mice homozygous for the Zic2 hypomorphic allele (Zic2 kd/kd) are reported to show holoprosencephaly (Nagai et al., 2000), which is analogous to the abnormality caused by human ZIC2 mutations (Brown et al., 1998). The Zic genes are expressed in the progenitors of the septum and cortical hem, and these two major sources of CR cells have been shown to be defective in the Zic2 and Zic1/3 mutant mice (Inoue et al., 2007a). Furthermore, we found that the Zic2 and Zic1/3 mutants possess altered cortical layer structures that have not yet been fully investigated.
In this study, we demonstrated that both meningeal cells and CR cells show expression of the Zic genes. Zic mutants showed reduced numbers of CR cells in the rostral cortex, with human type II lissencephaly-like cortical lamination defects, with disturbed migration of neurons, breakdown of the BM, and disorganization of the radial glial processes. Molecular and cellular analyses have revealed that the Zic genes play crucial roles in the proliferation of meningeal cells, which provide essential molecular cues for normal cortical development.
Materials and Methods
Animals.
All animal experiments were performed in accordance with the RIKEN guidelines for animal experiments. Every effort was made to minimize the number of animals used. Animals were maintained by the Research Resource Center, RIKEN Brain Science Institute. Embryonic day 0.5 (E0.5) was defined as the noon after the morning on which vaginal plugs were first observed.
C57BL/6J and BALB/c mice were purchased from Nihon SLC (Shizuoka, Japan). Mutant mice for Zic1 and Zic2 have been described previously (Aruga et al., 1998; Nagai et al., 2000) and were kept in a C57BL/6J background. Bent tail (Bn, Zic3Bn) mice were originally purchased from The Jackson Laboratory (Bar Harbor, ME). The spontaneous mutation, Bn, revealed to be a deletion of the X chromosome including Zic3 (Carrel et al., 2000; Klootwijk et al., 2000) were used for generating compound mutant mice lacking both Zic1 and Zic3 (Inoue et al., 2007a). The phenotypes of the Bn mice are very similar to those of the Zic3-deficient mice generated by a targeted mutation (Purandare et al., 2002; Aruga et al., 2004). Double-heterozygous females for Zic1 and Zic3 (Zic1+/− Zic3 Bn/+) and males (Zic1+/− Zic3 Bn/Y) were maintained on a C57BL/6J or mixed C57BL/6J × BALB/c background. Mating between (Zic1+/− Zic3 Bn/+) females and (Zic1+/− Zic3 Bn/Y) males generated compound mice (Zic1/3) lacking both Zic1 and Zic3 [males (Zic1−/− Zic3 Bn/Y) and females (Zic1−/− Zic3 Bn/Bn)]. Both C57BL/6J and BALB/c Bn mice were used for the initial characterization of the external appearance and histological abnormalities. Because there were no clear differences in the phenotype of the meninges or the CR cells in the compound mutant mice (data not shown), additional analyses of the Zic1/3 compound mice were performed in a mixed C57BL/6J × BALB/c background. Besides the Zic2 kd/kd and Zic1/3 mice, Zic1+/−, Zic 1−/−, Zic2 kd/+, Zic3 Bn/+, Zic3 Bn/Y, Zic3, Bn/Bn, and wild-type littermates were analyzed for characterization of the CR cell distribution, cortical layer, and meningeal phenotypes. There were no clear differences of the phenotypes among these genotypes. Zic1, Zic2, and Zic3 mutant alleles were genotyped according to the methods of Aruga et al. (1998), Nagai et al. (2000), and Franke et al. (2003), respectively. Reeler-Orleans (Relnrl-Orl) mice were obtained from The Jackson Laboratory and were studied at E15 and E18. The Relnrl-Orl allele produces a C-terminally truncated protein that is not secreted but can be detected using anti-reelin antibody G10 (de Bergeyck et al., 1997).
In situ hybridization.
For histological examination, the tissues were fixed in either Bouin's solution or 4% paraformaldehyde. After fixation, the tissues were dehydrated through a graded series of alcohols, embedded in paraffin, sectioned at a thickness of 8–10 μm, and stained with cresyl violet. In situ hybridization was performed using digoxygenin (DIG)-labeled RNA probes, as described previously (Nagai et al., 1997). RNA probes for GAD67, Reelin, Zic1, Zic2, and Zic3 have been described previously (Nagai et al., 1997; Inoue et al., 2007a,b). Probes for Calretinin, Cxcl12, Cxcr4, Dbx1, ER81, Fezl, Foxc1, Math2, Nid-1, p73, Rorβ, SCIP, SolLA, and Tbr1 were generated by reverse transcription (RT)-PCR. The sequences of the primers will be provided on request. Hybridization signals were detected with alkaline phosphatase-conjugated anti-DIG Fab fragments (Roche, Welwyn Garden City, UK), followed by color development with nitro blue tetrazolium and 5-bromo-4-chloro-3-indolyl-phosphate (Roche).
Immunofluorescence staining.
Immunofluorescence staining was performed as described previously (Aruga et al., 1998; Inoue et al., 2007a). Cryosections at 10–14 μm thickness were subjected to analysis. The sections were washed three times with PBS and preincubated for 30 min in PBS containing 5% normal goat serum and 0.1% Triton X-100 and then incubated overnight at 4°C in 1% normal goat serum and 0.1% Triton X-100 containing the primary antibodies. The primary antibodies were as follows: mouse anti-bromodeoxyuridine (BrdU) (Beckton Dickinson, Mountain View, CA), rat anti-BrdU (Abcam, Cambridge, UK), goat anti-Calretinin (Millipore, Billerica, MA), mouse anti-Collagen IV (Millipore), rabbit anti-laminin (Engelbreth-Holm-Swarm) (Sigma, St. Louis, MO), mouse anti-nestin (PharMingen, San Diego, CA), rabbit anti-phosphohistone H3 (Millipore), mouse anti-p73 (clone ER-15; Thermo Fisher Scientific, Waltham, MA,), mouse anti-ROR common (clone H3925; Perseus Proteomics, Tokyo, Japan), rabbit anti-Tbr1 (Millipore), mouse anti-reelin (clone G10; Millipore), and rabbit anti-pan-Zic (Inoue et al., 2007a,b). For the double- immunofluorescence detection of Zic with reelin or p73, frozen sections were immersed in 0.01 m citrate buffer, pH 6.0, and autoclaved at 105°C for 5 min to enable antigen retrieval. To detect these antibodies, cyanine 3-labeled (Jackson ImmunoResearch, West Grove, PA) or Alexa488- or Alexa594-labeled (Invitrogen, Carlsbad, CA) secondary antibodies were used. Fluorescence-labeled preparations were imaged under an Olympus (Tokyo, Japan) Fluoview FV300 confocal microscope or a Carl Zeiss (Jena, Germany) Axioskop2 plus microscope equipped with an AxioCam color CCD camera. All images were analyzed with Adobe Photoshop CS software (Adobe Systems, San Jose, CA). The number of p73-positive (p73+) CR cells at E16.5, postnatal day 0 (P0), and P6 was quantified by counting p73-immunoreactive nuclei in the cortical MZ/layer I of coronal sections of the rostral to the caudal forebrain, and Zic-positive nuclei were scored by capturing confocal digital images, assembling composite images spanning the entire cortical MZs. The organization of the cortical layer was examined by Tbr1 and Ror immunostaining in the E18.5 coronal sections (9 and 12 sections derived from three control and three Zic1/3 mutant embryos, respectively). Immunoreactive neurons were captured in the digital images. The grid overlay width was set 150 μm, and the cortical length was divided into five equal bins, with specific bin length proportional to cortical thickness of the sample. Numerical order of the bins corresponds to position of the cortical layers, with bin 1 defined as the most superficial layer and bin 5 as the deeper layer. The numbers of immunoreactive neurons were counted as numbers in 150-μm-wide windows of the coronal brain sections at E18.5.
Bromodeoxyuridine labeling.
For birthdating analysis to determine the distributions of the cells labeled with 50 mg/kg BrdU (Roche) in the neocortical wall of E18.5 mice, BrdU was injected intraperitoneally into pregnant mice at E13.5. The detection of BrdU was performed as described previously (Aruga et al., 1998). For the quantification, parasagittal sections at the level of the accessory olfactory bulb were used. At this level, 500-μm-wide radial stripes in the medial portions were divided into ∼40-μm-deep bins (12 horizontal bins both in wild-type and mutant cortex), and the position of each strongly and weakly labeled cell was assigned to a bin to generate histograms of the number of labeled cells against depth. Data from five sections from each of the wild-type (n = 4) and Zic1/3 mutant (n = 3) littermates were averaged to prepare the histograms. For examination of the cell proliferation in the meninges, pregnant female mice were injected intraperitoneally with BrdU (50 mg/kg) and killed 1 h later. At least three embryos for each genotype were analyzed at the indicated stages. BrdU-positive nuclei were counted only if they contained strong immunoreactivity and were localized in the meningeal layer directly attached to the telencephalon in coronal sections. We counted the number of BrdU-labeled cells in the meningeal layer from the dorsal interhemispheric fissure either to the level of the ventral tip of the fimbria in the caudal telencephalon or to the level of the PSB in the rostral telencephalon.
RNA extraction and RT-PCR.
Meningeal cell sheets were peeled off from the brain surface with fine forceps, and their RNAs were isolated using TRIzol reagent (Invitrogen). After DNaseI treatment, reverse transcription was performed with Superscript II reverse transcriptase (Invitrogen). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was detected to monitor RNA recovery. For quantification of the transcripts, semiquantitative RT-PCRs were performed after confirming that they were in the log-linear phase of the amplification curve at the indicated cycles. The PCR cycles, annealing temperature, and primer sequences were as follows: GAPDH, 25 cycles, 60°C, 5′-TCAAGATTGTCAGCAATGCATCCTG-3′ and 5′-GCAGGAGACAACCTGGTCCT-3′; Cxcl12, 27 cycles, 61°C, 5′-GCTAAGGTTTGCCAGCATAAAGACAC-3′ and 5′-GATAAAGGAGCCTCCCTCTGCC-3′; Foxc1, 28 cycles, 60°C, 5′-ACATGTTCGAGAACGGCAGCTTC-3′ and 5′-GCTCTGGCCCGGAGAGTAGG-3′; Laminin γ1, 25 cycles, 60°C, 5′-TGTGAGAACACGTACTCAAAGGCTAAC-3′ and 5′-GCACTTTTCACAGTCAACTCCGTATG-3′; Nid-1, 28 cycles, 62°C, 5′-CTCAGACTGGAAAGATTGAACGCCT-3′ and 5′-CTGTGCGAGAATCCTCCGGTT-3′; Perlecan, 28 cycles, 60°C, 5′-CCCCTCCACCCTGTGAGCCTAA-3′ and 5′-CCGAACACCATGCCACGCGA-3′.
Culture and immunostaining of primary meningeal fibroblasts.
Meningeal fibroblasts were isolated from wild-type, Zic1−/−, Zic3 Bn/Y, and Zic1/3 mutant embryonic cortex at E14.5. For the cell culture experiments, meningeal cell sheets were collected in HBSS supplemented with 6.5% glucose (Invitrogen). Meningeal cells were further dissociated by incubation in a solution containing 0.05% trypsin and 0.5 mg/ml collagenase and, after additional trituration, were seeded into dishes containing DMEM supplemented with 10% fetal bovine serum as described previously (Hartmann et al., 1998). The meningeal fibroblasts were cultured on glass coverslips coated with 1.0 mg/ml poly-d-lysine (Sigma) for up to 4 d before fixation in 4% paraformaldehyde for immunofluorescence staining. Glial contamination was undetectable in these cultures by immunofluorescence staining using anti-GFAP and anti-CD45 antibodies (data not shown). Double staining of the fibroblasts was performed by sequential incubation with anti-laminin antibody, Alexa488-labeled secondary antibody, and phalloidin-conjugated Alexa594 (Invitrogen).
Results
During corticogenesis, the meningeal cells and Cajal-Retzius cells in the cortical MZ expressed the Zic genes
To determine the roles of the Zic genes in cortical development, we first examined the distribution of the mRNAs and proteins from the preplate stage (E12) up to postnatal development (P6) (Figs. 1–3) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). During this period, Zic1, Zic2, and Zic3 mRNAs were expressed by the cells in the neural retina and medial neural tissues, such as the thalamus, preoptic area, septum, and cortical hem (Fig. 1A–C); abundant expression was not seen in the large portion of the cerebral cortex, which showed only very weak expression of Zic2 in the cortical VZ/SVZ (Fig. 1B, arrowheads) (Inoue et al., 2007a). Significant expression of the Zic genes was also detected in the marginal areas around the brain, in which Zic1 and Zic2 expression was stronger than that of Zic3 (Fig. 1A–C, asterisks). Examination at higher magnification at E16.5 revealed that the expression of Zic1, Zic2, and Zic3 in the marginal brain were similar to the expression of Nid-1, a gene implicated in the cross-linking of the BM (Mayer et al., 1993), and of Foxc1, the forkhead/winged helix transcription factor (Kume et al., 1998; Zarbalis et al., 2007) (Fig. 1F–J), suggesting that the Zic genes were expressed in the meninges, in addition to the expression of Zic1 and Zic2 in the cells of the cortical MZ (Fig. 1H,I, arrowheads). Examination of the distribution of the Zic proteins using a pan-Zic antibody also indicated that Zic proteins were produced in the marginal areas around the brain (Fig. 1D,E), and double immunostaining with collagen IV, one of the major components of the BM (Pöschel et al., 2004), revealed that the Zic proteins were localized in the nuclei in the meningeal cells (Fig. 1K) and several other cells in the cortical MZ (Fig. 1K, arrowheads). Immunostaining of meningeal cells in culture confirmed that the Zic-expressing cells were meningeal fibroblasts (Fig. 1K, inset). The expression of the Zic proteins in the meningeal cells continued until adulthood (data not shown).
Figure 1.
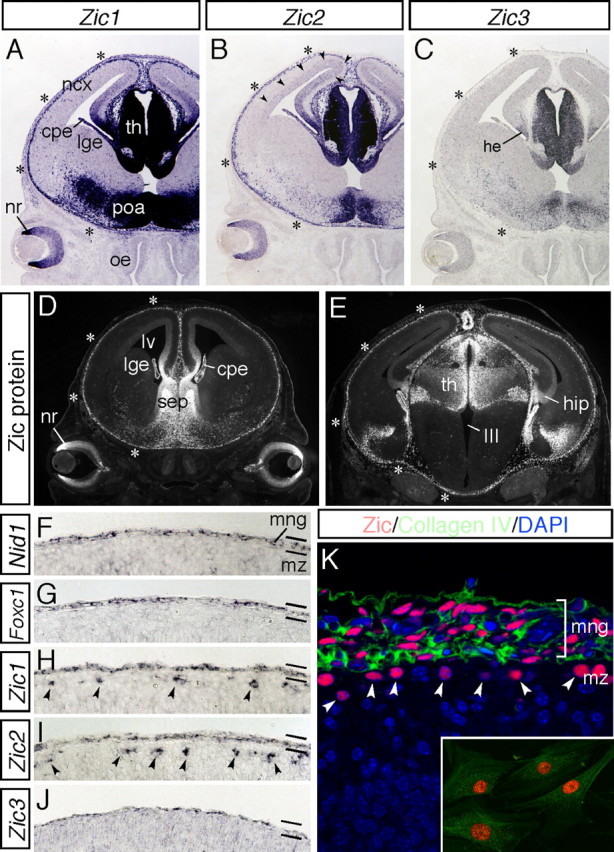
During corticogenesis, Zic genes are expressed by the meningeal cells. A–C, Expression of Zic1 (A), Zic2 (B), and Zic3 (C) mRNAs in coronal sections of the head at E14.5. Zic genes are abundantly expressed in the medial neural tissues and marginal brain. Strong expression of Zic1 and Zic2 in the marginal brain is indicated by asterisks. Expression of Zic2 in the dorsal VZ/SVZ is indicated by arrowheads. cpe, Choroid plexus; he, cortical hem; ncx, neocortex; lge, lateral ganglionic eminence; nr, neural retina; poa, preoptic area; oe, olfactory epithelium; th, thalamus. D, E, Distribution of the Zic proteins was examined using a pan-Zic antibody in coronal sections of the head at E14.5. Rostral (D) and caudal (E) sections are shown. Zic protein expression in the marginal brain is indicated by asterisks. cpe, Choroid plexus; hip, hippocampus; lge, lateral ganglionic eminence; lv, lateral ventricle; nr, neural retina; sep, septum; th, thalamus; III, third ventricle. F–J, Expression of Nid-1 (F), Foxc1 (G), Zic1 (H), Zic2 (I), and Zic3 (J) mRNAs in the marginal brain at E16.5. These genes are commonly expressed by the meningeal cells. Zic1 and Zic2 are additionally found to be expressed by the CR cells in the cortical MZ (arrowheads). mng, Meninges; mz, marginal zone. K, Distribution of the Zic proteins (red) and collagen IV (green) in the marginal brain at E16.5. The nuclei of the cells are stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). Zic-positive CR cells in the cortical MZ are indicated by arrowheads. Inset shows cultured meningeal cells from E16.5 brain. The meningeal fibroblasts are intensely positive for Zic (red) and collagen IV (green).
Figure 2.
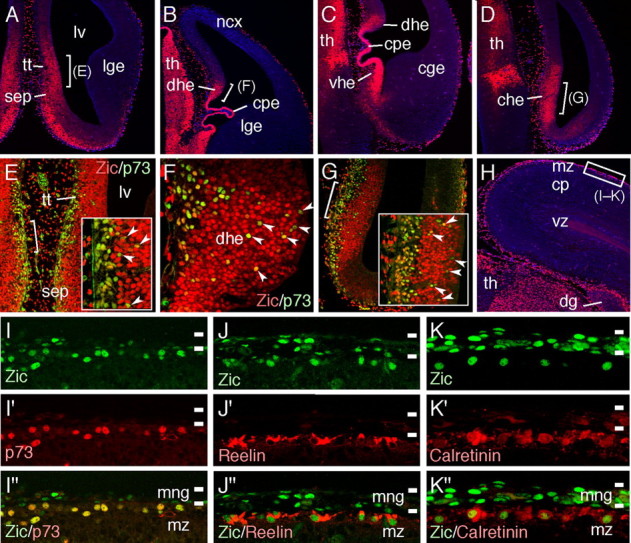
A–D, Distribution of Zic proteins in coronal sections at E12.5. Different rostrocaudal levels of the same brain are indicated (A is the most rostral and D is the most caudal). Zic proteins are abundantly expressed in the septum, choroid plexus, thalamus, dorsal, ventral, and caudal cortical hem. Sections are counterstained with DAPI (blue). cge, Caudal ganglionic eminence; che, caudal cortical hem; cpe, choroid plexus; dhe, dorsal cortical hem; lge, lateral ganglionic eminence; lv, lateral ventricle; ncx, neocortex; sep, septum; tt, tenia tecta; th, thalamus; vhe, ventral cortical hem. E–G, Double-immunofluorescence staining for Zic (red) and p73 (green). High-magnification views of the areas indicated in A, B, and D, respectively. Insets in E and G are higher-magnification views of the areas indicated in E and G, respectively. Typical colabeled cells are indicated by arrowheads. H, Distribution of the Zic proteins in the coronal brain sections at E16.5. The area shown in I–K is indicated by white squares. cp, Cortical plate; dg, dentate gyrus; mz, marginal zone; th, thalamus; vz, ventricular zone. I–K, Distribution of Zic proteins in the dorsal cortical MZ at E16.5 (green). Sections were subjected to costaining for p73 (I′–I″), reelin (J′–J″), and calretinin (K′–K″) (red). Marked overlapping of the Zic-producing nuclei in the cortical MZ with p73-positive (I″), reelin-positive (J″), and calretinin-positive (K″) CR cells. Zic proteins are also produced by the meningeal cells. mng, Meninges; mz, marginal zone.
Figure 3.
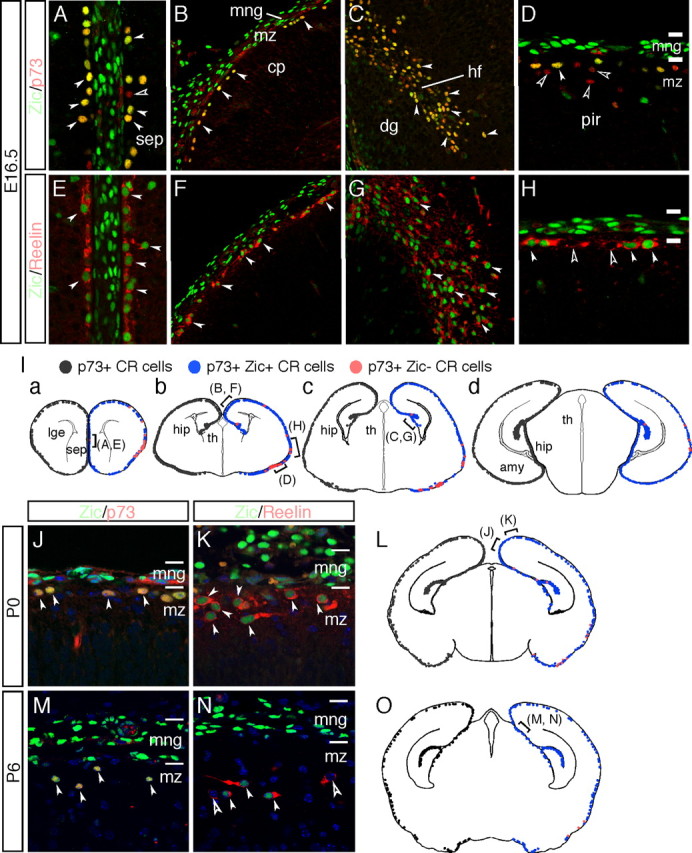
Zic proteins are produced by the majority of the CR cells in the embryonic and postnatal cortical MZs. A–H, Coronal sections of the septum (A, E), dorsal cortex (B, F), hippocampal fissure (C, G), and lateral cortex (D, H) at E16.5 are shown, with double-immunofluorescence staining for Zic (green), p73 (red in A–D), and reelin (red in E–H). dg, Dentate gyrus; hf, hippocampal fissure; ncx, neocortex; pir, piriform cortex; sep, septum. Marked overlapping of the distribution of the Zic+ cells with that of p73+ cells is observed (white arrowheads in A–D). Several p73+Zic−CR cells are observed in the lateral/piriform cortex (open arrowheads in D); reelin+Zic+ cells are also observed (white arrowheads in E–H). Several reelin+ Zic− cells are observed in the lateral/piriform cortex (open arrowheads in H). The locations of the images in the brain is indicated in the schema in I. I, Schematic representation of the distribution Zic-positive CR cells in coronal mouse brain sections from E16.5 mice at various rostral (a) to caudal (d) levels. p73+ CR cells are shown as black dots in the left half of each schema. On the right, p73+ Zic+ CR cells are represented as blue dots, and p73+ Zic− CR cells are represented by red dots. The majority of the p73+ CR cells express Zic proteins, and partial dissociation of Zic and p73 expression is observed in the lateral/piriform cortex at this age. amy, Amygdala; cp, cortical plate; dg, dentate gyrus; hf, hippocampal fissure; hip, hippocampus; lge, lateral ganglionic eminence; mng, meninges; pir, piriform cortex; sep, septum; th, thalamus. J–O, Expression of Zic proteins by CR cells in the cortical MZ at P0 (J–L) and P6 (M–O). Immunofluorescence staining for Zic proteins (green) with p73 (red in J, M) and reelin (red in K, N). The location of the images in the brain is indicated in the schema in L and O. Many reelin-expressing cells expressed Zic, but several cells were also negative for Zic expression (open arrowheads in N); these cells are reelin-expressing interneurons. mng, Meninges; mz, marginal zone of the cerebral cortex. L, O, Schematic representation of the distribution Zic-positive CR cells in P0 (L) and P6 (O) mouse brains at mediocaudal levels. Marked overlapping of expression of the Zic proteins with that of p73 at P0 and P6 (filled arrowheads) is shown. The notations for each cell type are the same as those indicated in I.
Next, the identity of the Zic-producing cells in the cortical MZ was investigated. It was shown previously that the Zic genes were abundantly expressed by the progenitors in the VZ and SVZ of the septum and cortical hem (Inoue et al., 2007a) and that these structures are partially defective in Zic2 hypomorphic (Nagai et al., 2000) and Zic1/3 (Inoue et al., 2007a) mutants. Because these regions are proposed as sources of CR cells (Meyer et al., 2002; Takiguchi-Hayashi et al., 2004; Bielle et al., 2005; Yoshida et al., 2006; García-Moreno et al., 2007), we examined the distributions of the Zic proteins and CR cell markers. For this purpose, the expressions of the Zic proteins were first compared with the expression of nuclear protein p73, which belongs to the p53 family of the tumor-suppressor proteins, known to be selectively expressed by CR cells (Meyer et al., 2002) (Figs. 2, 3). At E12.5, generations of CR cells were observed as identified by the high numbers of p73+ cells in the deep VZ of the tenia tecta, septum, and dorsal, ventral, and caudal cortical hem (Meyer et al., 2002; Takiguchi-Hayashi et al., 2004) (Fig. 2E–G) (supplemental Fig. 1A–D, arrowheads, available at www.jneurosci.org as supplemental material). Similarly, the progenitors in the tenia tecta, septum, and large region of the hems showed high expression levels of the Zic proteins and mRNAs (Fig. 2A–D) (supplemental Fig. 1E–H, arrowheads, available at www.jneurosci.org as supplemental material) (data not shown). The Zic genes were also abundantly expressed in the Dbx1-expressing septal area but not in the additional source of CR cells, namely, the PSB (supplemental Fig. 1I–L, available at www.jneurosci.org as supplemental material). Double-immunofluorescence staining for Zic and p73 in these regions showed overlapping expressions of the two proteins (Fig. 2E–G, arrowheads), suggesting that the CR cells originating in these regions were generated from Zic-expressing progenitors. Later, at E16.5, marked overlapping of p73-expressing nuclei with Zic-expressing nuclei was observed in the dorsal cortical MZs (Fig. 2H,I–I″), suggesting that the Zic-expressing cells in the cortical MZ were CR cells. Immunofluorescence staining for Zic together with that for the secreted extracellular glycoprotein reelin (Ogawa et al., 1995; Schiffmann et al., 1997; Meyer and Goffinet, 1998) and for the calcium-binding protein calretinin (Alcántara et al., 1998; Hevner et al., 2001) was also performed to confirm the presence of the Zic proteins in the CR cells. The results revealed marked overlapping of the reelin+ CR cells, which showed cytoplasmic distribution of reelin, with the Zic+ nuclei (Fig. 2J–J″), and most of the calretinin+ cells in the region were also positive for Zic expression (Fig. 2K–K″). These observations strongly support the previous observation that the Zic+ cells in the cortical MZ were CR cells.
We next estimated the proportions of Zic+ CR cells in the total population of CR cells at E16.5, P0, and P6 (Fig. 3). At E16.5, high numbers of p73+ Zic+ CR cells were distributed in the medial to dorsal and rostral to caudal cortical MZ (Fig. 3A–C), and >90% of the p73+ CR cells expressed Zic proteins (Fig. 3Ia–Id) (from rostral to caudal, 89.6 ± 3.6, 92.9 ± 6.1, 93.7 ± 7.3, and 98.6 ± 5.8, respectively, n = 3) (Fig. 3I). The analysis demonstrated that p73+ Zic-negative (Zic−) CR cells were distributed mostly around the lateral and piriform cortex (Fig. 3D,I). Distribution of p73+ Zic+ and p73+ Zic− cells in the MZ was similarly observed at P0 but decreased at P6 and thereafter (Fig. 3J,L,M,O) (data not shown). The distribution of the reelin+ Zic+ cells was also consistent with the above observations (Fig. 3E–H,K,N). Examination of Zic protein expression in the homozygous Relnrl-Orl cortex, which lacks functional reelin (de Bergeyck et al., 1997), at E15.5 and E18.5 showed a normal distribution of the Zic proteins as well as p73, along with mutated reelin, suggesting that functional reelin is not required for the production of Zic in the CR cells (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) (data not shown). Collectively, the above observations demonstrate that the Zic proteins are expressed by the meningeal cells, CR-generating progenitors in two of the major sources of the CR cells, and later in most of the CR cells, except in the lateral and piriform cortex region, during corticogenesis.
Reduction in number and abnormal distribution of CR cells in the cortical MZ of the Zic mutant mice
To know the effects of Zic mutations on the distribution of the CR cells, the numbers and positions of the CR cells were examined in the Zic1/3 double-mutant cortex (Fig. 4). We first analyzed the distribution of p73 mRNA at E13.0, just after the completion of generation of the CR cells (Fig. 4A–D). In the wild type, p73+ CR cells were widely distributed throughout the telencephalon from the rostral to the caudal MZs at E13.0 (Fig. 4A,B) (n = 6). However, in the Zic1/3 mutant, two of the sources of CR cells, the septum and dorsal hem, showed slight hypoplasia (Fig. 4C,D, asterisks), consistent with a previous report (Inoue et al., 2007a), and the number of p73-expressing cells was decreased in the marginal regions contiguous to these sources (Fig. 4C,D, arrowheads) (n = 5). The hypoplasia and reduction in the number of CR cells were observed in the septal area and in the rostral to intermediate dorsal hem/hippocampal primordium but were not always evident in the more caudal telencephalon (data not shown). When the distribution of CR cells was examined later at E15.5, the reduction in the number of the CR cells in the rostral regions of the cortices seemed milder than that observed at E13.0 but still evident (∼40% reduction) compared with that in the wild-type brains (Fig. 4E–T). When examined at higher magnifications, the wild-type embryos showed an almost continuous band of intensely labeled Reelin+, p73+, and Calretinin+ CR cells along the cortical MZ (Fig. 4I,K,M–O) (n = 6) whereas, in the Zic1/3 mutant, the CR cells were frequently separated by cell-free areas (Fig. 4J,L,R–T, brackets) (n = 5). Although such sparseness of the CR cells was observed in the rostral cortex, no significant differences were observed in the intermediate to caudal cortex between the wild-type and the Zic1/3 mutant animals (Fig. 4F,H) (data not shown).
Figure 4.

Reduced number of CR cells in the rostral cortical MZ of Zic1/Zic3 double-mutant mice. A–L, Morphology and marker analysis of the rostral (A, C, E, G), intermediate (B, D, F, H), cortical MZs at E13.0 (A–D) and E15.5 (E–L). In situ hybridization for p73 in the wild-type (A, B, E, F, I, K) and Zic1/3 double-mutant (C, D, G, H, J, L). Coronal sections are shown. I, K and J, L are higher-magnification images of the indicated areas in E and G, respectively. Dashed lines in E and G indicate the lateral ventricles. Reduced numbers of p73+ CR cells contiguous to their sources at E13.0 are indicated by arrowheads in C and D. In the rostral cortex of the Zic1/3 double-mutant mice at E15.5, the MZ has multiple areas lacking in p73+ CR cells (brackets in J and L). CA1, Hippocampal CA1 subfield; hippocampal CA3 subfield; dg, dentate gyrus; dg*, hypoplastic dentate gyrus; dth, dorsal thalamus; dth*, hypoplastic dorsal thalamus; lge, lateral ganglionic eminence; lv, lateral ventricle; mng, meninges; ncx, neocortex; pir, piriform cortex; sep, septum. M–V, In situ hybridization for Reelin (M, N, R, S), Calretinin (O, T), Cxcr4 (P, U), and Cxcl12 (Q, V) in the dorsal and medial regions of the rostral cortex. The regions in M and O–Q, R and T–V, N, and S correspond to the same regions indicated in K, L, I, and J, respectively. Cell-free areas without Reelin+, Calretinin+, or Cxcr4+ CR cells in the cortical MZ of the Zic1/3 mutant mice are indicated by brackets. The reduced expression of Cxcl12 in the meninges is indicated by asterisks in V.
In addition to the number of CR cells, the marginal distribution of the CR cells was also affected in the Zic1/3 mutant (Fig. 5). p73+ and Reelin+ CR cells were abnormally distributed in the upper half of the cortex (Fig. 5G–I) and hippocampal fields (Fig. 5J) in the embryos at E15.5 [0.08 ± 0.02 sites per section in control (84 sections derived from six embryos, n = 6); 0.82 ± 0.07 sites per section in Zic1/3 embryos (72 sections, n = 5) for p73 expression; p < 0.05, t test]. In contrast to the reduction in the number of CR cells observed in the rostral cortex of the Zic1/3 mutants, ectopic CR cells were observed throughout the cortex from E15.5 (Fig. 5G–J) to E18.5 (data not shown). The chemokine receptor Cxcr4, known to be expressed by CR cells (Stumm et al., 2003), and its ligand Cxcl12, which is expressed in the meninges, play crucial roles in the marginal distribution of the CR cells (Borrell and Marin 2006; Paredes et al., 2006). In the Zic1/3 mutant, the marginal expression of Cxcr4 was reduced in the rostral cortex (Fig. 4U). Interestingly, the expression of Cxcl12 in the meninges was also dramatically decreased (Fig. 4V, asterisks), and a reduced expression of Cxcl12 was widely observed around the cortex (Fig. 5K,L) (data not shown), suggesting that meningeal development was affected in the Zic1/3 mutant (discussed later).
Figure 5.

A–D, G–J, Abnormal distribution of CR cells in the cortical MZ of the Zic1/Zic3 double-mutant mice. Distribution of Reelin+ (A, G) and p73+ (B–D, H–J) CR cells in the rostromedial (A, G), rostrodorsal (B, H), and caudolateral (C, I) cortex and the caudal hippocampus (D, J) at E15.5. Distribution of CR cells in the coronal brain sections of the wild-type (A–D) and Zic1/3 mutant (G–J) embryos are shown. In the wild-type embryos, Reelin+ and p73+ CR cells are confined to the neocortical MZ (A–C) and hippocampal fissure (D). In the mutants, the CR cells are widely scattered and found in the cortical plate of the neocortex (G–I) and deeper positions in the hippocampal CA3 region (J) (arrowheads). CA1, Hippocampal field CA1; CA3, hippocampal field CA3; cp, cortical plate; dg, dentate gyrus; hf, hippocampal fissure; fm, fimbria; mng, meninges; mz, marginal zone. E, F, K, L, Expression of Cxcl12 in the wild-type mice (E, F) and Zic1/3 mutants (K, L) in the caudal hippocampus (E, K) and caudolateral cortex (F, L) at E15.5. E, F, K, and L are neighboring sections of D, C, J, and I, respectively. Reduced expression of Cxcl12 in the meningeal layer of the Zic1/3 mutants is indicated by asterisks in K and L.
Reduction in the number of p73+, Reelin+, and Calretinin+ CR cells in the dorsal and medial cortex were also observed in the rostral cortex of the Zic2 hypomorphic mutant from E15.5 to E18.5 (n = 7) (supplemental Fig. 3, available at www.jneurosci.org as supplemental material) (data not shown), although no reduction in the CR cell number was evident in the caudal cortex in these Zic2 mutants (data not shown). In addition, in the Zic2 mutants, reduced expression of Cxcl12 was widely observed throughout the cortex (supplemental Fig. 3L, asterisks, available at www.jneurosci.org as supplemental material) (data not shown). Overall, the abnormalities of the CR cells in the Zic2 mutants were similar to those in the Zic1/3 mutants.
Loss of the Zic genes results in cortical lamination defects
The finding of the abnormal CR cell distribution in the cortex in the Zic mutants raised the possibility that the altered distribution of CR cells might, in turn, affect the cortical structures. To determine whether a loss or reduced expression of the Zic genes might also affect the cortical layer structures, the expression of Tbr1, which distinguishes layers I, II/III, VI, subplate, and SVZ (Hevner et al., 2001), was examined at E18.5 (Fig. 6A–F). With slight thinning of the cortical layer especially in the Zic1/3 mutant (Inoue et al., 2007a) (ratios of the dorsolateral cortex thickness of Zic1/3 mutant to that of control littermates were as follows: rostral, 89 ± 6%; intermediate, 88 ± 5%; caudal, 69 ± 4%; n = 3), differences in Tbr1 expression were observed throughout the cortex in both the Zic2 and Zic1/3 mutants. Tbr1 staining of cortical layer VI and the subplate was winding and diffuse in both the Zic2 and Zic1/3 mutants compared with that in the wild-type cortex (Fig. 6B,C,E,F). Cell-poor marginal zones and Tbr1-positive layer VI were distinguishable in the Zic mutants, suggesting splitting of the preplate.
Figure 6.
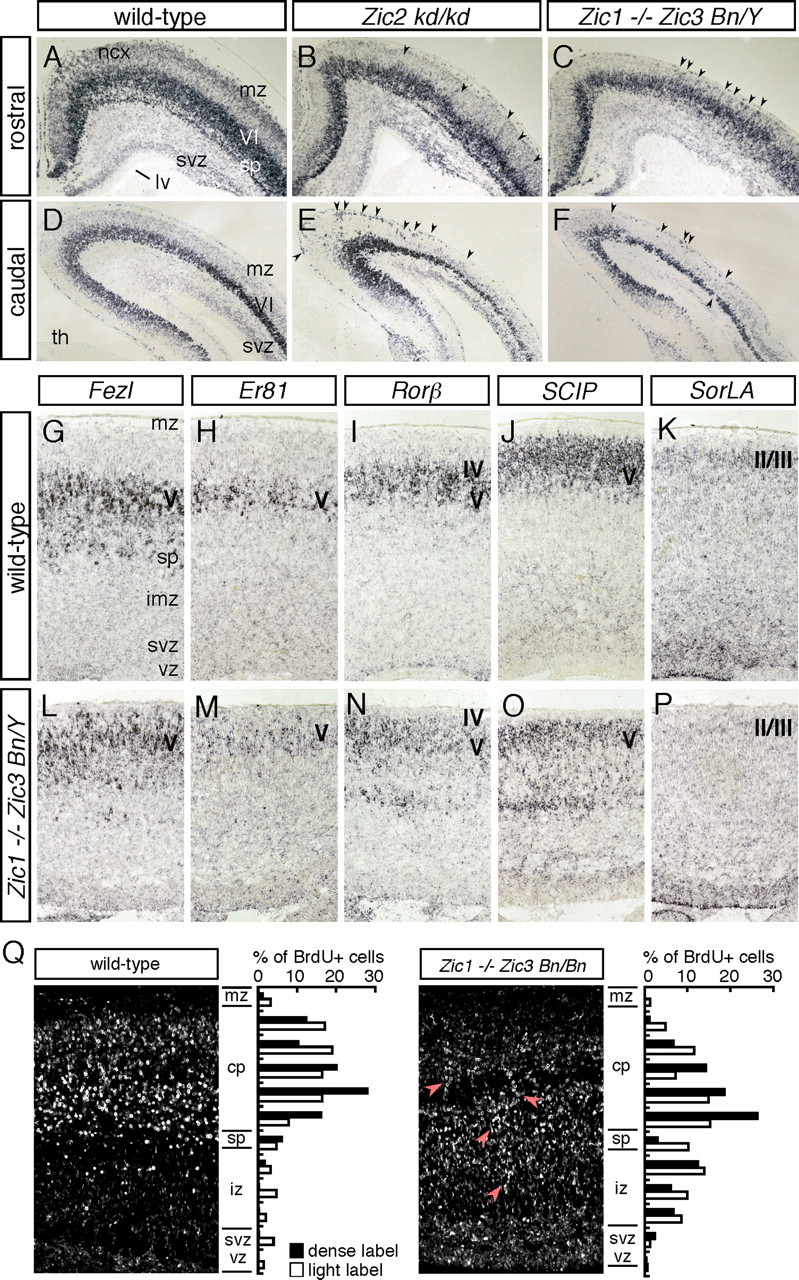
Loss and reduced expression of the Zic genes results in cortical lamination defects. A–F, In situ hybridization for Tbr1 in wild-type (A, D), Zic2 kd/kd (B, E), and Zic1/3 double-mutant (Zic1−/− Zic3 Bn/Y) (C, F) mice at E18.5. Coronal sections of the rostral (A–C) and caudal (D–F) brain are shown. In the wild type, Tbr1 is expressed in layer VI, the subplate, and the SVZ at this stage. In the Zic2 kd/kd and Zic1/3 mutants, the Tbr1 expression is disorganized and diffuse (arrowheads), suggesting the existence of cortical lamination defects in these mutants. lge, Lateral ganglionic eminence; lv, lateral ventricle; mz, marginal zones; ncx, neocortex; sp, subplate; th, thalamus; VI, layer VI. G–P, Expression of cortical-layer specific genes in the Zic1/3 mutant neocortex. In situ hybridization for Fezl (G, L), Er81 (H, M), Rorβ (I, N), SCIP (J, O), and SorLA (K, P) in coronal brain sections of E18.5 wild-type (G–K) and Zic1/3 mutant (L–P) mice. Q, Abnormal migration of Zic1/3 (Zic1−/− Zic3 Bn/Bn) mutant neurons. BrdU birthdating analysis reveals perturbed neuronal migration in the Zic1/3 mice. The final destination of the early-born neurons labeled by BrdU at E13.5 is shown in the sagittal sections of the wild-type (left) and Zic1/3 mutant (right) mice at E18.5. BrdU-positive nuclei were detected by immunohistochemistry. The arrows indicate neurons exhibiting aberrant migration. Bar graphs showing the radial distribution of heavily labeled cells (first generation at the time of the BrdU injection) and lightly labeled cells (the majority are second- and perhaps third-generation cells from subsequent progenitor cell divisions) in the neocortex of E18.5 wild-type (left) and Zic1/3 mutant (right) mice. cp, Cortical plate; iz, intermediate zone; mz, marginal zone; sp, subplate; svz, subventricular zone; vz, ventricular zone.
Examination of other layer-specific markers, such as Fezl, Er81, Rorβ, SCIP, and SorLA, which distinguish emerging layers (II/III, IV, and V), revealed that the expression of each of these genes appeared in the same pial-to-ventricular order in the cortex in both the Zic1/3 and Zic2 mutants (Fig. 6L–P) (data not shown); these findings indicate that the identity and organization of the cortical layers were not significantly altered in these mutant mice. The Zic mutant cortices did not show a typical Reeler phenotype, which is characterized by a failure of preplate splitting, disorganized cortical plate, and inverted layer structures (Caviness, 1982; Pinto-Lord et al., 1982). However, in the Zic1/3 cortex at E18.5, the layer-specific markers showed a diffuse distribution and an upper shift of the Fezl-, ER81-, Rorβ-, and SCIP-expressing layer V neurons and reduction in the number of SorLA-expressing layer II/III neurons, especially in the intermediate dorsal cortex (Fig. 6L–P), suggesting that neuronal migration was widely affected in the mutants. The results were also supported by quantification of Tbr1- and Rorpositive neurons (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). The positions of the Tbr1-labeled cells in cortical layer VI and Ror-labeled cells in layer IV/V were upper shifted, and the number of later-born neurons in cortical layer II–V were reduced. Changes in the expression of layer-specific markers in the Zic2 mutants were milder than those in the Zic1/3 mutants and were not always evident at E17.5–E18.5 (data not shown).
To directly determine whether the Zic1/3 double mutation might affect cortical lamination and neuronal migration, a quantitative birthdating analysis was performed by labeling the neurons at E13.5 with BrdU and then examining their final destination in the neocortex at E18.5 (Fig. 6Q). Most neurons generated at E13 were properly positioned in the cortical plate and subplate (Fig. 6Q, left). In the Zic1/3 mutant cortex, however, the BrdU-positive cells accumulated broadly in the intermediate zone and the subplate-like layers, in addition to being found in the deeper layers of the cortical plate (Fig. 6Q, right). These results support the contention that the migration dynamics of the neurons in the cortex is altered in the Zic1/3 mutants.
Lamination defects in the Zic mutants are associated with type II lissencephaly
The disorganization of the rostral cortex in the Zic2 and Zic1/3 mutants could be partly attributable to the changes in the number of CR cells. However, alternative explanations were also sought, because a disorganized cortical structure was observed even in the caudal cortices, in which no significant change in the CR cell number was observed. In addition, the slight reduction of CR cells may not be sufficient to change cortical organization, because a massive reduction of CR cells in the hem-ablated mice (Yoshida et al., 2006), mice lacking Dbx1-expressing progenitors in the septum and PSB (Bielle et al., 2005), and p73-deficient mice (Meyer et al., 2004) show anatomically correct neocortical layer. Furthermore, the ectopic distribution of the CR cells itself cannot explain the lamination defects observed in the Zic mutants, considering that the Cxcr4-deficient cortex, which shows marked ectopic localization of the CR cells in the deeper cortical layers (Borrell and Marin, 2006; Paredes et al., 2006), showed no obvious differences in the cortical layer formation (Lu et al., 2002). Because Zic proteins are also abundantly expressed by the meningeal cells and the expression of Cxcl12 in the meningeal layer was reduced widely throughout the cortex, we hypothesized that the abnormalities of the meningeal cells may contribute to the cortical disorganization observed in the Zic mutants.
To address this possibility, the cortical marginal structures in the Zic mutants were examined by in situ hybridization for Math2, a b-HLH gene expressed in the glutamatergic cortical neurons (Schuurmans et al., 2004) (Fig. 7A–L). Smooth marginal staining for Math2 was observed in the wild-type mice (Fig. 7A–D,I,J), whereas multiple regions of cell invasion into the MZ and subarachnoid space were observed throughout the cortical MZ in both the Zic2 (Fig. 7E–H) (n = 8) and Zic1/3 (Fig. 7K,L) (n = 7) mutants at E17.5. The ectopia in the cortical surfaces of the Zic mutants closely resembled the changes observed in human cobblestone lissencephaly (also known as type II lissencephaly) (Olson and Walsh, 2002). Staining with anti-neuronal-specific nuclear protein antibody also indicated that neurons were the major constituents of the ectopias (data not shown). Examination of neighboring sections in the Zic2 mutants revealed that Reelin+ and Calretinin+ CR cells were scarce in the regions of ectopia and extended beyond the MZ (Fig. 7R,S). GAD67+ interneurons were also ectopically localized (Fig. 7V), and the neuronal layer structures, as revealed by Tbr1 and SCIP expression, were distorted in these regions (Fig. 7T,U). These observations suggested that the lamination defects that were widespread in the cortices of the Zic mutants were associated with marginal extrusions of the cortical neurons.
Figure 7.
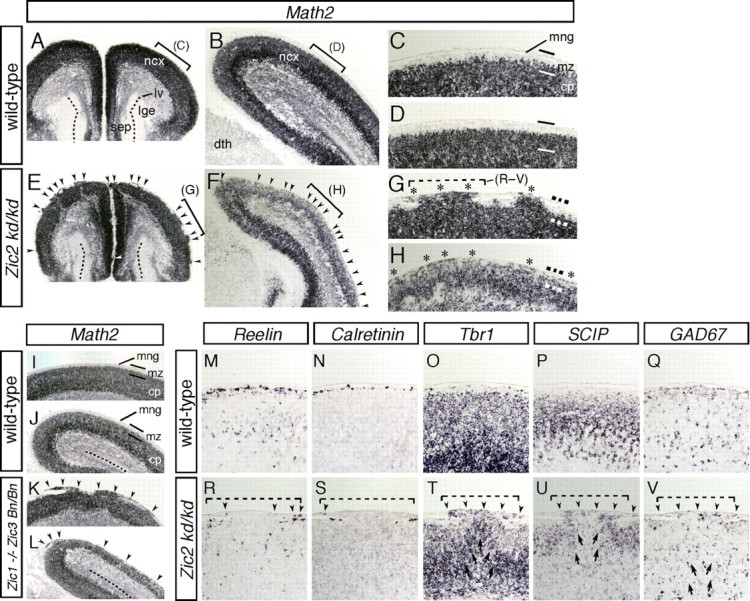
Reduced expression of Zic2 and loss of both Zic1 and Zic3 result in cortical lamination defects resembling those in cobblestone (type II) lissencephaly. A–H, In situ hybridization for Math2 in wild-type (A–D) and Zic2 mutant (E–H) brains at E17.5. Coronal sections of the rostral (A, C, E, G) and caudal (B, D, F, H) regions are shown. C, D, G, and H represent higher-magnification views of the indicated areas in A, B, E, and F, respectively. Wild-type brains showed a highly organized multilayered cortex with a hypocellular marginal zone at the surface of the brain (A–D). In contrast, Zic2 mutant brains showed severe disorganization of cortical lamination and neurons invading and obliterating the marginal zone (arrowheads in E, F; asterisk in G, H). cp, Cortical plate; dth, dorsal thalamus; lv, lateral ventricle; mng, meninges; mz, marginal zone of the cerebral cortex; ncx, neocortex; sep, septum. I–L, In situ hybridization for Math2 in wild-type (I, J) and Zic1/3 double-mutant (Zic1−/− Zic3 Bn/Bn) (K, L) mice at E17.5. Dorsal cortex in the rostral (I, K) and caudal (J, L) brain. M–V, In situ hybridization for Reelin (M, R), Calretinin (N, S), Tbr1 (O, T), SCIP (P, U), and GAD67 (Q, V) in wild-type (M–Q) and Zic2 mutant (R–V) mice at E17.5. Images in M–Q and R–V were taken from sequential sections in C and G, respectively. The area indicated by the dashed line in G corresponds to the indicated areas shown in R–V. Altered marginal expressions of the markers are indicated by arrowheads. Disorganized cortical layer structures beneath the marginal defects are indicated by arrows in T–V.
Defective pial BM and altered structure of the radial glial end feet in the Zic mutant neocortex
It has been shown that mice lacking BM components or proteins that play significant roles in cell adhesion and adhesion-dependent signaling events exhibit abnormal cortical development and focal marginal zone heterotopias (Georges-Labouesse et al., 1998; Graus-Porta et al., 2001; Halfter et al., 2002; Moore et al., 2002; Beggs et al., 2003). We therefore examined the meninges and pial BM in the Zic mutant mice. Immunofluorescence staining against pan-laminin, a major component of the meninges, revealed a thinner pial BM in the dorsal cortex, with several disruptions in the Zic1/3 mutant at E16.5 (Fig. 8B,D, arrowheads) (data not shown). Notably, a thinner and disrupted BM was observed in the cortex at all rostrocaudal levels from E15.5 to E18.5 (n = 9), suggesting that there were meningeal defects that might be responsible for abnormal mid-late embryonic cortical development in this mutant. An analysis of the meningeal transcripts quantified by semiquantitative RT-PCR at E15.5 showed reduction of Laminin γ1, Nid-1, Foxc1, and Perlecan, further supporting the finding of the defective assembly of the BM in the Zic1/3 mutant (Fig. 8E) (n = 3). In addition, analysis of the meningeal transcripts confirmed the reduction of Cxcl12 (Fig. 8E). Quantification of BrdU-labeled cells and of cells containing the phosphorylated form of histone H3 (PH3) in the meninges showed that the numbers of both BrdU- and PH3-positive proliferating cells in the meninges were significantly reduced, in both the rostral and caudal regions, in the Zic1/3 mutant, at E14.5 and E17.5 (Fig. 8F–I), whereas no significant changes in the cell death frequency in the meninges and cortical MZ were observed at these stages (data not shown).
Figure 8.
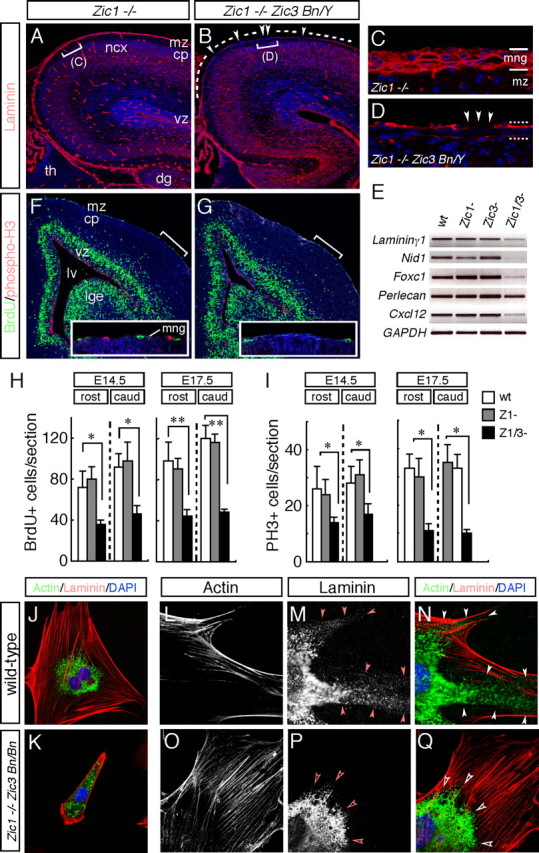
The pial BM is defective in the Zic1/3 double-mutant neocortex. A–D, Immunofluorescence detection of laminin (red) in coronal sections of the brain from the wild-type (A, C) and Zic1/3 mutant (B, D) mice at E16.5. The sections are counterstained with DAPI (blue). Control Zic1−/− show a smooth layer of basal lamina-associated laminin at the surface of the brain, as well as a basal lamina surrounding the blood vessels. In contrast, in the Zic1/3 mutant, the laminin layer at the surface of the brain is thinner compared with that in the Zic1−/− sections. C and D are high-magnification views of the indicated areas in A and B, respectively. E, Analysis for transcripts of the meningeal BM components and secreted factor. RT-PCR for Laminin γ1, Nid-1, Foxc1, Perlecan, and Cxcl12; GAPDH served as the internal control. Analysis was performed on cDNAs prepared from the meninges of wild-type (n = 6), Zic1−/− (n = 3), Zic3 Bn/Y (n = 4), and Zic1/3 (Zic1−/− Zic3 Bn/Y) (n = 3) mutants at E15.5. F, G, Decreased numbers of BrdU- and PH3-labeled proliferating meningeal cells in the Zic1/3 double-mutant mice. Proliferating meningeal cells in the S-phase were pulse labeled with BrdU for 1 h at E15.5. Immunohistochemical analysis showed decreased numbers of BrdU-labeled (green) and PH3-labeled (red) cells in the meningeal layer in the Zic1/3 mutants compared with those in the meningeal layer of the wild-type mice. The sections were counterstained with DAPI (blue). Comparable rostral areas in the Zic1/3 mutant and wild-type brains are shown. Insets are high-magnification views of the indicated areas. H, I, Quantification of the number of BrdU-labeled (H) and PH3-labeled (I) cells in the meningeal layers. BrdU- and PH3-labeled cells in the rostral and caudal regions were counted in the wild-type, Zic1−/−, and Zic1/3 mutant mice at E14.5 and E17.5. BrdU- and PH3-labeled meningeal cells were significantly decreased in the Zic1/3 mutants compared with those in the wild-type and Zic1−/− mice. The results are shown as the mean number of multiple comparable sections with SDs. *p < 0.05; **p < 0.01 by t test. J–Q, Primary meningeal fibroblasts from E14.5 wild-type (J, L–N) and Zic1/3 mutants (K, O–Q) were double stained with a laminin (J, K, M, N, P, Q) and F-actin (J, K, L, N, O, Q). N and Q are merged images of L, M and O, P, respectively. Meningeal fibroblasts were cultured for 8 h (J, K) and 3 d (L–Q). Fibroblasts from the Zic1/3 mutants spread less well and showed less proliferative activity (K). Fibrillar and punctate laminin is observed at the periphery in the wild-type meningeal fibroblasts (arrowheads in M, N), whereas it is absent from the meningeal fibroblasts of the Zic1/3 mutants (open arrowheads in P, Q). cp, Cortical plate; dg, dentate gyrus; lge, lateral ganglionic eminence; lv, lateral ventricle; mng, meninges; mz, marginal zone of the cerebral cortex; ncx, neocortex; th, thalamus; vz, ventricular zone; wt, wild type.
To further confirm that the meningeal fibroblasts of the Zic mutants exhibit altered production and/or organization of the BM components, immunofluorescence staining was performed to identify the expression of laminin in the primary meningeal fibroblast cultures of the wild-type (n = 8) and Zic1/3 mutant (n = 4) (Fig. 8J–Q) brains. Meningeal fibroblasts from the wild-type mice spread well 8 h after plating (Fig. 8J) and produced laminin that was arranged in a fibrillar and punctate pattern after 2–3 d in culture (Fig. 8M,N) (Beggs et al., 2003). Phalloidin staining of filamentous actin (F-actin) revealed the presence of stress fibers projecting into arrow-shaped focal adhesions (Fig. 8J,L,N). In contrast, fibroblasts from the Zic1/3 mutant were less spread and less proliferative, and 40% of the cells were still round-shaped 8 h after plating (data not shown). The remaining 60% of the cells had begun to spread by this time but lacked the typical focal adhesions and stress fibers (Fig. 8K). When the Zic1/3 mutant fibroblasts were cultured over 2 d, they still exhibited less spreading compared with the cells from the wild-type mice but formed focal adhesions and F-actin stress fibers (Fig. 8O,Q). Notably, the mutant meningeal fibroblasts showed a reduction in expression of punctate and fibrillar laminin at the cell periphery, although the overall expression level of laminin seemed to be comparable with that in the wild-type embryos (Fig. 8P,Q).
It has been demonstrated that the extending processes from the radial glial cells contact the BM at the pial surface and serve as structures guiding migrating neurons from their place of birth in the VZ/SVZ toward their final position in the CP (Rakic, 2003). It has also been suggested that proper anchorage of the end feet of the radial glial cells is dependent on the presence of an intact BM and a functioning cell adhesion system (Hartmann et al., 1998; Graus-Porta et al., 2001; Beggs et al., 2003; Niewmierzycka et al., 2005). Therefore, the morphologies of the radial glial processes and their end feet in the Zic mutant cortex were examined by immunofluorescence staining for nestin at E17.5. In the control Zic1−/− cortex, nestin immunoreactivity was seen in the radial glial end feet as a continuous band underlying the pial membrane, terminating in well defined end feet at the pial surface (Fig. 9A–D). However, in the Zic1/3 mutant, the nestin-positive band was interrupted in the regions of cortical ectopia (Fig. 9E, asterisks). Double staining with laminin and nestin revealed that, in the regions of cortical ectopia, the laminin-positive meninges were disrupted, and the nestin-positive radial glial processes showed no contact with the pial surface or retracted from the cortical surface (Fig. 9E–H). Occasionally, some of the radial glial processes aberrantly extended into the middle of an ectopic outgrowth (Fig. 8E, arrow). Nestin staining also revealed that, even in the cortical marginal areas without marked ectopias in the Zic1/3 mutant, the continuous band of nestin-positive radial glial end feet was thinner in most regions of the dorsal cortex (Fig. 8M,N,P). The altered nestin staining may be correlated with the broad thinning and disruptions of the laminin-positive BM overlying the cerebral cortex (Fig. 8O).
Figure 9.
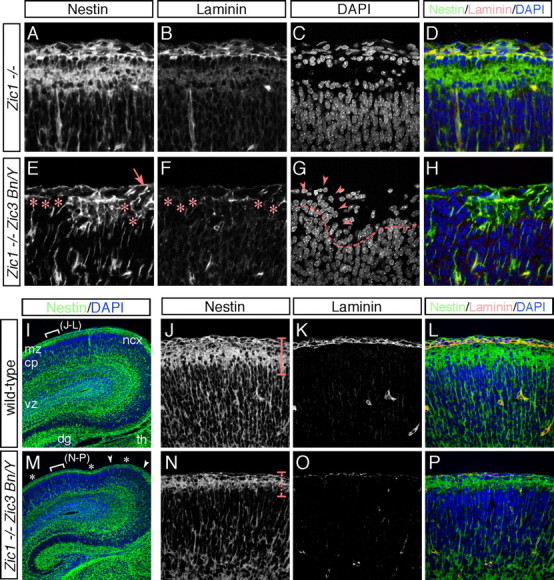
BM disruption and altered radial glial end feet in the Zic1/3 double-mutant neocortex. A–H, Immunofluorescence staining for nestin (A, E) and laminin (B, F) in the dorsal cortical MZ of the Zic1−/− (A–D) and Zic1/3 mutant (E–H) mice at E17.5. Sections were costained with DAPI (C, G). D and H are merged images of A–C and E–G, respectively. In the control Zic1−/− brain, radial glia showed a parallel arrangement of fibers that terminated in well defined end feet at the pial surface (A). In the Zic1/3 mutants, absence of the nestin-immunoreactive radial glial end feet (asterisks in E) was associated with breakdown of the surface laminin (asterisks in F) and neuronal ectopias (arrowheads in G). In the mutant brains, the radial glial end feet were occasionally found to be detached from the pial surface in the regions of the ectopias. These defects were seen scattered throughout the cortical MZ. I, M, Immunofluorescence staining for nestin in the dorsal cortex of the wild-type (I) and Zic1/3 (M) mice at E17.5. The sections were counterstained with DAPI (blue). The locations of the neuronal ectopias are shown in white arrowheads, and the thinner continuous band of nestin-positive radial glial end feet is indicated by asterisks in M. The locations indicated in J–L and N–P are indicated by white brackets in I and M, respectively. J–L, N–P, Higher magnification of the indicated areas in I and M. Immunofluorescence staining for nestin (J, N) and laminin (K, O) in the wild-type (J–L) and Zic1/3 mutant (N–P) mice. L and P are merged images of J,K and N,O, respectively. The sections were counterstained with DAPI (blue). The thinner continuous band of nestin-positive radial glial end feet in the Zic1/3 mutants is indicated by a dashed line in N.
Discussion
Expression of Zic genes in the CR cells and its distinct spatial origins
CR cells are transient early-born preplate neurons that are localized selectively in the outer MZ during development of the cortical plate (Marin-Padilla, 1998; Super et al., 1998; Meyer et al., 1999). The present study showed that Zic is expressed in the CR cells during the period of generation of the cortical layer structure. In the embryonic and perinatal MZs, the majority of the p73+, reelin+ CR neurons in the cortical MZ and hippocampus produced Zic proteins. The postnatal decrease of neocortical Zic-expressing cells is also consistent with the known temporal distribution of the CR cells (Alcántara et al., 1998; Zecevic and Rakic, 2001). Recently, Bielle et al. (2005) demonstrated that the CR cells that migrate into the rostromedial and lateral cortex arise from Dbx1-expressing progenitors in or near the septum and the PSB. Because most of the non-Zic-producing CR cells were distributed in the piriform and lateral cortex, a proportion of these cells may originate from the PSB. This idea is also supported by the finding that the CR cells generated in the hem and septum arise from the Zic-expressing progenitor zones and that virtually all the CR cells near these regions express Zic. Future experiments using genetic tracing will be required to determine the onset of appearance, final destinations, and molecular identity of the Zic-expressing CR cells.
Both Zic2 and Zic1/3 mutants show hypoplastic development of the septum and hem structures, and these defects have been shown to appear both during and after the CR cell production period (Inoue at al., 2007a). Because no significant changes were observed in the frequency of cell death in the cortical MZ from E12 to E18, the reduced number of CR cells in the rostral cortex in these Zic mutants would be caused by the smaller septal and hem progenitor zones, which might have produced lower numbers of CR cells. These ideas are supported by the observation that the number of CR cells was reduced near their sources, the hem and septum. However, we cannot fully exclude the slight contribution of cell death to the reduction in the number of CR cells, because meningeal loss of the Zic genes could indirectly influence their survival. Previous observations suggest that the selective degeneration of meningeal cells leads to secondary degeneration of the CR cells (Super et al., 1997), which indicates that meningeal cells may synthesize and secrete trophic factors for the CR cells, such as Bdnf, Nt-4, and Cxcl12 (Brunstrom et al., 1997; Ringstedt et al., 1998; Stumm et al., 2003; Alcántara et al., 2005).
The ectopic CR cells can be considered in the light of the result that the amount of the Cxcl12 transcript was severely reduced in the Zic mutant meninges. It has been shown recently that meningeal Cxcl12 controls the marginal distribution of the hem-derived Cxcr4-expressing CR cells and that the absence of Cxcr4 function results in derailment of the CR cells from a subpial position down to the CP (Borrell and Marin, 2006; Paredes et al., 2006). These studies also suggest that the integrity of the meninges is critical for the initial spread of the CR cells along the cortical surface and also for their subsequent marginal distribution. In the Zic1/3 mutant, the initial marginal spread of the CR cells and their meningeal organization was normal until E14.5 (data not shown), and the ectopic CR cells and reduction of Cxcl12 transcripts were noted only after E15.5. Because the ectopic distribution of the CR cells in the Zic mutants was similar to that in the Cxcr4 mutants, we can speculate that the CR cell mislocalization in the Zic mutants may be caused in part by reduced secretion of Cxcl12 by the meningeal cells, or more broadly, by reduced Cxcl12/Cxcr4 signaling.
Zic genes are required for the proliferation of meningeal fibroblasts and formation of the BM
Although Zic genes are unique in that they are expressed in both CR cells and meningeal cells during cortical development, the present analysis of the cortical phenotypes of the Zic mutants indicated the functional importance of the Zic genes expressed in the latter cells. Zic genes are abundantly produced by the meningeal cells, and the proliferation of meningeal cells was shown to be reduced in Zic1/3 mutants. These findings suggest that the Zic genes are involved in the expansion of the meningeal fibroblasts. Although the changes in the cell proliferation or laminin staining were not clear in the Zic2 mutants, it is still possible that Zic2 may have a role in the proliferation of the meningeal cells. Because the Zic2 hypomorphic mutant allele still produces intact proteins at the level of 20% of that in the wild type (Nagai et al., 2000), and the involvement in the cell proliferation has been reported in developing cerebella (Aruga et al., 2002a). Regulation of the cell cycle of the meningeal cells may also be affected by inhibiting their exit from the cell cycle, which is suggested for neural progenitors in the embryonic septum and hippocampus (Inoue et al., 2007a), cerebellum (Aruga et al., 2002a) and spinal cord (Aruga et al., 2002b). Zic may expand the cell population of the meningeal cells during massive increase in the cortical size.
The disturbed cell properties of the meningeal cells was a remarkable phenotype that appeared to be shared by the Zic1/3 and Zic2 mutant mice. Both mutants showed reduced Cxcl12 expression, and the Zic1/3 mutants showed reduced expression levels of the BM components and altered laminin organization in the cultured meningeal fibroblasts. These results indicate that the three Zic genes are critical not only for proliferation but also for maintaining the differentiated properties of the meningeal fibroblasts. Notably, reduction in punctate and fibrillar laminin in the cell periphery in the meningeal fibroblasts in the Zic1/3 mutants was closely similar to the phenotype in meningeal cultures of focal adhesion kinase (FAK) (Beggs et al., 2003) and EF65/EF65L1 (Guenette et al., 2006) mutants, both of which are associated with a cobblestone cortex. Because these proteins have also been indicated to be essential for cell adhesion and BM assembly, it would be tempting to speculate that some secretory factors and BM components essential for the functions of meningeal cells are possible downstream targets of transcriptional regulation by the Zic proteins.
Expression of the Zic genes in the meningeal fibroblasts are not limited to the cortical area but is widely observed in other parts of the CNS as well, such as the cerebellum and spinal cord (T. Inoue, M. Ogawa, and J. Aruga, unpublished observation). In this regard, Zic expression may represent a common property of meningeal cells. A recent study showed that human ZIC1 is upregulated in desmoid tumors and fibroproliferative disorders (Pourebrahim et al., 2007), raising the possibility that the role of the Zic proteins in fibroblast proliferation is not limited to the meningeal fibroblasts.
The role of Zic genes in corticogenesis
Because Zic1 and Zic3 are not significantly expressed in the cortical VZ, radial glia, and cortical neurons, lamination defects in the Zic1/3 mutant cortices may primarily reflect the defective properties of the meningeal cells. Meningeal fibroblasts secrete components of the BM, which then bind to receptors on the radial glial end feet of the glia limitans to assemble a functioning BM (Sievers et al., 1994; Shearer and Fawcett, 2001; Beggs et al., 2003; Niewmierzycka et al., 2005). Indeed, we found thin sheets of laminin-positive BM with frequent disruptions and alterations in the terminal morphologies of the radial glial cells in the regions with severe neuronal ectopias in the Zic1/3 mutant. Although the regionally altered CR cell distribution could partially contribute to the cortical lamination defects, changes in the pial radial glial end feet were more widespread than the area of abnormal distribution of the CR cells. Therefore, altered BM and marginal radial glial properties may represent a more clear account of the abnormalities in the Zic1/3 mutant cortex than the abnormal distribution of CR cells. This idea is also supported by the results of several studies of mutant mice. Mouse mutants for proteins involved in pial BM assembly, such as laminins, perlecan, dystroglycan, integrins, FAK, and integrin-linked kinase, cause BM abnormalities associated with neuronal migration defects (Georges-Labouesse et al., 1998; Costell et al., 1999; Graus-Porta et al., 2001; Halfter et al., 2002; Moore et al., 2002; Beggs et al., 2003; Niewmierzycka et al., 2005). The Zic mutants analyzed in the present study share many of the cortical phenotypes with these mutants, consistent with a model in which the radial glia disturbances attributable to the BM defects are primary cause of ectopia formation in these mutant cortices.
Marginal zone heterotopias and meningeal glioneuronal heterotopias are commonly found neuropathological abnormalities in human brains with cortical dysplasia (Hirano et al., 1992; Mischel et al., 1995). The cortical lamination defects in the Zic2 and Zic1/3 mutants closely resemble the cobblestone or type II lissencephaly observed in human congenital muscular dystrophy patients. Although it is not known whether human pathogenic mutations in the ZIC genes (Gebbia et al., 1997; Brown et al., 1998; Grinberg et al., 2004) produce any abnormalities in the meninges or cortical lamination, the current analysis in mouse mutants underscores the importance of clinical studies addressing the possible association between ZIC mutations and cortical abnormalities in humans.
Footnotes
This work was supported by RIKEN Brain Science Institute funds and a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan. We thank Kazunori Nakajima, Guendela Meyer, and Sam J. Pleasure for their helpful comments and Research Resource Center RIKEN Brain Science Institute for the technical assistance.
References
- Alcántara S, Ruiz M, D'Arcangelo G, Ezan F, de Lecea L, Curran T, Sotelo C, Soriano E. Regional and cellular patterns of reelin mRNA expression in the forebrain of the developing and adult mouse. J Neurosci. 1998;18:7779–7799. doi: 10.1523/JNEUROSCI.18-19-07779.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcántara S, Pozas E, Ibañez CF, Soriano E. BDNF-modulated spatial organization of Cajal-Retzius and GABAergic neurons in the marginal zone plays a role in the development of cortical organization. Cereb Cortex. 2005;16:487–499. doi: 10.1093/cercor/bhi128. [DOI] [PubMed] [Google Scholar]
- Aruga J. The role of Zic genes in neural development. Mol Cell Neurosci. 2004;26:205–221. doi: 10.1016/j.mcn.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Aruga J, Minowa O, Yaginuma H, Kuno J, Nagai T, Noda T, Mikoshiba K. Mouse Zic1 is involved in cerebellar development. J Neurosci. 1998;18:284–293. doi: 10.1523/JNEUROSCI.18-01-00284.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aruga J, Inoue T, Hoshino J, Mikoshiba K. Zic2 controls cerebellar development in cooperation with Zic1. J Neurosci. 2002a;22:218–225. doi: 10.1523/JNEUROSCI.22-01-00218.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aruga J, Tohmonda T, Homma S, Mikoshiba K. Zic1 promotes the expansion of dorsal neural progenitors in spinal cord by inhibiting neuronal differentiation. Dev Biol. 2002b;244:329–341. doi: 10.1006/dbio.2002.0598. [DOI] [PubMed] [Google Scholar]
- Aruga J, Ogura H, Shutoh F, Ogawa M, Franke B, Nagao S, Mikoshiba K. Locomotor and oculomotor impairment associated with cerebellar dysgenesis in Zic3-deficient (Bent tail) mutant mice. Eur J Neurosci. 2004;20:2159–2167. doi: 10.1111/j.1460-9568.2004.03666.x. [DOI] [PubMed] [Google Scholar]
- Beggs HE, Schahin-Reed D, Zang K, Goebbels S, Nave KA, Gorski J, Jones KR, Sretavan D, Reichardt LF. FAK deficiency in cells contributing to the basal lamina results in cortical abnormalities resembling congenital muscular dystrophies. Neuron. 2003;40:501–514. doi: 10.1016/s0896-6273(03)00666-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielle F, Griveau A, Narboux-Neme N, Vigneau S, Sigrist M, Arber S, Wassef M, Pierani A. Multiple origins of Cajal-Retzius cells at the borders of the developing pallium. Nat Neurosci. 2005;8:1002–1012. doi: 10.1038/nn1511. [DOI] [PubMed] [Google Scholar]
- Borrell V, Marin O. Meninges control tangential migration of hem-derived Cajal-Retzius cells via CXCL12/CXCR4 signaling. Nat Neurosci. 2006;9:1284–1293. doi: 10.1038/nn1764. [DOI] [PubMed] [Google Scholar]
- Brown SA, Warburton D, Brown LY, Yu CY, Roeder ER, Stengel-Rutkowski S, Hennekam RC, Muenke M. Holoprosencephaly due to mutations in ZIC2, a homologue of Drosophila odd-paired. Nat Genet. 1998;20:180–183. doi: 10.1038/2484. [DOI] [PubMed] [Google Scholar]
- Brunstrom JE, Gray-Swain MR, Osborne PA, Pearlman AL. Neuronal heterotopias in the developing cerebral cortex produced by neurotrophin-4. Neuron. 1997;18:505–517. doi: 10.1016/s0896-6273(00)81250-7. [DOI] [PubMed] [Google Scholar]
- Carrel T, Purandare SM, Harrison W, Elder F, Fox T, Casey B, Herman GE. The X-linked mouse mutation Bent tail is associated with a deletion of the Zic3 locus. Hum Mol Genet. 2000;9:1937–1942. doi: 10.1093/hmg/9.13.1937. [DOI] [PubMed] [Google Scholar]
- Caviness VS., Jr Neocortical histogenesis in normal and reeler mice: a developmental study based upon [3H]thymidine autoradiography. Brain Res. 1982;256:293–302. doi: 10.1016/0165-3806(82)90141-9. [DOI] [PubMed] [Google Scholar]
- Costell M, Gustafsson E, Aszódi A, Mörgelin M, Bloch W, Hunziker E, Addicks K, Timpl R, Fässler R. Perlecan maintains the integrity of cartilage and some basement membranes. J Cell Biol. 1999;147:1109–1122. doi: 10.1083/jcb.147.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- de Bergeyck V, Nakajima K, Lambert de Rouvroit C, Naerhuyzen B, Goffinet AM, Miyata T, Ogawa M. A truncated Reelin protein is produced but not secreted in the “Orleans” reeler mutation (Reln[rl-Orl]) Brain Res Mol Brain Res. 1997;50:85–90. doi: 10.1016/s0169-328x(97)00166-6. [DOI] [PubMed] [Google Scholar]
- Franke B, Klootwijk R, Hekking JW, de Boer RT, ten Donkelaar HJ, Mariman EC, van Straaten HW. Analysis of embryonic phenotype of Bent tail, a mouse model for X-linked neural tube defects. Anat Embryol. 2003;207:255–262. doi: 10.1007/s00429-003-0345-2. [DOI] [PubMed] [Google Scholar]
- Frotscher M. Cajal-Retzius cells, Reelin, and formation of layers. Cuur Opin Neurobiol. 1998;8:570–575. doi: 10.1016/s0959-4388(98)80082-2. [DOI] [PubMed] [Google Scholar]
- García-Moreno F, López-Mascaraque L, De Carlos A. Origins and migratory routes of murine Cajal-Retzius cells. J Comp Neurol. 2007;500:419–432. doi: 10.1002/cne.21128. [DOI] [PubMed] [Google Scholar]
- Gebbia M, Ferrero GB, Pilia G, Bassi MT, Aylsworth A, Penman-Splitt M, Bird LM, Bamforth JS, Burn J, Schlessinger D, Nelson DL, Casey B. X-linked situs abnormalities result from mutations in ZIC3. Nat Genet. 1997;17:305–308. doi: 10.1038/ng1197-305. [DOI] [PubMed] [Google Scholar]
- Georges-Labouesse E, Mark M, Messaddeq N, Gansmuller A. Essential role of alpha 6 integrins in cortical and retinal lamination. Curr Biol. 1998;8:983–986. doi: 10.1016/s0960-9822(98)70402-6. [DOI] [PubMed] [Google Scholar]
- Graus-Porta D, Blaess S, Senften M, Littlewood-Evans A, Damsky C, Huang Z, Orban P, Klein R, Schittny JC, Muller U. Beta1-class integrins regulate the development of laminae and folia in the cerebral and cerebellar cortex. Neuron. 2001;31:367–379. doi: 10.1016/s0896-6273(01)00374-9. [DOI] [PubMed] [Google Scholar]
- Grinberg I, Northrup H, Ardinger H, Prasad C, Dobyns WB, Millen KJ. Heterozygous deletion of the linked genes ZIC1 and ZIC4 is involved in Dandy-Walker malformation. Nat Genet. 2004;36:1053–1055. doi: 10.1038/ng1420. [DOI] [PubMed] [Google Scholar]
- Guenette S, Chang Y, Hiesberger T, Richardson JA, Eckman CB, Eckman EA, Hammer RE, Herz J. Essential roles for the FE65 amyloid precursor protein-interacting proteins in brain development. EMBO J. 2006;25:420–431. doi: 10.1038/sj.emboj.7600926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halfter W, Dong S, Yip YP, Willem M, Mayer U. A critical function of the pial basement membrane in cortical histogenesis. J Neurosci. 2002;22:6029–6040. doi: 10.1523/JNEUROSCI.22-14-06029.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann D, Schulze M, Sievers J. Meningeal cells stimulate and direct the migration of cerebellar external granule cells in vitro. J Neurocytol. 1998;27:395–409. doi: 10.1023/a:1006998609999. [DOI] [PubMed] [Google Scholar]
- Hevner RF, Shi L, Justice N, Hsueh Y, Sheng M, Smiga S, Bulfone A, Goffinet AM, Campagnoni AT, Rubenstein JLR. Tbr1 regulates differentiation of the preplate and layer 6. Neuron. 2001;29:353–366. doi: 10.1016/s0896-6273(01)00211-2. [DOI] [PubMed] [Google Scholar]
- Hirano S, Houdou S, Hasegawa M, Kamei A, Takashima S. Clinicopathologic studies on leptomeningeal glioneuronal heterotopia in congenital anomalies. Pediatr Neurol. 1992;8:441–444. doi: 10.1016/0887-8994(92)90006-k. [DOI] [PubMed] [Google Scholar]
- Inoue T, Ota M, Ogawa M, Mikoshiba K, Aruga J. Zic1 and Zic3 regulate medial forebrain development through expansion of neuronal progenitors. J Neurosci. 2007a;27:5461–5473. doi: 10.1523/JNEUROSCI.4046-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Ota M, Mikoshiba K, Aruga J. Zic2 and Zic3 synergistically control neurulation and segmentation of paraxial mesoderm in mouse embryo. Dev Biol. 2007b;306:669–684. doi: 10.1016/j.ydbio.2007.04.003. [DOI] [PubMed] [Google Scholar]
- Klootwijk R, Franke B, van der Zee CE, de Boer RT, Wilms W, Hol FA, Mariman EC. A deletion encompassing Zic3 in bent tail, a mouse model for X-linked neural tube defects. Hum Mol Genet. 2000;9:1615–1622. doi: 10.1093/hmg/9.11.1615. [DOI] [PubMed] [Google Scholar]
- Kume T, Deng KY, Winfrey V, Gould DB, Walter MA, Hogan BL. The forkhead/winged helix gene Mf1 is disrupted in the pleiotropic mouse mutation congenital hydrocephalus. Cell. 1998;93:985–996. doi: 10.1016/s0092-8674(00)81204-0. [DOI] [PubMed] [Google Scholar]
- Lu M, Grove EA, Miller RJ. Abnormal development of the hippocampal dentate gyrus in mice lacking the CXCR4 chemokine receptor. Proc Natl Acad Sci USA. 2002;99:7090–7095. doi: 10.1073/pnas.092013799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin-Padilla M. Cajal-Retzius cells and the development of the neocortex. Trends Neurosci. 1998;21:64–71. doi: 10.1016/s0166-2236(97)01164-8. [DOI] [PubMed] [Google Scholar]
- Mayer U, Nischt R, Poschl E, Mann K, Fukuda K, Gerl M, Yamada Y, Timpl R. A single RGF-like motif of laminin is responsible for high affinity nidogen binding. EMBO J. 1993;12:1879–1885. doi: 10.1002/j.1460-2075.1993.tb05836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer G, Goffinet AM. Prenatal development of reelin immunoreactive neurons in the human neocortex. J Comp Neurol. 1998;397:29–40. [PubMed] [Google Scholar]
- Meyer G, Goffinet AM, Fairen A. What is a Cajal-Retzius cell? A reassessment of a classical cell type based on recent observations in the developing neocortex. Cereb Cortex. 1999;9:765–775. doi: 10.1093/cercor/9.8.765. [DOI] [PubMed] [Google Scholar]
- Meyer G, Perez-Garcia CG, Abraham H, Caput D. Expression of p73 and Reelin in the developing human cortex. J Neurosci. 2002;22:4973–4986. doi: 10.1523/JNEUROSCI.22-12-04973.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer G, Cabrera Socorro A, Perez Garcia CG, Martinez Millan L, Walker N, Caput D. Developmental roles of p73 in Cajal-Retzius cells and cortical patterning. J Neurosci. 2004;24:9878–9887. doi: 10.1523/JNEUROSCI.3060-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mischel PS, Nguyen LP, Vinters HV. Cerebral cortical dysplasia associated with pediatric epilepsy. Review of neuropathologic features and proposal for a grading system. J Neuropathol Exp Neurol. 1995;54:137–153. doi: 10.1097/00005072-199503000-00001. [DOI] [PubMed] [Google Scholar]
- Moore SA, Saito F, Chen J, Michele DE, Henry MD, Messing A, Cohn RD, Ross-Barta SE, Westra S, Williamson RA, Hoshi T, Campbell KP. Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature. 2002;418:422–425. doi: 10.1038/nature00838. [DOI] [PubMed] [Google Scholar]
- Nagai T, Aruga J, Takada S, Gunther T, Sporle R, Schughart K, Mikoshiba K. The expression of the mouse Zic1, Zic2, and Zic3 gene suggests an essential role for Zic genes in body pattern formation. Dev Biol. 1997;182:299–313. doi: 10.1006/dbio.1996.8449. [DOI] [PubMed] [Google Scholar]
- Nagai T, Aruga J, Minowa O, Sugimoto T, Ohno Y, Noda T, Mikoshiba K. Zic2 regulates the kinetics of neurulation. Proc Natl Acad Sci USA. 2000;97:1618–1623. doi: 10.1073/pnas.97.4.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewmierzycka A, Mills J, St-Arnaud R, Dedhar S, Reichardt LF. Integrin-linked kinase deletion from mouse cortex results in cortical lamination defects resembling cobblestone lissencephaly. J Neurosci. 2005;25:7022–7031. doi: 10.1523/JNEUROSCI.1695-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa M, Miyata T, Nakajima K, Yagyu K, Seike M, Ikenaka K, Yamamoto H, Mikoshiba K. The reeler gene-associated antigen on Cajal-Retzius neurons is a crucial molecule for laminar organization of cortical neurons. Neuron. 1995;14:899–912. doi: 10.1016/0896-6273(95)90329-1. [DOI] [PubMed] [Google Scholar]
- Olson EC, Walsh CA. Smooth, rough and upside-down neocortical development. Curr Opin Genet Dev. 2002;12:320–327. doi: 10.1016/s0959-437x(02)00305-2. [DOI] [PubMed] [Google Scholar]
- Paredes MF, Li G, Berger O, Baraban SC, Pleasure SJ. Stromal-derived factor (CXCL12) regulates laminar position of Cajal-Retzius cells in normal and dysplastic brains. J Neurosci. 2006;26:9404–9412. doi: 10.1523/JNEUROSCI.2575-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto-Lord MC, Evrard P, Caviness VS., Jr Obstructed neuronal migration along radial glial fibers in the neocortex of the reeler mouse: a Golgi-EM analysis. Brain Res. 1982;256:379–393. doi: 10.1016/0165-3806(82)90181-x. [DOI] [PubMed] [Google Scholar]
- Pöschl E, Schlötzer-Schrehardt U, Brachvogel B, Saito K, Ninomiya Y, Mayer U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development. 2004;131:1619–1628. doi: 10.1242/dev.01037. [DOI] [PubMed] [Google Scholar]
- Pourebrahim R, Van Dam K, Bauters M, De Wever I, Sciot R, Cassiman JJ, Tejpar S. ZIC1 gene expression is controlled by DNA and histone methylation in mesenchymal proliferations. FEBS Lett. 2007;581:5122–5126. doi: 10.1016/j.febslet.2007.09.061. [DOI] [PubMed] [Google Scholar]
- Purandare SM, Ware SM, Kwan KM, Gebbia M, Bassi MT, Deng JM, Vogel H, Behringer RR, Belmont JW, Casey B. A complex syndrome of left-right axis, central nervous system and axial skeleton defects in Zic3 mutant mice. Development. 2002;129:2293–2302. doi: 10.1242/dev.129.9.2293. [DOI] [PubMed] [Google Scholar]
- Rakic P. Elusive radial glial cells: historical and evolutionary perspective. Glia. 2003;43:19–32. doi: 10.1002/glia.10244. [DOI] [PubMed] [Google Scholar]
- Rice DS, Curran T. Role of the reelin signaling pathway in central nervous system development. Annu Rev Neurosci. 2001;24:1005–1039. doi: 10.1146/annurev.neuro.24.1.1005. [DOI] [PubMed] [Google Scholar]
- Ringstedt T, Linnarsson S, Wagner J, Lendahl U, Kokaia Z, Arenas E, Ernfors P, Ibáñez CF. BDNF regulates reelin expression and Cajal-Retzius cell development in the cerebral cortex. Neuron. 1998;21:305–315. doi: 10.1016/s0896-6273(00)80540-1. [DOI] [PubMed] [Google Scholar]
- Schiffmann SN, Bernier B, Goffinet AM. Reelin mRNA expression during mouse brain development. Eur J Neurosci. 1997;9:1055–1071. doi: 10.1111/j.1460-9568.1997.tb01456.x. [DOI] [PubMed] [Google Scholar]
- Schuurmans C, Armant O, Nieto M, Stenman JM, Britz O, Klenin N, Brown C, Langevin LM, Seibt J, Tang H. Sequential phases of cortical specification involve Neurogenin-dependent and -independent pathways. EMBO J. 2004;23:2892–2902. doi: 10.1038/sj.emboj.7600278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearer MC, Fawcett JW. The astrocyte/meningeal cell interfacea–a barrier to successful nerve regeneration? Cell Tissue Res. 2001;305:267–273. doi: 10.1007/s004410100384. [DOI] [PubMed] [Google Scholar]
- Shinozaki K, Miyagi T, Yoshida M, Miyata T, Ogawa M, Aizawa S, Suda Y. Absence of Cajal-Retzius cells and subplate neurons associated with defects of tangential cell migration from ganglionic eminence in Emx1/2 double mutant cerebral cortex. Development. 2002;129:3479–3492. doi: 10.1242/dev.129.14.3479. [DOI] [PubMed] [Google Scholar]
- Sievers J, Pehlemann FW, Gude S, Berry M. Meningeal cells organize the superficial glia limitans of the cerebellum and produce components of both the interstitial matrix and the basement membrane. J Neurocytol. 1994;23:135–149. doi: 10.1007/BF01183867. [DOI] [PubMed] [Google Scholar]
- Stumm RK, Zhou C, Ara T, Lazarini F, Dubois-Dalcq M, Nagasawa T, Hollt V, Schulz S. CXCR4 regulates interneuron migration in the developing neocortex. J Neurosci. 2003;23:5123–5130. doi: 10.1523/JNEUROSCI.23-12-05123.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Super H, Martinez A, Soriano E. Degeneration of Cajal-Retzius cells in the developing cerebral cortex of the mouse after ablation of meningeal cells by 6-hydroxydopamine. Brain Res Dev Brain Res. 1997;98:15–20. doi: 10.1016/s0165-3806(96)00155-1. [DOI] [PubMed] [Google Scholar]
- Super H, Soriano E, Uylings HB. The function of the preplate in development and evolution of the neocortex and hippocampus. Brain Res Brain Res Rev. 1998;27:40–64. doi: 10.1016/s0165-0173(98)00005-8. [DOI] [PubMed] [Google Scholar]
- Takiguchi-Hayashi K, Sekiguchi M, Ashigaki S, Takamatsu M, Hasegawa H, Suzuki-Migishima R, Yokoyama M, Nakanishi S, Tanabe Y. Generation of reelin-positive marginal zone cells from the caudomedial wall of telencephalic vesicles. J Neurosci. 2004;24:2286–2295. doi: 10.1523/JNEUROSCI.4671-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissir F, Goffinet AM. Reelin and brain development. Nat Rev Neurosci. 2003;4:496–505. doi: 10.1038/nrn1113. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Assimacopoulos S, Jones KR, Grove EA. Massive loss of Cajal-Retzius cells does not disrupt neocortical layer order. Development. 2006;133:537–545. doi: 10.1242/dev.02209. [DOI] [PubMed] [Google Scholar]
- Zarbalis K, Siegenthaler JA, Choe Y, May SR, Peterson AS, Pleasure SJ. Cortical dysplasia and skull defects in mice with a Foxc1 allele reveal the role of meningeal differentiation in regulating cortical development. Proc Natl Acad Sci USA. 2007;104:14002–14007. doi: 10.1073/pnas.0702618104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecevic N, Rakic P. Development of layer I neurons in the primate cerebral cortex. J Neurosci. 2001;21:5607–5619. doi: 10.1523/JNEUROSCI.21-15-05607.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]