

Abstract
NMDA-only synapses, called silent synapses, are thought to be the initial step in synapse formation in several systems. However, the underlying mechanism and the role in circuit construction are still a matter of dispute. Using combined morphological and electrophysiological approaches, we searched for silent synapses at the level of the nucleus tractus solitarii (NTS), a brainstem structure that is a gateway for many visceral sensory afferent fibers. Silent synapses were detected at birth by using electrophysiological recordings and minimal stimulation protocols. However, anatomical experiments indicated that nearly all, if not all, NTS synapses had AMPA receptors. Based on EPSC fluctuation measurements and differential blockade by low-affinity competitive and noncompetitive glutamate antagonists, we then demonstrated that NTS silent synapses were better explained by glutamate spillover from neighboring fibers and/or slow dynamic of fusion pore opening. Glutamate spillover at immature NTS synapses may favor crosstalk between active synapses during development when glutamate transporters are weakly expressed and contribute to synaptic processing as well as autonomic circuit formation.
Keywords: silent synapses, brainstem, development, AMPA, spillover, glutamate
Introduction
Glutamatergic synapses are the primary source of excitatory synaptic transmission in the CNS. Their formation and refinement are critical in establishing neural networks as well as in the regulation of circuits by experience. Neural circuits are refined during development by activity-driven processes (Katz and Shatz, 1996; Aamodt and Constantine-Paton, 1999; Crair, 1999; Contestabile, 2000). Long-term changes in synaptic efficacy such as long-term depression and long-term potentiation (LTP) are thought to be involved in activity-dependent circuit development (Constantine-Paton et al., 1990; Constantine-Paton and Cline, 1998). Both processes are explained by recruitment or removal of AMPA receptors (AMPARs) (Malinow and Malenka, 2002). Stemming from this Hebbian framework, a leading hypothesis is that, during development, synapses only possessed NMDA receptors (NMDARs), rendering them silent at resting potential. LTP-like conversion of silent synapses to functional ones by AMPAR insertion allows appropriate connections to be established (Wu et al., 1996; Isaac et al., 1997; Rumpel et al., 1998; Zhu et al., 2000). Although silent synapses have been widely described, the underlying mechanism is still a matter of dispute (Poncer, 2003; Voronin and Cherubini, 2004). Depending on brain structures, presynaptic or postsynaptic mechanisms have been favored (see Discussion). The relative contribution of presynaptic and postsynaptic mechanisms may actually vary depending on the synapse “history.” In hippocampal neurons, repeated synapse activation leads to AMPAR silencing subsequently reversed by Hebbian induction just as silent synapses do (Montgomery and Madison, 2002; Xiao et al., 2004; Abrahamsson et al., 2005). In addition, the actual role of NMDAR-only synapses in circuit construction has also been questioned (Hohnke et al., 2000; Groc et al., 2003).
We recently described the development of excitatory synapses in the nucleus tractus solitarii (NTS) (Balland et al., 2006). This brainstem nucleus is a gateway for many primary afferents from visceral sensory receptors. This visceral information runs into different cranial nerves and triggers autonomic reflexes. We reported that, before birth, NTS synaptogenesis was an activity-independent process (Balland et al., 2006). However, visceral reflexes undergo maturational changes and NTS circuitry still develops after birth (Kasparov and Paton, 1997; Vincent and Tell, 1999; Lachamp et al., 2002; Moss, 2002; Rinaman, 2003; Vincent et al., 2004). Thus, environmental events during postnatal life can influence autonomic circuit formation (Card et al., 2005). We therefore extended our previous study by focusing on NTS synaptic development after birth, a period prone to activity-dependent alteration in autonomic circuitry (Gozal and Gozal, 2001; Vincent et al., 2004; Card et al., 2005). We found that silent synapses could be detected at birth by using electrophysiological recordings. However, anatomical experiments indicated that such silent synapses did not lack AMPARs. Based on EPSC fluctuation measurements and using different NMDAR antagonists, we demonstrated that reduced glutamate concentration arising from spillover from neighboring fiber terminals or slow dynamics in fusion pore are likely to be responsible for silent inputs. The relevance of these data to the construction of the autonomic circuits shall be discussed.
Materials and Methods
Animals.
Newborn Wistar rats of either sex were anesthetized first with halothane before slice preparation. All procedures were in agreement with the European Communities Council directive (86/609/EEC). The day of birth was considered as postnatal day 0 (P0).
Slice preparation.
Preparation of medullary slice developing Wistar rats was made as described previously (Lachamp et al., 2003; Balland et al., 2006). Slices were obtained rat pups at different postnatal ages (from P0 to P2 referred as P0 group and from P6 to P14 referred as P10 group). Animals were decapitated, and the brainstem was quickly removed, cooled to 4°C, and cut on a vibratome (VT1000 S; Leica Microsystems, Strasbourg, France), into 300-μm-thick medullary slices, in oxygenated (95% O2–5% CO2, pH 7.4) saline containing the following (in mm): 130 NaCl, 3 KCl, 0.5 CaCl2, 4 MgCl2, 26 NaHCO3, 1.25 KH2PO4, 10 glucose, 0.5 ascorbate, 2 pyruvate, and 3 myoinositol. Slices were then allowed to recover for 1 h in a similar saline containing 2 mm CaCl2 and 2 mm MgCl2 at room temperature.
Electrophysiology.
For recordings, slices were perfused in a chamber at 1–3 ml min-1 with a similar physiological solution containing 2 mm CaCl2, 2 mm MgCl2, and a mixture of GABAA receptor blockers (20 μm biccuculine and 50 μm picrotoxine) at 32–34°C. NMDA EPSCs were isolated in the presence of 10 μm 6,7-dinitroquinoxaline-2,3-dione acid (DNQX). Because the NTS is functionally divided into different subnuclei in the adult rat, we restricted our study to the medial NTS region on subpostremal slices. The dorsal motor nucleus of the vagus nerve and the tractus solitarius (TS) (a fiber bundle formed by visceral afferent fibers within the brainstem) (Andresen and Kunze, 1994) were taken as anatomical land marks to reproducibly target the recording region. Therefore, only one slice was selected per animal. Patch electrodes (2.5–5MΩ) contained the following (in mm): 120 caesium methane sulfonate, 10 NaCl, 1 MgCl2, 1 CaCl2, 10 EGTA, 2 ATP, 0.3 GTP, 10 glucose, 10 HEPES, 10 tetraethylammonium, and 4 4-AP, pH 7.4. The junction potential was ∼8 mV and was not corrected for. Stimulation of the TS (1–10 V; 50–100 μs duration) were applied through a tungsten bipolar electrode at low frequency (0.05–0.1 Hz). Excitatory synaptic responses were evoked by stimulating the TS in a saline containing a mixture of picrotoxine (100 μm) and biccuculine (20 μm). EPSCs were deemed monosynaptic by virtue of their short latencies and reduced jittering from stimulus onset (Doyle and Andresen, 2001; Lachamp et al., 2003; Balland et al., 2006). d-α-Amino-adipate (d-AA), 5,7-dichlorokynurenic acid (DCK), and DNQX were purchased from Tocris Cookson (Bristol, UK). Other chemicals were obtained from Sigma (St. Quentin Fallavier, France).
Minimal stimulation experiments.
In these series of experiments, two protocols were used. In the first protocol, stimulations were elicited as follow: initial stimulus intensity was kept low so that no synaptic responses were elicited. The stimulus intensity was then increased slowly to elicit a stable minimal response (a mixture of responses and failures, 40–50 consecutive stimulations, 0.05 Hz). As stimulus intensity was increased, responses appeared at a threshold stimulus and did not increase in amplitude over a relatively wide range of stimulus intensity, suggesting activation of only one TS fiber (Wu et al., 1996). Number of failure and successes were estimated at two different potentials (−70 and +40 mV) while keeping stimulation strength and rate constant.
In the second protocol, we used a direct demonstration of the existence of silent synapses by reducing, at hyperpolarized membrane potentials, the stimulation strength until no AMPA PSCs were observed. Inputs were considered to be silent if TS-evoked synaptic responses were never observed at any time at this holding potential. Routinely, 50 consecutive (range of 30–60) TS stimulations were delivered at low frequency (0.05 Hz) at negative holding potential of −70 mV. Then the neuron was held at +40 mV without changing stimulation parameters to check for the presence of NMDA-only responses. All failure traces obtained at −70 mV were averaged to ensure that no AMPA responses were hidden in the background noise.
Data acquisition and analysis.
Whole-cell patch-clamp recordings of NTS neurons were made with an Axopatch 200B (Molecular Devices, Sunnyvale, CA), filtered at 2 kHz, and digitized at 20 kHz. Series resistance was continuously monitored throughout the experiment by delivering a voltage step command at the end of each trace recorded. Neurons in which this parameter was >25 MΩ or not stable were discarded. The decay time constant (t) of NMDAR-mediated synaptic currents was obtained by fitting the decay phase (t = 0 at the peak) with double-exponential equations: F(t) = Afast exp(−t/tfast) + Aslow exp(−t/tslow) The weighted t (tw) was calculated according to: tw = (tfastAfast + tslowAslow)/(Afast + Aslow), where Afast and Aslow are the relative amplitudes of the two exponential components. Detection of failure was achieved by two methods: by visual discrimination and by doubling the number of positive or negative amplitudes at negative or positive membrane potentials, respectively (Liao et al., 1995). As a control, the visually selected failures were averaged, and the selection was resumed if the averaged traces contained stimulus-locked deflections different from that for the interstimulus period. AMPA and NMDA EPSC amplitudes were measured by averaging a 2–4 ms period coinciding with the peak of the average time course, subtracted from a baseline of similar duration immediately preceding the stimulation artifact. Potency was defined as the mean amplitude excluding failures.
For pharmacological experiments, EPSC amplitudes were plotted against the stimulus number, and linear fit were applied to the data. Only experiments in which this linear regression did not significantly deviate from zero slope were selected for additional analysis. The coefficient of variation (CV) was calculated for at least 40 consecutive trials as follows. The variance of EPSC peak were obtained. The variance of the background noise was measured from interleaved traces in which no stimuli were delivered and subtracted from the EPSC variance. Then the square root of this corrected variance was divided by the EPSC amplitude mean. Estimation of the percentage of silent synapses was made either by dividing the logarithmic values of failures obtained at the two potentials because failure rate equals (1 − p)n (p is the release probability, and n is the number of releasing sites and equals the number of nonsilent sites at rest and the number of nonsilent plus silent sites at +40 mV) or from CV differences between AMPA and NMDA EPSCs according to Xiao et al. (2001). All drugs were applied via the general perfusion system at a rate of 3–6 ml/min, allowing for a rapid wash-in of the slice. Drug effects generally stabilized in a few minutes.
Immunocytochemistry.
We used antigen retrieval by microwave irradiation to increase detection of synaptically located antigens (Fritschy et al., 1998) (see also Lachamp et al., 2003; Balland et al., 2006). Developing rat pups (P0–P30) were killed by decapitation under halothane anesthesia. Brainstems were removed and immediately frozen in cold isopentane (− 50°C). Coronal medullary sections (14 μm thick) were obtained on a cryostat, thaw mounted on gelatinized glass slides, and fixed by immersion in phosphate buffer (0.1 m), pH 7.4, containing 0.5% paraformaldehyde under microwave irradiation (45 s, 800 W). AMPARs were detected using a mouse antibody raised against the extracellular part of the AMPAR subunit glutamate receptor subunit 2 (GluR2) (MAB 397; 3 μg/ml; Millipore Bioscience Research Reagents, Temecula CA). A rabbit antibody recognizing the intracellular tail of the GluR2 subunit (AB 1768; Millipore Bioscience Research Reagents) was also used in some control experiments. The GluR2 subunit is the main subunit forming NTS AMPARs throughout development (Balland et al., 2006). Synaptic contacts were visualized using a mouse antibody that recognizes the CAZ protein Bassoon (VAM PS003; 3 μg/ml; Nventa Biopharmaceuticals, San Diego, CA). Glutamatergic axon terminals were identified using a mixture of rabbit antibodies raised against vesicular glutamate transporters (anti-VGLUT1, ant-iVGLUT2; 1:1000; Synaptic Systems, Goettingen, Germany) (Lachamp et al., 2005). Because two primary antibodies from the same species were used, triple fluorescent detection was performed by prelabeling the bassoon antibody with dye-coupled Fab fragments to prevent cross-reactions. Prelabeling was obtained by incubating the bassoon antibody with cyanine 5 (Cy5)-conjugated anti-mouse Fab fragments (Jackson ImmunoResearch, West Grove, PA) using a 6:1 molar ratio of labeled Fab to primary antibody. The reaction was stopped after 15 min by adding an excess of mouse Ig to the mixture. Triple immunofluorescent labeling was performed as follows. Slide-mounted cryostat sections were first blocked with normal goat serum and then incubated with a mixture of mouse anti-GluR2 and rabbit anti-VGLUTs (overnight) followed by a mixture of Alexa Fluor 488-conjugated goat anti-mouse and Alexa Fluor 546-conjugated goat anti-rabbit (1:200 each, 1 h; Invitrogen, Carlsbad, CA). Bassoon was detected by overnight incubation with the prelabeled antibody after a second blocking step with mouse Ig (Jackson ImmunoResearch). To check for a possible cross-reaction between Fab fragments and the GluR2 antibody, control experiments were performed by incubating some sections as described above except that the prelabeled bassoon antibody was replaced by Cy5-conjugated anti-mouse Fab fragments neutralized with an excess of mouse Ig. Confocal image acquisition was performed on a Leica TCS SP2 laser scanning microscope (Leica Microsystems, Heidelberg, Germany) using the 488 nm band of an argon laser for excitation of Alexa Fluor 488 (spectral detection, 500–535 nm) and the 543 and 633 nm bands of an helium–neon laser for excitation of Alexa Fluor 546 and Cy5, respectively (spectral detection, 565–620 and 650–790 nm). High-magnification images were acquired using a 63× oil immersion objective (numerical aperture 1.32). Pinhole size was set to “Airy one” to achieve the best possible resolution (theoretical lateral and axial limits, 165 and 330 nm, respectively) without reducing luminosity. Pixel size was set to 58 nm (i.e., approximately one-third of the theoretical lateral resolution) by adjusting the scanning zoom factor. For each channel, photomultiplier gains and offsets were adjusted to use full image dynamic range leaving minimal numbers of either “blind” or saturated pixels. Image deconvolution was performed on 2-μm-thick z-stacks (162 nm spacing) with Huygens Pro (Scientific Volume Imaging, Hilversum, The Netherlands), using the classic maximum likelihood estimation mode and point spread functions reconstructed from imaged 200 nm fluorescent latex microspheres (Invitrogen). Quantitative analysis was performed on deconvoluted two-dimensional images (one image from each stack) using the public domain NIH Image software and custom-made macros. Image editing was performed using Adobe Photoshop (Adobe Systems, Paris, France).
Statistical analysis.
Significance of the difference between means of two samples was computed with the nonparametric Wilcoxon's matched pairs test or the unpaired Mann–Whitney test. When at least three samples were compared, the variance of the whole population was first analyzed with the nonparametric Kruskhal–Wallis test. If the null hypothesis was rejected, the post hoc Dunn's test was then used to compare groups. Correlation was analyzed with the Spearman's rank-order test. Differences in distribution were assessed by the Kolgomorov–Smirnov test. Statistical data are given as mean ± SEM. The differences were considered significant at p < 0.05. Statistical tests were computed by using Kyplot or Instat (GraphPad Software, San Diego, CA) softwares.
Mathematical modeling.
See Appendix.
Results
Synaptic responses to minimal stimulations
We first assayed a protocol in which minimal stimulation was set to record small EPSCs that are attributable to the stochastic activation of a single or a small number of release sites and to distinguish them from synaptic failures. This protocol has been used previously to estimate the fraction of pure NMDAR synapses (Isaac et al., 1997; Hohnke et al., 2000; Bottjer, 2005; Cabezas and Buno, 2006). At P0, minimal stimulations evoked a mixture of successes and failures in NTS neurons (Fig. 1A). At a holding potential of −70 mV, the mean amplitude and the mean potency of synaptic responses were 9 ± 5 and 21 ± 7 pA, respectively (n = 10). For comparison, miniature EPSC amplitudes were 25.4 ± 2 pA (Balland et al., 2006), thus confirming that the minimal synaptic responses were attributable to transmission from only one or a few release sites. When neurons were held at +40 mV, failure rate was consistently reduced compared with the one obtained at −70 mV (n = 10) (Fig. 1A1–B). Subsequent addition of APV to the saline restored failure rate to control values (n = 6) (Fig. 1A1–B), indicating that additional responses recorded at depolarized potentials were solely mediated by NMDARs. Likewise, stepping back to control holding potential (−70 mV) without previous addition of APV restored failure rate to control value (data not shown; n = 5). The incidence of silent synapses can be estimated from the difference in failure rate between hyperpolarized and depolarized membrane potentials, assuming that the average release probability is similar at functional and silent synapses (Liao et al., 1995; Wu et al., 1996; Isaac et al., 1997; Rumpel et al., 1998). At P0, mean failure rate at −70 mV was 0.65 ± 0.07 (range, 0.35–0.97; n = 10). At +40 mV, it decreased to a mean value of 0.5 ± 0.09 (range, 0.07–0.86; n = 10). For six neurons, addition of APV restored mean failures rate to control values (−70 mV, 0.51 ± 0.03; +40 mV, 0.34 ± 0.04; APV at +40 mV, 0.56 ± 0.04) (Fig. 2B1–C). From these figures, the percentage of silent synapses at P0 was calculated to be ∼44 ± 8.5% (range, 17–84%).
Figure 1.

Silent inputs in the developing NTS. A1, Individual response amplitudes recorded at −70 and +40 mV holding potentials as a function of the stimulus number. Dashed line indicates the mean noise level. At −70 mV, responses are a mixture of successes and failures of synaptic transmission. Depolarization reduced the failure rate. Addition of APV, an NMDAR antagonist, restored failure rate to control values. A2, Superimposed individual traces from the experiment shown in A1 at −70 mV and +40 mV holding potentials. B, Summary graph of minimal stimulation experiments at P0 showing the different failure rate at hyperpolarized (n = 10) and depolarized (n = 10) membrane potentials and in the presence of APV (n = 6). Gray traces are mean values ± SEM. C, Example of a silent synapse at P0 showing exclusively NMDAR-mediated transmission. Right column, At +40 mV, TS stimulation did not induce synaptic response (bottom trace is the average of 40 traces, indicating that synaptic responses were undetectable because of a low signal-to-noise ratio). Left column, At −70 mV, TS stimulation revealed pure NMDA responses. Failures are indicated by asterisks, bottom trace is the average of 40 traces. D, Comparison of EPSC variance mediated by AMPARs and NMDARs. EPSC amplitudes are plotted against stimulus number, with holding potential and application of DNQX indicated schematically. Access resistance magnitude (Rs) is plotted against stimulus number. Top black traces represent average traces in the different conditions, and the gray traces are SDs. E, F, Plots of CVAMPA against CVNMDA at P0 (E) or P10 (F). The dotted lines are the identity lines. In both graphs, gray circles represent values for group 1 neurons (CVAMPA > CVNMDA) and black circles for group 2 neurons (CVAMPA ≤ CVNMDA).
Figure 2.
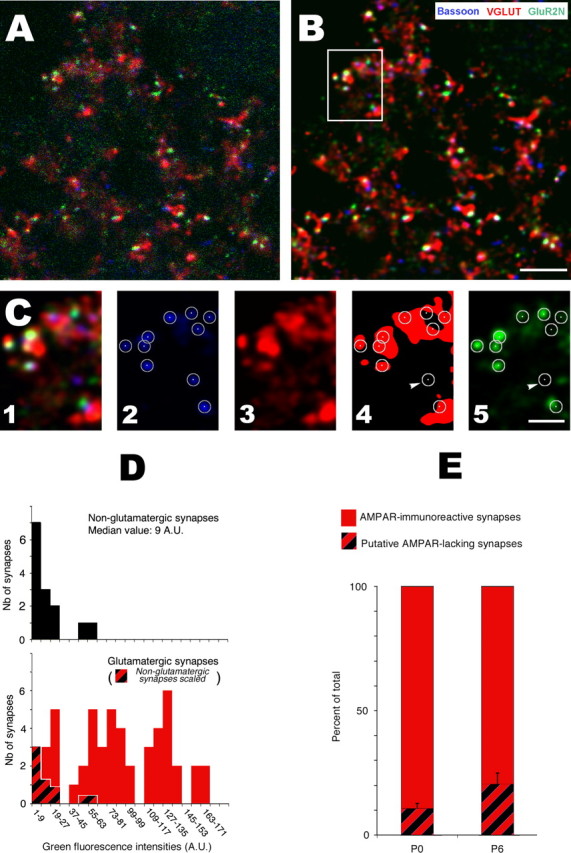
Quantifying AMPAR expression in NTS glutamatergic synapses. A, Single high-resolution confocal section from a P0 rat NTS showing simultaneous immunodetection of glutamatergic terminals (VGLUTs, red), presynaptic active zones (Bassoon, blue), and AMPARs (GluR2, green). B, Same optical section after deconvolution. C, High-magnification images of the boxed area in B illustrating the different steps used for quantitative analysis. C1, Composite image (3 channels). C2, Delineation of synapses by circles using a fixed radius (180 nm) and centers of bassoon puncta on the blue channel. C3, C4, Delineation of glutamatergic terminal by threshold segmentation of the red (VGLUTs) channel. Synapses (bassoon puncta from C2) were identified as glutamatergic or nonglutamatergic depending on their distance between their centers and the nearest VGLUT-immunoreactive terminal. Synapses were considered nonglutamatergic (one indicated by arrowhead) when centers were located more than one pixel (58 nm) away from VGLUT immunoreactivity. C5, Measurements of fluorescence levels on the GluR2 channel (green Alexa Fluor-488 emission) within synaptic circles. D, Distribution histograms of fluorescence intensities (green Alexa Fluor-488 emission) in glutamatergic (red, bottom) and nonglutamatergic (black, top) synapses. The proportion of putative AMPAR-lacking synapses (immunonegative synapses; red and black strips in the bottom) was estimated by scaling the distribution histogram of nonglutamatergic synapses against that of glutamatergic synapses using the first class as reference. A.U., Arbitrary units. E, Proportion of AMPA-immunoreactive synapses and putative AMPAR-lacking synapses in the NTS of P0 (n = 5) and P6 (n = 4) rats. Scale bars: B, 5 μm; C, 2 μm.
We next used a direct demonstration of the existence of silent synapses by reducing, at hyperpolarized membrane potentials, the stimulation strength until no AMPA PSCs were observed. Similar to previously studies (Rumpel et al., 1998; Choi et al., 2000; Gasparini et al., 2000), inputs were considered to be silent when 30–60 consecutive synaptic failures were obtained at a holding potential of −70 mV (Fig. 1C). At P0, changing the holding potential to +40 mV while keeping stimulation strength and rate constant revealed in some neurons (3 of 11) outward responses mediated by NMDARs (Fig. 1C). At P10, minimal stimulation of visceral fibers yielded to failure rates close to zero (5 of 15 neurons presented failures at a weak frequency <10%), precluding comparison of failure rates at different holding potentials. Mean response amplitude was 45 ± 4 pA (range, 28–80 pA; n = 15). As reported previously, miniature mean amplitude was 21 ± 2 pA (Balland et al., 2006). This result suggests that minimal stimulation activated more release sites or with a higher release probability than at birth. Accordingly, decreasing stimulation strength until no AMPA EPSC were observed never revealed NMDA outward responses at depolarized potential (n = 10). At first sight, this may suggest that silent synapses are present at birth and then rapidly disappear as the NTS develops. However, absence of pure NMDA responses at P10 may be attributable to the inability to activate a single release site rather than an actual absence of silent inputs. This may be particularly true at P10 because mature vagal afferents are characterized by large terminals with multiple release sites and high release probability (Bailey et al., 2006), rendering unlikely the observation of failures even with minimal protocol stimulation.
To circumvent this potential confounding problem, we used comparison of the CV between AMPA and NMDA EPSCs to probe for potential silent inputs (Kullmann, 1994; Selig et al., 1995; Xiao et al., 2001). At birth, TS stimulation evoked AMPAR-mediated EPSCs (mean value, 141 ± 21 pA; n = 13) with relatively large trial-to-trial amplitude variations (CVAMPA, 0.22 ± 0.04; n = 13) (Fig. 1D1–E). Addition of DNQX to the saline completely suppressed synaptic responses at negative holding potentials. Holding neurons at +40 mV revealed a slow APV-sensitive outward component (mean value, 71 ± 16 pA; n = 13) with a significantly smaller CV (0.14 ± 0.03; n = 13; p < 0.001). Plotting CVAMPA against CVNMDA revealed a CV decrease in most neurons (9 of 13) (Fig. 1E). According to this, neurons have been separated into two groups according to the CV change direction when comparing CVNMDA and CVAMPA (Fig. 1E,F). Group 1 corresponds to neurons for which CVNMDA was smaller that CVAMPA and group 2 to the remainders. We used the same approach to compare trial-to-trial amplitude fluctuation between AMPA and NMDA EPSCs in P10 neurons. AMPA and NMDA EPSC amplitudes amounted similar values than at birth (170 ± 30 and 96 ± 13 pA, respectively; n = 16). The CVAMPA was 0.27 ± 0.04 (n = 16). When neurons were subsequently held at +40 mV after addition of DNQX in the saline, CVNMDA was significantly smaller than CVAMPA (0.21 ± 0.03; n = 16) (Fig. 1F). Based on CV differences, 8 neurons of 16 fell into the group 1 (Fig. 1F). Thus, at both ages studied, approximately half of the neurons exhibited a larger quantal content for NMDA-mediated EPSC than for AMPA synaptic responses. Estimation of the percentage of putative silent input from differences in CV (Xiao et al., 2001) yielded to similar at P0 and P10 (45 ± 9% at P0; 32 ± 9% at P10; p = 0.2).
Expression of AMPARs in glutamatergic synapses
Having demonstrated that developing NTS neurons exhibited pure NMDA responses, we next investigated whether these responses were attributable to actual AMPAR-lacking synapses. Analysis was performed at the beginning of the first and second postnatal week (P0 and P6) by means of antigen retrieval, immunofluorescence, and high-resolution confocal imaging. Synapses were identified using the cytomatrix protein bassoon as a marker of the presynaptic active zone. Glutamatergic terminals were visualized by VGLUT immunoreactivity, and AMPARs were detected by immunolabeling of the GluR2 subunit. A typical experiment is depicted in Figure 2. Distinction between glutamatergic and nonglutamatergic synapses was performed by examining the relationship of bassoon immunoreactive puncta with VGLUT immunoreactivity (Fig. 2C4). Bassoon puncta with centers located within or close to VGLUT-immunoreactive patches were considered as representing glutamatergic synapses, whereas bassoon puncta with centers located more than one pixel (58 nm) away from VGLUT immunoreactivity were considered as nonglutamatergic (Fig. 2C, arrow). AMPAR expression at synapses (glutamatergic and nonglutamatergic) was evaluated by measuring Alexa Fluor-488 mean fluorescence intensities within circles (180 nm radii) centered on each bassoon puncta (Fig. 2C5). Whatever the experiment and the developmental stage, fluorescence levels measured at glutamatergic synapses (Fig. 2D, red columns) had a widespread distribution and partially overlapped fluorescence values measured at nonglutamatergic synapses (black columns). We next tried to obtain a conservative estimate of the population of glutamatergic synapses actually expressing AMPARs at each developmental stage. We made the assumption that nonglutamatergic synapses express no AMPARs and may thus serve as a reference for immunonegativity. Fluorescence found at these synapses was considered as being noise attributable to immunodetection and confocal imaging artifacts (such as variations in the levels of nonspecific labeling). We reasoned that, if AMPAR-lacking synapses existed among glutamatergic synapses, fluorescence levels measured at these AMPAR-lacking synapses should have a distribution similar to that found at nonglutamatergic (i.e., immunonegative) synapses. Thus, by scaling the distribution profile obtained at nonglutamatergic synapses against that obtained at glutamatergic synapses, it was possible to get an upper limit value for the proportion of glutamatergic synapses that should be considered as immunonegative in each experiment (Fig. 2D). Using this method, we obtained a high proportion of immunopositive glutamatergic synapses at both P0 (90 ± 4%; n = 5; range, 84–93%) and P6 (80 ± 10%; n = 4; range, 72–91%) (Fig. 2E).
We next performed control experiments to check for a possible contribution of subsynaptic accumulations of GluR2 subunits to glutamatergic synapse immunopositivity. These control experiments were based on the effects of microwave irradiation. Retrieval of synaptic antigens by microwaves is thought to be attributable to synaptic cleft widening and enhanced antibody access to intra-cleft epitopes (Fritschy et al., 1998). Thus, if immunopositivity was attributable to subsynaptic stocks of GluR2 subunits rather than to surface-inserted receptors, it should occur (1) without antigen retrieval and (2) whatever the localization of the epitope recognized by the GluR2 antibody. We first confirmed that no synaptic labeling occurred if antigen retrieval was omitted (data not shown). Then, we compared the labeling obtained after antigen retrieval using two different antibodies: the MAB 397 antibody raised against the N-terminal extracellular part of GluR2 (GluR2-N) and the AB 1768 rabbit antibody raised against a portion of the C-terminal intracellular tail of the receptor (GluR2-C). As shown in Figure 3, GluR2-N immunofluorescence was mainly associated with bassoon-immunoreactive spots, whereas GluR2-C immunolabeling had a more widespread localization. In addition, contrary to GluR2-N (Fig. 3A′), GluR2-C did not result in glutamatergic synapses immunopositivity, because it yielded similar fluorescence intensity distributions at glutamatergic and nonglutamatergic synapses (Fig. 3B′).
Figure 3.
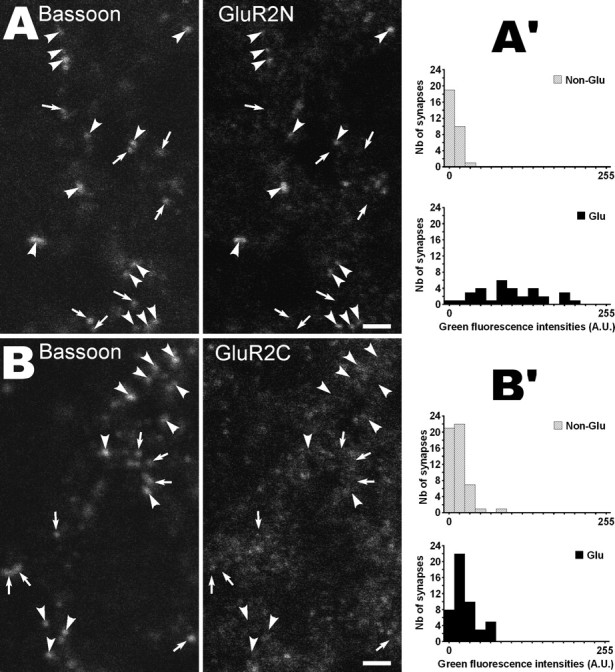
Effects of epitope location on AMPA receptor detection after antigen retrieval by microwaves (P0 rat NTS). A, B, Bassoon and GluR2 channels from two triple-labeled single confocal sections (VGLUT channels not shown). GluR2 detection was performed with an antibody recognizing the extracellular N-terminal part of the protein (GluR2N) in A and with an antibody directed against the intracellular C-terminal tail (GluR2C) in B. Arrowheads and arrows indicate glutamatergic and nonglutamatergic synapses, respectively (i.e., bassoon spots with or without VGLUT immunolabeling). Note that GluR2 immunofluorescence is mainly found at glutamatergic synapses in A but not in B. A′, B′, Distributions of fluorescence intensities produced by the GluR2N antibody (A′) and the GluR2C antibody (B′) in glutamatergic and nonglutamatergic synapses. Quantification was performed as described for Figure 2A. Significant difference between the two distributions was found in A′ (p < 0.02) but not in B′ (p > 0.99). Scale bars, 2 μm. A.U., Arbitrary units.
These finding indicate that the vast majority of glutamatergic synapses in the P0 and P6 NTS (90 and 80%, respectively) have surface-inserted AMPA receptors. The remaining apparently immunonegative contacts could be either synapses without AMPARs or synapses expressing AMPARs below detection levels. In the former case, the fraction of synapses devoid of AMPARs would amount to maximum values of 10% at P0 and 20% at P10. Actual values are certainly smaller than that and far from values calculated from electrophysiological experiments.
A competitive antagonist and a noncompetitive antagonist of NMDA differentially alter NMDA EPSC variability
Because anatomical data indicate that synapses lacking AMPARs are very rare in the neonate NTS, we searched for alternative mechanisms to explain our observations. Some synapses may appear silent because of a low probability of release (Pr) especially when recorded at low temperature (Gasparini et al., 2000). To guard against this possibility, all recordings were obtained at 32–34°C. Paired-pulse stimulation was consistently used to check for low Pr synapses in neonatal NTS neurons. In NTS neurons, this led to a consistent reduction of the EPSC amplitude to the second pulse (data not shown) (Balland et al., 2006), suggesting that the Pr was initially high (paired pulse ratio: 0.6 ± 0.07 at P0, n = 13; 0.5 ± 0.06 at P10, n = 11). Another concern could be that repeated stimulation converts conducting synapses to silent ones (Xiao et al., 2004; Abrahamsson et al., 2005). Amplitude and success rate remained stable for at least 30 stimulations at the highest stimulation frequency used in this experiment (0.1 Hz; −70 mV; n = 10) (Fig. 1A).
An alternative mechanism for silent synapses could be that some synapses encounter low glutamate concentration because glutamate spills from another distant synapse (Kullmann et al., 1996) or as a result of the fusion mode (Choi et al., 2000; Renger et al., 2001). If there are synapses activated by glutamate spillover in addition to synapses that signal via both ionotropic glutamate receptors, one would expect to record pure NMDA responses or reduction in failure rate at depolarizing potential using minimal stimulation protocol. Likewise, reduction of the trial-to-trial variation of the NMDAR EPSCs (expressed as the CV) when compared with that of the AMPAR EPSCs should be observed because of different quantal contents for AMPAR and NMDAR EPSCs (Kullmann, 1994; Gomperts et al., 1998).
We thus reasoned that, if NMDARs at some inactive synapses were activated by glutamate spillover, they would encounter a lower concentration of glutamate than those located in active synapses. Based on the binomial law, we then developed a mathematical model that takes into account synaptic sites directly or indirectly (“spillover sites”) activated by glutamate release (for mathematical modeling, see Appendix). Directly activated receptors could then be distinguished from low-glutamate-activated receptors by examining the action of an NMDAR competitive antagonist (Clements et al., 1992; Diamond and Jahr, 1997; Choi et al., 2000; Diamond, 2001). This model yields to several predictions. First, it predicts that, if NMDAR EPSCs are partially attributable to spillover, a competitive antagonist such as d-AA would increase NMDAR-mediated EPSC fluctuations. Conversely, if NMDAR EPSCs are partially attributable to actual NMDAR-only synapses, d-AA would lead to an unaltered CVNMDA. Second, noncompetitive antagonists, such as DCK, should block to the same extent NMDARs at all synaptic sites and thus would not alter the quantal content for NMDAR-mediated EPSCs.
Addition of d-AA (70 μm) (Fig. 4A) to the saline reduced the NMDA EPSC amplitude in both group (group 1, 58 ± 4.5% of control, n = 11; group 2, 61 ± 4.5% of control, n = 6). However, d-AA consistently increased trial-to-trial variation of NMDA current only in group 1 whatever the age (P0, n = 6; P10, n = 5) (Fig. 4B,C). When all group 1 neurons were pooled together, CVNMDA decreased to 55.5 ± 6% of CVAMPA (n = 11) and nearly reverted to AMPA values (85 ± 6%; n = 11) after d-AA application. Because this d-AA-induced CVNMDA increase is associated with a reduction of NMDA EPSC amplitude, it may be argued that CV measurement accuracy was altered because of a less favorable signal-to-noise ratio. Therefore, we performed experiments with DCK, a noncompetitive antagonist, the efficacy of which being independent of glutamate concentration. DCK application (3 μm) blocked the NMDAR EPSC to a similar extent than did d-AA (62 ± 7% of control, n = 12) (Fig. 5A). However, DCK did not alter the trial-to-trial variation of NMDA EPSC amplitude in group 1 neurons (n = 6) (B, C). As an additional control, neither d-AA nor DCK altered CVNMDA in group 2 neurons as predicted by our mathematical model (n = 6 for each group) (Fig. 5B,C).
Figure 4.

Competitive NMDA antagonist selectively restores CV. A, Comparison of EPSC variance mediated by AMPARs and NMDARs. EPSC amplitudes are plotted against stimulus number with holding potential and application of DNQX and d-AA indicated schematically. Access resistance magnitude is plotted against stimulus number. Top black traces represent average traces in the different conditions, and the gray traces are SDs. For this P0 neuron, CVAMPA was 0.6 and CVNMDA was 0.35. Application of d-AA decreased NMDA current and increased CV to 0.42. B, C, Bar histograms summarizing d-AA effect on CVNMDA relative to CVAMPA at P0 and P10 in group 1 neurons. For neurons whose CVNMDA was smaller than CVAMPA, application of d-AA nearly restored CVNMDA (DAA) to control values. *p < 0.05; **p < 0.01.
Figure 5.

Noncompetitive NMDA antagonist does not alter CVNMDA. A, Comparison of EPSC variance mediated by AMPARs and NMDARs. EPSC amplitudes are plotted against stimulus number with holding potential and application of DNQX and DCK indicated schematically. Access resistance magnitude is plotted against stimulus number. Top black traces represent average traces in the different conditions, and the gray traces are SDs. For this neuron, CVAMPA was 0.32, and CVNMDA was 0.25. Application of DCK decreased NMDA current but did not increase CVNMDA (0.23). B, Bar histogram summarizing DCK effect on CVNMDA relative to CVAMPA. In group 1 neurons (CVNMDA < CVAMPA), application of DCK did not alter CVNMDA (DCK). C, In group 2 neurons (CVNMDA ≥ CVAMPA), application of DCK or d-AA did not alter CVNMDA.
In previous studies, glutamate spillover was deemed to be responsible for prolonged decay in NMDA-induced synaptic currents (Diamond, 2001; Arnth-Jensen et al., 2002). As expected, d-AA decreased on average NMDA-induced current decay rates in NTS group 1 neurons (τ of 172 ± 12 and 138 ± 13 ms for NMDA and d-AA EPSC decay, respectively, n = 11; p = 0.001) (Fig. 6A) but not in group 2 neurons (τ of 185 ± 13 vs 197 ± 14 ms; n = 6) (Fig. 5A). In addition, DCK did not speed the decay of the NMDA EPSC in NTS neurons (group 1: τ of 170 ± 13 and 162 ± 18 ms for NMDA and DCK EPSC decay, respectively, n = 6; group 2, τ of 184 ± 18 and 191 ± 34 ms for NMDA and DCK EPSC decay, respectively, n = 6) (Fig. 6B).
Figure 6.

Competitive NMDA antagonist selectively speeds up NMDA EPSC deactivation kinetic. A, Left, Normalized average traces of NMDA EPSC before (gray trace) and after d-AA (DAA; black trace) application for a group 1 neuron. Note acceleration of the decay during d-AA application. Right, Bar histogram summarizing d-AA effect on NMDA current kinetics in both groups. Note that d-AA accelerated NMDA current decay in group 1 neurons (gray bars) but not in group 2 neurons (black bars). B, Left, Normalized average traces of NMDA EPSC before (gray trace) and after (black trace) DCK application. Note the unaltered kinetics during DCK application. Right, Bar histogram summarizing the absence of DCK effect on NMDA current kinetics in both groups.
Discussion
In this study, we describe the apparent existence of silent synapses between visceral afferent fibers and rat developing NTS neurons. However, we provide pharmacological and anatomical evidence that glutamate spillover from neighboring fiber terminals onto NMDARs is likely to be responsible for silent inputs.
Silent synapses were first detected and examined by using minimal stimulations and comparing failure rates at two potentials. This protocol was then refined by decreasing the stimulus intensity to a level at which no AMPAR-mediated synaptic transmission occurs. In some cases, this stimulation still excites one or few axons impinging on NMDAR-only synapses (Isaac et al., 1997; Rumpel et al., 1998). At birth, both methods allowed us to detect pure NMDAR responses. At P10, the picture was more complex. The “all-failure” protocol argues against NMDAR-only synapses, but the minimal stimulation strength may have been too weak to activate any fibers. If vagal fibers possessed at this age 3–4 release sites with a release probability of >0.5, it would be nearly impossible to obtain failures even when stimulating only one fiber. Therefore, some silent synapses may have been undetected in P10 neurons. To circumvent these problems, some experimenters have used simultaneous whole-cell recording from two connected cells or autaptic synapses (Gomperts et al., 1998; Montgomery et al., 2001). Both techniques were obviously not amenable to study synapses between vagal afferents and NTS neurons. Therefore, we used CV measurement to assess for changes in quantal content. We found, at both ages studied, a decrease in CV on depolarization for most NTS neurons, suggesting the presence of putative NMDAR-only synapses.
Therefore, we performed anatomical experiments to search for the presence of AMPAR-lacking glutamatergic synapses in the developing NTS. Several studies have attempted to correlate electrophysiological detection of silent synapses and anatomical presence of synapses actually devoid of AMPARs (Gomperts et al., 1998; Rao et al., 1998; Petralia et al., 1999). However, inability to label synaptic AMPARs may be caused by different reasons: actual absence of receptors, restricted access to synaptic epitope, or low sensitivity of immunoreaction. Postembedding immunolabeling experiments allow access to synaptic epitopes but suffers from low sensitivity of immunoreaction. Light microscope immunohistochemistry has a better sensitivity. Still, setting arbitrary threshold to distinguish between background and specific labeling may lead to false negative. We combined antigen retrieval allowing good access to synaptic AMPARs (Fritschy et al., 1998; Lachamp et al., 2003, 2005) and unbiased threshold setting because nonexcitatory synapses served as a reference for immunonegativity. With this approach, we observed that nearly all excitatory NTS synapses expressed AMPARs from birth. Glutamatergic synapses formed by primary afferent fibers develop before birth, whereas those made by central projections start to develop few days after birth (Rinaman and Levitt, 1993; Rinaman et al., 2000; Zhang and Ashwell, 2001; Lachamp et al., 2002; Rinaman, 2003). Thus, it is likely that synapses investigated in this study by electrophysiology as well as those analyzed by confocal microscopy belongs to the same population (i.e., primary afferent synapses). Thus, at birth, a maximum of 10% of synapses were potentially immunonegative for AMPARs, whereas electrophysiological data indicated that approximately half of them generated NMDAR-only responses. If AMPARs were present at most NTS synapses, NMDA-only responses could be explained in one of the two ways. (1) AMPARs were removed at some synapses by repetitive stimulations (Montgomery and Madison, 2002; Xiao et al., 2004; Abrahamsson et al., 2005). However, visceral fiber repetitive stimulations did not induce noticeable AMPAR EPSC depression, arguing against synapses silencing. (2) AMPARs appear silent because the peak of glutamate concentration is too low to activate low-affinity AMPARs. This latter possibility could result from either immature release machinery slowly releasing glutamate (Choi et al., 2000; Renger et al., 2001) or glutamate spillover coming from neighboring fiber terminals at presynaptic silent sites (Kullmann and Asztely, 1998). In both cases, only NMDARs could be activated because its affinity to glutamate is far higher than that of AMPARs. At both ages studied, CV of NMDA EPSCs was generally smaller than that of AMPA EPSCs. Application of d-AA selectively increased NMDA EPSC CV in neurons showing a reduced NMDA EPSC CV compared with that of AMPA EPSC (group 1 neurons). d-AA application nearly restored CV values observed at depolarized potential to those measured at rest, suggesting that addition of synaptic sites “seeing” low glutamate concentration primarily accounts for changes in CV. As reported previously for glutamate spillover (Diamond, 2001; Arnth-Jensen et al., 2002), NMDA EPSC decay was globally accelerated after d-AA treatment in this subgroup compared with the other. Altogether, our data indicate that the increase in CV can be explained by additional NMDA channels activated by lower glutamate concentration. Whether or not this is attributable to actual glutamate spillover from neighboring fibers or slow glutamate release at immature sites cannot be clearly distinguished.
A popular explanation for silent inputs is a lack of AMPARs at developing synapses. Many works have addressed this issue, but the strongest evidence to support this hypothesis was provided by experiments done on pairs of CA3 connected neurons in hippocampal slice cultures (Montgomery et al., 2001). Using simultaneous recordings from individual presynaptic and postsynaptic neurons, these authors showed that hippocampal neurons can be connected entirely by silent synapses. Increasing release probability or application of cyclothiazide did not produce responses from these silent synapses, strongly arguing for an actual lack of AMPARs. In a previous study, Choi et al. (2000) rather suggested changes in vesicular fusion modes (the whispering synapse hypothesis). These two studies hardly reconcile. Differences in temperature recordings (22°C vs 32°C), experimental models (acute vs cultured slices), stimulation frequency (0.5 vs 0.1 Hz), as well as minimal stimulation paradigms between these two studies (Choi et al., 2000 vs Montgomery et al., 2001, respectively) may have favored one mechanism to the other.
In both studies, silent synapses could be converted to functional synapses as described previously (Wu et al., 1996; Isaac et al., 1997; Rumpel et al., 1998; Zhu et al., 2000). Conversion is generally explained by de novo insertion of AMPARs via NMDA-induced biochemical cascades (Wu et al., 1996; Isaac et al., 1997; Rumpel et al., 1998; Zhu et al. 2000). In the NTS, silent synapses were never converted to functional ones by pairing visceral fiber stimulation and postsynaptic depolarization (5 min at 0 mV; data not shown). In addition, we demonstrated previously that early formation of synapses within the NTS involved a series of events different from that described in the neocortex or the hippocampus (Balland et al., 2006). For the latter, NMDARs seem predominant in the initial glutamatergic transmission, and their activation is required for the insertion of AMPARs (Wu et al., 1996; Isaac et al., 1997; Zhu et al., 2000). In the NTS, NMDAR and AMPAR insertion may occur independently because blockade of NMDARs in utero does not prevent AMPAR clustering in prenatal rats (Balland et al., 2006). Unlike the situation widely described in other systems, synaptic AMPARs undergo little changes, if any, in their molecular composition during prenatal and postnatal development (Balland et al., 2006). Present data are consistent with this view. The absence of synapses lacking AMPARs during a period in which excitatory NTS synapses still develop suggests that AMPA and NMDA receptor-mediated signaling emerge concomitantly rather than sequentially (Friedman et al., 2000; Groc et al., 2003). Absence of NMDAR-only synapses and LTP-like conversion extends previous conclusions on activity-independent NTS synapse formation (Balland et al., 2006) In a certain sense, NTS synapses are “born mature.” At the level of the NTS, the independence of synapse formation to synaptic activation suggests that circuit refinement rather occurs through synaptic elimination (Vincent et al., 2004) as suggested for other systems (Bear and Rittenhouse, 1999; Colonnese et al., 2005).
Whether glutamate spillover between NTS synapses may play a role in circuit formation remains an open question. In the hippocampus, glutamate spillover is dependent on active glutamate uptake presumably by glial cells (Asztely et al., 1997). Moreover, glutamate spillover appears to be developmentally regulated because of a low expression of glutamate transporters in young animals (Diamond and Jeffrey, 2005). Slower clearance in young animals would permit glutamate to diffuse greater distances. This may counterbalance the low synaptic density at birth by maximizing input–output gain of neurons, especially during burst discharge (Grebenyuk et al., 2004). More specifically related to synaptic formation, spillover may recruit extrasynaptic NMDARs or activate distant NMDAR clusters floating in the dendritic membrane (Friedman et al., 2000; Washbourne et al., 2002). For the adult NTS, vagal afferent terminals are generally large, and most synapses are part of synaptic glomeruli (Mrini and Jean, 1995; Hayakawa et al., 2003). This peculiar organization may favor crosstalk between active synapses during development and contribute to synaptic processing as well as autonomic circuit formation.
Appendix
Changes in CV have been extensively use to probe for presynaptic or postsynaptic changes in synaptic transmission (Faber and Korn, 1991). It relies on the assumption than CV does not depend on the quantal size of the synaptic current (q) because, mathematically, CV only depends of the release probability (p) and the number of synaptic sites (n). However, to be true, it implies that changes in synaptic transmission equally affect q. Increase in intersite heterogeneity for q would lead to CV increase despite constant values of p and n.
To predict changes in CV during application of NMDAR antagonists, we therefore took into account this potential caveat by deriving a simple mathematical model from the binomial model of synaptic transmission (Faber and Korn, 1991).
Assume that visceral fiber stimulation activates two types of release sites with equal probabilities (p). Some of these sites (n sites) signal through both AMPARs and NMDARs with quantal current of q, whereas the other sites (n′) signal through NMDARs only and have quantal currents, qspill, smaller or equal to q.
Mean synaptic current is equal to
 |
The global variance (VARg) is equal to the sum of variances for the different sites:
or
Let
 |
then
With the CV being equal to
 |
it follows that
 |
then
 |
The derivate of CV with respect to α is calculated to determine whether CV is increasing or decreasing with respect to α.
 |
then
 |
The denominator and p × (1 − p) are positive values whatever the α value:
 |
then
 |
then
 |
As stated before, qspill should be smaller than or equal to q. Hence, 0 < α < 1 and the derivative is a negative function with respect to α. If α decreases, then CV increases and vice versa.
d-AA should reduce α and increase CV if it reduces qspill more than q. This should occur if qspill represents the effect of lower glutamate concentrations as expected for spillover. If q and qspill are equally affected by d-AA and, as expected, if some synapses are NMDA only synapses, CV should not change.
A drug that does not reduce α while reducing I, such as the noncompetitive NMDAR antagonist DCK, should not alter CV.
Footnotes
This work was supported in part by a grant from the Ministère de la Recherche (Action Concertée Incitative Neurosciences Intégratives et Computationelles). We thank Dr. Dominique Debanne for critical reading of this manuscript.
References
- Aamodt SM, Constantine-Paton M. The role of neural activity in synaptic development and its implications for adult brain function. Adv Neurol. 1999;79:133–144. [PubMed] [Google Scholar]
- Abrahamsson T, Gustafsson B, Hanse E. Synaptic fatigue at the naive perforant path–dentate granule cell synapse in the rat. J Physiol (Lond) 2005;569:737–750. doi: 10.1113/jphysiol.2005.097725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andresen MC, Kunze DL. Nucleus tractus solitarius-gateway to neural circulatory control. Annu Rev Physiol. 1994;56:93–116. doi: 10.1146/annurev.ph.56.030194.000521. [DOI] [PubMed] [Google Scholar]
- Arnth-Jensen N, Jabaudon D, Scanziani M. Cooperation between independent hippocampal synapses is controlled by glutamate uptake. Nat Neurosci. 2002;5:325–331. doi: 10.1038/nn825. [DOI] [PubMed] [Google Scholar]
- Asztely F, Erdemli G, Kullmann DM. Extrasynaptic glutamate spillover in the hippocampus: dependence on temperature and the role of active glutamate uptake. Neuron. 1997;18:281–293. doi: 10.1016/s0896-6273(00)80268-8. [DOI] [PubMed] [Google Scholar]
- Bailey TW, Jin Y-H, Doyle MW, Smith SM, Andresen MC. Vasopressin inhibits glutamate release via two distinct modes in the brainstem. J Neurosci. 2006;26:6131–6142. doi: 10.1523/JNEUROSCI.5176-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balland B, Lachamp P, Kessler JP, Strube C, Tell F. Glutamatergic synapses in the rat nucleus tractus solitarii develop by direct insertion of calcium-impermeable AMPA receptors and without activation of NMDA receptors. J Physiol (Lond) 2006;574:245–261. doi: 10.1113/jphysiol.2006.108738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Rittenhouse CD. Molecular basis for induction of ocular dominance plasticity. J Neurobiol. 1999;41:83–91. doi: 10.1002/(sici)1097-4695(199910)41:1<83::aid-neu11>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Bottjer SW. Silent synapses in a thalamo-cortical circuit necessary for song learning in zebra finches. J Neurophysiol. 2005;94:3698–3707. doi: 10.1152/jn.00282.2005. [DOI] [PubMed] [Google Scholar]
- Cabezas C, Buno W. Distinct transmitter release properties determine differences in short-term plasticity at functional and silent synapses. J Neurophysiol. 2006;95:3024–3034. doi: 10.1152/jn.00739.2005. [DOI] [PubMed] [Google Scholar]
- Card JP, Levitt P, Gluhovsky M, Rinaman L. Early experience modifies the postnatal assembly of autonomic emotional motor circuits in rats. J Neurosci. 2005;25:9102–9111. doi: 10.1523/JNEUROSCI.2345-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Klingauf J, Tsien RW. Postfusional regulation of cleft glutamate concentration during LTP at “silent synapses.”. Nat Neurosci. 2000;3:330–336. doi: 10.1038/73895. [DOI] [PubMed] [Google Scholar]
- Clements JD, Lester RA, Tong G, Jahr CE, Westbrook GL. The time course of glutamate in the synaptic cleft. Science. 1992;258:1498–1501. doi: 10.1126/science.1359647. [DOI] [PubMed] [Google Scholar]
- Colonnese MT, Zhao J-P, Constantine-Paton M. NMDA receptor currents suppress synapse formation on sprouting axons in vivo. J Neurosci. 2005;25:1291–1303. doi: 10.1523/JNEUROSCI.4063-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantine-Paton M, Cline HT, Debski E. Patterned activity, synaptic convergence, and the NMDA receptor in developing visual pathways. Annu Rev Neurosci. 1990;13:129–154. doi: 10.1146/annurev.ne.13.030190.001021. [DOI] [PubMed] [Google Scholar]
- Constantine-Paton M, Cline HT. LTP and activity-dependent synaptogenesis: the more alike they are, the more different they become. Curr Opin Neurobiol. 1998;8:139–148. doi: 10.1016/s0959-4388(98)80017-2. [DOI] [PubMed] [Google Scholar]
- Contestabile A. Roles of NMDA receptor activity and nitric oxide production in brain development. Brain Res Brain Res Rev. 2000;32:476–509. doi: 10.1016/s0165-0173(00)00018-7. [DOI] [PubMed] [Google Scholar]
- Crair MC. Neuronal activity during development: permissive or instructive? Curr Opin Neurobiol. 1999;9:88–93. doi: 10.1016/s0959-4388(99)80011-7. [DOI] [PubMed] [Google Scholar]
- Diamond JS. Neuronal glutamate transporters limit activation of NMDA receptors by neurotransmitter spillover on CA1 pyramidal cells. J Neurosci. 2001;21:8328–8338. doi: 10.1523/JNEUROSCI.21-21-08328.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE. Transporters buffer synaptically released glutamate on a submillisecond time scale. J Neurosci. 1997;17:4672–4687. doi: 10.1523/JNEUROSCI.17-12-04672.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS, Jeffrey S. Deriving the glutamate clearance time course from transporter currents in CA1 hippocampal astrocytes: transmitter uptake gets faster during development. J Neurosci. 2005;25:2906–2916. doi: 10.1523/JNEUROSCI.5125-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle MW, Andresen MC. Reliability of monosynaptic sensory transmission in brain stem neurons in vitro. J Neurophysiol. 2001;85:2213–2223. doi: 10.1152/jn.2001.85.5.2213. [DOI] [PubMed] [Google Scholar]
- Faber DS, Korn H. Applicability of the coefficient of variation method for analyzing synaptic plasticity. Biophys J. 1991;60:1288–1294. doi: 10.1016/S0006-3495(91)82162-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman HV, Bresler T, Garner CC, Ziv NE. Assembly of new individual excitatory synapses: time course and temporal order of synaptic molecule recruitment. Neuron. 2000;27:57–69. doi: 10.1016/s0896-6273(00)00009-x. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Weinmann O, Wenzel A, Benke D. Synapse-specific localization of NMDA and GABA(A) receptor subunits revealed by antigen-retrieval immunohistochemistry. J Comp Neurol. 1998;390:194–210. [PubMed] [Google Scholar]
- Gasparini S, Saviane C, Voronin LL, Cherubini E. Silent synapses in the developing hippocampus: lack of functional AMPA receptors or low probability of glutamate release? Proc Natl Acad Sci USA. 2000;97:9741–9746. doi: 10.1073/pnas.170032297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomperts SN, Rao A, Craig AM, Malenka RC, Nicoll RA. Postsynaptically silent synapses in single neuron cultures. Neuron. 1998;21:1443–1451. doi: 10.1016/s0896-6273(00)80662-5. [DOI] [PubMed] [Google Scholar]
- Gozal E, Gozal D. Respiratory plasticity following intermittent hypoxia: developmental interactions. J Appl Phys. 2001;90:1995–1999. doi: 10.1152/jappl.2001.90.5.1995. [DOI] [PubMed] [Google Scholar]
- Grebenyuk SE, Lozovaya NA, Tsintsadze TS, Krishtal OA. Post-synaptic N-methyl-aspartate signalling in hippocampal neurons of rat: spillover increases the impact of each spike in a short burst discharge. Neurosci Lett. 2004;361:60–63. doi: 10.1016/j.neulet.2004.02.040. [DOI] [PubMed] [Google Scholar]
- Groc L, Gustafsson B, Hanse E. In vivo evidence for an activity-independent maturation of AMPA/NMDA signaling in the developing hippocampus. Neuroscience. 2003;121:65–72. doi: 10.1016/s0306-4522(03)00366-x. [DOI] [PubMed] [Google Scholar]
- Hayakawa T, Takanaga A, Tanaka K, Maeda S, Seki M. Ultrastructure of the central subnucleus of the nucleus tractus solitarii and the esophageal afferent terminals in the rat. Anat Embryol (Berl) 2003;206:273–281. doi: 10.1007/s00429-002-0288-z. [DOI] [PubMed] [Google Scholar]
- Hohnke CD, Oray S, Sur M. Activity-dependent patterning of retinogeniculate axons proceeds with a constant contribution from AMPA and NMDA receptors. J Neurosci. 2000;20:8051–8060. doi: 10.1523/JNEUROSCI.20-21-08051.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac JT, Crair MC, Nicoll RA, Malenka RC. Silent synapses during development of thalamocortical inputs. Neuron. 1997;18:269–280. doi: 10.1016/s0896-6273(00)80267-6. [DOI] [PubMed] [Google Scholar]
- Kasparov S, Paton JF. Changes in baroreceptor vagal reflex performance in the developing rat. Pflügers Arch. 1997;434:438–444. doi: 10.1007/s004240050418. [DOI] [PubMed] [Google Scholar]
- Katz LC, Shatz C. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- Kullmann DM. Amplitude fluctuations of dual-component EPSCs in hippocampal pyramidal cells: implications for long-term potentiation. Neuron. 1994;12:1111–1120. doi: 10.1016/0896-6273(94)90318-2. [DOI] [PubMed] [Google Scholar]
- Kullmann DM, Asztely F. Extrasynaptic glutamate spillover in the hippocampus: evidence and implications. Trends Neurosci. 1998;21:8–14. doi: 10.1016/s0166-2236(97)01150-8. [DOI] [PubMed] [Google Scholar]
- Kullmann DM, Erdemli G, Asztely F. LTP of AMPA and NMDA receptor-mediated signals: evidence for presynaptic expression and extrasynaptic glutamate spill-over. Neuron. 1996;17:461–474. doi: 10.1016/s0896-6273(00)80178-6. [DOI] [PubMed] [Google Scholar]
- Lachamp P, Tell F, Kessler JP. Successive episodes of synapses production in the developing rat nucleus tractus solitarii. J Neurobiol. 2002;52:336–342. doi: 10.1002/neu.10091. [DOI] [PubMed] [Google Scholar]
- Lachamp P, Balland B, Tell F, Crest M, Kessler JP. Synaptic localization of the glutamate receptor subunit GluR2 in the rat nucleus tractus solitarii. Eur J Neurosci. 2003;17:892–896. doi: 10.1046/j.1460-9568.2003.02494.x. [DOI] [PubMed] [Google Scholar]
- Lachamp P, Balland B, Tell F, Baude A, Strube C, Crest M, Kessler JP. Early expression of AMPA receptors and lack of NMDA receptors in developing rat climbing fibre synapses. J Physiol (Lond) 2005;564:751–763. doi: 10.1113/jphysiol.2005.084517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Montgomery J, Madison D. State-dependent heterogeneity in synaptic depression between pyramidal cell pairs. Neuron. 2002;33:765–777. doi: 10.1016/s0896-6273(02)00606-2. [DOI] [PubMed] [Google Scholar]
- Montgomery JM, Pavlidis P, Madison DV. Pair recordings reveal all-silent synaptic connections and the postsynaptic expression of long-term potentiation. Neuron. 2001;29:691–701. doi: 10.1016/s0896-6273(01)00244-6. [DOI] [PubMed] [Google Scholar]
- Moss IR. Maturation of respiratory control in the behaving mammal. Respir Physiol Neurobiol. 2002;132:131–144. doi: 10.1016/s1569-9048(02)00070-8. [DOI] [PubMed] [Google Scholar]
- Mrini A, Jean A. Synaptic organization of the interstitial subdivision of the nucleus tractus solitarii and of its laryngeal afferents in the rat. J Comp Neurol. 1995;355:221–236. doi: 10.1002/cne.903550206. [DOI] [PubMed] [Google Scholar]
- Petralia RS, Esteban JA, Wang Y-X, Partridge JG, Zhao H-M, Wenthold RJ, Malinow R. Selective acquisition of AMPA receptors over postnatal development suggests a molecular basis for silent synapses. Nat Neurosci. 1999;2:31–36. doi: 10.1038/4532. [DOI] [PubMed] [Google Scholar]
- Poncer JC. Hippocampal long term potentiation: silent synapses and beyond. J Physiol (Paris) 2003;97:415–422. doi: 10.1016/j.jphysparis.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Rao A, Kim E, Sheng M, Craig AM. Heterogeneity in the molecular composition of excitatory postsynaptic sites during development of hippocampal neurons in culture. J Neurosci. 1998;18:1217–1229. doi: 10.1523/JNEUROSCI.18-04-01217.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renger JJ, Egles C, Liu G. A developmental switch in neurotransmitter flux enhances synaptic efficacy by affecting AMPA receptor activation. Neuron. 2001;29:469–484. doi: 10.1016/s0896-6273(01)00219-7. [DOI] [PubMed] [Google Scholar]
- Rinaman L. Postnatal development of hypothalamic inputs to the dorsal vagal complex in rats. Physiol Behav. 2003;79:65–70. doi: 10.1016/s0031-9384(03)00105-7. [DOI] [PubMed] [Google Scholar]
- Rinaman L, Levitt P. Establishment of vagal sensorimotor circuits during fetal development in rats. J Neurobiol. 1993;24:641–659. doi: 10.1002/neu.480240509. [DOI] [PubMed] [Google Scholar]
- Rinaman L, Levitt P, Card JP. Progressive postnatal assembly of limbic-autonomic circuits revealed by central transneuronal transport of pseudorabies virus. J Neurosci. 2000;20:2731–2741. doi: 10.1523/JNEUROSCI.20-07-02731.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumpel S, Hatt H, Gottmann K. Silent synapses in the developing rat visual cortex: evidence for postsynaptic expression of synaptic plasticity. J Neurosci. 1998;18:8863–8874. doi: 10.1523/JNEUROSCI.18-21-08863.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selig DK, Hjelmstad GO, Herron C, Nicoll RA, Malenka RC. Independent mechanisms for long-term depression of AMPA and NMDA responses. Neuron. 1995;15:417–426. doi: 10.1016/0896-6273(95)90045-4. [DOI] [PubMed] [Google Scholar]
- Vincent A, Tell F. Postnatal development of rat nucleus tractus solitarius neurons: morphological and electrophysiological evidence. Neuroscience. 1999;93:293–305. doi: 10.1016/s0306-4522(99)00109-8. [DOI] [PubMed] [Google Scholar]
- Vincent A, Kessler JP, Baude A, Dipasquale E, Tell F. N-methyl-d-aspartate receptor activation exerts a dual control on postnatal development of nucleus tractus solitarii neurons in vivo. Neuroscience. 2004;126:185–194. doi: 10.1016/j.neuroscience.2004.03.012. [DOI] [PubMed] [Google Scholar]
- Voronin LL, Cherubini E. “Deaf, mute and whispering” silent synapses: their role in synaptic plasticity. J Physiol (Lond) 2004;557:3–12. doi: 10.1113/jphysiol.2003.058966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washbourne P, Bennett JE, McAllister AK. Rapid recruitment of NMDA receptor transport packets to nascent synapses. Nat Neurosci. 2002;5:751–759. doi: 10.1038/nn883. [DOI] [PubMed] [Google Scholar]
- Wu G, Malinow R, Cline HT. Maturation of a central glutamatergic synapse. Science. 1996;274:972–976. doi: 10.1126/science.274.5289.972. [DOI] [PubMed] [Google Scholar]
- Xiao MY, Niu YP, Dozmorov M, Wigstrom H. Comparing fluctuations of synaptic responses mediated via AMPA and NMDA receptor channels—implications for synaptic plasticity. Biosystems. 2001;62:45–56. doi: 10.1016/s0303-2647(01)00136-8. [DOI] [PubMed] [Google Scholar]
- Xiao MY, Wasling P, Hanse E, Gustafsson B. Creation of AMPA-silent synapses in the neonatal hippocampus. Nat Neurosci. 2004;7:236–243. doi: 10.1038/nn1196. [DOI] [PubMed] [Google Scholar]
- Zhang LL, Ashwell KW. The development of cranial nerve and visceral afferents to the nucleus of the solitary tract in the rat. Anat Embryol (Berl) 2001;204:135–151. doi: 10.1007/s004290100185. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Esteban JA, Hayashi Y, Malinow R. Postnatal synaptic potentiation: delivery of GluR4-containing AMPA receptors by spontaneous activity. Nat Neurosci. 2000;3:1098–1106. doi: 10.1038/80614. [DOI] [PubMed] [Google Scholar]