

Abstract
Sphingolipids containing 2-hydroxylated fatty acids are among the most abundant lipid components of the myelin sheath and therefore are thought to play an important role in formation and function of myelin. To prove this hypothesis, we generated mice lacking a functional fatty acid 2-hydroxylase (FA2H) gene. FA2H-deficient (FA2H−/−) mice lacked 2-hydroxylated sphingolipids in the brain and in peripheral nerves. In contrast, nonhydroxylated galactosylceramide was increased in FA2H−/− mice. However, oligodendrocyte differentiation examined by in situ hybridization with cRNA probes for proteolipid protein and PDGFα receptor and the time course of myelin formation were not altered in FA2H−/− mice compared with wild-type littermates. Nerve conduction velocity measurements of sciatic nerves revealed no significant differences between FA2H−/− and wild-type mice. Moreover, myelin of FA2H−/− mice up to 5 months of age appeared normal at the ultrastructural level, in the CNS and peripheral nervous system. Myelin thickness and g-ratios were normal in FA2H−/− mice. Aged (18-month-old) FA2H−/− mice, however, exhibited scattered axonal and myelin sheath degeneration in the spinal cord and an even more pronounced loss of stainability of myelin sheaths in sciatic nerves. These results show that structurally and functionally normal myelin can be formed in the absence of 2-hydroxylated sphingolipids but that its long-term maintenance is strikingly impaired. Because axon degeneration appear to start rather early with respect to myelin degenerations, these lipids might be required for glial support of axon function.
Keywords: axonal degeneration, fatty acid 2-hydroxylase, galactosylceramide, myelin, oligodendrocyte, Schwann cell, sphingolipids
Introduction
The myelin sheath is a highly specialized membrane, synthesized by oligodendrocytes in the central and Schwann cells in the peripheral nervous system (Baumann and Pham-Dinh, 2001). Synthesis of compacted myelin is essential for saltatory nerve conduction. To fulfill its function, myelin has a unique composition, in terms of both quality and quantity. Several abundant myelin-specific proteins, such as myelin basic protein (MBP) and proteolipid protein (PLP), and a high lipid content (>70%) are characteristic features of the myelin sheath. Galactosylceramide (GalC) and its sulfated derivative sulfatide belong to the most abundant lipids in myelin, making up ∼25% of myelin lipids (Norton and Cammer, 1984). More than 50% of GalC and sulfatide are hydroxylated at the 2-C atom of the amide linked fatty acid (Hoshi and Kishimoto, 1973). Thus, one can estimate that ∼25% of the lipids in the outer leaflet of the myelin membrane carry 2-hydroxylated fatty acids (HFA). In neurons HFA–sphingolipids are not detectable (Raghavan and Kanfer, 1972). The high HFA-content in myelin suggests, but does not prove, an important role of fatty acid 2-hydroxylation in the formation and/or function of the myelin sheath or the metabolism of oligodendrocytes. So far, however, no direct evidence for any specific function of 2-hydroxylated sphingolipids in the nervous system has been reported.
Indications for an important role of 2-hydroxylated sphingolipids in the myelin membrane came from studies of UDP-galactose:ceramide galactosyltransferase (CGT)-deficient (Bosio et al., 1996; Coetzee et al., 1996) and overexpressing mice (Fewou et al., 2005). CGT-deficient mice lack GalC and sulfatide (Bosio et al., 1996, 1998; Coetzee et al., 1996), which in part is compensated by the synthesis of HFA–glucosylceramide. The total amount of HFA–sphingolipids, however, is significantly reduced (Bosio et al., 1996; Coetzee et al., 1996). Maturation of myelin is delayed and myelin structure is abnormal in CGT-deficient mice (for review, see Marcus and Popko, 2002). Although cerebroside sulfotransferase (CST)-deficient mice, which lack only sulfatide but have normal GalC and almost normal HFA–sphingolipid levels, displayed a similar pathology, their phenotype is less severe when compared with CGT-deficient mice (Honke et al., 2002). The more severe phenotype in CGT-deficient mice might be attributable to the absence of GalC, but we hypothesized that it could also be caused by the substantially reduced HFA–sphingolipid levels.
Fatty acid 2-hydroxylase (FA2H) was recently identified as an enzyme involved in the synthesis of 2-hydroxylated sphingolipids (Alderson et al., 2004; Eckhardt et al., 2005). To prove the hypothesis that FA2H synthesizes HFA–sphingolipids in myelinating glial cells and to examine a possible role of HFA–sphingolipids in myelin formation and function, we generated mice deficient in FA2H (FA2H−/−). FA2H−/− mice lack HFA–GalC and HFA–sulfatide in brain and peripheral nerves. Unexpectedly, these mice developed structural and functional normal myelin up to early adulthood. Aged FA2H−/− mice, however, showed scattered axon and myelin sheath degeneration in the spinal cord and morphologically similar, but significantly more pronounced, lesions in peripheral nerves, indicating that FA2H is indispensable for long-term myelin sheath and axon maintenance.
Materials and Methods
Generation of FA2H-deficient mice.
A targeting vector was designed to replace part of the FA2H gene (149 bp of exon 3, intron 3, and exon 4, and 402 bp of intron 4) by a lacZ/neo cassette. The lacZ gene was inserted in frame into exon 3, leading to a potential transcript encoding a fusion protein of the cytochrome b5 domain of FA2H and β-galactosidase. FA2H genomic fragments were amplified by PCR from 129/Ola mouse HM-1 embryonic stem (ES) cell (Magin et al., 1992) genomic DNA. The 5′ homology arm (4.3 kb of intron 2 and 30 bp of exon 3) was amplified by PCR using primers FH1 (5′-CTTCAGTAAGGAGGACTAGAAGCTGG-3′), FH2 (5′-CAGCAGAGGTTTCTGCCAGTCC-3′), and Phusion DNA polymerase (Finnzymes). The PCR product was cloned into the SmaI site of pLRlacZpA/MCINeopA. The 3′ homologous region was a 1.8 kb PvuII fragment of intron 4, which was subcloned into the blunted SpeI site of the targeting vector. A HSV–TK cassette was isolated from the plasmid pHSV–TK by digestion with XhoI and XbaI and 5′ overhangs were filled in using Klenow enzyme. This fragment was subcloned into the SalI site downstream of the 3′ homology arm.
The FA2H targeting vector was linearized with SpeI and electroporated into HM-1 ES cells cultured on mitomycin C-treated feeder cells. After 48 h, the medium was supplemented with 350 μg/ml G418 and 2 μm ganciclovir (both from Sigma). ES cells surviving G418/ganciclovir selection were expanded, and genomic DNA was screened for homologous recombination by PCR. Positive clones were confirmed by Southern blot analysis. Therefore, genomic DNA was digested with BglII and analyzed by Southern blot hybridization with a [32P]-labeled 943 bp 3′ external probe. The probe was amplified by PCR using primers 5′-CCCGGAGCACCTGGAGACCTACTT-3′ and 5′-CTGGCTGGCTCATGCATCCTTCTC-3′. One targeted ES cell clone, exhibiting a normal karyotype, was injected into C57BL/6N (Charles River) blastocysts to obtain chimeric animals. Chimeric male mice with a high contribution of agouti coat color were bred to C57BL/6N females, and germ-line transmission of the disrupted FA2H allele was confirmed by Southern blotting of genomic tail DNA of agouti F1 mice. Subsequent genotyping was performed by PCR using the primers 5′-ATTCGCAGCGCATCGCCTTCTATC-3′, 5′-GTGCTGTACCTCAGCTGGTC-3′, and 5′-GCTCTTCTTCAAGAGCCATCC-3′, which amplified a 1045 bp fragment from the wild-type and a 685 bp fragment from the targeted allele.
Northern blot analysis.
Total RNA of murine brain was isolated using Trizol (Invitrogen) as described by the manufacturer. Total RNA (20 μg/lane) was separated in 1 m formaldehyde/1% agarose gels and transferred onto Hybond-N+ nylon membranes (GE Healthcare). A 568 bp fragment of the FA2H cDNA (corresponding to exons 3 and 4) was used to synthesize a [32P]-labeled probe by random priming using [α-32P]dCTP and Megaprime DNA labeling kit (GE Healthcare), following the instructions of the manufacturer. A β-actin probe (Stratagene) was used to control for equal loading. Hybridization was performed using standard procedures (Sambrook et al., 1989). Radioactivity signals were detected by autoradiography on x-ray films.
In situ hybridization.
Digoxigenin (DIG)-labeled cRNA probes were transcribed from mouse PLP cDNA and mouse platelet derived growth factor (PDGF) α receptor cDNA in pBluescript as described recently (Yaghootfam et al., 2005). A 568 bp mouse FA2H cDNA fragment (harboring the coding region of exons 3 and 4) was cloned into pBluescript. The plasmid was linearized with BamHI and XhoI, respectively, and digoxigenin-labeled antisense and sense cRNA probes were transcribed using SP6- and T7-RNA polymerases, respectively. Brains were removed and directly frozen in isopentane. Cryosections were cut at 12 μm and stored with dessicant at −80°C. Hybridization to digoxigenin-labeled cRNA probes was performed as described previously (Fewou et al., 2005). Briefly, sections were hybridized overnight at 70°C in 50% formamide, 1% Denhardt's solution, 0.2% SDS, 0.25 mg/ml salmon sperm DNA, 0.3 m NaCl, 10 mm PIPES, and 10 mm EDTA. Sections were washed twice with 2× SSC at 60°C for 5 and 30 min and once with 0.1× SSC for 45 min at 70°C. The sections were equilibrated in maleic acid buffer (100 mm maleic acid and 150 mm NaCl, pH 7.5) and incubated in maleic acid buffer containing 2% blocking reagent (Roche Diagnostics) for 1 h at room temperature. Bound RNA probes were detected using alkaline phosphatase-conjugated anti-digoxigenin Ig (Roche Diagnostics) at a dilution of 1:5000 in maleic acid buffer containing 2% blocking reagent at 4°C overnight in a humid chamber. Sections were washed with alkaline phosphatase buffer (100 mm Tris-HCl, 100 mm NaCl, and 50 mm MgCl2, pH 9.5) and stained using 0.33 mg/ml nitro-blue tetrazolium and 0.16 mg/ml 5-bromo-4-chloro-3-indolyl-phosphate in alkaline phosphatase buffer.
Western blot analysis.
Brains were homogenized in water containing 5 mm EDTA, 1 mm phenylmethylsulfonyl fluoride, and 1% SDS. Protein concentration was determined using the Bio-Rad DC protein assay. Samples of total homogenates were separated by SDS-PAGE on 12.5 or 10% gels, respectively, and transferred onto nitrocellulose membranes (Whatman Schleicher & Schuell) by semidry blotting (Harlow and Lane, 1988). Bound proteins were detected using the following primary antibodies: mouse anti-β-actin (1:5000; Sigma), rabbit anti-MBP (1:5000; Millipore Bioscience Research Reagents), mouse anti-2′,3′-cyclic nucleotide 3′-phosphohydrolase (CNP) (1:300; Millipore Bioscience Research Reagents), rat anti-neural cell adhesion molecule (NCAM) (clone H28, 1 μg/ml), and rabbit anti-large myelin-associated glycoprotein (L-MAG) (1:1000; kindly provided by Dr. Arthur Butt Center for Neuroscience Research, King's College London, London, UK). Secondary antibodies used were peroxidase-labeled anti-rabbit Ig and anti-rat Ig (Dianova). Bound antibodies were visualized by enhanced chemiluminescence. To this end, membranes were incubated for 1 min in 2.7 mm H2O2, 1.25 mm 3-amino-pthalhydrazid, 0.2 mm p-coumaric acid, and 100 mm Tris-HCl, pH 8.5, and exposed to x-ray films. In some experiments Western blot membranes were incubated with Alexa 680- or IR 800-conjugated secondary antibodies [anti-rat and anti-rabbit Alexa 680-conjugate (Invitrogen); anti-mouse IR-Dye 800 conjugate (Rockland/Biomol)], and bound antibodies were visualized using infrared detection (Odyssey System; Li-Cor Bioscience). Quantification of Western blots was done using a BAS-1800II BioImager (Fujifilm/Raytest).
Immunofluorescence.
Brains were quickly frozen in cold isopentane and stored at −80°C. Parasagittal sections (12 μm) were cut in a cryostat and postfixed in 4% paraformaldehyde in PBS. Sections were permeabilized with 0.5% Triton X-100 in PBS and blocked for 30 min with 1% BSA in PBS. Sections were incubated overnight at 4°C with rabbit anti-MBP (1:500; Millipore Bioscience Research Reagents). After washing with PBS, cyanine 2 (Cy2)-conjugated goat anti-rabbit Ig (1:400; Dianova) was used to detect primary antibodies. Nuclei were stained with Hoechst dye H33258 (Sigma). Micrographs were taken with an Axiovert 100M microscope (Carl Zeiss) and the Axiovision 4.5 program.
Optic nerves from adult FA2H−/− and FA2H+/+ mice were fixed for 30 min in 4% paraformaldehyde and cryoprotected by incubating in 20% sucrose/PBS at 4°C. Cryostat sections were cut at 12 μm and stored at −80°C. Sciatic nerves were isolated from adult FA2H−/− and FA2H+/+ mice and fixed in 4% paraformaldehyde. Teased sciatic nerve fibers were prepared using fine forceps and dried on glass slides overnight at room temperature and were then stored at −20°C. Immunofluorescence staining was performed as described by Altevogt et al. (2002) with minor modifications. Briefly, cryosections and teased fibers were postfixed and permeabilized by immersion in −20°C acetone for 10 min and blocked at room temperature for 60 min in 0.5% gelatin/0.5% Triton X-100 in PBS. Primary antibody incubation was performed in a wet chamber for ∼90 min with rabbit anti-Paranodin antiserum (1:1000; kindly provided by Dr. Jean-Antoine Girault, INSERM U536, Institut du Fer à Moulin, Paris, France) and mouse anti-sodium channel (pan) antibody (1:100; Sigma). Sections were rinsed thoroughly in PBS, followed by application of fluorescently labeled secondary antibodies (Cy2-conjugated goat anti-mouse and Cy3-conjugated goat anti-rabbit, diluted 1:400) for 60 min at room temperature. Finally, slides were washed consecutively in PBS, and nuclei were stained with 4′,6′-diamidino-2-phenylindole (1:500; Sigma) and mounted with polyvinyl alcohol medium (Fluka). Images were captured with an Axiovert 100M fluorescence microscope (Carl Zeiss, Jena, Germany) and analyzed with the Axiovision 4.5 program.
Real-time PCR and analysis of MAG splice variants.
Brain total RNA was isolated using Trizol (Invitrogen), following the instructions of the manufacturer. cDNA was synthesized from brain total RNA (2 μg) using Revert Aid First Strand cDNA Synthesis kit (Fermentas) and oligo-dT primers. Real-time PCR was done using a Bio-Rad i-cycler and SYBR Green ReadyMix (Sigma). The following primers were used to amplify PLP (5′-TGAGCGCAACGGTAACAGG-3′ and 5′-GGGAGAACACCATACATTCTGG-3′), MBP (5′-GACCATCCAAGAAGACCCCAC-3′ and 5′-GCCATAATGGGTAGTTCTCGTGT-3′), myelin and lymphocyte protein (MAL) (5′-TTTGTGAGTTTGTCTTTGGAGGC-3′ and 5′-CCGCCATGAGTACCAATTATGT-3′), and actin cDNA (5′-TCCATCATGAAGTGTGACGT-3′ and 5′-GAGCAATGATCTTGATCTTCAT-3′). PCR of short myelin-associated glycoprotein (S-MAG)/L-MAG splice variants was performed using primers MAG sense (5′-ATTGTGTGCTACATCACCCAGACG-3′) and MAG anti-sense (5′-GATCCCAGGCGCTGCTTCTCACT-3′), resulting in PCR products of 199 bp for S-MAG and 154 bp product for L-MAG. PCR conditions were as follows: 5 min at 94°C, followed by 32 cycles with 94°C for 30 s, 56°C for 30 s, and 72°C for 30 s. PCR products were separated in 2% agarose gels and visualized by ethidium bromide staining. Staining intensities were determined by densitometry using Advanced Image Data Analyzer (AIDA) program, and the ratio of S-MAG/L-MAG expression was calculated.
Lipid analysis and myelin purification.
Lipids were isolated from brain or sciatic nerves as described by van Echten-Deckert (2000) with minor modifications. Briefly, brains and sciatic nerves, respectively, were homogenized in water using an Ultra-turrax tissue homogenizer (Janke & Kunkel). Twenty volumes of chloroform/methanol (2:1; v/v) were added to the homogenate, and lipids were extracted for 4 h at 50°C under constant stirring. After centrifugation, the insoluble material was further extracted in chloroform/methanol (1:1; v/v) at 50°C for 4 h. Insoluble material was removed by filtration through glass wool. The two extracts were combined, dried at 50°C under a stream of nitrogen, and dissolved in chloroform/methanol (1:1; v/v). To remove glycerolipids, samples were treated with 10 mm NaOH in methanol for 2 h at 37°C and, thereafter, were desalted by reversed-phase chromatography using silica gel RP18 as described by van Echten-Deckert (2000).
Myelin was isolated from total brains of 10-week-old mice as described by Norton and Poduslo (1973). Tissues were homogenized in 10.5% (w/v) sucrose using an Ultra-turrax homogenizer (Janke & Kunkel). Homogenates were centrifuged for 45 min with 17,000 × g. The myelin containing upper part of the pellet was resuspended in 30% sucrose, overlaid with 10.5% sucrose, and centrifuged for 50 min with 68,000 × g. Myelin was recovered from the 30%/10.5% interphase, washed twice with water, and subjected to a second sucrose gradient to remove contaminating axolemma. Purified myelin was again washed twice with water, resuspended in water, and stored in aliquots at −80°C. Lipids were extracted from purified myelin by extraction in chloroform/methanol/water (4:8:3; v/v/v) for 4 h at 56°C under constant stirring. After centrifugation, the insoluble material was reextracted with chloroform/methanol (2:1; v/v) for 4 h at 56°C. The two extracts were combined, dried at 50°C under a stream of nitrogen, and dissolved in chloroform/methanol (1:1; v/v). Lipids corresponding to 1 mg of brain or 0.5 mg of sciatic nerve (wet weight), respectively, were loaded onto thin-layer chromatography (TLC) or HPTLC silica gel 60 plates (Merck) using an automatic TLC 4 sampler (CAMAG) and separated using chloroform/methanol/water (60:27:4; v/v/v) as solvent system. Lipids were visualized by spraying with 0.625 m copper sulfate in 8% (v/v) phosphoric acid, followed by heating at 180°C for 10 min (Yao and Rastetter, 1985). Serial dilutions (0.2–4 μg) of bovine GalC and sulfatide standards (Sigma) were applied onto the same TLC plate as the samples and used to construct a standard curve. After scanning, lipids were quantified by densitometry with the AIDA software (Raytest). Data were tested for significant differences using Student's t test.
Analysis of detergent-insoluble myelin fractions.
3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) extraction of purified myelin and Optiprep gradient centrifugation were performed as described by Simons et al. (2000). In brief, myelin was incubated in TNE buffer (25 mm Tris-HCl, pH 7.5, 150 mm NaCl, and 5 mm EDTA, supplemented with protease inhibitors) containing 20 mm CHAPS for 30 min at different temperatures (4, 25, or 37°C). Thereafter, all samples were placed on ice. The samples (250 μl) were mixed with 500 μl of 60% Optiprep, overlaid with 30% Optiprep in TNE buffer, and TNE buffer. Samples were centrifuged at 55,000 rpm in a TLS55 rotor. Centrifugation times were 2 or 16 h; both conditions gave similar results. Six fractions of 350 μl each were collected from top to bottom, and lipids were isolated according to Bligh and Dyer (1959). Lipids were analyzed by TLC as described above.
Matrix assisted-laser desorption ionization-time of flight mass spectrometry.
Regions containing the lipid of interest were cut out from TLC plates and extracted with chloroform/methanol (1:1; v/v), dried under nitrogen, and dissolved in chloroform/methanol (1:1; v/v). One microliter of the sample was mixed with 1 μl of 2,5-dihydroxy-benzoic acid (10 mg/ml in 70% acetonitrile) and loaded onto a well of a 100 well-sample plate. Mass spectra were recorded with a Voyager DE STR mass spectrometer (Applied Biosystems) in positive or negative ion mode (instrument settings: 25,000 V accelerating voltage and 75% grid voltage).
Electron microscopy, morphometry, and histology.
Mice were anesthetized and intracardially perfused with 6% glutaraldehyde. Tissues were processed and analyzed as described previously (Büssow, 1978). For the quantification of axon diameters and myelin sheath thickness, toluidine blue-stained cross sections were recorded on a Nikon 90i microscope using a 100×/numerical aperture 1.4 planapochromat objective and a calibrated 12 MP camera. Axons and the corresponding myelin sheaths were isolated by three-color segmentation of the recorded images and quantified using the Nikon NIS AR software package.
Tissue cryosections from mouse brains were fixed with 0.2% glutaraldehyde and stained for β-galactosidase activity using 100 mm phosphate buffer, pH 7.3, containing 2 mm MgCl2, 5 mm K4Fe(CN)6, 5 mm K3Fe(CN)6, and 1 mg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal). Sections were counterstained with eosin.
Behavioral examinations.
Behavioral analysis was performed with female FA2H+/+ (n = 24) and FA2H−/− (n = 25) mice on a mixed genetic C57BL/6 × 129/Ola background at 4 months of age. Open-field activity was measured using an open-field activity system (ENV-515–16; size, 44 × 44 cm) and Activity software (version 4.36; Med Associates). Mice were placed in the center of the open-field box, and activity was recorded for a period of 5 min. Rotarod tests of 4-month-old and 16- to 18-month-old mice at 6 and 12 rpm were done as described previously (Zöller et al., 2005).
Nerve conduction velocity measurement.
Sciatic nerves were isolated from female FA2H+/+ (seven animals) and FA2H−/− (12 animals) mice at 6–7 months of age. Nerves were collected in Tyrode's solution (in mm: 135 NaCl, 4 KCl, 1 MgCl2, 2 CaCl2, and 2 HEPES, pH 7.4). Recording of nerve conduction velocity was performed in a custom-made humid chamber that was heated to 36°C. In the humid chamber, the explanted nerve was placed on Ag/AgCl wire electrodes. Two electrodes were used for stimulation, and a third one connected the nerve to ground. Three pairs of recording electrodes were located along the nerve; each pair was 5 mm apart from its neighbors. The individual electrodes had a diameter of 0.5 mm, and the distance within a pair was 1 mm. Electrode pairs were connected to a differential amplifier and recorded action potentials (APs) externally. APs were elicited by stimuli of 200 μs, and the stimulus height was adjusted to achieve the maximal AP amplitude for each nerve, resulting in a stimulus of 1–2 V. The time at which the positive AP peak appeared at the first electrode pair was subtracted from the time at which the corresponding AP peak appeared at the second electrode pair. Based on this time difference, conduction velocity was calculated. The stimulation and the recordings were performed with a PowerLab 4/25T (ADInstruments) controlled by the software Scope 3.7 (ADInstruments) run on a Windows-based personal computer. Data were examined for a significant difference using the Kolmogorov–Smirnov test.
Results
Generation of FA2H-deficient mice
FA2H-deficient (FA2H−/−) mice were generated by homologous recombination in 129/Ola HM-1 ES cells (Fig. 1). Part of the FA2H gene, containing exons 3 and 4, were replaced by a β-galactosidase (lacZ) reporter gene, which was inserted in frame into exon 3 resulting in an encoded fusion protein of the N-terminal cytochrome b5 domain of FA2H and lacZ (Fig. 1A). ES cell clones harboring a homologous recombined FA2H gene locus were identified by PCR and confirmed by Southern blot analysis (Fig. 1B). Heterozygous (FA2H+/−) F1 mice were identified by Southern blotting and used to generate FA2H−/− mice (Fig. 1C). All experiments described in this report were done with mice on a mixed genetic 129/Ola × C57BL/6 background. Northern blot analysis of mouse brains with an exon 3/exon 4-specific probe showed expression of a 2.6 kb mRNA in FA2H+/+ and FA2H+/− mice, which was absent in FA2H−/− mice (Fig. 1D), as expected. Absence of the FA2H mRNA in FA2H−/− mice was further confirmed by in situ hybridization (Fig. 1E). FA2H+/− and FA2H−/− mice were born at the expected Mendelian frequency (Table 1) and did not show increased mortality up to an age of 1 year. Body weights of FA2H−/− mice were not significantly different from those of FA2H+/+ and FA2H+/− mice (data not shown). FA2H−/− mice did not show obvious behavioral abnormalities when observed in their home cage, and both genders were fertile. X-Gal staining of brain cryosections of FA2H+/− mice confirmed expression of the lacZ reporter gene in the white matter of the brain (Fig. 2). Furthermore, the lacZ expression pattern was very similar to the in situ hybridization signals obtained with an FA2H-specific cRNA antisense probe (Fig. 2h). Staining intensities increased during postnatal development (Fig. 2a–d), in agreement with the upregulation of FA2H gene expression during myelin formation (Eckhardt et al., 2005; Alderson et al., 2006). Compared with FA2H+/− mice, lacZ staining intensities were increased in FA2H−/− brains, as expected (Fig. 2f).
Figure 1.
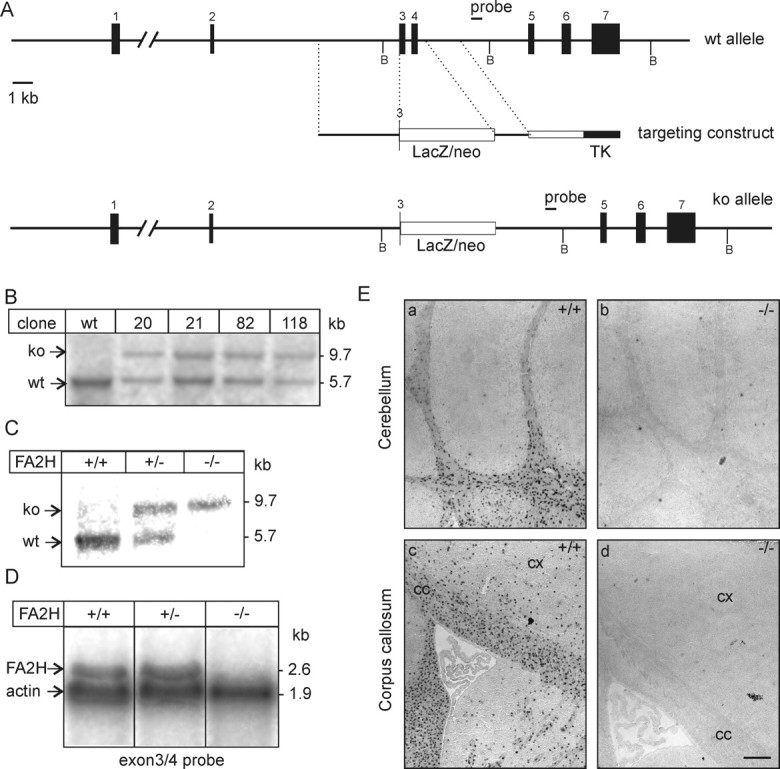
Generation of FA2H-deficient mice. A, Schematic representation of the FA2H wild-type allele, structure of the targeting vector, and targeted allele. Restriction sites for BglII (B) are indicated. B, Southern blot analysis of FA2H+/− ES cell clones, which survived G418/ganciclovir selection. Genomic DNA was digested with BglII and hybridized to the 3′ external probe, indicated in A. A nontargeted ES cell clone (wt) was included as control. ko, Knock-out. C, Southern blot analysis of FA2H alleles from mouse tail genomic DNA digested with BglII. D, Northern blot analysis of FA2H expression. Brain total RNA (20 μg/lane) of 6-week-old FA2H+/+, FA2H+/−, and FA2H−/− mice was separated in 1 m formaldehyde/1% agarose gels and transferred onto nylon membranes. Membranes were simultaneously hybridized to [32P]-labeled FA2H (derived from exons 3 and 4) and β-actin probes. Bound probes were visualized by autoradiography on x-ray films. E, In situ hybridization. Parasagittal sections of 4-week-old mouse brain of FA2H+/+ (a, c) and FA2H−/− (b, d) tissue sections were hybridized to a DIG-labeled FA2H-antisense probe. Shown are the hybridization signals in cerebellum (a, b) and forebrain (c, d). No FA2H mRNA-positive cells were observed in brain sections of FA2H−/− mice. cx, Cortex; cc, corpus callosum. Scale bar, 200 μm.
Table 1.
Genotype distribution in litters of heterozygous breedings
| Observed | Expected | |
|---|---|---|
| FA2H+/+ | 29 | 25.5 |
| FA2H+/− | 46 | 51 |
| FA2H−/− | 27 | 25.5 |
Figure 2.

β-Galactosidase expression in FA2H+/− mice. a, b, c, d, Parasagittal brain sections of brains from 1-week-old (a), 2-week-old (b), 3-week-old (c), and 4-week-old (d) FA2H+/− mice were stained for β-galactosidase using X-Gal. d, e, f, β-Galactosidase was absent from FA2H+/+ controls (e) and was increased in FA2H−/− mice (f) compared with FA2H+/− (d). h, Control in situ hybridization with DIG-labeled FA2H cRNA antisense probes gave identical expression patterns as the β-galactosidase. g, Control in situ hybridizations with sense probes gave no signals.
FA2H−/− mice lack 2-hydroxylated sphingolipids in brain and peripheral nerves
Lipids were isolated from brain (Fig. 3A,B), purified myelin (Fig. 3C,D), and sciatic nerves (Fig. 3E,F), and total lipids or lipids subjected to mild alkaline methanolysis were separated by TLC. These experiments showed that 2-hydroxylated sphingolipids were indeed absent from the CNS and peripheral nervous system of FA2H−/− mice (Fig. 3) and confirmed the hypothesis that FA2H is responsible for the synthesis of HFA–sphingolipids in the nervous system. In brain, myelin, and sciatic nerves of heterozygous FA2H+/− mice, the amount of HFA–GalC was significantly reduced compared with FA2H+/+ mice (Fig. 3B,D,F). Reduced HFA–GalC levels in heterozygous mice were compensated by an upregulation of nonhydroxy fatty acid (NFA)–GalC, and therefore the total amount of GalC in FA2H+/− mice was not significantly different from wild-type controls (data not shown). This also suggests that the amount of FA2H enzyme limits HFA–ceramide synthesis. In FA2H−/− mice, the increase in NFA–GalC did not fully compensate the loss of HFA–GalC, and the total amount of GalC was significantly reduced compared with wild-type and FA2H+/− mice (data not shown). Furthermore, HFA–sulfatide (lower band of the sulfatide double band in the TLCs shown in Fig. 3) was absent in FA2H−/− mice in brain, myelin, and sciatic nerves (Fig. 3A,C,E). However, because of an increase in NFA–sulfatide (upper band), total sulfatide levels were significantly reduced only in sciatic nerves (Fig. 1F). A separate quantification of NFA–sulfatide and HFA–sulfatide was not done because HFA–sulfatide and short-chain (C18:0/C20:0) NFA–sulfatide are in general not well separated by TLC. No significant changes in the concentration of gangliosides and sphingomyelin were observed. Furthermore, the protein profile of purified myelin as examined by silver staining of SDS polyacrylamide gels did not show differences between FA2H+/+ and FA2H−/− mice (data not shown).
Figure 3.
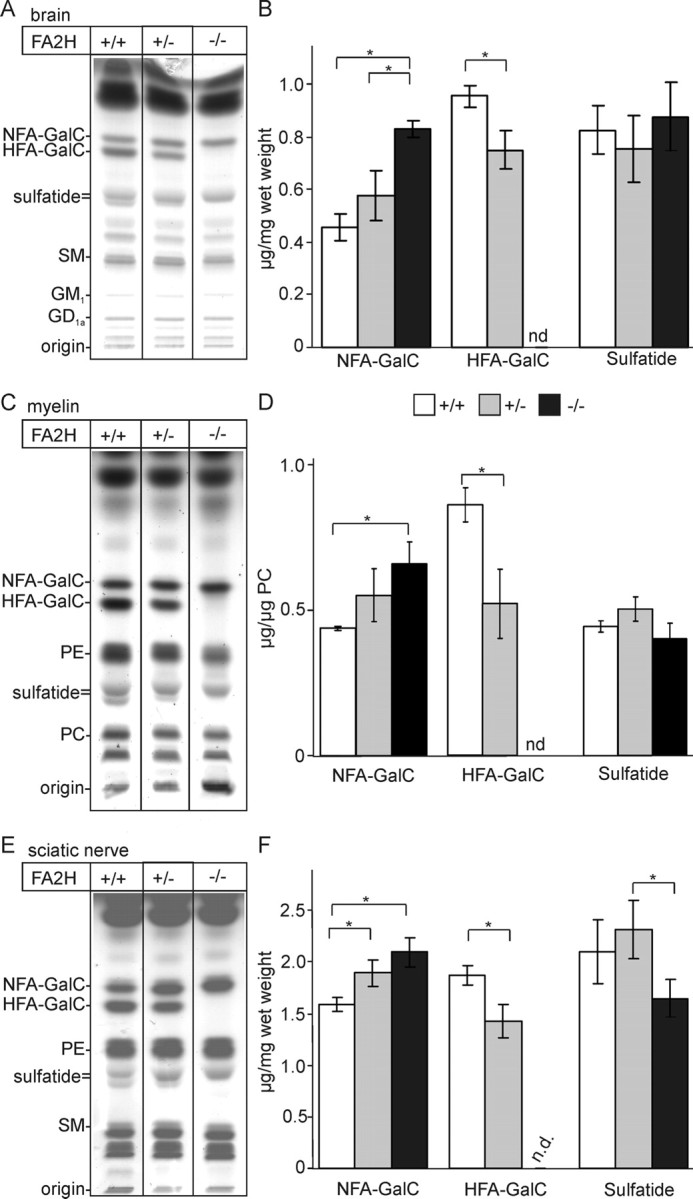
Lipid analysis. A, Lipids were isolated from total brain of FA2H+/+, FA2H+/−, and FA2H−/− mice at 28 d of age. Total lipids were subjected to mild alkaline methanolysis to remove phospholipids and separated by TLC on silica gel 60 TLC plates using chloroform/methanol/water (60:27:4; v/v/v) as solvent system (the amount of lipids corresponding to 1 mg wet weight of tissue was applied per lane). Lipids were visualized with cupric sulfate in aqueous phosphoric acid. B, NFA–GalC, HFA–GalC, and sulfatide were quantified by densitometry and are shown as microgram lipid per milligram wet weight (mean ± SD, n = 3). *p < 0.05, statistically significant difference (t test). C, Myelin was purified from total brains of FA2H+/+, FA2H+/−, and FA2H−/− mice at 10 weeks of age, and total lipids were isolated from the purified myelin. Lipids were separated by TLC (the amount of lipids corresponding to 0.5 mg of myelin was loaded) and quantified by densitometry. D, GalC and sulfatide levels were normalized to phosphatidyl choline (PC). E, Lipids were isolated from the sciatic nerves of FA2H+/+, FA2H+/−, and FA2H−/− mice at 28 d of age. F, Lipids were separated by TLC and quantified as described above (the amount of lipids corresponding to 0.5 mg wet weight of sciatic nerves was applied per lane). Positions of standard lipids run on the same TLC plate are indicated on the left (in A, C, E) (SM, sphingomyelin; gangliosides, GM1, GD1a; PE, phosphatidyl ethanolamine; PC, phosphatidyl choline). nd, Not detectable.
Absence of HFA–sphingolipids was further confirmed by matrix assisted-laser desorption ionization time of flight (MALDI-TOF) mass spectrometry (Fig. 4). Mass peaks corresponding to HFA–GalC (Fig. 4A, arrows) and HFA–sulfatide (Fig. 4C, arrows) were present in FA2H+/+ mice but were undetectable in FA2H−/− mice (Fig. 4B,D).
Figure 4.

MALDI-TOF mass spectrometry. A–D Alkaline stable lipids isolated from the brain of FA2H+/+ (A, C) and FA2H−/− (B, D) mice were subjected to MALDI-TOF mass spectrometry in negative (A, B) or positive (C, D) ion mode, to detect sulfatide (A, B) and GalC (C, D), respectively. Mass to charge ratios (m/z) of individual sulfatide and GalC species are shown in the insets. Hydroxylated sulfatide and GalC species were not detectable in FA2H−/− mice. Mass peaks that correspond to hydroxylated lipids and are missing in FA2H−/− mice are indicated by arrows in A and C. sb, Sphingosine base; fa, fatty acid; h, hydroxylated. Hydroxylated sphingolipids were undetectable in samples of FA2H−/− mice.
Normal oligodendrocyte differentiation in FA2H−/− mice
To detect deficits in oligodendrocyte differentiation or myelin formation, we examined expression of different myelin genes by means of immunofluorescence staining, Western blotting, in situ hybridization, and quantitative real-time reverse transcription (RT)-PCR in FA2H−/−, FA2H+/−, and wild-type littermates. MBP immunostaining of brain cryosections at postnatal day 10 (P10) and P19 did not show differences in staining intensities between FA2H−/− mice and wild-type littermates (data not shown). Furthermore, Western blot analysis revealed no significant differences in MBP protein levels at all examined time points between P10 and P70 (Fig. 5A,B). Similarly, no significant differences in the protein levels of other oligodendrocyte/myelin markers (CNP, L-MAG, and NCAM-120) were observed (Fig. 5C,D). As another sensitive marker of oligodendrocyte maturation, we determined the S-MAG/L-MAG mRNA ratio by RT-PCR. These two MAG variants are produced by alternative splicing and their ratio increases during oligodendrocyte differentiation (Fujita et al., 1998). No significant differences in the S-MAG/L-MAG ratio at P10 or P28 were detected between the three genotypes examined (Fig. 5E). The expression levels of the two major myelin genes, PLP and MBP, were examined by quantitative real-time RT-PCR at P10 and P28 and in adult (12-week-old) mice. No significant differences were observed in the expression level of PLP and MBP at all time points analyzed (Fig. 5F). In addition, we examined expression levels of the MAL protein, which has been found to be differentially expressed in several mouse lines exhibiting changes in the GalC and/or sulfatide level (Saravanan et al., 2004; Fewou et al., 2005). MAL expression levels were not significantly different between FA2H−/− and FA2H+/+ mice (Fig. 5F).
Figure 5.

Analysis of myelin markers. A, MBP Western blot analysis of total brain homogenates (50 μg/lane) from FA2H+/+, FA2H+/−, and FA2H−/− mice of different age groups. Membranes were stained with anti-MBP antiserum recognizing all MBP isoforms and anti-actin antibody to control for equal loading. B, Bound antibodies were detected by enhanced chemiluminescence. Densitometric quantification of MBP levels. Linear exposures of x-ray films were scanned, and the intensities of FA2H+/+ were set to 100%. Shown are the mean ± SD (n = 6 for P10; n = 3 for P19–P70). No significant differences between the three genotypes were observed. C, Western blot analysis of CNP. Brain homogenates of the indicated genotypes and ages were stained with anti-CNP antibody and anti-actin antibody as loading control. D, Western blot analysis of L-MAG and NCAM in 70-d-old brain homogenates. Relative protein levels (normalized to actin) of L-MAG and NCAM-120 are shown on the right (n = 3). E, Ratio of S-MAG and L-MAG expression levels at P10 and P28 was determined by RT-PCR. Intensities of ethidium bromide-stained PCR products were measured (n = 3). F, Quantitative real-time RT-PCR of MBP, PLP, and MAL expression (normalized to actin expression) at P10 and P28 and in adult FA2H−/− and FA2H+/+ mouse brains (12 weeks) showed upregulation of these myelin genes during the period of myelination, as expected. However, no statistically significant differences in their expression levels were observed at any time point examined (mean ± SEM; n = 3–5).
Differentiation of oligodendrocytes was further examined by in situ hybridization of parasagittal cryosections using DIG-labeled PDGFα receptor and PLP-specific cRNA probes (Fig. 6A,C) at P10 and P19. No significant differences in the number of oligodendrocyte progenitors (PDGFα receptor-positive) and mature oligodendrocytes (PLP-positive) in the forebrain, cerebellum, or brainstem were observed (Fig. 6B,D). We concluded that in vivo differentiation of oligodendrocytes was not significantly affected in FA2H−/− mice compared with wild-type controls.
Figure 6.
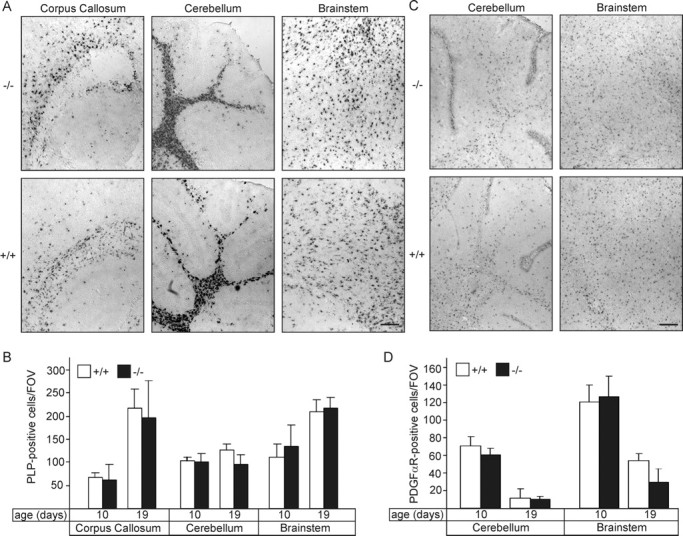
Oligodendrocyte differentiation in FA2H−/− mice. A, Parasagittal sections of brains from FA2H+/+ and FA2H−/− mice (10 and 19 d of age) were hybridized to a DIG-labeled PLP cRNA probe. Shown are representative stainings of the corpus callosum, cerebellum, and brainstem of 10-d-old mice. B, Quantification of PLP mRNA-positive cells [number of cells per field of view (FOV)] at P10 and P19 in the corpus callosum, cerebellum, and brainstem of FA2H+/+ and FA2H−/− mice (mean ± SEM; n = 3). No significant differences were found between FA2H+/+ and FA2H−/− mice. C, Parasagittal sections of brains from FA2H+/+ and FA2H−/− mice (10 and 19 d of age) were hybridized to a DIG-labeled PDGFα receptor cRNA probe. Shown are representative stainings of the cerebellum and brainstem of 10-d-old mice. D, Quantification of PDGFα receptor mRNA-positive cells (per field of view) at P10 and P19 in the cerebellum and the brainstem of FA2H+/+ and FA2H−/− mice (mean ± SEM; n = 3). No significant differences between genotypes were observed. Scale bars, 200 μm.
Myelin lacking 2-hydroxylated sphingolipids is resistant to extraction with CHAPS
It has been proposed that the 2-hydroxyl group of myelin sphingolipids could stabilize the myelin membrane. To test this possibility, purified myelin from 10-week-old FA2H−/− mice and wild-type controls was extracted with 20 mm CHAPS at 4°C followed by Optiprep density gradient (0, 30, 40%) centrifugation, as described by Simons et al. (2000). Gradient fractions were collected, and lipids were isolated and separated by TLC. The majority of myelin lipids, regardless of the presence or absence of 2-hydroxylated sphingolipids, was resistant to CHAPS extraction and floated at the 0–30% interphase (data not shown). Because higher temperatures should destabilize detergent-resistant membranes, the CHAPS extraction was repeated at higher temperatures (25 and 37°C). Again, the majority of myelin lipids was found in the CHAPS-insoluble membrane fractions (fractions 2 and 3, containing the 0–30% interphase) in both wild-type (Fig. 7A) and FA2H−/− (Fig. 7B) mice.
Figure 7.

Myelin lacking 2-hydroxylated sphingolipids is resistant to extraction with CHAPS. Purified myelin from 10-week-old wild-type (A) and FA2H−/− (B) mice was extracted with 20 mm CHAPS for 30 min at 37°C, followed by flotation in an Optiprep density gradient. Six fractions were collected from the top (fraction 1) to the bottom (fraction 6). Lipids were isolated from each fraction and analyzed by TLC. Lipids from myelin not extracted with CHAPS was loaded on the last lane (myelin). Shown is one representative experiment (of 5 independent experiments). Similar results were obtained when myelin was extracted at 4 or 25°C (data not shown). PE, Phosphatidylethanolamine; PC, phosphatidylcholine; SM, sphingomyelin.
Normal structure of the paranodal region in FA2H−/− mice
GalC and/or sulfatide are essential for stabilization of axon–glia contacts at the node of Ranvier (Dupree et al., 1998). Destabilization of these contact sites in CGT- and CST-deficient mice is accompanied by mislocalization of Caspr (Paranodin) (Dupree et al., 1998; Ishibashi et al., 2002). Because of the substantial changes in the GalC and sulfatide fatty acid composition, we examined whether the distribution of Caspr was altered in FA2H−/− mice. Caspr was examined in optic nerve cryosections (Fig. 8A) and teased fibers of sciatic nerves (Fig. 8C) of FA2H−/− mice (6–7 months of age) and wild-type littermates. The length and distance between pairs of Caspr clusters were measured. No significant differences were observed (Fig. 8B,D). In addition, clustering of sodium channels in FA2H−/− sciatic nerves was unaffected (Fig. 8C). These data suggest that the formation and maintenance of the paranodes in mice up to 6 months of age does not require 2-hydroxylated sphingolipids. Because of late-onset degeneration in aged FA2H−/− mice (see below), however, we cannot exclude structural changes of the paranodes in older FA2H−/− mice.
Figure 8.

Analysis of paranodal length of Paranodin/Caspr clusters. A, Longitudinal cryosections of optic nerves of FA2H+/+ and FA2H−/− mice (6–7 months of age) were stained for Paranodin/Caspr. B, The length and distance between pairs of Paranodin/Caspr clusters were measured. Shown are the mean ± SD (n = 20; 3 animals per genotype). No significant differences were observed. C, Teased fibers of sciatic nerves of FA2H+/+ and FA2H−/− mice were stained for Paranodin/Caspr (red) and sodium channel (green). D, The length and distance between pairs of Paranodin/Caspr clusters were measured. Shown are the mean ± SD (n = 7). No significant differences in the length and distance between Paranodin/Caspr clusters were detected. Scale bars: A, C, 5 μm.
Normal myelin structure in young mice but axon and myelin sheath degeneration in the spinal cord of aged FA2H−/− mice
Myelin ultrastructure of spinal cord white matter, optic nerves, and trigeminal and sciatic nerves were examined in FA2H−/−, FA2H+/−, and FA2H+/+ littermates at 4 weeks and 5 months of age (Fig. 9 and data not shown). Electron microscopy showed normal ultrastructure of myelin in the optic nerve (Fig. 9A,C) and peripheral nerves (Fig. 9D,H) of FA2H−/− mice compared with wild-type controls (Fig. 9B,E,G). Shown are electron micrographs of 5-month-old mice, but normal myelin structure was also found in 4-week-old FA2H−/− mice. Myelin was well compacted and displayed normal periaxonal spacing (Fig. 9C,F) and normal periodicity (Fig. 9C,H). It was also readily apparent that myelin thickness correlated with axon diameter in mice of all genotypes. Normal myelin structure was also observed in FA2H+/− mice (data not shown).
Figure 9.

Ultrastructural analysis of myelin. A, B, Electron microscopy of cross sections of the optic nerve of adult (5-month-old) FA2H−/− (A) and FA2H+/+ (B) mice. C, At higher magnification, normal structure of compacted myelin and periaxonal spacing were observed (C). D, E, Electron microscopy of the nervus trigeminus of 5-month-old FA2H−/− and FA2H+/+ mice. F, Myelinated axon of nervus ischiadicus of a FA2H−/− mouse displaying normal compacted myelin and periaxonal spacing. G, H, Normal compacted myelin in nervus trigeminus of FA2H−/− and FA2H+/+ mice, respectively. Scale bars: A, B, 1 μm; C, 100 nm; D, E, 10 μm; F–H, 200 nm.
Because other mouse mutants with deficiencies in myelin-specific genes exhibited late-onset degeneration (Griffiths et al., 1998; Lappe-Siefke et al., 2003), we examined aged FA2H−/− mice (18-month-old). Again, the overall phenotype was essentially normal in most brain regions (data not shown). Only in the spinal cord (with samples taken from the upper half of the cervical intumescence, which generally are less prone for artifacts attributable to suboptimal fixation) did we observe scattered profiles of degenerated myelin sheaths and axons (Fig. 10B,C,E). Morphologically, myelin sheaths in spinal cord featured signs of decompactation with accumulation of ill-defined material within the sheath wrappings. Axons appeared to collapse within even roughly preserved myelin sheaths and were often replaced by fibrillary material, which reacted with neither lipid stains nor alcian blue stains (Fig. 10D). Degeneration appear to predominantly affect large-caliber axons, but this should be taken with some caution because the degeneration process is likely to significantly distort axon and myelin size and shape. In addition, microscopically small tissue defects were observed in the white matter, without a clear boundary of, for example, astroglia, which could be the result of the loss of single or small groups of axons (Fig. 10F).
Figure 10.

Late-onset axonal degeneration in the spinal cord of FA2H−/− mice. A–F, The spinal cord white matter of 18-month-old mice features scattered degenerated myelin sheaths characterized by splitting of myelin lamellae (arrows in B and E), degenerated axons that often display accumulations of fibrillary material (circle in B, black asterisk in C) or collapsed axon cylinders (E, arrows), and microcystic spaces (B, white arrow in C, black asterisk in D and F; compare with controls in A; all photos taken close to the anterior horn). These microcystic spaces are sometimes surrounded by remnants of myelin sheaths (black asterisk in D), indicating that they represent leftovers of degenerated myelinated axons. Care was taken to document these pathologic changes only in the vicinity of well preserved myelin sheaths as an internal control for adequate fixation. F, “Microcysts” appear ultrastructurally as empty gaps without a discernable wall structure. Scale bars: A, 20 μm; B, C, 10 μm; D, E, 0.5 μm; F, 210 μm.
Late-onset axonal degeneration in the sciatic nerves of FA2H−/− mice
Axon diameters and myelin thickness were determined in toluidine blue-stained cross sections of sciatic nerves from 5-month-old FA2H−/− and FA2H+/− mice (Fig. 11A,B). The ratio of total fiber versus axonal cross-sectional area showed comparable values for both genotypes (FA2H+/−, 1.43 ± 0.04; FA2H−/−, 1.34 ± 0.03; mean ± SEM). Similarly, no significant differences were observed between FA2H−/− and FA2H+/+ mice in case of nervus trigeminus or nervus opticus (data not shown).
Figure 11.
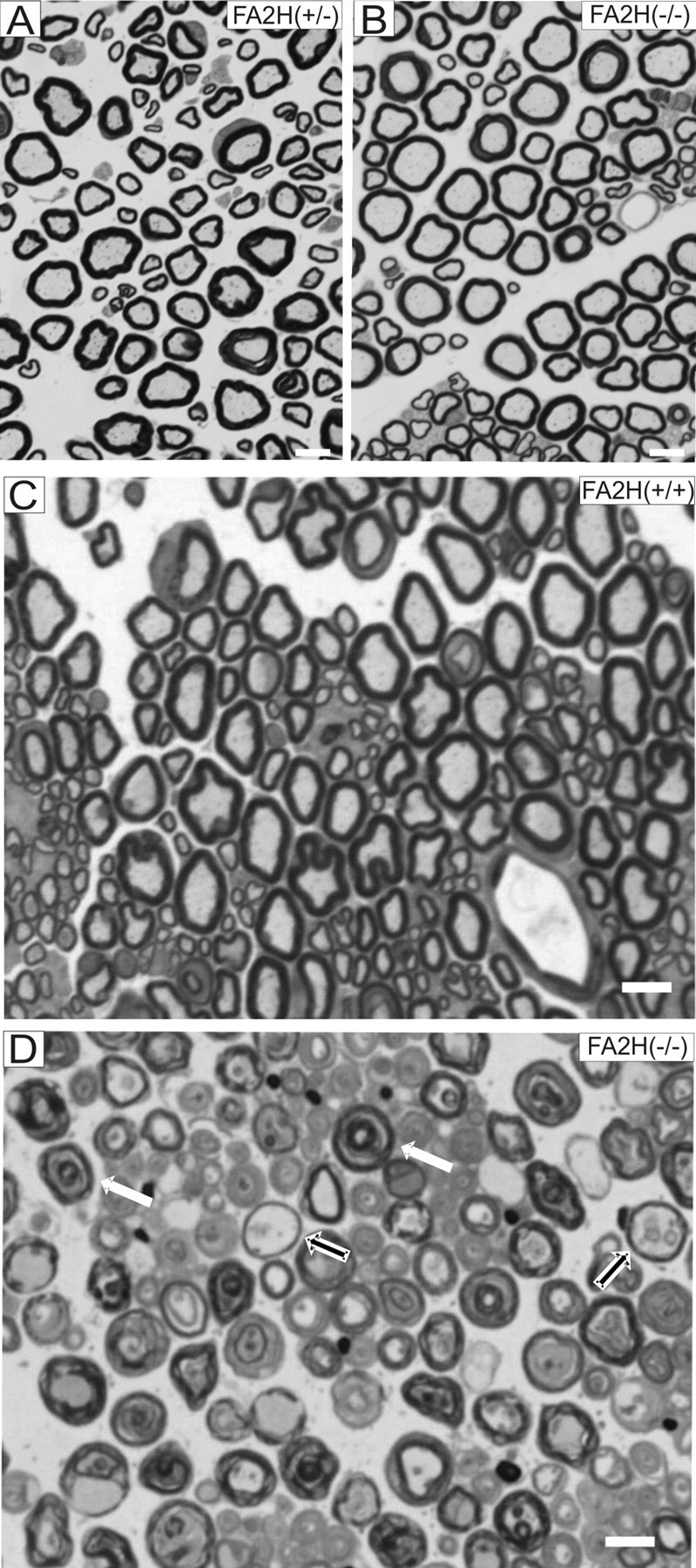
Late-onset axonal degeneration in sciatic nerves of FA2H−/− mice. Photomicrographs A and B show cross sections of the sciatic nerve of 5-month-old FA2H+/− (A) and FA2H−/− (B) mice, which were morphologically indistinguishable. Specifically, no evidence was found for alterations of the myelin sheaths; also unspecific signs for peripheral nerve damage such as macrophage invasion were absent. Sciatic nerves of late-adult FA2H−/− mice are characterized by a loss of stainability of myelin sheaths by toluidine blue (compare D with C) and, similar to the phenotype in the spinal cord, splitting of myelin lamellae into concentric rings (white arrows). Also, “empty” myelin sheaths as leftovers of preceding axon degeneration within the myelin sheaths (black arrows) are readily visible. Scale bars, 5 μm.
In contrast to the normal structure of myelinated axons of 5-month-old FA2H−/− mice, aged (18-month-old) FA2H−/− mice showed a massive axon and myelin sheath degeneration (Fig. 11D) with a notable loss of myelin stainability. Interestingly, neuronal muscle fiber lesions were still scant at that time, indirectly indicating that surviving axons within damaged myelin sheaths were still able to provide trophic support for muscle cells.
Behavioral studies and nerve conduction velocity measurement
Female FA2H−/− and wild-type mice at 4 months of age were examined by open-field and rotarod tests. Total activity and rearing activity, a measure for exploratory behavior (Van Meer and Raber, 2005), as well as the mean velocity were significantly reduced in FA2H−/− mice compared with wild-type controls (Table 2). In another set of experiments, we found a similar reduced activity of FA2H−/− mice compared with heterozygous FA2H+/− mice (data not shown). Rotarod experiments were performed to identify motor coordination deficits that would result from functional impairment of myelin. FA2H−/− and FA2H+/+ female mice were tested at 6 and 12 rpm for 1 min, and the number of cumulative falls was counted. This experiment revealed no statistically significant differences between FA2H−/− and wild-type controls (Table 2). However, FA2H−/− mice older than 22 months developed a progressive hindlimb paralysis and were unable to keep hold of the rotarod (data not shown), a finding in line with the late-onset myelin degeneration.
Table 2.
Summary of behavioral data
| FA2H+/+ | FA2H−/− | |
|---|---|---|
| Open field (5 min) | (n = 24) | (n = 25) |
| Distance traveled (cm) | 1358 ± 65a | 1053 ± 44* |
| Ambulatory time (s) | 45 ± 2 | 34 ± 2* |
| Rearing activity (s) | 14.3 ± 0.9 | 8.3 ± 0.7* |
| Time in center (s) | 105 ± 5 | 88 ± 6 (ns) |
| Mean velocity (m/s) | 33.4 ± 0.7 | 29.9 ± 0.7* |
| Rotarod (cumulative falls/min) | ||
| 4 months of age | (n = 11) | (n = 13) |
| 6 rpm | 0.3 ± 0.1 | 0.6 ± 0.3 (ns) |
| 12 rpm | 1.7 ± 0.6 | 2.7 ± 0.8 (ns) |
| 16–18 months of age | (n = 9) | (n = 9) |
| 6 rpm | 6.7 ± 2.3 | 3.4 ± 1.1 (ns) |
| 12 rpm | 6.8 ± 1.7 | 5.9 ± 1.2 (ns) |
ns, Not significant.
*p < 0.05, significantly different compared with wild type (ANOVA).
aMean ± SEM.
To confirm that the structural normal myelin in younger FA2H−/− mice also exhibited normal function, we measured nerve conduction velocities of isolated sciatic nerves. Compound action potentials were recorded from sciatic nerves isolated from adult (6- to 7-month-old) female FA2H−/− and FA2H+/+ mice (12 and 7 animals, respectively). Nerve conduction velocities were not significantly reduced in FA2H−/− mice (33.1 ± 2.0 m/s; n = 22) compared with wild-type controls (34.4 ± 2.9 m/s; n = 12). We therefore conclude that saltatory nerve conduction in mice does not require the presence of 2-hydroxylated sphingolipids in myelin, at least in mice up to an age of 7 months.
Discussion
In the nervous system, FA2H is specifically expressed in myelinating cells, and the postnatal upregulation of FA2H expression follows a similar time course as described for oligodendrocyte-specific genes involved in the synthesis of the myelin sheath, e.g., CGT and PLP (Eckhardt et al., 2005; Alderson et al., 2006). Furthermore, upregulation of the FA2H gene expression correlates with the increase in HFA during postnatal brain development (Alderson et al., 2006). FA2H−/− mice lack HFA–GalC and HFA–sulfatide in the brain and peripheral nerves, confirming the hypothesis that FA2H is responsible for HFA–sphingolipid synthesis in the nervous system. Oligodendrocytes lacking HFA–sphingolipids, however, differentiated normally in vivo, as shown by a normal time course of myelin gene expression during postnatal development and normal numbers of oligodendrocyte precursor cells and mature oligodendrocytes in the brain. Moreover, we found no structural abnormalities of myelin at the ultrastructural level in young FA2H−/− mice in both CNS and peripheral nervous system. Therefore, we conclude that HFA–sphingolipids are required for neither efficient oligodendrocyte differentiation nor the formation of normal compacted myelin. This finding is in agreement with the previous observation that lower vertebrates, such as urodeles, have normal compacted myelin independent of the presence or absence of HFA–sphingolipids (Ki et al., 1985; Kishimoto, 1986). Interestingly, however, urodeles lacking HFA–sphingolipids in myelin have reduced nerve conduction velocities, and, moreover, their myelin thickness appeared to be independent of the axon diameter (Ki et al., 1985), which led to the hypothesis that HFA–sphingolipids might be involved in regulating myelin thickness (Kishimoto, 1986). In contrast to this hypothesis, sciatic nerve conduction velocities were not significantly altered in FA2H−/− mice compared with wild-type controls, and we found a clear correlation between axon diameter and myelin thickness in mice lacking FA2H. Accordingly, the correlation between absence of HFA–sphingolipids and reduced nerve conduction velocity and axon diameter-independent myelin thickness in some lower vertebrates might be coincidental or attributable to species-specific functions of HFA–sphingolipids.
Mice overexpressing UDP-galactose:ceramide galactosyltransferase had reduced levels of HFA–GalC, whereas only NFA–GalC was increased (Fewou et al., 2005). Interestingly, in these mice, myelin was unstable and mice developed a progressive demyelination (Fewou et al., 2005). We previously hypothesized that this phenotype was attributable to the reduced HFA–GalC level. This, however, appears not to be the case. Particularly, FA2H+/− mice showed a similar reduction in HFA–GalC and change in the HFA–GalC/NFA-GalC ratio as CGT-transgenic mice without any detectable myelin abnormalities. Thus, myelin instabilities in CGT-transgenic mice might be attributable to an increased GalC turnover or the synthesis of other CGT products (e.g., monogalactosyl diglyceride).
Although FA2H−/− mice showed reduced activity in the open-field test at 4 months of age, we could not observe structural abnormalities in myelin of the CNS and peripheral nervous system at that age, which could explain this phenotype. It should be noted that HFA–sphingolipids are not detectable in neurons (Raghavan and Kanfer, 1972). Alderson et al. (2006) found low amounts of FA2H protein in microglia, but the FA2H protein was not detectable in primary cultured astrocytes (Alderson et al., 2006). It therefore seems not very likely that absence of FA2H in other cell types is responsible for the behavioral phenotype of FA2H−/− mice. Conversely, because FA2H is also expressed in other organs (e.g., in the intestinal tract and kidney), we cannot rule out the possibility that behavioral deficits are secondary to a functional impairment of one of these organs. At least the gross morphology of inner organs appeared to be normal in FA2H−/− mice (I. Zöller and M. Eckhardt, unpublished observation). In addition to the loss of HFA–sphingolipids, total GalC levels were significantly reduced in FA2H−/− mice compared with wild-type and FA2H+/− mice. Therefore, it is possible that the observed behavioral phenotype is attributable to reduced levels of GalC and not directly caused by the absence of HFA–sphingolipids. In that case, one should expect a related phenotype in other transgenic mice showing a similar reduction in HFA–sphingolipids but normal myelin structure, e.g., heterozygous CGT+/− mice (Bosio et al., 1996; Coetzee et al., 1996). Although no abnormal phenotype was observed in CGT+/− mice (Coetzee et al., 1996), behavioral studies have, to our knowledge, not been performed with heterozygous CGT mice.
A late-onset axonal degeneration, which we observed in aged FA2H−/− mice, has also been described in other mouse mutants deficient in myelin-specific genes, e.g., PLP (Griffiths et al., 1998; Edgar et al., 2004) and CNP (Lappe-Siefke et al., 2003). In contrast to these mouse mutants, we could, however, not find evidence for impaired axonal transport, i.e., axonal swellings resulting from accumulating organelles (Griffiths et al., 1998; Lappe-Siefke et al., 2003; Edgar et al., 2004). Immunostaining with antibodies directed against amyloid precursor protein did not detect abnormal axonal swellings in FA2H−/− mice (M. Meixner and M. Eckhardt, unpublished observation). Thus, axonal degeneration in aged FA2H−/− mice seems not to be caused by an impaired axonal transport.
At present, the physiological role of 2-hydroxylation remains unclear. Theoretically, 2-hydroxylation of sphingolipids might stabilize lateral interactions between sphingolipids and membrane–membrane interactions by presenting more hydroxyl groups for hydrogen bonds or complexation of ions. This hypothesis is supported by the observation that HFA–GalC and HFA–sulfatide form more stable calcium-dependent complexes than their nonhydroxylated counterparts (Menikh et al., 1997; Koshy et al., 1999). Biophysical studies on lipid monolayers showed that the phase behavior of ceramides is significantly affected by 2-hydoxylation (Singh et al., 1992). The galectin-4-dependent stabilization of so-called “super rafts,” detergent-resistant membranes that are detergent insoluble at 37°C (Braccia et al., 2003), appears to depend on the specific binding of galectin-4 to the 2-hydroxylated isoforms of GalC and sulfatide (Delacour et al., 2005). The potential stabilizing effect of 2-hydroxyl groups in sphingolipids, however, seems not to be important for stabilizing the myelin membrane, because FA2H−/− mice have normal compacted myelin. Furthermore, myelin lacking 2-hydroxylated sphingolipids was fully resistant to extraction with CHAPS, even at 37°C. Possibly a stabilizing effect of the additional hydroxyl group might be moderate and discernible only after challenging oligodendrocytes by additionally deleting other genes in FA2H−/− mice.
Deletion of oligodendrocyte-specific expressed genes can have a dramatic effect on axon maintenance and localization of neuronal proteins without strongly affecting myelin structure (Griffiths et al., 1998; Lappe-Siefke et al., 2003; Garcia-Fresco et al., 2006). Oligodendrocytes also are important for nodal sodium channel clustering, and this seems to be independent of myelin (Dupree et al., 2005). Combined deletion of FGF receptor-2 and CNP in oligodendrocytes led to massive hyperactivity, possibly as a result of an indirect effect of oligodendrocytes on neuronal function (Kaga et al., 2006). Similarly, impaired bidirectional signaling between neurons and oligodendrocytes could affect neuronal activity, leading to reduced activity of FA2H−/− mice in the open-field test, despite structural and functional normal myelin. Recent studies with membrane monolayers of a composition similar to the myelin outer leaflet showed that the size and number of membrane domains formed by GalC were significantly affected by 2-hydroxylation (Windschiegl and Steinem, 2006). This could potentially influence signaling between myelinating cells and neurons that depend on “lipid rafts.” The fibrillar material, which appears to accumulate in the myelin sheaths in the spinal cord of 18-month-old FA2H−/− mice, suggests that the transport and/or degradation of myelin components might be affected. This could, for example, affect the generation and release of myelin exosomes (Krämer-Albers et al., 2007; Trajkovic et al., 2008) and thereby the communication between myelinating cells and neurons. Because of the slow turnover of myelin components in adult mice, axonal degeneration and appearance of the fibrillar material might be the end point of a lengthy process that might already affect axonal function before morphological changes are apparent, leading to the behavioral changes observed in FA2H−/− mice.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft through Sonderforschungsbereich 645 of the University of Bonn. We thank Ivonne Becker, Birgit Rau, and M. Lindemann for expert technical assistance and Charlotte Schick for blastocysts injection. We thank Dr. Arthur Butt for MAG antibodies and Dr. Jean-Antoine Girault for the Paranodin antibody.
References
- Alderson NL, Rembiesa BM, Walla MD, Bielawska A, Bielawski J, Hama H. The human FA2H gene encodes a fatty acid 2-hydroxylase. J Biol Chem. 2004;279:48562–48568. doi: 10.1074/jbc.M406649200. [DOI] [PubMed] [Google Scholar]
- Alderson NL, Maldonado EN, Kern MJ, Bhat NR, Hama H. FA2H-dependent fatty acid 2-hydroxylation in postnatal mouse brain. J Lipid Res. 2006;47:2772–2780. doi: 10.1194/jlr.M600362-JLR200. [DOI] [PubMed] [Google Scholar]
- Altevogt BM, Kleopa KA, Postma FR, Scherer SS, Paul DL. Connexin29 is uniquely distributed within myelinating glial cells of the central and peripheral nervous systems. J Neurosci. 2002;22:6458–6470. doi: 10.1523/JNEUROSCI.22-15-06458.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann N, Pham-Dinh D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev. 2001;81:871–927. doi: 10.1152/physrev.2001.81.2.871. [DOI] [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Bosio A, Binczek E, Stoffel W. Functional breakdown of the lipid bilayer of the myelin membrane in central and peripheral nervous system by disrupted galactocerebroside synthesis. Proc Natl Acad Sci U S A. 1996;93:13280–13285. doi: 10.1073/pnas.93.23.13280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosio A, Büssow H, Adam J, Stoffel W. Galactosphingolipids and axono-glial interaction in myelin of the central nervous system. Cell Tissue Res. 1998;292:199–210. doi: 10.1007/s004410051051. [DOI] [PubMed] [Google Scholar]
- Braccia A, Villani M, Immerdal L, Niels-Christiansen LL, Nystrøm BT, Hansen GH, Danielsen EM. Microvillar membrane microdomains exist at physiological temperature. Role of galectin-4 as lipid raft stabilizer revealed by “superrafts.”. J Biol Chem. 2003;278:15679–15684. doi: 10.1074/jbc.M211228200. [DOI] [PubMed] [Google Scholar]
- Büssow H. Schwann cell myelin ensheating CNS axons in the nerve fiber layer of the cat retina. J Neurocytol. 1978;7:207–214. doi: 10.1007/BF01217919. [DOI] [PubMed] [Google Scholar]
- Coetzee T, Fujita N, Dupree J, Shi R, Blight A, Suzuki K, Suzuki K, Popko B. Myelination in the absence of galactocerebroside and sulfatide: normal structure with abnormal function and regional instability. Cell. 1996;86:209–219. doi: 10.1016/s0092-8674(00)80093-8. [DOI] [PubMed] [Google Scholar]
- Delacour D, Gouyer V, Zanetta JP, Drobecq H, Leteurtre E, Grard G, Moreau-Hannedouche O, Maes E, Pons A, André S, Le Bivic A, Gabius HJ, Manninen A, Simons K, Huet G. Galectin-4 and sulfatides in apical membrane trafficking in enterocyte-like cells. J Cell Biol. 2005;169:491–501. doi: 10.1083/jcb.200407073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupree JL, Coetzee T, Suzuki K, Popko B. Myelin abnormalities in mice deficient in galactocerebroside and sulfatide. J Neurocytol. 1998;27:649–659. doi: 10.1023/a:1006908013972. [DOI] [PubMed] [Google Scholar]
- Dupree JL, Mason JL, Marcus JR, Stull M, Levinson R, Matsushima GK, Popko B. Oligodendrocytes assist in the maintenance of sodium channel clusters independent of the myelin sheath. Neuron Glia Biol. 2005;1:1–14. doi: 10.1017/S1740925X04000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckhardt M, Yaghootfam A, Fewou SN, Zöller I, Gieselmann V. A mammalian fatty acid hydroxylase responsible for the formation of alpha-hydroxylated galactosylceramide in myelin. Biochem J. 2005;388:245–254. doi: 10.1042/BJ20041451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar JM, McLaughlin M, Yool D, Zhang SC, Fowler JH, Montague P, Barrie JA, McCulloch MC, Duncan ID, Garbern J, Nave KA, Griffiths IR. Oligodendroglial modulation of fast axonal transport in a mouse model of hereditary spastic paraplegia. J Cell Biol. 2004;166:121–131. doi: 10.1083/jcb.200312012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fewou SN, Büssow H, Schaeren-Wiemers N, Vanier MT, Macklin WB, Gieselmann V, Eckhardt M. Reversal of non-hydroxy:alpha-hydroxy galactosylceramide ratio and unstable myelin in transgenic mice overexpressing UDP-galactose:ceramide galactosyltransferase. J Neurochem. 2005;94:469–481. doi: 10.1111/j.1471-4159.2005.03221.x. [DOI] [PubMed] [Google Scholar]
- Fujita N, Kemper A, Dupree J, Nakayasu H, Bartsch U, Schachner M, Maeda N, Suzuki K, Popko B. The cytoplasmic domain of the large myelin-associated glycoprotein isoform is needed for proper CNS but not peripheral nervous system myelination. J Neurosci. 1998;18:1970–1978. doi: 10.1523/JNEUROSCI.18-06-01970.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Fresco GP, Sousa AD, Pillai AM, Moy SS, Crawley JN, Tessarollo L, Dupree JL, Bhat MA. Disruption of axo-glial junctions causes cytoskeletal disorganization and degeneration of Purkinje neuron axons. Proc Natl Acad Sci U S A. 2006;103:5137–5142. doi: 10.1073/pnas.0601082103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths I, Klugmann M, Anderson T, Yool D, Thomson C, Schwab MH, Schneider A, Zimmermann F, McCulloch M, Nadon N, Nave KA. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science. 1998;280:1610–1613. doi: 10.1126/science.280.5369.1610. [DOI] [PubMed] [Google Scholar]
- Harlow E, Lane D. Antibodies. A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1988. [Google Scholar]
- Honke K, Hirahara Y, Dupree J, Suzuki K, Popko B, Fukushima K, Fukushima J, Nagasawa T, Yoshida N, Wada Y, Taniguchi N. Paranodal junction formation and spermatogenesis require sulfoglycolipids. Proc Natl Acad Sci U S A. 2002;99:4227–4232. doi: 10.1073/pnas.032068299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi M, Kishimoto Y. Synthesis of cerebronic acid from lignoceric acid by rat brain preparation. Some properties and distribution of the alpha-hydroxylation system. J Biol Chem. 1973;248:4123–4130. [PubMed] [Google Scholar]
- Ishibashi T, Dupree JL, Ikenaka K, Hirahara Y, Honke K, Peles E, Popko B, Suzuki K, Nishino H, Baba H. A myelin galactolipid, sulfatide, is essential for maintenance of ion channels on myelinated axon but not essential for initial cluster formation. J Neurosci. 2002;22:6507–6514. doi: 10.1523/JNEUROSCI.22-15-06507.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaga Y, Shoemaker WJ, Furusho M, Bryant M, Rosenbluth J, Pfeiffer SE, Oh L, Rasband M, Lappe-Siefke C, Yu K, Ornitz DM, Nave KA, Bansal R. Mice with conditional inactivation of fibroblast growth factor receptor-2 signaling in oligodendrocytes have normal myelin but display dramatic hyperactivity when combined with Cnp1 inactivation. J Neurosci. 2006;26:12339–12350. doi: 10.1523/JNEUROSCI.3573-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ki PF, Kishimoto Y, Lattman EE, Stanley EF, Griffin JW. Structure and function of urodele myelin lacking alpha-hydroxy fatty acid-containing galactosphingolipids: slow nerve conduction and unusual myelin thickness. Brain Res. 1985;345:19–24. doi: 10.1016/0006-8993(85)90831-5. [DOI] [PubMed] [Google Scholar]
- Kishimoto Y. Phylogenetic development of myelin glycosphingolipids. Chem Phys Lipids. 1986;42:117–128. doi: 10.1016/0009-3084(86)90047-2. [DOI] [PubMed] [Google Scholar]
- Koshy KM, Wang J, Boggs JM. Divalent cation-mediated interaction between cerebroside sulfate and cerebrosides: an investigation of the effect of structural variations of lipids by electrospray ionization mass spectrometry. Biophys J. 1999;77:306–318. doi: 10.1016/S0006-3495(99)76891-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krämer-Albers EM, Bretz N, Tenzer S, Winterstein C, Möbius W, Berger H, Nave KA, Schild H, Trotter J. Oligodendrocytes secrete exosomes containing major myelin and stress-protective proteins: trophic support for axons? Proteomics Clin Appl. 2007;1:1446–1461. doi: 10.1002/prca.200700522. [DOI] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet. 2003;33:366–374. doi: 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- Magin TM, McWhir J, Melton DW. A new mouse embryonic stem cell line with good germ line contribution and gene targeting frequency. Nucleic Acids Res. 1992;20:3795–3796. doi: 10.1093/nar/20.14.3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus J, Popko B. Galactolipids are molecular determinants of myelin development and axo-glial organization. Biochim Biophys Acta. 2002;1573:406–413. doi: 10.1016/s0304-4165(02)00410-5. [DOI] [PubMed] [Google Scholar]
- Menikh A, Nyholm PG, Boggs JM. Characterization of the interaction of Ca2+ with hydroxy and non-hydroxy fatty acid species of cerebroside sulfate by Fourier transform infrared spectroscopy and molecular modeling. Biochemistry. 1997;36:3438–3447. doi: 10.1021/bi961869q. [DOI] [PubMed] [Google Scholar]
- Norton W, Cammer W. Isolation and characterization of myelin. In: Morell P, editor. Myelin. New York: Plenum; 1984. pp. 147–180. [Google Scholar]
- Norton WT, Poduslo SE. Myelination in rat brain: method of myelin isolation. J Neurochem. 1973;21:749–757. doi: 10.1111/j.1471-4159.1973.tb07519.x. [DOI] [PubMed] [Google Scholar]
- Raghavan S, Kanfer JN. Ceramide galactoside of enriched neuronal and glial fractions from rat brain. J Biol Chem. 1972;247:1055–1056. [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- Saravanan K, Schaeren-Wiemers N, Klein D, Sandhoff R, Schwarz A, Yaghootfam A, Gieselmann V, Franken S. Specific downregulation and mistargeting of the lipid raft associated protein MAL in a glycolipid storage disorder. Neurobiol Dis. 2004;16:396–406. doi: 10.1016/j.nbd.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Simons M, Krämer EM, Thiele C, Stoffel W, Trotter J. Assembly of myelin by association of proteolipid protein with cholesterol- and galactosylceramide-rich membrane domains. J Cell Biol. 2000;151:143–154. doi: 10.1083/jcb.151.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh D, Jarrell HC, Barber KR, Grant CW. Glycosphingolipids: 2H NMR study of the influence of ceramide fatty acid characteristics on the carbohydrate headgroup in phospholipid bilayers. Biochemistry. 1992;31:2662–2669. doi: 10.1021/bi00125a005. [DOI] [PubMed] [Google Scholar]
- Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, Schwille P, Brügger B, Simons M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science. 2008;319:1244–1247. doi: 10.1126/science.1153124. [DOI] [PubMed] [Google Scholar]
- van Echten-Deckert G. Sphingolipid extraction and analysis by thin-layer chromatography. Methods Enzymol. 2000;312:64–79. doi: 10.1016/s0076-6879(00)12900-3. [DOI] [PubMed] [Google Scholar]
- Van Meer P, Raber J. Mouse behavioural analysis in systems biology. Biochem J. 2005;389:593–610. doi: 10.1042/BJ20042023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windschiegl B, Steinem C. Influence of alpha-hydroxylation of glycolipids on domain formation in lipid monolayers. Langmuir. 2006;22:7454–7457. doi: 10.1021/la060146x. [DOI] [PubMed] [Google Scholar]
- Yaghootfam A, Gieselmann V, Eckhardt M. Delay of myelin formation in arylsulfatase A deficient mice. Eur J Neurosci. 2005;21:711–720. doi: 10.1111/j.1460-9568.2005.03891.x. [DOI] [PubMed] [Google Scholar]
- Yao JK, Rastetter GM. Microanalysis of complex tissue lipids by high-performance thin-layer chromatography. Anal Biochem. 1985;150:111–116. doi: 10.1016/0003-2697(85)90447-6. [DOI] [PubMed] [Google Scholar]
- Zöller I, Büssow H, Gieselmann V, Eckhardt M. Oligodendrocyte-specific ceramide galactosyltransferase (CGT) expression phenotypically rescues CGT-deficient mice and demonstrates that CGT activity does not limit brain galactosylceramide level. Glia. 2005;52:190–198. doi: 10.1002/glia.20230. [DOI] [PubMed] [Google Scholar]