

Abstract
Hypoxia inducible factor-1α (HIF-1α) is a key regulator of oxygen homeostasis, because it is responsible for the regulation of genes involved in glycolysis, erythropoiesis, angiogenesis, and apoptosis. In the CNS, HIF-1α is stabilized by insults associated with hypoxia and ischemia. Because its many target genes mediate both adaptive and pathological processes, the role of HIF-1α in neuronal survival is debated. Although neuronal HIF-1α function has been the topic of several studies, the role of HIF-1α function in astrocytes has received much less attention. To characterize the role of HIF-1α in neurons and astrocytes, we induced loss of HIF-1α function specifically in neurons, astrocytes, or both cell types in neuron/astrocyte cocultures exposed to hypoxia. Although loss of HIF-1α function in neurons reduced neuronal viability during hypoxia, selective loss of HIF-1 function in astrocytes markedly protected neurons from hypoxic-induced neuronal death. Although the pathological processes induced by HIF-1α in astrocytes remain to be defined, induction of inducible nitric oxide synthase likely contributes to the pathological process. This study delineates, for the first time, a cell type-specific action for HIF-1α within astrocytes and neurons.
Keywords: HIF-1, astrocyte, neuron, hypoxia, ischemia, neurotoxicity
Introduction
Hypoxic inducible factor 1α (HIF-1α), a master regulator of the cellular response to hypoxia, is a transcription factor that is stabilized and activated by hypoxia (Semenza, 2000). During hypoxia, HIF-1α protein is stabilized by a decrease in activity of prolyl hydroxylases, binds to its Per-Arnt-Sim binding partner HIF-1β, and translocates to the nucleus to bind to hypoxia-responsive cis-elements (HREs) (Bruick and McKnight, 2001; Jaakkola et al., 2001). HIF-1 activates multiple genes, including glycolytic enzymes, vascular endothelial growth factor, and the proapoptotic proteins Nip 3 and Nix (Bruick, 2000; Semenza, 2000). The role of HIF-1α in altering neuronal survival during hypoxia is controversial, because HIF-1α function likely mediates both adaptive and pathological functions. For example, a study examining the effects of HIF-1α overexpression in a hippocampal cell line suggests a pathological role for HIF-1α during oxidative stress, whereas HIF-1α overexpression suppressed cell death during DNA damage or endoplasmic reticulum stress (Aminova et al., 2004). Loss of HIF-1α function in the brain reduces neuronal death in the cortex and CA1 region of the hippocampus induced by hypoxia or transient global ischemia (Helton et al., 2005) but augments delayed neuronal death after stroke (Baranova et al., 2007). Although HIF-1α is expressed at higher levels in neurons than astrocytes (Chavez et al., 2006), HIF-1α induces multiple astrocyte-specific targets during hypoxia (Chavez et al., 2006; Rempe et al., 2007), implicating a role for HIF-1α in astrocytes as well as neurons.
To date, much effort has been expended to elucidate the molecular mechanisms within neurons that mediate neuronal death during stroke and hypoxia. Although this approach certainly has merit, astrocytes also play critical roles in health and disease. Multiple studies demonstrate that astrocytes are active partners with neurons and brain vasculature participating in two-way communication that alters neuronal physiology and survival (Chen and Swanson, 2003; Nedergaard et al., 2003). The influence of the astrocyte on neuronal survival during disease is complex, involving both adaptive and pathological functions.
The present study has focused on defining a cell type-specific role for HIF-1α during hypoxia in neurons and astrocytes. Using conditional loss of HIF-1α function in cocultures of neurons and astrocytes, we removed HIF-1α function in neurons, astrocytes, or both cell types. In neuron/astrocyte cocultures, transient exposure of cocultures to severe hypoxia induced delayed death selectively in neurons. Surprisingly, selective loss of HIF-1α function in astrocytes profoundly inhibits hypoxia-induced neuronal death, whereas loss of HIF-1α function in neurons increased neuronal susceptibility to hypoxia-induced damage. These results suggest the novel finding that the adaptive and pathological functions of HIF-1α are cell type specific.
Materials and Methods
Astrocyte and coculture preparation.
Astrocyte cultures were prepared from Po neonatal mice as detailed previously (Rempe et al., 2007). Briefly, the brain was removed and the cortex and hippocampus were dissected away from the brainstem. The telencephalon was placed into MEM (Invitrogen, San Diego, CA) supplemented with 6 g/L glucose, 1 mm sodium pyruvate, 6% horse serum, and 4% FBS and mechanically dissociated. In preparation for the experiments, astrocytes were plated (6.0 × 104 cells/well) into 12-well dishes and allowed to become confluent after which AraC was added to prohibit microglial proliferation. At 3 d in vitro (DIV), the astrocyte media was changed to serum free N2.1. Neurons were isolated from embryonic day 14 (E14) to E15 embryos. After removal of the meninges, the telencephalon was placed into trypsin containing EDTA for 20 min, washed three times in DMEM, and dispersed in N2.1. The neurons were then directly plated on the astrocytes (astrocyte 4–5 DIV) at 1.75 × 105 neurons/well. On neuronal DIV 4, cocultures were exposed for 48 h to tamoxifen (100 nm). In a subset of cultures, astrocyte cultures were prepared from CXCR3-green fluorescent protein (GFP) mice, which label microglia with GFP. In our astrocyte cultures, microglia composed <1% of the cells. When preparing cocultures, in some cases, neurons were obtained from GFP-mice (Jax), in which GFP is ubiquitously expressed (Okabe et al., 1997). Neurons composed 96% of GFP(+) cells in the cocultures (Fig. S1, available at www.jneurosci.org as supplemental material).
Transgenic mice.
Because loss of HIF-1α function is embryonic lethal, transgenic mice, that harbor a floxed exon2 of HIF-1α (HIF-1α f+/f+) gene, were used previously (Ryan et al., 2000). These HIF-1α f+/f+ mice were bred against tamoxifen-inducible B6.Cg-Tg(cre/Esr1) 5Amc mice (referred to as “ESRCre”; The Jackson Laboratory, Bar Harbor, ME), in which cre recombinase is activated by tamoxifen (Hayashi and McMahon, 2002).
Hypoxia treatment paradigms/assessing neuronal viability.
On neuronal DIV 9, cocultures were exposed to hypoxia in a triple gas incubator that replaces oxygen with nitrogen to achieve 0.5% oxygen (Thermo Electron, Waltham, MA). After hypoxia, neuronal viability was assessed by counting (>2000 cells/data point) phase bright (live neurons) to those labeled with propidium iodide (PI) (2 μg/ml) to mark dead cells. In some cases, media was transferred from cocultures after hypoxia and 6 h of reperfusion onto cocultures that had not experienced hypoxia. Only 80% of the media was exchanged, because neuronal death was induced with complete volume exchanges.
Protein isolation/Western blots.
As detailed previously (Rempe et al., 2007), enriched fractions of nuclear proteins were isolated using a rapid method (Andrews and Faller, 1991). Samples were stored at −80°C and proteins separated under denaturing conditions on PAGE gels, transferred to polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA) for Western blotting (HIF-1α, Novus NB100–449, 1/500).
RNA purification, cDNA, quantitative PCR, and genomic DNA PCR screen.
We followed the manufacturer directions for RNA extraction (Qiagen, Valencia, CA), cDNA synthesis (super script III), and quantitative PCR (TaqMan primer-probe; 7300; Applied Biosystems, Foster City, CA) as detailed previously (Rempe et al., 2007). In particular, we used primer/probe sets that were specific for hexokinase 2 (HKII) or exon 2 of HIF-1α (supplemental Table 1, available at www.jneurosci.org as supplemental material) to quantify HKII or HIF-1α transcript. To examine for recombination of loxP sites in cocultures, genomic DNA was extracted, and primer sets (supplemental Table 2, available at www.jneurosci.org as supplemental material) were used to screen for loss of the floxed DNA.
Results
Neurons in coculture with astrocytes die in a delayed manner after severe hypoxia exposure
The principal goal of this study is to evaluate the impact of loss of HIF-1α function selectively within neurons or astrocytes on hypoxia-induced neuronal death. First, we examined the time course of neuronal death during/after hypoxia in cocultures of neurons and astrocytes. Neuronal viability was quantified after different intervals of “reperfusion” (normoxia) after an exposure to hypoxia (0.5%) (Fig. 1A). Transient exposure to hypoxia induced HIF-1α protein and a delayed neuronal death (Fig. 1B–D), whereas astrocytes remained viable (Fig. S2, available at www.jneurosci.org as supplemental material). Immediately after hypoxia, only a small portion of neurons had died (20%), whereas the majority died 12–24 h after hypoxia (Fig. 1C). HIF-1α protein abundance was markedly induced in cocultures immediately after the hypoxia exposure (Fig. 1D) but returned to baseline by 6 h of reperfusion.
Figure 1.
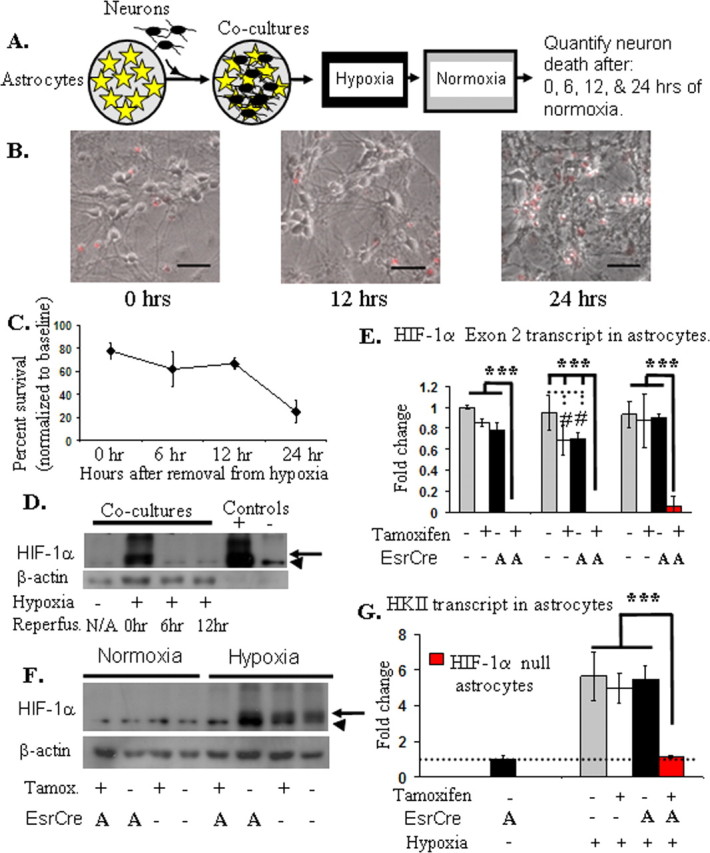
A, Neurons cocultured with astrocytes die in a delayed manner after transient hypoxia: schematic diagram of experimental protocol. Neurons (black cells) are plated directly on the monolayer of astrocytes (yellow stars) and allowed to mature for 9 d before being subjected to hypoxia. After hypoxia (12 h, 0.5% oxygen), the cultures were placed in normoxia, and cell death was quantified by counting PI (+) neurons (red fluorescence). B, C, Superimposed phase contrast/fluorescent images (B) demonstrate that most neurons are viable immediately after hypoxia (scale bars, 20 μm), yet profound neuronal death is appreciated 24 h after the exposure to hypoxia (C). Hypoxia-induced HIF-1α protein in cocultures by Western blot (D, F; arrow, hypoxia-induced HIF-1α; arrowhead, nonspecific band; Reperfus., reperfusion). D, HIF-1α protein is increased immediately after hypoxia but returns to baseline after 6 h of reperfusion (normoxia). HN33 (a transformed neuronal cell line) cell lysates (+) or (−) cobalt served as controls. E–G, The addition of tamoxifen (100 nm) to astrocyte cultures derived from HIF-1α f+/f+::ESRCre mice markedly decreases HIF-1α transcript (E) and protein (F) and inhibits hypoxia-induced expression of HKII (G). Three separate replicates (3 samples/replicate) of exon 2 transcript abundance are shown in E (ANOVA; ***p < 0.001; #p < 0.01). In this and subsequent figures: A, astrocyte; N, neuron as cell type for EsrCre expression; error bars are SD, and flat bars above bar graphs indicate no significant difference.
Inducing selective loss of HIF-1α function in astrocytes or neurons in primary cocultures
Mice with a floxed HIF-1α allele were crossed with EsrCre mice allowing for conditional loss of HIF-1α function when exposed to tamoxifen. In HIF-1α f+/f+::ESRCre astrocyte cultures, tamoxifen induced >95% loss of HIF-1α exon 2 transcript (Fig. 1E), and HIF-1α protein was markedly reduced (Fig. 1F). Loss of HIF-1α function was confirmed by the fact that hypoxia-induced hexokinase 2 (HKII) transcript abundance, which is dependent on HIF-1 function (Rempe et al., 2007), was eliminated in HIF-1α null cultures (Fig. 1G). In contrast, tamoxifen had minimal effect on HIF-1α abundance or function in astrocyte cultures derived from HIF-1α f+/f+ mice, which lack EsrCre. Similar to what we demonstrated in primary astrocyte cultures, marked loss of HIF-1α protein expression is produced in primary neuronal cultures derived from HIF-1α f+/f+::ESRCre mice (Fig. S3A,B, available at www.jneurosci.org as supplemental material).
Selective loss of HIF-1α function was achieved in either neurons or astrocytes in coculture by combining cells of different genotypes. For example, selective loss of HIF-1α in neurons was provided by culturing HIF-1αf+/f+::ESRCre neurons with C57BL/6J astrocytes and treating with tamoxifen (Fig. 2A). To ensure that tamoxifen induced recombination of the floxed allele in our bigenic cultures, we used PCR to screen for recombination of the floxed exon 2 of HIF-1α. Because the smaller (recombined) DNA product is preferentially produced during PCR, this was a sensitive means to ensure that DNA recombination occurred within the bigenic cultures in a single-cell type (Fig. S3C,D, available at www.jneurosci.org as supplemental material).
Figure 2.
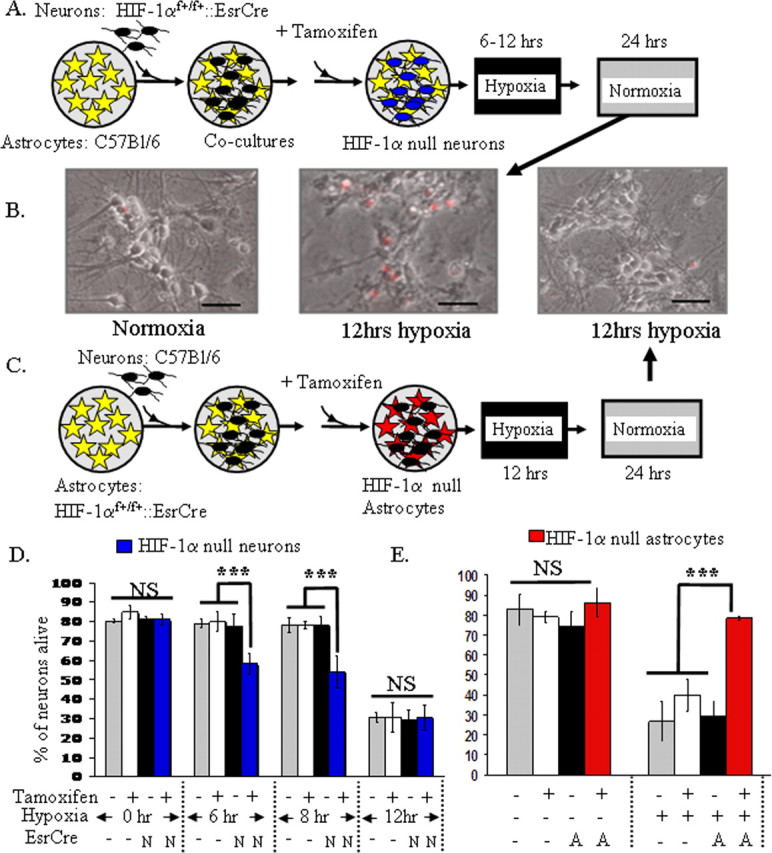
Loss of HIF-1α function in astrocytes and neurons has opposite effects on hypoxia-induced neuronal death. A, A schematic diagram illustrating that neurons derived from HIF-1αf+/f+::ESRCre embryos were plated directly onto a monolayer of astrocytes, which were derived from C57BL/6J mice. Using this approach, tamoxifen induced loss of HIF-1 function selectively in neurons in cocultures (indicated by blue cells). B, As can be appreciated in the phase contrast images, hypoxia (12 h, 0.5% O2) induced death in the vast majority of neurons. Hypoxia for shorter time periods demonstrated that loss of HIF-1 in neurons enhances neuronal susceptibility to hypoxia-induced neuronal death (D; ***p < 0.001, ANOVA). Scale bars, 20 μm. C, A schematic diagram illustrating that tamoxifen induces loss of HIF-1 function selectively in astrocytes (red cells) in cocultures in which astrocytes are derived from HIF-1αf+/f+::EsrCre mice. Loss of astrocyte HIF-1α markedly protected the overlying neurons from hypoxia-induced cell death (B, E) (***p < 0.001, ANOVA; NS, nonsignificant difference). These experiments with selective loss of HIF-1 in neurons or astrocytes are representative examples of the replicates.
Loss of neuronal HIF-1α increases neuronal susceptibility to hypoxia-induced death
Studies suggest both pathological and adaptive roles for HIF-1α in neurons (Halterman et al., 1999; Aminova et al., 2004; Helton et al., 2005). Thus, we wanted to examine whether selective loss of HIF-1α function in neurons alters hypoxia-induced neuronal death. Loss of HIF-1α function in neurons elicited neuronal death with 6–8 h of hypoxic exposure, which elicited no discernable neuronal death in controls (Fig. 2D). When comparing data from the 6 and 8 h time points (two independent experiments), loss of HIF-1 function resulted in an average increase in neuronal death of 23% (±6%). When exposure to hypoxia was extended to 12 h, marked neuronal death was observed in all conditions without any change in viability with loss of HIF-1 function (five independent experiments).
Loss of HIF-1α within astrocytes markedly protects neurons from hypoxia-induced cell death
Surprisingly, the selective loss of HIF-1 function in astrocytes had a profound effect on neuronal viability. Cocultures were prepared similar to that detailed above except that astrocytes were derived from HIF-1α f+/f+::ESRCre mice, whereas neurons were obtained from C57BL/6J mice (Fig. 2C). In this paradigm, tamoxifen induced selective loss of HIF-1 function in astrocytes. After exposure to transient hypoxia, neurons in cocultures with astrocytes lacking HIF-1α function were markedly protected from hypoxia-induced cell death (Fig. 2B,E) (seven independent experiments). This neuronal protection was not mediated by tamoxifen as the application of tamoxifen in the absence of EsrCre expression was not protective. Because HIF-1α induces multiple glycolytic enzymes, we examined media pH and glucose concentrations in the absence of HIF-1α function in astrocytes. Media pH (7.2–7.3) and glucose concentration (data not shown) were not altered in cocultures by loss of HIF-1α function in astrocytes.
Astrocyte HIF-1 likely mediates neuronal death by inducible nitric oxide synthase-dependent and -independent pathways
An HIF-1 target in astrocytes that may induce neuronal death is inducible nitric oxide synthase (iNOS). In astrocyte cultures, iNOS transcript is not detected in normoxia but markedly increased by hypoxia in an HIF-1-dependent process (Fig. 3A). Similar to treatment with the nonspecific NOS inhibitor N-ω-nitro-l-arginine methyl ester (l-NAME), the specific iNOS inhibitor aminoguanidine (AG; 25–100 μm) significantly attenuated hypoxia-induced neuronal death in cocultures (Fig. 3B). Because microglia constitute <1% of the cells in our cultures (see Materials and Methods), the iNOS is not derived from microglia but instead is likely released from astrocytes.
Figure 3.

A, Astrocytic HIF-1α induces neuronal death by both iNOS-dependent and -independent mechanisms. iNOS was greatly induced by hypoxia in astrocytes, which was essentially eliminated with loss of HIF-1 function (ANOVA; ***p < 0.001; #p < 0.05). B, AG, a selective iNOS inhibitor, protects neurons from hypoxia-induced cell death (***p < 0.001; ANOVA) in cocultures in a dose-dependent manner with a similar efficacy as l-NAME, a nonselective NOS inhibitor. Greater neuronal protection is provided by 50–100 μm AG and l-NAME compared with 25 μm AG (###p < 0.001; ANOVA). Compared across multiple independent experiments, loss of HIF-1 in astrocytes is more protective compared with treatment with l-NAME (D) (p < 0.01; comparing y-intercepts following linear regression analysis). To control for a variable amount of hypoxia-induced neuronal death between experiments, the percentages of live neurons in the test groups (red diamonds, HIF-1 loss; blue box, l-NAME) were plotted as a function of neuronal death observed in hypoxia controls (black triangles). C, Experimental methods for media transfer experiments. After hypoxia and reperfusion for 6 h, media from cocultures, which in some cases lacked astrocyte HIF-1 function (red stars/astrocytes), was placed onto normoxia wild-type cocultures, and neuronal death was measured (C). E, Modest (∼20%) cell death was induced by media that was obtained from hypoxia/reperfusion cultures, which was eliminated by loss of HIF-1 function in astrocytes (***p < 0.001; ANOVA).
To evaluate the efficacy of iNOS inhibition versus loss of astrocyte HIF-1 function in attenuating hypoxia-induced neuronal death, we compared the percentage of neuronal death (normalized to normoxia controls) between the two “treatment” groups. In each independent experiment, both normoxia and hypoxia nontreatment controls were examined. The percentage of neurons viable in normoxia (normoxia control) was not significantly different between the astrocyte HIF-1 null group (72 ± 5.7%; n = 7 independent experiments) and the l-NAME group (79 ± 9%; n = 3 independent experiments) (p = 0.15; independent t test), suggesting comparable health of the cocultures. The degree of hypoxia-evoked neuronal death varied somewhat among experiments. The greater the neuronal death induced by hypoxia, the less effective the intervention at reducing neuronal death. Thus, to control for degree of neuronal death, we plotted the percentage of neuronal survival for each treatment as a function of the neuronal death evoked in hypoxia controls (Fig. 3D). When graphed using this approach, the two groups are clearly distinct. Although both interventions protected neurons, loss of astrocyte HIF-1 function was more effective compared with treatment with l-NAME (p < 0.01; linear regression analysis comparing y-intercepts of two lines).
Because nitric oxide has a short half-life in media, media exchanges were performed to examine whether other HIF-1 inducible factors are released by astrocytes and contribute to neuronal death. After exposure to hypoxia, media from cocultures, which did or did not lack HIF-1α function in astrocytes, was placed onto wild-type control cocultures (C57BL/6J) (Fig. 3C). After hypoxia and 6 h of reperfusion (normoxia), media was transferred from the test cocultures to wild-type cocultures that were cultured only in normoxia. Modest cell death was induced by the media that was obtained from hypoxia/reperfusion cultures, which was attenuated by loss of HIF-1 function in astrocytes (Fig. 3E).
Protective role of HIF-1 in neurons remains during severe prolonged hypoxia
As stated above, when cocultures are exposed to hypoxia for 12 h, marked neuronal death was observed in the cocultures, and loss of HIF-1α function in neurons did not alter neuronal viability (Fig. 2). Yet, it was unclear whether HIF-1 loses its protective role for neurons under these conditions because of enhanced proapoptotic function or whether HIF-1-dependent protection cannot be appreciated because of the overwhelming neuronal death induced by this paradigm. To evaluate this question, we examined neuronal death when HIF-1 function was selectively lost in astrocytes, or lost in both astrocytes and neurons using similar methodology as detailed above. After exposure to transient hypoxia, neurons in cocultures with loss of HIF-1α function in both astrocytes and neurons were protected compared with controls. However, the protection was significantly less (19.9 ± 4% normalized to controls) than that observed with loss of HIF-1α function in astrocytes alone (Fig. 4C). Thus, HIF-1α maintains its protective role in neurons under conditions of prolonged hypoxia.
Figure 4.
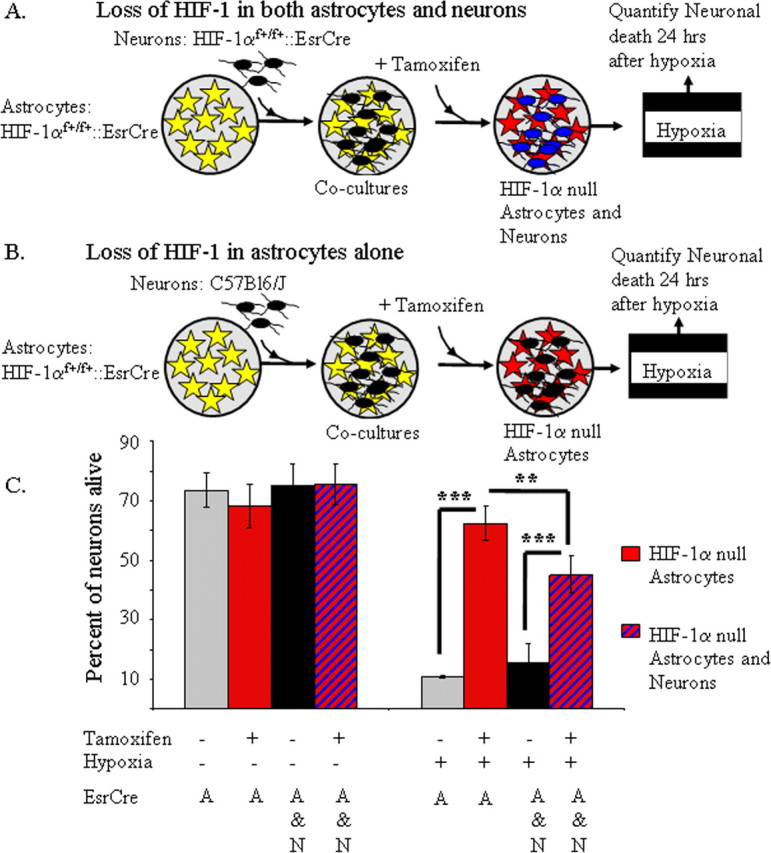
Loss of HIF-1α function in both astrocytes and neurons reveals an adaptive role for HIF-1α function in neurons during hypoxic stress. A, A schematic diagram illustrating that neurons derived from HIF-1αf+/f+::ESRCre embryos were plated directly onto a monolayer of astrocytes, which were also derived from HIF-1αf+/f+::ESRCre mice. Using this approach, tamoxifen induced loss of HIF-1 function in both neurons (blue) and astrocytes (red). B, A schematic diagram of the approach used to induce selective loss of HIF-1 function in astrocytes. C, In agreement with the data in Figure 2, loss of astrocyte HIF-1α markedly protected the overlying neurons from hypoxia-induced cell death (***p < 0.001; ANOVA). Although loss of HIF-1 in both neurons and astrocytes (red and blue striped) also attenuated neuronal death, neuronal survival was significantly less than the loss of HIF-1α in astrocytes alone (red; C) (**p < 0.01; ANOVA). The experiment with loss of HIF-1α in both cell types is a representative example of three independent separate replicates.
Discussion
The role of HIF-1α as an adaptive versus pathological modifier of neuronal survival is likely dependent on the insult that induces neuronal death, and the cell type in which it is induced. Although the role of HIF-1α in neurons has been addressed, its role in astrocytes remains primarily undefined. We examined the impact of selective loss of HIF-1α function in neurons or astrocytes on hypoxia-induced neuronal death in astrocyte/neuron cocultures. First, we determined that transient hypoxia induced delayed neuronal death in cocultures. Interestingly, selective loss of HIF-1α function in astrocytes markedly reduced hypoxia-induced neuronal death in cocultures, whereas loss of HIF-1α function in neurons increased susceptibility to hypoxia-induced neuronal death.
Recent work by two different groups found divergent effects on neuronal viability with loss of HIF-1α function in the brain (Helton et al., 2005; Baranova et al., 2007). In both cases, we used floxed HIF-1 mice crossed with mice expressing cre recombinase driven by a calcium/calmodulin-dependent kinase promoter, CAMCRE. In these mice, cre recombinase is reported to be expressed primarily in neurons. Therefore, in theory, these mice offer the opportunity to determine neuronal specific loss of HIF-1 function and its impact on hypoxia and ischemia-induced neuronal death. In the first study, mice with loss of HIF-1 function had less neuronal death in the cortex and CA1 subfield of the hippocampus after being subjected to exposure to hypoxia or transient global ischemia (Helton et al., 2005). Moreover, this study identified several new putative proapoptotic HIF-1 targets that may have contributed to this effect. The second study examined the loss of HIF-1 function in stroke (Baranova et al., 2007). Interestingly, loss of HIF-1 function in neurons resulted in increased infarct volume when examined 3–4 d after stroke but not 24 h after stroke. Thus, HIF-1 function in neurons appeared to protect against delayed neuronal death in the peri-infarct tissue after stroke. Do our in vitro observations address these divergent observations? Interestingly, the study describing a pathological role for HIF-1 reported >92% excision of the floxed DNA based on a Southern blot (Helton et al., 2005). Moreover, they describe changes in gene expression with loss of HIF-1 function in aquaporin 4, which is specifically expressed in astrocytes. In contrast, in the study describing a protective role for HIF-1, the degree of recombination of the floxed DNA was more modest and loss of HIF-1 function was specific for neurons (Baranova et al., 2007). Therefore, it is plausible that loss of HIF-1 function involved non-neuronal cell types, perhaps astrocytes, in the study by Helton et al. (2005), which may have resulted in an overall pathological role for HIF-1. Moreover, our observation of an adaptive role for HIF-1 in neurons agrees with a protective neuronal function for HIF-1 during stroke (Baranova et al., 2007). Yet, admittedly, several other factors could account for the differences between the above studies, including the difference in pathological stimuli, known in cells lines to induce divergent roles of HIF-1 (Aminova et al., 2004). In vivo studies using similar hypoxic and ischemic insults with selective loss of function in each cell type will be required to definitively address this question.
The finding that loss of HIF-1 function in astrocytes so markedly protected neurons from hypoxia is consistent with a growing body of literature that suggests that astrocyte function is an important contributor to neuronal survival in disease. For example, recent work demonstrated a pathological role for astrocytes in the neurodegenerative disease amyotrophic lateral sclerosis (Nagai et al., 2007). The mechanism by which astrocyte HIF-1 function induces neuronal death remains primarily undetermined. One of the most important determinants of neuronal viability during ischemia is the ability of astrocytes to maintain glutamate transport. Reversal of glutamate transport out of astrocytes into the extracellular space occurs in conditions of severe ischemia when energy stores in astrocytes are severely compromised (Anderson and Swanson, 2000). However, in the present study, glucose remained available to the cells, and astrocyte viability was not compromised, conditions under which glutamate uptake is only modestly reduced (Swanson, 1992). Because glycolytic targets are induced by hypoxia in a HIF-1-dependent manner in astrocytes (Rempe et al., 2007), loss of HIF-1 function may impede glycolysis in astrocytes providing more glucose to neurons and producing less lactic acid secretion and acidosis. Yet, the pH in astrocyte cultures remained stable regardless of the status of HIF-1 function, and the experiments were performed in an excess of glucose, making these possibilities unlikely.
A putative HIF-1 target could induce neuronal death through the induction of nitric oxide by iNOS. In fact, iNOS was induced in astrocytes by a HIF-1-dependent mechanism, and hypoxia-induced neuronal death was reduced by AG and l-NAME (Fig. 3). Yet, inhibiting iNOS was less effective at reducing neuronal death compared with loss of HIF-1 function in astrocytes, suggesting that other factors also contribute to HIF-1-dependent neuronal death. Media exchange from cocultures after hypoxia/reperfusion also induced modest neuronal death in control cocultures in an HIF-1-dependent manner. Nitric oxide is unlikely to have significantly contributed to cell death induced by media exchange because of its short half-life in media. Thus, multiple HIF-1 regulated targets in astrocytes likely contribute to the neuronal toxicity. Regardless, this unique finding identifies an important new molecular mechanism by which astrocytes modify neuronal viability during pathological states.
Footnotes
This work was supported in part by National Institute of Neurological Disorders and Stroke Grants 5K08NS046633, 1R01NS054192, and 1P01NS050315, and American Heart Association Predoctoral Award 0615696T (G.V.). We thank Dr. Randall Johnson for providing the floxed HIF-1α mice. We thank Drs. Marc Halterman and Maiken Nedergaard for helpful comments regarding this manuscript. Finally, we thank Maria Jepson, Landa Prifti, and Rita Giuliano for their excellent technical assistance.
References
- Aminova LR, Chavez JC, Lee J, Ryu H, Kung A, Lamanna JC, Ratan RR. Prosurvival and prodeath effects of hypoxia-inducible factor-1alpha stabilization in a murine hippocampal cell line. J Biol Chem. 2004;280:3996–4003. doi: 10.1074/jbc.M409223200. [DOI] [PubMed] [Google Scholar]
- Anderson CM, Swanson RA. Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia. 2000;32:1–14. [PubMed] [Google Scholar]
- Andrews NC, Faller DV. A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucl Acids Res. 1991;19:2499. doi: 10.1093/nar/19.9.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranova O, Miranda LF, Pichiule P, Dragatsis I, Johnson RS, Chavez JC. Neuron-specific inactivation of the hypoxia inducible factor 1α increases brain injury in a mouse model of transient focal cerebral ischemia. J Neurosci. 2007;27:6320–6332. doi: 10.1523/JNEUROSCI.0449-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruick R. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad Sci USA. 2000;97:9082–9087. doi: 10.1073/pnas.97.16.9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- Chavez JC, Baranova O, Lin J, Pichiule P. The transcriptional activator hypoxia inducible factor 2 (HIF-2/EPAS-1) regulates the oxygen-dependent expression of erythropoietin in cortical astrocytes. J Neurosci. 2006;26:9471–9481. doi: 10.1523/JNEUROSCI.2838-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Swanson RA. Astrocytes and brain injury. J Cereb Blood Flow Metab. 2003;23:137–149. doi: 10.1097/01.WCB.0000044631.80210.3C. [DOI] [PubMed] [Google Scholar]
- Halterman MW, Miller CC, Federoff HJ. Hypoxia-inducible factor 1a mediates hypoxia induced delayed neuronal death that involves p53. J Neurosci. 1999;19:6818–6824. doi: 10.1523/JNEUROSCI.19-16-06818.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244:305–318. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- Helton R, Cui J, Scheel JR, Ellison JA, Ames C, Gibson C, Blouw B, Ouyang L, Dragatsis I, Zeitlin S, Johnson RS, Lipton SA, Barlow C. Brain-specific knock-out of hypoxia-inducible factor-1α reduces rather than increases hypoxic-ischemic damage. J Neurosci. 2005;25:4099–4107. doi: 10.1523/JNEUROSCI.4555-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10:615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard M, Ransom B, Goldman SA. New roles for astrocytes: redefining the functional architecture of the brain. Trends Neurosci. 2003;26:523–530. doi: 10.1016/j.tins.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y. “Green mice” as a source of ubiquitous green cells. FEBS Lett. 1997;407:313–319. doi: 10.1016/s0014-5793(97)00313-x. [DOI] [PubMed] [Google Scholar]
- Rempe DA, Lelli KM, Vangeison G, Johnson RS, Federoff HJ. In cultured astrocytes, p53 and MDM2 do not alter hypoxia-inducible factor-1(alpha) function regardless of the presence of DNA damage. J Biol Chem. 2007;282:16187–16201. doi: 10.1074/jbc.M702203200. [DOI] [PubMed] [Google Scholar]
- Ryan HE, Poloni M, McNulty W, Elson D, Gassmann M, Arbeit JM, Johnson RS. Hypoxia-inducible factor-1a is a positive factor in solid tumor growth. Cancer Res. 2000;60:4010–4015. [PubMed] [Google Scholar]
- Semenza GL. HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol. 2000;88:1474–1480. doi: 10.1152/jappl.2000.88.4.1474. [DOI] [PubMed] [Google Scholar]
- Swanson RA. Astrocyte glutamate uptake during chemical hypoxia in vitro. Neurosci Lett. 1992;147:143–146. doi: 10.1016/0304-3940(92)90580-z. [DOI] [PubMed] [Google Scholar]