

Abstract
Endocannabinoids and endocannabinoid-related compounds (ERCs) are involved in many physiological processes. They are released on demand from phosphoinositide and N-acylphosphatidyl ethanolamine (NAPE) precursors and comprise 2-monoacylglycerols (2-MGs) and FA ethanolamides (FEAs). Despite the abundance of advanced quantitative methods, however, their determined concentrations in blood plasma are inconsistent because 2-MGs and FEAs undergo artifactual de novo formation, chemical isomerization, and degradation during sample collection and storage. For a comprehensive survey of these compounds in blood and plasma, we have developed and validated an ultra-HPLC-MS/MS method to quantify 24 endocannabinoids, ERCs, and their phospholipid precursors. Immediate acidification of EDTA-blood to pH 5.8 blocked artifactual FEA formation for at least 4 h on ice. The 2-MGs were stabilized after plasma harvest with 0.5 M potassium thiocyanate at pH 4.7. FEA and MG plasma concentrations in six healthy volunteers ranged between 0.04–3.48 and 0.63–6.18 ng/ml, respectively. Interestingly, only 1–5% of circulating FEAs were present in their free form, while the majority was bound to NAPEs. Similarly, 97% of 2-arachidonoylglycerol (2-AG) was bound to a potential phosphoinositide pool. The herein-described stabilization and extraction methods may now be used to reliably and comprehensively quantify endocannabinoids, ERCs, and their phospholipid precursors in clinical studies.
Keywords: fatty acid ethanolamide, 2-arachidonoylglycerol, 2-monoacylglycerol, N-acylphosphatidyl ethanolamine, phosphatidyl ethanolamine, arachidonoyl phosphoinositide, Orlistat, blood, lipases, phospholipases
The endocannabinoid system is a versatile modulator of endocrine and neuronal functions that plays an important role in many physiological processes and pathological conditions (1). Its components include the G-protein coupled receptors (GPRs) CB1 and CB2, the endocannabinoid-related receptors GPR55, GPR119, GPR18, PPARα, and their endogenous ligands and biosynthesizing and degrading enzymes (2–5). The classical endocannabinoids arachidonoyl ethanolamide (AEA) and 2-arachidonoylglycerol (2-AG) activate CB1 and CB2 (4). In addition to AEA and 2-AG, several endocannabinoid-related compounds (ERCs) have been identified that predominantly activate endocannabinoid-related receptors. Equal to AEA and 2-AG, ERCs are either FA ethanolamides (FEAs) or 2-monoacylglycerols (2-MGs), which are esterified at the sn-2 position of glycerol. Saturated and monounsaturated FEAs include conjugates of myristic acid ethanolamide (MEA), palmitic acid ethanolamide [PEA; 2-palmitoylglycerol (2-PG)], and stearic acid ethanolamide (SEA) FAs and conjugates of palmitoleic acid ethanolamide (POEA), and oleic acid ethanolamine [OEA; 2-oleoylglycerol (2-OG)] FAs. Among those, PEA, OEA, POEA, and 2-OG bind to GPR55 and GPR119 (4, 6). In addition, OEA is a potent ligand of PPARα (5). FEAs and 2-MGs of PUFAs are synthesized from essential FAs and include conjugates of the ω-6-derived linoleic acid (LEA; 2-LG), γ-linolenic acid (GLEA), dihomo-γ-linolenic acid (DGLEA), and ethanolamides of the ω-3 derived α-linolenic acid (ALEA), dihomo-α-linolenic acid (DALEA), eicosatetraenoic acid (ETEA), eicosapentaenoic acid (EPEA), and docosahexaenoic acid (DHEA) (1). Compared with AEA, DHEA and EPEA show similar affinities to CB1 and CB2 receptors and act as partial agonists (1, 7). Finally, 2-arachidonoylglycerol ether [noladin ether (NE)] binds potently to CB1, while N-acylarachidonoylglycine (NAGly) is a ligand of the GPR18 receptor (3, 4).
The biosynthesis of endocannabinoids and ERCs comprises multiple steps and starts with the synthesis of glycerophospholipids from the FA pool. Phosphatidylinositols constitute precursors for 2-MG biosynthesis. They are hydrolyzed by phospholipase C (PLC) to form diacylglycerols, which in turn are deacetylated by sn-1-specific diacylglycerol lipases (DAGLs α and β) to yield 2-MGs (8). Circulating 2- and 1-MGs may additionally form during the digestion of dietary lipids or from lipoprotein and endothelial lipases (9–11). MGs are subsequently degraded via hydrolysis of the ester bond by MG lipase (MAGL) or FA amide hydrolase (FAAH) (8, 12). The main precursors for FEA biosynthesis are phosphatidylcholines. After transferring the sn-1 acyl group to the amino group of phosphatidylethanolamine, the resulting N-acylphosphatidyl ethanolamine (NAPE) is subsequently hydrolyzed by NAPE-specific phospholipase D (NAPE-PLD) to yield the free FEA, which, in turn, is degraded by FAAH (8). Endocannabinoids and ERCs are synthesized in various organs, predominantly in the gastrointestinal tract, adipose tissue, and the CNS (13). They can be released “on demand” from the membranes of specialized cells within these tissues, where they subsequently bind to their respective receptors (1).
Among other processes, endocannabinoid signaling is involved in the regulation of food intake and macronutrient use. It modulates the metabolism of muscle, liver, and adipose cells, mediates the communication between the CNS and the gastrointestinal tract, and acts directly within the CNS (1, 5). Its role within this so-called gut-brain axis has been extensively studied in animal models, in which tissue extracts for the quantification of endocannabinoids and ERCs were used (1, 13). Because tissue biopsies are not readily available in human subjects, clinical studies mostly rely on serum and plasma samples. Because it has been suggested that circulating endocannabinoids and ERCs originate, at least partly, as a spillover from tissues of the gut-brain axis, their monitoring in circulation is used to indirectly assess signaling events in said tissues (13). Although some clinical studies have indeed documented specific changes of circulating endocannabinoids and ERCs in response to food intake, more studies are needed to establish their use as specific biomarkers for ingestive behavior (14). Moreover, hardly anything is known about the concentrations of NAPE and 2-MG-phosphoinositide (2-MG-PI) precursors in plasma. Both have been detected in rodent and human circulation, but despite the evidence that they are synthesized and secreted by the gastrointestinal tract in response to food intake, studies so far only quantified postprandial changes of free endocannabinoids and ERCs (14–16). Because NAPEs and 2-MG-PIs might serve as substrates for endocannabinoid signaling within the gut-brain axis, their quantification in circulation could offer a more direct method to assess the state and signaling potential of the endocannabinoid system (16).
The use of endocannabinoids and their related compounds as specific biomarkers is further limited by the fact that their determined concentrations strongly depend on sample-collection conditions and time required for processing. One particular problem is that blood cell membranes, due to their intrinsic FAAH and NAPE-PLD activity, are able to both release and degrade FEAs ex vivo after sampling (17, 18). It has been shown that prolonged storage of blood prior to plasma harvest resulted in an overestimation of endogenous FEAs and that FEA concentrations in EDTA-free serum were even higher than those in EDTA plasma (19, 20). Quantification of 2-MGs is complicated by the fact that blood cells are able to generate 2-MGs after sampling, which likewise may result in an overestimation of their concentrations (21, 22). There is also evidence that 2-MGs are rapidly degraded at physiological pH or undergo isomerization to their inactive 1-MG isomers (23). Although a partial inhibition of these processes was successful with hydrolase inhibitors like PMSF and Orlistat, immediate sample workup so far provides the only reliable means to minimize all of the above mentioned processes (18, 22).
Circulating endocannabinoids and ERCs are typically measured in lipid extracts of serum or plasma, using LC/MS with positive ESI (LC/ESI/MS/MS) with stable isotope dilution. In many reported cases, extraction of endocannabinoids and ERCs was performed by protein precipitation with methanol, acetonitrile, or acetone, followed by solvent evaporation and reconstitution of the extract, or further workup by solid-phase extraction (20, 22, 24–27). These methods, however, reportedly favor the isomerization of 2-MGs to 1-MGs, especially during evaporation of protic solvents (28). NAPEs have been quantified indirectly by liberating their bound FEAs during sample workup and subsequent LC/MS/MS analysis. For this purpose, NAPEs were extracted, purified, and subsequently hydrolyzed with NAPE-PLD or with KOH in methanol after protein precipitation (16, 29).
Finally, studies that focus on ingestive behavior usually rely on a longitudinal design, where repeated sampling in short intervals is necessary (14). With the current analytical protocols, which require 100–500 µl of plasma, such experiments cannot be easily applied to rodent models due their limited amount of blood. Consequently, animal models remain difficult to compare with human studies.
The goal of the present study was therefore to develop, validate and apply an ultra-HPLC (UHPLC)-MS/MS method for the simultaneous quantification of endocannabinoids, ERCs, and their phospholipid precursors in blood plasma. Because the various endocannabinoids and ERCs interact with different receptors and with different efficacy, a range of 24 metabolites was included in this study (Fig. 1). A particular focus was set on the sample workup, which was systematically improved with the aim to suppress artificial degradation and de novo formation of endogenous analytes and to minimize the required sample volume.
Fig. 1.

Overview of the FEAs, FA glycerol esters, arachidonoyl glycerol ether and arachidonoyl N-glycine amide included in the study. The abbreviation of the respective endocannabinoid is shown beside the structure.
MATERIALS AND METHODS
Chemicals
Chemicals of at least analytical quality were purchased from Sigma-Aldrich. Methanolic or ethanolic solutions of unlabeled and deuterated analytical standards (purity >99%) were ordered from Cayman Chemical and Sigma-Aldrich, including N-(2-hydroxyethyl)-cis-5,8,11,14-eicosatetraenamide-5,6,8,9,11,12,14,15-d8 (AEA-d8, >99% atomic D); 1,3-dihydroxypropan-2-yl-cis-5,8,11,14-eicosatetraenoic-5,6,8,9,11,12,14,15-d8 ester (2-AG-d8, >99% atomic D); N-(1-oxo-cis-5,8,11,14-eicosatetraenyl)-glycine-5,6,8,9,11,12,14,15-d8 (NAGly-d8, >99% atomic D); N-(2-hydroxyethyl-1,1’,2,2’-d4)-octadeca-cis-9,12,15-trienamide (LEA-d4, >99% atomic D); N-(1-oxo-cis-5,8,11,14-eicosa-tetraenyl)-glycine (NAGly); 2-[(cis-5,8,11,14)-5,8,11,14-icosatetraen-1-yloxy]1,3-propanediol (NE); 2-AG; 1,3-dihydroxy-2-propanyl-cis-9,12-octadecadienoate (2-LG); and 1,3-dihydroxypropan-2-yl-cis-9-octadecenoate (2-OG). Honeywell Fluka supplied methanol, 2-propanol, 1-butanol, toluene, acetonitrile, formic acid, and acetic acid (all LC/MS grade). Water was taken from a Millipore Synergy 185 lab-water system (Merck Chemicals).
The blood stabilization solution consisted of 0.98 g of MES in 3 ml of water and 5 ml of acetic acid (>99%), brought to a volume of 10 ml with water and stored at room temperature.
For NAPE hydrolysis, a fresh solution of guanidine hydroxide was prepared by mixing methanolic solutions of 1 M potassium hydroxide and 1 M guanidine hydrochloride (1:1, v/v). After removing the precipitate with a paper filter, the remaining solution of 0.5 M guanidine hydroxide was stored at room temperature.
Synthesis and purity of FEAs
Eighteen FEA reference standards were synthesized as described before (30) using methyl esters of the FAs as starting material, namely, tetradecanoic acid (MEA), hexadecanoic acid (PEA), octadecanoic acid (SEA), cis-9-hexadecenoic acid (POEA), cis-9-octadecenoic acid (OEA), cis-11-octadecenoic acid (vaccenic acid ethanolamide, VEA), trans-9-octadecenoic acid (elaidic acid ethanolamide; EEA), trans-11-octadecenoic acid (tVEA), trans-6-octadecenoic acid (trans-petroselinic acid ethanolamide, tPeEA), cis-5,8,11,14-eicosatetraenoic acid (AEA), cis-5,8,11,14,17-eicosapentaenoic acid (EPEA), cis-4,7,10,13,16,19-docosahexaenoic acid (DHEA), cis-9,12-octadecadienoic acid (LEA), cis-6,9,12-octadecatrienoic acid (ALEA), cis-6,9,12-octadecatrienoic acid (GLEA), cis-11,14,17-eicosatrienoic acid (DALEA), cis-8,11,14-eicosatrienoic acid (DGLEA), and cis-8,11,14,17-eicosatetraenoic acid (ETEA). Deuterated internal standards (ISs) of OEA-d4, POEA-d4, PEA-d4, and SEA-d4 were synthesized in a smaller scale, with the respective acyl methyl ester and 1,1,2,2-d4 ethanolamine (>99%, >98 atomic % D) as reaction components. GC/MS analysis as described before confirmed a purity >99% for all synthesized FEAs (supplemental Fig. S1) (30). The isotopic purity of deuterated FEAs was >98% D.
Detection and prevention of laboratory artifacts
Contamination by laboratory artifacts such as plasticizers from laboratory material and by compounds from human origin can occur during liquid handling and extraction procedures with organic solvents (31, 32). In our laboratory, MEA, PEA, SEA, OEA, 2-PG, 1-PG, and 1-OG were frequently detected during analysis. To minimize contamination with these substances, we worked with nitrile gloves only. Glass equipment was washed with pure methanol and pipette tips (low retention, 2–200 µl; Brand) were rinsed three times with pure methanol prior to use. Toluene was measured by a glass syringe (250 µl; SGE Analytical Science). To remove plastic contaminants, 0.5 ml Eppendorf tubes and Supelco screw caps were stirred for 4 h in a toluene/1-butanol solution (20:1) at 40°C. Afterward, each item was rinsed twice in toluene/1-butanol (20:1 v/v) using a pair of tweezers. The kit for NAPE hydrolysis consisted of 2 ml opening glass vials, screw caps with PTFE/Silicone septa, and flat-bottom 350 µl glass inserts (Supelco). The glass inserts were panned twice in acetone and air-dried before use.
The above provisions prevented any detectable contaminations in our standards. Compared with endogenous FEA levels, contaminations in plasma extracts were less than 5% (0.13 ng/ml) PEA, 10% (0.2 ng/ml) SEA, and 1% (0.01 ng/ml) OEA. Contaminations in NAPE extracts were less than 4% (13 ng/ml) PEA, 6% (10 ng/ml) SEA, and 1% (1.2 ng/ml) OEA. Contaminations with MEA (0.05 ng/ml), 2-PG (2 ng/ml), 1-PG (>40 ng/ml), and 1-OG (>0.6 ng/ml) exceeded 50% of endogenous levels in all extracts during validation experiments. These substances were thus excluded from the study.
Preparation of standard solutions
Stock solutions were prepared in methanol containing 0.05% acetic acid and stored at −80°C or −20°C (in case of working stock solutions). All other dilution steps and the solubilization of extracts were done in methanol/water (70:30 v/v, 0.05% acetic acid), resulting in standards and extracts stable against degradation or isomerization. The deuterated IS solution contained 0.83 ng/ml LEA-d4, 1.67 ng/ml AEA-d8, 1.67 ng/ml PEA-d4, 1.67 ng/ml SEA-d4, 1.67 ng/ml OEA-d4, 0.17 ng/ml POEA-d4, 33 ng/ml 2-AG-d8, and 8.3 ng/ml NAGly-d8. The chosen concentrations of deuterated FEAs were close to endogenous analyte concentrations. Concentrations of 2-AG-d8 and NAGly-d8 were spiked 30 and 15 times higher of endogenous levels, which ensured better accuracy and precision. The stock solution for calibration contained 600 ng/ml each 2-lineoylglycerol (2-LG) and 2-OG; 300 ng/ml each PEA, OEA, VEA, and 2-AG; 150 ng/ml each LEA, AEA, and NAGly; 75 ng/ml each DHEA and NE; 60 ng/ml SEA, 30 ng/ml each of POEA, EEA, tVEA, tPeEA, EPEA, DALEA, DGLEA, ALEA, and GLEA; and 10 ng/ml ETEA. To set up a calibration curve, the stock solution was diluted four times by a serial 1:5 dilution. Volumes of 10, 15, 25, and 40 µl of each resulting solution were added to 20 µl of IS, and the mixture was brought to 60 µl with methanol/water (70:30 v/v, 0.05% acetic acid). Supplemental Table S1 lists the resulting concentrations.
Blood sampling and workup
The ethics committee of Friedrich-Alexander-Universität Erlangen-Nürnberg has approved the present study. Human venous blood was collected under informed consent from the forearm of healthy donors. The protocol was optimized and validated with pooled plasma of unfasted blood, because studies on ingestive behavior typically quantify endocannabinoids and ERCs in different postprandial states. The protocol thus reflects the expected sample type of future studies better than an optimized protocol with fasted blood. For the application of the method, blood was drawn at 11:30 AM from six healthy volunteers, who could eat and drink ad libitum until 2 h prior to the experiment.
The workflow of the sampling, stabilization, storage, and workup procedures is summarized in Fig. 2. Venous blood was first drawn into vacutainer tubes containing K2-EDTA (Monovette, Sarstedt), inverted multiple times, and placed on ice. The blood samples were immediately stabilized by pipetting 1 ml of blood into precooled 2 ml Eppendorf tubes containing 7 µl of blood stabilization solution (50% acetic acid and 0.5 M MES), inverted multiple times, and placed on ice. After centrifuging the tubes at 2,000 g for 10 min at 4°C, approximately 350 µl of plasma was taken from the upper layer. For analysis of free metabolites and lipid-bound MGs, 25 µl of plasma was pipetted into 500 µl Eppendorf tubes, flash-frozen in liquid nitrogen, and stored at −80°C until extraction. For NAPE analysis, aliquots of 25 µl were pipetted into 500 µl Eppendorf tubes containing 75 µl of pure water and 100 µl of 4 M guanidine thiocyanate. The mixture was vortexed for 5 s, flash-frozen in liquid nitrogen, and stored at −80°C until extraction.
Fig. 2.

Overview of the entire protocol including sample collection, stabilization, storage, and the three analytical protocols used for quantification of free and lipid-bound FEAs and MGs.
For blood incubation experiments (Table 1), aliquots of 1 ml of fresh blood were either left untreated, acidified to pH 5.8 with 7 µl of blood stabilization solution, or spiked with Orlistat and PMSF to a final concentration of 5 µM and 2.5 mM, respectively (to a total concentration of 1% DMSO in blood). Aliquots of 100 µl were then transferred to 0.5 ml tubes and incubated at 30°C under shaking at 600 rpm or placed on ice inversing the tubes repeatedly every 10 min. Plasma was obtained by centrifugation at 2,000 g for 10 min at 4°C after the appropriate incubation time and extracted according to the protocol. To check the stability of IS in blood, the samples were spiked in duplicate prior to incubation. To check the stability of endogenous endocannabinoids, the IS was spiked in triplicate after plasma harvest. The experiments were reproduced once.
TABLE 1.
Stability of endocannabinoids in blood, assessed by incubation of untreated, acidified, and inhibitor-treated (2.5 mM PMSF, 5 µM Orlistat) full-blood EDTA samples
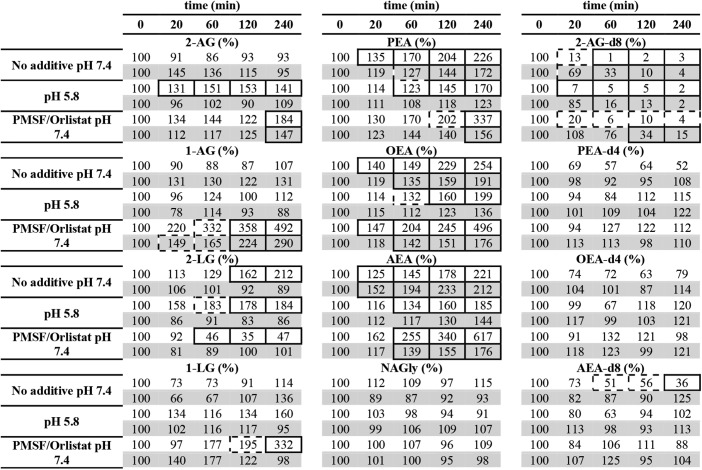 |
Incubations were performed at 30°C (white fields) and on ice (gray fields). To quantify endogenous endocannabinoid levels, deuterated IS was spiked after plasma harvest (n = 3). To assess the stability of IS, EDTA blood was spiked prior to incubation (n = 2). Statistically significant differences at different time points relative to time point zero were tested with one-way ANOVA with repeated measures and Bonferroni´s post hoc correction. Samples with P < 0.05 are shown in a dashed box; those with P < 0.01 are highlighted in a full box.
Plasma incubation experiments (Table 2) were performed in triplicate with 25 µl of plasma. Plasma was either left untreated, acidified with 80 µl of water and 60 µl of 0.25% acetic acid, or stabilized with 80 µl of water, 60 µl of 0.25% acetic acid, and 25 µl of potassium thiocyanate (KSCN; 4 M). Samples were incubated at 25°C under shaking at 600 rpm. The reaction of untreated and acidified plasma was stopped by adding 25 µl of KSCN (4 M). To determine the stability of IS in plasma, samples were spiked prior to incubation. Changes of endogenous analytes were determined by spiking IS after incubation. The experiments were reproduced once.
TABLE 2.
Stability of endocannabinoids in plasma, assessed by incubation experiments of untreated, acidified, and stabilized plasma samples
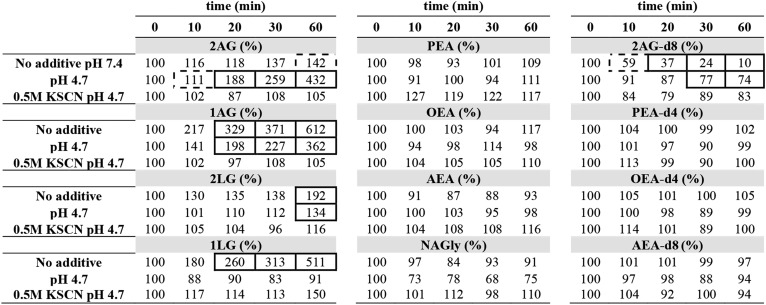 |
All incubations were performed at 25°C. To assess the stability of deuterated IS, samples were spiked prior to incubation (n = 3). To quantify endogenous metabolite levels, IS was spiked after incubation (n = 3). Statistical testing relative to time point zero was performed with one-way ANOVA with repeated measures, with Bonferroni´s post hoc correction. Samples with P < 0.05 are shown in dashed boxes; those with P < 0.01 are highlighted in a full box.
Extraction of circulating endocannabinoids and ERCs
Flash-frozen plasma (25 µl) was thawed on ice, mixed with a solution of 80 µl of water, 60 µl of 0.25% acetic acid, and 25 µl of KSCN (4 M), and vortexed for 5 s. After adding 10 µl of IS, samples were vortexed briefly and rested at room temperature for 5 min. After adding 210 µl of toluene/1-butanol (20:1 v/v), the mixture was vortexed at maximum speed for 1 min in a VTX-3000L mixer (Uzusio, LMS). The homogenate was centrifuged at 12,000 g for 10 min at 25°C. The upper layer (190 µl) was pipetted into a 200 µl glass insert, evaporated to dryness in a speedvac concentrator (Savant SPD121P, Thermo Scientific), and stored in sealed glass vials at −20°C. Prior to analysis, the extracts were solved in 30 µl of methanol/water (70:30, 0.05% acetic acid) and vortexed for 30 s.
Extraction of NAPEs
After thawing diluted, flash-frozen plasma on ice and vortexing, aliquots of 20 µl were pipetted into flat-bottom 350 µl glass inserts containing 160 µl of guanidine hydroxide (0.5 M) and 20 µl of IS in pure methanol. Each sample was placed into a 2 ml glass vial, sealed with a gas-tight septum, vortexed for 5 s, and heated to 60°C in a shaking incubator for 120 min, at 1,400 rpm. The reaction was stopped by adding 80 µl of pure water and cooling on ice. After removing the methanol in a speedvac concentrator, 210 µl of toluene/1-butanol (20:1 v/v) was added, and the sample was vortexed in a VTX-3000L mixer at maximum speed for 1 min. The homogenate was centrifuged at 8,000 g for 10 min at 25°C by placing the glass inserts into closed 2 ml Eppendorf tubes. The upper organic phase (190 µl) was pipetted into 200 µl glass inserts, evaporated to dryness in a speedvac concentrator, and stored in sealed glass vials at −20°C. For analysis, the extracts were solved in 60 µl of methanol/water (70:30 v/v, 0.05% acetic acid) and vortexed for 30 s.
Extraction of lipid-bound MGs
Frozen plasma samples were thawed on ice, mixed with 60 µl of 0.25% acetic acid and 80 µl of water, vortexed, spun down, and placed into a shaking incubator at 25°C for 300 min, at 600 rpm. After stopping the reaction with 25 µl of KSCN (4 M), 10 µl of IS and 210 µl of toluene/1-butanol solution (20:1 v/v) were added to the sample and vortexed at maximum speed for 1 min in a VTX-3000L mixer. After centrifuging the mixture at 12,000 g for 10 min at 25°C, 190 µl of the upper layer was pipetted into a 200 µl glass insert, evaporated to dryness in a speedvac concentrator, and stored in sealed glass vials at −20°C. For analysis, the extracts were solved in 30 µl of methanol/water (70:30 v/v, 0.05% acetic acid) and vortexed for 30 s.
UHPLC-ESI/MS/MS analysis
Analysis was carried out with an API 4000 QTRAP mass spectrometer (AB Sciex) with an ESI source (Applied Biosystems) coupled to a pump for direct infusion (Harvard Apparatus 11plus) and a UHPLC system (Dionex UltiMate 3000 RS, Thermo Fisher Scientific) equipped with a Kinetex Core Shell C18 UHPLC column (Phenomenex, 1.7 µm, 150×2.1 mm). To maximize chromatographic resolution of all analytes, the column compartment was set to 15°C. The temperature of the autosampler was maintained at 5°C. Eluent A consisted of 65% water, 35% 2-propanol, and 0.05% acetic acid and eluent B of acetonitrile with 0.05% acetic acid. After injecting 24 µl of extract (corresponding to an equivalent of 20 µl of plasma), the following gradient was run: 0–1 min 45% B, 1–19 min to 70% B, 19–24 min to 90% B, and 24–24.5 min to 100% B at 0.25 ml/min. Parameters of the ESI interface were: nebulizer gas (GS1) 30 psig, desolvation gas (GS2) 65 psig, temperature 550°C, ion spray voltage (positive mode) 4,500 V, and curtain gas 40 psig. The vertical adjustment for the ESI capillary was set to 4 mm.
Molecular mass, fragmentation spectra, and tuning of the multiple reaction monitoring (MRM) transitions of all standards were assessed by direct infusion of 1 µg/ml stock solutions at a flow rate of 20 µl/min. MS parameters and mass transitions of [M+H]+ parent ions are summarized in supplemental Table S2.
Data was recorded with Analyst 1.5.1 software in scheduled MRM (sMRM) mode. For this, the mass transitions of a given analyte were successively recorded at their respective retention time in a window of 140 s. The cycle time was set to 1.8 s. The parameters of the quantification tool were set to peak smoothing (3), base noise (50%), and peak split ratio (2). All transitions were integrated automatically and checked visually. Near the limit of detection (LOD), the signals were integrated manually.
Calculation of endocannabinoid concentrations
The analytes were matched with deuterated standards according to chemical similarity and to their retention times (supplemental Table S2). Endocannabinoid concentrations were calculated by linear regression from external calibration curves (9–11 points) interpolating the peak area ratio of endogenous analytes and their respective IS to the respective external calibration by the formula endocannabinoid (ng/ml) = [(peak area ratio − intercept)/slope], taking the dilution of the samples during reconstitution into account.
Method validation
A single batch of pooled plasma was used for validation experiments. Plasma was harvested from stabilized blood at pH 5.8. The linearity of the method was assessed by spiking plasma in triplicate with 11 calibrator solutions (10 µl, threefold concentrated; supplemental Table S1). Recoveries were calculated by subtracting endogenous endocannabinoid levels from the total quantified values.
LOD and limit of quantification (LOQ) were determined in triplicate using six calibrators at low concentrations (supplemental Table S1). Mathematical LOD and LOQ were calculated by linear regression using the parameters “standard deviation of the residuals” (σ) and the slope of the regression line (s) with LOD = (3*σ)/s and LOQ = (10*σ)/s. Calculated LOD and LOQ were confirmed by spiking experiments of diluted IS estimating LOD with a signal to noise ratio (S/N) = 3, and LOQ with S/N = 10. LOQ was calculated with the linear regression of the quantifier transition (Q) and LOD with the regression of the qualifier transition (q). Accordingly, LODs of some analytes were higher than their respective LOQs, which were subsequently matched to the LOD of the respective analyte (Table 3).
TABLE 3.
Validation of the UHPLC-MS/MS-sMRM method to quantify free endocannabinoids and ERCs in human plasma
| Recovery (%) n = 6 | Intraday CV (%) n = 6 | Interday CV (%) 3x n = 4 | Literature | |||||||||||||
| Amount spiked | Q | q | Q | q | ||||||||||||
| Low | Med | High | Unspiked Plasma | Low | Med | High | Unspiked NAPEs | Unspiked Plasma | Low | Med | High | LOQ | LOD | LOQ | LOD | |
| 2-AG | 91 | 92 | 92 | 3 | 4 | 4 | 5 | < LOD | 8 | 9 | 7 | 4 | 0.27 | 0.1 | 0.5-3.2a,d,e,f | 0.3a |
| 1-AG | 107 | 101 | 96 | 8 | 7 | 1 | 5 | < LOD | 15 | 21 | 5 | 7 | 0.27 | 0.1 | ||
| 2-LG | 107 | 93 | 95 | 9 | 9 | 6 | 4 | < LOD | 8 | 10 | 4 | 4 | 0.62 | 0.24 | 2.5-29.3d,e | — |
| 1-LG | 98 | 96 | 99 | 7 | 7 | 5 | 4 | < LOD | 10 | 10 | 6 | 5 | 0.62 | 0.24 | ||
| 2-OG | 82 | 86 | 88 | 9 | 11 | 9 | 6 | < LOD | 11 | 12 | 7 | 10 | 1.82 | 0.36 | 2.5e | — |
| NE | 93 | 95 | 98 | < LOD | 7 | 5 | 3 | < LOD | < LOD | 13 | 8 | 4 | 0.07 | 0.07 | 1.8-3.2a,d | 3.2a |
| PEA | 96 | 98 | 100 | 5 | 3 | 2 | 2 | 6 | 10 | 9 | 5 | 2 | 0.21 | 0.06 | 0.17-3.2a,c,d,e | 0.5a |
| SEA | 101 | 107 | 103 | 9 | 4 | 3 | 1 | 6 | 22 | 12 | 14 | 21 | 0.66 | 0.66 | 0.27-8a,c,d,e | 0.5a |
| POEA | 112 | 97 | 102 | 5 | 4 | 1 | 4 | 4 | 4 | 6 | 3 | 3 | 0.08 | 0.08 | 0.02-1.6d,e | — |
| OEA | 91 | 100 | 101 | 7 | 5 | 1 | 2 | 5 | 6 | 6 | 2 | 2 | 0.15 | 0.15 | 0.12-0.5a,c,d,e | 0.2a |
| VEA | 128 | 102 | 112 | 7 | 5 | 3 | 4 | 9 | 6 | 5 | 6 | 5 | 0.15 | 0.14 | — | — |
| tVEA | 113 | 103 | 108 | 14 | 6 | 5 | 7 | 5 | 16 | 10 | 13 | 10 | 0.04 | 0.04 | — | — |
| EEA | 93 | 97 | 103 | < LOD | 9 | 4 | 4 | < LOD | < LOD | 13 | 15 | 11 | 0.04 | 0.04 | — | — |
| tPeEA | 87 | 85 | 90 | < LOD | 13 | 3 | 6 | < LOD | < LOD | 12 | 5 | 6 | 0.02 | 0.01 | — | — |
| ALEA | 98 | 97 | 99 | 9 | 7 | 4 | 4 | 10 | 9 | 9 | 5 | 4 | 0.04 | 0.04 | 0.11c | — |
| GLEA | 96 | 96 | 96 | < LOD | 5 | 5 | 3 | 11 | < LOD | 7 | 4 | 3 | 0.02 | 0.02 | — | — |
| DALEA | 99 | 92 | 98 | < LOD | 10 | 4 | 7 | < LOD | < LOD | 9 | 6 | 6 | 0.12 | 0.12 | — | — |
| DGLEA | 109 | 97 | 98 | 7 | 6 | 3 | 4 | 7 | 10 | 8 | 4 | 4 | 0.02 | 0.02 | 0.02e | — |
| LEA | 108 | 100 | 102 | 3 | 3 | 1 | 2 | 3 | 5 | 5 | 3 | 2 | 0.08 | 0.06 | 0.1-0.24a,c,d,e | 0.6a |
| AEA | 98 | 95 | 98 | 3 | 5 | 2 | 2 | 5 | 6 | 5 | 4 | 3 | 0.06 | 0.02 | 0.02-0.36a,c,d,e,f | 0.1a |
| ETEA | 95 | 93 | 97 | < LOQ | 6 | 3 | 3 | 9 | < LOQ | 5 | 3 | 3 | 0.01 | 0.01 | — | — |
| EPEA | 95 | 99 | 96 | 6 | 6 | 6 | 4 | 6 | 14 | 6 | 4 | 4 | 0.01 | 0.01 | 0.02-0.36c,e | — |
| DHEA | 105 | 94 | 94 | 6 | 5 | 1 | 3 | 6 | 7 | 6 | 3 | 3 | 0.03 | 0.01 | 0.02-2.6a,d | 2.6a |
| NAGly | 108 | 99 | 102 | 5 | 8 | 5 | 3 | < LOD | 9 | 8 | 5 | 4 | 0.12 | 0.12 | 0.32-0.73b,d | 0.63b |
Accuracy (recovery, %) was determined by spiking three calibrator solutions at the lower and limits of quantification (LOQ), in an intermediate and in a high concentration range as indicated in supplemental Table S3. Interday and intraday precision (CV %) was assessed with unspiked plasma and with three different concentrations of spiked analytes. Precision of NAPE hydrolysis was determined with unspiked plasma. LOQ of the quantifier transition Q and LOD of the qualifier transition q are given in ng/ml. References: a (24), b (29), c (27), d (26), e (22), and f (28).
For quality control, plasma was spiked with IS only (unspiked plasma) or with a calibrator mix with low, medium, and high concentrations (supplemental Table S1, boxes). The recovery was assessed in six replicates and was calculated by the quotient between the observed concentration and the nominal concentration after subtraction of the endogenous levels. Intraday precision was determined by six replicates within 1 day and is given as the coefficient of variation (CV, %). Interday precision was determined by four replicates on 3 days, respectively, spiking thawed samples on each day. To assess matrix effects on ESI efficiency, unspiked plasma extracts were reconstituted in methanol/water (70:30 v/v, 0.05% acetic acid) containing half, singly, and doubly concentrated IS.
The stability during long-term storage was determined for plasma at pH 5.8. All samples were flash-frozen in liquid nitrogen and stored at −80°C. Three independent samples each were analyzed at storage days 0, 7, 35, and 56 (supplemental Table S3).
Statistical analysis
Statistical analysis was performed with the software package XLSTAT. Significance between the incubation times relative to time point zero was calculated using one-way ANOVA with repeated measures, followed by Bonferroni’s post hoc test.
RESULTS AND DISCUSSION
Analysis of 24 endocannabinoids and ERCs by UHPLC-MS/MS sMRM
The goal of the present study was to develop and validate an analytical method that allows the quantification of a wide range of FEAs and 2-MGs in parallel. For this purpose, a highly sensitive UHPLC-ESI-MS/MS sMRM method with high-chromatographic resolution was developed that required minimal sample volumes. Besides the previously analyzed FEAs, 2- and 1-MGs, NE, and NAGly, the protocol included GLEA and DALEA for the first time. Extending a UHPLC method to analyze the regio-isomers OEA and VEA (30), the present study focused on the separation of the regio-isomers ALEA/GLEA and DGLEA/DALEA, because these analytes have identical m/z ratios and fragmentation spectra and can only be differentiated chromatographically.
Based on the chromatographic separation of all analytes in plasma (Fig. 3), we developed an sMRM method allowing for the identification and quantification of all endocannabinoids and ERCs by their retention time, as well as their quantifier and qualifier transitions (supplemental Table S2). ALEA and GLEA could be separated clearly. The method also distinguished between DALEA and DGLEA, although no baseline separation could be achieved. Close to the POEA signal, two further isobaric transitions could be observed (Fig. 3, arrows labeled “?”). We hypothesize that these peaks represent the ethanolamides of 6-cis-hexadecenoic and 9-trans-hexadecenoic acids because their retention behavior was similar to the isomers VEA, OEA, and tVEA (30). Regardless of their identity, this result shows that a thorough chromatographic resolution is needed to quantify POEA correctly. NE was not detectable in plasma at the retention time of the standard. However, a signal with an isobaric transition appeared shortly afterward (Fig. 3, arrow labeled “?”). Further studies are required to identify these compounds.
Fig. 3.

Chromatograms of endogenous endocannabinoids and ERCs and ISs in plasma displaying the quantifier transition in counts per second. An equivalent of 20 µl of plasma was injected. Isobaric of POEA and NE quantifiers are marked with a question mark (“?”).
Optimization of the plasma workup
It is well established that 2-MGs are vulnerable to degradation and isomerization to 1-MGs during plasma extraction and due to its high physiological pH and the presence of albumin. Additionally, coextraction of phospholipids reduces ionization efficiency during LC/MS analysis and thus results in strong matrix effects (28). To minimize these problems, toluene and methyl tert-butyl ether were suggested as extraction solvents for pure plasma (22, 28). In our study, extraction with pure toluene resulted in low recovery rates of spiked ISs [FEA-d4 below 60%, 2-AG-d8 at 10%, and NAGly-d8 at 4% (data not shown)]. To increase recovery, the extraction solvent was first replaced with a toluene/1-butanol solution (20:1 v/v). Second, plasma was acidified to pH 4.7 prior to extraction, because acyl migration of 2-AG to 1-arachidonoylglycerol (1-AG), and thus of 2-AG-d8 to 1-AG-d8, has been shown to be blocked below pH 5 in solution (23). These workup conditions decisively increased the yield of NAGly-d8 to 87% and of 2-AG-d8 to 71%. Isomerization of 2-AG-d8 to 1-AG-d8 was also inhibited as the observed peak area ratio after extraction improved from about 50% 2-AG-d8 and 50% 1-AG-d8 to about 84% 2-AG-d8 and 16% 1-AG-d8, which was similar to the ratio of pure standard (88% 2-AG-d8 and 12% 1-AG-d8). However, these conditions introduced a high variability of extraction efficiency between different individuals. Because albumin has been reported to form insoluble aggregates under acidic conditions (33), we hypothesized that this effect might originate from a coprecipitation of albumin and plasma lipids in different postprandial states. To circumvent this process, the chaotropic salt sodium thiocyanate was added to the extraction mix, which reportedly solubilizes albumin and prevents aggregation (34). SCN− concentration and SCN− counterions were further optimized with regard to recovery and matrix effect. The final protocol with 0.5 M KSCN at pH 4.7 showed very high IS recovery rates with 78–99% for FEA-d4, 84% for NAGly-d8, and 77% for 2-AG-d8 standards, which was reproducible for six independent unfasted human samples (supplemental Table S4). Ion suppression of coextracted matrix was equal to or below 10% for all ISs, with the exception of SEA-d4 that showed a 15% reduction in signal response at low concentrations. The signal of 2-AG-d8 was reduced by 10–19% in all concentrations (supplemental Table S4). Compared with studies using toluene or methyl tert-butyl ether for extraction, the present yield of IS was equal or higher, whereas matrix effects were generally lower (22, 28).
Using this optimized protocol, the required plasma volume could be reduced to 25 µl without losing sensitivity (Table 3). Thus, the method can be easily transferred to rodent experiments, which can now be directly compared with human studies.
Sample workup for the analysis of NAPE-bound FEAs
It has been suggested before that NAPEs can be indirectly quantified by analyzing the respective FEAs that were liberated after hydrolysis with NAPE-PLD or with KOH in methanol (16, 29). In the present study, hydrolysis with KOH was very slow and required long incubation times. The replacement of KOH by guanidine hydroxide, which shows strong noncovalent interaction with phosphate ions, greatly improved the hydrolysis rate. To minimize the required sample volume and the risk of contamination with plasticizers, sample workup was done in a one-pot procedure, where lipid hydrolysis took place in a homogeneous solution with all plasma components and was followed by liquid-liquid extraction. Fig. 4A illustrates the time-dependent release of FEAs during hydrolysis of NAPEs. Hydrolysis was complete after 120 min and revealed high NAPE concentrations in plasma of up to 600 ng/ml. Stability of FEAs during hydrolysis was confirmed by spiking IS prior to incubation, resulting in a recovery of FEA-d4 standards of 95–105% (supplemental Table S4). The recovery of NAGly-d8 under these conditions decreased to 78%, presumably due to a reduced extraction efficiency of its deprotonated form. 2-AG-d8, and other MGs were not detectable after hydrolysis. This served as a positive control of the protocol, because all esters were supposed to be hydrolyzed under these conditions. The influence of matrix components on ionization efficiency ranged from −18% to 29% for all ISs (supplemental Table S4). Whereas previously published methods required plasma volumes of 100 µl or more, the newly developed method achieved the indirect quantification of NAPE-bound FEAs with an equivalent of only 2.5 µl of plasma (16, 29).
Fig. 4.

A: Time-dependent release of FAEs from their NAPE precursors in pooled plasma during hydrolysis by guanidine hydroxide (0.5 M in methanol) at 60°C (n = 3). B: Time-dependent release of glycerol esters during incubation of pooled plasma at pH 4.7 and 25°C (n = 3). C: Incubation of pooled plasma at pH 4.7 and 25°C in presence of 5 µM Orlistat (n = 3).
Quantification of ex vivo generation and degradation of endocannabinoids and ERCs and their stabilization in blood
As elaborated in the introduction, the quantification of endocannabinoids and ERCs in blood plasma is complicated by various ex vivo processes. Recent studies successfully used enzymatic inhibitors such as PMSF and Orlistat to block FAAH-mediated FEA degradation, triacylglycerol lipase and DAGL-mediated 2-MG generation and MAGL-dependent degradation of 1- and 2-MGs in blood (18, 22). The established inhibitors, however, were insufficient to suppress FEA accumulation during storage (18). Moreover, degradation and acyl migration of 2-MGs to 1-MGs still occurred at physiological pH (22). Because blood cells tolerate a pH of 5.8 without hemolysis, we tested whether these processes could be simultaneously blocked at a blood pH of 5.8 (35). Furthermore, the stabilizing effects of the unspecific FAAH inhibitor PMSF (2.5 mM) and the unspecific lipase inhibitor Orlistat (5 µM) were reproduced from previous studies. Table 1 summarizes the results of all incubations. The selected analytes are representative for the substance class and the concentration range.
Incubation of deuterated FEA standards in untreated blood revealed FAAH activity, which significantly degraded AEA-d8 to 51% at 30°C after 60 min. The activity of FAAH could be suppressed by storing the samples on ice, in the presence of PMSF and at pH 5.8 at 30°C. With regard to endogenous metabolites, we observed that FEAs significantly accumulated within 20 min and continued to rise during the incubation of untreated blood at 30°C. As expected, reduced FAAH activity in inhibitor-treated samples resulted in a faster and higher accumulation of FEAs. The highest effect was observed for AEA, which accumulated to 617% after 240 min at 30°C. Storing the samples on ice slowed the release of FEAs considerably in untreated and inhibitor-treated samples, but failed to prevent significant accumulation at all tested time points. Acidification of blood was likewise insufficient to block FEA release at 30°C. Storing acidified samples on ice, however, suppressed all ex vivo activity, as no significant accumulation of FEAs was observed during the incubation time of 240 min. Our results confirm that the storage of untreated blood on ice prevents FEA degradation, but not their ex vivo generation by NAPE-PLD (17, 18, 36). This process could only be blocked under acidic conditions on ice, supposedly by further lowering the specific enzymatic activity at this pH as reported before (37).
The 2-AG-d8 appeared to be extremely unstable under all tested conditions. Storage at pH 5.8 on ice did not improve its stability, whereas storage in the presence of inhibitors and on ice was sufficient to stabilize this molecule for at least 60 min. We thus conclude that the stability of 2-AG-d8 in blood is more dependent on MAGL activity and less on blood pH.
Quantification of endogenous 2-LG in untreated blood revealed a significant accumulation to 162% after 120 min at 30°C. Treatment with inhibitors resulted in a significant decrease of 2-LG to 46% after 60 min at 30°C. This result was surprising, because treatment with Orlistat and PMSF was supposed to inhibit both DAGL and MAGL and, thus, to stabilize 2-LG levels. Because a significant accumulation of its isomer 1-LG to 195% was detected after 120 min in inhibitor-treated samples, we hypothesize that both enzymes were indeed inhibited during incubation and that the initially present 2-LG isomerized to 1-LG. In support of this hypothesis, 1-LG did not accumulate to significant levels in acidified samples. In contrast to 2-LG, 2-AG did not accumulate in untreated blood at 30°C. Moreover, 2-AG levels in inhibitor-treated samples increased to 184% after 240 min at 30°C. The only similarity between both molecules was that 1-AG accumulated to significant levels in inhibitor-treated blood. These observations suggest that 2-AG and 2-LG are released by different enzymatic mechanisms, while both compounds undergo degradation by MAGL and isomerization at physiological pH. Because 2-AG accumulated to significant levels in inhibitor-treated blood, we further conclude that 2-AG is released by enzymes that are unresponsive to Orlistat. Under acidic conditions, 2-LG and 2-AG accumulated significantly at 30°C, whereas their 1-MG isomers remained unchanged. This result supports the assumption that both molecules are constantly released by lipases and isomerize to their 1-MG counterparts at physiological pH.
Because only acidic conditions and treatment on ice could suppress the described processes, we suggest that blood samples are immediately stabilized at pH 5.8 and stored on ice. Thus, initial FEA and 2-MG concentrations can be preserved for up to 4 h. Storage experiments revealed that all analytes were stable for 35 days under these conditions (supplemental Table S3).
Inhibition of MG formation in plasma ex vivo
Similar to the ex vivo processes observed in blood, EDTA-plasma reportedly showed an accumulation of 2-MGs and an isomerization to inactive 1-MGs during storage (22). Therefore, we assessed their stability of FEAs and MGs in untreated, acidified (pH 4.7), and stabilized plasma (0.5 M KSCN at pH 4.7) at 25°C (Table 2). The selected analytes are representative for the substance class and the concentration range.
Contrary to blood, incubation with FEA-d4 standards did not disclose FAAH activity, as all compounds remained unchanged during incubation. Endogenous FEAs likewise remained unchanged in all tested treatments, which allows for the conclusion that only blood cells, but not plasma, contain FAAH and NAPE-PLD enzymes.
The stability of spiked 2-AG-d8 in untreated plasma was very poor, with a significant reduction to 10% after 60 min of incubation. At pH 4.7, 2-AG-d8 was more stable, but still degraded significantly to 74% after 60 min, indicating that unspecific chemical hydrolysis and isomerization of the ester were the major processes in plasma at physiological pH. Because 2-AG-d8 appeared to be further stabilized in the presence of KSCN, presumably by denaturating the tertiary structure of enzymes (38), residual MAGL activity in plasma cannot be excluded.
With regard to acylglycerols, a significant accumulation of 2-MGs could be observed in untreated plasma, which was accompanied by a strong increase of the respective 1-MG isomers. This result confirmed that lipases generate 2-MGs ex vivo, which in turn isomerize at physiological pH. As observed with 2-LG and 1-LG isomers, this process could be sufficiently suppressed at pH 4.7. In contrast to 2-LG, however, 2-AG was released to more than 43 times of its initial concentration during the incubation at pH 4.7. This effect appeared in multiple independent experiments and in different plasma samples and thus suggests that 2-AG precursors and respective metabolic enzymes are present in plasma. This effect also underlines that rigorous pH control is of utmost importance to avoid artifact 2-AG formation prior to extraction. Conveniently, the extraction mix of the present study (0.5 M KSCN at pH 4.7) intrinsically blocks all ex vivo activity and may thus be used to stabilize plasma up to 2 h prior to extraction.
Quantification and possible origin of lipid-bound 2-MGs
As shown above, 2-AG heavily accumulated during the incubation of plasma at pH 4.7. To further assess the release of 2-AG, we incubated acidified plasma samples for up to 5 h at 25°C prior to extraction (Fig. 4B). After 4 h, the endpoint of the reaction was reached, and 2-AG levels remained nearly unchanged for the following hour. The 2-AG accumulated 24-fold within 5 h of incubation, followed by 1-AG that increased 8-fold, whereas 2-LG, 1-LG, and 2-OG increased 3-, 2-, and 2-fold, respectively. We speculated that lipases were responsible for this process and tested whether the increase could be prevented by adding the unspecific lipase inhibitor Orlistat to the initial mixture. As a result, 2-LG, 2-OG, and 1-LG remained unaffected during incubation (Fig. 4C), indicating that these molecules are mostly generated by diacylglycerol and triacylglycerol lipases in blood, which is in accordance with previous studies (9, 11, 22). The 2-AG levels, however, still increased 5-fold and 1-AG levels increased 3.5-fold. These results were consistent with the previous incubation experiments and strongly suggest that 2-AG is released by different mechanisms than the other acylglycerols. Because acidification of plasma to pH 3.8 reduced the release of 2-AG to 131% after 60 min compared with 885% at pH 4.7 (supplemental Fig. S2A), an acid-catalyzed effect can be excluded. Further considering the observation that ex vivo activity in plasma was blocked completely in presence of KSCN, presumably by destabilizing enzymes or lipid complexes in plasma (38, 39), this finding suggests that the release of 2-AG is a specific process that is controlled by enzymatic activity. Therefore, we hypothesize that 2-AG is released by the activity of phosphatidylinositol-specific PLC (PI-PLC). This enzyme primarily converts PI to diacylglycerols, which, in turn, release 2-AG by the action of the sn-1-selective DAGLs DAGLα and/or DAGLβ (40). This assumption is supported by the fact that PI from mammalian tissue primarily consists of stearic acid esterified at the sn-1 position and arachidonic acid esterified at the sn-2 position (41). Furthermore, PIs were identified in lipoproteins such as LDL and HDL2 in human circulation (42). In addition, the PI-PLC-γ enzyme is reportedly present in circulation and is induced at pH 5 through conformational changes (43). These findings in total may explain the accelerated release of 2-AG at pH 4.7. Because a small fraction of 2-AG was still released in presence of Orlistat, we cannot exclude that PLA1 produces sn-2-lysophosphatidylinositides that are subsequently hydrolyzed by PI-PLC (8). It could also be observed that 1-AG accumulated to some extent in the presence of Orlistat at pH 4.7 (Fig. 3C). It is difficult to state whether its release is an enzymatic process or the result of isomerization of 2-AG; however, because the levels of 1-AG remained unchanged at pH 3.8 within 60 min (supplemental Fig. S2B), we argue that 1-AG is generated by isomerization of accumulating 2-AG at pH 4.7. Consequently, the total 2-AG-PI levels might be more correctly represented as the sum of released 2- and 1-AG.
In summary, this method may offer a new and simple way to quantify circulating 2-AG-PI levels with minimum amount of plasma (25 µl). To the best of our knowledge, 2-AG-PI was so far only quantified from tissue and cell extracts using LC/MS/MS. Because of the low abundance of PIs in mammalian cells and due to the high complexity of their metabolome, PIs are difficult to analyze and require multiple purification steps prior to analysis. By using our indirect approach, we were able to quantify 2-AG-PI with comparable sensitivity to methods that analyzed phospholipids directly (44). However, although our method may be an easy and inexpensive approach, the direct quantification of PIs is preferable.
Validation of the UHPLC-MS/MS method
For validation of the method, linearity, LOD, LOQ, recovery, and intraday as well as interday precision were assessed. Spiked calibrators in the low concentration range were at or close to the LOQ of most analytes (supplemental Table S1) and thus were suitable to assess accuracy and precision at the calculated LOQs. Linearity of the method was determined in pure solvent and in spiked human plasma. Linear regression analysis showed consistent results in both matrices with correlation coefficients (R2) >0.995 for all quantifier transitions and R2 > 0.98 for all qualifier transitions (supplemental Table S5). Mean recovery rates of spiked calibrators in low, medium, and high concentrations were 90–110% of the nominal value for most analytes, disclosing a high accuracy of the method (Table 3). At all spiked concentrations, the observed recovery of 2-OG was more than 10% lower than the nominal values, indicating that the 2-OG response is probably reduced by a matrix effect and that 2-OG should be quantified with a deuterated analog. The intraday CV (%) was about 5% for most endogenous and spiked analytes, whereas interday measurements revealed CVs of 10–20% for 1-AG, SEA, tVEA, EEA, and EPEA in unspiked plasma and plasma spiked with low concentrations. With the exception of SEA, tVEA, and EEA, all other spiking experiments revealed a good precision with CVs of about 5%. NAPE hydrolysis of unspiked plasma also showed a CV of about 5%. Table 3 includes the calculated LODs and LOQs of all analytes, which are generally comparable with studies in which similar LC/MS equipment was used.
Quantification of circulating endocannabinoids, NAPEs, and lipid-bound MGs in six healthy volunteers
The optimized and validated UHPLC-ESI/MS/MS sMRM method was applied to quantify circulating lipids in six unfasted human volunteers (Table 4). Saturated and monounsaturated PEA, OEA, SEA, and VEA constituted the largest fraction in plasma with 82% of total circulating FEAs. Their concentrations (mean ± SD) were between 2.51 ± 0.39 and 0.81 ± 0.20 ng/ml. POEA was present at 0.11 ± 0.05 ng/ml. The remaining FEA fraction consisted of conjugates of ω-6 and ω-3 FAs, with contents of LEA of 0.49 ± 0.15 ng/ml, AEA of 0.17 ± 0.03 ng/ml, and DHEA of 0.26 ± 0.09 ng/ml, respectively. ALEA, DGLEA, and EPEA were detected at concentrations below 0.1 ng/ml. GLEA and DALEA were present above LOQ at 0.02 and 0.12 ng/ml, respectively, but were not quantified due to the absence of their qualifier transition. All other FEAs were below their respective LOD between 0.01 and 0.04 ng/ml. In total, the concentrations measured in this study were in accordance to literature data.
TABLE 4.
Circulating endocannabinoids and ERCs and their NAPE and PI-2-AG (lipid-bound) precursors in plasma of six healthy human volunteers (n = 6)
| Free (ng/ml) | Lipid-bound (ng/ml) | Literature values (ng/ml) | ||
| Free | Lipid-bound | |||
| PEA | 2.51 ± 0.39 | 330 ± 53.8 | 0.48 (a), 1.5 (b), 2.1 (c), 4 (d), 4.3 (e) | 50.92 (b) |
| SEA | 2.03 ± 1.21 | 167 ± 30.3 | 0.62 (b), 1.2 (c) 2.1 (d), 3.3 (a) | 34.31 (b) |
| POEA | 0.11 ± 0.05 | 6.52 ± 0.9 | 0.17 (c), 20.9 (a) | — |
| VEA | 0.81 ± 0.20 | 26.1 ± 4.67 | 2.33 (f) | — |
| OEA | 1.24 ± 0.25 | 123 ± 20.5 | 0.54 (d), 1 (a), 1.5 (e), 1.98 (b), 3.1 (c) | 22.66 (b) |
| tVEA | <LOD | 4.3 ± 1.4 | — | — |
| EEA | <LOD | <LOD | — | — |
| tPeEA | <LOD | <LOD | — | — |
| ALEA | 0.05 ± 0.02 | 2.73 ± 0.77 | 0.04 (c), 0.21 (d) | — |
| DALEA | <LOD | <LOD | — | — |
| ETEA | <LOD | 0.47 ± 0.11 | — | — |
| EPEA | 0.02 ± 0.01 | 1.91 ± 0.93 | 0.03 (c), 0.1 (a) | — |
| DHEA | 0.26 ± 0.09 | 13.7 ± 6.7 | 0.41 (b), 0.44 (c), 0.46 (a) | 3.99 (b) |
| LEA | 0.49 ± 0.15 | 39.4 ± 9.79 | 1.23 (c), 2.54 (a), 17.97 (d) | — |
| GLEA | <LOD | 1 ± 0.4 | — | — |
| DGLEA | 0.04 ± 0.01 | 4.66 ± 1.58 | 0.12 (c) | — |
| AEA | 0.17 ± 0.03 | 22.1 ± 6.08 | 0.23 (d), 0.34 (e), 0.37 (b), 0.39 (c), 0.41, 0.5 (a) | 8.07 (b) |
| 2-AG | 0.63 ± 0.21 | 18.6 ± 5.1 | 0.57 (e), 0.72 (h), 0.89 (c), 9.4 (a), 1.1 (g) | — |
| 1-AG | 0.09 ± 0.07 | 1.58 ± 0.43 | 0.18 (e) | — |
| 2-LG | 2.31 ± 0.80 | 9.24 ± 2.92 | 7.8 (c), 15.7 (a), 27.6 (g) | — |
| 1-LG | 1.00 ± 0.29 | 1.61 ± 0.31 | — | — |
| 2-OG | 6.18 ± 3.95 | 15.7 ± 10.4 | 8.93 (c), 17.8 (g) | — |
| NE | <LOD | <LOD | — | — |
| NAGly | 0.25 ± 0.09 | <LOD | 0.61 (a) | — |
Our results further show that the majority of FEAs were bound to their respective NAPE precursors, and only 1–5% of circulating FEAs were present in their free form. The most abundant compounds were NAPEs of PEA, SEA, and OEA with concentrations of 330 ± 53.8 to 123 ± 20.5 ng/ml, followed by LEA, VEA, AEA, and DHEA between 39.4 ± 9.79 and 13.7 ± 6.7 ng/ml. In contrast to their free form, NAPE-bound tVEA, GLEA, and ETEA were detected at concentrations between 2.82 ± 1.00 and 0.47 ± 0.11 ng/ml, indicating that a wide range of FAs may be utilized for NAPE biosynthesis, including trans-FAs. NAGly was not detectable after hydrolysis, indicating that this compound is not bound to other lipids. The presently measured NAPE concentrations were generally by multiple times higher than those reported before (29). The reason for this discrepancy may be that the previous study precipitated proteins prior to hydrolysis in methanol, whereas our method used a homogeneous methanolic solution for hydrolysis. Because no phospholipid standards were used during protein precipitation, it cannot be excluded that a fraction of lipids coprecipitated with the protein pellet. Moreover, we monitored several time points over the course of 180 min during hydrolysis (Fig. 4A) and observed that quantitative hydrolysis required 120 min at 60°C. Therefore, the hydrolysis time of 45 min at 60°C applied in the previous study might simply have been too short.
The present concentrations of 2- and 1-AG (0.63 ± 0.21 and 0.09 ± 0.07 ng/ml) were comparable to previous studies. In agreement with published data, the concentration of 2-AG amounted to 87.5% of the sum of 2- and 1-AG, indicating minimal isomerization during our sampling and sample workup procedures (28). Although the protocol of the present study provides a reliable snapshot of endogenous 2- and 1-AG levels, it is difficult to assess whether 1-AG is the product of enzymatic processes or the product of chemical isomerization. We suggest that 1-AG is continuously formed via isomerization in blood, which appears to be in an equilibrium with degradative processes. Concentrations of 2-OG were comparable to a study in which Orlistat was used to stabilize 2-MGs in plasma, whereas the content of 2-LG was much lower (22). Compared with studies without stabilization steps, 2-LG and 2-OG levels of the present study were much lower, indicating that other studies might have overestimated the concentrations due to ex vivo processes (26, 45). In support of this interpretation, the concentrations of lipid-bound 2-LG and 2-OG in the present study were close to concentrations of free circulating 2-LG and 2-OG of other studies, stressing once more that blood stabilization is crucial to quantify these compounds correctly (26, 45). With 18.6 ± 5.1 ng/ml, the content of lipid-bound 2-AG was about 30-fold higher than its free circulating form. To our knowledge, quantitative data of circulating 2-AG-PI has not been published before. One study focusing on phospholipids, however, qualitatively determined that 2-AG-PI is present in abundance in the lipoprotein fraction of human plasma (42).
As elaborated in the introduction, endocannabinoids and ERCs may partially originate as a spillover from signaling events in endocrine tissues. During their diffusion through the lymph into circulation, a fraction of these molecules might be degraded by their catabolic enzymes FAAH and MAGL. Because of this, their concentrations in plasma may be of limited use to assess the endocannabinoid tone within tissues (13). The finding that NAPEs and 2-AG-PI are present in high concentrations in plasma, however, is physiologically important. Because FAEs and 2-AG are released from these precursors, the quantification of NAPEs and 2-AG-PI may be used to calculate the released amounts of FEAs and 2-AG during signaling events. The relative abundance of the different NAPE structures may moreover provide a direct measure of the metabolic flux of the EC system and allow for estimating the signaling potential at the different cannabinoid receptors. For instance, the result that NAPEs of AEA and DHEA are present in a similar concentration range is important, because both AEA and DHEA are known to bind with similar affinities to the CB1 receptor (EC50 of 31 and 50 nM, respectively). Because DHEA is a much weaker agonist at the CB1 receptor, with about one-third of the efficacy of AEA (4, 7), DHEA was suggested to reduce the endocannabinoid tone by competing with AEA. This hypothesis has been verified in vitro and in animal experiments with the result that supplementation with DHEA increased insulin sensitivity and glucose uptake in muscle cells and decreased the accumulation of fat mass in adipose cells in a CB1-dependent manner (46, 47). Thus, the ratio of NAPE-AEA and NAPE-DHEA may potentially serve as a biomarker for the endocannabinoid tone in different tissues and provide insights into the mechanisms that contribute to the metabolic syndrome (1). The quantities of NAPEs might also be used to study the EC system in circulation, rather than in tissue extracts. This idea is substantiated by the fact that NAPEs were observed to be in equilibrium between circulation and lymphatic fluids. One study, for instance, has shown that radiolabeled NAPEs of PEA accumulated in the hypothalamus of mice that were injected with NAPE ip (16). The same study also observed that NAPE-PEA dose dependently reduced food intake in animals, suggesting a functional relationship between circulating NAPEs and central regulation of energy balance.
In summary, we have employed an analytical method, which allows the comprehensive quantification of free and phospholipid-bound endocannabinoids and ERCs in a minimum amount of plasma. We further assessed their stability in detail after sampling and during extraction and have established a protocol which reliably quantifies their endogenous concentrations. The methods can now be applied in human and rodent studies to comprehensively investigate their role in the regulation of food intake, energy metabolism, substance dependence, and inflammation (1, 13, 45).
Supplementary Material
Acknowledgments
The authors thank Christine Meissner for proofreading the manuscript.
Footnotes
Abbreviations:
- 2-AG
- 2-arachidonoylglycerol
- 1-AG
- 1-arachidonoylglycerol
- AEA
- arachidonoyl ethanolamide
- ALEA
- α-linolenic acid
- CV
- coefficient of variation
- DAGL
- diacylglycerol lipase
- DALEA
- dihomo-α-linolenic acid
- DGLEA
- dihomo-γ-linolenic acid
- DHEA
- docosahexaenoic acid ethanolamide
- EEA
- elaidic acid ethanolamide
- EPEA
- eicosapentaenoic acid ethanolamide
- ERC
- endocannabinoid-related compound
- ETEA
- eicosatetraenoic acid ethanolamide
- FAAH
- fatty acid amide hydrolase
- FEA
- fatty acid ethanolamide
- GLEA
- γ-linolenic acid
- GPR
- G-protein coupled receptor
- IS
- internal standard
- KSCN
- potassium thiocyanate
- LG
- lineoylglycerol
- LOD
- limit of detection
- LOQ
- limit of quantification
- MAGL
- monoacylglycerol lipase
- MEA
- myristic acid ethanolamide
- MG
- monoacylglycerol
- NAGly
- N-arachidonylglycine
- NAPE
- N-acylphosphatidyl ethanolamine
- NE
- noladin ether
- OEA
- oleic acid ethanolamine
- OG
- oleoylglycerol
- PEA
- palmitic acid ethanolamide
- PG
- palmitoylglycerol
- PI
- phosphoinositide
- PL
- phospholipase
- POEA
- palmitoleic acid ethanolamide
- SEA
- stearic acid ethanolamide
- sMRM
- scheduled multiple reaction monitoring
- tPeEA
- trans-petroselinic acid ethanolamide
- UHPLC
- ultra-HPLC
- VEA
- vaccenic acid ethanolamide
This study is part of the Neurotrition Project that was supported by the Friedrich-Alexander-Universität Erlangen-Nürnberg Emerging Fields Initiative. It was also supported by Deutsche Forschungsgemeinschaft and Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU) within the funding programme Open Access Publishing.The authors declare that they have no conflict of interest.
The online version of this article (available at http://www.jlr.org) contains a supplement.
REFERENCES
- 1.Watkins B. A., and Kim J.. 2015. The endocannabinoid system: directing eating behavior and macronutrient metabolism. Front. Psychol. 5: 1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown A. J. 2007. Novel cannabinoid receptors. Br. J. Pharmacol. 152: 567–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McHugh D., Hu S. S. J., Rimmerman N., Juknat A., Vogel Z., Walker J. M., and Bradshaw H. B.. 2010. N-arachidonoyl glycine, an abundant endogenous lipid, potently drives directed cellular migration through GPR18, the putative abnormal cannabidiol receptor. BMC Neurosci. 11: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ryberg E., Larsson N., Sjogren S., Hjorth S., Hermansson N. O., Leonova J., Elebring T., Nilsson K., Drmota T., and Greasley P. J.. 2007. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 152: 1092–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fu J., Gaetani S., Oveisi F., Lo Verme J., Serrano A., Rodríguez de Fonseca F., Rosengarth A., Luecke H., Di Giacomo B., Tarzia G., et al. 2003. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-α. Nature. 425: 90. [DOI] [PubMed] [Google Scholar]
- 6.Hansen K. B., Rosenkilde M. M., Knop F. K., Wellner N., Diep T. A., Rehfeld J. F., Andersen U. B., Holst J. J., and Hansen H. S.. 2011. 2-Oleoyl glycerol is a GPR119 agonist and signals GLP-1 release in humans. J. Clin. Endocrinol. Metab. 96: E1409–E1417. [DOI] [PubMed] [Google Scholar]
- 7.Brown I., Cascio M. G., Wahle K. W., Smoum R., Mechoulam R., Ross R. A., Pertwee R. G., and Heys S. D.. 2010. Cannabinoid receptor-dependent and -independent anti-proliferative effects of omega-3 ethanolamides in androgen receptor-positive and -negative prostate cancer cell lines. Carcinogenesis. 31: 1584–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsuboi K., Uyama T., Okamoto Y., and Ueda N.. 2018. Endocannabinoids and related N-acylethanolamines: biological activities and metabolism. Inflamm. Regen. 38: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mead J. R., Irvine S. A., and Ramji D. P.. 2002. Lipoprotein lipase: structure, function, regulation, and role in disease. J. Mol. Med. (Berl.). 80: 753–769. [DOI] [PubMed] [Google Scholar]
- 10.Mu H., and Høy C. E.. 2004. The digestion of dietary triacylglycerols. Prog. Lipid Res. 43: 105–133. [DOI] [PubMed] [Google Scholar]
- 11.Yu J. E., Han S. Y., Wolfson B., and Zhou Q.. 2018. The role of endothelial lipase in lipid metabolism, inflammation, and cancer. Histol. Histopathol. 33: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patricelli M. P., and Cravatt B. F.. 1999. Fatty acid amide hydrolase competitively degrades bioactive amides and esters through a nonconventional catalytic mechanism. Biochemistry. 38: 14125–14130. [DOI] [PubMed] [Google Scholar]
- 13.Hillard C. J. 2018. Circulating endocannabinoids: from whence do they come and where are they going? Neuropsychopharmacology. 43: 155–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Monteleone A. M., Di Marzo V., Monteleone P., Dalle Grave R., Aveta T., Ghoch M. E., Piscitelli F., Volpe U., Calugi S., and Maj M.. 2016. Responses of peripheral endocannabinoids and endocannabinoid-related compounds to hedonic eating in obesity. Eur. J. Nutr. 55: 1799–1805. [DOI] [PubMed] [Google Scholar]
- 15.Artmann A., Petersen G., Hellgren L. I., Boberg J., Skonberg C., Nellemann C., Hansen S. H., and Hansen H. S.. 2008. Influence of dietary fatty acids on endocannabinoid and N-acylethanolamine levels in rat brain, liver and small intestine. Biochim. Biophys. Acta. 1781: 200–212. [DOI] [PubMed] [Google Scholar]
- 16.Gillum M. P., Zhang D., Zhang X. M., Erion D. M., Jamison R. A., Choi C., Dong J., Shanabrough M., Duenas H. R., Frederick D. W., et al. 2008. N-acylphosphatidylethanolamine, a gut- derived circulating factor induced by fat ingestion, inhibits food intake. Cell. 135: 813–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmidt A., Brune K., and Hinz B.. 2006. Determination of the endocannabinoid anandamide in human plasma by high-performance liquid chromatography. Biomed. Chromatogr. 20: 336–342. [DOI] [PubMed] [Google Scholar]
- 18.Jian W., Edom R., Weng N., Zannikos P., Zhang Z., and Wang H.. 2010. Validation and application of an LC-MS/MS method for quantitation of three fatty acid ethanolamides as biomarkers for fatty acid hydrolase inhibition in human plasma. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 878: 1687–1699. [DOI] [PubMed] [Google Scholar]
- 19.Hillard C. J., Weinlander K. M., and Stuhr K. L.. 2012. Contributions of endocannabinoid signaling to psychiatric disorders in humans: genetic and biochemical evidence. Neuroscience. 204: 207–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lam P. M., Marczylo T. H., and Konje J. C.. 2010. Simultaneous measurement of three N-acylethanolamides in human bio-matrices using ultra performance liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 398: 2089–2097. [DOI] [PubMed] [Google Scholar]
- 21.Prescott S. M., and Majerus P. W.. 1983. Characterization of 1,2-diacylglycerol hydrolysis in human platelets. Demonstration of an arachidonoyl-monoacylglycerol intermediate. J. Biol. Chem. 258: 764–769. [PubMed] [Google Scholar]
- 22.Pastor A., Farre M., Fito M., Fernandez-Aranda F., and de la Torre R.. 2014. Analysis of ECs and related compounds in plasma: artifactual isomerization and ex vivo enzymatic generation of 2-MGs. J. Lipid Res. 55: 966–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rouzer C. A., Ghebreselasie K., and Marnett L. J.. 2002. Chemical stability of 2-arachidonylglycerol under biological conditions. Chem. Phys. Lipids. 119: 69–82. [DOI] [PubMed] [Google Scholar]
- 24.Gachet M. S., Rhyn P., Bosch O. G., Quednow B. B., and Gertsch J.. 2015. A quantitative LC-MS/MS method for the measurement of arachidonic acid, prostanoids, endocannabinoids, N-acylethanolamines and steroids in human plasma. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 976–977: 6–18. [DOI] [PubMed] [Google Scholar]
- 25.Marchioni C., de Souza I. D., Grecco C. F., Crippa J. A., Tumas V., and Queiroz M. E. C.. 2017. A column switching ultrahigh-performance liquid chromatography-tandem mass spectrometry method to determine anandamide and 2-arachidonoylglycerol in plasma samples. Anal. Bioanal. Chem. 409: 3587–3596. [DOI] [PubMed] [Google Scholar]
- 26.Gouveia-Figueira S., Spath J., Zivkovic A. M., and Nording M. L.. 2015. Profiling the oxylipin and endocannabinoid metabolome by UPLC-ESI-MS/MS in human plasma to monitor postprandial inflammation. PLoS One. 10: e0132042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ottria R., Ravelli A., Gigli F., and Ciuffreda P.. 2014. Simultaneous ultra-high performance liquid chromathograpy-electrospray ionization-quadrupole-time of flight mass spectrometry quantification of endogenous anandamide and related N-acylethanolamides in bio-matrices. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 958: 83–89. [DOI] [PubMed] [Google Scholar]
- 28.Zoerner A. A., Batkai S., Suchy M. T., Gutzki F. M., Engeli S., Jordan J., and Tsikas D.. 2012. Simultaneous UPLC-MS/MS quantification of the endocannabinoids 2-arachidonoyl glycerol (2AG), 1-arachidonoyl glycerol (1AG), and anandamide in human plasma: minimization of matrix-effects, 2AG/1AG isomerization and degradation by toluene solvent extraction. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 883–884: 161–171. [DOI] [PubMed] [Google Scholar]
- 29.Balvers M. G., Wortelboer H. M., Witkamp R. F., and Verhoeckx K. C.. 2013. Liquid chromatography-tandem mass spectrometry analysis of free and esterified fatty acid N-acyl ethanolamines in plasma and blood cells. Anal. Biochem. 434: 275–283. [DOI] [PubMed] [Google Scholar]
- 30.Röhrig W., Waibel R., Perlwitz C., Pischetsrieder M., and Hoch T.. 2016. Identification of the oleic acid ethanolamide (OEA) isomer cis-vaccenic acid ethanolamide (VEA) as a highly abundant 18:1 fatty acid ethanolamide in blood plasma from rats and humans. Anal. Bioanal. Chem. 408: 6141–6151. [DOI] [PubMed] [Google Scholar]
- 31.Tsikas D. 2010. Identifying and quantifying contaminants contributing to endogenous analytes in gas chromatography/mass spectrometry. Anal. Chem. 82: 7835–7841. [DOI] [PubMed] [Google Scholar]
- 32.Angelini R., Argueta D. A., Piomelli D., and DiPatrizio N. V.. 2017. Identification of a widespread palmitoylethanolamide contamination in standard laboratory glassware. Cannabis Cannabinoid Res. 2: 123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vetri V., Librizzi F., Leone M., and Militello V.. 2007. Thermal aggregation of bovine serum albumin at different pH: comparison with human serum albumin. Eur. Biophys. J. 36: 717–725. [DOI] [PubMed] [Google Scholar]
- 34.Bagger H. L., Ogendal L. H., and Westh P.. 2007. Solute effects on the irreversible aggregation of serum albumin. Biophys. Chem. 130: 17–25. [DOI] [PubMed] [Google Scholar]
- 35.Dodge J. T., Mitchell C., and Hanahan D. J.. 1963. The preparation and chemical characteristics of hemoglobin-free ghosts of human erythrocytes. Arch. Biochem. Biophys. 100: 119–130. [DOI] [PubMed] [Google Scholar]
- 36.Fanelli F., Di Lallo V. D., Belluomo I., De Iasio R., Baccini M., Casadio E., Gasparini D. I., Colavita M., Gambineri A., Grossi G., et al. 2012. Estimation of reference intervals of five endocannabinoids and endocannabinoid related compounds in human plasma by two dimensional-LC/MS/MS. J. Lipid Res. 53: 481–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsuboi K., Okamoto Y., Ikematsu N., Inoue M., Shimizu Y., Uyama T., Wang J., Deutsch D. G., Burns M. P., Ulloa N. M., et al. 2011. Enzymatic formation of N-acylethanolamines from N-acylethanolamine plasmalogen through N-acylphosphatidylethanolamine-hydrolyzing phospholipase D-dependent and -independent pathways. Biochim. Biophys. Acta. 1811: 565–577. [DOI] [PubMed] [Google Scholar]
- 38.Bye J. W., Baxter N. J., Hounslow A. M., Falconer R. J., and Williamson M. P.. 2016. Molecular mechanism for the Hofmeister effect derived from NMR and DSC measurements on barnase. ACS Omega. 1: 669–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cunningham B. A., Tamuralis W., Lis L. J., and Collins J. M.. 1989. Thermodynamic properties of acyl chain and mesophase transitions for phospholipids in KSCN. Biochim. Biophys. Acta Biomembranes. 984: 109–112. [Google Scholar]
- 40.Astarita G., and Piomelli D.. 2009. Lipidomic analysis of endocannabinoid metabolism in biological samples. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 877: 2755–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Michell R. H. 2008. Inositol derivatives: evolution and functions. Nat. Rev. Mol. Cell Biol. 9: 151–161. [DOI] [PubMed] [Google Scholar]
- 42.Dashti M., Kulik W., Hoek F., Veerman E. C., Peppelenbosch M. P., and Rezaee F.. 2011. A phospholipidomic analysis of all defined human plasma lipoproteins. Sci. Rep. 1: 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jones G. A., and Lazarchic M.. 2000. Phosphatidylinositol-specific phospholipase C-gamma1 undergoes pH-induced activation and conformational change. Biochim. Biophys. Acta. 1487: 209–221. [DOI] [PubMed] [Google Scholar]
- 44.Haag M., Schmidt A., Sachsenheimer T., and Brügger B.. 2012. Quantification of signaling lipids by nano-electrospray ionization tandem mass spectrometry (nano-ESI MS/MS). Metabolites. 2: 57–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.García Marchena N., Araos P., Pavón F. J., Ponce G., Pedraz M., Serrano A., Arias F., Romero-Sanchiz P., Suárez J., Pastor A., et al. 2016. Psychiatric comorbidity and plasma levels of 2-acyl-glycerols in outpatient treatment alcohol users. Analysis of gender differences. Adicciones. 29: 83–96. [DOI] [PubMed] [Google Scholar]
- 46.Kim J., Carlson M. E., and Watkins B. A.. 2014. Docosahexaenoyl ethanolamide improves glucose uptake and alters endocannabinoid system gene expression in proliferating and differentiating C2C12 myoblasts. Front. Physiol. 5: 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim J., Carlson M. E., Kuchel G. A., Newman J. W., and Watkins B. A.. 2016. Dietary DHA reduces downstream endocannabinoid and inflammatory gene expression and epididymal fat mass while improving aspects of glucose use in muscle in C57BL/6J mice. Int. J. Obes. (Lond.). 40: 129–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.