

In this perspective, Antonova et al. determine the evolutionary history of all TFIID subunits and place them in a functional context to understand their diversification. This analysis of TFIID evolution exemplifies how phylogenetic protein interrogation aids in uncovering existing structures, drawing parallels between related complexes and challenges offered by genome expansions that can be countered by exploiting chromatin modifications.
Keywords: basal transcription, phylogenetic analyses, SAGA, TFIID
Abstract
The basal transcription factor TFIID is central for RNA polymerase II-dependent transcription. Human TFIID is endowed with chromatin reader and DNA-binding domains and protein interaction surfaces. Fourteen TFIID TATA-binding protein (TBP)-associated factor (TAF) subunits assemble into the holocomplex, which shares subunits with the Spt–Ada–Gcn5–acetyltransferase (SAGA) coactivator. Here, we discuss the structural and functional evolution of TFIID and its divergence from SAGA. Our orthologous tree and domain analyses reveal dynamic gains and losses of epigenetic readers, plant-specific functions of TAF1 and TAF4, the HEAT2-like repeat in TAF2, and, importantly, the pre-LECA origin of TFIID and SAGA. TFIID evolution exemplifies the dynamic plasticity in transcription complexes in the eukaryotic lineage.
The complexity of eukaryotic organisms requires tightly regulated and fine-tuned gene expression programs for the adaptation to intracellular and extracellular challenges (López-Maury et al. 2008; Rosanova et al. 2017). The basal transcription factor TFIID is critical for gene transcription by RNA polymerase II (Pol II), as it is the first protein complex to recognize core promoters and nucleate preinitiation complex assembly (Gupta et al. 2016). Comprised of TATA-binding protein (TBP) and 13–14 TBP-associated factors (TAFs), the TFIID complex includes a number of domains essential for its core promoter recognition function (Fig. 1; Chalkley and Verrijzer 1999; Vermeulen et al. 2007; Gupta et al. 2016). Several TFIID subunits are shared with the Spt–Ada–Gcn5–acetyltransferase (SAGA) coactivator complex (Fig. 1; Spedale et al. 2012). SAGA is a multimeric complex consisting of several functional modules carrying histone acetyltransferase (HAT) or deubiquitination (DUB) functions (Helmlinger and Tora 2017). The evolutionary link between SAGA and TFIID is evident by shared and paralogous subunits, which resulted from gene duplication and subfunctionalization events (Spedale et al. 2012). However, it is unclear when the ancestral subunits of TFIID and SAGA emerged and how they should be placed on the evolutionary tree of eukaryotes (Fig. 1). Insights into the timing of these duplications helps to understand the subfunctionalization and redundancy of TAFs and TFIID and might also provide a better understanding of the idiosyncrasies of transcription regulation across the whole domain of eukarya.
Figure 1.

Structural variation between human (h) and yeast (y) TFIID and SAGA complexes. Shared TAFs between TFIID and SAGA may reflect a common ancestral origin for the two complexes (here “ancestor?”). Reduction of shared TAFs between TFIID and SAGA in human versus yeast Saccharomyces cerevisiae as well as loss of epigenetic domains in S. cerevisiae (e.g., TAF1 BrDs [bromodomains] and TAF3 PHD) indicate divergence in TFIID and SAGA adaptation to transcriptional requirements across different eukaryotic branches (Matangkasombut et al. 2000; Gangloff et al. 2001a; Spedale et al. 2012). Unique and shared subunits as well as epigenetic reader domains are color-coded as indicated.
Here, we determine the evolutionary history of all TFIID subunits by examining the occurrence and structure of their genes over a time span of almost 2 billion years. TFIID and SAGA subunits are placed in a functional context to understand their diversification. We address the following questions: What is the origin of TFIID? Are functional domains conserved throughout gene duplication events in TAFs? Which functional domains of TAFs are highly dynamic across eukaryotic evolution, and which ones are relatively stable? When did SAGA and TFIID duplicate and diverge? How did TFIID diversify in structure and function to meet the growing morphological complexity across evolving species?
These questions are examined by phylogenetic comparisons and by profile searches across a set of well-annotated genomes representative of the eukaryotic kingdom (Supplemental Fig. S1). The results are organized per sets of functionally similar TAFs. First, we start by examining the three TAFs implicated in chromatin binding (TAF1, TAF2, and TAF3). Second, we determine the relationships between TAF8, TAF3, and the SAGA subunit SPT7. Third, we describe the evolutionary rather invariant subunits TAF5, TAF6, TAF7, TAF9, and TAF10, including their paralogs. Fourth, the relationships of the TAF4 and TAF12 pair are analyzed with respect to the ADA1 subunit of SAGA. Finally, we propose models for the origin of TAF11, TAF13, and their SAGA paralog, SPT3. Our combined results strongly support a pre-LECA (last eukaryotic common ancestor) origin for the complete TFIID complex, comprising the full ensemble of TAFs. Later lineage-specific duplications resulted in TAF1L, TAF3, TAF4B, TAF4x, TAF7L, TAF9B, and the TAF12 paralog, EER4, which allowed subfunctionalizations to support increasingly complex multicellularity. Highly sensitive profile searches in a representative set of eukaryotic proteomes indicate a dynamic distribution of the bromodomain (BrD) and plant homeodomain (PHD) epigenetic domains within TFIID evolution. These dynamic domains are in sharp contrast to the invariable histone fold (HF), WD40, and HEAT domains, whose conservation reflects their central role in the complex integrity (Kolesnikova et al. 2018; Patel et al. 2018). Additionally, besides TAF paralogous subfunctionalizations, we characterize a stable ancestral repertoire of TFIID subunits combined with a persistent and invariable structure across the entire eukaryotic lineage.
Evolutionary dynamics of the basal transcription machinery
TFIID uses TBP to recognize the TATA element of core Pol II promoters, and this has been well studied (Tora and Timmers 2010). Stable binding of TBP to TATA-boxes involves insertion of two highly conserved phenylalanine pairs of TBP into the DNA, which results in an ∼80° angle. In vitro binding of TATA by TBP displays a long half-life, which is countered by the NC2 and BTAF1/MOT1 regulators of TBP activity. These proteins are required for the dynamic behavior of TBP in vivo (Tora and Timmers 2010). Phylogenetic comparisons revealed that these two phenylalanine pairs in TBP coevolved with genes encoding NC2 and BTAF1/MOT1 (Koster et al. 2015). TBP variants binding less stably to TATA elements do not seem to require NC2 and BTAF1/MOT1, indicating that the Pol II basal transcription machinery can adapt to evolutionary pressures. All eukarya contain at least a single gene for TBP, but TBP homologs can also be found in certain archaeal lineages. However, the genes encoding NC2, BTAF1, or the TFIID TAFs are unique to eukarya (Koster et al. 2015; our unpublished results) and absent from currently available archaeal genomes. It has been shown that several TAFs are duplicated in eukaryotic evolution, which suggests that functional and structural divergence correlates with increased transcriptional complexity. In metazoa, TBP and TAF paralogs are actively involved in promoting development and differentiation as well as maintaining cell and tissue identity (Frontini et al. 2005; D'Alessio et al. 2009; Pijnappel et al. 2013; Zhou et al. 2013, 2014).
Domain analysis of TAF1 and TAF2 reveals BrD dynamics in fungi
The two largest TFIID subunits are represented by the TAF1 and TAF2 proteins. TAF1 is characterized by a variety of domains, which together classify it as the most structurally diverse subunit of TFIID. This complexity is reflected by the large size of the protein, its neuronal-specific alternative splicing, and its functions in both chromatin binding and complex stabilization (Chalkley and Verrijzer 1999; Gupta et al. 2016). The tandem BrDs of metazoan TAF1 are central to TFIID function as a transcription regulatory complex. BrDs mediate binding to acetylated lysines on histones H3 and H4, which are hallmarks of active promoters and transcription (Jacobson et al. 2000). Interestingly, human TAF1 is localized on the X chromosome, and missense mutations in the BrD region have been identified in male patients with intellectual disability phenotypes (O'Rawe et al. 2015). In the plant Arabidopsis thaliana (see the Glossary for species, clades, etc.), TAF1 only has a single BrD, and Saccharomyces cerevisiae TAF1 lacks both BrDs (Matangkasombut et al. 2000; Bertrand et al. 2005). These observations prompted us to investigate in more detail at which point during evolution (see Supplemental Fig. S1 for an evolutionary time tree) the BrDs were lost or acquired.
Analysis of TAF1 domain organization among orthologs revealed a secondary loss of the BrD in the ancestor of dikarya, a subkingdom of fungi that contains ascomycota and basidiomycota (Fig. 2A). Notably, ascomycota include model organisms such as S. cerevisiae, Kluyveromyces lactis, Neurospora crassa, and Schizosaccharomyces pombe. Furthermore, the BrD has an overall patchy (or irregular) occurrence in fungal species and points toward independent loss in at least five lineages, including mucoromycota, chytridiomycota, and the early fungal relative fonticula (Supplemental Figs. S1, S2). All metazoans, on the other hand, possess a second BrD acquired in their common ancestor. The presence of a BrD is predicted to have direct implications for TAF1 binding to acetylated nucleosomes in the vicinity of transcription regulatory elements (Jacobson et al. 2000). The absence of BrDs in fungal lineages suggests that acetylated nucleosomes do not serve as anchoring points for fungal TFIID.
Figure 2.

Inferred evolutionary history of TAF1 and TAF2. (A) TAF1 is duplicated in the Old World monkeys. BrD is gained in the ancestor of metazoa and lost in dikarya. Streptophyta acquired a ubiquitin-like domain. (B) TAF2 contains previously unrecognized HEAT2-like repeats. Various BrDs were acquired early in fungal evolution and subsequently lost late in fungi. Duplications are represented as red arrows; gradient domains are not predicted in all species of that respective (super)group.
In contrast to TAF1, TAF2 domain analysis revealed highly dynamic BrD distribution across fungi (Fig. 2B). While TAF2 from ascomycota does not possess any BrD (similarly to TAF1), differential loss and gain of a variable number of BrDs (ranging from one to six) was observed in TAF2 from members of the basidiomycota, mucoromycota, and blastocladiomycota. The dynamic nature of the TAF2 BrDs is emphasized by a differential occurrence in related fungi. For example, the two closely related species of Mucor circinelloides and Phycomyces blakesleeanus (members of mucoromycotina) (Supplemental Fig. S1) contains six BrDs or no BrD, respectively (Supplemental Fig. S3). Mortierellomycetes (closely related to mucoromycotina) contain four BrDs. As indicated, TAF2 from ascomycota consistently lacks a BrD, but TAF2 from their closest basidiomycota contains BrDs (Supplemental Figs. S1, S3). These BrDs were likely acquired in a fungal ancestor, since their emergence is only specific for the fungal branch of the eukaryotic tree.
Altogether, the analysis of BrD occurrence across TAFs shows that this domain is present only in TAF1 and TAF2. Due to its direct involvement in epigenetic regulation via acetylated histone recognition, the overall BrD dynamics observed in TAF1 and TAF2 emphasize differential transcription regulation in diverse eukaryotic lineages. Notably, in fungal TFIID, BrDs were differentially acquired and have been independently lost multiple times during evolution. Several fungal species seem to compensate for the absence of a TAF1 BrD by the presence of multiple BrDs in TAF2. Transfer of the BrD from TAF1 to TAF2 could influence TFIID conformation on the core promoter but could also reflect different histone acetylation patterns between species.
Beside BrDs, TAF1 structure is characterized by an N-terminal TBP-binding domain (TAND) (Burley and Roeder 1998), a central domain for dimerization with TAF7 (Bhattacharya et al. 2014), and a zinc finger region (zf-CCHC_6) or Zn knuckle only recently described as involved in core promoter DNA binding (Curran et al. 2018). Previous work indicated that TAF1 can bind to the INR element of core promoters (Chalkley and Verrijzer 1999), but this function has not been mapped to a TAF1 domain yet. Examination of domain dynamics of the TAND and Zn knuckle regions across species is limited by the low sequence conservation of these regions, resulting in their patchy (or irregular) distributions across the phylogenetic tree (Fig. 2A). However, the observation that both domains occur in numerous common ancestors indicates that they have been present in an ancient eukaryotic progenitor.
In A. thaliana, a TAF1 ubiquitin-like module has been proposed (Bertrand et al. 2005). Our analysis indicates that this module was acquired as early as the ancestor of the streptophyta, which contains land plants and related eukaryotic algae (Fig. 2A). Klebsormidium flaccidum is the earliest branching species that contains a ubiquitin-like domain, which is retained up to A. thaliana (Supplemental Figs. S1, S2). The domain is inserted into the TAF7 interaction domain (Bertrand et al. 2005; Wang et al. 2014). The existence of a conserved ubiquitin-like domain within TAF1 in streptophyta is suggestive of regulatory processes involving ubiquitin-binding modules, but the exact link with transcription remains to be discovered. Finally, our analysis did not reveal any HAT or kinase domain within TAF1, which has been suggested previously (Matangkasombut et al. 2000).
Besides domain rearrangements, TAF1 evolution is further marked by a duplicative retrotransposition event in the hominoid lineage. This TAF1L gene was first identified in Old World monkeys (cercopithecidae) using intron-spanning primers, and protein expression is only observed in the testis (Wang and Page 2002). Adding Old and New World monkeys to the data set and analyzing TAF1 paralogs confirmed this duplication timing, as there is no paralogous TAF1 gene in Callithrix jacchus, a New World monkey, while the Old World monkey genomes of Hylobates leucogenys and Papio anubus contain TAF1L (Fig. 2A; Supplemental Figs. S1, S2). Furthermore, TAF1L of these species clusters with human TAF1L in the gene tree (Supplemental Fig. S2), which reflects the close relationship of Homo sapiens with Old World monkeys.
Domain interrogation highlights a HEAT2-like repeat region in TAF2
TAF2 is characterized by the N-terminal aminopeptidase M1-like domain, homologous to the catalytic domain of leukotriene A4 hydrolase (LTA4H), a member of the structurally conserved M1 aminopeptidase family of proteins (Papai et al. 2009; Drinkwater et al. 2017). Despite this homology, the signature exopeptidase motif of M1 aminopeptidase, GxMxN, is not present in any of the TAF2 orthologs (data not shown), indicating that the protein lacks peptidase activity. However, the zinc-binding motif of the catalytic site, HExxHx18E, is present in a number of TAF2 orthologs, including human (Hosein et al. 2010). Domain investigation of TAF2 confirmed the consistent presence of peptidase M1-like across all eukaryotes (Fig. 2B; Supplemental Fig. S3), indicating a pre-LECA origin.
Interestingly, analysis using the Conserved Domain Database (CDD; NCBI) in the TAF2 orthologous group revealed a HEAT2-like repeat region (data not shown). The presence of a HEAT structure in TAF2 is consistent with recent cryo-EM results of human TFIID, which revealed a density in TAF2 with architecture resembling an armadillo fold (Louder et al. 2016). The study used human endoplasmic reticulum aminopeptidase1 (ERAP1), also a member of the M1 aminopeptidase family, for homology-based modeling of TAF2 into the structure of TFIID. Notably, atomic structure analysis of ERAP1 revealed eight atypical HEAT repeats at its C terminus (Nguyen et al. 2011). To enhance the sensitivity for detection of HEATs in our TAF2 orthologous group, the identified HEAT region was included in the tree analysis, since the best predictors for a HEAT repeat identity are protein internal repeats (Yoshimura and Hirano 2016). Indeed, two pairs of HEAT2-like repeats were identified and are ubiquitously present in TAF2 orthologs across all eukaryotes (Fig. 2B). The first pair of HEAT repeat resides in all TAF2 orthologs, while the second repeat is present mainly in metazoa and has a patchy distribution across the rest of the eukaryotic tree. However, even with the enhanced detection sensitivity, the sequence divergence in HEAT repeat sequences was quite large, and, outside the regions of HEAT2 homology, the exact architecture and number of the expected repeats could not be determined. Based on our analyses and the ERAP1 structure, we propose that human TAF2 contains a HEAT2-like region spanning amino acids 646–976 and likely consisting of eight repeats (Fig. 2B).
In conclusion, our analyses showed that, unlike their epigenetic domains, the remaining structures of TAF1 and TAF2 are invariable across eukaryotes and most likely have a pre-LECA origin. A notable exception is the ubiquitin-like domain insertion in TAF1, originating in the plant branches of eukaryotic evolution. TAF2 in LECA contained the N-terminal aminopeptidase M1-like domain followed by a region of HEAT2-like repeats.
The TAF3 PHD finger is dynamic in the eukaryotic lineage
Generation of the orthologous tree for TAF3 was complicated by consistent cross-identification of TAF8 and the SPT7 (SUPT7L in metazoa) subunit of SAGA in the profile search. Notably, all three proteins are shown to form a HF pair with TAF10 (Gangloff et al. 2001b). Therefore, the proteins were combined in a single group. Investigation of their evolutionary relationship indicated that they share a highly conserved HF domain (HFD), which is the reason for their cross-identification. In addition to the HFD, TAF8 is characterized by a proline-rich region, which is invariable across species (Fig. 3). This allowed separation of TAF3 and SPT7 to examine their domain evolution. In contrast to TAF8, both TAF3 and SPT7 have undergone substantial reorganization across species, which is discussed separately.
Figure 3.

Inferred evolutionary history of TAF3, TAF8, and SPT7. (A) TAF3 arises from a duplication of a shared ancestor of TAF8 in opisthokonta. TAF3 acquired a PHD, which is secondarily lost in late fungi. (B,C) SPT7 duplicated either in the ancestor of the amoebozoa (B) or pre-LECA (C), implying differential loss. Metazoan SPT7 lost its BrD. Duplications are represented as red arrows.
Given the highly similar domain architectures of the TAF3 N-terminal HFD and the C-terminal PHD finger within early branches of fungal mucoromycotina (e.g., in M. circinelloides) and metazoans, it seems that TAF3 emerged in the opisthokonta (animal and fungal lineages and their unicellular relatives but not plants) ancestor through a duplication of TAF8 followed by acquisition of a PHD finger (Fig. 3A; Supplemental Figs. S1, S4). Within opisthokonta, TAF3 is highly variable. As such, human TAF3 includes the C-terminally acquired PHD finger central to H3K4me3 recognition and TFIID association with active promoters (Vermeulen et al. 2007). S. cerevisiae TAF3 lacks this chromatin reader domain (Gangloff et al. 2001a), which is consistent with observations that H3K4me3 modifications are less important for gene transcription in this yeast (Howe et al. 2017). Interrogation of the timing of PHD loss indicates a secondary loss early in fungal evolution based on the presence of a C-terminal PHD finger in mucoromycotina (e.g., M. circinelloides) and metazoa (Fig. 3A; Supplemental Figs. S1, S4). In contrast, fungi branching after the split of mucoromycotina (mainly dikarya) lack a PHD finger and are characterized solely by the HF (Fig. 3A; Supplemental Figs. S1, S4). Interestingly, there is significant overlap between fungal members that lack both the TAF1 BrD and the TAF3 PHD finger, which strengthens the hypothesis of a smaller contribution of chromatin modifications to transcription regulation in these organisms. On the other hand, the gain of TAF2 BrDs in some species may reflect an alternative mechanism for chromatin binding by TFIID or an intermediate stage of adaptation.
Outside of the opisthokonta, most proteins in the gene tree contain a canonical TAF8 or SPT7 but not TAF3, indicating that TAF3 is not present in nonopisthokonta eukaryotic branches such as plants. Earlier work using a yeast two-hybrid approach in A. thaliana could not identify TAF3, supporting its absence in archaeplastida (including also land plants) (Lawit et al. 2007). The absence of TAF3 in archaeplastida suggests that TAF8 may be present in two copies in the TFIID complex in this supergroup in order to match two proposed two-copy stoichiometry of TAF10 within the complex (Bieniossek et al. 2013).
The hypothesis of TAF8 duplication at opisthokonta and the resultant TAF3 is evident only from the domain analysis and not from ortholog clustering (Fig. 3A; Supplemental Fig. S4). Indeed, several TAF3 proteins in ascomycota (fungi) partly cluster close to their TAF8 counterpart, which could be interpreted as independent duplication and convergent evolution of TAF3 in fungi. Furthermore, TAF3 paralogs in mucoromycotina (fungi) cluster together with TAF8 in the supergroup of SAR (stramenopiles, alveolates, and rhizaria), which suggests the presence of one common protein (TAF8) rather than a separate TAF3 protein (Supplemental Fig. S4). In addition, no holozoa (single-celled organisms closely resembling animals) TAF3s are present in the gene tree (Fig. 3A). A possible explanation for such clustering inconsistencies comes from the relatively short sequences of TAF3, TAF8, and SPT7 HFDs used as a baseline for our tree, which likely lacks sufficient evolutionary information. Since all three proteins interact differently with their common interaction partner TAF10 (Gangloff et al. 2001b), significant sequence divergence is also likely to play a role in the observed clustering inconsistencies. Consequently, our TAF3 origin and evolution hypothesis is based mostly on domain organization analysis and not clustering in the gene tree.
In conclusion, TAF8 is present invariantly across the entire eukaryotic lineage and has a pre-LECA origin. Subsequent duplication in opisthokonta most likely gave rise to TAF3. Subsequently, TAF3 acquired a PHD finger, which is retained in metazoa and early fungi (mucoromycotina) and is subsequently lost later in fungal evolution. Similar to TAF1 and TAF2, TAF3 PHD evolution demonstrates the dynamic nature of epigenetic readers within TFIID across the eukaryotic lineage.
Comparative evolutionary analysis of TAF8 and SPT7 reveals BrD gains in SAGA
SPT7 is a SAGA-specific subunit that interacts via its HFD with TAF10, which is shared between SAGA and TFIID across species (Spedale et al. 2012). Domain characterization revealed that in amoebozoa and opisthokonta, ancestral SPT7 includes an N-terminal BrD followed by an HFD (Fig. 3B). Interestingly, in metazoa, SPT7 (hSUPT7L) lacks a BrD, which results from secondary loss in the animal ancestor, as is evidenced by the presence of a BrD in unicellular holozoan SPT7 (Fig. 3B; Supplemental Fig. S4). None of the early animals, such as Nematostella vectensis, retained this BrD, which suggests a functional reduction of metazoan SAGA in binding acetylated lysines (Fig. 3B; Supplemental Figs. S1, S4). This may be compensated for in metazoan SAGA through a BrD in the GCN5 subunit of the HAT module (Hassan et al. 2002). The GCN5 BrD is essential for SAGA chromatin recognition and transcriptional activation (Syntichaki et al. 2000). The SPT7 BrD has been shown to anchor SAGA to acetylated chromatin but was dispensable for the function of the complex in S. cerevisiae (Hassan et al. 2002). Structural interrogation of BrDs suggests conserved core residues involved in the recognition of acetylated lysines surrounded by a target-specific cavity, which differs between individual domains (Josling et al. 2012). As such, while dispensable for SAGA function, the BrD of fungal SPT7 seems to add versatility to the molecular mechanisms of chromatin recognition by SAGA, and this function has been lost in animals. This highlights the diverging functions of SAGA between fungi and metazoa and mimics in reverse TFIID evolution, in which animals, but not fungi, increase their epigenetic dependency through acquisition of relevant domains within the complex.
Similarly to TAF3, timing the origin of SPT7 is challenging, but domain analysis indicates that SPT7 resulted from a duplication event of TAF8. This event occurred either in the ancestor of amoebozoa and opisthokonta (Fig. 3B) or pre-LECA (Fig. 3C). Orthologs of SPT7 are present across all amoebozoa and opisthokonta, which suggests an origin in their ancestor. Nevertheless, there are two proteins in the plant, including archaeplastida that cluster together with SPT7—one from Cyanophora paradoxa (a glaucophyte), which only has the HFD, and another from K. flaccidum, which contains a BrD followed by an HFD (Supplemental Fig. S4). The presence of an SPT7-like sequence outside of amoebozoa and opisthokonta could be due to (1) SPT7 originating pre-LECA and differential loss in the respective supergroups (Fig. 3C); (2) horizontal gene transfer (HGT) to these species, which is a rare eukaryotic event (Leger et al. 2018); or (3) technical difficulties in domain sequence alignment and low conservation. Based on this, the precise timing and origins of SPT7 remains unclear.
In conclusion, we propose that SPT7 originates from either amoebozoa or a pre-LECA TAF8-like ancestor. In the latter case, the SPT7/TAF8 ancestor was probably a subunit of both SAGA and TFIID. A duplication event resulted in ancestral TAF8 and SPT7 proteins, both of which subfunctionalized to TFIID and SAGA, respectively. After this duplication, SPT7 acquired a BrD in the amoebozoa–opisthokonta ancestor and some archaeplastida, which has been lost subsequently in animals. This dynamic gain (in fungi) and loss (in metazoa) of BrDs is reminiscent of their variable occurrence in TAF1 and TAF2 between these two kingdoms, which supports plasticity in binding acetylated lysines. This appears to be a common theme in the dynamic evolution of TFIID and SAGA complexes.
The invariable ancestral repertoire of TFIID
The dynamic domain variations in TAF1, TAF2, and TAF3 are contrasted by relatively stable domain organization of the other TAF subunits. These include the core TFIID subunits TAF5, TAF6, and TAF9 (Bieniossek et al. 2013) as well as TAF10 and TAF7. The TAF4 and TAF12 core subunits appear to have followed a distinct evolutionary pathway in plants and therefore are discussed separately. In addition, while TAF11 and TAF13 maintain their simple HFD-only structure, these proteins are discussed separately in light of their evolutionary link with the SAGA subunit SPT3 (hSUPT3H).
The evolution of TAF5 and TAF6 shares a common theme of duplication into paralogs that subfunctionalized toward TFIID or SAGA. Previous studies speculated on animal-specific timing of duplication, as both paralogs are present in Drosophila melanogaster (Spedale et al. 2012). The earliest detection of TAF5 and TAF6 paralogs in our orthologous gene tree is in N. vectensis, which confirms duplication of TAF5 and TAF6 in an ancestor of metazoa (Fig. 4A,B; Supplemental Figs. S1, S5, S6). This analogous evolution fits well with the close interaction of the proteins in core TFIID and their central role in overall complex integrity (Bieniossek et al. 2013). In addition, a simultaneous occurrence of the SAGA-specific TAF5L and TAF6L paralogs is indicative of the combinatorial structural basis for discrimination between TFIID and SAGA in terms of architecture, assembly, and function.
Figure 4.

Evolutionary history of the relative invariable TFIID subunits. (A) TAF5 duplicated in the ancestor of animals and contains seven WD40 repeats. (B) TAF6 duplicated in the ancestor of animals. TAF5 and TAF6 paralogs subfunctionalized to either SAGA or TFIID. (C) TAF9 duplicated in placentalia but did not subfunctionalize to SAGA. Duplications are represented as red arrows; gradient domains are not predicted in all species of that respective (super)group.
Domain analysis of TAF5 and TAF6 revealed an overall stable organization. TAF5 has been characterized by the presence of a Lis homology domain (LisH) followed by the N-terminal domain 2 (NTD2) and WD40 repeats (Bieniossek et al. 2013; Malkowska et al. 2013). Assessing the domain organization of TAF5 indicated seven WD40 repeats, which are widely spread across all eukaryotes (Fig. 4A; Supplemental Fig. S5). The first and last repeat diverged more compared with the other repeats, which complicated identification using the canonical WD40 model and required optimization of repeat detection. In SAR and excavata (supergroup containing unicellular organisms), the prediction accuracy was insufficient, resulting in a variable number of WD40 repeats ranging from one in Blastocystis hominis to seven in Aplanochytrium kerguelense (Supplemental Fig. S5). Moreover, the sequence length separating the repeats increases, indicating possible structural divergence, which likely contributed to the occasional patchiness of the repeats in our tree (Fig. 4A; Supplemental Fig. S5). The LisH domain also exhibited patchy distribution in the alignments due to low conservation of these sequences.
TAF6 has been characterized previously by the presence of a N-terminal HFD-mediating TAF9 interaction, which is followed by a region of HEAT repeats (Bieniossek et al. 2013). Domain analysis revealed no innovations for TAF6 among eukaryotes (Fig. 4B; Supplemental Fig. S6). Notably, the publicly available TAF6_C_HEAT (Pfam: PF07571) model does not cover the entire HEAT repeat and starts only from helix 2 in repeat 3 to helix 1 in repeat 5 (Scheer et al. 2012). The entire HEAT region in human TAF6 spans from 218 to 477 (Scheer et al. 2012). The individual HEAT repeats have diverged significantly in sequence, which prevents the determination of possible gains or losses in HEAT repeats.
Similar to its TAF6 interaction partner, TAF9 is present across the entire eukaryotic lineage (Fig. 4C; Supplemental Fig. S7). As TAF9 has been duplicated into TAF9b in mammals (Frontini et al. 2005), additional mammals were included in the representative eukaryotic tree to determine the timing of this duplication event. This revealed that gene duplication occurred in the ancestor of placental mammals, as a single TAF9 protein was detected within Ornithorhynchus anatinus (platypuses). Meanwhile, two TAF9 proteins are present within Loxodonta africana (African elephants), each of which clusters with TAF9 and TAF9b of H. sapiens and Mus musculus, respectively (Fig. 4C; Supplemental Figs. S1, S7). Furthermore, Macropus eugenii (wallabies) or Monodelphis domestica (opossums) do not have a TAF9 duplication. These two organisms belong to the marsupialia and are the closest relatives of the placentalia (Deakin 2012). Hence, TAF9 was duplicated later than TAF5 and TAF6. The timing difference suggests distinct functional outcomes for the three duplication events. Indeed, neither TAF9 nor TAF9b subfunctionalized toward SAGA. The structural invariability and functional conservation of TAF9 possibly reflects on its role in complex integrity of both TFIID and SAGA, while the TAF9 interaction partner TAF6 and its associated partner, TAF5, provide context-dependent variability in animals.
The pattern of invariability in one interaction partner while the other is continuously evolving is also observed for other TAFs. A highly conserved structural organization of TAF10 and TAF7 is observed across species, but their interaction partners (TAF3 and TAF8 or TAF1, respectively) are characterized by a dynamic domain organization. In short, TAF10 contains only an HFD and maintains this basic fold across the entire tree of eukaryotes (Supplemental Figs. S8A, S9). TAF7 is characterized by the presence of an NTD (Pfam; TAFII55_N), essential for the interaction with TAF1 (Bhattacharya et al. 2014). TAF7 structure is conserved across all eukaryotes (Supplemental Figs. S8A, S10). The TAF7 paralog TAF7L is essential for spermatogenesis in mice (Cheng et al. 2007), which is striking in light of testis-specific expression of the TAF1 paralog TAF1L (Wang and Page 2002). Timing the duplication for this paralog was challenging due to a low sequence conservation. In the vertebrate ohnolog database, TAF7L has an intermediate confidence for being duplicated in the vertebrate whole-genome duplication (WGD) event (Singh et al. 2015). Addition of vertebrate species to the database does not support this prediction and reveals a likely origin of TAF7L in mammals, as two copies of TAF7 were detected in O. anatinus (Supplemental Figs. S1, S8B, S10)—a part of the monotremes (egg-laying mammals) that branched early in mammalian evolution and has a striking combination of mammalian and reptilian features (Luo et al. 2011).
In summary, TAF5 and TAF6 duplicated in the ancestor to metazoa and are otherwise present across the eukaryotic lineage as a single-copy gene, which stresses the pre-LECA origin of both TAFs. Metazoan paralogs subfunctionalized to localize to either TFIID or SAGA. No domain innovations have been found for either protein. Duplications of TAF7 and TAF9 were specific for later animal branching events (TAF7 in mammalia and TAF9 in placentailia), but none of them is linked to SAGA-specific subfunctionalization. Together with the Old World monkey appearance of TAF1L, these duplications are indicative of animal-specific TFIID subfunctionalization events, which may be linked in part to mammalian reproduction. The presence of TAF5, TAF6, TAF7, TAF9, and TAF10 across the different eukaryotic supergroups implies that these subunits have a pre-LECA origin, since no specific eukaryotic origin could be identified within the early branching events.
Duplications and plant-specific variations for TAF4, Ada1, and TAF12 interaction partners
TAF4 is widespread across eukaryotes and mostly lacking in SAR (with the exception of Oxytricha trifallax) (Supplemental Fig. S11). Still, the TAF12 HFD partner of TAF4 is widespread in SAR (Supplemental Fig. S12), indicating that TAF4's absence is due to poor genome quality or gene prediction. TAF4 is characterized by the presence of a highly disordered N-terminal region, which is followed by an NHR1-binding (or TAFH-binding) motif in animals or an RST-binding motif in plants and an HFD (Gangloff et al. 2001b). The RST motif is found within RCD1, SRO, and TAF4 proteins and is a binding interface for multiple transcription factors (TFs) (Jaspers et al. 2010). RST is proposed to be a streptophytan invention and identified in TAF4 by sequence similarity (Jaspers et al. 2010). Indeed, the motif first appeared in the streptophyta and is not present in green algae or other archaeplastida (Fig. 5A; Supplemental Figs. S1, S11). In animals, NHR1 has a position similar to plant RST, but they do not share any homology (by HHSearch; data not shown). The NHR1 domain was acquired in the ancestor of animals, which is indicated by its presence in N. vectensis and absence in holozoa or amoebozoa (Fig. 5A; Supplemental Figs. S1, S11). Functional analysis of the NHR1 motif showed that it interacts with TFs and is associated with ETO (eight twenty-one) oligomerization (Wei et al. 2007). The functional similarity of RST and NHR1 motifs points toward convergent evolution of TF-binding interfaces within TAF4 and that this TFIID subunit acts as a target for tissue- and lineage-specific regulation of transcription.
Figure 5.

Inferred evolutionary history of TAF4/Ada1 and the TAF12 HF partner. (A) TAF4 duplicated in the ancestor of vertebrates through a WGD. Afterward, an additional small-scale duplication took place, named TAF4x, which is lost in tetrapoda. The RST domain is acquired in the ancestor of streptophyta, while the NHR1 domain is acquired in animals specifically. (B) TAF12 duplicated in the angiosperm through a WGD. (C) TAF4 and Ada1 emerged through a pre-LECA duplication and subfunctionalized to either SAGA or TFIID. WGD events are represented as blue arrows.
TAF12 is conserved across species and consists of a single HFD (Fig. 5B). It is present as a single copy across eukaryotes, except for a gene duplication event yielding EER4 in plants. TAF12 duplication is proposed to have occurred with angiosperm (flowering plant) WGD (Jiao et al. 2011). Streptophyta such as Physcomitrella patens, which branches earlier than angiosperm, do not contain EER4, which confirms the time of duplication (Fig. 5B; Supplemental Figs. S1, S12). This plant-specific innovation in TAF12 mirrors the RST motif variation observed in plant TAF4.
Besides TAF4, TAF12 also interacts via its HFD with the SAGA subunit ADA1 (hTADA1 in humans) (Spedale et al. 2012), which suggests a possible evolutionary link between TAF4 and ADA1. The SAGA-Tad1 domain in ADA1 (Pfam: 12767), which includes the HFD, was used to generate a TAF4/ADA1 tree (Supplemental Fig. S13). This showed that ADA1 duplicated several times within streptophyta, which is consistent with previously recognized duplications in archaeplastida (Srivastava et al. 2015). This plant-specific event again points toward lineage-specific variations within TAF4/TAF12/ADA1 interactions and suggests that the proteins are intimately linked in structure and function. Analysis of the timing of TAF4/ADA1 subfunctionalization showed that both proteins form monophyletic groups in the gene tree, which points toward a duplication and complete subfunctionalization before the emergence of eukaryotes (Supplemental Fig. S13). It seems that TAF4 and ADA1 likely share a pre-LECA ancestor, which resided in both the TFIID and SAGA complexes (Fig. 5C). The duplication event freed this ancestor for specialization toward a single complex.
TAF4 underwent additional duplications as a TFIID subunit in vertebrates. The best known is TAF4B, which emerged after the vertebrate WGD based on the vertebrate ohnolog database (Singh et al. 2015) and additional vertebrate species in our eukaryotic tree (Fig. 5A; Supplemental Figs. S1, S11). Strikingly, we found an additional TAF4 duplication within Latimeria chalumnae (coelacanths), a sarcopterygii (lobe-finned fish) closely related to the tetrapoda (four-limbed vertebrates) (Supplemental Fig. S1; Amemiya et al. 2013). The clustering with other vertebrates confirmed the existence of a paralog next to TAF4 and TAF4B, which we named TAF4x (Fig. 5A). It seems that TAF4x has been lost in tetrapoda. These results are in line with the 2R hypothesis of the vertebrate WGD, pointing toward two back-to-back WGDs followed by differential loss of this TAF4 paralog (Kasahara 2007). Notably, the model organism Danio rerio (a ray-finned fish that belongs to actinopterygii) also still contains TAF4x (Fig. 5A; Supplemental Figs. S1, S11).
Altogether, our data indicated that an ancestral TAF4/ADA1 protein existed pre-LECA, which had undergone duplication and subfunctionalization, resulting in TFIID-specific TAF4 and SAGA-specific ADA1. TAF4 had undergone additional duplications after WGD events in vertebrates, leading to the TAF4B and the fish-specific TAF4x paralogs. TAF4, ADA1, and TAF12 have all undergone plant-specific innovations, indicating differential evolution of these interaction partners within specific eukaryotic plant branches.
Common evolution for TAF11, TAF13, and SPT3 proteins
TAF11 and TAF13 are TFIID-specific HFD interaction partners with a simple organization of a single HFD (Gupta et al. 2017). Notably, the SPT3 subunit of SAGA contains two HFDs in tandem. The HFD of SPT3 at the N-terminal half resembles TAF13, while the one in the C-terminal half is homologous to TAF11 (Gangloff et al. 2001b). TBP has been shown to interact with both the TAF11/TAF13 dimer and SPT3 (Eisenmann et al. 1992; Mengus et al. 1995). This raises questions about the evolutionary relationship between the three proteins. To examine this, the HFDs of SPT3 were separated in order to infer a phylogenetic tree of SPT3-N, SPT3-C, TAF11, and TAF13, which suggest a pre-LECA origin of these proteins (Supplemental Fig. S14). The tree showed two clear clusters—one containing mainly the TAF13 and SPT3-N HFDs, while the other contained the TAF11 and SPT3-C HFDs. Within these clusters, additional separation is also observed between the TAFs and SPT3 sequences, which stresses their subfunctionalization in TFIID and SAGA.
TAF11 and TAF13 are widespread across the entire eukaryotic tree with a few exceptions in SAR species (namely, Albugo laibachii and Bigelowiella natans), which contain a single HFD cluster with the SPT3-N HFD (Supplemental Fig. S14). Due to the HFD sequence similarity, it remains possible that these are TAF13 proteins in reality. Essentially, all opisthokonta contain SPT3, with the most notable exception of Thecamonas trahens (part of apusozoa), which is an early branching sister group of amoebozoa (Supplemental Fig. S1; Paps et al. 2013). In other supergroups, SPT3 is seemingly lost, with the exception of Naegleria gruberi (excavates), Acanthamoeba castellanii (amoebozoa), Galdieria sulphuraria, and Cyanidioschyzon merolae (red algae) (Supplemental Fig. S14). The differential loss of SPT3 outside of opisthokonta suggests the existence of SAGA lacking SPT3 in those organisms or sharing TAF11 and TAF13 with TFIID. This could be resolved by biochemical analysis of SAGA complexes from organisms lacking SPT3.
The TAF11/TAF13/SPT3 gene tree points toward two hypotheses for the origin of these proteins. (1) SPT3 is the ancestral protein (Fig. 6A). This pre-LECA ancestor (aSPT3) would have duplicated, and TAF11 and TAF13 then arose as the result of a gene split. (2)TAF11 and TAF13 were the ancestral proteins (Fig. 6B), both of which were duplicated before fusing into SPT3 in a pre-LECA genome. Irrespective of the exact scenario, the duplication allowed subfunctionalization toward either SAGA (SPT3) or TFIID (TAF11 and TAF13), while the ancestor was likely functional in both complexes. The aSPT3 hypothesis describes the more evolutionarily simple process, which requires only two events (duplication followed by fission). In contrast, the TAF11/TAF13 hypothesis requires two independent duplications followed by a specific fusion between SPT3-N and SPT3-C.
Figure 6.

Inferred evolutionary history of TAF11/TAF13/SPT3. (A) SPT3 is the ancestral protein that gave rise to TAF11 and TAF13 through a duplication followed by a gene fission. (B) TAF11 and TAF13 are ancestral and gave rise to SPT3 through independent duplications followed by gene fusion. WGD events are shown in blue arrows.
In summary, the analysis of the TAF11, TAF13, and SPT3 orthologous groups revealed their common ancestry and pre-LECA roots. Our results reveal that duplication and subfunctionalization differentiated the proteins in TFIID- and SAGA-specific subunits.
Discussion
This work reconstructs the evolutionary history of TAF subunits forming the basal transcription complex TFIID, which is central to all Pol II transcription. A common theme emerging is a pre-LECA origin for all TFIID subunits, with the later duplications resulting in TAF3, TAF4B, TAF4x, TAF7L, TAF9B, and TAF1L. Most likely, an almost complete—as compared with human TFIID—complex existed in pre-LECA ancestors (Fig. 7). Our analysis of the eukaryotic lineage revealed that most of the TAF duplication events occurred predominantly in opisthokonta branches. Large expansions of TF and cofactor families in metazoan evolution have been suggested to support increased morphological and genome complexity (Cheatle Jarvela and Hinman 2015). The observations with TFIID match well with a versatile transcriptional regulation in opisthokonta. The only other clade in which TFIID is duplicating, albeit it to a lesser extent, is plants. Other examples of evolutionary expansions in major cellular complexes are observed in ribosomes, spliceosomes, and proteasomes (Vosseberg and Snel 2017).
Figure 7.
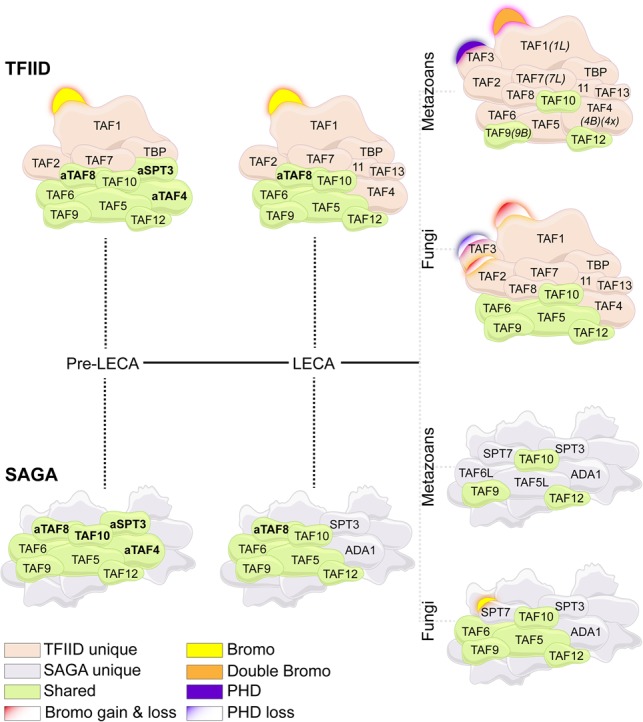
Model of TFIID and SAGA evolutionary divergence from pre-LECA until fungal and metazoan ancestors. In a pre-LECA, the ancestral repertoire (green) of TFIID and SAGA was completely shared. Through duplication and subfunctionalization of the resulting paralogs, the complexes diverged to share fewer subunits throughout eukaryotic evolution (pink and gray). Metazoan TFIID acquired several lineage-specific paralogs (e.g., TAF1L, TAF4B, TAF4x, TAF7L, and TAF9B). Epigenetic domains are differentially gained and lost in metazoan and fungal TFIID and SAGA: Metazoan TFIID acquired epigenetic domains (double BrDs in TAF1 and a PHD in TAF3), while metazoan SAGA lost BrD in SUPT7L (retained in fungal SAGA); in contrast, fungal TFIID gradually lost the TAF3 PHD and carries only one BrD in TAF1 (in some late fungi, the BrDs are completely lost). Additionally, fungal TAF2 displays dynamic gains and losses of numerous BrDs, in contrast to metazoan TAF2. Unique and shared subunits as well as dynamics in epigenetic reader domains are color-coded as indicated.
A salient feature in TFIID evolution is the extensive dynamics of chromatin reader and TF-binding domains between the TAFs in opisthokonta and streptophyta. Notably, highly dynamic chromatin reader domains occur only in the TAF1, TAF2, and TAF3 subunits (Fig. 7). TAF3 was duplicated from TAF8 early in opisthokonta evolution and acquired a PHD finger, which was lost subsequently in later branching fungi (such as dikarya). In striking similarity, metazoan TAF1 acquired a second BrD, while TAF1 in dikarya (branching in fungi) has lost its BrD. In early fungi, highly dynamic BrDs are present in TAF2, which could compensate for the loss in TAF1 BrDs in some fungal species. Ascomycota (part of dikarya) subsequently lost BrDs from TAF2. Interestingly, all common yeast models are included in ascomycota, which suggests that research in S. cerevisiae and S. pombe focuses on an intriguing exception of the TFIID complex. In these model systems, TFIID is entirely deprived of chromatin reader domains as compared with TFIID complexes across the rest of the eukaryotic lineage. This is consistent with previous work in S. cerevisiae, which shows a reduced association of TAFs with chromatin regulators (Huisinga and Pugh 2004). Notably, S. cerevisiae has been characterized by loss of components of other cellular machineries, including the spliceosome and RNA-modifying and protein-folding complexes (Aravind et al. 2000; Vosseberg and Snel 2017). Complexity reduction in evolutionary terms often indicates alternative (and beneficial) functional adaptations of the living organism. Such benefits are exemplified by the lack of an RNAi pathway in S. cerevisiae, which allows for its symbiotic coexistence with the dsRNA killer virus, which is highly toxic for other fungal species (Drinnenberg et al. 2011). With respect to transcription, the loss of epigenetic domains indicates that TFIID becomes less dependent on chromatin marks for targeting to promoter regions during the course of fungal evolution. How this correlates with SAGA fungal evolution, where SPT7 has gained a BrD, remains to be tested. During plant evolution, TAF1 acquired a ubiquitin-like domain in streptophyta, and TAF4 has gained nonhomologous TF-binding interface RST (as opposed to the metazoan NHR1 domain). This indicates that TFIID is a direct TF target in archaeplastida. Furthermore, TAF4 and TAF12 duplications in the plant kingdom indicate possible roles in driving specific lineage programs. The domain analysis of TAF2 revealed the presence of a highly conserved HEAT2-like repeat region. HEAT repeats are commonly present in a wide range of eukaryotic proteins. TAF6 also has a HEAT repeat region, which has been proposed as highly flexible (Yoshimura and Hirano 2016). In TAF1, we also confirmed the presence of a Zn knuckle structure (Curran et al. 2018), which represents a highly conserved Zn finger involved in directing TFIID promoter binding (Curran et al. 2018).
The phylogenetic analysis of TAFs stresses the evolutionary linkage of TFIID with SAGA (Fig. 7). We propose that at least eight invariable subunits (ancestral TAF4, TAF5, TAF6, TAF8, TAF9, TAF10, TAF11/13, and TAF12) were shared between the two complexes and that their divergence already started at a pre-LECA stage (Fig. 7). Probably TAF4/ADA1 and TAF11/TAF13/SPT3 (and possibly TAF8/SPT7) were the first shared members to duplicate and subfunctionalize toward each of the complexes, indicating their core role in TFIID and SAGA structural discrimination. This facilitated functional separation of the TFIID and SAGA complexes. In contrast, TAF5L and TAF6L are more recent SAGA-specific subfunctionalizations. In animals, TFIID shares only three subunits (TAF9, TAF10, and TAF12) with SAGA (Fig. 7). Interestingly, TFIID-specific subfunctionalizations are also evident among metazoa, including TAF4B in vertebrates and TAF4x in fish, mammalian TAF7L, placental-specific TAF9B, and the Old World monkey-specific TAF1L (Fig. 7). The high rate of TAF subfunctionalization coinciding with increased morphological complexity implies a selection for functional divergence of TFIID and SAGA, which started in the pre-LECA era. Our orthologous trees provide a framework for evolutionary reconstruction of the structural changes underlying TAF subfunctionalization through paleostructural biology. From a broader perspective, it is clear that the analysis of TFIID evolution exemplifies how phylogenetic protein interrogation aids in uncovering existing structures, drawing parallels between related complexes, and challenges offered by genome expansions can be countered by exploiting chromatin modifications.
Materials and methods
Phylogenetic analysis of the TFIID complex members
Species and genome selection
To reconstruct the evolution of the TFIID subunits across the eukaryotic tree of life, a selected reference set of species was chosen such that it was large enough to reliably reconstruct TFIID subunit dynamics across the eukaryotic tree of life but small enough for manual curation and inspection of protein phylogenies (Supplemental Table S1). Predicted proteomes for these species were downloaded from diverse sources (Supplemental Table S1), and protein identifiers were changed to allow manual annotation of duplications and losses in the protein trees. For a subset of TFIID subunits, the addition of specific proteins from phylogenetically informative species was essential to accurately time the duplications and losses. These protein-specific additions included primates and placental mammals for TAF1, nontetrapod vertebrates for TAF4, streptophytes for TAF12, and early branching mammals for TAF7 as well as TAF9.
Sequence analysis and alignment
Protein domains were identified using Pfam version 29.0 (Finn et al. 2016) or CDD (Marchler-Bauer et al. 2015) or were based on literature-proposed domains (Supplemental Fig. S15). Orthologous groups for each TAF were acquired using Pfam's gathering cutoffs or manual curation when new HMMER models were made. Sequences were aligned using MAFFT version 7.294 einsi or linsi based on the domain organization of the proteins (Katoh and Standley 2013). linsi was used mostly for orthologous groups where a single domain or excised domains were aligned, while einsi was used for groups with complex domain organizations. Alignments were visualized using Jalview (Waterhouse et al. 2009). After manual inspection, alignments were curated with the trimal option automated if the alignment contained few gaps or gappyout if the alignment was patchy (Capella-Gutierrez et al. 2009). Curated alignments of selected species were visualized using ESPript 3.0, and conserved residues at >70% threshold were marked.
Phylogenetic reconstruction and annotation
Phylogenetic trees were reconstructed with default Phyml version 3.0 settings (LG model of evolution) (Lefort et al. 2017) using the curated alignments (Supplemental Fig. S15). Visualization was done in interactive Tree Of Life (iTOL) (Letunic and Bork 2007). A custom Python script was developed to provide a file for iTOL to color the sequences according to which eukaryotic supergroup the species belong and where the proteins came from (Burki 2014). A second custom python script was developed to provide a file for iTOL to delineate and color domain organization of each protein, as inferred from Pfam searches as described above. The resulting phylogenetic trees were reconciled with the species tree using phylogenetic as well as domain considerations to infer timing of gene duplications and losses. The results of these reconciliations are shown in Figures 2–5 and Supplemental Figures S2–S7 and S9–S14.
Data availability
The results from all intermediate steps as well as all final trees are available at https://bioinformatics.bio.uu.nl/snel/TFIID. These results include custom HMMER models to search for domains, FASTA files of orthologs, selected protein domain alignments (both the FASTA files and the imagery representation), and annotated protein trees. Graphical representations of the domain and protein alignments for selected species are in Supplemental Figures S16–S32.
Glossary
Note: With recent advances in phylogenetics, the classical taxonomy of the eukaryotic tree of life has undergone extensive revisions. As a result, there is a current lack of uniform taxonomic nomenclature for eukaryotes. This glossary aims to familiarize the readers in general terms with the species and names used throughout the study. For further reading on the different classifications, we suggest several reviews (Burki 2014; Brown et al. 2018).
Acanthamoeba castellanii: genus in amoebozoa.
Actinopterygii: ray-finned fish, in which skin webs of the fins are connected by bony spines; kingdom of metazoa.
Albugo laibachii: species belonging to the supergroup of SAR (stramenopiles, alveolates, and rhizaria); pathogens of A. thaliana.
Alveolates: a taxonomic group of primarily single-celled eukaryotes, characterized by the presence of sacs underneath their cell membranes; forms the “A” in the eukaryotic supergroup SAR.
Amoebozoa: a taxonomic group of primarily single-celled eukaryotes, characterized by the presence of pseudopodia and movement through internal cytoplasmic flow.
Angiosperm: a large group in the kingdom of plantae, which includes flowering land plants.
Aplanochytrium kerguelense: a genus included in the eukaryotic supergroup of SAR; a common marine microorganism.
Apusozoa: or obazoa, is an early branching group in eukarya, which includes opisthokonta (also known as fungi and animals but not plants) but excludes amoebozoa.
Arabidopsis thaliana: flowering plant (plantae kingdom); a model organism commonly used in laboratory settings.
Archaeplastida: a taxonomic classification that includes viridiplantae (e.g., land plants and green algae) as well as rhodophytae (e.g., red algae).
Ascomycota: phylum in the fungal subkingdom of dikarya, which includes the commonly used yeast model organisms (e.g., S. cerevisiae, K. lactis, N. crassa, and S. pombe).
Basidiomycota: phylum in the fungal subkingdom of dikarya, which includes mushrooms.
Bigelowiella natans: flagellated species in SAR with a marine lifestyle; model organism in laboratory settings.
Blastocystis hominis: a genus belonging to the eukaryotic supergroup of SAR; contains unicellular parasites capable of infecting humans.
Blastocladiomycota: phylum in the kingdom of fungi; parasitic lifestyle; includes model organisms Allomyces macrogynus and Blastocladiella emersonii.
Callithrix jacchus: common marmoset, a New World monkey; a model organism used in laboratory settings.
Chytridiomycota: division in the kingdom of fungi, characterized by the unique (for fungi) ability to lead a motile lifestyle due to presence of posterior flagellum; a parasite among plants and amphibians.
Cyanidioschyzon merolae: unicellular extremophile adapted to sulphur-rich hot spring environments; red algae; a model organism with minimalist cell structure, used for studying organelle and cellular organization.
Danio rerio: or zebrafish, is a ray-finned fish (skin webs of the fins are connected by bony spines) in the kingdom of metazoa; commonly used model organism in research and popular in aquarium trade.
Dikarya: subkingdom of fungi, also known as “higher fungi.”
Excavata: eukaryotic supergroup, including flagellated unicellular organisms.
Fonticula: a genus with lifestyle similar to clime mold; includes unicellular organisms capable of assembling into multicellular structures; relative of fungi.
Galdieria sulphuraria: species of red algae; a thermoacidophile, suggested to have acquired its extremophilic adaptations through rare horizontal gene transfer events from archaea and bacteria.
Hylobates leucogenys: or Nomascus leucogenys, white-cheeked gibbon; species of Old World monkey.
Holozoa: taxonomic group within opisthokonta that includes animals and closely related unicellular organisms but excludes fungal branches.
Klebsormidium flaccidum: a species of fresh-water filamentous green algae; kingdom of plantae.
Kluyveromyces lactis: a species of Saccharomycetes class (ascomycota division); part of fungi kingdom; commonly used model organism in yeast studies.
Loxodonta africana: or African savanna elephant; mammal; kingdom of metazoa.
Latimeria chalumnae: species of coelacanth (living fossil), lobe-finned fish; fins are supported on a fleshy lobe-like structure connected to the body in a way similar to tetrapod limbs; more closely related to tetrapods than to ray-finned fish, kingdom of metazoa.
LECA: last eukaryotic common ancestor; proposed and reconstructed unicellular organism with nucleus.
Mucor circinelloides: species of mucormycota division; fungi kingdom; frequently infecting farm animals.
Monodelphis domestica: (laboratory) opossum, mammal in the marsupial cohort; metazoa kingdom; model organism.
Macropus eugenii: wallaby, mammal in the marsupial cohort; metazoa kingdom; model organism.
Mammalia: all animals nursing their young with milk; metazoa kingdom.
Marsupialia: cohort of mammals, carrying their young in pouch; metazoa kingdom.
Metazoa: kingdom of animals.
Mortierellomycetes: fungal order, belongs to mucoromycota phylum; fungi kingdom.
Mucoromycota: a lineage in the fungal kingdom, separate from dikarya; includes common bread mold.
Mus musculus: house mouse, mammal in the order rodentia; metazoa kingdom; commonly used model organism.
Naegleria gruberi: species belonging to excavata, capable of changing from amoeba to flagellated unicellular organism with cytoskeletal structure.
Nematostella vectensis: or starlet sea anemone, a species of sea anemone; metazoa kingdom; model organism, holding position at the base of the animal tree; predatory lifestyle.
Neurospora crassa: species of ascomycota (dikarya lineage); fungal kingdom; model organism.
New World monkeys: includes families of primates, distinguished from Old World monkeys and apes in the nasal structure, among others; metazoa kingdom.
Ornithorhynchus anatinus: or platypus, is an egg-laying mammal; metazoa kingdom.
Oxytricha trifallax: species in SAR; ciliated model organism.
Old World monkey: family of primates, more closely related to hominoid lineages than New World monkeys; metazoa kingdom.
Opisthokonta: group of eukarya, which includes animal, fungal lineages, and their unicellular relatives but not plants.
Papio anubis: or olive baboon, member of Old World Monkeys; metazoa kingdom.
Phycomyces blakesleeanus: filamentous fungal species, belongs to mucoromycota phylum; fungi kingdom.
Physcomitrella patens: earth moss, species in the kingdom of plantae; model organism.
Placentalia: cohort of mammals, carrying their young in womb; metazoa kingdom.
Protozoa: unicellular heterotrophic eukaryotes.
Rhizaria: taxonomic group of mostly unicellular organisms, which forms the “R” in the eukaryotic supergroup of SAR.
Saccharomyces cerevisiae: species of ascomycota (dikarya lineage); fungal kingdom; common model organism.
SAR: taxonomic supergroup of primarily single-celled eukaryotes (includes stramenopiles, alveolates, and rhizaria groups).
Sarcopterygii: a class of lobe-finned fish, including coelacanths and closely related to tetrapoda; kingdom of metazoa.
Schizosaccharomyces pombe: species of ascomycota (dikarya lineage); fungal kingdom; common model organism.
Stramenopiles: diverse group of eukaryotes, including plant pathogenic oomycetes, photosynthetic diatoms, and brown algae such as kelp; forms the S in eukaryotic supergroup SAR.
Streptophyta: a branching in the kingdom of plantae that includes land plants and green algae and excludes red algae.
Thecamonas trahens: genus of apusozoa.
Tetrapoda: includes four-limbed vertebrates; kingdom of metazoa.
Supplementary Material
Acknowledgments
We thank Tanja Bhuiyan, Laszlo Tora, and Imre Berger for discussions and critical reading of the manuscript. This research was financially supported by the SFB850 and SFB992 networks of the Deutsche Forschungsgemeinschaft (to H.T.M.T.) and Netherlands Organization for Scientific Research (NWO) grants 022.004.019 (to S.V.A.) and 016.160.638 Vici (to B.S.) as well as ALW820.02.013 (to H.T.M.T.). We apologize to our colleagues whose primary findings could not be cited due to space constraints.
Author contributions: H.T.M.T. and B.S. conceived the study with input from S.V.A. and J.B. J.B. carried out the bioinformatic analysis, assisted by B.S. Data interpretation and presentation were carried out by S.V.A., J.B., H.T.M.T., and B.S. S.V.A. and H.T.M.T. wrote the manuscript together with input from J.B. and B.S.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.300475.117.
References
- Amemiya CT, Alföldi J, Lee AP, Fan S, Philippe H, Maccallum I, Braasch I, Manousaki T, Schneider I, Rohner N, et al. 2013. The African coelacanth genome provides insights into tetrapod evolution. Nature 496: 311–316. 10.1038/nature12027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L, Watanabe H, Lipman DJ, Koonin EV. 2000. Lineage-specific loss and divergence of functionally linked genes in eukaryotes. Proc Natl Acad Sci 97: 11319–11324. 10.1073/pnas.200346997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand C, Benhamed M, Li YF, Ayadi M, Lemonnier G, Renou JP, Delarue M, Zhou DX. 2005. Arabidopsis HAF2 gene encoding TATA-binding protein (TBP)-associated factor TAF1, is required to integrate light signals to regulate gene expression and growth. J Biol Chem 280: 1465–1473. 10.1074/jbc.M409000200 [DOI] [PubMed] [Google Scholar]
- Bhattacharya S, Lou X, Hwang P, Rajashankar KR, Wang X, Gustafsson JA, Fletterick RJ, Jacobson RH, Webb P. 2014. Structural and functional insight into TAF1–TAF7, a subcomplex of transcription factor II D. Proc Natl Acad Sci 111: 9103–9108. 10.1073/pnas.1408293111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieniossek C, Papai G, Schaffitzel C, Garzoni F, Chaillet M, Scheer E, Papadopoulos P, Tora L, Schultz P, Berger I. 2013. The architecture of human general transcription factor TFIID core complex. Nature 493: 699–702. 10.1038/nature11791 [DOI] [PubMed] [Google Scholar]
- Brown MW, Heiss AA, Kamikawa R, Inagaki Y, Yabuki A, Tice AK, Shiratori T, Ishida KI, Hashimoto T, Simpson AGB, et al. 2018. Phylogenomics places orphan protistan lineages in a novel eukaryotic super-group. Genome Biol Evol 10: 427–433. 10.1093/gbe/evy014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burki F. 2014. The eukaryotic tree of life from a global phylogenomic perspective. Cold Spring Harb Perspect Biol 6: a016147 10.1101/cshperspect.a016147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burley SK, Roeder RG. 1998. TATA box mimicry by TFIID: autoinhibition of pol II transcription. Cell 94: 551–553. 10.1016/S0092-8674(00)81596-2 [DOI] [PubMed] [Google Scholar]
- Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25: 1972–1973. 10.1093/bioinformatics/btp348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalkley GE, Verrijzer CP. 1999. DNA binding site selection by RNA polymerase II TAFs: a TAF(II)250-TAF(II)150 complex recognizes the initiator. EMBO J 18: 4835–4845. 10.1093/emboj/18.17.4835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheatle Jarvela AM, Hinman VF. 2015. Evolution of transcription factor function as a mechanism for changing metazoan developmental gene regulatory networks. Evodevo 6: 3 10.1186/2041-9139-6-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Buffone MG, Kouadio M, Goodheart M, Page DC, Gerton GL, Davidson I, Wang PJ. 2007. Abnormal sperm in mice lacking the Taf7l gene. Mol Cell Biol 27: 2582–2589. 10.1128/MCB.01722-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran EC, Wang H, Hinds TR, Zheng N, Wang EH. 2018. Zinc knuckle of TAF1 is a DNA binding module critical for TFIID promoter occupancy. Sci Rep 8: 4630 10.1038/s41598-018-22879-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Alessio JA, Wright KJ, Tjian R. 2009. Shifting players and paradigms in cell-specific transcription. Mol Cell 36: 924–931. 10.1016/j.molcel.2009.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deakin JE. 2012. Marsupial genome sequences: providing insight into evolution and disease. Scientifica 2012: 543176 10.6064/2012/543176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drinkwater N, Lee J, Yang W, Malcolm TR, McGowan S. 2017. M1 aminopeptidases as drug targets: broad applications or therapeutic niche? FEBS J 284: 1473–1488. 10.1111/febs.14009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drinnenberg IA, Fink GR, Bartel DP. 2011. Compatibility with killer explains the rise of RNAi-deficient fungi. Science 333: 1592 10.1126/science.1209575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenmann DM, Arndt KM, Ricupero SL, Rooney JW, Winston F. 1992. SPT3 interacts with TFIID to allow normal transcription in Saccharomyces cerevisiae. Genes Dev 6: 1319–1331. 10.1101/gad.6.7.1319 [DOI] [PubMed] [Google Scholar]
- Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, et al. 2016. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res 44: D279–D285. 10.1093/nar/gkv1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frontini M, Soutoglou E, Argentini M, Bole-Feysot C, Jost B, Scheer E, Tora L. 2005. TAF9b (formerly TAF9L) is a bona fide TAF that has unique and overlapping roles with TAF9. Mol Cell Biol 25: 4638–4649. 10.1128/MCB.25.11.4638-4649.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff YG, Pointud JC, Thuault S, Carre L, Romier C, Muratoglu S, Brand M, Tora L, Couderc JL, Davidson I. 2001a. The TFIID components human TAFII140 and Drosophila BIP2 (TAFII155) are novel metazoan homologues of yeast TAFII47 containing a histone fold and a PHD finger. Mol Cell Biol 21: 5109–5121. 10.1128/MCB.21.15.5109-5121.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff YG, Romier C, Thuault S, Werten S, Davidson I. 2001b. The histone fold is a key structural motif of transcription factor TFIID. Trends Biochem Sci 26: 250–257. 10.1016/S0968-0004(00)01741-2 [DOI] [PubMed] [Google Scholar]
- Gupta K, Sari-Ak D, Haffke M, Trowitzsch S, Berger I. 2016. Zooming in on transcription preinitiation. J Mol Biol 428: 2581–2591. 10.1016/j.jmb.2016.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta K, Watson AA, Baptista T, Scheer E, Chambers AL, Koehler C, Zou J, Obong-Ebong I, Kandiah E, Temblador A, et al. 2017. Architecture of TAF11/TAF13/TBP complex suggests novel regulation properties of general transcription factor TFIID. Elife 6: e30395 10.7554/eLife.30395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan AH, Prochasson P, Neely KE, Galasinski SC, Chandy M, Carrozza MJ, Workman JL. 2002. Function and selectivity of bromodomains in anchoring chromatin-modifying complexes to promoter nucleosomes. Cell 111: 369–379. 10.1016/S0092-8674(02)01005-X [DOI] [PubMed] [Google Scholar]
- Helmlinger D, Tora L. 2017. Sharing the SAGA. Trends Biochem Sci 42: 850–861. 10.1016/j.tibs.2017.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosein FN, Bandyopadhyay A, Peer WA, Murphy AS. 2010. The catalytic and protein–protein interaction domains are required for APM1 function. Plant Physiol 152: 2158–2172. 10.1104/pp.109.148742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe FS, Fischl H, Murray SC, Mellor J. 2017. Is H3K4me3 instructive for transcription activation? Bioessays 39: 1–12. 10.1002/bies.201670013 [DOI] [PubMed] [Google Scholar]
- Huisinga KL, Pugh BF. 2004. A genome-wide housekeeping role for TFIID and a highly regulated stress-related role for SAGA in Saccharomyces cerevisiae. Mol Cell 13: 573–585. 10.1016/S1097-2765(04)00087-5 [DOI] [PubMed] [Google Scholar]
- Jacobson RH, Ladurner AG, King DS, Tjian R. 2000. Structure and function of a human TAFII250 double bromodomain module. Science 288: 1422–1425. 10.1126/science.288.5470.1422 [DOI] [PubMed] [Google Scholar]
- Jaspers P, Overmyer K, Wrzaczek M, Vainonen JP, Blomster T, Salojärvi J, Reddy RA, Kangasjärvi J. 2010. The RST and PARP-like domain containing SRO protein family: analysis of protein structure, function and conservation in land plants. BMC Genomics 11: 170 10.1186/1471-2164-11-170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, Wickett NJ, Ayyampalayam S, Chanderbali AS, Landherr L, Ralph PE, Tomsho LP, Hu Y, Liang H, Soltis PS, et al. 2011. Ancestral polyploidy in seed plants and angiosperms. Nature 473: 97–100. 10.1038/nature09916 [DOI] [PubMed] [Google Scholar]
- Josling GA, Selvarajah SA, Petter M, Duffy MF. 2012. The role of bromodomain proteins in regulating gene expression. Genes 3: 320–343. 10.3390/genes3020320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara M. 2007. The 2R hypothesis: an update. Curr Opin Immunol 19: 547–552. 10.1016/j.coi.2007.07.009 [DOI] [PubMed] [Google Scholar]
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30: 772–780. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolesnikova O, Ben-Shem A, Luo J, Ranish J, Schultz P, Papai G. 2018. Molecular structure of promoter-bound yeast TFIID. Nat Commun 9: 4666 10.1038/s41467-018-07096-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster MJ, Snel B, Timmers HT. 2015. Genesis of chromatin and transcription dynamics in the origin of species. Cell 161: 724–736. 10.1016/j.cell.2015.04.033 [DOI] [PubMed] [Google Scholar]
- Lawit SJ, O'Grady K, Gurley WB, Czarnecka-Verner E. 2007. Yeast two-hybrid map of Arabidopsis TFIID. Plant Mol Biol 64: 73–87. 10.1007/s11103-007-9135-1 [DOI] [PubMed] [Google Scholar]
- Lefort V, Longueville JE, Gascuel O. 2017. SMS: smart model selection in PhyML. Mol Biol Evol 34: 2422–2424. 10.1093/molbev/msx149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leger MM, Eme L, Stairs CW, Roger AJ. 2018. Demystifying eukaryote lateral gene transfer. Bioessays 40: e1700242 10.1002/bies.201700242 [DOI] [PubMed] [Google Scholar]
- Letunic I, Bork P. 2007. Interactive tree of life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23: 127–128. 10.1093/bioinformatics/btl529 [DOI] [PubMed] [Google Scholar]
- López-Maury L, Marguerat S, Bähler J. 2008. Tuning gene expression to changing environments: from rapid responses to evolutionary adaptation. Nat Rev Genet 9: 583–593. 10.1038/nrg2398 [DOI] [PubMed] [Google Scholar]
- Louder RK, He Y, López-Blanco JR, Fang J, Chacón P, Nogales E. 2016. Structure of promoter-bound TFIID and model of human pre-initiation complex assembly. Nature 531: 604–609. 10.1038/nature17394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo ZX, Yuan CX, Meng QJ, Ji Q. 2011. A Jurassic eutherian mammal and divergence of marsupials and placentals. Nature 476: 442–445. 10.1038/nature10291 [DOI] [PubMed] [Google Scholar]
- Malkowska M, Kokoszynska K, Rychlewski L, Wyrwicz L. 2013. Structural bioinformatics of the general transcription factor TFIID. Biochimie 95: 680–691. 10.1016/j.biochi.2012.10.024 [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI, et al. 2015. CDD: NCBI's conserved domain database. Nucleic Acids Res 43: D222–D226. 10.1093/nar/gku1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matangkasombut O, Buratowski RM, Swilling NW, Buratowski S. 2000. Bromodomain factor 1 corresponds to a missing piece of yeast TFIID. Genes Dev 14: 951–962. [PMC free article] [PubMed] [Google Scholar]
- Mengus G, May M, Jacq X, Staub A, Tora L, Chambon P, Davidson I. 1995. Cloning and characterization of hTAFII18, hTAFII20 and hTAFII28: three subunits of the human transcription factor TFIID. EMBO J 14: 1520–1531. 10.1002/j.1460-2075.1995.tb07138.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TT, Chang SC, Evnouchidou I, York IA, Zikos C, Rock KL, Goldberg AL, Stratikos E, Stern LJ. 2011. Structural basis for antigenic peptide precursor processing by the endoplasmic reticulum aminopeptidase ERAP1. Nat Struct Mol Biol 18: 604–613. 10.1038/nsmb.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rawe JA, Wu Y, Dörfel MJ, Rope AF, Au PY, Parboosingh JS, Moon S, Kousi M, Kosma K, Smith CS, et al. 2015. TAF1 variants are associated with dysmorphic features, intellectual disability, and neurological manifestations. Am J Hum Genet 97: 922–932. 10.1016/j.ajhg.2015.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papai G, Tripathi MK, Ruhlmann C, Werten S, Crucifix C, Weil PA, Schultz P. 2009. Mapping the initiator binding Taf2 subunit in the structure of hydrated yeast TFIID. Structure 17: 363–373. 10.1016/j.str.2009.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paps J, Medina-Chacón LA, Marshall W, Suga H, Ruiz-Trillo I. 2013. Molecular phylogeny of unikonts: new insights into the position of apusomonads and ancyromonads and the internal relationships of opisthokonts. Protist 164: 2–12. 10.1016/j.protis.2012.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AB, Louder RK, Greber BJ, Grünberg S, Luo J, Fang J, Liu Y, Ranish J, Hahn S, Nogales E. 2018. Structure of human TFIID and mechanism of TBP loading onto promoter DNA. Science 362: eaau8872 10.1126/science.aau8872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pijnappel WW, Esch D, Baltissen MP, Wu G, Mischerikow N, Bergsma AJ, van der Wal E, Han DW, Bruch H, Moritz S, et al. 2013. A central role for TFIID in the pluripotent transcription circuitry. Nature 495: 516–519. 10.1038/nature11970 [DOI] [PubMed] [Google Scholar]
- Rosanova A, Colliva A, Osella M, Caselle M. 2017. Modelling the evolution of transcription factor binding preferences in complex eukaryotes. Sci Rep 7: 7596 10.1038/s41598-017-07761-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheer E, Delbac F, Tora L, Moras D, Romier C. 2012. TFIID TAF6–TAF9 complex formation involves the HEAT repeat-containing C-terminal domain of TAF6 and is modulated by TAF5 protein. J Biol Chem 287: 27580–27592. 10.1074/jbc.M112.379206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh PP, Arora J, Isambert H. 2015. Identification of ohnolog genes originating from whole genome duplication in early vertebrates, based on synteny comparison across multiple genomes. PLoS Comput Biol 11: e1004394 10.1371/journal.pcbi.1004394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spedale G, Timmers HT, Pijnappel WW. 2012. ATAC-king the complexity of SAGA during evolution. Genes Dev 26: 527–541. 10.1101/gad.184705.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava R, Rai KM, Pandey B, Singh SP, Sawant SV. 2015. Spt–Ada–Gcn5–acetyltransferase (SAGA) complex in plants: genome wide identification, evolutionary conservation and functional determination. PLoS One 10: e0134709 10.1371/journal.pone.0134709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syntichaki P, Topalidou I, Thireos G. 2000. The Gcn5 bromodomain co-ordinates nucleosome remodelling. Nature 404: 414–417. 10.1038/35006136 [DOI] [PubMed] [Google Scholar]
- Tora L, Timmers HT. 2010. The TATA box regulates TATA-binding protein (TBP) dynamics in vivo. Trends Biochem Sci 35: 309–314. 10.1016/j.tibs.2010.01.007 [DOI] [PubMed] [Google Scholar]
- Vermeulen M, Mulder KW, Denissov S, Pijnappel WW, van Schaik FM, Varier RA, Baltissen MP, Stunnenberg HG, Mann M, Timmers HT. 2007. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell 131: 58–69. 10.1016/j.cell.2007.08.016 [DOI] [PubMed] [Google Scholar]
- Vosseberg J, Snel B. 2017. Domestication of self-splicing introns during eukaryogenesis: the rise of the complex spliceosomal machinery. Biol Direct 12: 30 10.1186/s13062-017-0201-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang PJ, Page DC. 2002. Functional substitution for TAF(II)250 by a retroposed homolog that is expressed in human spermatogenesis. Hum Mol Genet 11: 2341–2346. 10.1093/hmg/11.19.2341 [DOI] [PubMed] [Google Scholar]
- Wang H, Curran EC, Hinds TR, Wang EH, Zheng N. 2014. Crystal structure of a TAF1-TAF7 complex in human transcription factor IID reveals a promoter binding module. Cell Res 24: 1433–1444. 10.1038/cr.2014.148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. 2009. Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25: 1189–1191. 10.1093/bioinformatics/btp033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Liu S, Lausen J, Woodrell C, Cho S, Biris N, Kobayashi N, Wei Y, Yokoyama S, Werner MH. 2007. A TAF4-homology domain from the corepressor ETO is a docking platform for positive and negative regulators of transcription. Nat Struct Mol Biol 14: 653–661. 10.1038/nsmb1258 [DOI] [PubMed] [Google Scholar]
- Yoshimura SH, Hirano T. 2016. HEAT repeats - versatile arrays of amphiphilic helices working in crowded environments? J Cell Sci 129: 3963–3970. 10.1242/jcs.185710 [DOI] [PubMed] [Google Scholar]
- Zhou H, Grubisic I, Zheng K, He Y, Wang PJ, Kaplan T, Tjian R. 2013. Taf7l cooperates with Trf2 to regulate spermiogenesis. Proc Natl Acad Sci 110: 16886–16891. 10.1073/pnas.1317034110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Wan B, Grubisic I, Kaplan T, Tjian R. 2014. TAF7L modulates brown adipose tissue formation. Elife 3: e02811 10.7554/eLife.02811 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The results from all intermediate steps as well as all final trees are available at https://bioinformatics.bio.uu.nl/snel/TFIID. These results include custom HMMER models to search for domains, FASTA files of orthologs, selected protein domain alignments (both the FASTA files and the imagery representation), and annotated protein trees. Graphical representations of the domain and protein alignments for selected species are in Supplemental Figures S16–S32.