

In this study, Sun et al. investigated the role of the orphan nuclear receptor SHP, a well-known transcriptional corepressor of bile acid and lipid metabolism in the liver, in other tissues. They report that SHP functions as a regulator of ER stress in the exocrine pancreas, specifically via the regulation of XBP1s stability.
Keywords: SHP, exocrine pancreas, XBP1s, ER stress, Cullin3, ubiquitination
Abstract
The orphan nuclear receptor SHP (small heterodimer partner) is a well-known transcriptional corepressor of bile acid and lipid metabolism in the liver; however, its function in other tissues is poorly understood. Here, we report an unexpected role for SHP in the exocrine pancreas as a modulator of the endoplasmic reticulum (ER) stress response. SHP expression is induced in acinar cells in response to ER stress and regulates the protein stability of the spliced form of X-box-binding protein 1 (XBP1s), a key mediator of ER stress response. Loss of SHP reduces XBP1s protein level and transcriptional activity, which in turn attenuates the ER stress response during the fasting–feeding cycle. Consequently, SHP-deficient mice also are more susceptible to cerulein-induced pancreatitis. Mechanistically, we show that SHP physically interacts with the transactivation domain of XBP1s, thereby inhibiting the polyubiquitination and degradation of XBP1s by the Cullin3–SPOP (speckle-type POZ protein) E3 ligase complex. Together, our data implicate SHP in governing ER homeostasis and identify a novel posttranslational regulatory mechanism for the key ER stress response effector XBP1.
Small heterodimer partner (SHP; NR0B2) is an atypical orphan member of the nuclear receptor family that is best known for its role in hepatic bile acid and lipid metabolism (Lee et al. 2007; Chanda et al. 2008). SHP lacks a canonical DNA-binding domain (DBD) and functions as a transcriptional repressor by interacting directly with other nuclear receptors and transcription factors (Båvner et al. 2005). In hepatocytes, Shp expression is induced by the bile acid-activated nuclear receptor FXR (NR1H4), and, in turn, SHP interacts with nuclear receptors liver receptor homolog-1 (LRH-1; NR5H2) and hepatocyte nuclear factor 4α (HNF4α; NR2A1) to repress the expression of enzymes mediating bile acid synthesis (Goodwin et al. 2000; Lu et al. 2000; Kir et al. 2012). In addition, SHP regulates macrophage inflammation, enteric cholesterol absorption, and gonadal steroidogenesis and suppresses tumorigenesis (Volle et al. 2007; Zhang et al. 2008; Yuk et al. 2011; Kim et al. 2019). Interestingly, while Shp mRNA has been detected in the pancreas (Sanyal et al. 2002), its function there remains unclear.
A key step in nutrient digestion is the synthesis and secretion of digestive enzymes by acinar cells in the exocrine pancreas. The endoplasmic reticulum (ER) is the site for protein synthesis and folding of these digestive enzymes (Logsdon and Ji 2013). During the cycle of fasting and feeding, acinar cells produce more protein than any other cell type in the body. Acinar cells adapt to this dynamic demand in protein synthesis through a universal and highly conserved quality control mechanism known as the unfolded protein response (UPR) to reset ER folding capacity and homeostasis (Walter and Ron 2011). In mammals, the UPR is initiated through the activation of three ER-resident sensors: inositol-requiring enzyme 1α (IRE1α), activating transcription factor 6 (ATF6), and protein kinase R (PKR)-like ER kinase (PERK) (Logsdon and Ji 2013).
IRE1α initiates the most conserved UPR signaling pathway. Upon sensing the accumulation of misfolded proteins in the ER, activated IRE1α splices 26 nucleotides (nt) from the unspliced X-box-binding protein 1 (Xbp1u) mRNA, leading to the generation of an active transcription factor, XBP1s (spliced XBP1) (Logsdon and Ji 2013). XBP1s contains a C-terminal transactivation domain (TAD), allowing it to translocate to the nucleus and induce transcription of genes involved in protein folding and degradation in the ER (Logsdon and Ji 2013). Studies in animal models have revealed the essential role of XBP1s in the exocrine pancreas. The IRE1α–XBP1s pathway is constitutively activated in the exocrine pancreas and is further activated during the fasting–feeding cycle (Yang et al. 2010). Mice deficient in Xbp1 exhibit severe exocrine pancreatic insufficiency, leading to early postnatal lethality (Lee et al. 2005). Xbp1 heterozygous mice are prone to alcohol-induced pancreatic damage (Lugea et al. 2011). Together, these results establish the crucial role of the IRE1α–XBP1s pathway in maintaining ER homeostasis and physiological functions of the exocrine pancreas.
XBP1s transcriptional activity can be regulated further by posttranslational modifications such as phosphorylation, acetylation, and sumoylation (Chen and Qi 2010; Wang et al. 2011; Liu et al. 2016). Moreover, a recent study showed that XBP1s protein stability can also be modulated in natural killer cells in response to interleukin-15 signaling (Wang et al. 2019). How XBP1s protein is degraded in other cell types or tissues remains unclear. Here, we report a positive feedback regulatory mechanism between SHP and XBP1s, wherein Shp expression is induced by XBP1s in response to ER stress. SHP in turn stabilizes XBP1s protein and thereby enhances its activity by inhibiting the Cullin3–SPOP (speckle-type POZ protein) E3 ligase complex that degrades XBP1s. Our findings reveal an unexpected role for SHP as an important regulator of the ER stress response in the exocrine pancreas and identify an E3 ligase complex responsible for XBP1s protein turnover.
Results
SHP expression in the pancreas
Determining in which tissues SHP is expressed has been severely hampered by the lack of SHP-specific antibodies. To circumvent this issue, we generated a knock-in mouse model using CRISPR/Cas9 technology that expresses three Flag tags in tandem at the N terminus of SHP (Fig. 1A). In addition to the liver, SHP protein was highly expressed in the pancreas (Fig. 1A). As a previous two-hybrid screen identified a physical interaction between SHP and XBP1 (Ravasi et al. 2010) and since XBP1s is a key regulator of ER homeostasis in the pancreas (Lee et al. 2005), we hypothesized that SHP may be involved in the regulation of ER homeostasis via XBP1s in the pancreas.
Figure 1.

SHP expression in pancreatic acinar cells is induced by ER stress in an IRE1α–XBP1s-dependent manner. (A) Genomic tagging of endogenous Shp gene loci with 3XFlag in the 3XFlag-Shp knock-in (KI) mice (top panel) and Western blot analysis of SHP expression in different tissues (bottom panel). (Panc) Pancreas; (WAT) white adipose tissue; (BAT) brown adipose tissue; (WT) wild-type mice for negative control; (βactin) a loading control. (B) Quantitative PCR (qPCR) analysis of pancreatic Shp mRNA levels in WT mice treated with dithiothreitol (DTT) at 0.75 mmol per kilogram of body weight for 6 h. (C,D) qPCR analysis of Shp mRNA levels in AR42J acinar cells (C) or primary acinar cells (D) treated with vehicle (Veh), 100 nM thapsigargin (Tg), or 2 mM DTT for 4 h. (E,F) qPCR analysis of mRNA levels of Shp (E) and Erdj4 (F) in AR42J cells treated with vehicle, 30 µM IRE1α inhibitor 4µ8C, and/or 2 mM DTT for 4 h. (G,H) qPCR analysis of mRNA levels of Shp (G) and Erdj4 (H) in Xbp1 knockout AR42J cells (KO-1 and KO-2) or control cells (CON) with a nonspecific targeting guide, treated with vehicle or 2 mM DTT for 4 h. (I) Alignment of the human, rat, and mouse Shp promoters highlighting the putative XBP1s-binding site (underlined). (J,K) Luciferase assays using the mouse Shp promoter (from −492 to +36) showing that Shp is a direct transcriptional target of XBP1s. (J) Sequence of mouse Shp promoter with a mutated (mut) or deleted (del) putative XBP1s-binding site. (K) Cotransfection assay in HEK293T cells expressing luciferase reporter plasmids driven by Shp promoters as indicated and an XBP1s-expressing plasmid. (L) Chromatin immunoprecipitation (ChIP) and qPCR analysis showing enrichment of XBP1s at the Shp and BiP promoters. 266-6 pancreatic acinar cells were transfected with an XBP1s-Flag-expressing plasmid or empty plasmid (control [CON]). ChIP was performed with normal rabbit IgG (IgG) or anti-Flag antibody. The positions of qPCR amplicons relative to the transcription initiation site of each locus are indicated. Representative data of two experiments are shown. In B–K, data are represented as mean ± SEM. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001 by unpaired two-tailed Student's t-test for two-group comparison or by two-way ANOVA analysis for multigroup comparison. See also Supplemental Figure S1.
XBP1s induces Shp gene transcription in pancreatic acinar cells
ER stress-inducing agents such as thapsigargin (Tg) and dithiothreitol (DTT) strongly stimulated Shp expression in the pancreas in vivo and in AR42J acinar cells and primary acinar cells in vitro (Fig. 1B–D). We next determined how ER stress induces Shp. The expression of Shp in acinar cells during ER stress was attenuated by treatment with the IRE1α-specific inhibitor 4µ8C (Fig. 1E; Supplemental Fig. S1A; Cross et al. 2012). As expected, 4µ8C treatment also down-regulated the expression of the XBP1s target genes Erdj4, Hrd1, and BiP but not the PERK downstream target Chop (Fig. 1F; Supplemental Fig. S1B). Deletion of Xbp1 in AR42J cells (Supplemental Fig. S1C) abolished the induction of Shp expression by ER stress (Fig. 1G). Xbp1 deletion also reduced the expression of other IRE1α–XBP1s target genes such as Erdj4, Hrd1, and BiP, but not Chop, in response to ER stress (Fig. 1H; Supplemental Fig. S1D). Taken together, these data suggest that the IRE1α–XBP1s signaling pathway directly regulates Shp expression in response to ER stress.
We next determined whether XBP1s directly regulates Shp gene transcription. Computational analysis of the mouse Shp gene promoter revealed a putative XBP1s-binding site (Acosta-Alvear et al. 2007) at nucleotide −193 upstream of the transcription initiation site that is conserved in rats and humans (Fig. 1I). To establish that XBP1s acts through this site, we constructed mouse Shp promoter (−492 to +36 bp) luciferase reporters with wild-type (WT), mutant, or deleted XBP1s-binding sites (Fig. 1J) and performed cotransfection assays in HEK293T cells expressing XBP1s. Overexpression of XBP1s enhanced luciferase expression driven by the WT Shp promoter, which was significantly attenuated when the XBP1s-binding site was mutated or deleted (Fig. 1J,K). Chromatin immunoprecipitation (ChIP) experiments showed enrichment of XBP1s binding around this element (−190 bp) but not at control sites either upstream of (−2600 bp) or downstream from (+2400 bp) the Shp promoter. As a positive control, we detected XBP1s binding to the Bip promoter at −100 bp but not −2800 bp as reported previously (Yoshida et al. 1998). Collectively, these data demonstrate that Shp is transcriptionally induced by ER stress in pancreatic acinar cells via the IRE1α–XBP1s signaling pathway.
SHP regulates the ER stress response during fasting/refeeding
We next investigated whether SHP plays a physiologic role in UPR signaling in the exocrine pancreas. To that end, we examined the induction of pancreatic UPR in Shp knockout mice during fasting and refeeding. In line with previous studies (Yang et al. 2010), UPR genes were induced in the pancreas of WT mice after refeeding for 2 h following an overnight fast (Fig. 2A,B). Notably, the induction of XBP1s target genes, including Hrd1, BiP, Pdia6, and Erdj4, was either significantly reduced or abolished in the pancreata of Shp knockout mice (Fig. 2A). Shp deficiency had no effect on the PERK signaling pathway, as evidenced by the lack of effect of Shp deficiency on Chop gene expression (Fig. 2B).
Figure 2.

SHP regulates XBP1s protein level and activity during ER stress. WT (Shp+/+), Shp knockout (Shp−/−), and heterozygous (Shp+/−) littermates were fasted overnight (F) and refed (R) for 2 h. Pancreatic tissues were analyzed for mRNA levels of XBP1s target genes (A) and Chop (B), Xbp1 mRNA splicing by RT-PCR (C; quantification at the right), and XBP1s protein levels (D; quantification at the right). Data are represented as mean ± SEM. (**) P < 0.01; (***) P < 0.001 by two-way ANOVA analysis. n = 4 mice of each genotype. In C and D, each lane represents one mouse. Representative data of two experiments are shown. See also Supplemental Figure S2.
To dissect how Shp deficiency down-regulates the IRE1α–XBP1 pathway during ER stress, we analyzed the splicing of Xbp1 mRNA and its protein level. While Xbp1 mRNA splicing by IRE1α in response to feeding was comparable between WT and Shp knockout mice (Fig. 2C), the induction of XBP1s protein levels after refeeding was abolished in the Shp knockout pancreas (Fig. 2D). Shp heterozygous mice were indistinguishable from their WT littermates, suggesting that one copy of Shp is sufficient to maintain XBP1s protein level and activity (Fig. 2A–C). Together, these data indicate that SHP modulates ER homeostasis in the pancreas by posttranslationally regulating XBP1s protein levels.
Because SHP also is highly expressed in the liver (Fig. 1A), we examined whether SHP similarly regulates hepatic XBP1s signaling. Intraperitoneal injection of tunicamycin (Tuni) strongly induced the ER stress response in the liver (Supplemental Fig. S2A,B). Similar to the pancreas, the induction of XBP1s target genes, but not Chop, was attenuated in Shp knockout livers during ER stress (Supplemental Fig. S2A,B). While IRE1α-mediated Xbp1 mRNA splicing was comparable between WT and Shp knockout mice, the induction of XBP1s protein was markedly reduced in the Shp knockout liver in response to ER stress (Supplemental Fig. S2C,D). These results suggest a broader role for SHP in regulating XBP1s protein level and activity.
Shp-deficient mice are prone to cerulein-induced pancreatitis (CIP)
The IRE1α–XBP1s pathway is important for not only maintaining the normal digestive function of the pancreas but also protecting against pancreatitis (Lee et al. 2005; Lugea et al. 2011). To test whether SHP plays a role in this protection, pancreatitis was induced in mice by six hourly injections of cerulein followed by measurement of pancreatitis markers. Relative to WT mice, Shp knockout mice were sensitized to CIP, as evidenced by elevated plasma levels of amylase (Fig. 3A), increased transcription of inflammatory markers and Chop (Fig. 3B), and aggravated edema and cell death in the pancreas (Fig. 3C). Note that the elevated expression of Chop is likely secondary to elevated tissue inflammation and cell death during CIP; thus, it was not observed in the previous experiments (Fig. 2B; Supplemental Fig. S2B). These data show that SHP plays an important role in protecting the pancreas from cellular stress and inflammation during pancreatitis.
Figure 3.
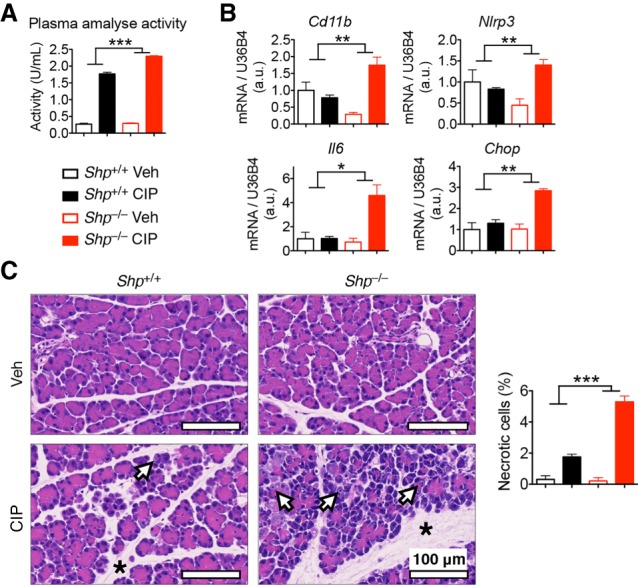
SHP protects mice from CIP. WT (Shp+/+) and SHP knockout (Shp−/−) littermates were treated with cerulein to induce CIP. Mice were analyzed for plasma amylase activity (A), pancreatic mRNA levels of inflammatory genes (B), and pancreatic morphology by H&E staining (C) showing edema (asterisks) and necrotic cells (arrows). The percentage of necrotic cells is quantified at the right. Data are represented as mean ± SEM. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001 by two-way ANOVA analysis. n = 3 for vehicle (Veh); n = 5 for CIP mice of each genotype. Representative data of two experiments are shown.
SHP interacts directly with XBP1s and attenuates its degradation
To investigate how SHP regulates XBP1s protein and activity, we first examined whether the two proteins physically interact. In transfected HEK293T cells, XBP1s was readily immunoprecipitated by HA-tagged SHP (Fig. 4A). Using the 3XFlag-Shp knock-in mice, we also observed that SHP immunoprecipitated XBP1s in the pancreas under refed conditions (Fig. 4B). To map the domain in XBP1s that mediates its interaction with SHP, we generated XBP1s truncations by deleting either the N-terminal DBD or portions of the C-terminal TAD (Fig. 4C; Yoshida et al. 2001; Lee et al. 2002). Interestingly, the last 70 amino acids (301–371) of the C-terminal TAD of XBP1s were required for its interaction with SHP (Fig. 4D).
Figure 4.

SHP interacts with the C-terminal TAD of XBP1s. (A) Western blot analysis of immunoprecipitates of Flag-agarose in HEK293T cells transfected with the indicated plasmids, showing the interaction between XBP1s and SHP. (B) Western blot analysis of immunoprecipitates of Flag-agarose in pancreatic lysates from WT and 3XFlag-SHP knock-in (Flag-ShpKI) mice after an overnight fast and/or 4 h of refeeding, showing the interaction between endogenous XBP1s and SHP under a physiological setting. (C) Truncated mutants of XBP1s used in D. (D) Western blot analysis of immunoprecipitates of Flag-agarose in HEK293T cells transfected with the indicated plasmids, showing the interaction between variants of XBP1s and SHP. Numbers indicate amino acid positions.
Given that XBP1s is a highly unstable protein (Calfon et al. 2002), we speculated that SHP might regulate XBP1s protein levels by modulating its degradation. Indeed, overexpression of SHP markedly inhibited XBP1s polyubiquitination in transfected HEK293T cells (Fig. 5A). To directly measure the XBP1s protein turnover rate, we induced endogenous XBP1s by Tuni injection with or without the protein translation inhibitor cycloheximide (CHX). XBP1s was degraded faster in Shp knockout than in WT mice (Fig. 5B). Thus, SHP interacts with and stabilizes XBP1s protein by attenuating its ubiquitination.
Figure 5.
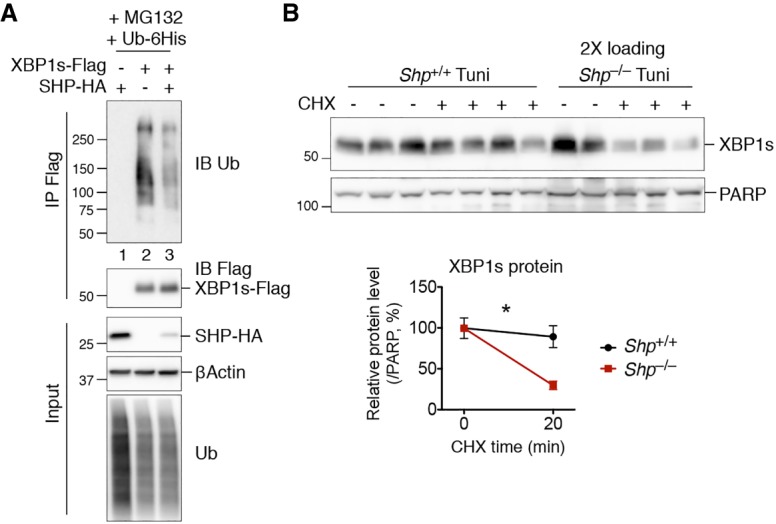
SHP regulates XBP1s protein stability by attenuating its ubiquitination. (A) Western blot analysis of immunoprecipitates of Flag-agarose in HEK293T cells transfected with the indicated plasmids showing the inhibition of XBP1s ubiquitination by SHP. Cells were treated with proteasomal inhibitor MG132 for the last 3 h prior to immunoprecipitation. (B) Western blot analysis of XBP1s protein half-life in mouse livers from WT (Shp+/+) and SHP knockout (Shp−/−) littermates treated with Tuni at 1 mg per kilogram of body weight for 4 h followed by CHX at 2 mg per kilogram of body weight for the last 20 min. Twice the amount of protein lysate was loaded for Shp−/− samples to allow comparable protein levels without CHX. Quantification is shown below. Data are represented as mean ± SEM. (*) P < 0.05 by two-way ANOVA analysis. n = 2–4 mice of each group. Representative data of two experiments are shown.
The Cullin3–SPOP E3 ligase complex mediates XBP1s ubiquitination
We next explored how SHP regulates XBP1s ubiquitination. Treatment with the proteasomal inhibitor MG132 stabilized XBP1s and led to its accumulation, as expected (Fig. 6A, lanes 1–3). Intriguingly, treating cells with MLN4924, an inhibitor targeting the Cullin-based family of E3 ligases (Brownell et al. 2010; Liao et al. 2011), also stabilized XBP1s protein (Fig. 6A, lanes 4–6), although not to the same extent as MG132. These results suggest that Cullins may be one class of E3 ligases that can mediate XBP1s degradation.
Figure 6.

Cullin3 is an XBP1s E3 ligase. (A) Western blot analysis of XBP1s in HEK293T cells transfected with an XBP1s-Flag-expressing plasmid and treated with 10 µM MG132 or 1 µM Cullin inhibitor MLN4924 for 0, 2, or 4 h. Quantification of XBP1s protein levels is shown at the right. Data are represented as mean ± SEM. (B) Western blot analysis of immunoprecipitates of Flag-agarose in HEK293T cells transfected with the indicated plasmids, showing the interaction between XBP1s and Cullin family members. (C,D) Western blot analysis of immunoprecipitates of Myc-agarose in HEK293T cells transfected with the indicated plasmids, showing XBP1s ubiquitination by Cullin3 alone (C, lane 3) or Cullin3 and SPOP (D, lane 3). In C, the level of XBP1s ubiquitination is quantified below the blot. (E) Western blot analysis of endogenous XBP1s decay in Cullin3 knockout 266-6 acinar cells (knockout [KO]) or control cells (CON) with a nonspecific targeting guide, treated with 2.5 nM Tg for 2.5 h followed by 150 μM CHX for 1 h. Quantification is shown at the right. Data are represented as mean ± SEM. (F) Western blot analysis of immunoprecipitates of Flag-agarose in 266-6 cells transfected with XBP1s-Flag-expressing plasmid showing the interaction between XBP1s and endogenous Cullin3 and SPOP. In B–D and F, cells were treated with proteasomal inhibitor MG132 for the last 3 h prior to immunoprecipitation.
The Cullin-based E3 ligases comprise the largest known family of ubiquitin ligases. Cullin proteins function as scaffolds for the assembly of the multiunit E3 ligase complexes, within which specific adaptor proteins confer substrate specificity (Petroski and Deshaies 2005). In order to identify Cullin family members involved in XBP1s ubiquitination, we examined which Cullins interact physically with XBP1s. Among the seven Cullin members, Cullin1, Cullin3, and Cullin4B strongly interacted with XBP1s in transfected HEK293T cells (Fig. 6B). Further analysis revealed that Cullin3 alone induced some polyubiquitination of XBP1s (Fig. 6C). Notably, however, the addition of SPOP, an adaptor protein that recruits protein substrates to Cullin3 (Zhuang et al. 2009), markedly enhanced Cullin3-mediated XBP1s ubiquitination (Fig. 6D). Collectively, these results establish that the Cullin3–SPOP E3 ligase complex can mediate XBP1s ubiquitination.
To establish that Cullin3 mediates XBP1s degradation, we measured endogenous XBP1s protein decay in cells with or without Cullin3. Deletion of Cullin3, but not Cullin1 or Cullin4B, in both HEK293T cells and 266-6 pancreatic acinar cells attenuated XBP1s protein degradation (Fig. 6E; Supplemental Fig. S3A,B). In addition, XBP1s physically interacted with both endogenous Cullin3 and SPOP proteins in both cell types (Fig. 6F; Supplemental Fig. S3C). Thus, the Cullin3–SPOP complex mediates XBP1s protein ubiquitination and degradation.
SHP attenuates Cullin3-mediated XBP1s ubiquitination
Given that SHP inhibits and Cullin3–SPOP induces XBP1s ubiquitination, we next asked whether SHP inhibits the activity of Cullin3/SPOP. First, we analyzed the subcellular localization of these proteins using fractionation and immunocytochemistry. XBP1s, SHP, Cullin3, and SPOP all localized in the nucleus (Fig. 7A; Supplemental Fig. S4). By immunostaining, SHP, Cullin3, and SPOP all colocalized with XBP1s to varying degrees (Fig. 7B; Supplemental Fig. S4). Next, we investigated whether SHP inhibits Cullin3–SPOP-mediated XBP1s ubiquitination. Overexpression of SHP strongly attenuated Cullin3–SPOP-induced XBP1s polyubiquitination (Fig. 7C). Interestingly, similar to SHP, the Cullin3–SPOP complex physically interacted with amino acid residues 301–371 of the C-terminal TAD of XBP1s (Fig. 7D), suggesting that SHP and Cullin3–SPOP compete for XBP1s binding. Indeed, overexpression of SHP effectively inhibited the interaction between XBP1s and Cullin3–SPOP (Fig. 7E). In contrast, Cullin3–SPOP had no effect on the interaction between XBP1s and SHP (Fig. 7E). These data suggest that SHP directly blocks the Cullin3–SPOP E3 ligase complex from interacting with XBP1s, thereby stabilizing XBP1s.
Figure 7.

SHP attenuates Cullin3-mediated XBP1s ubiquitination. (A) Western blot analysis of pancreatic tissues from WT and 3XFlag-SHP knock-in (Flag-ShpKI) mice demonstrating the localization of various proteins in the cytosolic (c) and nuclear (n) fractions. PARP and HSP90 were used as controls for nuclear and cytosolic fractions. (B) Representative confocal images of immunofluorescent staining of HEK293T cells transfected with plasmids expressing the indicated proteins. The level of colocalization is quantified by the Pearson's correlation coefficient (r). Original images as well as negative controls are in Supplemental Figure S4. (C–E) Western blot analysis of immunoprecipitates of Flag-agarose in HEK293T cells transfected with the indicated plasmids, showing that Cullin3/SPOP-induced XBP1s ubiquitination is attenuated by SHP (C), the interaction between truncated variants of XBP1s (see Fig. 4C) and Cullin3/SPOP (D), and that the interaction between XBP1s and Cullin3/SPOP is inhibited by SHP (E). Cells were treated with proteasomal inhibitor MG132 for the last 3 h prior to immunoprecipitation. (F) Model of the positive regulatory loop between XBP1s and SHP that maintains ER homeostasis.
Discussion
In this study, we identified a feedback regulatory loop between SHP and XBP1s that governs ER stress in the pancreas (Fig. 7F). We demonstrate that Shp is a direct transcriptional target of XBP1s in the exocrine pancreas, which in turn regulates the protein stability of XBP1s by inhibiting its polyubiquitination. We further show that this positive regulatory loop is an important part of the normal physiological response to the cycle of fasting and refeeding and that it protects against the pathogenesis of pancreatitis.
XBP1s plays a central role in maintaining ER homeostasis and physiological functions of the exocrine pancreas. Xbp1 heterozygous mice, which have half the amount of XBP1s protein in the pancreas, develop more severe alcohol-induced pancreatitis associated with elevated expression of the proapoptotic gene Chop (Lugea et al. 2011). In this study, we demonstrated that Shp knockout mice were also prone to CIP associated with elevated Chop expression (Fig. 3). The phenotype of Shp knockout mice is consistent with the known prosurvival function of XBP1s (Romero-Ramirez et al. 2004; Gupta et al. 2010) and with the phenotype of Xbp1 heterozygous mice. Interestingly, under basal conditions, Shp knockout mice appear normal in terms of XBP1s protein level (Fig. 2), pancreatic function, and inflammation (Fig. 3), pointing to additional transcriptional or translational mechanisms regulating XBP1s level. Nevertheless, our findings demonstrate that a major role of SHP in the exocrine pancreas is to govern stress-induced XBP1s function. A previous report using single-cell sequencing analysis suggested that SHP is expressed in both the exocrine pancreas and the endocrine pancreas (Segerstolpe et al. 2016). Thus, it is likely that SHP may have additional XBP1s-independent actions in the pancreas.
The XBP1s protein is known to be unstable (Calfon et al. 2002); however, the underlying mechanism remains poorly understood. A recent study suggested that PGC1α represses XBP1s protein level in the liver by promoting its ubiquitination and degradation (Lee et al. 2018). Interestingly, this study mapped amino acid residues 227–252 of XBP1s as required for its interaction with PGC1α (Lee et al. 2018), distinct from residues 301–371, which are required to interact with SHP. Very recently, another study showed that XBP1s ubiquitination and protein stability in natural killer cells are modulated in response to interleukin-15 signaling (Wang et al. 2019). An outstanding question has been the nature of the E3 ligase that mediates XBP1s degradation. Here, we show that the Cullin3/SPOP complex is a strong candidate for one of the E3 ligase activities. Future studies are required to determine whether the Cullin3/SPOP-mediated degradation of XBP1s is regulated by physiological and pathological signals such as fasting, feeding, and inflammation.
We showed previously that Shp is transcriptionally induced by the FXR/RXR nuclear receptor heterodimer in response to bile acid stimulation in the liver, where it functions as a repressor in the feedback regulatory loop that governs bile acid synthesis (Goodwin et al. 2000; Lu et al. 2000). In this study, we report the presence of a conserved XBP1s response element that is distinct from the bile acid/FXR response element in the SHP promoter, suggesting a distinct mode of regulation in response to ER stress. Thus, it is intriguing to compare the differences between bile acid and XBP1s signaling in the exocrine pancreas. Bile acid reflux into the pancreatic duct is proposed to be a triggering event of biliary pancreatitis (Lerch and Aghdassi 2010). It has been shown that bile acids induce acinar cell injury by eliciting calcium release from the ER into the cytosol, which also triggers ER stress (Kim et al. 2002; Voronina et al. 2002; Mekahli et al. 2011). Notably, however, we failed to induce Shp transcription or UPR in vitro by treating AR42J acinar cells with bile acids or the FXR agonist GW4064 (data not shown), further suggesting that the two pathways are distinct. In the future, it will be worth investigating whether bile acids can coordinate with UPR signaling to regulate Shp expression and modulate ER homeostasis in normal pancreatic physiology and during pancreatitis.
In summary, our study establishes the importance of the orphan nuclear receptor SHP in modulating ER homeostasis in vivo. Shp is transcriptionally induced by XBP1s in response to ER stress and in turn regulates the stability of XBP1s by inhibiting the activity of the Cullin3–SPOP E3 ligase complex. We further established the physiological importance of this regulatory mechanism for maintaining normal pancreatic physiology and protecting against the pathogenesis of pancreatitis. This work expands the tissue-specific actions of SHP and the regulation of XBP1 and highlights the cross-talk between these two important signaling pathways.
Materials and methods
Mice
Shp knockout mice (Kerr et al. 2002) and WT littermates were maintained on a C57BL/6J background. For fasting–refeeding experiments, mice were fasted overnight and refed for 2 h with standard chow diet. To induce XBP1s and measure its decay in vivo, mice were injected intraperitoneally with DTT (Sigma, D9779) at 0.75 mmol per kilogram of body weight for 6 h, Tuni (Tocris, 3516) at 1 mg per kilogram of body weight for 4 h, and/or CHX (Millipore, 508739) at 2 mg per kilogram of body weight for 20 min. Acute pancreatitis was induced by the administration of six hourly intraperitoneal injections of cerulein (Tocris, 6264) at 50 mg per kilogram of body weight each time, and tissues were collected 1 h after the last injection. Plasma amylase activity was measured using amylase assay kit per the manufacturer's instructions (Abcam, ab102523). For histology, the pancreas was fixed in 10% neutral-buffered formalin overnight, paraffin-embedded, sectioned, and H&E-stained. Images were acquired with a Zeiss Axioscan Z1 slide scanner, and necrotic cell percentages were quantified by counting the number of necrotic cells and total cells from three random fields of the tissue section of each mouse.
3XFlag-Shp knock-in mice were generated with the CRISPR/Cas9 method (Yang et al. 2013). A 66-nt sequence (5′-GACTACAAGGACCACGACGGTGACTACAAGGACCACGACATCGACTACAAGGACGACGACGACAAG-3′) encoding a 3XFlag tag was inserted in-frame into the 5′ end of the endogenous SHP-coding region right after the translation start codon. A single guide RNA with sequence 5′-TGGGCAGACCCCTGACTGGC-3′ was microinjected into C57BL/6J zygotes in conjunction with the Cas9 mRNA (Trilink Bio) and a single-stranded homology-directed repair (HDR) template containing the 66-nt 3XFlag-coding sequence flanked by the left (66-nt) and right (65-nt) arms of homologous sequence (IDT Ultramer). The injected zygotes were implanted into pseudopregnant female C57BL/6J mice. Mice born from these microinjections were screened by PCR from tail DNA to verify HDR template insertion, and germline transmission was confirmed in the second generation of breeding with WT C57BL/6J mice to eliminate any potential mosaicism. For this study, male homozygous Shp knock-in mice and their WT littermates were used.
Male mice were used for all experiments. All animal experiments were approved by the Institutional Animal Care and Research Advisory Committee of the University of Texas Southwestern Medical Center. Mice were housed in a pathogen-free and temperature-controlled environment with 12-h light/dark cycles (6 am–6 pm) and fed standard irradiated rodent chow (Harlan Teklad, 2916).
Isolation of primary acinar cells
Primary mouse acinar cells were prepared as described previously (Coate et al. 2017). Briefly, pancreata were dissected, rinsed, and cut in ice-cold Hank's buffered salt solution (HBSS) into 1- to 2-mm pieces. The pancreas pieces were then digested with digestion medium (0.75 mg/mL collagenase type I-A [Sigma, C2674], 0.1 mg/mL soybean trypsin inhibitor [Sigma, T6522], 1% BSA, 10% FBS in DMEM) for 35 min at 37°C with constant mixing using serological pipets. Cells were then washed three times with washing buffer (10% FBS, 1% penicillin–streptomycin in DMEM), resuspended in incubation medium (0.1 mg/mL soybean trypsin inhibitor, 1% BSA, 10% FBS, 1% penicillin–streptomycin in DMEM), and filtered through a 100-μm strainer. Cells were plated in incubation medium and allowed to recover for 30 min before the 2 mM DTT treatment for 4 h.
Cell culture
Rat AR42J acinar cells were cultured in DMEM (Gibco) containing 20% heat-inactivated FBS (VWR) and 1% penicillin–streptomycin (Gibco). For RNA or protein analysis, cells were treated with 100 nM Tg (Tocris, 1138), 2 mM DTT, and/or 30 μM 4μ8C (Millipore. 412512) for 4 h. For CRISPR knockouts of Xbp1, Lenti-X 293T cells (Clontech) were transfected with plasmids LentiCRISPR-v2 (Addgene, 52961), psPAX2 (Addgene, 12260), and pMD2 (Addgene, 12259) using Fugene HD reagent (Promega, E2311) to generate lentivirus carrying a nontargeting control guide (5′-GATCGTTTCCGCTTAACGGC-3′) or guides targeting rat Xbp1 (5′-CGCGGGCCCGGAACCATGAG-3′ and 5′-TTCCGGGCCCGCGAGCCGCA-3′). AR42J cells were transduced with lentivirus in the presence of 8 μg/mL polybrene (Millipore, TR-1003-G), selected with 2 μg/mL puromycin for 2 d, and analyzed 8 d after transduction.
HEK293T cells were cultured in DMEM (Gibco) containing 10% FBS and 1% penicillin–streptomycin and transfected with various plasmids using 80 μg/mL polyethylenimine (PEI; Sigma, 408727). After transfection, HEK293T cells were treated with 10 μM MG132 (Millipore, 474790) or 1 μM MLN4924 (Cayman Chemical, 15217) before collection, as indicated in the figure legends. Plasmids used for transfection were as follows (“h” denotes human genes, and “m” denotes mouse genes): pcDNA3-mShp-HA, pcDNA3-Flag-mXBP1s, pcDNA3-Myc-mXBP1s, pcDNA3-6His-mUb, pcDNA3-mSpop-Flag, pcDNA3-mSpop-Myc, and pcDNA3-mCullin3-Myc. mXBP1s truncation mutants (1–161, 1–300, and 140–371) were generated using site-directed mutagenesis from pcDNA3-Flag-mXBP1s. Plasmids pEBB-Flag-hCullin1∼7 were kind gifts from Dr. Ezra Burstein at University of Texas Southwestern. For CRISPR knockouts of Cullin1, Cullin3, and Cullin4B, HEK293T cells were transduced with lentivirus carrying LentiCRISPR-v2 construct with a nontargeting control guide (5′-GATCGTTTCCGCTTAACGGC-3′) or guides targeting human Cullin1 (5′-GTTCGCCGTGAATGTGACGA-3′ and 5′-GCATCCAGCAGGTGTACACA-3′), Cullin3 (5′-ATGATTGCAAGAGAGCGGAA-3′ and 5′-GGAAGAACCAATTGTAAAGG-3′), or Cullin4B (5′-GCTTCTTCTGTATCGGTACG-3′ and 5′-GAATCCCTGGGTTGTAAAGG-3′) and then selected with 1 μg/mL puromycin for 2 d. To analyze XBP1s degradation, 8 d after transduction, cells were treated with 2.5 nM Tg (Tocris, 1138) for 2.5 h followed by vehicle or 150 μM CHX (Millipore, 508739) for 1 h.
Mouse 266-6 acinar cells were cultured in DMEM (Gibco) containing 10% heat-inactivated FBS (VWR), 1 mM sodium pyruvate (Gibco), and 1% penicillin–streptomycin (Gibco). For CRISPR knockouts of Cullin3, cells were transduced with lentivirus carrying LentiCRISPR-v2 construct with a nontargeting control guide (5′-GATCGTTTCCGCTTAACGGC-3′) or a guide targeting mouse Cullin3 (5′- ATGATTGCCAGAGAGCGGAA-3′), selected with 0.5 μg/mL puromycin for 2 d, and analyzed 10 d after transduction.
Luciferase reporter assay
The promoter region of mouse Shp (−492 to +36 bp) was cloned from mouse liver genomic DNA of C57BL/6J mice and ligated into pGL4.23[Luc2/minP] luciferase reporter vector (Promega). The XBP1s-binding site was mutated or deleted by site-directed mutagenesis. For luciferase reporter assay, HEK293T cells were transfected with a luciferase reporter plasmid, a pGL4.75[hRluc/CMV] for normalization, and a pcDNA3-Flag-mXBP1s or empty plasmid using Fugene HD reagent (Promega E2311). Luciferase activity was measured using dual-luciferase reporter assay system (Promega, E1910). The firefly luciferase activity was normalized to the renilla luciferase activity.
Immunoprecipitation
For immunoprecipitation, cells or tissue were lysed by sonication in a buffer containing 150 mM NaCl, 50 mM Tris–HCl (pH 7.5), 1 mM EDTA, 1% NP-40, protease inhibitors, and 5 mM N-ethylmaleimide. A total of 2–5 mg of protein lysates was precleared with protein G agarose for 2 h at 4°C and then incubated with anti-Flag M2 agarose (Sigma, A2220) or anti-Myc agarose (Cell Signaling, 3400) overnight at 4°C with gentle rocking. Immunocomplexes were washed four times in the lysis buffer and eluted by boiling for 5 min at 95°C in SDS sample buffer.
Western blot
Western blot was performed using 10–30 μg of total protein lysates or 20 μg of nuclear extracts. The following antibodies were used in this study: XBP1s (Cell Signaling, 83418), CHOP (Cell Signaling, 2895), PARP (Cell Signaling, 9532), ubiquitin (Cell Signaling, 3933), Cullin1 (Cell Signaling, 4995), Cullin3 (Cell Signaling, 2759), Cullin4B (Proteintech, 12916-1-AP), SPOP (Proteintech, 16750-1-AP), Flag (Sigma, F1804 and F7425), Myc (Cell Signaling, 2278 and 2276), HA (Sigma, H3663; and Cell Signaling, 3724), βActin-HRP (Abcam, ab49900), and HSP90 (Santa Cruz Biotechnology, sc7947). Secondary antibodies—donkey antirabbit or mouse IgG HRP (Jackson ImmunoResearch)—were used at 1:6000 dilution. Western blot membranes were developed using the Clarity Western ECL substrate (Bio-Rad), and signal was detected with an ImageQuant LAS4000 luminescent imager (General Electric). Quantification was performed using ImageQuantTL software (General Electric).
Nuclear fractionation
Nuclear fractionation was performed as described previously (Sha et al. 2009). Briefly, liver or pancreatic tissues were lysed in a hypotonic buffer (10 mM HEPES at pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, protease inhibitors) with dounce tissue grinders (DWK Life Sciences, 885303-0002) and allowed to swell for 15 min on ice followed by the addition of 10% NP-40 to a final concentration of 0.6%. After being vortexed for 15 sec, the lysate was centrifuged at 12,000g for 5 min at 4 °C. Supernatant was collected as the cytosolic fraction. Nuclear pellets were resuspended in a high-salt buffer (20 mM HEPES at pH 7.9, 0.4 M NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT) and vortexed vigorously for 15 sec every 5 min for a total of 20 min. Extracts were centrifuged, and the supernatant was collected as the nuclear fraction.
RNA extraction, RT-PCR for Xbp1 mRNA splicing, and quantitative real-time PCR
Pancreatic RNA was isolated as described previously (Sun et al. 2014; Coate et al. 2017). Briefly, mice were anesthetized, and pancreas tissues were perfused locally with RNAlater reagent (Qiagen, 76104). Pancreatic tissues were then excised and soaked in RNAlater on ice for stabilization. Liver tissues were snap-frozen in liquid nitrogen for RNA extraction. Total RNA from tissues and cultured cells was extracted using RNA-Stat 60 (IsoTex Diagnostics) and RNA purification kit (Omega Biotek, R683402) with DNase I digestion (Roche). RNA quality was determined by measuring the OD260/280 and visualization on an agarose gel. cDNA was generated using the high-capacity cDNA reverse transcription kit (Life Technologies). RT-PCR for Xbp1 mRNA splicing was performed as described previously (Sha et al. 2009) using GoTaq Green master mix (Promega) and an annealing temperature of 58°C for 35 cycles. PCR products were separated by electrophoresis on a 2% agarose gel. The percentage of Xbp1 mRNA splicing, defined as the ratio of XBP1s level to total Xbp1 (Xbp1u + XBP1s) level, was quantified using ImageQuantTL software (General Electric). Quantitative PCR (qPCR) was performed by the SYBR Green method as described (Bookout et al. 2006). U36b4 was used as the reference. The primer sequences used for RT-PCR were rat Xbp1 (TGCCCTGGTTACTGAAGAGG and CCATGGGAAGATGTTCTGGG), rat U36b4 (CACCTTCCCACTGGCTGAA and TCCTCCGACTCTTCCTTTGC), mouse Xbp1 (ACACGCTTGGGAATGGACAC and CCATGGGAAGATGTTCTGGG), and mouse U36b4 (CGTCCTCGTTGGAGTGACA and CGGTGCGTCAGGGATTG). The primer sequences used for qPCR were rat Shp (GGAGCAGCCCTCGTCTCA and ACACTGTATGCAAACCGAGGAA), rat Erdj4 (GCCAAAGTCAGCCTCAGAGA and GGCATCCGAGAGTGTTTCAT), rat Bip (AAGAAGGTCACCCATGCAGTT and ATGACATTCAGTCCAGCAATAGTG), rat Hrd1 (CATGCTGCAAATCAACCAGT and GAGGCTTTTCAGCTTCAGGA), rat Chop (TATCTCATCCCCAGGAAACG and AGGTGCTTGTGACCTCTGCT), mouse Shp (CGATCCTCTTCAACCCAGATG and GGGCTCCAAGACTTCACACA), mouse Hrd1 (AGCTACTTCAGTGAACCCCACT and CTCCTCTACAATGCCCACTGAC), mouse Bip (CAAGGATTGAAATTGAGTCCTTCTT and GGTCCATGTTCAGCTCTTCAAA), mouse Pdia6 (TGGTTCCTTTCCTACCATCACT and ACTTTCACTGCTGGAAAACTGC), mouse Erdj4 (CAGAATTAATCCTGGCCTCC and ACTATTGGCATCCGAGAGTG), mouse Chop (CCAGAAGGAAGTGCATCTTCA and ACTGCACGTGGACCAGGTT), mouse Cd11b (ATCAACACAACCAGAGTGGATTC and GTTCCTCAAGATGACTGCAGAAG), mouse Nlrp3 (GTGGTGACCCTCTGTGAGGT and TCTTCCTGGAGCGCTTCTAA), and mouse Il6 (TCGTGGAAATGAGAAAAGAGTTG and AGTGCATCATCGTTGTTCATACA).
ChIP
266-6 acinar cells were transfected with pcDNA3-Flag-mXBP1s or empty plasmid using Lipofectamine 2000 reagent (Thermo Fisher, 11668019). Twenty hours after transfection, cells were cross-linked by 1% formaldehyde for 10 min at room temperature. ChIP was performed using SimpleChIP plus enzymatic ChIP kit with magnetic beads (Cell Signaling, 9005) following the protocol provided. Immunoprecipitation was performed with normal rabbit IgG (Cell Signaling, 2729) or ChIP-grade anti-Flag antibody (Cell Signaling, 14793). The primer sequences used for qPCR analysis were as follows: Shp, −190 bp (CAATGGCCACTTCATTGACTAA and ATACACACACACAATGCATACACG), Shp −2600 bp (AGTGTACGCTGAATAAACCCTTTC and CAGTGTCTTAGTCGGGGTTTCTAT), Shp +2400 bp (GGTTCTGAGCAAAAGAACCTCTTA and ACTGCCACCTTCATTATTTACCAT), Bip −100 bp (AACGAGTAGCGACTTCACCAAT and AAGTGTCCAGGTCAGTGTTGTCT), and Bip +2800 bp (AGGGAAGAAAGGTACAGTGATGAG and CCACACACACTTTAGGAAAAATGA).
Immunocytochemistry
HEK293T cells were plated on poly-L-lysine-coated coverslips and transfected with pcDNA3-mShp-HA, pcDNA3-mSpop-Myc, pcDNA3-mCullin3-Myc, and pcDNA3-Flag-mXBP1s plasmids. Eighteen hours after transfection, cells were fixed with paraformaldehyde, permeabilized with 1% Triton-100, and blocked with 200 mM glycine and then 5% normal donkey serum. Cells were then labeled with anti-Flag (mouse; 1:200; Sigma, F1804), anti-Myc (rabbit; 1:200; Cell Signaling, 2278), or anti-HA (rabbit; 1:200; Cell Signaling, 3724) primary antibodies followed by donkey antimouse-488 and donkey antirabbit-594 secondary antibodies from Jackson ImmunoResearch and mounted with Prolong Gold antifade reagent with DAPI (Invitrogen). Fluorescent images were taken under a Zeiss LSM700 confocal microscope. The level of colocalization was quantified by the Pearson's correlation coefficient (r) using ImageJ Fiji software with Costes autothreshold determination method for colocalization analysis.
Statistical analyses
Unpaired two-tailed Student's t-test was used for two-group analyses. Two-way ANOVA analysis was used for multigroup analyses (GraphPad Prism). Data are presented as the mean ± SEM; P < 0.05 was considered significant.
Supplementary Material
Acknowledgments
We thank Dr. Joshua Mendell and Ryan Golden for help with the lentiviral CRISPR system, Dr. Ezra Burstein and Dr. Petro Starokadomskyy for sharing hCullin plasmids, and members of the Mangelsdorf–Kliewer laboratory for comments and technical assistance. This work was supported by the Helen Hay Whitney Foundation (S.S.), the National Institutes of Health (grant R01DK067158 to S.A.K. and D.J.M.), the Robert A. Welch Foundation (grants I-1558 to S.A.K. and I-1275 to D.J.M.), and the Howard Hughes Medical Institute (D.J.M.).
Author contributions: S.S. designed, performed, and analyzed experiments and wrote the paper; S.K. assisted with the generation of 3XFlag-Shp knock-in mice; and S.A.K. and D.J.M. supervised the project and revised the manuscript. All authors commented on and approved the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.326868.119.
References
- Acosta-Alvear D, Zhou Y, Blais A, Tsikitis M, Lents NH, Arias C, Lennon CJ, Kluger Y, Dynlacht BD. 2007. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol Cell 27: 53–66. 10.1016/j.molcel.2007.06.011 [DOI] [PubMed] [Google Scholar]
- Båvner A, Sanyal S, Gustafsson JA, Treuter E. 2005. Transcriptional corepression by SHP: molecular mechanisms and physiological consequences. Trends Endocrinol Metab 16: 478–488. 10.1016/j.tem.2005.10.005 [DOI] [PubMed] [Google Scholar]
- Bookout AL, Cummins CL, Mangelsdorf DJ, Pesola JM, Kramer MF. 2006. High-throughput real-time quantitative reverse transcription PCR. Curr Protoc Mol Biol 73: 15.8.1–15.8.28. 10.1002/0471142727.mb1508s73 [DOI] [PubMed] [Google Scholar]
- Brownell JE, Sintchak MD, Gavin JM, Liao H, Bruzzese FJ, Bump NJ, Soucy TA, Milhollen MA, Yang X, Burkhardt AL, et al. 2010. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8–AMP mimetic in situ. Mol Cell 37: 102–111. 10.1016/j.molcel.2009.12.024 [DOI] [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. 2002. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415: 92–96. 10.1038/415092a [DOI] [PubMed] [Google Scholar]
- Chanda D, Park JH, Choi HS. 2008. Molecular basis of endocrine regulation by orphan nuclear receptor small heterodimer partner. Endocr J 55: 253–268. 10.1507/endocrj.K07E-103 [DOI] [PubMed] [Google Scholar]
- Chen H, Qi L. 2010. SUMO modification regulates the transcriptional activity of XBP1. Biochem J 429: 95–102. 10.1042/BJ20100193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coate KC, Hernandez G, Thorne CA, Sun S, Le TDV, Vale K, Kliewer SA, Mangelsdorf DJ. 2017. FGF21 is an exocrine pancreas secretagogue. Cell Metab 25: 472–480. 10.1016/j.cmet.2016.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross BC, Bond PJ, Sadowski PG, Jha BK, Zak J, Goodman JM, Silverman RH, Neubert TA, Baxendale IR, Ron D, et al. 2012. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc Natl Acad Sci 109: E869–E878. 10.1073/pnas.1115623109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, Galardi C, Wilson JG, Lewis MC, Roth ME, et al. 2000. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell 6: 517–526. 10.1016/S1097-2765(00)00051-4 [DOI] [PubMed] [Google Scholar]
- Gupta S, Deepti A, Deegan S, Lisbona F, Hetz C, Samali A. 2010. HSP72 protects cells from ER stress-induced apoptosis via enhancement of IRE1α–XBP1 signaling through a physical interaction. PLoS Biol 8: e1000410 10.1371/journal.pbio.1000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr TA, Saeki S, Schneider M, Schaefer K, Berdy S, Redder T, Shan B, Russell DW, Schwarz M. 2002. Loss of nuclear receptor SHP impairs but does not eliminate negative feedback regulation of bile acid synthesis. Dev Cell 2: 713–720. 10.1016/S1534-5807(02)00154-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Kim KH, Lee JA, Namkung W, Sun AQ, Ananthanarayanan M, Suchy FJ, Shin DM, Muallem S, Lee MG. 2002. Transporter-mediated bile acid uptake causes Ca2+-dependent cell death in rat pancreatic acinar cells. Gastroenterology 122: 1941–1953. 10.1053/gast.2002.33617 [DOI] [PubMed] [Google Scholar]
- Kim YC, Byun S, Seok S, Guo G, Xu HE, Kemper B, Kemper JK. 2019. Small heterodimer partner and fibroblast growth factor 19 inhibit expression of NPC1L1 in mouse intestine and cholesterol absorption. Gastroenterology 156: 1052–1065. 10.1053/j.gastro.2018.11.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kir S, Zhang Y, Gerard RD, Kliewer SA, Mangelsdorf DJ. 2012. Nuclear receptors HNF4α and LRH-1 cooperate in regulating Cyp7a1 in vivo. J Biol Chem 287: 41334–41341. 10.1074/jbc.M112.421834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T, Yoshida H, Mori K, Kaufman RJ. 2002. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev 16: 452–466. 10.1101/gad.964702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AH, Chu GC, Iwakoshi NN, Glimcher LH. 2005. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. Embo J 24: 4368–4380. 10.1038/sj.emboj.7600903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Chanda D, Sim J, Park YY, Choi HS. 2007. Structure and function of the atypical orphan nuclear receptor small heterodimer partner. Int Rev Cytol 261: 117–158. 10.1016/S0074-7696(07)61003-1 [DOI] [PubMed] [Google Scholar]
- Lee J, Salazar Hernández MA, Auen T, Mucka P, Lee J, Ozcan U. 2018. PGC-1α functions as a co-suppressor of XBP1s to regulate glucose metabolism. Mol Metab 7: 119–131. 10.1016/j.molmet.2017.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerch MM, Aghdassi AA. 2010. The role of bile acids in gallstone-induced pancreatitis. Gastroenterology 138: 429–433. 10.1053/j.gastro.2009.12.012 [DOI] [PubMed] [Google Scholar]
- Liao H, Liu XJ, Blank JL, Bouck DC, Bernard H, Garcia K, Lightcap ES. 2011. Quantitative proteomic analysis of cellular protein modulation upon inhibition of the NEDD8-activating enzyme by MLN4924. Mol Cell Proteomics 10: M111.009183 10.1074/mcp.M111.009183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Ibi D, Taniguchi K, Lee J, Herrema H, Akosman B, Mucka P, Salazar Hernandez MA, Uyar MF, Park SW, et al. 2016. Inflammation improves glucose homeostasis through IKKβ–XBP1s interaction. Cell 167: 1052–1066.e18. 10.1016/j.cell.2016.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logsdon CD, Ji B. 2013. The role of protein synthesis and digestive enzymes in acinar cell injury. Nat Rev Gastroenterol Hepatol 10: 362–370. 10.1038/nrgastro.2013.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J, Mangelsdorf DJ. 2000. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell 6: 507–515. 10.1016/S1097-2765(00)00050-2 [DOI] [PubMed] [Google Scholar]
- Lugea A, Tischler D, Nguyen J, Gong J, Gukovsky I, French SW, Gorelick FS, Pandol SJ. 2011. Adaptive unfolded protein response attenuates alcohol-induced pancreatic damage. Gastroenterology 140: 987–997.e8. 10.1053/j.gastro.2010.11.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekahli D, Bultynck G, Parys JB, De Smedt H, Missiaen L. 2011. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb Perspect Biol 3: a004317 10.1101/cshperspect.a004317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petroski MD, Deshaies RJ. 2005. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol 6: 9–20. 10.1038/nrm1547 [DOI] [PubMed] [Google Scholar]
- Ravasi T, Suzuki H, Cannistraci CV, Katayama S, Bajic VB, Tan K, Akalin A, Schmeier S, Kanamori-Katayama M, Bertin N, et al. 2010. An atlas of combinatorial transcriptional regulation in mouse and man. Cell 140: 744–752. 10.1016/j.cell.2010.01.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Ramirez L, Cao H, Nelson D, Hammond E, Lee AH, Yoshida H, Mori K, Glimcher LH, Denko NC, Giaccia AJ, et al. 2004. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res 64: 5943–5947. 10.1158/0008-5472.CAN-04-1606 [DOI] [PubMed] [Google Scholar]
- Sanyal S, Kim JY, Kim HJ, Takeda J, Lee YK, Moore DD, Choi HS. 2002. Differential regulation of the orphan nuclear receptor small heterodimer partner (SHP) gene promoter by orphan nuclear receptor ERR isoforms. J Biol Chem 277: 1739–1748. 10.1074/jbc.M106140200 [DOI] [PubMed] [Google Scholar]
- Segerstolpe A, Palasantza A, Eliasson P, Andersson EM, Andréasson AC, Sun X, Picelli S, Sabirsh A, Clausen M, Bjursell MK, et al. 2016. Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab 24: 593–607. 10.1016/j.cmet.2016.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha H, He Y, Chen H, Wang C, Zenno A, Shi H, Yang X, Zhang X, Qi L. 2009. The IRE1α–XBP1 pathway of the unfolded protein response is required for adipogenesis. Cell Metab 9: 556–564. 10.1016/j.cmet.2009.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Shi G, Han X, Francisco AB, Ji Y, Mendonça N, Liu X, Locasale JW, Simpson KW, Duhamel GE, et al. 2014. Sel1L is indispensable for mammalian endoplasmic reticulum-associated degradation, endoplasmic reticulum homeostasis, and survival. Proc Natl Acad Sci 111: E582–E591. 10.1073/pnas.1318114111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volle DH, Duggavathi R, Magnier BC, Houten SM, Cummins CL, Lobaccaro JM, Verhoeven G, Schoonjans K, Auwerx J. 2007. The small heterodimer partner is a gonadal gatekeeper of sexual maturation in male mice. Genes Dev 21: 303–315. 10.1101/gad.409307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voronina S, Longbottom R, Sutton R, Petersen OH, Tepikin A. 2002. Bile acids induce calcium signals in mouse pancreatic acinar cells: implications for bile-induced pancreatic pathology. J Physiol 540: 49–55. 10.1113/jphysiol.2002.017525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Ron D. 2011. The unfolded protein response: from stress pathway to homeostatic regulation. Science 334: 1081–1086. 10.1126/science.1209038 [DOI] [PubMed] [Google Scholar]
- Wang FM, Chen YJ, Ouyang HJ. 2011. Regulation of unfolded protein response modulator XBP1s by acetylation and deacetylation. Biochem J 433: 245–252. 10.1042/BJ20101293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang Y, Yi P, Dong W, Nalin AP, Zhang J, Zhu Z, Chen L, Benson DM, Mundy-Bosse BL, et al. 2019. The IL-15–AKT–XBP1s signaling pathway contributes to effector functions and survival in human NK cells. Nat Immunol 20: 10–17. 10.1038/s41590-018-0265-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Xue Z, He Y, Sun S, Chen H, Qi L. 2010. A Phos-tag-based approach reveals the extent of physiological endoplasmic reticulum stress. PLoS One 5: e11621 10.1371/journal.pone.0011621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. 2013. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154: 1370–1379. 10.1016/j.cell.2013.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Haze K, Yanagi H, Yura T, Mori K. 1998. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J Biol Chem 273: 33741–33749. 10.1074/jbc.273.50.33741 [DOI] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. 2001. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107: 881–891. 10.1016/S0092-8674(01)00611-0 [DOI] [PubMed] [Google Scholar]
- Yuk JM, Shin DM, Lee HM, Kim JJ, Kim SW, Jin HS, Yang CS, Park KA, Chanda D, Kim DK, et al. 2011. The orphan nuclear receptor SHP acts as a negative regulator in inflammatory signaling triggered by Toll-like receptors. Nat Immunol 12: 742–751. 10.1038/ni.2064 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xu P, Park K, Choi Y, Moore DD, Wang L. 2008. Orphan receptor small heterodimer partner suppresses tumorigenesis by modulating cyclin D1 expression and cellular proliferation. Hepatology 48: 289–298. 10.1002/hep.22342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang M, Calabrese MF, Liu J, Waddell MB, Nourse A, Hammel M, Miller DJ, Walden H, Duda DM, Seyedin SN, et al. 2009. Structures of SPOP–substrate complexes: insights into molecular architectures of BTB-Cul3 ubiquitin ligases. Mol Cell 36: 39–50. 10.1016/j.molcel.2009.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.