

In this review, Yu et al. discuss the recent advances in our knowledge of the Polycomb-repressive complex 2 (PRC2) in mammalian systems. They also discuss important discoveries on Polycomb function derived from model organisms such as plants, worms, flies, and some yeast strains in the context of understanding mammalian PRC2 function.
Keywords: chromatin, epigenetics, PRC2, Polycomb
Abstract
As the process that silences gene expression ensues during development, the stage is set for the activity of Polycomb-repressive complex 2 (PRC2) to maintain these repressed gene profiles. PRC2 catalyzes a specific histone posttranslational modification (hPTM) that fosters chromatin compaction. PRC2 also facilitates the inheritance of this hPTM through its self-contained “write and read” activities, key to preserving cellular identity during cell division. As these changes in gene expression occur without changes in DNA sequence and are inherited, the process is epigenetic in scope. Mutants of mammalian PRC2 or of its histone substrate contribute to the cancer process and other diseases, and research into these aberrant pathways is yielding viable candidates for therapeutic targeting. The effectiveness of PRC2 hinges on its being recruited to the proper chromatin sites; however, resolving the determinants to this process in the mammalian case was not straightforward and thus piqued the interest of many in the field. Here, we chronicle the latest advances toward exposing mammalian PRC2 and its high maintenance.
Past studies have developed a rich description of Drosophila body plan segmentation during development, which entails appropriate, spatially, and temporally regulated Hox gene expression. The Polycomb group (PcG) genes were found to be integral to this process along with the Trithorax group (TrxG) genes that function in opposition. PcG proteins safeguard the repressive state of gene expression through a self-perpetuating mechanism that has not yet been demonstrated for TrxG proteins that are functionally coupled with and downstream from transcription factors for the active state of gene expression (Reinberg and Vales 2018).
Key breakthroughs clarified the requirement for PcG proteins in maintaining the gene repression pattern once established early in development by the Gap proteins (e.g., Kruppel and Hunchback) (Pelegri and Lehmann 1994; Beuchle et al. 2001). As levels of these Gap proteins subside very early in development, it is the PcG proteins that preserve this established gene repression profile throughout development and into adulthood. Moreover, it is the PcG proteins that are pivotal for perpetuating the appropriate gene repression patterns in daughter cells after cell division, requisite for propagating a given cell lineage. PcG proteins do not function as DNA sequence-specific transcription factors as do many of the Gap proteins. Instead, PcG proteins are recruited to chromatin and facilitate its compaction to maintain appropriate gene repression profiles.
Similar to the case in Drosophila, mammalian PcG proteins assemble into two major multisubunit enzymatic complexes, Polycomb-repressive complex 1 (PRC1) and PRC2. Each of them harbors distinct enzymatic activities directed toward specific histone substrates, with PRC1 catalyzing monoubiquitination of Lys119 of histone H2A (H2AK119ub) and PRC2 catalyzing the monomethylation, dimethylation, and trimethylation of Lys27 of histone H3 (H3K27me1/me2/me3) (Fig. 1A). PRC1 can compact chromatin independently of its catalytic activity as can a specific version of PRC2 (see “EZH1 and EZH2, PRC2 Writers with Different Functions”). While Drosophila and mammalian PRC2 share many functional similarities, a key distinction is their mode of recruitment to chromatin, which entails interaction with sequence-specific DNA-binding proteins and a requirement for cis-acting DNA elements termed Polycomb response elements (PREs) in the case of Drosophila PRC2 (Coleman and Struhl 2017; Laprell et al. 2017). This mode of recruitment requires the constant presence of PRE for maintenance and propagation of H3K27 methylation passed to daughter cells after each round of DNA replication and cell division, which is not the case in mammalian cells (see “Features of Mammalian PRC2 Recruitment”). PRC1 and PRC2 often target the same genomic loci, functioning together to maintain gene silencing such that a deficiency in either complex results in defects in lineage differentiation and thus disruptions to normal development in fruit flies, mammals, and plants (Margueron and Reinberg 2011). Mammalian PcG proteins are also involved in establishing X-chromosome dosage compensation as well as “parent of origin” gene imprinting (Plath et al. 2003; Inoue et al. 2017). Interestingly, in nematodes, PcG proteins appear to be dispensable for normal development but are involved in X-chromosome repression during germline development (Gaydos et al. 2014). Dysregulation of PcG proteins has been implicated in human developmental diseases, type II diabetes as well as neurodegenerative diseases (Chen et al. 2009; Seong et al. 2010; Gibson et al. 2012; von Schimmelmann et al. 2016; Imagawa et al. 2017; Lu et al. 2018). PcG genes are also found to be mutated in some types of human cancer, including either gain-of-function or loss-of-function mutations (Laugesen et al. 2016). Moreover, a naturally occurring mutant of the PRC2 substrate, a lysine-to-methionine substitution at Lys27 (K27M) in a small subset of histone H3 is functionally dominant, inhibiting wild-type PRC2 activity in a specific subtype of pediatric glioma, diffuse intrinsic pontine glioma (DIPG) (Schwartzentruber et al. 2012; Sturm et al. 2012; Lewis et al. 2013). How cancer cells hijack these dysregulated pathways to facilitate their progression is being rigorously investigated. Of late, PRC2 enzymatic activity has been recognized as a potential pharmacological target and is being clinically assessed for the treatment of different types of cancer.
Figure 1.
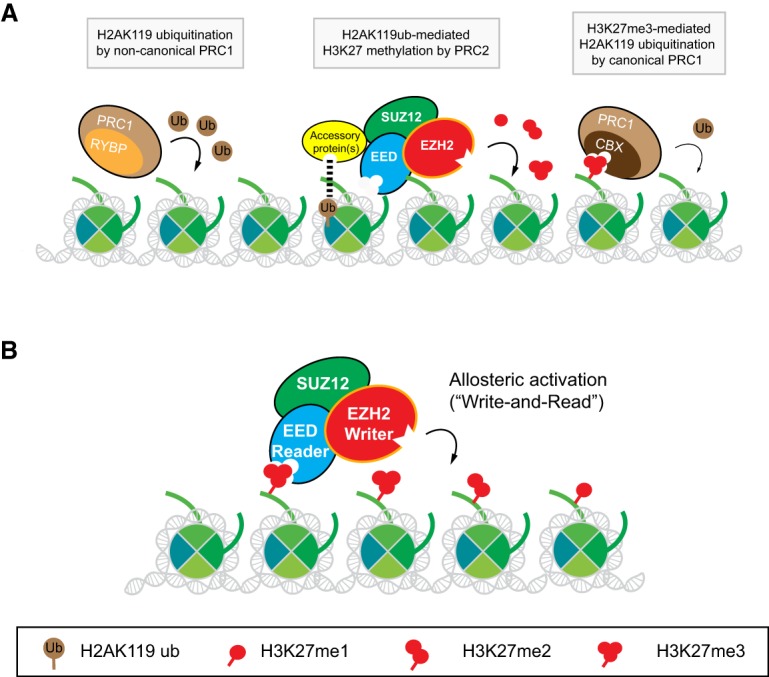
Interplay between Polycomb proteins: PRC1 and PRC2. (A) A model for reciprocal recruitment between PRC1 and PRC2. RYBP-containing PRC1 (noncanonical PRC1) catalyzes H2AK119ub, which in turn putatively recruits PRC2 through its accessory protein (i.e., JARID2) interaction with H2AK119ub. The catalysis of H3K27me3 by PRC2 recruits CBX-containing PRC1 (canonical PRC1). CBX-containing PRC1 harbors an intrinstic activity for chromatin compaction and a relatively low activity for the catalysis of H2AK119ub. (B) Schematic illustration of allosteric activation of PRC2 involving its “write and read” mechanism.
In the past decade, a number of seminal breakthroughs have greatly expanded our grasp of the regulation and biology of the PRC complexes. This review focuses on the ample progress made in the field regarding the properties of PRC2 in mammalian systems, including insights gained from structural analyses, the multilayered regulation of PRC2 activity, mechanisms of its recruitment, and its dysregulation in disease states. However, we describe important discoveries on Polycomb function derived from model organisms such as plants, worms, flies, and some yeast strains in the context of understanding mammalian PRC2 function.
Interplay between PRC1 and PRC2
PRC1 and PRC2 are present in all the metazoans. However, PRC1 is yet to be found in unicellular eukaryotes, whereas PRC2 exists in some, such as Tetrahymena thermophila, marine diatom Phaeodactylum tricornutum, and yeast strains Chaetomium thermophilum and Cryptococcus neoformans, but not in the common model organisms Saccharomyces cerevisiae and Saccharomyces pombe (Veluchamy et al. 2015; Pirrotta 2017). The yeast C. neoformans harbors H3K27 methylation only at subtelomeric regions in a partial overlap with H3K9-methylated constitutive heterochromatin, suggesting that PRC2 might have evolved from redundant gene silencing functions arising from the H3K9me pathway (Lhuillier-Akakpo et al. 2014; Dumesic et al. 2015). Importantly, the evolved partnership between PRC1 and PRC2 in metazoans suggests that H2AK119 ubiquitinated and/or H3K27-methylated facultative heterochromatin at gene-rich loci was acquired to regulate complex transcriptional networks and define cell type-specific gene expression profile
Extensive genetic, functional and molecular studies demonstrate a tight link between PRC1 and PRC2. Mammalian PRC1 complexes are numerous, being classified as canonical and noncanonical, with some components being mutually exclusive (Gao et al. 2012; Tavares et al. 2012). Similar to the case in Drosophila, canonical versions contain one of several different CBX subunits that binds to H3K27me2/me3 through its chromodomain (CD), suggesting that PRC2 fosters PRC1 recruitment. Apparently, the chromatin-associated catalytic product of PRC1, H2AK119ub (Wang et al. 2004; Cao et al. 2005), can also facilitate recruitment of PRC2 during X chromosome inactivation in female mice, yet the mechanism and its generality remains controversial (Blackledge et al. 2014; Cooper et al. 2014; Kalb et al. 2014; Żylicz et al. 2019). Since PRC1 also has noncanonical versions that lack CBX proteins but remain catalytically active for H2AK119ub, it is conceivable that additional pathways also regulate this cascade recruitment of noncanonical PRC1/PRC2. Several previous and excellent reviews on PRC1 and/or PRC2 put the models forward to account for a PRC1/PRC2 joint recruitment pathway (Fig. 1A; Cooper et al. 2016; Brockdorff 2017; Holoch and Margueron 2017), yet the existence of different PRC1 complexes complicates our understanding of PRC1 recruitment in mammals and a clear picture of this process is still underdeveloped. What is clear is that a subset of PRC1 can functionally compact chromatin upon binding to H3K27me2/me3 through their CBX subunits. Also, it is clear that such compaction is independent of the PRC1 catalytic activity (Francis et al. 2004; Eskeland et al. 2010; Illingworth et al. 2015; Pengelly et al. 2015). Since all of the studies investigating PRC1–PRC2 interplay thus far involve steady-state analyses, the design of dynamic experimental systems that can follow de novo Polycomb domain formation as PRC1 and PRC2 are recruited to chromatin (see “De Novo Mammalian PRC2 Recruitment to Chromatin”), might pave the way to sorting out their functional connection in generating facultative heterochromatin.
Chromatin regions comprising methylated H3K27
In mammals, PRC2 is the sole histone methyltransferase (HMT) that catalyzes the three different methylation states of H3K27: H3K27me1, H3K27me2, and H3K27me3. The levels of these products are inversely related to the degree of methylation in vitro, with me1 being most favored and me3 least favored kinetically (Zheng et al. 2012). As described below, PRC2 is subject to an allosteric activation (Fig. 1B) that elevates the levels of the kinetically unfavored trimethylated product in vitro and in vivo. Additionally, distinct PRC2-associated factors facilitate the deposition of H3K27me3 (see “Features of the PRC2 Holoenzyme Regulated by Distinct Accessory Factors”). Extensive genomic studies detail the distinct localization of each of these states of H3K27 methylation. In mouse embryonic stem cells (mESCs), H3K27me1 is generally associated with active genes, although its functional importance is not yet understood. It is unlikely that the presence of H3K27me1 is due to inefficient demethylation by H3K27 demethylases (Ferrari et al. 2014). Instead, as transcription can evict PRC2 and subsequently nucleosomes modified with H3K27me2 or H3K27me3 (Riising et al. 2014; Braun et al. 2017; Kadoch et al. 2017) and as nascent RNA transcripts can compete with chromatin for PRC2 binding (Kaneko et al. 2013; Beltran et al. 2016), the transient nature of PRC2 interaction with active chromatin likely thwarts higher states of methylation. H3K27me1 at active genes might be deposited to facilitate a rapid PRC2-mediated conversion to H3K27me2 or H3K27me3 once target gene expression is shut off, as in the case during lineage commitment. In Drosophila and mice, H3K27me2 fills the gaps of large intragenic regions toward the boundary of euchromatic regions (Ferrari et al. 2014; Lee et al. 2015a; Streubel et al. 2018). As acetylated versions of histones H3 and H4 are associated with active transcription, H3K27me2 is proposed to inhibit pervasive transcription by antagonizing inappropriate histone acetylation on enhancers (Ferrari et al. 2014).
In mESCs, H3K27me3 occupies promoters of repressed genes serving as a docking site for canonical PRC1 to compact chromatin (see “Interplay Between PRC1 and PRC2”; Margueron and Reinberg 2011). Notably, H3K27me3 is intermixed with a histone modification associated with transcription activation (H3K4me3) within bivalent promoters (Bernstein et al. 2006; Voigt et al. 2013). These latter promoters are presumably poised for a specific transcriptional outcome to be implemented by master regulators (DNA sequence-specific transcription factors) during lineage commitment. Some of the genes associated with bivalent promoters will become transcriptionally active, a process inhibitory to PRC2. Others will be subject to transcription repression such that PRC2 can now spread H3K27me2/me3 throughout the promoter and the gene body, being driven by a positive feedback loop (Fig. 1B) as discussed below.
Structural studies of PRC2
The mammalian PRC2 core complex comprises EZH1/2 (EZH1 and/or EZH2), EED, and SUZ12, which associate with one of the histone-binding proteins, RBAP46/48 (Fig. 2A). EZH1/2 are the catalytic subunits of PRC2 each containing a SET domain through which a methyl moiety from the cofactor S-adenosyl-L-methionine (SAM) is transferred to Lys27 of histone H3 (H3K27). However, PRC2 comprising EZH1 versus EZH2 does exhibit several distinguishing features (discussed below). Interestingly, PRC2 containing EZH1/2 exhibits an automethylation activity at two main residues within their respective unstructured loop between the SANT2L and CXC domains (Fig. 2A; Lee et al. 2018a; Wang et al. 2018b), the function of which is currently being investigated but likely serves to positively regulate PRC2.
Figure 2.

PRC2 architecture. (A) Schematic representation of the PRC2 complex. (Left) Displayed are the composition of core PRC2 and association of its subunits derived from structural and biochemical studies. The aromatic cage of EED and the catalytic sites of EZH1/2 (substrate-binding pocket and SAM-binding pocket) are indicated. (Right) Domains within PRC2 core subunits are indicated. (B) Schematic representation of the PRC2 states: autoinhibitory (left), basal (middle), and “allosterically” stimulated (right). For simplicity, only key features of the PRC2 structure are illustrated. (Middle) The EZH2-SET domain is catalytically inactive and this autoinhibitory state is released by forming the EZH2-EED-SUZ12 complex. In this basal state, the stimulatory-responsive motif (SRM) is disordered (middle; dashed red line) but can align with SET-I when H3K27me3 is recognized by the aromatic cage of EED (right).
The first crystal structure of a characteristic SET domain (the Rubisco large subunit methyltransferase) reveals two pore entry sites: one for substrate (H3K27me0, H3K27me1, or H3K27me2 in the case of PRC2) and the other for SAM (Fig. 2A). Interestingly, crystal structures of the EZH2 SET domain in isolation reveal an autoinhibitory conformation: The EZH2 post-SET domain and SET-inserted (SET-I) domain block the binding channel for its substrate (H3K27), and the binding pocket for the cofactor (SAM) exhibits a suboptimal configuration (Fig. 2B; Antonysamy et al. 2013; Jiao and Liu 2015; Justin et al. 2016). Indeed, biochemical studies demonstrate that EZH2 alone is catalytically inactive and requires minimally an interaction with EED and SUZ12 for HMT activity (Cao et al. 2002; Kuzmichev et al. 2002). Since then, many groups have attempted to solve the structure of the PRC2 core complex.
Electron microscopy analyses at a low resolution (21 Å) provide an important first step toward elucidating the organization of the core PRC2 (Ciferri et al. 2012), revealing the important physical contacts within human PRC2 comprising an accessory protein, AEBP2 (Cao et al. 2002). Soon after, details of the physical interactions between the minimal core PRC2 subunits required to form an active complex: EZH2, EED, and the VEFS domain of SUZ12 (Fig. 2B, right) were discovered and, notably, how these interactions change the conformation of the EZH2 SET domain to relieve its autoinhibition. These structural studies include thermophilic fungus Chaetomium thermophilum PRC2 (CtPRC2) (Jiao and Liu 2015), human PRC2 (Justin et al. 2016), and a chimeric PRC2 consisting of human EED and SUZ12 together with EZH2 from the American chameleon Anolis carolinensis (Hs/Ac PRC2) (Brooun et al. 2016). Comparison between the structures of the EZH2 SET domain and the PRC2 core complex indicate that EZH2 interaction with EED and the VEFS domain of SUZ12 conveys stability to an active configuration of the SET domain. EZH2 autoinhibition is relieved through rotation of its SET-I domain and realignment of its post-SET domain, thereby generating the full cofactor-binding pocket and unblocking its substrate channel (Fig. 2B).
‘Write and read’ mechanism
PRC2 comprises both a “writer” and a “reader” module that catalyzes and binds to methylated H3K27, respectively. These functions segregate into two different subunits, EZH1/2 and EED, respectively. The PRC2 terminal enzymatic product, H3K27me3, is recognized with an apparently higher affinity relative to H3K27me2 and its unmodified counterpart by the aromatic cage of EED (“reader”), which sits atop a β-propeller structure formed by seven WD40 repeated domains (Fig. 2A,B; Margueron et al. 2009). EED–H3K27me3 interaction results in an induced conformational change in PRC2 and most importantly, in EZH2 (“writer”) that further stabilizes its catalytic SET domain, thereby markedly enhancing its HMT activity (Fig. 2B; Jiao and Liu 2015; Justin et al. 2016). This allosteric activation of the complex arising from its interaction with the product of its catalysis fosters a positive feedback loop (Fig. 1B) such that H3K27me2/me3 can spread into large genomic regions, forming extended repressive domains. This “write and read” mechanism can also account for the inheritance of H3K27me3-repressive domains by daughter cells during cell division (see “Restoring H3K27me3-Repressive Domains After DNA Replication”; Hansen et al. 2008; Oksuz et al. 2018; Reinberg and Vales 2018).
The overall changes elicited upon allosteric activation of PRC2 are evidenced by the structure of CtPRC2 with and without an H3K27me3 peptide bound to EED (Jiao and Liu 2015). One of the most striking differences is that the stimulatory-responsive motif (SRM) (Fig. 2A, right) of EZH2 is unstructured and disordered in the absence of the H3K27me3 (“basal state”) but ordered upon binding (“stimulated state”) (Fig. 2B, middle and right). The trimethyl-lysine is directly recognized by the aromatic cage of EED, and the SRM of EZH2 interacts with the other side of the H3K27me3 tail, generating a sandwich-like structure with H3K27me3 positioned in the middle (Fig. 2B, right). In addition, the SRM forms hydrophobic interactions with SET-I thereby stabilizing the SET domain and promoting the HMT activity of PRC2. Of note, prior to these structural studies of PRC2, the SET-I reorientation was proposed as being key to the stable conformation of the SET domain, which fosters proper HMT activity for SET-domain containing proteins (Southall et al. 2009). Therefore, the SRM and SET-I interaction is considered a hallmark of allosteric PRC2 activation. These findings are substantiated by the studies of human PRC2 cocrystalized with the JARID2-K116me3 peptide (Justin et al. 2016), a similar allosteric activator of PRC2 (see “Features of the PRC2 Holoenzyme Regulated by Distinct Accessory Factors”), highlighting the evolutionary conservation of this activation mechanism from fungi to humans. Although these studies demonstrate the molecular basis of H3K27me3- or JARID2-K116me3-mediated allosteric activation of PRC2, these crystal structures were generated by inhibiting the SET domain using either a peptide containing a naturally occurring methionine substitution at Lys27 (H3K27M) (see “The H3K27M Oncohistone”; Jiao and Liu 2015; Justin et al. 2016) or a PRC2 inhibitor that binds closely to the pocket that recognizes the cofactor SAM (Brooun et al. 2016). Therefore, the full spectrum of conformational changes during allosteric activation still remain elusive. This issue was partly addressed through cryo-EM structural studies of PRC2 in complex with two of its partner proteins, AEBP2 and JARID2 (Kasinath et al. 2018a), indicating that multiple activation states exist during allosteric activation. These active states include the conformational change not only in the SRM but also in the SBD, EBD, and SANT1L domain within EZH2 (Fig. 2A, right; Kasinath et al. 2018b).
Similar to the case with PRC2, the two histone H3K9 methyltransferases, SUV39H1/2, responsible for H3K9me2/3 in Drosophila and mammals, along with CLR4 in fission yeast, exhibit a “write and read” mechanism, but, in these cases, the mechanism is self-contained within a single polypeptide. The CD of SUV39H1/2/CLR4 forms a cage that recognizes H3K9me3 (Fig. 3A; Rea et al. 2000; Zhang et al. 2008). Mutation of this CD blocks the positive feedback loop, leading to a large reduction in HMT activity (Muller et al. 2016). That the “writer” and “reader” modules are segregated into two proteins in the case of PRC2 (Fig. 3B) and that its allosteric activation differs as a function of the presence of EZH1 versus EZH2 (see “EZH1 and EZH2, PRC2 Writers with Different Functions”) (Lee et al. 2018c), suggests that this HMT activity is modulated by the ratio of EZH1 and EZH2 in a cell context-dependent manner. Such a difference between SUV39H1/2 and PRC2 might also reflect a more dynamic temporospatial regulation inherent to facultative heterochromatin that comprises H3K27me2/me3 relative to constitutive heterochromatin that comprises H3K9me2/me3 (Fig. 3). Moreover, PRC2 can form homodimers or heterodimers with EZH1 and/or EZH2 in vitro (Davidovich et al. 2014) and while being ectopically overexpressed in cells (Son et al. 2013), which could provide additional layers of PRC2 regulation (Fig. 3B). Thus far, such PRC2/EZH1 and PRC2/EZH2 homodimers and heterodimers are detected only in solution but not yet through structural studies. These observations remain to be further investigated in the context of distinct cell types and most importantly, throughout the process of differentiation by analyzing homogenous (single) populations of cells.
Figure 3.

Reader and writer modules within the PRC2 complex and SUV39H1/2 (yeast CLR4). The “writer” and “reader” modules comprise one protein in the case of SUV39H1/2 (yeast CLR4) (A) but are segregated into two distinct subunits (EZH1/EZH2 and EED, respectively) in the case of PRC2 (B, left). (B, right) PRC2 comprises one of two distinct “writers.” (Middle) EZH1 or EZH2 and can form homodimers or heterodimers, likely providing unknown regulatory roles that are cell type-specific.
Features of the PRC2 holoenzyme regulated by distinct accessory factors
Recent structural studies revealed that SUZ12 functions as a structural platform that coordinates not only the assembly of core PRC2 but also most of its interactions with accessory proteins that define distinct PRC2 subcomplexes. These interactions could occur “transiently,” generating dynamic PRC2 holoenzyme complexes (Fig. 4; Chen et al. 2018; Kasinath et al. 2018a). We hypothesize that the formation of different PRC2 complexes is likely tissue-specific and important during differentiation to specific lineages (Son et al. 2013; A. Schaefer, unpubl.). While SUZ12 interacts with EZH2 and EED through its C-terminal VEFS domain, essential for the catalytic activity of PRC2, the N-terminal region of SUZ12 interacts with the histone-binding proteins RBAP46/48 as well as other PRC2 accessory factors, including AEBP2, the PCL family of proteins (PHF1/PCL1, MTF2/PCL2, and PHF19/PCL3), JARID2, EPOP, and other recently described proteins with ill-defined functions (Fig. 4). Among these factors, AEBP2 and the individual PCL proteins interact with the C2 domain of SUZ12 in a mutually exclusive manner (Fig. 4; Grijzenhout et al. 2016; Chen et al. 2018). Similarly, JARID2 and EPOP compete for binding to the ZnB-Zn domain of SUZ12 (Chen et al. 2018). Intriguingly, through their binding to disparate domains of SUZ12, AEBP2 and JARID2 cooperatively interact and stabilize the holoenzyme complex (Fig. 4; Chen et al. 2018; Kasinath et al. 2018a) and functionally stimulate catalysis through different mechanisms (Sanulli et al. 2015; Wang et al. 2017b; Lee et al. 2018c).
Figure 4.
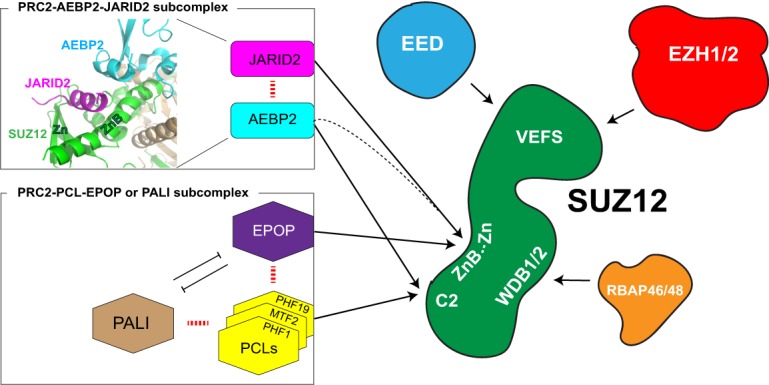
Assembly of core PRC2 and accessory proteins. Schematic representation illustrating the means by which SUZ12 (green) functions as a structural platform. PRC2 subunits and accessory proteins interact with different domains of SUZ12, as indicated (see arrows). (Left) Two major PRC2 subcomplexes are illustrated (square boxes). Interactions and antagonisms among accessory factors are illustrated by dashed red lines and blocked arrows, respectively. (Top left) Structure showing AEBP2 and JARID2 cooperative interaction with SUZ12. AEBP2 and SUZ12 (ZnB and Zn domains) create a groove that fits the TR (transrepression) domain of JARID2 (modified from Protein Data Bank: 5WAI). This three-way junction formed by JARID2, AEBP2, and SUZ12 creates a stable and unique PRC2 subcomplex PRC2–AEBP2–JARID2. Note that AEBP2 interacts with both the C2 and ZnB domains. Although AEBP2 binds to the ZnB domain, it cooperatively interacts with JARID2 (dashed black arrow), while competing with PCLs.
Biochemical approaches directed at analyzing a specific function of PRC2 accessory proteins, including AEBP2, JARID2, and the PCL family of proteins, show that these proteins regulate PRC2 activity in part by increasing its affinity for nucleosomes and/or DNA (Son et al. 2013; Wang et al. 2017b; Lee et al. 2018c). Among them, AEBP2 stabilizes the PRC2 complex by bridging the EZH2 SET domain, SUZ12, and RBAP46/48 (Kasinath et al. 2018a) and enhances PRC2 binding to nucleosomes through its KR motif (Fig. 5A,B), thereby stimulating the HMT activity (Lee et al. 2018c). AEBP2 binds DNA through its Zn finger domain (Kim et al. 2009) and apparently confers to PRC2 a preference for binding methylated DNA in vitro (Wang et al. 2017b). It remains unclear as to whether this DNA-binding activity is required for PRC2 stimulation or PRC2 recruitment to chromatin (Fig. 5A,B), as methylated CpG-containing DNA appears to inhibit PRC2 binding (Li et al. 2017). However, this surprising observation might become important in the coexistence of PRC2 and imprinted genes. Similar to AEBP2, JARID2 also stimulates PRC2 activity and does so in part by contributing to nucleosome binding through a region in the middle of the protein that includes its JmjN domain (Son et al. 2013). Moreover, JARID2 also binds to H2AK119ub through its ubiquitin-interacting motif (UIM) and exhibits DNA-binding activity through its C terminus, including the AT-rich interaction domain (ARID) (Li et al. 2010; Cooper et al. 2016), each of which fosters PRC2 recruitment. The PCL proteins bind to unmethylated CpG islands (CGIs) (see below) in the context of a reduced helical twist arising from their conserved extended homologous (EH) regions that fold into a winged-helix structure (Fig. 5A,B; Li et al. 2017; Perino et al. 2018). This interaction with DNA is critical for PRC2 recruitment to developmentally regulated genes and importantly, increases its residence time on chromatin, likely facilitating catalysis of the trimethylated state of H3K27 (Sarma et al. 2008; Choi et al. 2017; Li et al. 2017).
Figure 5.

Biochemical characterization of PRC2 accessory factors. (A) Domains within PRC2 accessory proteins are indicated. SUZ12 binding domains are highlighted by gray dots. (Bottom) Sequence alignment of the Tudor domains within three mammalian PCL proteins. The critical aromatic residues that can form a cage are highlighted by red squares. The cage within the PCLtudor are bound to H3K36me3 (and, to a lesser extent, H3K36me2) in vitro. (B) PRC2 accessory proteins regulate its activity through several means: increasing its affinity for nucleosome binding (e.g., AEBP2KR motif and JARID2JmjN) and DNA (e.g., AEBP2Zn, JARID2C-term, and PCLEH), interacting with histone posttranslational modifications (hPTMs) (e.g., JARID2UIM [H2AK119ub] and PCLTudor [H3K36me2/me3]), and/or regulating its allosteric activation (e.g., JARID2-K116me3). (C2B) C2-binding domain; (TR) transrepression; (RBR) RNA-binding region; (JmjN) Jumonji N; (JmjC) Jumonji C; (PHD1/2): plant homeodomain 1/2.
Moreover, PCLs contain a Tudor domain that recognizes H3K36me2/me3 in vitro, two modifications that antagonize PRC2 activity (Ballare et al. 2012; Brien et al. 2012; Musselman et al. 2012, 2013; Cai et al. 2013). Indeed, PHF19/PCL3 facilitates the recruitment of both PRC2 and an H3K36me3 demethylase (e.g., NO66 or KDM2B) to active loci during their transition to a Polycomb-repressed state (Ballare et al. 2012; Brien et al. 2012). Although the three mammalian PCL proteins have highly conserved functional domains (Fig. 5B), their respective role in PRC2 regulation might be nonredundant. Interestingly, the binding affinity of MTF2 to an H3K36me3 peptide is much lower than that of PHF1 due to loss of a key aromatic residue (Fig. 5A; Gatchalian et al. 2015; Li et al. 2017). Thus, while all PCLs stimulate PRC2 activity, we speculate that MTF2 mediates PRC2 recruitment to sites devoid of H3K36me2/me3, while PHF1 and PHF19 stabilize PRC2 on sites occupied by H3K36me2/me3. In this scenario, an H3K36 demethylase recruited by PHF19 could remove H3K36 methylation, thereby facilitating methylated-H3K27 deposition (Ballare et al. 2012).
JARID2 stands alone in stimulating PRC2 activity through its “write and read” mechanism (Sanulli et al. 2015). In addition to H3K27, PRC2 methylates the Lys116 residue of JARID2 and similar to H3K27me3, JARID2-K116me3 allosterically activates PRC2 (Sanulli et al. 2015). Cryo-EM studies of PRC2–AEBP2–JARID2 (Kasinath et al. 2018a) show that unmethylated JARID2 and JARID2-K116me3 can reside at the EZH2 active site and the EED aromatic cage, respectively. JARID2-K116me3 might “jump-start” the positive feedback loop inherent to PRC2 even before its initial recruitment to chromatin and without its encountering H3K27me3, and this property might facilitate the de novo deposition of H3K27me3 during differentiation (see “PRC2 Dynamics in ESCs and Early Development”).
While recently identified PRC2 factors are found associated with the core complex (e.g., EPOP and PALI), their functions are unknown (Zhang et al. 2011; Conway et al. 2018) and, in some instances, contradictory. EPOP, previously known as C17ORF96 and esPRC2p48, is a mammalian PRC2-associated protein (Zhang et al. 2011; Alekseyenko et al. 2014; Liefke and Shi 2015; Beringer et al. 2016; Liefke et al. 2016) that interacts with PRC2 through its C-terminal region. However, conflicting studies exist regarding both the function and types of complexes formed by EPOP. Using immunoprecipitation techniques, one study finds the presence of a trimeric complex composed of EPOP, PRC2, and the Elongin BC, a submodule of the Elongin complex (Beringer et al. 2016; Liefke et al. 2016), which apparently functions to maintain a low level of gene expression from PRC2 genomic targets (Beringer et al. 2016). However, gel filtration experiments lead another group to conclude that EPOP forms two distinct complexes: one with Elongin BC and another with PRC2 (Liefke et al. 2016) and that EPOP functions as a transcriptional activator independent of PRC2. Thus, EPOP function in the context of PRC2 remains to be defined. On the other hand, PALI1/2 (PRC2-associated LCOR isoform 1/2) is a vertebrate-specific PRC2 interacting protein. The LCOR locus exhibits five alternative splicing variants, one of which encodes both LCOR and C10ORF12, generating the LCOR/C10ORF12 fusion protein PALI1/2 (Conway et al. 2018). PALI1 competes with AEBP2, JARID2, and EPOP for PRC2 interaction but can form a complex with PRC2 comprising the PCLs. PALI1 stimulates PRC2 activity in vitro and its knockout in mice leads to a global loss in H3K27me2/me3 (Conway et al. 2018), pointing to PALI being a positive regulator of PRC2.
These studies of PRC2 interaction with different factors point to PRC2 being dynamically regulated in vivo, most likely in a cell type-specific manner. The limitation of the studies with PRC2 interactors thus far is that most, if not all, have been performed at steady-state levels in cultured cells. Given the stochastic nature of biomolecules, studies using cell populations provide a static and average readout of PRC2 subcomplexes that likely obscure the exact distribution and dynamics of PRC2 subcomplexes at a single-cell level. Since PRC2 is a complex that maintains rather than establishes patterns of gene repression, ascertaining its function during development and differentiation in an in vivo context is paramount. For example, studies in some types of neurons are starting to shed light on the complexity of PRC2, as some neurons mainly harbor either PRC2/EZH1 or PRC2/EZH2, while others have both (A. Schaefer, pers. comm.). Whether the dimeric state of PRC2 (i.e., PRC2/EZH1–PRC2/EZH2 heterodimer) is relevant during the course of neural development remains unknown. These recent observations also point to the presence of JARID2 in some neurons. This finding is unexpected given previous reports of JARID2 expression being limited to uncommitted cells and lost during lineage differentiation, as in the case of myogenesis: JARID2 is high in ESCs, declines in myoblasts, and disappears in myotubes (Son et al. 2013). The latter observations led to the hypothesis that JARID2 function is essential during cell lineage commitment, but once a lineage is established, JARID2 as well as EZH2 are no longer required such that the PRC2–EZH1 complex “passively” maintains the repressed pattern of gene expression in a cell type-specific manner (Son et al. 2013). Indeed, JARID2 and EZH2 are essential to actively maintain the previously instructed pattern of gene repression upon the initial steps of differentiation to a specific lineage (Son et al. 2013). We look forward to future advancements in imaging and single-cell analysis technology for a more in-depth investigation of PRC2-regulated processes.
PRC2 and its chromatin substrates
PRC2 activity is regulated by the chromatin structure. Early studies show that PRC2 favors dinucleosome and oligonucleosome substrates over mononucleosomes, octamers, histone H3, or H3-derived peptides in vitro (Cao and Zhang 2004; Kuzmichev et al. 2004; Martin et al. 2006), suggesting that neighboring nucleosomes stimulate its activity. Indeed, dense nucleosome arrays activate Drosophila PRC2 activity, which corroborates its functional role in facultative heterochromatin formation (Yuan et al. 2012). In addition, PRC2 is significantly stimulated by a specific length of the DNA linking two nucleosomes (Wang et al. 2017b; Lee et al. 2018c), with 40 bp being the optimal linker length in vitro (Lee et al. 2018c). These results suggest that PRC2 binds dinucleosomes with a preferred orientation such that EED binding to one nucleosome containing H3K27me3 would position the EZH2 SET domain in close proximity to the histone H3 tail from the second, unmodified nucleosome (Fig. 6A). Indeed, cryo-EM of PRC2–AEBP2 complexed with such heterodinucleosomes shows that H3K27me3 resides in the EED aromatic cage, while unmethylated H3K27 is at the EZH2 active site (Poepsel et al. 2018). In this case, the SBD and SANT1L domains of EZH2 are in contact with the H3K27me3-containing nucleosome and the CXC domain of EZH2 interacts with the unmethylated nucleosome, thereby bridging the two (Fig. 6A).
Figure 6.

Interaction between the PRC2 complex and chromatin. (A) Schmatic representation of the PRC2 complex bound to a dinucleosome of which one comprises trimethylated H3K27 and the other unmodified H3K27. (Left) The SBD and SANT1L domains of EZH2 are in contact with the H3K27me3-containing nucleosome and the CXC and SET domains of EZH2 interact with the unmethylated nucleosome, thereby bridging the two (big arrows). H3K27me3 resides in the EED aromatic cage (left), while unmethylated H3K27 is at the EZH2 active site (right). RBAP46/48 interacts with the histone H3 and H4 tails (small arrows). (B) Schematic representation of RBAP46/48 interacting with the histone H3 and H4 tails. These interactions are competitive with RBAP46/48 interaction with SUZ12 and AEBP2.
Structural studies of the RBAP46/48 component of PRC2 reveal that the histone H4 tail binds to a pocket on the side of the WD40 domain, with the histone H3 tail being positioned atop (Fig. 6B; Schmitges et al. 2011). Lys4 of histone H3 interacts with the WD40 domain, but this interaction is significantly weakened when H3K4 is methylated (Schmitges et al. 2011), stressing that H3K4 methylation that is associated with active transcription is antagonistic to PRC2 activity. However, such histone-binding activity of RBAP46/48 might actually be negatively regulated in the context of PRC2 given that SUZ12 and AEBP2 overlap with its H3 and H4 tail-binding region (Fig. 6B; Chen et al. 2018; Kasinath et al. 2018a). We speculate that the RBAP46/48-nucleosome interaction might be dynamic based on the structural flexibility of either SUZ12 or AEBP2. Of note, a small population of PRC2–AEBP2 in complex with heterodinucleosomes (H3K27me3–H3K27) indicates that RBAP48 is in contact with the nucleosomes (Poepsel et al. 2018), supporting that RBAP48 contributes to PRC2 stabilization on chromatin. Thus, RBAP46/48 likely enhances PRC2 affinity for its target sites as previously proposed (Margueron and Reinberg 2011), thereby promoting PRC2 activity. The significance of the interaction between RBAP46/48 and the histone H3 or H4 tail under physiological conditions warrants further investigation; however, it is important to stress that RBAP46/48 are components of a large number of chromatin associated complexes (Margueron and Reinberg 2011; Millard et al. 2016; Moody et al. 2018), and their function is likely providing stable binding to chromatin, perhaps in a dynamic way.
De novo mammalian PRC2 recruitment to chromatin
Some histone modifications, including H3K27 methylation, are reset during early development in mice (Heard and Martienssen 2014; Liu et al. 2016; Zheng et al. 2016). For example, H3K27me3 is depleted from promoters in preimplantation embryos, and de novo H3K27me3 is deposited in the epiblast, illustrating a massive epigenetic reprogramming (Zheng et al. 2016). In agreement, upon neuronal differentiation of mESCs, novel targets of PRC2 repression surface at both the progenitor and the terminal neuron states, which requires a dynamic regulation of PRC2 recruitment to chromatin (Mohn et al. 2008). Tremendous efforts have been extended to home in on exactly how PRC2 achieves its targeting of different sets of genes specific to individual cellular lineages in mammals.
Features of mammalian PRC2 recruitment
In mammals, PRC2 acts as a general factor to maintain repressed states of distinct gene types: active protein-coding genes that are shut off, imprinted genes, and developmentally regulated genes. Accordingly, its recruitment to chromatin in these cases is regulated distinctively (Margueron and Reinberg 2011; Riising et al. 2014). At active protein-coding genes, PRC2 is sequestered by RNA in ESCs (Kaneko et al. 2013; Beltran et al. 2016), wherein its concentration is much higher than in other cell types analyzed (Stafford et al. 2018). However, at imprinted genes it is likely that binding of PRC2 to noncoding RNA (ncRNA) originating from the imprinted locus is important in its recruitment to aid in attaining an allele-specific repressed state. At developmentally regulated genes, PRC2 recruitment is more complex, requiring the participation of accessory factors (Oksuz et al. 2018).
Active protein-coding genes
Seemingly paradoxical, PRC2 is sequestered by RNA transcribed from active protein-coding genes as its EZH2 core subunit interacts with the 5′ region of nascent RNAs (Fig. 7A; Davidovich et al. 2013; Kaneko et al. 2013; Zhang et al. 2019). Of note, SET domain-containing proteins in general interact with ssDNA and ssRNA (Krajewski et al. 2005). In the case of PRC2 however, RNA interaction inhibits its methyltransferase activity in vitro and in cells (Cifuentes-Rojas et al. 2014; Kaneko et al. 2014b). However, as RNA does not impede PRC2-intrinsic automethylation activity (Wang et al. 2017b), its interaction with PRC2 is later ascribed to an RNA “decoy” effect, essentially blocking stable PRC2 interactions with chromatin (Wang et al. 2017b). Another hypothesis is that RNA interferes with the allosteric activation of PRC2 by interacting with the SRM of EZH2 (Zhang et al. 2019). These two hypotheses need not be mutually exclusive given that the RNA-binding sites within EZH2 identified by many groups are indeed critical for both nucleosome/DNA contact and PRC2 activity (He et al. 2016; Long et al. 2017). In accordance, the major RNA-binding sites are within the SRM and the N terminus of the CXC domain (Long et al. 2017; Zhang et al. 2019), which are key to allosteric activation and nucleosome binding, respectively. Although PRC2 and its accessory subunit, JARID2, interact with multitudinous RNAs without sequence specificity (Davidovich et al. 2013; Cifuentes-Rojas et al. 2014; Kaneko et al. 2014a), the activity of PRC2/JARID2 appears to be regulated by phosphorylation (Kaneko et al. 2014a) and is also inhibited by poly(G) RNA, which forms a G-quadruplex structure, but not by an unfolded poly(A) RNA (Kaneko et al. 2014b). Indeed, PRC2 exhibits a high-affinity binding for one type of G-quadruplex RNA (Wang et al. 2017a). Although current technologies are limited in detecting G-quadruplex structures in cells, sequence analysis suggests their abundance in the human transcriptome (Wang et al. 2017a). PRC2 association with the 5′ end of nascent RNA weakens its binding to chromatin and potentially keeps PRC2 in close proximity to the promoters of active protein-coding genes (Kaneko et al. 2013, 2014b; Beltran et al. 2016). Should nascent transcripts be shut off by DNA sequence-specific transcription repressors, PRC2 would now be free to associate with chromatin and deposit H3K27me2/me3, pointing to its general role in maintaining gene repression (Kaneko et al. 2014b; Riising et al. 2014); such a scenario is observed in cells when premature transcription termination is promoted by inserting a poly(A) site near the 5′ end of a gene (Kaneko et al. 2014b).
Figure 7.

Three modes of PRC2 recruitment to chromatin in mammals. (A) At active protein coding genes, the 5′ region of the nascent RNA recruits PRC2 through interaction with its core EZH2 subunit. This interaction precludes PRC2 activity. (B) At imprinted genes, long ncRNAs (lncRNAs) produced from the same loci recruit PRC2 in cis. PRC2 deposits H3K27me3 on these transcriptionally silent genes. (C) At developmental genes, promoter architecture and specific CGIs recruit PRC2. (Panel i) Erk1/2 localizes to GC-/GA-rich regions on the genome, mediating nucleosome turnover as well as phosphorylation of RNAPII at its C-terminal domain (CTD)-Ser5 residue. These events provide a promoter architecture conducive to PRC2/JARID2 recruitment. (Panel ii) Through its low-affinity interactions with chromatin, PRC2 can recognize nucleation sites that contain hypomethylated CGIs with GA-rich and/or GCN tandem repeat motifs via a “hit and run” mechanism, but its on rate is lower than its off rate. The on rate is increased by PRC2 interaction with MTF2/PCL2 and/or JARID2. (D) Nucleation sites (both strong and weak) are enriched for dense CGIs, which have a high number of CG dinucleotides within the island (blue boxes). Both types of nucleation sites are enriched for “GA” and/or “GCN” tandem repeat motifs; however, strong nucleation sites have longer GCN tandem repeats (red boxes) and a different distribution of GA content (light-green and dark-green boxes) compared with weak ones.
Imprinted genes
PRC2 is recruited to the transcriptionally silent alleles of imprinted genes by long ncRNAs (lncRNAs), apparently generated from the same loci in cis (Fig. 7B). As RNA-binding inhibits the activity of PRC2, how does PRC2 establish repression in cis upon the intial recruitment by lncRNAs? We speculate that, unlike the active loci, the lack of transcriptional machinery as well as active histone modifications (e.g., H3K4me or H3K36me) at imprinted genes likely allows PRC2 to slowly deposit H3K27me3 across several cell cycles in spite of the low activity in association with lncRNA. As RNA-mediated inhibition of PRC2 activity is antagonistic to its allosteric activation, RNA likely serves as an initial recruiter of PRC2 to such imprinted genes, but its inhibitory effect is gradually alleviated by the accumulation of H3K27me3, eventually leading to stable establishment of repression. The RNA-mediated recruiting process was first detected in the case of PRC2 occupancy of the inactive X chromosome (Xi) in female mESCs upon X-chromosome inactivation (XCI), directed by its interaction with the Xi-specific transcript (Xist) (Zhao et al. 2008; da Rocha et al. 2014). Xist RNA can apparently mediate PRC2 recruitment to the Xi through its binding to EZH2, JARID2, and a chromatin remodeler, ATRX (Zhao et al. 2008; da Rocha et al. 2014; Sarma et al. 2014; Almeida et al. 2017). Since this recruitment is mediated by RNA, whether G-quadruplex structures proposed to bind PRC2 with high affinity (Wang et al. 2017a) are involved is unknown. As well, the domains on Xist RNA that mediate recruitment (direct or indirect) of PRC2 remain controversial (Kohlmaier et al. 2004; Zhao et al. 2008). Nevertheless, a recent study indicates that the RepB domain of Xist mediates the interdependent recruitment between Xist, PRC1, and PRC2 (Colognori et al. 2019). Surprisingly, some studies suggest that PRC2 recruitment in response to XCI at the mouse female Xi is accomplished by one of the noncanonical PRC1 complexes through its catalysis of H2AK119ub (Almeida et al. 2017; Pintacuda et al. 2017), which appears to precede H3K27me3 deposition upon experimental induction of Xist (Żylicz et al. 2019). Of note, JARID2 mediates the interaction between H2AK119ub and PRC2 through its conserved ubiquitin interaction motif during XCI (Figs. 1A, 5B; Cooper et al. 2016). While these ongoing studies are intriguing, they have been performed in steady-state systems. To clarify the sequence of events and determinants to this process, experimental systems designed to follow de novo Polycomb domain formation as PRC1 and PRC2 are recruited to the Xi would be highly beneficial. Another example is the recruitment of PRC2 to the KCNQ1-overlapping transcript 1 (Kcnq1ot1) locus on the paternally imprinted region by lncRNAs transcribed from this locus in mice (Pandey et al. 2008). These results highlight the role of PRC2 in maintaining transcriptional inactivity at imprinted genes, consistent with a recent report demonstrating that some imprinted genes use an H3K27me3-dependent pathway, in lieu of DNA methylation (Inoue et al. 2017).
Developmental genes
Exactly how PRC2 is recruited to establish bivalent promoters (see “PRC2 Dynamics in ESCs and Early Development”; Bernstein et al. 2006) or retain repressed expression at its target developmental genes has been challenging to identify in the mammalian case relative to that in Drosophila (Schuettengruber et al. 2009; Voigt et al. 2013). While Drosophila is devoid of bivalent domains (Schuettengruber et al. 2009), its PRC2 accesses developmental genes through the more familiar mode of high-affinity interactions with DNA sequence-specific transcription factors (Brown et al. 1998; Grimaud et al. 2006; Geisler and Paro 2015), for which there is little evidence in the case of mammalian PRC2. Nonetheless, many low-affinity chromatin interactions exhibited by both PRC2 and its associated factors can facilitate PRC2 recruitment to and maintenance at its targets (Margueron and Reinberg 2011). Importantly, within the last decade, extensive studies by many in the field have expanded and clarified the multiple, key criteria inherent to this process.
Promoter architecture
The promoters of mammalian developmental genes are associated with CpG-rich DNA (see below) and are bivalent, containing both H3K27me3 and H3K4me3 within the same nucleosome but on different H3 tails (Voigt et al. 2012, 2013). The following evidence supports that modulation of the promoter architecture can set the stage for promoter bivalency at developmental genes in mESCs (Voigt et al. 2013). Upon its activation, the extracellular signal-regulated kinase (Erk1/2) binds to and colocalizes with a subset of PRC2 target genes enriched in GC-/GA-rich motifs in vivo (Fig. 7C, panel i). Activated Erk1/2 enhances local nucleosome turnover, facilitating the access of PRC2 to target genes; whether this access applies to MLL family members that catalyze H3K4me3 is not known. Surprisingly, Erk1/2 also promotes the phosphorylation of the C-terminal domain (CTD) of RNA polymerase II at Ser5, a modification primarily catalyzed by Cdk7, the conventional kinase found in TFIIH. Such phosphorylation fosters promoter “disengagement” whereby RNA polymerase II breaks its interaction with most of the general transcription factors necessary for its recruitment to promoters (Usheva et al. 1992; Komarnitsky et al. 2000; Tee et al. 2014). This swapping of Cdk7 activity is novel and likely arises from the promoter architecture and its G-C content (Parvin and Sharp 1993; Goodrich and Tjian 1994); accordingly, transcription from these developmental genes is independent of Cdk7 in vitro and in vivo (Tee et al. 2014).
Indeed, CGIs (see below) correlate with low nucleosome occupancy, and PRC2 is recruited to CGIs in response to transcription inhibition in mESCs (Deaton and Bird 2011; Fenouil et al. 2012; Riising et al. 2014). Earlier studies in Drosophila demonstrate that homeotic gene clusters, which are targets of PcG and TrxG proteins, show rapid histone turnover and low nucleosome density (Mito et al. 2007; Deal et al. 2010).
CGIs
The most obvious feature of PRC2-binding sites at developmental genes is the presence of CGIs, ∼1- to 2-kb DNA elements comprising CpG-rich DNA near the transcriptional start sites (TSSs) of promoters (Illingworth and Bird 2009; Mendenhall et al. 2010; Lynch et al. 2012; Long et al. 2013). They are generally found at promoters lacking DNA methylation (Fig. 7C, panel ii). Integration of DNA elements containing CpG-rich sequences to an exogenous locus is sufficient to nucleate PRC2 in vivo (Mendenhall et al. 2010; Lynch et al. 2012; Jermann et al. 2014). However, and of importance, these CGIs must be devoid of transcription-activating motifs (Mendenhall et al. 2010). In accordance, transcription inhibition leads to an increased occupancy of PRC2 at CGIs in mESCs in keeping with its role in maintaining gene silencing (Riising et al. 2014). Collectively, these results point to CGIs as being critical to mammalian PRC2 recruitment. This scenario is somewhat similar to the case in Drosophila, where PRC2 is recruited to PREs that contain high-affinity DNA-binding sites for sequence-specific DNA-binding proteins with which PRC2 interacts (Grimaud et al. 2006; Geisler and Paro 2015). However, in the case of mammalian PRC2, interaction with CGIs is direct and a consequence of many low-affinity interactions with some specificity in the DNA sequence, as described below. These low-affinity sequence-specific interactions are exhibited by a subset of PRC2 accessory proteins (e.g., JARID2 and MTF2) (see below) and are stabilized by interactions with nucleosomes and DNA (e.g., through RBAP48, JARID2, and AEBP2) (see Figs. 5B, 6B; “Features of the PRC2 Holoenzyme Regulated by Distinct Accessory Factors”).
Given that the majority of mammalian gene promoters do contain CGIs, those that are targeted by PRC2 should have additional features (Illingworth and Bird 2009; Long et al. 2013). Indeed, a recent report finds them to be distinguished by an enrichment in “GA” and/or “GCN” tandem repeat motifs in mESCs (Fig. 7C, panel ii; Oksuz et al. 2018). This study devised a system in mESCs to follow the kinetics of initial PRC2 recruitment and its subsequent establishment de novo of extensive chromatin domains with H3K27me. PRC2 integrity is first disrupted through an EED knockout such that H3K27me2/me3 is depleted from the cell, and then PRC2 is rescued by an inducible expression of EED. By tracking the de novo PRC2 occupancy on chromatin, PRC2 sites of “nucleation” are identified and grouped as strong or weak to reflect the levels of PRC2 and H3K27me3 deposition (Oksuz et al. 2018). Although the majority of nucleation sites are enriched for “GA”- and/or “GCN”-rich tandem repeat motifs, the distribution of GA content is slightly different and in the case of weak sites, the GCN tandem repeats are shorter (Fig. 7D). Importantly, these nucleation sites correspond to bivalent promoters in mESCs (see below). H3K27me3 also exhibits a delayed deposition at genomic locales termed “spreading” sites that initially comprise a stretch of H3K27me2 (see below). Consistent with the established correlation between CGIs and PRC2 binding, the nucleation sites are enriched with those CGIs, comprising a higher average CpG count relative to CGIs genome-wide and those within spreading sites. This study points to the key role of nucleation sites in de novo PRC2 targeting (Oksuz et al. 2018) and is consistent with a recent study suggesting, not surprisingly, that H3K27me is not the primary recruitment factor for PRC2 (Hojfeldt et al. 2018).
DNA methylation status
Another feature of CGI targets is their hypomethylation. Genome-wide studies demonstrate that cytosine DNA methylation at CGIs adversely affects PRC2 binding (Holoch and Margueron 2017). However, PRC2 and DNA hypermethylation do coexist in the case of mouse XCI and/or other imprinted genes (Brockdorff 2017). Importantly, the mechanism for PRC2 recruitment to the Xi is still under debate and is likely to differ from that at developmental genes, perhaps being CGI-independent (see “Imprinted Genes”). As AEBP2 appears to mediate binding of PRC2 to methylated DNA in vitro (see “Features of the PRC2 Holoenzyme Regulated by Distinct Accessory Factors”; Wang et al. 2017b), AEBP2 might exert a role in recruiting PRC2 to the mouse Xi and perhaps to a small set of defined genomic loci and/or transposons in vivo.
Partners aiding mammalian PRC2 recruitment to developmental genes
What then is the molecular link between PRC2 and CGIs? Intriguingly, JARID2 and PCL proteins that associate with PRC2 also bind directly to unmethylated CGIs at a subset of promoters (Li et al. 2010, 2017). Importantly, the knockout of either JARID2 or MTF2 (the predominant PCL protein in mESCs) in steady-state mESCs abolishes stable PRC2 binding to chromatin but is ineffectual with respect to the extent of H3K27me3 deposition (Shen et al. 2009; Landeira et al. 2010; Li et al. 2010, 2017; Pasini et al. 2010; Casanova et al. 2011). Nonetheless, some detectable H3K27me3 foci do become evident in EED rescue experiments performed in cells devoid of both JARID2 and MTF2, albeit considerably later, suggesting that PRC2 can still access its targets in a weak and unstable manner (Fig. 7C, panel ii), as core PRC2 does bind to chromatin via its many low-affinity interactions (Fig. 6; Margueron and Reinberg 2011; Poepsel et al. 2018). In accordance, a recent report demonstrates preferential but transient interaction of PRC2 to GC-rich DNA in vitro (Wang et al. 2017b). There appears then to be a redundancy of factors affecting PRC2 recruitment and its stability on chromatin. While PRC2 cannot be readily detected on chromatin by ChIP (chromatin immunoprecipitation) assays using a population of cultured cells, H3K27me3 eventually recovers during G1 and G2–M phase as its turnover mainly occurs at the S phase of the cell cycle. Importantly, in fast-developing early stage embryos, a more stable association of PRC2 with chromatin is likely a necessity to deposit H3K27me3 expeditiously.
Given that core PRC2, JARID2, and MTF2 can interact independently with GC-rich DNA, we propose that PRC2 recruitment can occur via a “hit and run” mechanism whereby core PRC2 transiently interacts with the nucleation sites and inefficiently catalyzes H3K27me3 on chromatin (Oksuz et al. 2018). Indeed, recent live-cell single-molecule imaging studies demonstrate the dynamic interaction of PRC2 with chromatin in human osteosarcoma cells (U2OS) (Youmans et al. 2018). Still, in complex with JARID2/MTF2, PRC2 is stabilized at the nucleation sites leading to efficient H3K27me3 catalysis. The newly identified PRC2-interacting factors such as EPOP, PALI1/2, and others, can also affect PRC2 interaction with chromatin, even in a way to dislodge PRC2 from target sites (Beringer et al. 2016; Holoch and Margueron 2017; Conway et al. 2018). Furthermore, although not required for initial PRC2 recruitment, H3K27me3 does stabilize its binding on chromatin, as initial structural and kinetic studies demonstrate that PRC2 displays a higher binding affinity toward those nucleosomes containing H3K27me3 relative to unmodified ones (Margueron et al. 2009; Hojfeldt et al. 2018; Oksuz et al. 2018). Such increased PRC2 stability on chromatin as a function of the presence of H3K27me3 has direct implications for H3K27me3 transmission during DNA replication (see below; Hansen et al. 2008; Margueron et al. 2009).
Taken together and in agreement with our previously proposed model (Margueron and Reinberg 2011), the stability of PRC2 on chromatin is reflected by the sum of its chromatin interactions that vary in affinity, as evidenced by: PRC2 core constituents, PRC2 accessory subunits, EED–H3K27me3 interaction, and PRC2–RNA interactions. In isolation, the low-affinity interactions might synergize to enable detectable PRC2 catalysis on chromatin. Notably, some PRC2 accessory factors, including MTF2 and JARID2, are expressed at very low levels in most differentiated cells (Li et al. 2010; Zhang et al. 2011; Son et al. 2013). In these cases, the remaining low-affinity interactions might be sufficient to replenish H3K27me levels. For example, EZH1 maintains targeting of PRC2 in terminally differentiated myoblasts that do not express JARID2 or EZH2 (see “EZH1 and EZH2, PRC2 Writers with Different Functions”; Son et al. 2013). Alternatively, cell type-specific PRC2-interacting factors that have yet to be identified might facilitate PRC2 recruitment in different cell lineages.
Spreading of PRC2 activity
Following its recruitment to nucleation sites, PRC2 generates extensive regions of H3K27me2/me3 domains across the genome that range in size from a few kilobases to >100 kb, as found across the Hox clusters (Boyer et al. 2006). Though distant with respect to each other on the linear genome, these domains often colocalize within the nucleus, forming a network of intrachromosomal and interchromosomal interactions in mESCs and in Drosophila (Bantignies et al. 2011; Tolhuis et al. 2011; Denholtz et al. 2013; Joshi et al. 2015; Schoenfelder et al. 2015; Vieux-Rochas et al. 2015; Ogiyama et al. 2018). Interestingly, these long-range interactions are not essentially dependent on PRC2, as many are retained in the absence of EED and H3K27me2/me3, but they do require PRC1 (Denholtz et al. 2013; Schoenfelder et al. 2015; Kundu et al. 2017; Oksuz et al. 2018). Importantly, the identified nucleation sites are enriched within spatially interacting Polycomb targets, wherein initial H3K27me3 catalysis is observed as foci by immunofluorescence (Fig. 8A; Oksuz et al. 2018) Thus, not all, but specific, CGIs participate in a network of interactions that function to nucleate PRC2. How, then, are the large H3K27me2 or H3K27me3 domains established and maintained after PRC2 recruitment to a nucleation site? Also, what is the contribution of genomic clustering (through long-range interactions) of polycomb targets in this process?
Figure 8.

Spreading of PRC2 activity after initial recruitment. (A) PRC2 targets are engaged in a network of interactions wherein nucleation sites are concentrated, forming Polycomb foci. Following the nucleation event, PRC2 spreads H3K27me2/3 domains across the genome proximally as well as distally via long-range 3D contacts, all within Polycomb foci. (B) Detailed mechanism by which PRC2 spreads the products of its catalysis. PRC2 first catalyzes H3K27me2 at the nucleation sites (strong or weak), which are then converted to H3K27me3 once PRC2 reaches sufficient concentrations. Through binding to H3K27me3, PRC2 is allosterically stimulated and rapidly spreads H3K27me2 to adjacent chromatin. H3K27me2 is then converted to H3K27me3, and, as PRC2 moves further from the nucleation sites, its stability on chromatin decreases such that H3K27me3 domains remain proximal and H3K27me2 domains remain distal to its nucleation sites. The strong and weak nucleation sites engage in long-range interactions.
H3K27me3 could spread to neighboring regions on the genome via its “write and read” mechanism and form large domains of H3K27me3 (Reinberg and Vales 2018). Based on results from the in vivo EED rescue experiments, we propose the following two-step mechanism (nucleation and spreading) to portray the formation of large repressive chromatin domains via PRC2 (Oksuz et al. 2018). PRC2 in complex with accessory proteins (such as JARID2 and/or MTF2 or other factors depending on the cell type) recognizes and stably binds to the nucleation sites wherein H3K27me2 is deposited initially and converted to H3K27me3 once a “critical” PRC2 concentration is attained. From this initial nucleation event, PRC2 binds to its own product, H3K27me3; undergoes an allosteric stimulation; and rapidly spreads H3K27me2 domains. H3K27me2 is then converted to H3K27me3 proximally as well as distally via long-range 3D contacts, all within Polycomb foci. The mechanistic basis for H3K27me2 rather than H3K27me3 being spread initially is currently unclear but likely reflects the slower kinetics of trimethyl catalysis, in keeping with the necessity for allosteric activation of PRC2. Of note, these initial tracks of H3K27me2 do exhibit peaks of H3K27me3. In some cases, these peaks correspond to those nucleation sites designated as “weak” based on their CGI sequence (see above), while others might arise from long-range 3D contacts with other nucleation sites. As PRC2 “travels” even further from its nucleation sites, its residency time on chromatin decreases resulting in a more pronounced deposition of H3K27me2 than H3K27me3 (Fig. 8A,B).
The nucleation and spreading model point to the importance of genome organization in the establishment and maintenance of repressive H3K27me domains. In Drosophila, a transgenic locus containing a PRE and its endogenous counterpart located in a different chromosome engage in physical contact mediated by long-range chromosomal interaction (Ciabrelli et al. 2017). This interaction promotes a stable epigenetic state that nonetheless could be stably reverted by disrupting the chromatin contacts, underscoring the significance of chromatin architecture in the formation of epigenetically inherited repressive H3K27me chromatin domains (Ciabrelli et al. 2017; Ogiyama et al. 2018).
Boundaries to spreading
How, then, does H3K27me2/me3 spreading eventually stop? The classic phenomenon of position effect variegation (PEV) in Drosophila arises from the spreading of a translocated region of H3K9me3-type constitutive heterochromatin into adjacent euchromatic genes, with resultant silencing (Talbert and Henikoff 2006). A similar scenario might be expected in the case of H3K27me3-rich domains. However, the following findings indicate the opposite.
The HOXA cluster of genes has a number of CTCF cognate DNA-binding sites, several of which border the promoters of specific HoxA genes that are repressed in ESCs but temporally expressed upon induction to differentiation (Mazzoni et al. 2013). The DNA sequence-specific transcription factor CTCF is well-known to function as an insulator and to engage in the formation of topologically associated domains (TADs) in vivo (Phillips-Cremins and Corces 2013). mESCs exhibit repression of the HOX cluster, and these repressed genes are contained within independent single TADs, all decorated with H3K27me2/me3 (Narendra et al. 2015). Upon differentiation into cervical motor neurons, the HOXa1 to HOXa6 genes are expressed in the midst of active chromatin features such as H3K4me3 and RNA polymerase II and now are within a new TAD delineated by preexisting CTCF sites. Under these conditions, the proximal HOXa7 gene normally remains repressed. However, upon deletion of the CTCF site caudal to the HOXa7 promoter, the HOXa7 gene is inappropriately activated in both cell culture and mice, resulting in homeotic transformations (Narendra et al. 2015, 2016). As such, it is the actual process of transcription that results in the loss of H3K27me2/me3 domains with the concomitant gain of histone modifications associated with active transcription, such as H3K4me3, giving rise to expression of the Hoxa7 gene and its relocation to the TAD containing the Hoxa1 to Hoxa6 genes. Thus, in contrast to PEV in Drosophila, an active chromatin domain can invade the repressive H3K27me3-domain, the integrity of which depends on CTCF binding to its cognate DNA site (Fig. 9A,B). Given that the stability of CTCF on chromatin requires its RNA binding activity (Hansen et al. 2018; Saldana-Meyer et al. 2019), we suspect that disrupting this latter activity would give rise to similar developmental defects.
Figure 9.
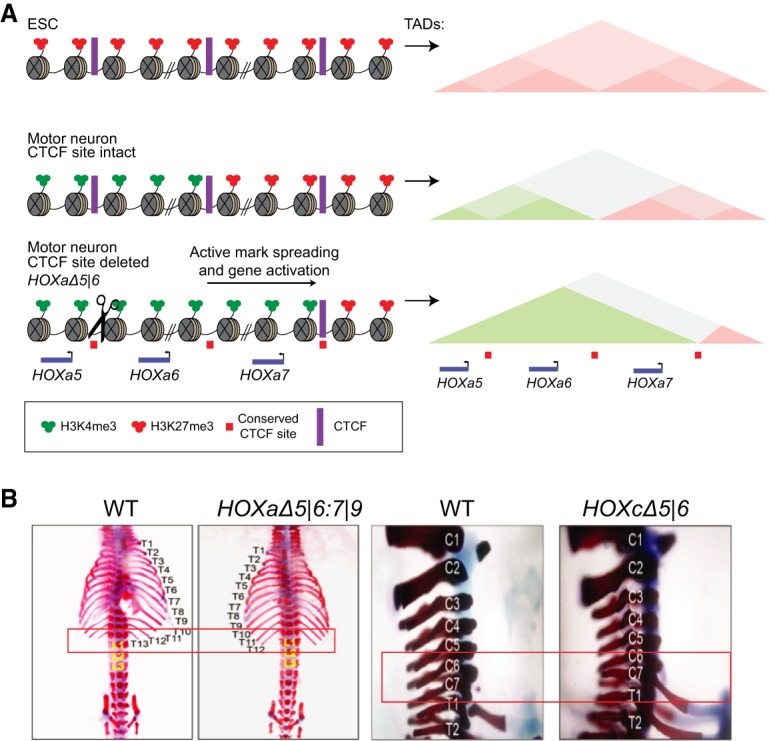
How does PRC2 spreading stop? (A) Upon differentiation of ESCs to motor neurons, a tight boundary between transcriptionally active domains and PRC2-mediated repressive domains is maintained by CTCF binding to its cognate sites in the HoxA cluster. In this case, active loci are sequestered in a TAD independent from that sequestering repressed domains. (B) Deletion of CTCF-binding sites result in homeotic transformation in mice. (Left) Proper ribs do not protrude from the T13 position. (Right) An extra rib aberrantly protrudes from the C7 position (adapted from Narendra et al. 2016).
In keeping with the homeotic transformations that arise upon loss of H3K27me2/me3 domains in the HOXA cluster, disruption to chromatin compaction mediated by the CBX2 component of a class of canonical PRC1 complexes also disturbs body patterning in mice (Lau et al. 2017). Mutation of the CBX2 residues responsible for chromatin compaction leads to homeotic transformation. These findings with PRC2 and PRC1 underscore their joint roles in maintaining gene repression such that the curtailment of either undermines the endurance of the developmental process.
PRC2 dynamics in ESCs and early development
Perhaps the considerable fascination with PRC2 rests on its regulation in ESCs, given that 95% of EZH2 occupies the promoters of developmentally regulated genes that are bivalent and silent or expressed at low levels (Bernstein et al. 2006). However, as ESCs differentiate, most of these genes lose their bivalency and are subsequently activated or remain stably repressed depending on the lineage pathway (Voigt et al. 2013). In ESCs, although loss of PRC2 increases expression from some target bivalent genes, it does not prevent them from being maintained in the “ground state” (i.e., not activated or repressed by DNA sequence-specific transcription factors, as occurs during differentiation) (Boyer et al. 2006; Shan et al. 2017). Instead, PRC2-deficient ESCs manifest a more drastic and aberrant activation of bivalent genes during induction to differentiation (Boyer et al. 2006). Thus, PRC2 is essential to securing repression after bivalent genes are silenced during differentiation. Bivalent promoters are also present in adult stem cells, albeit being fewer than and distinctive to those in ESCs (Jadhav et al. 2016). However, loss of PRC2 in adult stem cells mainly derepresses these tissue-restricted bivalent genes (Jadhav et al. 2016).
While bivalent promoters are crucial in developing and adult tissues, how are the active and repressive histone modifications spatially organized? Mass spectrometry studies reveal that H3K4me3 usually coexists with H3K27me3 asymmetrically at the two histone H3 tails within a histone octamer of each nucleosome (Voigt et al. 2012). In accordance, biochemical studies show that H3K4me3 directly inhibits PRC2 activity, preventing H3K27me3 catalysis in cis (Schmitges et al. 2011). Likewise, H3K27me3 inhibits H3K4 methyltransferases (Kim et al. 2013a), as described above (“PRC2 and Its Chromatin Substrates”). Together, these features constitute the basis of gene bivalency and demonstrate the antagonistic nature of these active and repressive histone marks while being compatible at a bivalent gene. How bivalent genes acquire this balance and coordinate the coexistence between these two marks at distinct genomic loci in a cell type-dependent manner remains largely unknown. Of note, most current studies on gene bivalency use populations of cultured cells and as such, the stochastic variation of histone modifications should be considered using single-cell analysis tools in the future.
The identification of PRC2 nucleation sites within developmental genes raises the question of how cells adapt to use different nucleation sites upon changing their cellular state, especially given that not all “GA”-rich and/or “GCN” tandem repeat motifs within CGIs serve as nucleation sites in mESCs. For example, there are ∼2700 CGIs with “GCN” tandem repeat motifs in mESCs, but only a subset of these (17%) act as nucleation sites for PRC2 (Oksuz et al. 2018). Certainly, these other candidates might function as nucleation sites in other cell types. For example, during differentiation, a subset of PRC2 target genes are selectively activated, while others are subject to repression. Once an active gene containing a PRC2-unoccupied nucleation site is silenced by DNA sequence-specific transcription repressors during differentiation and all the active chromatin features are cleared, PRC2 would be able to nucleate this GCN motif within a CGI near the TSS. PRC2 would then maintain transcriptional repression through its catalysis of H3K27me3 (Fig. 10). On the other hand, some of the nucleation sites for PRC2 in mESCs might be overriden by transcriptional activators and in this case, PRC2 is evicted as seen in the case of the HoxA cluster during differentiation, whereupon the dynamics of transcription clear the H3K27me3-containing chromatin domains as described above (“Boundaries to Spreading”). In accordance, studies in Drosophila show that the TrxG proteins, which antagonize Polycomb-dependent silencing, can also bind to PREs (Steffen and Ringrose 2014).
Figure 10.
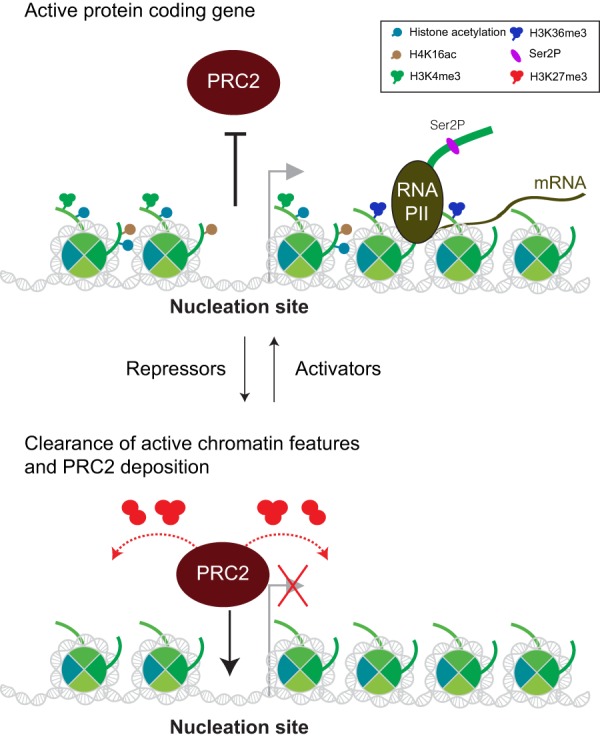
Establishment of PRC2 on chromatin during cellular state transitions. When a given nucleation site is occupied by active chromatin modifications within an actively transcribing gene, PRC2 binding is precluded. Should this gene be bound by transcriptional repressors in response to a change in the cellular state, histone deacetylases and demethylases would then clear all of the active chromatin features. PRC2 can now bind to the cleared nucleation site and maintain repression of this gene in this specific cellular lineage. This process is conceivably reversible, as binding of transcriptional activatiors and histone-modifiying enzymes such as histone acetyltransferases and methyltransferases could evict PRC2 and reactivate this gene in response to a reversal of the cellular state.
PRC2 recruitment to appropriate targets during differentiation requires JARID2 nucleosome-binding activity and JARID2K116me3-mediated allosteric stimulation of PRC2 activity (Sanulli et al. 2015). While the loss of JARID2 or a K116 alanine substitution mutant (JARID2-K116A) has little impact on the level and distribution of H3K27me3 in the ground state of mESCs, the pattern of H3K27me3 deposition is significantly altered upon differentiation in JARID2 knockout cells (Sanulli et al. 2015). H3K27me3 is deposited aberrantly in intergenic regions and nongenic regions of the genome with mis-targeted regions skewed toward CGI-containing regions in the case of JARID2K116A, which retains nucleosome-binding activity. This finding not only substantiates the role of JARID2 in the appropriate recruitment of PRC2 but suggests that during differentiation, PRC2 activation by methylated JARID2 is important in stabilizing its occupancy at the appropriate sites. However, as JARID2 and EZH2 expression decline upon differentiation, PRC2/EZH1 must somehow compensate (see “EZH1 and EZH2, PRC2 Writers with Different Functions”; Son et al. 2013).
EZH1 and EZH2, PRC2 writers with different functions
At this juncture, the properties of the two catalytic homologs of mammalian PRC2 are contrasted to showcase their disparate expression levels and distinctive roles as a function of development. EZH1 and EZH2 likely arose during evolution from the duplication of the single catalytic subunit of PRC2, E(z), found in Drosophila. EZH1 and EZH2 share a highly conserved (94% identical) SET domain and lesser conservation in other regions (64% identical) (Fig. 11A). However, this apparent redundancy belies their seemingly specialized roles during development. Deletion of EZH2 in mESCs impairs cell differentiation due to unscheduled gene expression, and deletion of both EZH1 and EZH2 results in more significant differentiation defects (Shen et al. 2008). However, an EZH2 null mutation results in lethality at early stages of mouse development, while EZH1 null mice are viable (O'Carroll et al. 2001; Ezhkova et al. 2011). However, EZH1 is required for hematopoietic stem cell maintenance, hair follicle homeostasis, and protection from neurodegeneration in the absence of EZH2, indicating its important role(s) in adult tissues (Ezhkova et al. 2009, 2011; Hidalgo et al. 2012; von Schimmelmann et al. 2016; Vo et al. 2018). Intriguingly, while EZH1 is dispensable during heart development, EZH1, but not EZH2, is required for efficient neonatal heart regeneration in an experimental mouse model (Ai et al. 2017).
Figure 11.
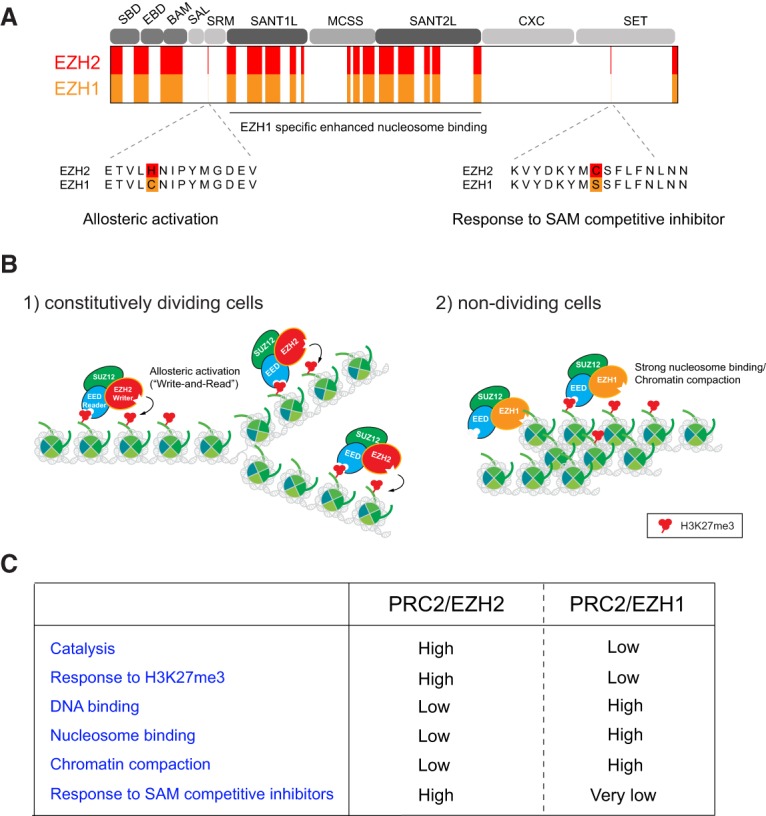
Two distinct catalytic subunits, EZH1 and EZH2. (A) Schematic representation depicting the residues/regions that distinguish EZH2 and EZH1. While the SAL, SRM, CXC, and SET domains are well conserved, the SBD, EBD, BAM, SANT1L, and SANT2L domains are less conserved (highlighted in red and pink). Distinct residues within the SRM and SET domains are indicated. The EZH1-specific nucleosome-binding regions (SANT1L, MCSS, and SANT2L) (Son et al. 2013) are poorly conserved with EZH2. (B) The distinct functions of PRC2/EZH2 and PRC2/EZH1 during development are indicated. (C) A summary of the comparsion between PRC2/EZH2 and PRC2/EZH1 activities.
How do these two catalytic subunits differ? First, PRC2 comprising EZH2 (PRC2/EZH2) exhibits a markedly higher level of catalytic activity than PRC2/EZH1 (Margueron et al. 2008; Son et al. 2013). Second, allosteric activation of PRC2/EZH1 by H3K27me3 is much less efficient than that of PRC2/EZH2 (Lee et al. 2018c), at least partially due to a difference in a key residue within their SRM domains (Fig. 11A). In accordance, recently discovered EED inhibitors that target allosteric activation are less effective against PRC2/EZH1 than PRC2/EZH2 (He et al. 2017; Qi et al. 2017; Lee et al. 2018c). Third, although PRC2/EZH1 contains lower catalytic activity, it does exhibit a higher intrinsic nucleosome/DNA-binding affinity relative to PRC2/EZH2 (Son et al. 2013; Lee et al. 2018c). Last, only PRC2/EZH1 can compact chromatin robustly in vitro and in vivo and does so independently of its catalytic activity (Margueron et al. 2008). Whether this property is due to its strong nucleosome-binding activity is yet to be determined. Nonetheless, these intrinsic properties of PRC2/EZH1 and PRC2/EZH2 point to their differing roles as a function of cell differentiation and development (Fig. 11B,C).
EZH2 activity and expression levels predominate over EZH1 in ESCs, and EZH2 expression is associated with actively proliferating cells (Bracken et al. 2003). The molecular understanding of its elevated catalytic activity and proficient use of its “write and read” mechanism (Lee et al. 2018b,c) underscores why PRC2/EZH2 is most suitable for robustly maintaining H3K27me3-repressive domains after cell division in constitutively dividing cells. Moreover, JARID2/PRC2/EZH2 can drive cells early in differentiation with high levels of methylated H3K27 at appropriate genomic regions given its strong HMT activity and JARID2-mediated allosteric activation and recruitment of PRC2. However, as ESCs differentiate, EZH2 and JARID2 levels decline rapidly in some types of cells, including myotubes (Son et al. 2013) and some neurons (von Schimmelmann et al. 2016; A Schaefer, pers. comm.), while EZH1 is rather stable in all cell types, including terminally differentiated resting cells (Shen et al. 2008), or might even show increased mRNA and protein expression in the postnatal brain (von Schimmelmann et al. 2016). The higher intrinsic nucleosome-binding activity of PRC2/EZH1 could compensate for the loss of JARID2 in differentiated cells, with respect to accessing nucleosomes. As repressive domains are already established in nondividing cells, its lower catalytic activity likely functions to maintain/restore the levels of H3K27me2/me3 in nondividing cells after DNA damage or other injuries to the PRC2-repressed genomic regions. Additionally, the unique property of EZH1 to compact chromatin as well as the presence of PRC2 accessory proteins could also compensate for its low catalytic activity in functionally maintaining repression.
Restoring H3K27me3-repressive domains after DNA replication
During DNA replication, parental nucleosomes containing the histone isoforms H3.1 and H3.2 are disassembled into intact H3–H4 tetramers and H2A–H2B dimers (Yamasu and Senshu 1990; Xu et al. 2010). Nucleosomes reassemble onto newly synthesized daughter DNA from a pool of histones, which consist of parentally modified octamers as well as newly synthesized naïve histones (Fig. 12). The twofold dilution of parental histone posttranslational modifications (hPTMs) are fully restored in daughter cells after DNA replication and/or cell division (Alabert et al. 2015). This arrangement contrasts with the idea that some histone modifications are erased and then re-established in each cell division (Petruk et al. 2012, 2013, 2017a,b). In agreement with the preservation of parental hPTMs during cell division, two repressive hPTMs (H3K27me3 and H3K9me3) persist on chromatin in the absence of their respective modifying enzymatic activity and, as expected, are diluted during subsequent rounds of cell division (Hansen et al. 2008; Zhang et al. 2008; Gaydos et al. 2014; Audergon et al. 2015; Coleman and Struhl 2017; Laprell et al. 2017). For daughter cells to maintain parental cellular identity through appropriate gene expression profiles, the levels of some hPTMs and their parental-specific localization on the genome must be fully restored in daughter cells. The parental levels of either H3K9me2/me3 or H3K27me3 could be attained in daughter cells through the “write and read” mechanism inherent to SUV39H1/2-CLR4 (for fission yeast) or PRC2, respectively (Reinberg and Vales 2018). However, do these parentally “modified” nucleosomes segregate dependably to the same chromatin domain in daughter cells after DNA replication?
Figure 12.
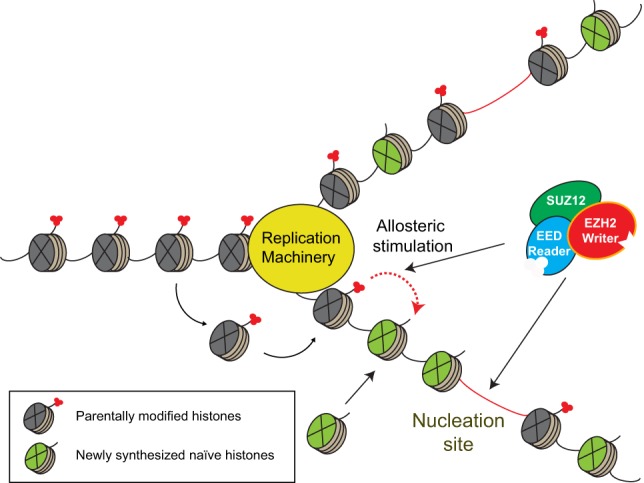
Maintenance of Polycomb repression after DNA replication. Following replication of an H3K27me3-modified region, parentally modified (dark gray) and newly synthesized naïve octamers (green) are randomly deposited to daughter DNA strands. The EED subunit of PRC2 can recognize H3K27me3-modified nucleosomes, with resultant allosteric activation of PRC2. This “write and read” mechanism stimulates catalysis of H3K27me3 to adjacent nucleosomes. In parallel, PRC2 could recognize a nucleation site and spread the modification accordingly.
Indeed, there is evidence from in vitro studies to support local segregation of histones (Madamba et al. 2017). Moreover, a recent report suggests that repressive H3K27me3 as well as active histone marks such as H3K4me3, H3K36me3, and H3K79me3 are locally redeposited on newly synthesized DNA by histone recycling (Reveron-Gomez et al. 2018). However, whether the active histone marks are derived from recycled parental ones or are newly and rapidly synthesized after naïve nucleosome deposition is not distinguished. Another recent report describes an in vivo experimental system capable of formally testing local or distal (dispersed) segregation of nucleosomes/octamers (Escobar et al. 2018). In this case, biotin-tagged parental histones within single genes of active versus repressed chromatin domains are followed temporally during DNA replication. Their local redeposition does occur at the repressive but not at the active chromatin, suggesting that only repressive chromatin domains transmit epigenetic information to daughter cells. In line with this model, H3K27me3 contributes to the transmission of repressive chromatin domains to daughter cells in mESCs (Hansen et al. 2008) as well as across organismal generations in Caenorhabditis elegans (see Box 1, “PRC2 in Transgenerational Inheritance”; Gaydos et al. 2014). In mESCs, transient recruitment of PRC2 and the deposition of H3K27me2/me3 to a reporter system is sufficient to maintain H3K27me3 levels and repression of the reporter for several cell divisions (Hansen et al. 2008), suggesting that, once established, H3K27me3 can provide a platform for PRC2 to maintain the mark using its self-sustaining “write and read” mechanism (Fig. 12). Moreover, that H3K27me2/me3 deposition is limited to nucleation sites when PRC2 contains an EED cage mutant that cannot interact with nor be allosterically activated by H3K27me3, demonstrates the importance of these latter properties for propagating H3K27me2/me3 domains (Oksuz et al. 2018).
Box 1. PRC2 in transgenerational inheritance
Similar to mammalian PRC2, the core components of PRC2 are conserved, albeit duplicated, in plants. In Arabidopsis thaliana, there are 12 homologs for the three core proteins, and they assemble into three forms of holoenzymes: EMF-PRC2, VRN-PRC2, and FIS-PRC2 (Derkacheva and Hennig 2014). However, unlike mammalian PRC2 that catalyzes all states of H3K27 methylation, plants have two distinct H3K27 monomethyltransferases: ATRX5 and ATXR6 (Jacob et al. 2009). While ATXR5/6 and the three PRC2 complexes are crucial for plant development at distinct phase transitions, one of the most compelling processes involving PRC2 is vernalization, the cold-induced flowering. During vernalization, the floral repressor FLOWERING LOCUS C (FLC) is transcriptionally silenced, and, subsequently, PRC2 is recruited de novo to the FLC locus by an antisense noncoding transcript from the locus that acts in cis, resulting in H3K27me3 deposition (Bastow et al. 2004; Swiezewski et al. 2009; Liu et al. 2010). The PRC2-dependent FLC repression is maintained throughout the lifetime in Arabidopsis but undergoes an epigenetic resetting and FLC reactivation during germline development (Choi et al. 2009). Strikingly, a loss-of-function mutation in ELF6, a plant H3K27me3 demethylase, results in an incomplete erasure of the vernalization memory at FLC, allowing a transgenerational inheritance of the vernalized state to subsequent generations (Crevillén et al. 2014).
In the case of C. elegans, PRC2 is dispensable for most developmental processes except for XCI during germline development (Gaydos et al. 2014). C. elegans comprise males with one X chromosome (XO) and hermaphrodites with two X chromosomes (XX). The X chromosome in germ cells is globally repressed except during late oogenesis. Thus, the X chromosome is silent in sperms but unsilenced in oocytes. In PRC2-deficient male (XO) worms, those that inherit an X chromosome from a paternal origin are mostly fertile, whereas those with an X chromosome of maternal origin exhibit a germline defect and are sterile (Gaydos et al. 2014). Interestingly, the PRC2-deficient males use H3K9me2 as an alternative mechanism to maintain the repressed X state, suggesting an evolutionary redundancy in these pathways. In the same study, the paternal H3K27me is transmitted to daughter chromatids through several rounds of cell division in the absence of maternal PRC2, while, in its presence, H3K27me is restored on the “gamete of origin” chromosomes throughout embryogenesis (Gaydos et al. 2014). These findings are consistent with an epigenetic memory being transmitted to the next generation as a function of the presence of the PRC2-H3K27me self-sustaining “write and read” mechanism.
While these unique modes of epigenetic memory regulated by PRC2 have been elegantly demonstrated in plants and worms, the role of PRC2 in mammalian transgenerational inheritance is still under debate and warrants further investigation.
Interestingly, after H3K27me3 is established at a reporter gene in Drosophila, removal of the PRE causes inefficient copying of H3K27me3, promoting its replication-coupled serial dilution (Coleman and Struhl 2017; Laprell et al. 2017). Since Drosophila and mammalian PRC2 both exhibit the “write and read” mechanism, this discrepancy in their long-term maintenance of H3K27me3 is likely due to their distinct recruitment mechanisms. A recent study identified allele-specific inheritance of H3K27me3 domains at imprinted genes that are not regulated by DNA methylation but harbor maternal allele-specific deposition of H3K27me3 in mice (Inoue et al. 2017). Strikingly, ectopic removal of H3K27me3 by overexpressing an H3K27me3-specific demethylase, led to increased expression of these maternal-specific alleles. These H3K27me3 domains are likely maintained through the PRC2-H3K27me3 self-sustaining mechanism as both alleles have the same DNA sequence but only the premodified allele propagates these domains (Inoue et al. 2017).
Aberrant PRC2 regulation during development and in disease
Underscoring its critical role during early development, a genetic deficiency in any core PRC2 component results in embryonic lethality in mice, except for EZH1 due to the presence of the more potent and redundant EZH2 ortholog (Margueron et al. 2008). Interestingly, EZH2 heterozygous mutant mice are viable and fertile but are born at a sub-Mendelian ratio (O'Carroll et al. 2001; Simon et al. 2012). Heterozygous EED or SUZ12 mutations in hematopoietic progenitors impair the integrity of hematopoiesis, and germline SUZ12 heterozygous mutant mice manifest diverse malformations of the brain and neural tube (Miró et al. 2009; Lee et al. 2015b; Ikeda et al. 2016; Yu et al. 2017). These genetic analyses indicate the haploinsufficiency of PRC2 in regulating the proper gene expression program during mouse development. As the levels of PRC2 core subunits, with the exception of EZH1 (see below), decline during development, it has been noted that appropriate levels of PRC2 are needed at specific developmental stages of the nervous system to control the balance between self-renewal and lineage differentiation as well as the switch in cell fate (Hirabayashi et al. 2009; Pereira et al. 2010). Thus, disrupting the dynamic expression of PRC2 during the natural course of development can lead to several developmental defects in humans and mice.
In humans, genetic and nongenetic dysregulation of PRC2 can occur in postzygotic or adult stem cell tissues, leading to the pathogenesis of developmental diseases as well as cancer (Table 1). A case in point is the germline or de novo postzygotic loss-of-function and heterozygous mutations in EZH2 or EED found in patients affected by Weaver syndrome (Gibson et al. 2012; Imagawa et al. 2017). Weaver patients exhibit an overgrowth of various tissues and are also more susceptible to hematopoietic malignancies in which PRC2 loss-of-function mutations are frequently found (Imagawa et al. 2017). While the effect of a haploinsufficiency of PRC2 on development is well-described in mice and humans, the timing and exact mechanism remain obscure. The molecular basis of PRC2 haploinsufficiency likely rests on the antagonism between PRC2 and TrxG proteins such that a disturbance in their appropriate balance leads to a loss in gene bivalency and the firing of undesired promoters.
Table 1.
Genetic mutations altering PRC2 activity in human cancer

While there is no known genetic alteration in PRC2 to date in diabetic patients, a reduction in H3K27me3 is found in insulin-producing pancreatic β-cell islets compared with healthy individuals (Lu et al. 2018). Conditional knockout mouse models for EED or EZH2 in pancreatic β cells suggest that PRC2 activity is required to maintain repression of genes associated with diabetes pathogenesis (Chen et al. 2009; Lu et al. 2018). However, a dedifferentiation phenotype of pancreatic β cells is observed only in a mouse model having a conditional knockout of EED but not of EZH2, while a drastic loss in H3K27me3 is seen in both models (Chen et al. 2009; Lu et al. 2018). This discrepancy suggests that while inefficient in H3K27me3 catalysis, the PRC2/EZH1 complex is still partly safeguarding cell identity, possibly through its high nucleosome-binding activity. Although the data in mice are highly indicative, how PRC2 activity is dampened during the natural course of diabetes progression still remains unclear.
While the function of PRC2 has been rigorously investigated in mESCs and many adult stem cells and differentiation systems, it is also largely unclear as to whether PRC2 remains operational in terminally differentiated, nonproliferative cells. For example, in the first conditional EZH2 knockout mouse model, its ablation in B-cell progenitors led to severe deficiencies in B-cell development, whereas its depletion in peripheral B cells showed no overt functional defect (Su et al. 2003). As most neurogenesis is completed during the embryonic stages and gives rise to neurons that manifest extremely limited proliferative or regenerative potential, the role of PRC2 throughout adulthood merits investigation. A recent report demonstrates that H3K27me3 accumulates in striatal neurons in an age-dependent manner, while, surprisingly, EZH2 expression declines, and PRC2-EZH1 becomes dominant during this process (von Schimmelmann et al. 2016). Thus, despite its lower catalytic activity, PRC2-EZH1 might contribute more than maintenance of presilenced genes in this system. The codepletion of EZH1 and EZH2 in medium spiny neurons (MSNs) leads to a slow but progressive derepression of a specific group of bivalent developmental genes that encode transcription factors with autoregulatory activities and eventually, a neurodegenerative phenotype in mice (von Schimmelmann et al. 2016). Thus, PRC2-mediated suppression of specific genes is critical in differentiated cells, as, once activated, the gene products positively regulate further expression. Notably, PRC1 or DNA methylation cannot compensate for this PRC2-dependent maintenance (von Schimmelmann et al. 2016). As distinct types of neurons express EZH1 and/or EZH2 as well as JARID2 (see “EZH1 and EZH2, PRC2 Writers with Different Functions”), future investigations are warranted to probe the dynamic regulation of PRC2 subcomplexes in the nervous system. These findings together with early elegant genetic experiments in Drosophila demonstrate that PRC2 is required not only during development but also during adulthood to maintain cell identity.
Dysregulation of PRC2 in cancer
Cancer genetics of PRC2
The explosive advancement in high-throughput sequencing technologies during the past decade has greatly benefitted surveillance of the cancer genome. A large number of mutations (Fig. 13A), deletions, amplifications, and translocations in chromatin-modifying enzymes, including those in different subunits of PRC2 are found in various types of human cancer. For example, nonsense and inactivating mutations in EED or SUZ12 are found in 70%–90% of malignant peripheral nerve sheath tumors (MPNSTs); in EZH2, EED, or SUZ12 in 25% of T-cell acute lymphoblastic leukemia (T-ALL) (Ntziachristos et al. 2012; Lee et al. 2014); and in PRC2 core subunits in other cancer types with lower frequencies, such as myelodysplastic syndrome (MDS) and other myeloid malignancies (Sashida et al. 2014; Ueda et al. 2016). Although the functions of many other sense mutations in core PRC2 subunits remain undetermined, most of them are likely to be loss of function, as mutations tend to be deleterious rather than to evolve new functions. However, a few exceptions include the hot spot mutations at Tyr646 (Y646) of human EZH2 and, to a lesser extent, mutations at Ala682 and Ala692 (A682 and A692) (Morin et al. 2010; Bödör et al. 2013). The EZH2-Y646 mutations, including tyrosine to serine, asparagine, phenylalanine, cysteine, or histidine (EZH2Y646X; X = S, N, F, C, or H), are gain-of-function “kinetic mutants” (Fig. 13B; see “EZH2Y646X Mutant Tumors”) and are found in ∼20% of diffuse large B-cell lymphoma (DLBCL), follicular lymphoma (FL), and ∼1% of cutaneous melanoma (Morin et al. 2010; Harms et al. 2014). Intriguingly, recent reports indicate that the EZH1Q571R mutation exhibits a gain-of-function activity in ∼27% of autonomous thyroid adenomas (ATA) (Calebiro et al. 2016). However, the underlying mechanism and biological function of this hyperactive mutation remain largely unknown. Overall, cancer genetic data demonstrate that PRC2 exhibits a pleiotropic role in human cancer. Depending on the cellular context, tumor cells have hijacked PRC2 to facilitate transcriptional programs favorable to their progression.
Figure 13.
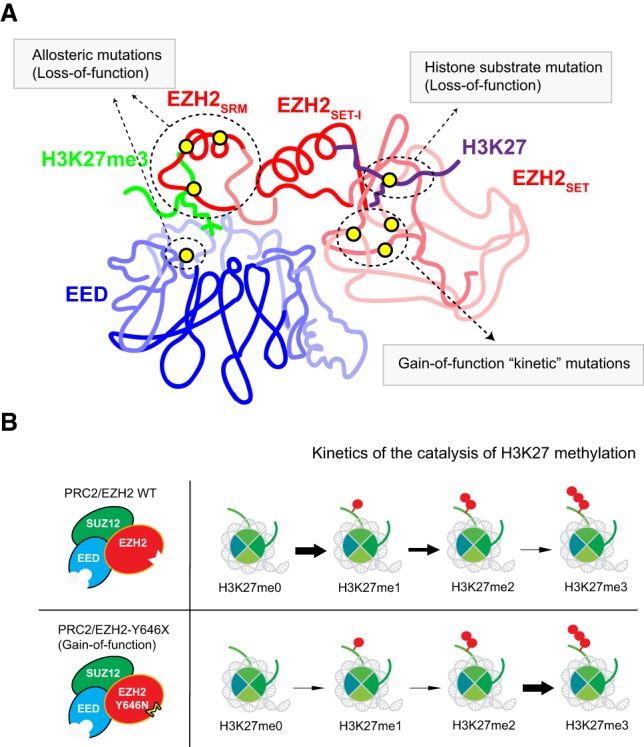
Mutations of PRC2 and its substrate in cancer. (A) Major groups of mutations that influence PRC2 function in cancer. Mutations that are found in the EED cage (EED I363M) (Ueda et al. 2016) and SRM domain of EZH2 (P132S, D142V, and F145L) inhibit allosteric activation of PRC2 (Lee et al. 2018c). Mutations that are found in the catalytic SET domain (Y646X [X = S, N, F, C, or H], A682G, A682V, and A692V) are gain-of-function “kinetic” mutations. Histone H3K27M is a dominant-negative substrate mutation that globally inhibits PRC2 activity. (B) Illustration depicting the kinetics of catalysis of each H3K27 methylation state. EZH2 mutants in Y646 specifically promote the catalysis of H3K27me3 from H3K27me2 but dampen catalysis of the lower methylation states.
Another interesting genetic alteration of PRC2 is the translocation of juxtaposed with another Zn finger protein 1 (JAZF1) generating the JAZF1-SUZ12 fusion protein, hallmarking 50%–80% of endometrial stromal sarcomas (ESSs) (Hrzenjak 2016). Initially, the JAZF1-SUZ12 fusion protein was thought to disrupt PRC2 assembly (Ma et al. 2017). However, a recent study demonstrated that the JAZF1-SUZ12 fusion protein incorporates into PRC2 and precludes the binding of accessory factors JARID2 and EPOP (Chen et al. 2018). Overall, the de novo function and mechanism of JAZF1-SUZ12 fusion protein in ESS oncogenesis remain to be determined. In addition, other gene fusion events involving PRC2 core and accessory proteins have been reported, such as MEAF6-PHF1, EPC1-PHF1, and JAZF1-PHF1 (Micci et al. 2006, 2014), and, similarly, their functions remain unclear.
Although the presence of overexpressed EZH2 in many types of cancers led to speculation as to its role as an oncogene, recent evidence challenges this view (Wassef et al. 2016). As EZH2 expression is coupled to cell cycle progression by the Rb–E2F pathway (Bracken et al. 2003), tumors with high proliferative potential tend to express more EZH2 in order to restore H3K27 methylation during cell division. However, little or no association of EZH2 expression levels with either tumor progression or poor patient survival is evidenced by the proliferation gene signature in metastatic prostate cancer data sets (Bracken et al. 2003). Of note, an increasing amount of literature suggests that EZH2 might exert a PRC2-independent role through other interacting partners (Kim and Roberts 2016). However, most evidence relies on EZH2 immunoprecipitation results, and it remains unclear whether such interactions are actually stable or physiologically relevant. Moreover, given its autoinhibitory state (see “Structural Studies of PRC2”), it is unlikely that EZH2 can methylate target proteins independently of its PRC2 context. Below is an overview of cancers harboring genetic alterations in PRC2.
PRC2-inactivated tumors
PRC2-inactivating homozygous or heterozygous mutations are found frequently in hematopoietic malignancies as well as in MPNST. In a mouse model of T-ALL, MX1-Cre-driven homozygous deletion of EZH2 in the bone marrow is sufficient to potentiate initial oncogenesis, whereas mice with hemizygous EZH2 are protected (Simon et al. 2012). In a mouse model of MPNST, hemizygous deletion of either SUZ12 or NF1 is not sufficient to drive tumorigenesis but together exhibit a synergistic effect on tumor initiation (Lee et al. 2014). Thus, PRC2 appears to act as a tumor suppressor in a dosage-dependent manner. A partial or full elimination of PRC2 along with H3K27me2/me3 would allow transcription factors access to previously silenced target genes. Depending then on the availability of such transcription and other chromatin factors, some genes can be derepressed, whereas some might remain poised. In the NF1 mutant MPNST, partial or full depletion of EED or SUZ12 augments the transcriptional output downstream from the NF1–RAS–MAPK pathway (De Raedt et al. 2014). Similarly, silencing of SUZ12 or EZH2 results in an enriched NOTCH1 gene expression signature genome-wide in T-ALL (Ntziachristos et al. 2012). In EED- or SUZ12-deficient MPNST, two independent studies demonstrate that ectopic expression of EED or SUZ12, respectively, functionally suppresses cancer cell proliferation (De Raedt et al. 2014; Lee et al. 2014). This outcome is surprising as once PRC2 is aberrantly inactivated, genes that allow access to transcriptional machineries and HMTs that deposit H3K4me3 and H3K36me2/me3 would inhibit, in principle, subsequent PRC2 recruitment and activity. Thus, further studies are warranted to examine the chromatin dynamics in PRC2-rescued MPNST cells. Overall, in these tumors, PRC2-mediated gene repression is a key antitumor mechanism protecting against aberrant gene activation downstream from oncogenic signal transduction and transcription during the course of cancer progression.
EZH2Y646X mutant tumors
EZH2Y646X mutations were initially reported as loss of function due to an inability to catalyze H3K27 methylation in vitro using an unmethylated H3 tail peptide as substrate (Morin et al. 2010). Of note, the Y646 residue of full-length human EZH2 is equivalent to Y641 of a shorter EZH2 isoform found in both humans and mice. Further biochemical studies demonstrate that EZH2Y646X mutants do fail to efficiently catalyze H3K27me1 and H3K27me2 but manifest a strikingly enhanced conversion of H3K27me2 to H3K27me3 (Yap et al. 2011). Such findings correlate with the high levels of H3K27me3 found in EZH2Y646X mutant tumors in vivo, and an excess of H3K27me3 at certain tumor suppressors and lineage differentiation genes facilitates oncogenesis (Velichutina et al. 2010; Yap et al. 2011; Béguelin et al. 2013). Of note, the EZH2A682 and EZH2A692 mutations behave similarly to EZH2Y646X (Bödör et al. 2013). These mutations are more precisely “kinetic mutants” (Fig. 13A,B) and are always found to be heterozygous as the wild-type EZH2 allele must provide the H3K27me2 substrate (Morin et al. 2010; Bödör et al. 2013). Mechanistically, Y646, A682, and A692 are present within the substrate-binding pocket of the SET domain of EZH2 such that the pocket size and substrate orientation is altered to selectively favor H3K27me3 (McCabe et al. 2012a). Surprisingly, the conditional expression of EZH2Y646N in lymphoid progenitors of mouse models is sufficient to drive lymphomagenesis, indicating that EZH2Y646N is a bona fide oncogene in B-cell lymphoma (Velichutina et al. 2010; Béguelin et al. 2013). More importantly, in human DLBCL and FL, tumor cells harboring EZH2Y646X exhibit a dependency on PRC2 activity, and the use of PRC2 inhibitors is currently undergoing rigorous clinical development.
Similarly, accumulating evidence indicates that tumors harboring loss-of-function mutations in the negative or antagonistic regulators of PRC2 and H3K27me3 can also be hypersensitive to PRC2 inhibitors. These mutations are found in the H3K27 demethylases (UTX and JMJD3), the H2AK119 deubiquitinase (BAP1), and MLL3 (one of the H3K4 methyltransferases that functions specifically at enhancers, yet its enzymatic activity is dispensable) (van Haaften et al. 2009; LaFave et al. 2015; Dorighi et al. 2017; Wang et al. 2018a). Of note, some of these mutations leading to excessive PRC2 activity appear to be context- and cancer type-dependent, at least in the case of BAP1 (LaFave et al. 2015; Schoumacher et al. 2016). While conceptually reasonable to target the augmented PRC2 activity in these tumors, the therapeutic index of PRC2 inhibitors in these contexts remains to be investigated.
PRC2 inhibitors
Many pharmaceutical companies have invested vigorously in the development of inhibitors to tackle PRC2-addicted human cancers (Gulati et al. 2018). An initial inhibitor, 3-deazaneplanocin A (DZNep), that blocks S-adenosylhomocysteine (SAH) synthesis was developed. However, DZNep functions by reducing the intracellular levels of SAM and consequently is a nonspecific inhibitor targeting different HMTs (Miranda et al. 2009). Thereafter, high-throughput screening efforts yielded pyridone-containing competitive inhibitors of SAM (SAM-competitive inhibitors), such as El1 (Qi et al. 2012), UNC1999 (Konze et al. 2013), GSK-126 (McCabe et al. 2012b), tazemetostat (Knutson et al. 2014), CPI-1205 (Vaswani et al. 2016), and DS-3201, an EZH1/2 dual inhibitor (Honma et al. 2017; Fujita et al. 2018). While many SET domain-containing HMTs have a SAM-binding pocket, pyridone-based inhibitors specifically block SAM binding to PRC2/EZH2 (Brooun et al. 2016). In addition, residues within EED and the SAL and SET domains of EZH2 anchor this inhibitor, further explaining its specificity for PRC2. However, those residues are conserved in EZH1. A few residues are different between the highly conserved SET domains of EZH1 and EZH2 and one of these (Fig. 11A) is determinant to their respective specificity with pyridone-based inhibitors (Bratkowski et al. 2018; Lee et al. 2018c).
On the other hand, studies that reveal the critical role of the EED aromatic cage in PRC2 allosteric activation (Margueron et al. 2009) have led to the development of a novel type of PRC2 inhibitor that blocks a key mechanism of allosteric activation rather than targeting the catalytic site. While SAM-competitive inhibitors completely abrogate the catalytic activity of PRC2, an EED inhibitor could target only allosteric activation, likely preserving some PRC2 basal activity. These structure-guided EED inhibitors, including A-395, EED-226, and UNC5115, directly bind to the aromatic cage of EED, inhibiting its interaction with H3K27me3 (Barnash et al. 2017; He et al. 2017; Huang et al. 2017; Qi et al. 2017), and are quite effective on cells with acquired resistance to SAM-competitive inhibitors (He et al. 2017; Qi et al. 2017). A molecule derived from the EED-226 compound, MAK683, recently entered phase I clinical testing.
Another potential strategy against PRC2 activity involves targeting an interaction interface within the PRC2 complex, including that of EZH2/EED, EZH2-SRM/EZH2-SET-I, or EZH2-SRM/EED (Kim et al. 2013b; Lee et al. 2018b). For example, the α-helical EED-binding domain (EBD) of EZH2 interacts with the opposite side of the EED aromatic cage (Han et al. 2007), and an EBD mimic peptide specifically inhibits EZH2/EED interaction and impairs PRC2 activity in vivo (Kim et al. 2013b). Indeed, using structure-based virtual screening (SBVS), astemizole was identified as an inhibitor of EZH2/EED interaction (Kong et al. 2014). Astemizole destabilizes the PRC2 core subunits, thereby suppressing its activity and arresting the proliferation of PRC2-driven DLBCL. While astemizole is a Food and Drug Administration-approved drug in the treatment of seasonal allergic rhinitis through its histamine H1 antagonist activity, it also causes rare, severe cardiotoxicity by inhibiting the hERG potassium channel (Zhou et al. 1999). Therefore, further studies are required to redevelop new structural analogs.
At this time, there are three major targets of PRC2 inhibitors: the SAM-binding pocket of PRC2/EZH2, the aromatic cage of EED, and protein–protein interaction interfaces (PPIs). Despite the challenges, targeting specific interfaces within the PRC2 complex could streamline the disruption to PRC2 functions. For instance, targeting the interface between PRC2 and accessory factors (Fig. 4) or between PRC2 accessory factors and nucleosomes (Fig. 5B) could inhibit specific PRC2 subcomplexes. Recent structural studies revealing these interfaces (Jiao and Liu 2015; Chen et al. 2018; Kasinath et al. 2018a; Poepsel et al. 2018; Youmans et al. 2018) provide the grist for discovering PPI inhibitors that could be beneficial alternative treatments for PRC2-associated cancer.
The H3K27M oncohistone
Similar to the disease-associated variations in PRC2 itself, a mutation in its histone substrate, H3K27, has given unique insight into PRC2 function. This mutation is a lysine-to-methionine substitution at residue 27 on one copy of H3.1, H3.2, or H3.3 variants (H3K27M) and characterizes ∼80% of DIPG (Schwartzentruber et al. 2012; Wu et al. 2012). H3K27M has been dubbed an “oncohistone” as recent studies suggest that it is likely an initial mutational event in DIPG (Mackay et al. 2017; Filbin et al. 2018; Vinci et al. 2018). Other histone mutations surrounding the H3K27 residue have recently been described, such as H3K36M and H3G34R/V in other malignancies. However, we focus on H3K27M due to the dramatic loss in H3K27me2/me3, a surprising result given that only ∼1%–15% of the total H3 pool in DIPG contains H3K27M (Lewis et al. 2013). Our understanding of how H3K27M impacts PRC2 has revealed subtleties in the dynamic nature of PRC2 interaction with its substrate as well as the lasting impact of aberrant H3K27 substrates on PRC2 function and downstream effects on other histone modifications.
Converging lines of research have been particularly instructive with respect to the repercussions sustained by PRC2 upon its interaction with H3K27M. The initial event is a high-affinity interaction between H3K27M and the EZH2 SET domain (Jiao and Liu 2015, 2016; Justin et al. 2016) that depends on the presence of SAM in vitro (Justin et al. 2016), similar to the interaction of other K-to-M mutants (H3K9M and H3K36M) with their respective SET domains (Jayaram et al. 2016; Yang et al. 2016). This initial interaction is largely transient, as numerous studies indicate that PRC2 and H3K27M do not colocalize on chromatin, as evidenced by ChIP-seq (ChIP combined with high-throughput sequencing)-based (Herz et al. 2014; Piunti et al. 2017) and imaging-based (Hetey et al. 2017; Tatavosian et al. 2018) methodologies. A recent kinetic analysis confirms that H3K27M–PRC2 is not stable on chromatin and that their interaction is transient (Stafford et al. 2018). Following their initial interaction, the steady-state outcome is that the vast majority of PRC2 is redistributed to loci that are mutually exclusive of H3K27M such that PRC2 must be largely released from H3K27M at some point (Fig. 14; Piunti et al. 2017; Fang et al. 2018; Stafford et al. 2018). How PRC2 is evicted is not yet clear but likely relates to a combination of competing factors at the region of H3K27M deposition, such as antagonistic histone modifications, active transcription, and modulation of PRC2 itself (see below). Nonetheless, these studies help reconcile discrepant findings in the literature by showing that H3K27M does indeed recruit PRC2 but transiently, thereby accounting for its higher affinity for H3K27M and the loss of their colocalization after long periods of H3K27M expression.
Figure 14.

Diverse modes of PRC2 inhibition by H3K27M. (Top) When H3K27M is first expressed in a cell, PRC2 becomes trapped due to its higher affinity for nucleosomes comprising H3K27M relative to the wild-type case. (Middle) Interaction between PRC2 and H3K27M is transient, but freed PRC2 is compromised, being less active. The less active PRC2 is then recruited to loci independent of H3K27M, where it exhibits altered activity. (Bottom) The decrease in H3K27me2/3 deposition in H3K27M cells results in a progressive gain in the chromatin deposition of histone posttranslational modifications (hPTMs), such as bulk acetylated histones, including H3K27 acetylation and H3K36me2. Such hPTMs have the potential to directly repel PRC2. Thus, the gain in these hPTMs in H3K27M cells lead to wide-scale effects on PRC2 activity and the epigenetic landscape of chromatin. On the other hand, “protected” PRC2 target loci that are largely devoid of H3K27M exhibit focal gains of H3K27me2/3.
One implication of this dynamic H3K27M–PRC2 interaction is that, despite its transient nature, H3K27me2/me3 is lost in a widespread manner, and this phenomenon might arise from a lasting inhibitory effect of H3K27M on PRC2. Indeed, a recent study shows that PRC2 purified from H3K27M cells displays deficits in its activity (Stafford et al. 2018). Results from a fully reconstituted system comprising highly purified core PRC2 and recombinant oligonucleosome arrays further demonstrate that PRC2 exhibits a persistent inhibition after its interaction with and release from H3K27M (Fig. 14). This lasting effect might relate to PRC2 occupancy being uncoupled from its catalytic activity, as observed with PRC2 allosteric mutants (Lee et al. 2018b). The mechanistic basis for how PRC2 retains a functional “memory” upon its contact with H3K27M is an active area of investigation but might entail a direct conformational change in PRC2 as observed in other cases (e.g., prions and MAD2) (Telling et al. 1996; Hara et al. 2015) and/or a posttranslational modification of PRC2, such as EZH2 automethylation (Lee et al. 2018a; Wang et al. 2018b). Future studies as to how PRC2 activity is changed following its interaction with H3K27M chromatin might provide novel insights into polycomb function as well as how proteins retain a memory of their substrate.
Remarkably, a few select foci in H3K27M-DIPG cells exhibit sharp gains in H3K27me3 despite the large-scale loss in H3K27me2/me3, offering novel insights into how PRC2 establishes repressive domains. Importantly, these remaining H3K27me2/me3 loci are important in DIPG biology given that they associate with alterations in various cell-cycle pathways (Mohammad et al. 2017; Piunti et al. 2017). Beyond the disease relevance, basic insights into this counterintuitive observation can be gleaned from elegant studies in S. pombe. Here, a gain in H3K9me3 and occupancy of its methyltransferase (Clr4) at H3K9me3 recruitment sites is observed in cells that express an analogous K-to-M substitution at H3K9 (H3K9M). Of note, H3K9me3 can no longer be spread from that recruitment site to form a repressive domain (Shan et al. 2016). This scenario is similar to the H3K27M case in that there is a loss in H3K27me3 spreading around regions that show its focal gain (Bender et al. 2013; Chan et al. 2013; Funato et al. 2014; Mohammad et al. 2017; Stafford et al. 2018). As regions showing focal H3K27me3 gains are largely devoid of H3K27M, it is not likely that H3K27M directly traps PRC2 or influences its activity at those regions (Mohammad et al. 2017; Piunti et al. 2017; Fang et al. 2018). Instead, H3K27M is more likely affecting the allosteric activity of PRC2 given that H3K27me3-activated PRC2 displays an increased affinity and heightened sensitivity to the inhibitory effects of H3K27M (Stafford et al. 2018). In addition, strong polycomb targets remain “protected” from H3K27M as H3K27me3 domains and polycomb targets form very dense long-range intrachromosomal and interchromosomal interacting domains that are extremely robust (Denholtz et al. 2013; Schoenfelder et al. 2015; Vieux-Rochas et al. 2015; Oksuz et al. 2018). Thus, the focal gains in H3K27me3 appear to arise from a failure of PRC2 to allosterically activate and spread H3K27me3 within higher-order chromatin domains, leading to the delimited accumulation of H3K27me3 at strong PRC2 targets (Fig. 14).
An emerging theme of particular relevance to therapeutic approaches for H3K27M DIPG is that the suboptimal PRC2 function leads to other downstream alterations in the epigenome. One of the most striking is an increase in deposition of euchromatic marks (e.g., acetylation of histone H3 and H4 together with dimethylation of H3K36), which occur in concert with their binders (e.g., BRDs) (Bender et al. 2013; Chan et al. 2013; Pathak et al. 2015; Piunti et al. 2017; Stafford et al. 2018). These combined factors lead to a cascade that not only further impacts PRC2 occupancy but redistributes a host of histone modifications resulting in an altered chromatin landscape. Each of the direct and indirect consequences of H3K27M on chromatin is beginning to reveal potential therapeutic vulnerabilities in DIPG. These strategies include the direct targeting of H3K27me3 itself and the abnormal function of PRC2 generated by H3K27M (Hashizume et al. 2014; Grasso et al. 2015; Wiese et al. 2016; Mohammad et al. 2017). Recent strategies focus on the abnormalities in histone acetylation by targeting either histone deacetylases, bromodomain proteins or a combination thereof (Grasso et al. 2015; Taylor et al. 2015; Nagaraja et al. 2017; Piunti et al. 2017). Other histone marks and chromatin factors that are also perturbed in H3K27M–DIPG (e.g., H3K36me2) (Stafford et al. 2018), represent viable, yet to be exploited vulnerabilities. While it remains to be seen how any of these approaches will fare in the clinic, H3K27M–DIPG will continue to provide a unique opportunity to study basic biology that will surely impact this devastating disease.
Concluding remarks
Through the elegant genetic screens designed to identify genes important for early development in Drosophila, two gene families with antagonistic functions were discovered: the Trithorax and Polycomb group (PcG) genes. Most important to the studies discussed in this review is the seminal finding that these PcG genes maintain rather than establish patterns of gene repression. As such, their misregulation during development or postdevelopment are expected to result in homeotic and/or cellular transformation, respectively; predictions that are confirmed by many studies in multicellular organisms. In this review, we summarize important findings related to the PcG genes that maintain a silent state of gene expression, specifically PRC2. As PRC2 functions tightly with PRC1, we highlight their functional interrelationship as well as their differences, focusing primarily on the mammalian complexes. From the studies described here, it is clear that mammalian PRC2 is subject to multiple types of regulation, some of which also operate in Drosophila, but we attempted to stress their functional differences. PRC2 regulation is manifested at the structural level as well as by its multiple interactors. The most striking feature is its regulation through an allosteric activation manifested by important structural changes in the complex induced upon interaction of one of its subunits with the product of its catalysis, H3K27me3.
While mammalian PRC2 can form complexes with two different catalytic subunits, EZH1 or EZH2, and EZH1 displays a deficiency in response to allosteric activation due to a specific amino acid substitution, only one catalytic subunit, E(z), exists in Drosophila. Thus, the regulation of the mammalian and Drosophila complexes are clearly different, particularly in differentiated cells as EZH1 expression predominates over that of EZH2 in the mammalian case. However, one of their most important differences lies in their mode of recruitment to target genes. In Drosophila, PRC2 is recruited through its interactions with proteins that display high-affinity binding for specific DNA elements present in the so-called PREs. PREs are absent in mammals, and those proteins binding to PREs are either absent or display different functions in mammals and do not recruit PRC2. These differences relate directly to the respective mechanism of propagation of the repressed state of target genes (H3K37me2/3). If PREs are deleted in Drosophila, the repressed state of target genes will be propagated through only a few cell divisions, operating through the “write and read”-induced allosteric activation of PRC2. However, as cells continue to divide without an efficient mode to recruit the complex to maintain the histone modification, the epigenetic effect is lost as the H3K27me modification is diluted. In contrast, mammalian PRC2 is recruited to CGI elements distributed throughout its target genes and independent of proteins displaying high affinity for specific DNA elements.
Whether the different PRC2 interacting proteins form transient or more stable associations with the core complex, their roles within defined programs during differentiation or in differentiated, postmitotic cells are most pivotal. Recent advances point to some of these roles but also engender further questions on the dynamic regulation of distinct PRC2 subcomplexes in tissue-specific contexts during the course of development and in disease states. A full appreciation of the biological relevance of these different PRC2 complexes and the contributing functions of the various associated proteins will require the development of assays that can detect PRC2 in action. The introduction of single-cell gene expression profiles in conjunction with the dynamic association of core PRC2 with different polypeptides within different differentiation programs will be highly informative. Indeed, increasing the sensitivity of mass spectrometry to identify the interactomes in single cells will be extremely valuable, an ambitious goal, but with multiple efforts in new technology, we expect this to be possible in the future. The combination of such new technology with the existing PRC2 structural studies and the rich biochemistry described (and to be described) will provide an expansive resource for future studies. One could ask why is it so important to elucidate the precise cellular programs controlled by PRC2? In our opinion, any combination of genes that function together to maintain a gene expression profile dictated early in development by master regulators and a program that incurs alterations through naturally occurring mutations calls for such comprehensive studies.
Competing interest statement
D.R. is a cofounder of Constellation Pharmaceuticals and Fulcrum Therapeutics. All other authors declare that they have no competing interests.
Acknowledgments
We thank Dr. L. Vales for critical reading, advice, and revision of the manuscript. We are also most grateful to Dr. K.-J. Armache, Dr. R. Bonasio, Dr. C. Kadoch, Dr. S.J. Gamblin, Dr. R. Margueron, Dr. A. Schaefer, Dr. A. Tarakhovsky, Dr. P. Trojer, and Dr. J.R. Wilson for their critical comments. D.R.’s laboratory is supported by the National Institutes of Health (R01CA199652 and R01NS100897) and the Howard Hughes Medical Institute. J.-R.Y. is supported by the American Cancer Society (PF-17-035-01). J.M.S. was supported by the Simon Foundation and is currently supported by the National Institutes of Health (K99AA024837). D.R. is grateful to Dr. Edith Heard for so many fruitful discussions in the early stages of this review as well as for the support from Les Chaires Internationales de Research Blaise Pascal, where this review was initially conceived during sabbatical at the Curie Institute in Paris, France.
Footnotes
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.325050.119.
References
- Ai S, Yu X, Li Y, Peng Y, Li C, Yue Y, Tao G, Li C, Pu WT, He A. 2017. Divergent requirements for EZH1 in heart development versus regeneration. Circ Res 121: 106–112. 10.1161/CIRCRESAHA.117.311212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alabert C, Barth TK, Reveron-Gomez N, Sidoli S, Schmidt A, Jensen ON, Imhof A, Groth A. 2015. Two distinct modes for propagation of histone PTMs across the cell cycle. Genes Dev 29: 585–590. 10.1101/gad.256354.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseyenko AA, Gorchakov AA, Kharchenko P V, Kuroda MI. 2014. Reciprocal interactions of human C10orf12 and C17orf96 with PRC2 revealed by BioTAP-XL cross-linking and affinity purification. Proc Natl Acad Sci 111: 2488–2493. 10.1073/pnas.1400648111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida M, Pintacuda G, Masui O, Koseki Y, Gdula M, Cerase A, Brown D, Mould A, Innocent C, Nakayama M, et al. 2017. PCGF3/5-PRC1 initiates Polycomb recruitment in X chromosome inactivation. Science 356: 1081–1084. 10.1126/science.aal2512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonysamy S, Condon B, Druzina Z, Bonanno JB, Gheyi T, Zhang F, MacEwan I, Zhang A, Ashok S, Rodgers L, et al. 2013. Structural context of disease-associated mutations and putative mechanism of autoinhibition revealed by X-ray crystallographic analysis of the EZH2-SET domain. PLoS One 8: e84147 10.1371/journal.pone.0084147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audergon PN, Catania S, Kagansky A, Tong P, Shukla M, Pidoux AL, Allshire RC. 2015. Epigenetics. Restricted epigenetic inheritance of H3K9 methylation. Science 348: 132–135. 10.1126/science.1260638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballare C, Lange M, Lapinaite A, Martin GM, Morey L, Pascual G, Liefke R, Simon B, Shi Y, Gozani O, et al. 2012. Phf19 links methylated Lys36 of histone H3 to regulation of Polycomb activity. Nat Struct Mol Biol 19: 1257–1265. 10.1038/nsmb.2434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantignies F, Roure V, Comet I, Leblanc B, Schuettengruber B, Bonnet J, Tixier V, Mas A, Cavalli G. 2011. Polycomb-dependent regulatory contacts between distant Hox loci in Drosophila. Cell 144: 214–226. 10.1016/j.cell.2010.12.026 [DOI] [PubMed] [Google Scholar]
- Barnash KD, The J, Norris-Drouin JL, Cholensky SH, Worley BM, Li F, Stuckey JI, Brown PJ, Vedadi M, Arrowsmith CH, et al. 2017. Discovery of peptidomimetic ligands of EED as allosteric inhibitors of PRC2. ACS Comb Sci 19: 161–172. 10.1021/acscombsci.6b00174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastow R, Mylne JS, Lister C, Lippman Z, Martienssen RA, Dean C. 2004. Vernalization requires epigenetic silencing of FLC by histone methylation. Nature 427: 164–167. 10.1038/nature02269 [DOI] [PubMed] [Google Scholar]
- Béguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, Shen H, Yang SN, Wang L, Ezponda T, et al. 2013. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 23: 677–692. 10.1016/j.ccr.2013.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran M, Yates CM, Skalska L, Dawson M, Reis FP, Viiri K, Fisher CL, Sibley CR, Foster BM, Bartke T, et al. 2016. The interaction of PRC2 with RNA or chromatin is mutually antagonistic. Genome Res 26: 896–907. 10.1101/gr.197632.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DT, Kool M, Zapatka M, Northcott PA, Sturm D, Wang W, et al. 2013. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 24: 660–672. 10.1016/j.ccr.2013.10.006 [DOI] [PubMed] [Google Scholar]
- Beringer M, Pisano P, Di Carlo V, Blanco E, Chammas P, Vizán P, Gutiérrez A, Aranda S, Payer B, Wierer M, et al. 2016. EPOP functionally links elongin and polycomb in pluripotent stem cells. Mol Cell 64: 645–658. 10.1016/j.molcel.2016.10.018 [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. 2006. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125: 315–326. 10.1016/j.cell.2006.02.041 [DOI] [PubMed] [Google Scholar]
- Beuchle D, Struhl G, Müller J. 2001. Polycomb group proteins and heritable silencing of Drosophila Hox genes. Development 128: 993–1004. [DOI] [PubMed] [Google Scholar]
- Blackledge NP, Farcas AM, Kondo T, King HW, McGouran JF, Hanssen LLP, Ito S, Cooper S, Kondo K, Koseki Y, et al. 2014. Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell 157: 1445–1459. 10.1016/j.cell.2014.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bödör C, Grossmann V, Popov N, Okosun J, O'Riain C, Tan K, Marzec J, Araf S, Wang J, Lee AM, et al. 2013. EZH2 mutations are frequent and represent an early event in follicular lymphoma. Blood 122: 3165–3168. 10.1182/blood-2013-04-496893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, et al. 2006. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441: 349–353. 10.1038/nature04733 [DOI] [PubMed] [Google Scholar]
- Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. 2003. EZH2 is downstream of the pRB–E2F pathway, essential for proliferation and amplified in cancer. EMBO J 22: 5323–5335. 10.1093/emboj/cdg542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratkowski M, Yang X, Liu X. 2018. An evolutionarily conserved structural platform for PRC2 inhibition by a class of Ezh2 inhibitors. Sci Rep 8: 9092 10.1038/s41598-018-27175-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun SMG, Kirkland JG, Chory EJ, Husmann D, Calarco JP, Crabtree GR. 2017. Rapid and reversible epigenome editing by endogenous chromatin regulators. Nat Commun 8: 560 10.1038/s41467-017-00644-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brien GL, Gambero G, O'Connell DJ, Jerman E, Turner SA, Egan CM, Dunne EJ, Jurgens MC, Wynne K, Piao L, et al. 2012. Polycomb PHF19 binds H3K36me3 and recruits PRC2 and demethylase NO66 to embryonic stem cell genes during differentiation. Nat Struct Mol Biol 19: 1273–1281. 10.1038/nsmb.2449 [DOI] [PubMed] [Google Scholar]
- Brockdorff N. 2017. Polycomb complexes in X chromosome inactivation. Philos Trans R Soc L B Biol Sci 372: 20170021 10.1098/rstb.2017.0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooun A, Gajiwala KS, Deng Y-L, Liu W, Bolaños B, Bingham P, He Y-A, Diehl W, Grable N, Kung P-P, et al. 2016. Polycomb repressive complex 2 structure with inhibitor reveals a mechanism of activation and drug resistance. Nat Commun 7: 11384 10.1038/ncomms11384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JL, Mucci D, Whiteley M, Dirksen ML, Kassis JA. 1998. The Drosophila Polycomb group gene pleiohomeotic encodes a DNA binding protein with homology to the transcription factor YY1. Mol Cell 1: 1057–1064. 10.1016/S1097-2765(00)80106-9 [DOI] [PubMed] [Google Scholar]
- Cai L, Rothbart SB, Lu R, Xu B, Chen WY, Tripathy A, Rockowitz S, Zheng D, Patel DJ, Allis CD, et al. 2013. An H3K36 methylation-engaging tudor motif of polycomb-like proteins mediates PRC2 complex targeting. Mol Cell 49: 571–582. 10.1016/j.molcel.2012.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calebiro D, Grassi ES, Eszlinger M, Ronchi CL, Godbole A, Bathon K, Guizzardi F, de Filippis T, Krohn K, Jaeschke H, et al. 2016. Recurrent EZH1 mutations are a second hit in autonomous thyroid adenomas. J Clin Invest 126: 3383–3388. 10.1172/JCI84894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R, Zhang Y. 2004. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED–EZH2 complex. Mol Cell 15: 57–67. 10.1016/j.molcel.2004.06.020 [DOI] [PubMed] [Google Scholar]
- Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. 2002. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298: 1039–1043. 10.1126/science.1076997 [DOI] [PubMed] [Google Scholar]
- Cao R, Tsukada Y, Zhang Y. 2005. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol Cell 20: 845–854. 10.1016/j.molcel.2005.12.002 [DOI] [PubMed] [Google Scholar]
- Casanova M, Preissner T, Cerase A, Poot R, Yamada D, Li X, Appanah R, Bezstarosti K, Demmers J, Koseki H, et al. 2011. Polycomblike 2 facilitates the recruitment of PRC2 Polycomb group complexes to the inactive X chromosome and to target loci in embryonic stem cells. Development 138: 1471–1482. 10.1242/dev.053652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K-M, Fang D, Gan H, Hashizume R, Yu C, Schroeder M, Gupta N, Mueller S, James CD, Jenkins R, et al. 2013. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev 27: 985–990. 10.1101/gad.217778.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Gu X, Su I, Bottino R, Contreras JL, Tarakhovsky A, Kim SK. 2009. Polycomb protein Ezh2 regulates pancreatic β-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev 23: 975–985. 10.1101/gad.1742509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Jiao L, Shubbar M, Yang X, Liu X. 2018. Unique structural platforms of Suz12 dictate distinct classes of PRC2 for chromatin binding. Mol Cell 69: 802–819.e1. 10.1016/j.molcel.2018.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Hyun Y, Kang M-J, In Yun H, Yun J-Y, Lister C, Dean C, Amasino RM, Noh B, Noh Y-S, et al. 2009. Resetting and regulation of FLOWERING LOCUS C expression during Arabidopsis reproductive development. Plant J 57: 918–931. 10.1111/j.1365-313X.2008.03776.x [DOI] [PubMed] [Google Scholar]
- Choi J, Bachmann AL, Tauscher K, Benda C, Fierz B, Müller J. 2017. DNA binding by PHF1 prolongs PRC2 residence time on chromatin and thereby promotes H3K27 methylation. Nat Struct Mol Biol 24: 1039–1047. 10.1038/nsmb.3488 [DOI] [PubMed] [Google Scholar]
- Ciabrelli F, Comoglio F, Fellous S, Bonev B, Ninova M, Szabo Q, Xuereb A, Klopp C, Aravin A, Paro R, et al. 2017. Stable Polycomb-dependent transgenerational inheritance of chromatin states in Drosophila. Nat Genet 49: 876–886. 10.1038/ng.3848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciferri C, Lander GC, Maiolica A, Herzog F, Aebersold R, Nogales E. 2012. Molecular architecture of human polycomb repressive complex 2. Elife 1: e00005 10.7554/eLife.00005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes-Rojas C, Hernandez AJ, Sarma K, Lee JT. 2014. Regulatory interactions between RNA and polycomb repressive complex 2. Mol Cell 55: 171–185. 10.1016/j.molcel.2014.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman RT, Struhl G. 2017. Causal role for inheritance of H3K27me3 in maintaining the OFF state of a Drosophila HOX gene. Science 356: eaai8236 10.1126/science.aai8236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colognori D, Sunwoo H, Kriz AJ, Wang C-Y, Lee JT. 2019. Xist deletional analysis reveals an interdependency between Xist RNA and Polycomb complexes for spreading along the inactive X. Mol Cell 74: 101–117.e10. 10.1016/j.molcel.2019.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway E, Jerman E, Healy E, Ito S, Holoch D, Oliviero G, Deevy O, Glancy E, Fitzpatrick DJ, Mucha M, et al. 2018. A family of vertebrate-specific Polycombs encoded by the LCOR/LCORL genes balance PRC2 subtype activities. Mol Cell 70: 408––421.e8. 10.1016/j.molcel.2018.03.005 [DOI] [PubMed] [Google Scholar]
- Cooper S, Dienstbier M, Hassan R, Schermelleh L, Sharif J, Blackledge NP, De Marco V, Elderkin S, Koseki H, Klose R, et al. 2014. Targeting polycomb to pericentric heterochromatin in embryonic stem cells reveals a role for H2AK119u1 in PRC2 recruitment. Cell Rep 7: 1456–1470. 10.1016/j.celrep.2014.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper S, Grijzenhout A, Underwood E, Ancelin K, Zhang T, Nesterova TB, Anil-Kirmizitas B, Bassett A, Kooistra SM, Agger K, et al. 2016. Jarid2 binds mono-ubiquitylated H2A lysine 119 to mediate crosstalk between Polycomb complexes PRC1 and PRC2. Nat Commun 7: 13661 10.1038/ncomms13661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crevillén P, Yang H, Cui X, Greeff C, Trick M, Qiu Q, Cao X, Dean C. 2014. Epigenetic reprogramming that prevents transgenerational inheritance of the vernalized state. Nature 515: 587–590. 10.1038/nature13722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Rocha ST, Boeva V, Escamilla-Del-Arenal M, Ancelin K, Granier C, Matias NRR, Sanulli S, Chow J, Schulz E, Picard C, et al. 2014. Jarid2 is implicated in the initial Xist-induced targeting of PRC2 to the inactive X chromosome. Mol Cell 53: 301–316. 10.1016/j.molcel.2014.01.002 [DOI] [PubMed] [Google Scholar]
- Davidovich C, Zheng L, Goodrich KJ, Cech TR. 2013. Promiscuous RNA binding by Polycomb repressive complex 2. Nat Struct Mol Biol 20: 1250–1257. 10.1038/nsmb.2679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidovich C, Goodrich KJ, Gooding AR, Cech TR. 2014. A dimeric state for PRC2. Nucleic Acids Res 42: 9236–9248. 10.1093/nar/gku540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deal RB, Henikoff JG, Henikoff S. 2010. Genome-wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science 328: 1161–1164. 10.1126/science.1186777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaton AM, Bird A. 2011. CpG islands and the regulation of transcription. Genes Dev 25: 1010–1022. 10.1101/gad.2037511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denholtz M, Bonora G, Chronis C, Splinter E, de Laat W, Ernst J, Pellegrini M, Plath K. 2013. Long-range chromatin contacts in embryonic stem cells reveal a role for pluripotency factors and polycomb proteins in genome organization. Cell Stem Cell 13: 602–616. 10.1016/j.stem.2013.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Raedt T, Beert E, Pasmant E, Luscan A, Brems H, Ortonne N, Helin K, Hornick JL, Mautner V, Kehrer-Sawatzki H, et al. 2014. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 514: 247–251. 10.1038/nature13561 [DOI] [PubMed] [Google Scholar]
- Derkacheva M, Hennig L. 2014. Variations on a theme: Polycomb group proteins in plants. J Exp Bot 65: 2769–2784. 10.1093/jxb/ert410 [DOI] [PubMed] [Google Scholar]
- Dorighi KM, Swigut T, Henriques T, Bhanu N V, Scruggs BS, Nady N, Still CD, Garcia BA, Adelman K, Wysocka J. 2017. Mll3 and Mll4 facilitate enhancer RNA synthesis and transcription from promoters independently of H3K4 monomethylation. Mol Cell 66: 568–576.e4. 10.1016/j.molcel.2017.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumesic PA, Homer CM, Moresco JJ, Pack LR, Shanle EK, Coyle SM, Strahl BD, Fujimori DG, Yates JR III, Madhani HD, et al. 2015. Product binding enforces the genomic specificity of a yeast polycomb repressive complex. Cell 160: 204–218. 10.1016/j.cell.2014.11.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar T, Oksuz O, Descostes N, Bonasio R, Reinberg D. 2018. Precise re-deposition of nucleosomes on repressive chromatin domains sustain epigenetic inheritance during DNA replication. bioRxiv 10.1101/418707 [DOI] [Google Scholar]
- Eskeland R, Leeb M, Grimes GR, Kress C, Boyle S, Sproul D, Gilbert N, Fan Y, Skoultchi AI, Wutz A, et al. 2010. Ring1B compacts chromatin structure and represses gene expression independent of histone ubiquitination. Mol Cell 38: 452–464. 10.1016/j.molcel.2010.02.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezhkova E, Pasolli HA, Parker JS, Stokes N, Su IH, Hannon G, Tarakhovsky A, Fuchs E. 2009. Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell 136: 1122–1135. 10.1016/j.cell.2008.12.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezhkova E, Lien W-H, Stokes N, Pasolli HA, Silva JM, Fuchs E. 2011. EZH1 and EZH2 cogovern histone H3K27 trimethylation and are essential for hair follicle homeostasis and wound repair. Genes Dev 25: 485–98. 10.1101/gad.2019811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang D, Gan H, Cheng L, Lee JH, Zhou H, Sarkaria JN, Daniels DJ, Zhang Z. 2018. H3.3K27M mutant proteins reprogram epigenome by sequestering the PRC2 complex to poised enhancers. Elife 7: e36696 10.7554/eLife.36696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenouil R, Cauchy P, Koch F, Descostes N, Cabeza JZ, Innocenti C, Ferrier P, Spicuglia S, Gut M, Gut I, et al. 2012. CpG islands and GC content dictate nucleosome depletion in a transcription-independent manner at mammalian promoters. Genome Res 22: 2399–2408. 10.1101/gr.138776.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari KJ, Scelfo A, Jammula S, Cuomo A, Barozzi I, Stutzer A, Fischle W, Bonaldi T, Pasini D. 2014. Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol Cell 53: 49–62. 10.1016/j.molcel.2013.10.030 [DOI] [PubMed] [Google Scholar]
- Filbin MG, Tirosh I, Hovestadt V, Shaw ML, Escalante LE, Mathewson ND, Neftel C, Frank N, Pelton K, Hebert CM, et al. 2018. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 360: 331–335. 10.1126/science.aao4750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis NJ, Kingston RE, Woodcock CL. 2004. Chromatin compaction by a polycomb group protein complex. Science 306: 1574–1577. 10.1126/science.1100576 [DOI] [PubMed] [Google Scholar]
- Fujita S, Honma D, Adachi N, Araki K, Takamatsu E, Katsumoto T, Yamagata K, Akashi K, Aoyama K, Iwama A, et al. 2018. Dual inhibition of EZH1/2 breaks the quiescence of leukemia stem cells in acute myeloid leukemia. Leukemia 32: 855–864. 10.1038/leu.2017.300 [DOI] [PubMed] [Google Scholar]
- Funato K, Major T, Lewis PW, Allis CD, Tabar V. 2014. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science 346: 1529–1533. 10.1126/science.1253799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Zhang J, Bonasio R, Strino F, Sawai A, Parisi F, Kluger Y, Reinberg D. 2012. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol Cell 45: 344–356. 10.1016/j.molcel.2012.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatchalian J, Kingsley MC, Moslet SD, Rosas Ospina RD, Kutateladze TG. 2015. An aromatic cage is required but not sufficient for binding of Tudor domains of the Polycomblike protein family to H3K36me3. Epigenetics 10: 467–473. 10.1080/15592294.2015.1042646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaydos LJ, Wang W, Strome S. 2014. Gene repression. H3K27me and PRC2 transmit a memory of repression across generations and during development. Science 345: 1515–1518. 10.1126/science.1255023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler SJ, Paro R. 2015. Trithorax and Polycomb group-dependent regulation: a tale of opposing activities. Development 142: 2876–2887. 10.1242/dev.120030 [DOI] [PubMed] [Google Scholar]
- Gibson WT, Hood RL, Zhan SH, Bulman DE, Fejes AP, Moore R, Mungall AJ, Eydoux P, Babul-Hirji R, An J, et al. 2012. Mutations in EZH2 cause weaver syndrome. Am J Hum Genet 90: 110–118. 10.1016/j.ajhg.2011.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich JA, Tjian R. 1994. Transcription factors IIE and IIH and ATP hydrolysis direct promoter clearance by RNA polymerase II. Cell 77: 145–156. 10.1016/0092-8674(94)90242-9 [DOI] [PubMed] [Google Scholar]
- Grasso CS, Tang Y, Truffaux N, Berlow NE, Liu L, Debily M-A, Quist MJ, Davis LE, Huang EC, Woo PJ, et al. 2015. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat Med 21: 555–559. 10.1038/nm.3855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grijzenhout A, Godwin J, Koseki H, Gdula MR, Szumska D, McGouran JF, Bhattacharya S, Kessler BM, Brockdorff N, Cooper S. 2016. Functional analysis of AEBP2, a PRC2 Polycomb protein, reveals a Trithorax phenotype in embryonic development and in ESCs. Development 143: 2716–2723. 10.1242/dev.123935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaud C, Negre N, Cavalli G, Nè N, Cavalli G. 2006. From genetics to epigenetics: the tale of Polycomb group and trithorax group genes. Chromosom Res 14: 363–375. 10.1007/s10577-006-1069-y [DOI] [PubMed] [Google Scholar]
- Gulati N, Béguelin W, Giulino-Roth L. 2018. Enhancer of zeste homolog 2 (EZH2) inhibitors. Leuk Lymphoma 59: 1574–1585. 10.1080/10428194.2018.1430795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Z, Xing X, Hu M, Zhang Y, Liu P, Chai J. 2007. Structural basis of EZH2 recognition by EED. Structure 15: 1306–1315. 10.1016/j.str.2007.08.007 [DOI] [PubMed] [Google Scholar]
- Hansen KH, Bracken AP, Pasini D, Dietrich N, Gehani SS, Monrad A, Rappsilber J, Lerdrup M, Helin K. 2008. A model for transmission of the H3K27me3 epigenetic mark. Nat Cell Biol 10: 1291–1300. 10.1038/ncb1787 [DOI] [PubMed] [Google Scholar]
- Hansen AS, Hsieh T-HS, Cattoglio C, Pustova I, Darzacq X, Tjian R. 2018. An RNA-binding region regulates CTCF clustering and chromatin looping. bioRxiv 10.1101/495432v2 [DOI] [Google Scholar]
- Hara M, Ozkan E, Sun H, Yu H, Luo X. 2015. Structure of an intermediate conformer of the spindle checkpoint protein Mad2. Proc Natl Acad Sci 112: 11252–11257. 10.1073/pnas.1512197112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms PW, Hristov AC, Kim DS, Anens T, Quist MJ, Siddiqui J, Carskadon S, Mehra R, Fullen DR, Johnson TM, et al. 2014. Activating mutations of the oncogene EZH2 in cutaneous melanoma revealed by next generation sequencing. Hum Pathol Case Reports 1: 21–28. 10.1016/j.ehpc.2014.07.002 [DOI] [Google Scholar]
- Hashizume R, Andor N, Ihara Y, Lerner R, Gan H, Chen X, Fang D, Huang X, Tom MW, Ngo V, et al. 2014. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat Med 20: 1394–1396. 10.1038/nm.3716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Sidoli S, Warneford-Thomson R, Tatomer DC, Wilusz JE, Garcia BA, Bonasio R. 2016. High-resolution mapping of RNA-binding regions in the nuclear proteome of embryonic stem cells. Mol Cell 64: 416–430. 10.1016/j.molcel.2016.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Selvaraju S, Curtin ML, Jakob CG, Zhu H, Comess KM, Shaw B, The J, Lima-Fernandes E, Szewczyk MM, et al. 2017. The EED protein–protein interaction inhibitor A-395 inactivates the PRC2 complex. Nat Chem Biol 13: 389–395. 10.1038/nchembio.2306 [DOI] [PubMed] [Google Scholar]
- Heard E, Martienssen RA. 2014. Transgenerational epigenetic inheritance: myths and mechanisms. Cell 157: 95–109. 10.1016/j.cell.2014.02.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz HM, Morgan M, Gao X, Jackson J, Rickels R, Swanson SK, Florens L, Washburn MP, Eissenberg JC, Shilatifard A. 2014. Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science 345: 1065–1070. 10.1126/science.1255104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetey S, Boros-Olah B, Kuik-Rozsa T, Li Q, Karanyi Z, Szabo Z, Roszik J, Szaloki N, Vamosi G, Toth K, et al. 2017. Biophysical characterization of histone H3.3 K27M point mutation. Biochem Biophys Res Commun 490: 868–875. 10.1016/j.bbrc.2017.06.133 [DOI] [PubMed] [Google Scholar]
- Hidalgo I, Herrera-Merchan A, Ligos JM, Carramolino L, Nuñez J, Martinez F, Dominguez O, Torres M, Gonzalez S. 2012. Ezh1 is required for hematopoietic stem cell maintenance and prevents senescence-like cell cycle arrest. Cell Stem Cell 11: 649–662. 10.1016/j.stem.2012.08.001 [DOI] [PubMed] [Google Scholar]
- Hirabayashi Y, Suzki N, Tsuboi M, Endo TA, Toyoda T, Shinga J, Koseki H, Vidal M, Gotoh Y. 2009. Polycomb limits the neurogenic competence of neural precursor cells to promote astrogenic fate transition. Neuron 63: 600–613. 10.1016/j.neuron.2009.08.021 [DOI] [PubMed] [Google Scholar]
- Hojfeldt JW, Laugesen A, Willumsen BM, Damhofer H, Hedehus L, Tvardovskiy A, Mohammad F, Jensen ON, Helin K. 2018. Accurate H3K27 methylation can be established de novo by SUZ12-directed PRC2. Nat Struct Mol Biol 25: 225–232. 10.1038/s41594-018-0036-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holoch D, Margueron R. 2017. Mechanisms regulating PRC2 recruitment and enzymatic activity. Trends Biochem Sci 42: 531–542. 10.1016/j.tibs.2017.04.003 [DOI] [PubMed] [Google Scholar]
- Honma D, Kanno O, Watanabe J, Kinoshita J, Hirasawa M, Nosaka E, Shiroishi M, Takizawa T, Yasumatsu I, Horiuchi T, et al. 2017. Novel orally bioavailable EZH1/2 dual inhibitors with greater antitumor efficacy than an EZH2 selective inhibitor. Cancer Sci 108: 2069–2078. 10.1111/cas.13326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrzenjak A. 2016. JAZF1/SUZ12 gene fusion in endometrial stromal sarcomas. Orphanet J Rare Dis 11: 15 10.1186/s13023-016-0400-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Zhang J, Yu Z, Zhang H, Wang Y, Lingel A, Qi W, Gu J, Zhao K, Shultz MD, et al. 2017. Discovery of first-in-class, potent, and orally bioavailable embryonic ectoderm development (EED) inhibitor with robust anticancer efficacy. J Med Chem 60: 2215–2226. 10.1021/acs.jmedchem.6b01576 [DOI] [PubMed] [Google Scholar]
- Ikeda K, Ueda T, Yamasaki N, Nakata Y, Sera Y, Nagamachi A, Miyama T, Kobayashi H, Takubo K, Kanai A, et al. 2016. Maintenance of the functional integrity of mouse hematopoiesis by EED and promotion of leukemogenesis by EED haploinsufficiency. Sci Rep 6: 29454 10.1038/srep29454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illingworth RS, Bird AP. 2009. CpG islands–’a rough guide’. FEBS Lett 583: 1713–1720. 10.1016/j.febslet.2009.04.012 [DOI] [PubMed] [Google Scholar]
- Illingworth RS, Moffat M, Mann AR, Read D, Hunter CJ, Pradeepa MM, Adams IR, Bickmore WA. 2015. The E3 ubiquitin ligase activity of RING1B is not essential for early mouse development. Genes Dev 29: 1897–1902. 10.1101/gad.268151.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imagawa E, Higashimoto K, Sakai Y, Numakura C, Okamoto N, Matsunaga S, Ryo A, Sato Y, Sanefuji M, Ihara K, et al. 2017. Mutations in genes encoding polycomb repressive complex 2 subunits cause Weaver syndrome. Hum Mutat 38: 637–648. 10.1002/humu.23200 [DOI] [PubMed] [Google Scholar]
- Inoue A, Jiang L, Lu F, Suzuki T, Zhang Y. 2017. Maternal H3K27me3 controls DNA methylation-independent imprinting. Nature 547: 419–424. 10.1038/nature23262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob Y, Feng S, LeBlanc CA, Bernatavichute Y V, Stroud H, Cokus S, Johnson LM, Pellegrini M, Jacobsen SE, Michaels SD. 2009. ATXR5 and ATXR6 are H3K27 monomethyltransferases required for chromatin structure and gene silencing. Nat Struct Mol Biol 16: 763–768. 10.1038/nsmb.1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadhav U, Nalapareddy K, Saxena M, Yuan G-C, Orkin SH, Shivdasani RA. 2016. Acquired tissue-specific promoter bivalency is a basis for PRC2 necessity in adult cells. Cell 165: 1389–1400. 10.1016/j.cell.2016.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaram H, Hoelper D, Jain SU, Cantone N, Lundgren SM, Poy F, Allis CD, Cummings R, Bellon S, Lewis PW. 2016. S-adenosyl methionine is necessary for inhibition of the methyltransferase G9a by the lysine 9 to methionine mutation on histone H3. Proc Natl Acad Sci 113: 6182–6187. 10.1073/pnas.1605523113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jermann P, Hoerner L, Burger L, Schubeler D. 2014. Short sequences can efficiently recruit histone H3 lysine 27 trimethylation in the absence of enhancer activity and DNA methylation. Proc Natl Acad Sci 111: E3415–E3421. 10.1073/pnas.1400672111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao L, Liu X. 2015. Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science 350: 291 10.1126/science.aab2713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao L, Liu X. 2016. Response to comment on ‘Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2’. Science 354: 1543 10.1126/science.aaj2335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi O, Wang S-Y, Kuznetsova T, Atlasi Y, Peng T, Fabre PJ, Habibi E, Shaik J, Saeed S, Handoko L, et al. 2015. Dynamic reorganization of extremely long-range promoter-promoter interactions between two states of pluripotency. Cell Stem Cell 17: 748–757. 10.1016/j.stem.2015.11.010 [DOI] [PubMed] [Google Scholar]
- Justin N, Zhang Y, Tarricone C, Martin SR, Chen S, Underwood E, De Marco V, Haire LF, Walker PA, Reinberg D, et al. 2016. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat Commun 7: 11316 10.1038/ncomms11316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C, Williams RT, Calarco JP, Miller EL, Weber CM, Braun SMG, Pulice JL, Chory EJ, Crabtree GR. 2017. Dynamics of BAF–Polycomb complex opposition on heterochromatin in normal and oncogenic states. Nat Genet 49: 213–222. 10.1038/ng.3734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalb R, Latwiel S, Baymaz HI, Jansen PWTC, Müller CW, Vermeulen M, Müller J. 2014. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat Struct Mol Biol 21: 569–571. 10.1038/nsmb.2833 [DOI] [PubMed] [Google Scholar]
- Kaneko S, Son J, Shen SS, Reinberg D, Bonasio R. 2013. PRC2 binds active promoters and contacts nascent RNAs in embryonic stem cells. Nat Struct Mol Biol 20: 1258–1264. 10.1038/nsmb.2700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S, Bonasio R, Saldaña-Meyer R, Yoshida T, Son J, Nishino K, Umezawa A, Reinberg D. 2014a. Interactions between JARID2 and noncoding RNAs regulate PRC2 recruitment to chromatin. Mol Cell 53: 290–300. 10.1016/j.molcel.2013.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S, Son J, Bonasio R, Shen SS, Reinberg D. 2014b. Nascent RNA interaction keeps PRC2 activity poised and in check. Genes Dev 28: 1983–1988. 10.1101/gad.247940.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasinath V, Faini M, Poepsel S, Reif D, Feng XA, Stjepanovic G, Aebersold R, Nogales E. 2018a. Structures of human PRC2 with its cofactors AEBP2 and JARID2. Science 359: 940–944. 10.1126/science.aar5700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasinath V, Poepsel S, Nogales E. 2018b. Recent structural insights into PRC2 regulation and substrate binding. Biochemistry 58: 346–354. 10.1021/acs.biochem.8b01064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Roberts CWM. 2016. Targeting EZH2 in cancer. Nat Med 22: 128–134. 10.1038/nm.4036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Kang K, Kim J. 2009. AEBP2 as a potential targeting protein for Polycomb repression complex PRC2. Nucleic Acids Res 37: 2940–2950. 10.1093/nar/gkp149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D-H, Tang Z, Shimada M, Fierz B, Houck-Loomis B, Bar-Dagen M, Lee S, Lee S-K, Muir TW, Roeder RG, et al. 2013a. Histone H3K27 trimethylation inhibits H3 binding and function of SET1-like H3K4 methyltransferase complexes. Mol Cell Biol 33: 4936–4946. 10.1128/MCB.00601-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Bird GH, Neff T, Guo G, Kerenyi MA, Walensky LD, Orkin SH. 2013b. Targeted disruption of the EZH2–EED complex inhibits EZH2-dependent cancer. Nat Chem Biol 9: 643–650. 10.1038/nchembio.1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang K-C, Xiao Y, Kadowaki T, Uesugi M, Kuznetsov G, Kumar N, et al. 2014. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol Cancer Ther 13: 842–854. 10.1158/1535-7163.MCT-13-0773 [DOI] [PubMed] [Google Scholar]
- Kohlmaier A, Savarese F, Lachner M, Martens J, Jenuwein T, Wutz A. 2004. A chromosomal memory triggered by Xist regulates histone methylation in X inactivation. PLoS Biol 2: E171 10.1371/journal.pbio.0020171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarnitsky P, Cho EJ, Buratowski S. 2000. Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev 14: 2452–2460. 10.1101/gad.824700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong X, Chen L, Jiao L, Jiang X, Lian F, Lu J, Zhu K, Du D, Liu J, Ding H, et al. 2014. Astemizole arrests the proliferation of cancer cells by disrupting the EZH2–EED interaction of polycomb repressive complex 2. J Med Chem 57: 9512–9521. 10.1021/jm501230c [DOI] [PubMed] [Google Scholar]
- Konze KD, Ma A, Li F, Barsyte-Lovejoy D, Parton T, MacNevin CJ, Liu F, Gao C, Huang XP, Kuznetsova E, et al. 2013. An orally bioavailable chemical probe of the lysine methyltransferases EZH2 and EZH1. ACS Chem Biol 8: 1324–1334. 10.1021/cb400133j [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajewski WA, Nakamura T, Mazo A, Canaani E. 2005. A motif within SET-domain proteins binds single-stranded nucleic acids and transcribed and supercoiled DNAs and can interfere with assembly of nucleosomes. Mol Cell Biol 25: 1891–1899. 10.1128/MCB.25.5.1891-1899.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu S, Ji F, Sunwoo H, Jain G, Lee JT, Sadreyev RI, Dekker J, Kingston RE. 2017. Polycomb repressive complex 1 generates discrete compacted domains that change during differentiation. Mol Cell 65: 432–446.e5. 10.1016/j.molcel.2017.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. 2002. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev 16: 2893–2905. 10.1101/gad.1035902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmichev A, Jenuwein T, Tempst P, Reinberg D. 2004. Different EZH2-containing complexes target methylation of histone H1 or nucleosomal histone H3. Mol Cell 14: 183–193. 10.1016/S1097-2765(04)00185-6 [DOI] [PubMed] [Google Scholar]
- LaFave LM, Béguelin W, Koche R, Teater M, Spitzer B, Chramiec A, Papalexi E, Keller MD, Hricik T, Konstantinoff K, et al. 2015. Loss of BAP1 function leads to EZH2-dependent transformation. Nat Med 21: 1344–1349. 10.1038/nm.3947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landeira D, Sauer S, Poot R, Dvorkina M, Mazzarella L, Jorgensen HF, Pereira CF, Leleu M, Piccolo FM, Spivakov M, et al. 2010. Jarid2 is a PRC2 component in embryonic stem cells required for multi-lineage differentiation and recruitment of PRC1 and RNA Polymerase II to developmental regulators. Nat Cell Biol 12: 618–624. 10.1038/ncb2065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprell F, Finkl K, Muller J. 2017. Propagation of Polycomb-repressed chromatin requires sequence-specific recruitment to DNA. Science 356: 85–88. 10.1126/science.aai8266 [DOI] [PubMed] [Google Scholar]
- Lau MS, Schwartz MG, Kundu S, Savol AJ, Wang PI, Marr SK, Grau DJ, Schorderet P, Sadreyev RI, Tabin CJ, et al. 2017. Mutation of a nucleosome compaction region disrupts Polycomb-mediated axial patterning. Science 355: 1081–1084. 10.1126/science.aah5403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laugesen A, Højfeldt JW, Helin K. 2016. Role of the Polycomb Repressive Complex 2 (PRC2) in Transcriptional Regulation and Cancer. Cold Spring Harb Perspect Med 6: a026575 10.1101/cshperspect.a026575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Teckie S, Wiesner T, Ran L, Prieto Granada CN, Lin M, Zhu S, Cao Z, Liang Y, Sboner A, et al. 2014. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet 46: 1227–1232. 10.1038/ng.3095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H-G, Kahn TG, Simcox A, Schwartz YB, Pirrotta V. 2015a. Genome-wide activities of Polycomb complexes control pervasive transcription. Genome Res 25: 1170–1181. 10.1101/gr.188920.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SCW, Miller S, Hyland C, Kauppi M, Lebois M, Di Rago L, Metcalf D, Kinkel SA, Josefsson EC, Blewitt ME, et al. 2015b. Polycomb repressive complex 2 component Suz12 is required for hematopoietic stem cell function and lymphopoiesis. Blood 126: 167–175. 10.1182/blood-2014-12-615898 [DOI] [PubMed] [Google Scholar]
- Lee C-H, Granat J, Yu J-R, LeRoy G, Stafford J, Reinberg D. 2018a. Automethylation of PRC2 fine-tunes its catalytic activity on chromatin. bioRxiv 10.1101/349449 [DOI] [Google Scholar]
- Lee C-H, Yu J-R, Kumar S, Jin Y, LeRoy G, Bhanu N, Kaneko S, Garcia BA, Hamilton AD, Reinberg D. 2018b. Allosteric activation dictates PRC2 activity independent of its recruitment to chromatin. Mol Cell 70: 422–434.e6. 10.1016/j.molcel.2018.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C-H, Holder M, Grau D, Saldaña-Meyer R, Yu J-R, Ganai RA, Zhang J, Wang M, LeRoy G, Dobenecker M-W, et al. 2018c. Distinct stimulatory mechanisms regulate the catalytic activity of Polycomb repressive complex 2. Mol Cell 70: 435–448.e5. 10.1016/j.molcel.2018.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PW, Müller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD. 2013. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340: 857–861. 10.1126/science.1232245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lhuillier-Akakpo M, Frapporti A, Denby Wilkes C, Matelot M, Vervoort M, Sperling L, Duharcourt S. 2014. Local effect of enhancer of Zeste-like reveals cooperation of epigenetic and cis-acting determinants for zygotic genome rearrangements. PLoS Genet 10: e1004665 10.1371/journal.pgen.1004665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Margueron R, Ku M, Chambon P, Bernstein BE, Reinberg D. 2010. Jarid2 and PRC2, partners in regulating gene expression. Genes Dev 24: 368–380. 10.1101/gad.1886410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Liefke R, Jiang J, Kurland JV, Tian W, Deng P, Zhang W, He Q, Patel DJ, Bulyk ML, et al. 2017. Polycomb-like proteins link the PRC2 complex to CpG islands. Nature 549: 287–291. 10.1038/nature23881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liefke R, Shi Y. 2015. The PRC2-associated factor C17orf96 is a novel CpG island regulator in mouse ES cells. Cell Discov 1: 15008 10.1038/celldisc.2015.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liefke R, Karwacki-Neisius V, Shi Y, Alekseyenko AA, Gorchakov AA, Kharchenko PV, Kuroda MI, Aso T, Lane WS, Conaway JW, et al. 2016. EPOP interacts with elongin BC and USP7 to modulate the chromatin landscape. Mol Cell 64: 659–672. 10.1016/j.molcel.2016.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Marquardt S, Lister C, Swiezewski S, Dean C. 2010. Targeted 3′ processing of antisense transcripts triggers Arabidopsis FLC chromatin silencing. Science 327: 94–97. 10.1126/science.1180278 [DOI] [PubMed] [Google Scholar]
- Liu X, Wang C, Liu W, Li J, Li C, Kou X, Chen J, Zhao Y, Gao H, Wang H, et al. 2016. Distinct features of H3K4me3 and H3K27me3 chromatin domains in pre-implantation embryos. Nature 537: 558–562. 10.1038/nature19362 [DOI] [PubMed] [Google Scholar]
- Long HK, Blackledge NP, Klose RJ. 2013. ZF-CxxC domain-containing proteins, CpG islands and the chromatin connection. Biochem Soc Trans 41: 727–740. 10.1042/BST20130028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long Y, Bolanos B, Gong L, Liu W, Goodrich KJ, Yang X, Chen S, Gooding AR, Maegley KA, Gajiwala KS, et al. 2017. Conserved RNA-binding specificity of polycomb repressive complex 2 is achieved by dispersed amino acid patches in EZH2. Elife 6: e31558 10.7554/eLife.31558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu TT, Heyne S, Dror E, Casas E, Leonhardt L, Boenke T, Yang CH, Sagar, Arrigoni L, Dalgaard K, et al. 2018. The Polycomb-dependent epigenome controls β cell dysfunction, dedifferentiation, and diabetes. Cell Metab 27: 1294–1308.e7. 10.1016/j.cmet.2018.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch MD, Smith AJH, De Gobbi M, Flenley M, Hughes JR, Vernimmen D, Ayyub H, Sharpe JA, Sloane-Stanley JA, Sutherland L, et al. 2012. An interspecies analysis reveals a key role for unmethylated CpG dinucleotides in vertebrate Polycomb complex recruitment. EMBO J 31: 317–329. 10.1038/emboj.2011.399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Wang J, Wang J, Ma CX, Gao X, Patriub V, Sklar JL. 2017. The JAZF1-SUZ12 fusion protein disrupts PRC2 complexes and impairs chromatin repression during human endometrial stromal tumorogenesis. Oncotarget 8: 4062–4078. 10.18632/oncotarget.13270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR, Bjerke L, Clarke M, Vinci M, Nandhabalan M, et al. 2017. Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell 32: 520–537 e5. 10.1016/j.ccell.2017.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madamba EV, Berthet EB, Francis NJ. 2017. Inheritance of histones H3 and H4 during DNA Replication in Vitro. Cell Rep 21: 1361–1374. 10.1016/j.celrep.2017.10.033 [DOI] [PubMed] [Google Scholar]
- Margueron R, Reinberg D. 2011. The Polycomb complex PRC2 and its mark in life. Nature 469: 343–349. 10.1038/nature09784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Li G, Sarma K, Blais A, Zavadil J, Woodcock CL, Dynlacht BD, Reinberg D. 2008. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell 32: 503–518. 10.1016/j.molcel.2008.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ, Voigt P, Martin SR, Taylor WR, De Marco V, et al. 2009. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 461: 762–767. 10.1038/nature08398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin C, Cao R, Zhang Y. 2006. Substrate preferences of the EZH2 histone methyltransferase complex. J Biol Chem 281: 8365–8370. 10.1074/jbc.M513425200 [DOI] [PubMed] [Google Scholar]
- Mazzoni EO, Mahony S, Peljto M, Patel T, Thornton SR, McCuine S, Reeder C, Boyer LA, Young RA, Gifford DK, et al. 2013. Saltatory remodeling of Hox chromatin in response to rostrocaudal patterning signals. Nat Neurosci 16: 1191–1198. 10.1038/nn.3490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe MT, Graves AP, Ganji G, Diaz E, Halsey WS, Jiang Y, Smitheman KN, Ott HM, Pappalardi MB, Allen KE, et al. 2012a. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc Natl Acad Sci 109: 2989–2994. 10.1073/pnas.1116418109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A, Diaz E, et al. 2012b. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492: 108–112. 10.1038/nature11606 [DOI] [PubMed] [Google Scholar]
- Mendenhall EM, Koche RP, Truong T, Zhou VW, Issac B, Chi AS, Ku M, Bernstein BE. 2010. GC-rich sequence elements recruit PRC2 in mammalian ES cells. PLoS Genet 6: e1001244 10.1371/journal.pgen.1001244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micci F, Panagopoulos I, Bjerkehagen B, Heim S. 2006. Consistent rearrangement of chromosomal band 6p21 with generation of fusion genes JAZF1/PHF1 and EPC1/PHF1 in endometrial stromal sarcoma. Cancer Res 66: 107–112. 10.1158/0008-5472.CAN-05-2485 [DOI] [PubMed] [Google Scholar]
- Micci F, Gorunova L, Gatius S, Matias-Guiu X, Davidson B, Heim S, Panagopoulos I. 2014. MEAF6/PHF1 is a recurrent gene fusion in endometrial stromal sarcoma. Cancer Lett 347: 75–78. 10.1016/j.canlet.2014.01.030 [DOI] [PubMed] [Google Scholar]
- Millard CJ, Varma N, Saleh A, Morris K, Watson PJ, Bottrill AR, Fairall L, Smith CJ, Schwabe JWR. 2016. The structure of the core NuRD repression complex provides insights into its interaction with chromatin. Elife 5: e13941 10.7554/eLife.13941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda TB, Cortez CC, Yoo CB, Liang G, Abe M, Kelly TK, Marquez VE, Jones PA. 2009. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Ther 8: 1579–1588. 10.1158/1535-7163.MCT-09-0013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miró X, Zhou X, Boretius S, Michaelis T, Kubisch C, Alvarez-Bolado G, Gruss P. 2009. Haploinsufficiency of the murine polycomb gene Suz12 results in diverse malformations of the brain and neural tube. Dis Model Mech 2: 412–418. 10.1242/dmm.001602 [DOI] [PubMed] [Google Scholar]
- Mito Y, Henikoff JG, Henikoff S. 2007. Histone replacement marks the boundaries of cis-regulatory domains. Science 315: 1408–1411. 10.1126/science.1134004 [DOI] [PubMed] [Google Scholar]
- Mohammad F, Weissmann S, Leblanc B, Pandey DP, Højfeldt JW, Comet I, Zheng C, Johansen JV, Rapin N, Porse BT, et al. 2017. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat Med 23: 483–492. 10.1038/nm.4293 [DOI] [PubMed] [Google Scholar]
- Mohn F, Weber M, Rebhan M, Roloff TC, Richter J, Stadler MB, Bibel M, Schubeler D. 2008. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol Cell 30: 755–766. 10.1016/j.molcel.2008.05.007 [DOI] [PubMed] [Google Scholar]
- Moody RR, Lo M-C, Meagher JL, Lin C-C, Stevers NO, Tinsley SL, Jung I, Matvekas A, Stuckey JA, Sun D. 2018. Probing the interaction between the histone methyltransferase/deacetylase subunit RBBP4/7 and the transcription factor BCL11A in epigenetic complexes. J Biol Chem 293: 2125–2136. 10.1074/jbc.M117.811463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, et al. 2010. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet 42: 181–185. 10.1038/ng.518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller MM, Fierz B, Bittova L, Liszczak G, Muir TW, Müller MM, Fierz B, Bittova L, Liszczak G, Muir TW. 2016. A two-state activation mechanism controls the histone methyltransferase Suv39h1. Nat Chem Biol 12: 188–193. 10.1038/nchembio.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musselman CA, Avvakumov N, Watanabe R, Abraham CG, Lalonde M-E, Hong Z, Allen C, Roy S, Nuñez JK, Nickoloff J, et al. 2012. Molecular basis for H3K36me3 recognition by the Tudor domain of PHF1. Nat Struct Mol Biol 19: 1266–1272. 10.1038/nsmb.2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musselman CA, Gibson MD, Hartwick EW, North JA, Gatchalian J, Poirier MG, Kutateladze TG. 2013. Binding of PHF1 Tudor to H3K36me3 enhances nucleosome accessibility. Nat Commun 4 10.1038/ncomms3969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraja S, Vitanza NA, Woo PJ, Taylor KR, Liu F, Zhang L, Li M, Meng W, Ponnuswami A, Sun W, et al. 2017. Transcriptional dependencies in diffuse intrinsic pontine glioma. Cancer Cell 31: 635–652.e6. 10.1016/j.ccell.2017.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra V, Rocha PP, An DS, Raviram R, Skok JA, Mazzoni EO, Reinberg D. 2015. CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science 347: 1017–1021. 10.1126/science.1262088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra V, Bulajic M, Dekker J, Mazzoni EO, Reinberg D. 2016. CTCF-mediated topological boundaries during development foster appropriate gene regulation. Genes Dev 30: 2657–2662. 10.1101/gad.288324.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntziachristos P, Tsirigos A, Van Vlierberghe P, Nedjic J, Trimarchi T, Flaherty MS, Ferres-Marco D, da Ros V, Tang Z, Siegle J, et al. 2012. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat Med 18: 298–301. 10.1038/nm.2651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Carroll D, Erhardt S, Pagani M, Barton SC, Surani MA, Jenuwein T. 2001. The polycomb-group gene Ezh2 is required for early mouse development. Mol Cell Biol 21: 4330–4336. 10.1128/MCB.21.13.4330-4336.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiyama Y, Schuettengruber B, Papadopoulos GL, Chang JM, Cavalli G. 2018. Polycomb-dependent chromatin looping contributes to gene silencing during Drosophila development. Mol Cell 71: 73–88 e5. 10.1016/j.molcel.2018.05.032 [DOI] [PubMed] [Google Scholar]
- Oksuz O, Narendra V, Lee C-H, Descostes N, LeRoy G, Raviram R, Blumenberg L, Karch K, Rocha PP, Garcia BA, et al. 2018. Capturing the onset of PRC2-mediated repressive domain formation. Mol Cell 70: 1149–1162.e5. 10.1016/j.molcel.2018.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey RR, Mondal T, Mohammad F, Enroth S, Redrup L, Komorowski J, Nagano T, Mancini-Dinardo D, Kanduri C. 2008. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell 32: 232–246. 10.1016/j.molcel.2008.08.022 [DOI] [PubMed] [Google Scholar]
- Parvin JD, Sharp PA. 1993. DNA topology and a minimal set of basal factors for transcription by RNA polymerase II. Cell 73: 533–540. 10.1016/0092-8674(93)90140-L [DOI] [PubMed] [Google Scholar]
- Pasini D, Cloos PAC, Walfridsson J, Olsson L, Bukowski J-P, Johansen J V, Bak M, Tommerup N, Rappsilber J, Helin K. 2010. JARID2 regulates binding of the Polycomb repressive complex 2 to target genes in ES cells. Nature 464: 306–310. 10.1038/nature08788 [DOI] [PubMed] [Google Scholar]
- Pathak P, Jha P, Purkait S, Sharma V, Suri V, Sharma MC, Faruq M, Suri A, Sarkar C. 2015. Altered global histone-trimethylation code and H3F3A-ATRX mutation in pediatric GBM. J Neurooncol 121: 489–497. 10.1007/s11060-014-1675-z [DOI] [PubMed] [Google Scholar]
- Pelegri F, Lehmann R. 1994. A role of polycomb group genes in the regulation of gap gene expression in Drosophila. Genetics 136: 1341–1353. 10.1016/0168-9525(90)90006-R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pengelly AR, Kalb R, Finkl K, Müller J. 2015. Transcriptional repression by PRC1 in the absence of H2A monoubiquitylation. Genes Dev 29: 1487–1492. 10.1101/gad.265439.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira JD, Sansom SN, Smith J, Dobenecker M-WW, Tarakhovsky A, Livesey FJ. 2010. Ezh2, the histone methyltransferase of PRC2, regulates the balance between self-renewal and differentiation in the cerebral cortex. Proc Natl Acad Sci 107: 15957–15962. 10.1073/pnas.1002530107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perino M, van Mierlo G, Karemaker ID, van Genesen S, Vermeulen M, Marks H, van Heeringen SJ, Veenstra GJC. 2018. MTF2 recruits Polycomb repressive complex 2 by helical-shape-selective DNA binding. Nat Genet 50: 1002–1010. 10.1038/s41588-018-0134-8 [DOI] [PubMed] [Google Scholar]
- Petruk S, Sedkov Y, Johnston DM, Hodgson JW, Black KL, Kovermann SK, Beck S, Canaani E, Brock HW, Mazo A. 2012. TrxG and PcG proteins but not methylated histones remain associated with DNA through replication. Cell 150: 922–933. 10.1016/j.cell.2012.06.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petruk S, Black KL, Kovermann SK, Brock HW, Mazo A. 2013. Stepwise histone modifications are mediated by multiple enzymes that rapidly associate with nascent DNA during replication. Nat Commun 4: 2841 10.1038/ncomms3841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petruk S, Cai J, Sussman R, Sun G, Kovermann SK, Mariani SA, Calabretta B, McMahon SB, Brock HW, Iacovitti L, et al. 2017a. Delayed accumulation of H3K27me3 on nascent DNA is essential for recruitment of transcription factors at early stages of stem cell differentiation. Mol Cell 66: 247–257 e5. 10.1016/j.molcel.2017.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petruk S, Mariani SA, De Dominici M, Porazzi P, Minieri V, Cai J, Iacovitti L, Flomenberg N, Calabretta B, Mazo A. 2017b. Structure of nascent chromatin is essential for hematopoietic lineage specification. Cell Rep 19: 295–306. 10.1016/j.celrep.2017.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips-Cremins JE, Corces VG. 2013. Chromatin insulators: linking genome organization to cellular function. Mol Cell 50: 461–474. 10.1016/j.molcel.2013.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pintacuda G, Wei G, Roustan C, Kirmizitas BA, Solcan N, Cerase A, Castello A, Mohammed S, Moindrot B, Nesterova TB, et al. 2017. hnRNPK recruits PCGF3/5-PRC1 to the Xist RNA B-repeat to establish Polycomb-mediated chromosomal silencing. Mol Cell 68: 955–969.e10. 10.1016/j.molcel.2017.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirrotta V. 2017. Polycomb group proteins. Academic Press, New York. [Google Scholar]
- Piunti A, Hashizume R, Morgan MA, Bartom ET, Horbinski CM, Marshall SA, Rendleman EJ, Ma Q, Takahashi Y, Woodfin AR, et al. 2017. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat Med 23: 493–500. 10.1038/nm.4296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H, de la Cruz CC, Otte AP, Panning B, Zhang Y. 2003. Role of histone H3 lysine 27 methylation in X inactivation. Science 300: 131–135. 10.1126/science.1084274 [DOI] [PubMed] [Google Scholar]
- Poepsel S, Kasinath V, Nogales E. 2018. Cryo-EM structures of PRC2 simultaneously engaged with two functionally distinct nucleosomes. Nat Struct Mol Biol 25: 154–162. 10.1038/s41594-018-0023-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, Zeng J, Li M, Fan H, Lin Y, et al. 2012. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci 109: 21360–21365. 10.1073/pnas.1210371110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi W, Zhao K, Gu J, Huang Y, Wang Y, Zhang H, Zhang M, Zhang J, Yu Z, Li L, et al. 2017. An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat Chem Biol 13: 381–388. 10.1038/nchembio.2304 [DOI] [PubMed] [Google Scholar]
- Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtier K, Ponting CP, Allis CD, et al. 2000. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406: 593–599. 10.1038/35020506 [DOI] [PubMed] [Google Scholar]
- Reinberg D, Vales LD. 2018. Chromatin domains rich in inheritance. Science 361: 33–34. 10.1126/science.aat7871 [DOI] [PubMed] [Google Scholar]
- Reveron-Gomez N, Gonzalez-Aguilera C, Stewart-Morgan KR, Petryk N, Flury V, Graziano S, Johansen J V, Jakobsen JS, Alabert C, Groth A. 2018. Accurate recycling of parental histones reproduces the histone modification landscape during DNA replication. Mol Cell 72: 239–249 e5. 10.1016/j.molcel.2018.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riising EM, Comet I, Leblanc B, Wu X, Johansen JV, Helin K. 2014. Gene silencing triggers Polycomb repressive complex 2 recruitment to CpG islands genome wide. Mol Cell 55: 347–360. 10.1016/j.molcel.2014.06.005 [DOI] [PubMed] [Google Scholar]
- Saldana-Meyer R, Rodriguez-Hernaez J, Nishana M, Jacome-Lopez K, Nora EP, Bruneau BG, Furlan-Magaril M, Skok J, Reinberg D. 2019. RNA interactions with CTCF are essential for its proper function. bioRxiv 10.1101/530014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanulli S, Justin N, Teissandier AA, Ancelin K, Portoso M, Caron M, Michaud A, Lombard BB, da Rocha ST, Offer J, et al. 2015. Jarid2 methylation via the PRC2 complex regulates H3K27me3 deposition during cell differentiation. Mol Cell 57: 769–783. 10.1016/j.molcel.2014.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarma K, Margueron R, Ivanov A, Pirrotta V, Reinberg D. 2008. Ezh2 requires PHF1 to efficiently catalyze H3 lysine 27 trimethylation in vivo. Mol Cell Biol 28: 2718–2731. 10.1128/MCB.02017-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarma K, Cifuentes-Rojas C, Ergun A, Del Rosario A, Jeon Y, White F, Sadreyev R, Lee JT. 2014. ATRX directs binding of PRC2 to Xist RNA and Polycomb targets. Cell 159: 869–883. 10.1016/j.cell.2014.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sashida G, Harada H, Matsui H, Oshima M, Yui M, Harada Y, Tanaka S, Mochizuki-Kashio M, Wang C, Saraya A, et al. 2014. Ezh2 loss promotes development of myelodysplastic syndrome but attenuates its predisposition to leukaemic transformation. Nat Commun 5: 4177 10.1038/ncomms5177 [DOI] [PubMed] [Google Scholar]
- Schmitges FW, Prusty AB, Faty M, Stützer A, Lingaraju GM, Aiwazian J, Sack R, Hess D, Li L, Zhou S, et al. 2011. Histone methylation by PRC2 is inhibited by active chromatin marks. Mol Cell 42: 330–341. 10.1016/j.molcel.2011.03.025 [DOI] [PubMed] [Google Scholar]
- Schoenfelder S, Sugar R, Dimond A, Javierre BM, Armstrong H, Mifsud B, Dimitrova E, Matheson L, Tavares-Cadete F, Furlan-Magaril M, et al. 2015. Polycomb repressive complex PRC1 spatially constrains the mouse embryonic stem cell genome. Nat Genet 47: 1179–1186. 10.1038/ng.3393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoumacher M, Le Corre S, Houy A, Mulugeta E, Stern M-H, Roman-Roman S, Margueron R. 2016. Uveal melanoma cells are resistant to EZH2 inhibition regardless of BAP1 status. Nat Med 22: 577–578. 10.1038/nm.4098 [DOI] [PubMed] [Google Scholar]
- Schuettengruber B, Ganapathi M, Leblanc B, Portoso M, Jaschek R, Tolhuis B, van Lohuizen M, Tanay A, Cavalli G. 2009. Functional anatomy of polycomb and trithorax chromatin landscapes in Drosophila embryos. PLoS Biol 7: e13 10.1371/journal.pbio.1000013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzentruber J, Korshunov A, Liu X-Y, Jones DTW, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang D-AK, Tönjes M, et al. 2012. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482: 226–231. 10.1038/nature10833 [DOI] [PubMed] [Google Scholar]
- Seong IS, Woda JM, Song J-J, Lloret A, Abeyrathne PD, Woo CJ, Gregory G, Lee J-M, Wheeler VC, Walz T, et al. 2010. Huntingtin facilitates polycomb repressive complex 2. Hum Mol Genet 19: 573–583. 10.1093/hmg/ddp524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan CM, Wang J, Xu K, Chen H, Yue JX, Andrews S, Moresco JJ, Yates JR, Nagy PL, Tong L, et al. 2016. A histone H3K9M mutation traps histone methyltransferase Clr4 to prevent heterochromatin spreading. Elife 5: e17903 10.7554/eLife.17903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Y, Liang Z, Xing Q, Zhang T, Wang B, Tian S, Huang W, Zhang Y, Yao J, Zhu Y, et al. 2017. PRC2 specifies ectoderm lineages and maintains pluripotency in primed but not naïve ESCs. Nat Commun 8: 672 10.1038/s41467-017-00668-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Liu Y, Hsu YJ, Fujiwara Y, Kim J, Mao X, Yuan GC, Orkin SH. 2008. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol Cell 32: 491–502. 10.1016/j.molcel.2008.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Kim W, Fujiwara Y, Simon MD, Liu Y, Mysliwiec MR, Yuan GC, Lee Y, Orkin SH. 2009. Jumonji modulates polycomb activity and self-renewal versus differentiation of stem cells. Cell 139: 1303–1314. 10.1016/j.cell.2009.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon C, Chagraoui J, Krosl J, Gendron P, Wilhelm B, Lemieux S, Boucher G, Chagnon P, Drouin S, Lambert R, et al. 2012. A key role for EZH2 and associated genes in mouse and human adult T-cell acute leukemia. Genes Dev 26: 651–656. 10.1101/gad.186411.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son J, Shen SS, Margueron R, Reinberg D. 2013. Nucleosome-binding activities within JARID2 and EZH1 regulate the function of PRC2 on chromatin. Genes Dev 27: 2663–2677. 10.1101/gad.225888.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southall SM, Wong P-S, Odho Z, Roe SM, Wilson JR. 2009. Structural basis for the requirement of additional factors for MLL1 SET domain activity and recognition of epigenetic marks. Mol Cell 33: 181–191. 10.1016/j.molcel.2008.12.029 [DOI] [PubMed] [Google Scholar]
- Stafford JM, Lee C-H, Voigt P, Descostes N, Saldana-Meyer R, Yu J-R, Leroy G, Oksuz O, Chapman JR, Suarez F, et al. 2018. Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma. Sci Adv 4: eaau5935 10.1126/sciadv.aau5935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen PA, Ringrose L. 2014. What are memories made of? How Polycomb and Trithorax proteins mediate epigenetic memory. Nat Rev Mol Cell Biol 15: 340–356. 10.1038/nrm3789 [DOI] [PubMed] [Google Scholar]
- Streubel G, Watson A, Jammula SG, Scelfo A, Fitzpatrick DJ, Oliviero G, McCole R, Conway E, Glancy E, Negri GL, et al. 2018. The H3K36me2 methyltransferase Nsd1 demarcates PRC2-mediated H3K27me2 and H3K27me3 domains in embryonic stem cells. Mol Cell 70: 371–379.e5. 10.1016/j.molcel.2018.02.027 [DOI] [PubMed] [Google Scholar]
- Sturm D, Witt H, Hovestadt V, Khuong-Quang D-A, Jones DTW, Konermann C, Pfaff E, Tönjes M, Sill M, Bender S, et al. 2012. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22: 425–437. 10.1016/j.ccr.2012.08.024 [DOI] [PubMed] [Google Scholar]
- Su I, Basavaraj A, Krutchinsky AN, Hobert O, Ullrich A, Chait BT, Tarakhovsky A. 2003. Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat Immunol 4: 124–131. 10.1038/ni876 [DOI] [PubMed] [Google Scholar]
- Swiezewski S, Liu F, Magusin A, Dean C. 2009. Cold-induced silencing by long antisense transcripts of an Arabidopsis Polycomb target. Nature 462: 799–802. 10.1038/nature08618 [DOI] [PubMed] [Google Scholar]
- Talbert PB, Henikoff S. 2006. Spreading of silent chromatin: inaction at a distance. Nat Rev Genet 7: 793–803. 10.1038/nrg1920 [DOI] [PubMed] [Google Scholar]
- Tatavosian R, Duc HN, Huynh TN, Fang D, Schmitt B, Shi X, Deng Y, Phiel C, Yao T, Zhang Z, et al. 2018. Live-cell single-molecule dynamics of PcG proteins imposed by the DIPG H3.3K27M mutation. Nat Commun 9: 2080 10.1038/s41467-018-04455-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavares L, Dimitrova E, Oxley D, Webster J, Poot R, Demmers J, Bezstarosti K, Taylor S, Ura H, Koide H, et al. 2012. RYBP-PRC1 complexes mediate H2A ubiquitylation at polycomb target sites independently of PRC2 and H3K27me3. Cell 148: 664–678. 10.1016/j.cell.2011.12.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor IC, Hutt-Cabezas M, Brandt WD, Kambhampati M, Nazarian J, Chang HT, Warren KE, Eberhart CG, Raabe EH. 2015. Disrupting NOTCH slows diffuse intrinsic pontine glioma growth, enhances radiation sensitivity, and shows combinatorial efficacy with bromodomain inhibition. J Neuropathol Exp Neurol 74: 778–790. 10.1097/NEN.0000000000000216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tee WW, Shen SS, Oksuz O, Narendra V, Reinberg D. 2014. Erk1/2 activity promotes chromatin features and RNAPII phosphorylation at developmental promoters in mouse ESCs. Cell 156: 678–690. 10.1016/j.cell.2014.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, Mastrianni J, Lugaresi E, Gambetti P, Prusiner SB. 1996. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 274: 2079–2082. 10.1126/science.274.5295.2079 [DOI] [PubMed] [Google Scholar]
- Tolhuis B, Blom M, Kerkhoven RM, Pagie L, Teunissen H, Nieuwland M, Simonis M, de Laat W, van Lohuizen M, van Steensel B. 2011. Interactions among Polycomb domains are guided by chromosome architecture. PLoS Genet 7: e1001343 10.1371/journal.pgen.1001343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda T, Nakata Y, Nagamachi A, Yamasaki N, Kanai A, Sera Y, Sasaki M, Matsui H, Honda Z-I, Oda H, et al. 2016. Propagation of trimethylated H3K27 regulated by polycomb protein EED is required for embryogenesis, hematopoietic maintenance, and tumor suppression. Proc Natl Acad Sci 113: 10370–10375. 10.1073/pnas.1600070113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usheva A, Maldonado E, Goldring A, Lu H, Houbavi C, Reinberg D, Aloni Y. 1992. Specific interaction between the nonphosphorylated form of RNA polymerase II and the TATA-binding protein. Cell 69: 871–881. 10.1016/0092-8674(92)90297-P [DOI] [PubMed] [Google Scholar]
- van Haaften G, Dalgliesh GL, Davies H, Chen L, Bignell G, Greenman C, Edkins S, Hardy C, O'Meara S, Teague J, et al. 2009. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat Genet 41: 521–523. 10.1038/ng.349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaswani RG, Gehling VS, Dakin LA, Cook AS, Nasveschuk CG, Duplessis M, Iyer P, Balasubramanian S, Zhao F, Good AC, et al. 2016. Identification of (R)-N-((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1-(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (CPI-1205), a potent and selective inhibitor of histone methyltransferase EZH2, suitable for phase I clinical trials for B-cell lymphomas. J Med Chem 59: 9928–9941. 10.1021/acs.jmedchem.6b01315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velichutina I, Shaknovich R, Geng H, Johnson NA, Gascoyne RD, Melnick AM, Elemento O. 2010. EZH2-mediated epigenetic silencing in germinal center B cells contributes to proliferation and lymphomagenesis. Blood 116: 5247–5255. 10.1182/blood-2010-04-280149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veluchamy A, Rastogi A, Lin X, Lombard B, Murik O, Thomas Y, Dingli F, Rivarola M, Ott S, Liu X, et al. 2015. An integrative analysis of post-translational histone modifications in the marine diatom Phaeodactylum tricornutum. Genome Biol 16: 102 10.1186/s13059-015-0671-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieux-Rochas M, Fabre PJ, Leleu M, Duboule D, Noordermeer D. 2015. Clustering of mammalian Hox genes with other H3K27me3 targets within an active nuclear domain. Proc Natl Acad Sci 112: 4672–4677. 10.1073/pnas.1504783112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinci M, Burford A, Molinari V, Kessler K, Popov S, Clarke M, Taylor KR, Pemberton HN, Lord CJ, Gutteridge A, et al. 2018. Functional diversity and cooperativity between subclonal populations of pediatric glioblastoma and diffuse intrinsic pontine glioma cells. Nat Med 24: 1204–1215. 10.1038/s41591-018-0086-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo LT, Kinney MA, Liu X, Zhang Y, Barragan J, Sousa PM, Jha DK, Han A, Cesana M, Shao Z, et al. 2018. Regulation of embryonic haematopoietic multipotency by EZH1. Nature 553: 506–510. 10.1038/nature25435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt P, LeRoy G, Drury WJ, Zee BM, Son J, Beck DB, Young NL, Garcia BA, Reinberg D. 2012. Asymmetrically modified nucleosomes. Cell 151: 181–193. 10.1016/j.cell.2012.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt P, Tee WW, Reinberg D. 2013. A double take on bivalent promoters. Genes Dev 27: 1318–1338. 10.1101/gad.219626.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Schimmelmann M, Feinberg PA, Sullivan JM, Ku SM, Badimon A, Duff MK, Wang Z, Lachmann A, Dewell S, Ma'ayan A, et al. 2016. Polycomb repressive complex 2 (PRC2) silences genes responsible for neurodegeneration. Nat Neurosci 19: 1321–1330. 10.1038/nn.4360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wang L, Erdjument-Bromage H, Vidal M, Tempst P, Jones RS, Zhang Y. 2004. Role of histone H2A ubiquitination in Polycomb silencing. Nature 431: 873–878. 10.1038/nature02985 [DOI] [PubMed] [Google Scholar]
- Wang X, Goodrich KJ, Gooding AR, Naeem H, Archer S, Paucek RD, Youmans DT, Cech TR, Davidovich C. 2017a. Targeting of polycomb repressive complex 2 to RNA by short repeats of consecutive guanines. Mol Cell 65: 1056–1067 e5. 10.1016/j.molcel.2017.02.003 [DOI] [PubMed] [Google Scholar]
- Wang X, Paucek RD, Gooding AR, Brown ZZ, Ge EJ, Muir TW, Cech TR. 2017b. Molecular analysis of PRC2 recruitment to DNA in chromatin and its inhibition by RNA. Nat Struct Mol Biol 24: 1028–1038. 10.1038/nsmb.3487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zhao Z, Ozark PA, Fantini D, Marshall SA, Rendleman EJ, Cozzolino KA, Louis N, He X, Morgan MA, et al. 2018a. Resetting the epigenetic balance of Polycomb and COMPASS function at enhancers for cancer therapy. Nat Med 24: 758–769. 10.1038/s41591-018-0034-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Long Y, Paucek RD, Gooding AR, Lee T, Cech TR. 2018b. Regulation of histone methylation by automethylation of PRC2. bioRxiv 10.1101/343020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassef M, Michaud A, Margueron R. 2016. Association between EZH2 expression, silencing of tumor suppressors and disease outcome in solid tumors. Cell Cycle 15: 2256–2262. 10.1080/15384101.2016.1208872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiese M, Schill F, Sturm D, Pfister S, Hulleman E, Johnsen SA, Kramm CM. 2016. No significant cytotoxic effect of the EZH2 inhibitor tazemetostat (EPZ-6438) on pediatric glioma cells with wildtype histone 3 or mutated histone 3.3. Klin Padiatr 228: 113–117. 10.1055/s-0036-1593548 [DOI] [PubMed] [Google Scholar]
- Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, et al. 2012. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 44: 251–253. 10.1038/ng.1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Long C, Chen X, Huang C, Chen S, Zhu B. 2010. Partitioning of histone H3-H4 tetramers during DNA replication-dependent chromatin assembly. Science 328: 94–98. 10.1126/science.1178994 [DOI] [PubMed] [Google Scholar]
- Yamasu K, Senshu T. 1990. Conservative segregation of tetrameric units of H3 and H4 histones during nucleosome replication. J Biochem 107: 15–20. 10.1093/oxfordjournals.jbchem.a122999 [DOI] [PubMed] [Google Scholar]
- Yang S, Zheng X, Lu C, Li GM, Allis CD, Li H. 2016. Molecular basis for oncohistone H3 recognition by SETD2 methyltransferase. Genes Dev 30: 1611–1616. 10.1101/gad.284323.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap DB, Chu J, Berg T, Schapira M, Cheng SWG, Moradian A, Morin RD, Mungall AJ, Meissner B, Boyle M, et al. 2011. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 117: 2451–2459. 10.1182/blood-2010-11-321208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youmans DT, Schmidt JC, Cech TR. 2018. Live-cell imaging reveals the dynamics of PRC2 and recruitment to chromatin by SUZ12-associated subunits. Genes Dev 32: 794–805. 10.1101/gad.311936.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Zhang F, Wang S, Fu Y, Chen J, Liang X, Le H, Pu WT, Zhang B. 2017. Depletion of polycomb repressive complex 2 core component EED impairs fetal hematopoiesis. Cell Death Dis 8: e2744 10.1038/cddis.2017.163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W, Wu T, Fu H, Dai C, Wu H, Liu N, Li X, Xu M, Zhang Z, Niu T, et al. 2012. Dense chromatin activates Polycomb repressive complex 2 to regulate H3 lysine 27 methylation. Science 337: 971–975. 10.1126/science.1225237 [DOI] [PubMed] [Google Scholar]
- Zhang K, Mosch K, Fischle W, Grewal SIS. 2008. Roles of the Clr4 methyltransferase complex in nucleation, spreading and maintenance of heterochromatin. Nat Struct Mol Biol 15: 381–388. 10.1038/nsmb.1406 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Jones A, Sun CW, Li C, Chang CW, Joo HY, Dai Q, Mysliwiec MR, Wu LC, Gou Y, et al. 2011. PRC2 complexes with JARID2, MTF2, and esPRC2p48 in ES cells to modulate ES cell pluripotency and somatic cell reprograming. Stem Cells 29: 229–240. 10.1002/stem.578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, McKenzie NJ, Warneford-Thomson R, Gail EH, Flanigan SF, Owen BM, Lauman R, Levina V, Garcia BA, Schittenhelm RB, et al. 2019. RNA exploits an exposed regulatory site to inhibit the enzymatic activity of PRC2. Nat Struct Mol Biol 26: 237–247. 10.1038/s41594-019-0197-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Sun BK, Erwin JA, Song J-J, Lee JT. 2008. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322: 750–756. 10.1126/science.1163045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng YP, Sweet SMM, Popovic R, Martinez-Garcia E, Tipton JD, Thomas PM, Licht JD, Kelleher NL. 2012. Total kinetic analysis reveals how combinatorial methylation patterns are established on lysines 27 and 36 of histone H3. Proc Natl Acad Sci 109: 13549–13554. 10.1073/pnas.1205707109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Huang B, Zhang B, Xiang Y, Du Z, Xu Q, Li Y, Wang Q, Ma J, Peng X, et al. 2016. Resetting epigenetic memory by reprogramming of histone modifications in mammals. Mol Cell 63: 1066–1079. 10.1016/j.molcel.2016.08.032 [DOI] [PubMed] [Google Scholar]
- Zhou Z, Vorperian VR, Gong Q, Zhang S, January CT. 1999. Block of HERG potassium channels by the antihistamine astemizole and its metabolites desmethylastemizole and norastemizole. J Cardiovasc Electrophysiol 10: 836–843. 10.1111/j.1540-8167.1999.tb00264.x [DOI] [PubMed] [Google Scholar]
- Żylicz JJ, Bousard A, Žumer K, Dossin F, Mohammad E, da Rocha ST, Schwalb B, Syx L, Dingli F, Loew D, et al. 2019. The implication of early chromatin changes in X chromosome inactivation. Cell 176: 182–197.e23. 10.1016/j.cell.2018.11.041 [DOI] [PMC free article] [PubMed] [Google Scholar]