

Abstract
The conceptual understanding of hippocampal function has been challenged recently by the finding that new granule cells are born throughout life in the mammalian dentate gyrus (DG). The number of newborn neurons is dynamically regulated by a variety of factors. Kainic acid-induced seizures, a rodent model of human temporal lobe epilepsy, strongly induce the proliferation of DG neurogenic progenitor cells and are also associated with long-term cognitive impairment. We show here that the antiepileptic drug valproic acid (VPA) potently blocked seizure-induced neurogenesis, an effect that appeared to be mainly mediated by inhibiting histone deacetylases (HDAC) and normalizing HDAC-dependent gene expression within the epileptic dentate area. Strikingly, the inhibition of aberrant neurogenesis protected the animals from seizure-induced cognitive impairment in a hippocampus-dependent learning task. We propose that seizure-generated granule cells have the potential to interfere with hippocampal function and contribute to cognitive impairment caused by epileptic activity within the hippocampal circuitry. Furthermore, our data indicate that the effectiveness of VPA as an antiepileptic drug may be partially explained by the HDAC-dependent inhibition of aberrant neurogenesis induced by seizure activity within the adult hippocampus.
Keywords: valproic acid, neurogenesis, hippocampus, seizure, object recognition, learning, histone deacetylase
Introduction
The hippocampal subgranular zone contains cells that continue to divide and differentiate into neurons throughout adulthood (Gage, 2000). Adult neurogenesis can be enhanced by a number of extrinsic factors, including exercise, enriched environment, and learning (Kempermann et al., 1997; Gould et al., 1999; Van Praag et al., 1999). Experimental insults, such as status epilepticus (SE), can also cause a marked increase in adult neurogenesis (Bengzon et al., 1997; Parent et al., 1997; Scott et al., 1998). It has been speculated that seizure-induced neurogenesis contributes to the deficits in hippocampal learning and memory that are associated with SE (Stafstrom et al., 1993; Holmes, 1997; Parent, 2002; Parent and Lowenstein, 2002).
Valproic acid (VPA) (2-propylpentanoic acid) is a well known anticonvulsant and mood stabilizer (Henry, 2003). Recently, VPA has been shown to directly inhibit histone deacetylases (HDACs) (Gottlicher et al., 2001; Phiel et al., 2001). HDACs belong to a family of enzymes that modify the N-terminal tails of histones, altering the interaction between histones and DNA and serving as an epigenetic regulator of gene expression. Huang et al. (2002) reported that changes in glutamate receptor 2 (GluR2) and brain-derived neurotrophic factor (BDNF) after pilocarpine-induced SE were associated with increased histone acetylation in a promoter-specific manner. Both GluR2 and BDNF contain a neuron-restrictive silencing element (NRSE), which is a consensus 21 bp DNA binding site for the neuron-restrictive silencing factor [NRSF, also called repressor element 1 (RE1)-silencing transcription factor (REST)] (Chong et al., 1995; Schoenherr and Anderson, 1995). NRSF transcripts are rapidly induced in the hippocampus after seizures (Palm et al., 1998). Recent work from our laboratory has shown that NRSF also functions as an activator of gene expression in adult neural stem cells to induce neurogenesis (Kuwabara et al., 2004). In undifferentiated neural progenitors or in glial cells, NRSF can associate with repressor proteins such as HDACs and/or methyl-DNA binding proteins (such as methyl-CpG binding protein 2) to repress neuronal gene expression (Naruse et al., 1999; Ballas et al., 2001). However, in neuroblasts, NRSF interacts with a noncoding RNA with the same sequence as the NRSE to activate gene expression and promote neuronal differentiation (Kuwabara et al., 2004). We previously reported that VPA blocked the proliferation and induced neuronal differentiation of adult hippocampal neural progenitor cells in vitro and in vivo (Hsieh et al., 2004). This effect of VPA was associated with a hyperacetylation of histones and an upregulation of NRSF-regulated neuronal genes (Hsieh et al., 2004).
Here we examined the effects of the HDAC inhibitor VPA on kainic acid (KA)-induced seizures. We found that VPA inhibited seizure-induced neurogenesis and reduced abnormal morphological changes of newborn neurons to the level seen in control animals. The ability of VPA to inhibit seizure-induced neurogenesis was associated with increase in NRSF mRNA as well as altered expression levels of several NRSF-regulated neuronal genes. VPA treatment potently protected epileptic animals from hippocampus-dependent cognitive impairment after KA-induced seizures. The data presented support the notion that increased neurogenesis after SE is essentially aberrant and might contribute to pathological changes associated with epileptic activity within the hippocampal circuitry.
Materials and Methods
Animals.
Sixty adult female Fisher 344 rats (180–200 g) were used in this study. To induce SE, rats were injected with KA (12 mg/kg, i.p.). Seizure activity was monitored behaviorally, and only animals exhibiting 3 h of continuous convulsive SE were included in these experiments. Five hours after the onset of seizures, rats in the VPA groups received their first VPA injection (150 mg/kg twice daily, i.p.) that has been shown previously to inhibit HDACs in vivo (Shen et al., 2005; Winkler et al., 2005). Control animals received injections of identical volumes of saline. The day after seizure induction, all rats received daily injections of bromodeoxyuridine (BrdU) (50 mg/kg, i.p.) for 7 d. Rats in the proliferation group were killed 1 d after the last BrdU injection (controls, n = 8; KA, n = 11). Rats in the survival group survived for 4 more weeks (controls, n = 8; KA, n = 10). VPA was injected twice daily throughout the entire length of the experiment (7 d in the proliferation group and 5 weeks in the survival group).
In a separate experiment, six adult female Fisher 344 rats (180–200 g) were chronically implanted with electroencephalographic/electromyographic (EEG/EMG) electrodes and allowed to recover from surgery for 3 weeks (see below). After habituation, rats were injected with KA, and, 5 h after the onset of SE, animals were injected with either VPA (300 mg/kg, i.p.) (n = 3) or saline vehicle controls (n = 3) and we began EEG/EMG monitoring, which continued for 3 d. During the following 2 d, VPA (300 mg/kg, i.p.) or saline was injected once a day.
To obtain hippocampal RNA for reverse transcription (RT)-PCR (see below), eight adult female Fisher rats (180–200 g) were injected with KA alone or with KA after a “preinjection” with VPA (300 mg/kg, i.p.) or trichostatin A (TSA) (1 mg/kg, i.p.) 1 h before the induction of SE. Rats were killed 3 or 24 h after the onset of seizures. In a subsequent experiment, VPA, TSA, or valpromide (100 mg/kg, i.p.) was given 5 h after KA-induced seizure onset (plus saline and KA only controls), and the hippocampi were harvested 24 h later. An additional six rats was used for the KA time course RT-PCR (see Fig. 4A) and eight rats for the KA time course Western blots (see Fig. 4B).
Figure 4.

VPA inhibits seizure-mediated neurogenesis by normalizing changes in gene expression. A–D, RT-PCR and Western blot analysis of DG and CA3 from saline-injected (Cont) and KA-injected (KA) rats from 1 h to 3 d (A, B). RT-PCR of DG and CA3 from control (Cont) and seizure (KA) rats from 1–24 h (A). Representative Western blots of DG and CA3 from control (Cont) and seizure (KA) animals from 3 h to 3 d (B). RT-PCR of DG from saline-injected (Cont) and KA-injected animals receiving either VPA (K/V) or TSA (K/T) (1 h before treatment) and RNA harvested 3 and 24 h after onset of seizures (C). RT-PCR of DG from control (Cont) and KA-treated rats with VPA (K/V), TSA (K/T), or valpromide (K/M) given 5 h after seizure onset, and RNA was harvested 24 h later (D). GAPDH and/or β-actin (data not shown) were used as normalization controls. Data are representative of at least three independent experiments.
Preparation of hippocampal tissue, RT-PCR, and Western blots.
The hippocampus of both hemispheres was dissected out and chopped into 1-mm-thick coronal slices (McIlwain tissue chopper). The dentate gyrus (DG), CA3, and CA1 were carefully isolated using a dissection microscope. RNA isolation from the obtained tissue and cDNA synthesis were performed as described previously (Hsieh et al., 2004). Primer sequences are available on request. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and β-actin were used as normalization controls. Western blot analysis was performed essentially as described previously with slight modifications (Hsieh et al., 2004). Protein lysates from the obtained tissue were solubilized in standard radioimmunoprecipitation assay buffer and homogenized by doing three pulses for 10 s each on ice at setting 1 (Tissue Tearor; Biospec, Bartlesville, OK). Supernatants were normalized for total protein concentration using a BCA protein assay kit (Pierce, Rockford, IL) and separated by SDS-PAGE (NuPage 7% Tris–acetate gel; Invitrogen, Carlsbad, CA). Primary antibodies were rabbit anti-REST (1:500; Upstate Biotechnology, Lake Placid, NY), rabbit anti-GluR2/3 (1:500; Millipore, Temecula, CA), and mouse anti-GAPDH (1:2500; Millipore). Secondary antibodies were anti-rabbit (1:750) or anti-mouse (1:1000) horseradish peroxidase from Cell Signaling Technology (Beverly, MA), and chemiluminescence signals were detected with ECL plus detection kit (GE Healthcare, Little Chalfont, UK) and exposed to film.
Immunohistochemistry.
Animals were anesthetized with a ketamine/xylazine mixture and perfused with 0.9% NaCl solution, followed by 4% paraformaldehyde (PFA) in 0.1 m phosphate buffer, pH 7.4. Brains were postfixed in 4% PFA for 24 h and then transferred into 30% sucrose. Forty micrometer coronal section series were cut using a sliding microtome. All staining was done with free-floating sections. For BrdU detection, the sections were pretreated with 2N HCl for 30 min at 37°C and washed in 0.1 m borate buffer, pH 8.5, for 10 min.
To determine the absolute number of BrdU-labeled cells and to visualize the morphology of doublecortin (DCX)-positive cells, the peroxidase method (ABC system; Vectastain; Vector Laboratories, Burlingame, CA) with biotinylated anti-rat and anti-goat antibodies and nickel-intensified diaminobenzidine (Sigma, St. Louis, MO) as chromogen was used. Immunofluorescent triple labeling was done as described previously (Jessberger and Kempermann, 2003). As primary antibodies, we used rat anti-BrdU (1:500; Harlan Sera-Lab, Loughborough, UK), mouse anti-neuronal neuclei (NeuN) (1:100; Millipore), goat anti-doublecortin (1:250; Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-S100β (1:1000; Swant, Bellinzona, Switzerland), rabbit anti-Ki67 (1:250; Novocastra Laboratories, Newcastle upon Tyne, UK), and rabbit-anti-REST (1:100; Upstate Biotechnology). To determine the extension of basal dendrites arising from new neurons, we captured 12 images of the granule cell layer (GCL) spanning 250 μm in length for each animal. Images were exported into Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA), and three zones starting from the inner border of the GCL and stretching 20 μm apart from each other were outlined. All processes in each zone were counted.
For Fluoro-Jade B staining, sections were mounted on coated slides and dried for 24 h at room temperature. The sections were rehydrated, washed for 2 min in ddH20, blocked in 0.06% potassium permanganate, and incubated for 30 min in the staining solution with 0.0008% Fluoro-Jade B (Millipore).
Cell counts.
The total number of BrdU-positive cells was counted using every 12th section (480 μm apart) from all animals. BrdU-positive cells were counted throughout the rostrocaudal extent of the granule cell layer using a 20× objective and a conventional bright-field microscope (Eclipse E800; Nikon, Tokyo, Japan) as described previously (Kempermann and Gage, 1999). Derived numbers were multiplied by 12 to obtain total cell numbers per GCL.
One-in-12 series of sections from animals from the proliferation group and the survival group (see Fig. 1) were double stained for Ki67/DCX or BrdU and NeuN. Fluorescent signals were detected using a spectral confocal microscope (Radiance 2100; Bio-Rad, Hercules, CA). All analyses were performed in sequential scanning mode, and double labeling was confirmed by three-dimensional reconstructions of z-series. From each animal, 50 Ki67-positive (proliferation group) or BrdU-positive (survival group) cells were randomly picked throughout the GCL and analyzed. Images were processed with Adobe Photoshop 7.0, and only general contrast enhancements and color level adjustments were performed.
Figure 1.
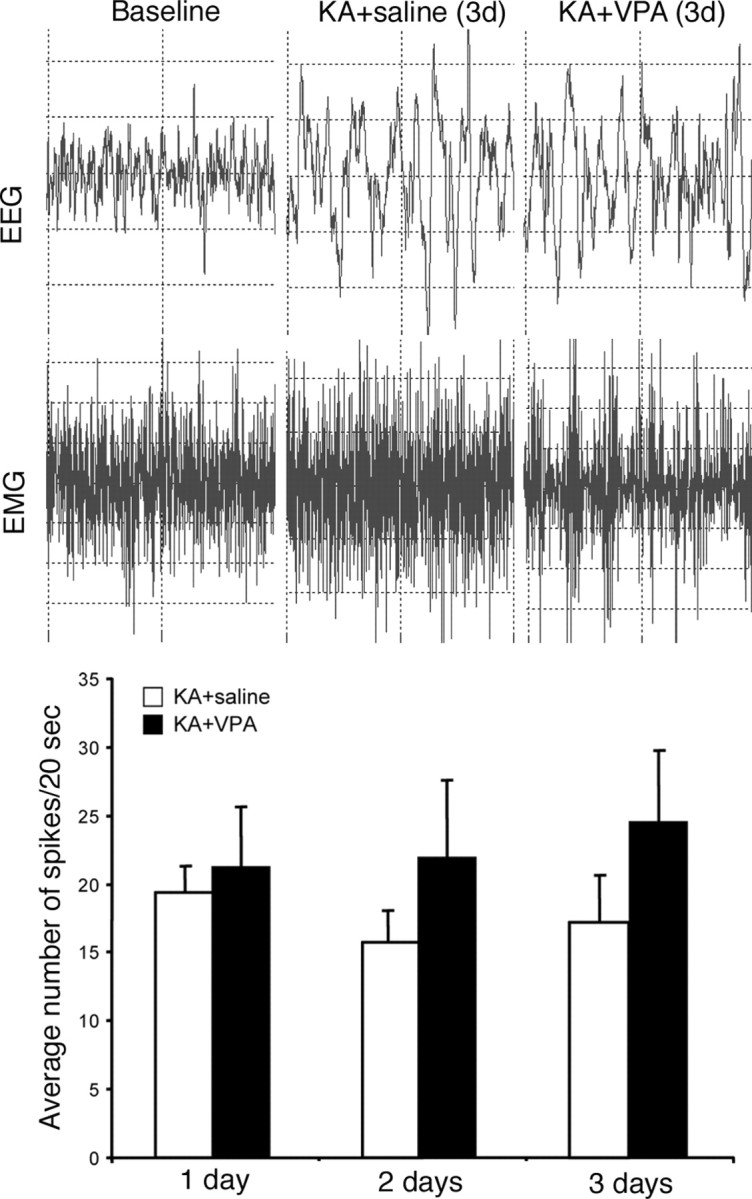
Representative examples of EEG monitoring at baseline and spike-wave activity characteristic of seizures in KA-treated animals receiving saline or VPA injections after 3 d. Quantification of the average number of spikes per 20 s time period between VPA- and saline-treated seizure animals at 1, 2, and 3 d, showing no significant difference between the two groups.
Stereological counting of the hippocampal subfields was done as described previously (Kempermann et al., 1997). StereoInvestigator software (MicroBrightField, Williston, VT) controlling an epifluorescence microscope (Nikon Eclipse E600) was used. The grid size for the granule cell layer was 150 μm2 and for the CA fields was 250 μm2. The counting frame size for the GCL was 10 μm2 and for the CA fields was 15 μm2. The coefficient of error (Gundersen) was between 0.07 and 0.15 for the GCL and 0.08 and 0.31 for the CA fields.
Behavioral analysis.
All animals in the survival groups were tested in a hippocampus-dependent object recognition task (controls, n = 8; KA, n = 10). Rats were habituated to the testing room (45 min) and the empty testing chamber (5 min) for 3 d. The testing chamber was an opaque plastic chamber (61 × 42 × 37 cm) with regular rodent bedding. On testing day, the animals were set into the testing chamber for 1 min to rehabituate. Then two identical objects were placed into the testing chamber, and the rat was allowed to explore the objects for 15 min (“familiarization”). After a 3 h delay, the rat was replaced into the testing chamber again with one object identical to one used in the familiarization phase and a new object (“testing”). The rats were given 15 min to explore the familiar and new objects during the testing phase. The familiarization and testing phases were filmed with a digital video camera for off-line analysis. The analysis was done using the software developed by Robert E. Clark (University of California, San Diego, La Jolla, CA) using a handheld game controller (Clark et al., 2000). The complete exploration time during the familiarization was scored. During the testing phase, the first 30 s of cumulative exploration time was scored. Two main criteria had to be fulfilled to be scored as exploratory behavior: (1) the animal's nose had to point toward the object not farther away than one rat head's length, and (2) there had to be a movement of the whiskers. The testing chamber and used objects were washed with 70% ethanol before the next rat was tested. All of the objects used were made of plastic and did not differ in overall size but differed in shape and color. Objects and positions in the testing chamber were systematically counterbalanced between groups.
Recording of the EEG and EMG.
EEG/EMG recordings were performed as described previously (Beuckmann et al., 2004). Female Fisher 344 rats (180–200 g; n = 3 per group) were anesthetized (50 mg/kg ketamine, 5 mg/kg xylazine, and 1 mg/kg acepromazine, i.p.), held in a stereotaxic apparatus, and surgically implanted under sterile conditions with a miniaturized prefabricated block of recording electrodes. The electrode block was lowered stereotaxically until the electrodes just penetrated the skull and just touched the dura. This depth was critical to ensure that the lower end of the electrode made electrical contact with the dura but did not penetrate it. The electrode block was then affixed to the skull using a glass ionomer dental cement (Ketac Cem, ESPE; 3M, St. Paul, MN). EEG signals were recorded unilaterally from a fronto-occipital electrode pair, positioned 1.5 mm rostral and 1.45 mm laterally from bregma, and 6.1 mm caudal and 1.45 mm lateral from bregma. EMG signals were recorded concurrently from two flexible wires, insulated except at the tips, and implanted bilaterally by blunt dissection into the nuchal musculature. All rats recovered from the surgery and were habituated to the recording conditions for 3 weeks before recording commenced. Throughout the recording, the rats were tethered via a lightweight cable that was attached to a counterbalanced arm (SMCLA; Instech Laboratories, Plymouth Meeting, PA) mounted to the side of a standard shoebox cage, with the top cut away. EEG/EMG signals were recorded under controlled conditions (12 h light/dark cycle; 24 ± 1°C) for 3 consecutive days. Signals were digitized at 250 Hz, recorded to hard disk, and subsequently archived to optical media for off-line analysis. Concurrently with EEG/EMG recording, the behavior of the rats was videotaped for 4 h. Using customized display software, each 24 h EEG record was visually screened in 20 s epochs for seizure epochs, the timing and duration of each seizure was noted, and comparison was made against the behavior of the rat from the synchronized video record during the initial period. The display was in terms of 20 s epochs. Seizures were characterized as a spike-wave pattern on the EEG, usually accompanied by atonic periods or sustained rhythmic contractions on the EMG. Repetitive spike-wave patterns were frequently observed, sometimes accompanied by rhythmic slow activity. Fast waves of increasing amplitude and decreasing frequency could also be seen. Subclinical seizures were typically evident on the EEG record as a lower-amplitude abnormal wave pattern without change to the EMG. Each seizure lasting for 2 s or more was noted, and those that occurred within 5 s of each other were considered the same seizure. Analysis was in terms of the total number and of the mean duration of seizures during the 12 h dark and light periods.
Statistics.
All statistical analyses were performed using Statview 5.0.1 (SAS Institute, Cary, NC). For all comparisons, ANOVA was performed followed by Fisher's post hoc test, when appropriate. Differences were considered statistically significant at p < 0.05.
Results
KA-induced subclinical seizures persist with VPA treatment
We used KA to induce SE in Fisher 344 rats, because they show relatively little variance in their response to KA (Golden et al., 1995). Seizures could be detected behaviorally within 1 h after KA injection and lasted for at least 2–3 h. To examine whether VPA affected seizure activity, we injected rats with either VPA or saline 5 h after initial seizure onset and started EEG monitoring. Both groups showed similar, pronounced spike-wave epileptiform discharges consistent with behavioral seizures during the first 24 h (Fig. 1). Even after overt behavioral seizures subsided after the first day, both seizure groups still showed spike wave activity characteristic of subclinical seizures for up to 3 d of EEG/EMG monitoring. Thus, VPA treatment in seizure animals did not significantly affect seizure strength or frequency in the first 3 d after KA injection compared with untreated seizure control animals.
VPA inhibits seizure-induced neurogenesis in the adult dentate GCL
To examine the potential effects of VPA on seizure-induced neurogenesis, we used the thymidine analog BrdU to label dividing cells and their progeny. The animals were separated into two groups, an early group receiving 7 d of BrdU injections and killed 1 d after the last BrdU injection (proliferation group) and a late group receiving 7 d of BrdU injections and killed 4 weeks later (survival group). Consistent with previous reports (Bengzon et al., 1997; Parent et al., 1997; Scott et al., 1998), seizure activity induced a strong increase in BrdU-positive cells in the DG of animals in the early group compared with animals injected with saline or VPA only (Fig. 2A–E) (p < 0.01). Animals receiving daily VPA injections after KA-induced seizures displayed reduced levels of BrdU-positive cells compared with seizure animals without VPA (Fig. 2A–E).
Figure 2.
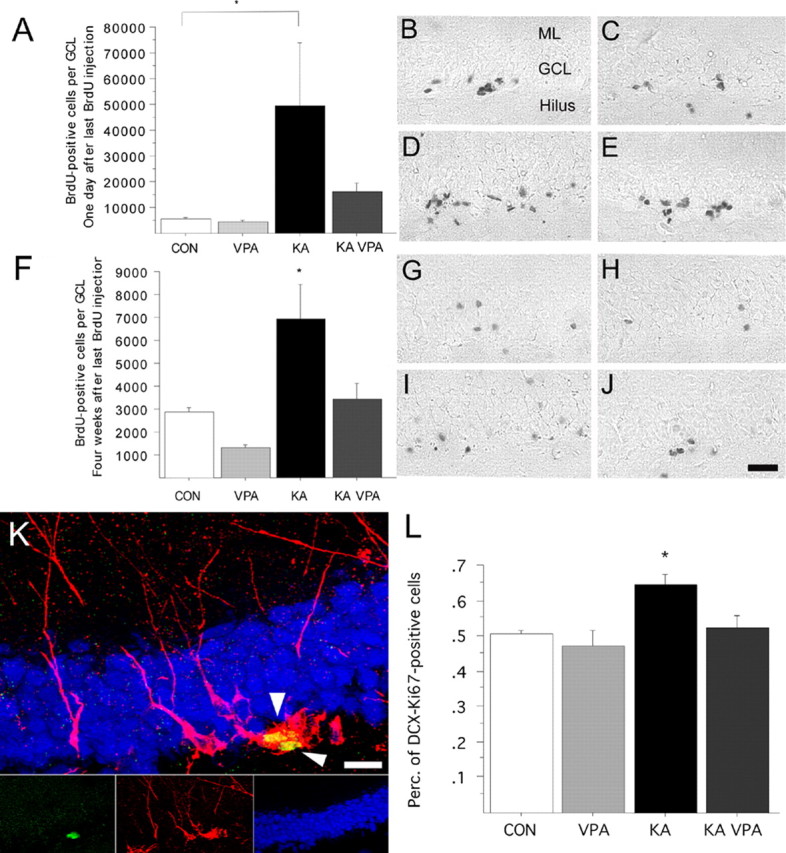
VPA treatment blocks seizure-induced neurogenesis and prevents aberrant DCX-expressing progenitor cell activation. A–E, Number of BrdU-positive cells 1 d after the last BrdU injection, indicative of proliferative activity after SE (A). Representative BrdU staining of saline (B), VPA-treated (C), KA-injected (D), and KA-injected plus VPA-treated (E) animals killed 1 d after last BrdU injection. F–J, Number of BrdU-positive cells 4 weeks after the last BrdU injection, indicative of stable cell genesis after SE (F). Representative BrdU staining of saline (G), VPA-treated (H), KA-injected (I), and KA-injected plus VPA-treated (J) animals killed 4 weeks after last BrdU injection. K, Example of two DCX (red)/Ki-67 (green) colabeled cells (arrowheads) in the subgranular zone (blue, NeuN) 8 d after SE. L, The percentage of DCX/Ki-67 colabeled cells is increased 8 d after SE compared with controls. VPA treatment prevents aberrant DCX-positive progenitor cell activation. Scale bars: (in J) B–J, 100 μm; K, 20 μm. *p < 0.01.
To determine the phenotype of dividing cells mediating the KA-induced increase in cell proliferation in the presence or absence of VPA, we analyzed proliferating progenitors (using Ki67 as a marker of cycling cells) (Gerdes et al., 1983; Kee et al., 2002) for their coexpression of DCX, a marker for dividing neuroblasts and immature neurons (Brown et al., 2003; Couillard-Despres et al., 2005; Jessberger et al., 2005) by confocal microscopy. The number of Ki67-positive colabeling with DCX increased in animals treated with KA (Fig. 2K,L) (p < 0.01). However, KA-injected animals treated with VPA did not display an increase in DCX–Ki67 double-positive cells and showed levels similar to those in control and VPA-treated only animals (Fig. 2L).
We next examined the number of BrdU-labeled cells in the DG of animals in the survival group. Reflecting the strong increase in proliferation soon after SE, there was a significant increase in the number of BrdU-positive cells in KA-treated animals. Strikingly, the seizure-induced increase in net neurogenesis was significantly reduced with VPA treatment (Fig. 2F–J). To identify the phenotype of BrdU-labeled cells 4 weeks after the last BrdU injection, we determined the percentage of cells that were positive for BrdU and positive for NeuN, a marker of mature neurons or S100β, a Ca2+-binding protein expressed by astrocytes. We did not find any significant differences in relative neuronal or glial differentiation of newborn cells (data not shown). However, the absolute number of BrdU–NeuN double-positive cells in the KA-treated group was substantially increased compared with that in control and VPA-alone animals (Fig. 3A–C). Notably, seizure animals that had received VPA injections showed numbers of newborn neurons that were comparable with those in control animals, showing that VPA potently inhibited seizure-induced neurogenesis in the adult hippocampus.
Figure 3.
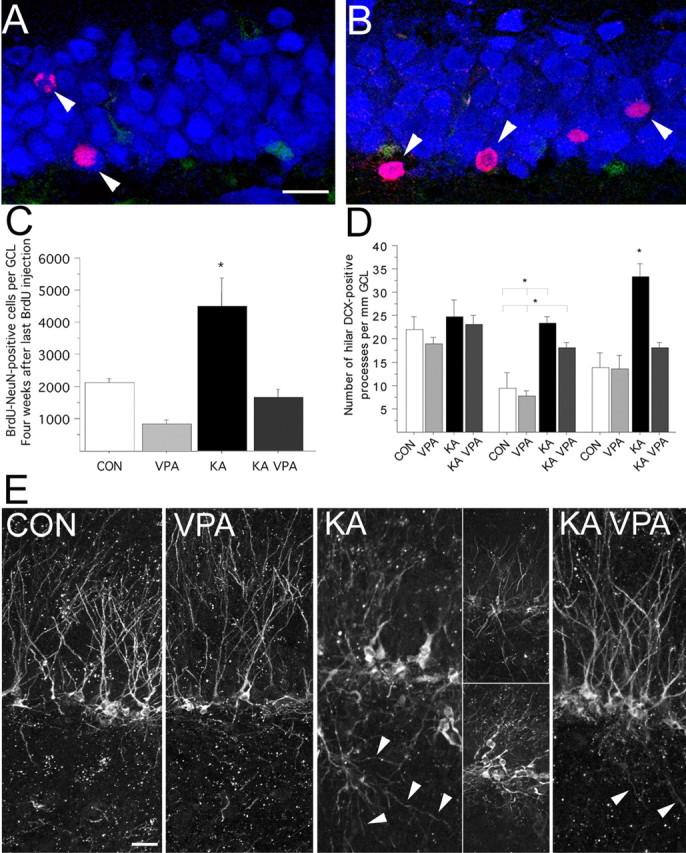
VPA treatment abates the number of seizure-generated neurons and decreases the formation of hilar basal dendrites. A–C, Compared with control animals (A), the number of new neurons (arrowheads) 4 weeks after the last BrdU injection is dramatically increased after KA injection (B; red, BrdU; blue, NeuN; green, S100β). VPA treatment significantly reduced the number of new neurons after SE (C). D, E, VPA treatment inhibited the formation of basal dendrites reaching into the polymorphic cell layer (D). Representative examples of DCX-immunoreactive cells in saline (CON), VPA-treated (VPA), KA-injected (KA), and KA-injected plus VPA-treated (KA VPA) animals killed 8 d after SE (E). Arrowheads point toward hilar basal dendrites. Scale bars: A, E, 20 μm. *p < 0.01.
VPA normalizes the morphology of newly generated dentate granule cells after seizures
One of the characteristic changes seen after SE is the presence of hilar basal dendrites arising from granule cells that fail to retract and extend their processes deep into the hilus (Buckmaster and Dudek, 1999; Dashtipour et al., 2003; Shapiro and Ribak, 2006). The presence of these hilar basal dendrites on dentate granule cells as well as ectopic hilar granule cells after seizures could lead to a recurrent excitatory circuitry (Ribak et al., 2000; Scharfman et al., 2000). Therefore, we analyzed the extent of DCX-positive basal dendrites reaching into the hilus. Confirming previous results (Dashtipour et al., 2003; Shapiro and Ribak, 2006), we found a significant increase in the length and number of dendritic processes reaching into the hilus 8 d after KA-induced seizures compared with controls (Fig. 3E). VPA treatment of seizure animals almost completely prevented the formation of hilar basal dendrites and normalized the morphology of newborn granule cells similar to controls (Fig. 3E).
VPA inhibits seizure-mediated neurogenesis by regulating changes in gene expression
The observation that there was a decrease in the number of newborn neurons and normalized morphology after seizures with VPA treatment led us to investigate how VPA acts to block the detrimental effects of seizure-induced neurogenesis. The effect of KA-induced seizures on NRSF transcripts has been studied in the adult brain (Palm et al., 1998). The researchers found increased expression of NRSF in granular neurons of the DG 4 h after KA treatment and increased expression in the pyramidal layers CA1–CA4 24 h after KA treatment. In our experiments, a time course RT-PCR after KA treatment was performed (Fig. 4A). Indeed, NRSF mRNA levels were significantly increased in both the DG and CA3 as early as 6 h after KA injection and remained elevated throughout the course of the experiment (Fig. 4A). In other experiments, NRSF levels returned to baseline at 3 d after KA treatment, suggesting that there may be a critical period initially after SE that NRSF regulation of direct downstream target genes is occurring. NRSF protein levels appeared relatively unchanged in control and KA-treated animals at various time points (3 h to 3 d), although there may be slightly increased NRSF protein levels at the 1 d time point with KA treatment in both DG and CA3 (Fig. 4B).
KA-induced seizures have been shown to upregulate the expression of some NRSF target genes, such as BDNF (Zafra et al., 1990; Tongiorgi et al., 2004) and NaCh II (Gastaldi et al., 1997), and downregulate the expression of other genes, such as GluR2 (Grooms et al., 2000; Sanchez et al., 2001). We found that there are gene-specific differences among various NRSF target genes (i.e., GluR2, BDNF, synapsin I) with respect to their upregulation or downregulation after KA in the DG at 3 and 24 h (Fig. 4C). We also observed GluR2 protein levels were down by 3 d with KA treatment in both DG and CA3, which has been suggested previously to be one of the critical genes downregulated after seizures (Fig. 4B) (Pollard et al., 1993; Friedman et al., 1994). Interestingly, animals receiving both KA and either VPA (K/V) or TSA (K/T) displayed high levels of NRSF-regulated gene expression as early as 3 h (as for BDNF and synapsin I) and, in some cases, up to 24 h (as for BDNF). In the case of GluR2, there was downregulation of the message after KA induction by 24 h, which was prevented with VPA or TSA treatment (Fig. 4C). This finding is consistent with results from Huang et al. (2002), who demonstrated that TSA prevented the downregulation of GluR2 in CA3 after pilocarpine-induced seizures (Huang et al., 2002). As for BDNF (at 3 and 24 h) and synapsin I (at 3 h), VPA or TSA treatment in KA-injected animals kept the mRNA levels high; however, the level of synapsin I dropped back down to baseline by 24 h. These results suggest that NRSF and a subset of NRSF target genes may be preferentially dysregulated after KA-induced seizures and that VPA, through HDAC inhibition, may function to normalize these aberrant changes in gene expression.
In the above experiments, animals were first pretreated with VPA or TSA for 1 h before seizure induction to maximize the levels of HDAC inhibitors during SE (as described previously by Huang et al., 2002). In a follow-up study, we treated seizure rats with VPA (K/V), TSA (K/T), or valpromide (K/M) 5 h after onset of SE, which is identical to the paradigm for the neurogenesis and behavioral studies with the addition of a second HDAC inhibitor (Fig. 4D). Similar to the pretreatment scheme, VPA and TSA (although to a lesser extent) prevented the KA-induced downregulation of GluR2. Interestingly, the antiepileptic valpromide (K/V), which is an amide analog of VPA but is not an HDAC inhibitor (Gottlicher et al., 2001), does not block the downregulation of GluR2 after KA treatment. Together, these data suggest that VPA blocks seizure-induced neurogenesis via inhibiting HDACs, possibly through modulation of NRSF activity and neuronal target genes.
Neuronal cell death does not appear to be affected by VPA
These results indicated that VPA potently inhibited seizure-induced, aberrant neurogenesis by normalizing HDAC-regulated gene expression. Recent studies have suggested that VPA has a neuroprotective effect after traumatic brain injury (Morland et al., 2004; Ren et al., 2004). Because KA-induced seizures result in a considerable amount of neuronal cell death (Ben-Ari and Cossart, 2000; Bengzon et al., 2002), we next analyzed whether VPA had a neuroprotective effect after KA-induced seizures, in addition to its inhibition of seizure-induced neurogenesis. We stained sections from all rats of the survival groups with Fluoro-Jade B, which labels degenerating neurons (Schmued and Hopkins, 2000). Careful visual analysis by two blinded investigators did not reveal any significant differences in the amount of neuronal cell death, observed throughout the brains of seizure animals that did or did not receive VPA injections (Fig. 5A–D). Nevertheless, we sought unbiased, quantitative numbers of surviving neurons. Therefore, we stereologically assessed cell densities of the hippocampus in all rats that were killed 5 weeks after seizure induction. Confirming previous results, we found decreased neuronal cell densities within the DG in all rats that had experienced SE (Fig. 5E) (Holtkamp et al., 2005). Importantly, the VPA-treated animals showed a comparable degree of neuronal degeneration in the dentate area as the untreated KA-injected animals (Fig. 5E,F). In addition, we did not detect any neuroprotective effect of VPA on pyramidal cells in the cornu ammonis (data not shown). In summary, VPA treatment did not result in a substantial neuroprotective effect after KA-induced seizures in the adult rat.
Figure 5.

Seizure-induced cell death in the dentate area is not affected by VPA treatment. A–D, Fluoro-Jade B staining of saline (A), VPA-treated (B), KA-injected (C), and KA-injected plus VPA-treated (D) animals killed 5 weeks after SE shows degenerating neuronal cells in the hilus in the KA-injected animals. E, F, Cell density and DG volume in control animals and seizure animals 5 weeks after SE. Scale bar, 100 μm.
Improved hippocampal function in VPA-treated, KA-injected animals
Prolonged seizure activity eventually leads to severe impairment in cognition and in learning and memory in humans but also in rodent models of temporal lobe epilepsy (TLE) (Stafstrom et al., 1993; Holmes, 1997; Helmstaedter, 2002; Stefan and Pauli, 2002; Elger et al., 2004). VPA treatment blocked aberrant neurogenesis after KA-induced SE and thus inhibited the excess addition of seizure-generated granule cells into the preexisting dentate circuitry. Given this finding, we hypothesized that VPA treatment may protect KA-treated animals from cognitive impairment by inhibiting the addition of new, aberrant neurons to the adult GCL. We tested all rats in the survival groups 5 weeks after seizure induction in a hippocampus-dependent object recognition test (Clark et al., 2000). All groups showed comparable levels of exploratory behavior (Fig. 6A), and we found no preference for one of the objects used in this experiment (data not shown). After a 3 h delay, a new object was introduced into the testing chamber together with an identical copy of the familiar object. Control animals showed a strong preference to explore the new object within the first 30 s of exploration (Fig. 6B). In clear contrast, rats that had experienced KA-induced seizures spent the same amount of time exploring the familiar object as they did exploring the new object, indicating impairment in hippocampus-dependent object memory (Fig. 6B). Strikingly, this phenotype was completely prevented in the VPA-treated KA-injected animals, which showed a preference toward the novel object similar to controls (Fig. 6B) (p < 0.001; F = 27.3). Given this finding, VPA treatment protected KA-injected animals from the cognitive impairment that was detectable in untreated KA-injected animals in a hippocampus-dependent object recognition test. Because previous studies had shown that the increase in proliferative activity occurred mainly during the first 14 d after SE (Parent et al., 1997) and to strengthen the association between seizure-induced neurogenesis and subsequent memory impairment, we restricted, in a separate experiment, the duration of VPA injections to the first 2 weeks after KA injection. Paralleling our first experiment, we found a robust inhibition of seizure-induced neurogenesis by short-term VPA treatment (in this experiment, the animals were killed 2 and 4 weeks after SE). Strikingly, treatment for 14 d after SE with VPA also prevented seizure-associated memory impairment (tested 4 weeks after SE) at a comparable level with the long-term VPA treatment (data not shown).
Figure 6.

Seizure-associated cognitive impairment is prevented by VPA treatment. A, B, Although the total exploration time was comparable between all groups (A), KA-challenged animals did not show a preference for the new object 3 h after the presentation of the initial objects (B). VPA treatment prevented this phenotype, and KA-challenged animals that received VPA injections showed the same level of preference for the new objects as controls did. *p < 0.01.
Discussion
Here we show that aberrant neurogenesis induced by seizure activity within the hippocampal circuitry was potently blocked by the antiepileptic drug VPA. The efficacy of VPA in decreasing the neurogenic response to seizure activity appeared to be mediated by VPA-induced inhibition of HDACs and subsequent normalization of HDAC-dependent gene expression. Strikingly, animals that had experienced SE, but received VPA treatment, did not show seizure-associated cognitive impairment in a hippocampus-dependent object recognition test.
After decades of controversy, the finding that new neurons are added into the hippocampal circuitry throughout life is now broadly accepted (Gage, 2002). However, the functional significance of adult neurogenesis remains unclear (Kempermann, 2002; Schinder and Gage, 2004). Supporting a functional role for physiologically generated new granule cells is the finding that ablation of new neurons with cytostatic drugs or irradiation correlates with impairment in some hippocampus-dependent learning tasks (Shors et al., 2001, 2002; Snyder et al., 2005), although the specificity of the above-mentioned ablation techniques (Monje et al., 2002) is subject to controversy within the field of adult neurogenesis (Dupret et al., 2005). Interestingly, seizure activity within the hippocampal formation induces the formation of new granule cells, apparently without having a beneficial effect on learning and memory. Given this finding, it has been speculated that new neurons generated in response to epileptic activity may contribute to the pathological consequences of SE rather than represent an attempt by the injured brain to replace lost cells (Parent, 2002; Parent and Lowenstein, 2002). What distinguishes seizure-induced neurogenesis from physiological neurogenesis? Furthermore, might certain antiepileptic drugs modulate seizure-induced neurogenesis?
Seizure activity leads to increased proliferative activity of DCX-positive dividing cells, presumably representing committed neuroblasts (Jessberger et al., 2005). Currently, the consequences of increased activity of DCX-positive cells remain to be characterized (Jessberger et al., 2005). Nevertheless, this abnormal activation of late progenitors was completely absent in rats treated with the antiepileptic drug VPA. Furthermore, the hilar extension of basal dendrites arising from seizure-generated granule cells was prevented by VPA treatment of KA-injected animals. Previous studies suggested that extensive basal dendrites might contribute to recurrent excitatory circuits interfering with normal synaptic transmission (Ribak et al., 2000; Dashtipour et al., 2003; Overstreet-Wadiche et al., 2006; Shapiro and Ribak, 2006).
In striking contrast to the KA and pilocarpine rat and mouse model of TLE that are all associated with dramatic morphologic changes of seizure-induced granule cells (Parent et al., 1997; Scharfman et al., 2000; Overstreet-Wadiche et al., 2006; Shapiro et al., 2006), Jakubs et al. (2006) recently reported that granule cells that are born after SE induced by electrical stimulation are less excitable than granule cells born in running rats. Jakubs and colleagues speculated that decreased excitability of seizure-induced cells might be an attempt of the injured brain to restore brain homeostasis. The reasons for the discrepancy between their findings (regular morphology) and other reports (extensive changes in granule cell morphology after SE) remain unknown but might be explained by the different seizure models used in the respective studies.
Besides the normalization of seizure-associated morphological changes after KA-induced seizures, we found a significant reduction in the number of seizure-generated neurons in VPA-treated animals. This effect of VPA appeared to be mainly mediated by inhibition of seizure-induced cell proliferation. Indeed, the cyclin-dependent inhibitor p21WAF1/CIP1 is a common target induced by short-chain fatty acid HDAC inhibitors such as VPA and sodium butyrate (Gurvich et al., 2004) and is rapidly induced with VPA treatment after KA treatment (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Previously, our group reported that VPA and other HDAC inhibitors have the ability to block adult hippocampal neural progenitor cell proliferation and promote neuronal and suppress glial differentiation, respectively (Hsieh et al., 2004). The mechanism of VPA as an antiepileptic has remained elusive (Owens and Nemeroff, 2003). Two possible models to explain the nature of its effects on blocking aberrant neurogenesis include a direct versus indirect mode of action. An example of an indirect course of action is that VPA blocks seizure activity, possibly through the modulation of GABA levels, and the reduction of seizure activity and/or recurrent seizures leads to the decrease in aberrant neurogenesis. We think that several arguments speak against the explanation that VPA injections simply reduce initial seizure activity. First of all, we injected the first VPA dose 5 h after the onset of SE. At that time, behavioral seizure activity had already been terminated. Previous studies also showed that even milder seizures induce the maximum amount of neurogenic cell proliferation (Mohapel et al., 2004). Second, our EEG data suggested that VPA does not significantly alter seizure strength or frequency in seizure rats compared with saline controls early after KA injection.
Thus, we favor the alternative model: the actions of VPA directly inhibit HDACs and normalize changes in gene expression after seizures. Among the numerous genes that change after seizure induction (Elliott et al., 2003; Majores et al., 2004), it was reported recently that, after pilocarpine-induced SE, changes in two NRSF-regulated neuronal genes, GluR2 and BDNF, could be prevented by the HDAC inhibitor TSA (Huang et al., 2002). Because there is also a rapid induction of NRSF mRNAs in the hippocampus after seizures (Palm et al., 1998), NRSF may function as a context-dependent regulator of gene expression in both neurons and non-neuronal cells. Immunohistochemistry experiments revealed detectable levels of NRSF in the dentate granule cell layer of adult rat hippocampus (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The specific cell types that differentially express NRSF after seizure activity remain unknown. Nevertheless, modulation of NRSF expression and/or activity [i.e., by small noncoding RNAs matching the same sequence as the NRSF binding site called NRSE smRNAs (Kuwabara et al., 2004)] could be a critical event in seizure-mediated neurogenesis. Additional experiments are needed to determine whether NRSF gene expression and/or NRSF protein activity become dysregulated after SE and whether VPA and other HDAC inhibitors act on the expression of NRSF itself, which in turn might affect downstream target genes or act directly on the expression of NRSE-containing neuronal genes. In fact, a recent study has found that VPA treatment could significantly reduce seizure-induced expression of two immediate early genes, c-fos and c-Jun, reinforcing the idea that VPA belongs to a class of antiepileptic drugs that normalizes the expression of seizure-induced genes (Szot et al., 2005). What might be the result of aberrant seizure-induced gene expression? Simplified, two features reflecting human pathology are associated with the KA rodent model of human TLE: epileptogenesis (the occurrence of spontaneous seizures after a latent period) and cognitive impairment (Helmstaedter et al., 2003). Altered gene expression within mature hippocampal neurons but also aberrant gene expression that results in disturbed neurogenesis may be responsible for these two consequences of prolonged epileptic activity within the hippocampal circuitry. Here we report that VPA treatment robustly prevented seizure-induced neurogenesis and protected animals that had experienced SE from seizure-associated impairment in a hippocampus-dependent learning task. To assess hippocampal function, we used an object recognition task with a time interval between the presentation of a new object relative to the familiarization that depends on intact hippocampal function in rodents (Clark et al., 2000). Rats that had experienced KA-induced SE showed a severe impairment in this test. This finding was in striking contrast to the performance of SE animals that had received VPA injections. Interestingly, it has been reported previously that patients with TLE display deficits in recognition memory (for review, see Elger et al., 2004). In addition, a previous study showed the prevention of cognitive impairment after KA-induced seizures by VPA treatment using the Morris water maze (Bolanos et al., 1998). Therefore, the inhibition of aberrant seizure-induced neurogenesis and the morphological normalization of neurons that were generated in response to SE by VPA treatment might at least partially explain the observed protection from seizure-associated cognitive impairment. At this point, we cannot determine the relative impact of the increased numbers or altered morphology of seizure-generated neurons on cognitive performance after SE.
Given the variety of cellular and molecular changes after SE, it is clear that KA-induced aberrant neurogenesis is not the sole cause of cognitive impairment associated with seizure activity. Therefore, ongoing studies will address the functional consequences of seizure-generated neurons on a single-cell level and the effects of aberrant neurons on hippocampal synaptic transmission. In addition, future ablation studies will further characterize the relative contribution of seizure-induced neurogenesis to impaired learning and memory after SE.
Footnotes
S.J. was supported in part by the Deutsche Forschungsgemeinschaft (Je297/1-1) and the American Epilepsy Society. J.H. was supported in part by The Esther A. and Joseph Klingenstein Fund, The Ellison Medical Foundation, and The Advanced Research Program, Texas Higher Education Coordinating Board. Additional support was provided by grants from the National Institute of Neurological Disorders and Stroke/National Institute on Aging, the Max Planck Research Award Program funded by the German Ministry for Education, Science, Research, and Technology, and the Pritzker Research Consortium (F.H.G.). We thank members of the Gage laboratory for helpful discussions and M. L. Gage for editorial comments. We also thank Nicola J. Broadbent and Robert E. Clark (University of California, San Diego, La Jolla, CA) for help with the object recognition testing.
References
- Ballas N, Battaglioli E, Atouf F, Andres ME, Chenoweth J, Anderson ME, Burger C, Moniwa M, Davie JR, Bowers WJ, Federoff HJ, Rose DW, Rosenfeld MG, Brehm P, Mandel G. Regulation of neuronal traits by a novel transcriptional complex. Neuron. 2001;31:353–365. doi: 10.1016/s0896-6273(01)00371-3. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Cossart R. Kainate, a double agent that generates seizures: two decades of progress. Trends Neurosci. 2000;23:580–587. doi: 10.1016/s0166-2236(00)01659-3. [DOI] [PubMed] [Google Scholar]
- Bengzon J, Kokaia Z, Elmér E, Nanobashvili A, Kokaia M, Lindvall O. Apoptosis and proliferation of dentate gyrus neurons after single and intermittent limbic seizures. Proc Natl Acad Sci USA. 1997;94:10432–10437. doi: 10.1073/pnas.94.19.10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengzon J, Mohapel P, Ekdahl CT, Lindvall O. Neuronal apoptosis after brief and prolonged seizures. Prog Brain Res. 2002;135:111–119. doi: 10.1016/S0079-6123(02)35011-8. [DOI] [PubMed] [Google Scholar]
- Beuckmann CT, Sinton CM, Williams SC, Richardson JA, Hammer RE, Sakurai T, Yanagisawa M. Expression of a poly-glutamine-ataxin-3 transgene in orexin neurons induces narcolepsy-cataplexy in the rat. J Neurosci. 2004;24:4469–4477. doi: 10.1523/JNEUROSCI.5560-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolanos AR, Sarkisian M, Yang Y, Hori A, Helmers SL, Mikati M, Tandon P, Stafstrom CE, Holmes GL. Comparison of valproate and phenobarbital treatment after status epilepticus in rats. Neurology. 1998;51:41–48. doi: 10.1212/wnl.51.1.41. [DOI] [PubMed] [Google Scholar]
- Brown JP, Couillard-Despres S, Cooper-Kuhn CM, Winkler J, Aigner L, Kuhn HG. Transient expression of doublecortin during adult neurogenesis. J Comp Neurol. 2003;467:1–10. doi: 10.1002/cne.10874. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Dudek FE. In vivo intracellular analysis of granule cell axon reorganization in epileptic rats. J Neurophysiol. 1999;81:712–721. doi: 10.1152/jn.1999.81.2.712. [DOI] [PubMed] [Google Scholar]
- Chong JA, Tapia-Ramirez J, Kim S, Toledo-Aral JJ, Zheng Y, Boutros MC, Altshuller YM, Frohman MA, Kraner SD, Mandel G. REST: a mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell. 1995;80:949–957. doi: 10.1016/0092-8674(95)90298-8. [DOI] [PubMed] [Google Scholar]
- Clark RE, Zola SM, Squire LR. Impaired recognition memory in rats after damage to the hippocampus. J Neurosci. 2000;20:8853–8860. doi: 10.1523/JNEUROSCI.20-23-08853.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couillard-Despres S, Winner B, Schaubeck S, Aigner R, Vroemen M, Weidner N, Bogdahn U, Winkler J, Kuhn HG, Aigner L. Doublecortin expression levels in adult brain reflect neurogenesis. Eur J Neurosci. 2005;21:1–14. doi: 10.1111/j.1460-9568.2004.03813.x. [DOI] [PubMed] [Google Scholar]
- Dashtipour K, Wong AM, Obenaus A, Spigelman I, Ribak CE. Temporal profile of hilar basal dendrite formation on dentate granule cells after status epilepticus. Epilepsy Res. 2003;54:141–151. doi: 10.1016/s0920-1211(03)00082-2. [DOI] [PubMed] [Google Scholar]
- Dupret D, Montaron MF, Drapeau E, Aurousseau C, Le Moal M, Piazza PV, Abrous DN. Methylazoxymethanol acetate does not fully block cell genesis in the young and aged dentate gyrus. Eur J Neurosci. 2005;22:778–783. doi: 10.1111/j.1460-9568.2005.04262.x. [DOI] [PubMed] [Google Scholar]
- Elger CE, Helmstaedter C, Kurthen M. Chronic epilepsy and cognition. Lancet Neurol. 2004;3:663–672. doi: 10.1016/S1474-4422(04)00906-8. [DOI] [PubMed] [Google Scholar]
- Elliott RC, Miles MF, Lowenstein DH. Overlapping microarray profiles of dentate gyrus gene expression during development- and epilepsy-associated neurogenesis and axon outgrowth. J Neurosci. 2003;23:2218–2227. doi: 10.1523/JNEUROSCI.23-06-02218.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman LK, Pellegrini-Giampietro DE, Sperber EF, Bennett MV, Moshe SL, Zukin RS. Kainate-induced status epilepticus alters glutamate and GABAA receptor gene expression in adult rat hippocampus: an in situ hybridization study. J Neurosci. 1994;14:2697–2707. doi: 10.1523/JNEUROSCI.14-05-02697.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage F. Mammalian neural stem cells. Science. 2000;287:1433–1438. doi: 10.1126/science.287.5457.1433. [DOI] [PubMed] [Google Scholar]
- Gage FH. Neurogenesis in the adult brain. J Neurosci. 2002;22:612–613. doi: 10.1523/JNEUROSCI.22-03-00612.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gastaldi M, Bartolomei F, Massacrier A, Planells R, Robaglia-Schlupp A, Cau P. Increase in mRNAs encoding neonatal II and III sodium channel alpha-isoforms during kainate-induced seizures in adult rat hippocampus. Brain Res Mol Brain Res. 1997;44:179–190. doi: 10.1016/s0169-328x(96)00199-4. [DOI] [PubMed] [Google Scholar]
- Gerdes J, Schwab U, Lemke H, Stein H. Production of a mouse monoclonal antibody reactive with a human nuclear antigen associated with cell proliferation. Int J Cancer. 1983;31:13–20. doi: 10.1002/ijc.2910310104. [DOI] [PubMed] [Google Scholar]
- Golden GT, Smith GG, Ferraro TN, Reyes PF. Rat strain and age differences in kainic acid induced seizures. Epilepsy Res. 1995;20:151–159. doi: 10.1016/0920-1211(94)00079-c. [DOI] [PubMed] [Google Scholar]
- Gottlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG, Heinzel T. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001;20:6969–6978. doi: 10.1093/emboj/20.24.6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould E, Beylin A, Tanapat P, Reeves A, Shors TJ. Learning enhances adult neurogenesis in the hippoampal formation. Nat Neurosci. 1999;2:260–265. doi: 10.1038/6365. [DOI] [PubMed] [Google Scholar]
- Grooms SY, Opitz T, Bennett MV, Zukin RS. Status epilepticus decreases glutamate receptor 2 mRNA and protein expression in hippocampal pyramidal cells before neuronal death. Proc Natl Acad Sci USA. 2000;97:3631–3636. doi: 10.1073/pnas.050586497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurvich N, Tsygankova OM, Meinkoth JL, Klein PS. Histone deacetylase is a target of valproic acid-mediated cellular differentiation. Cancer Res. 2004;64:1079–1086. doi: 10.1158/0008-5472.can-03-0799. [DOI] [PubMed] [Google Scholar]
- Helmstaedter C. Effects of chronic epilepsy on declarative memory systems. Prog Brain Res. 2002;135:439–453. doi: 10.1016/S0079-6123(02)35041-6. [DOI] [PubMed] [Google Scholar]
- Helmstaedter C, Kurthen M, Lux S, Reuber M, Elger CE. Chronic epilepsy and cognition: a longitudinal study in temporal lobe epilepsy. Ann Neurol. 2003;54:425–432. doi: 10.1002/ana.10692. [DOI] [PubMed] [Google Scholar]
- Henry TR. The history of valproate in clinical neuroscience. Psychopharmacol Bull. 2003;37(Suppl 2):5–16. [PubMed] [Google Scholar]
- Holmes GL. Epilepsy in the developing brain: lessons from the laboratory and clinic. Epilepsia. 1997;38:12–30. doi: 10.1111/j.1528-1157.1997.tb01074.x. [DOI] [PubMed] [Google Scholar]
- Holtkamp M, Matzen J, van Landeghem F, Buchheim K, Meierkord H. Transient loss of inhibition precedes spontaneous seizures after experimental status epilepticus. Neurobiol Dis. 2005;19:162–170. doi: 10.1016/j.nbd.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Hsieh J, Nakashima K, Kuwabara T, Mejia E, Gage FH. Histone deacetylase inhibition-mediated neuronal differentiation of multipotent adult neural progenitor cells. Proc Natl Acad Sci USA. 2004;101:16659–16664. doi: 10.1073/pnas.0407643101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Doherty JJ, Dingledine R. Altered histone acetylation at glutamate receptor 2 and brain-derived neurotrophic factor genes is an early event triggered by status epilepticus. J Neurosci. 2002;22:8422–8428. doi: 10.1523/JNEUROSCI.22-19-08422.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubs K, Nanobashvili A, Bonde S, Ekdahl CT, Kokaia Z, Kokaia M, Lindvall O. Environment matters: synaptic properties of neurons born in the epileptic adult brain develop to reduce excitability. Neuron. 2006;52:1047–1059. doi: 10.1016/j.neuron.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Jessberger S, Kempermann G. Adult-born hippocampal neurons mature into activity-dependent responsiveness. Eur J Neurosci. 2003;18:2707–2712. doi: 10.1111/j.1460-9568.2003.02986.x. [DOI] [PubMed] [Google Scholar]
- Jessberger S, Romer B, Babu H, Kempermann G. Seizures induce proliferation and dispersion of doublecortin-positive hippocampal progenitor cells. Exp Neurol. 2005;196:342–351. doi: 10.1016/j.expneurol.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Kee N, Sivalingam S, Boonstra R, Wojtowicz JM. The utility of Ki-67 and BrdU as proliferative markers of adult neurogenesis. J Neurosci Methods. 2002;115:97–105. doi: 10.1016/s0165-0270(02)00007-9. [DOI] [PubMed] [Google Scholar]
- Kempermann G. Why new neurons? Possible functions for adult hippocampal neurogenesis. J Neurosci. 2002;22:635–638. doi: 10.1523/JNEUROSCI.22-03-00635.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempermann G, Gage FH. Experience-dependent regulation of adult hippocampal neurogenesis: effects of long-term stimulation and stimulus withdrawal. Hippocampus. 1999;9:321–332. doi: 10.1002/(SICI)1098-1063(1999)9:3<321::AID-HIPO11>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Kuhn HG, Gage FH. More hippocampal neurons in adult mice living in an enriched environment. Nature. 1997;386:493–495. doi: 10.1038/386493a0. [DOI] [PubMed] [Google Scholar]
- Kuwabara T, Hsieh J, Nakashima K, Taira K, Gage FH. A small modulatory dsRNA specifies the fate of adult neural stem cells. Cell. 2004;116:779–793. doi: 10.1016/s0092-8674(04)00248-x. [DOI] [PubMed] [Google Scholar]
- Majores M, Eils J, Wiestler OD, Becker AJ. Molecular profiling of temporal lobe epilepsy: comparison of data from human tissue samples and animal models. Epilepsy Res. 2004;60:173–178. doi: 10.1016/j.eplepsyres.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Mohapel P, Ekdahl CT, Lindvall O. Status epilepticus severity influences the long-term outcome of neurogenesis in the adult dentate gyrus. Neurobiol Dis. 2004;15:196–205. doi: 10.1016/j.nbd.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Monje ML, Mizumatsu S, Fike JR, Palmer TD. Irradiation induces neural precursor-cell dysfunction. Nat Med. 2002;8:955–962. doi: 10.1038/nm749. [DOI] [PubMed] [Google Scholar]
- Morland C, Boldingh KA, Iversen EG, Hassel B. Valproate is neuroprotective against malonate toxicity in rat striatum: an association with augmentation of high-affinity glutamate uptake. J Cereb Blood Flow Metab. 2004;24:1226–1234. doi: 10.1097/01.WCB.0000138666.25305.A7. [DOI] [PubMed] [Google Scholar]
- Naruse Y, Aoki T, Kojima T, Mori N. Neural restrictive silencer factor recruits mSin3 and histone deacetylase complex to repress neuron-specific target genes. Proc Natl Acad Sci USA. 1999;96:13691–13696. doi: 10.1073/pnas.96.24.13691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet-Wadiche LS, Bromberg DA, Bensen AL, Westbrook GL. Seizures accelerate functional integration of adult-generated granule cells. J Neurosci. 2006;26:4095–4103. doi: 10.1523/JNEUROSCI.5508-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens MJ, Nemeroff CB. Pharmacology of valproate. Psychopharmacol Bull. 2003;37(Suppl 2):17–24. [PubMed] [Google Scholar]
- Palm K, Belluardo N, Metsis M, Timmusk T. Neuronal expression of zinc finger transcription factor REST/NRSF/XBR gene. J Neurosci. 1998;18:1280–1296. doi: 10.1523/JNEUROSCI.18-04-01280.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM. The role of seizure-induced neurogenesis in epileptogenesis and brain repair. Epilepsy Res. 2002;50:179–189. doi: 10.1016/s0920-1211(02)00078-5. [DOI] [PubMed] [Google Scholar]
- Parent JM, Lowenstein DH. Seizure-induced neurogenesis: are more new neurons good for an adult brain? Prog Brain Res. 2002;135:121–131. doi: 10.1016/S0079-6123(02)35012-X. [DOI] [PubMed] [Google Scholar]
- Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci. 1997;17:3727–3738. doi: 10.1523/JNEUROSCI.17-10-03727.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 2001;276:36734–36741. doi: 10.1074/jbc.M101287200. [DOI] [PubMed] [Google Scholar]
- Pollard H, Heron A, Moreau J, Ben-Ari Y, Khrestchatisky M. Alterations of the GluR-B AMPA receptor subunit flip/flop expression in kainate-induced epilepsy and ischemia. Neuroscience. 1993;57:545–554. doi: 10.1016/0306-4522(93)90004-y. [DOI] [PubMed] [Google Scholar]
- Ren M, Leng Y, Jeong M, Leeds PR, Chuang DM. Valproic acid reduces brain damage induced by transient focal cerebral ischemia in rats: potential roles of histone deacetylase inhibition and heat shock protein induction. J Neurochem. 2004;89:1358–1367. doi: 10.1111/j.1471-4159.2004.02406.x. [DOI] [PubMed] [Google Scholar]
- Ribak CE, Tran PH, Spigelman I, Okazaki MM, Nadler JV. Status epilepticus-induced hilar basal dendrites on rodent granule cells contribute to recurrent excitatory circuitry. J Comp Neurol. 2000;428:240–253. doi: 10.1002/1096-9861(20001211)428:2<240::aid-cne4>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Sanchez RM, Koh S, Rio C, Wang C, Lamperti ED, Sharma D, Corfas G, Jensen FE. Decreased glutamate receptor 2 expression and enhanced epileptogenesis in immature rat hippocampus after perinatal hypoxia-induced seizures. J Neurosci. 2001;21:8154–8163. doi: 10.1523/JNEUROSCI.21-20-08154.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, Goodman JH, Sollas AL. Granule-like neurons at the hilar/CA3 border after status epilepticus and their synchrony with area CA3 pyramidal cells: functional implications of seizure-induced neurogenesis. J Neurosci. 2000;20:6144–6158. doi: 10.1523/JNEUROSCI.20-16-06144.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinder AF, Gage FH. A hypothesis about the role of adult neurogenesis in hippocampal function. Physiology (Bethesda) 2004;19:253–261. doi: 10.1152/physiol.00012.2004. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ. Fluoro-Jade: novel fluorochromes for detecting toxicant-induced neuronal degeneration. Toxicol Pathol. 2000;28:91–99. doi: 10.1177/019262330002800111. [DOI] [PubMed] [Google Scholar]
- Schoenherr CJ, Anderson DJ. The neuron-restrictive silencer factor (NRSF): a coordinate repressor of multiple neuron-specific genes. Science. 1995;267:1360–1363. doi: 10.1126/science.7871435. [DOI] [PubMed] [Google Scholar]
- Scott BW, Wang S, Burnham WM, De Boni U, Wojtowicz JM. Kindling-induced neurogenesis in the dentate gyrus of the rat. Neurosci Lett. 1998;248:73–76. doi: 10.1016/s0304-3940(98)00355-3. [DOI] [PubMed] [Google Scholar]
- Shapiro LA, Ribak CE. Newly born dentate granule neurons after pilocarpine-induced epilepsy have hilar basal dendrites with immature synapses. Epilepsy Res. 2006;69:53–66. doi: 10.1016/j.eplepsyres.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Shen S, Li J, Casaccia-Bonnefil P. Histone modifications affect timing of oligodendrocyte progenitor differentiation in the developing rat brain. J Cell Biol. 2005;169:577–589. doi: 10.1083/jcb.200412101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shors TJ, Miesegaes G, Beylin A, Zhao M, Rydel T, Gould E. Neurogenesis in the adult is involved in the formation of trace memories. Nature. 2001;410:372–376. doi: 10.1038/35066584. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Townsend DA, Zhao M, Kozorovitskiy Y, Gould E. Neurogenesis may relate to some but not all types of hippocampal-dependent learning. Hippocampus. 2002;12:578–584. doi: 10.1002/hipo.10103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder JS, Hong NS, McDonald RJ, Wojtowicz JM. A role for adult neurogenesis in spatial long-term memory. Neuroscience. 2005;130:843–852. doi: 10.1016/j.neuroscience.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Stafstrom CE, Chronopoulos A, Thurber S, Thompson JL, Holmes GL. Age-dependent cognitive and behavioral deficits after kainic acid seizures. Epilepsia. 1993;34:420–432. doi: 10.1111/j.1528-1157.1993.tb02582.x. [DOI] [PubMed] [Google Scholar]
- Stefan H, Pauli E. Progressive cognitive decline in epilepsy: an indication of ongoing plasticity. Prog Brain Res. 2002;135:409–417. doi: 10.1016/S0079-6123(02)35038-6. [DOI] [PubMed] [Google Scholar]
- Szot P, White SS, Shen DD, Anderson GD. Valproic acid, but not lamotrigine, suppresses seizure-induced c-fos and c-Jun mRNA expression. Brain Res Mol Brain Res. 2005;135:285–289. doi: 10.1016/j.molbrainres.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Tongiorgi E, Armellin M, Giulianini PG, Bregola G, Zucchini S, Paradiso B, Steward O, Cattaneo A, Simonato M. Brain-derived neurotrophic factor mRNA and protein are targeted to discrete dendritic laminas by events that trigger epileptogenesis. J Neurosci. 2004;24:6842–6852. doi: 10.1523/JNEUROSCI.5471-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Praag H, Kempermann G, Gage FH. Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat Neurosci. 1999;2:266–270. doi: 10.1038/6368. [DOI] [PubMed] [Google Scholar]
- Winkler I, Blotnik S, Shimshoni J, Yagen B, Devor M, Bialer M. Efficacy of antiepileptic isomers of valproic acid and valpromide in a rat model of neuropathic pain. Br J Pharmacol. 2005;146:198–208. doi: 10.1038/sj.bjp.0706310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafra F, Hengerer B, Leibrock J, Thoenen H, Lindholm D. Activity dependent regulation of BDNF and NGF mRNAs in the rat hippocampus is mediated by non-NMDA glutamate receptors. EMBO J. 1990;9:3545–3550. doi: 10.1002/j.1460-2075.1990.tb07564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]