

Abstract
The vesicle priming protein Munc13-1 is regulated by diacylglycerol (DAG) and is therefore hypothesized to play a role in the control of neurotransmitter release by phospholipase C (PLC)-coupled receptors. We combined voltage-clamp recordings of voltage-gated Ca2+ channels (VGCCs) and high-resolution capacitance measurements to investigate the mechanism of receptor-mediated modulation of exocytosis in bovine chromaffin cells. Activation of endogenous H1 Gq-protein-coupled receptors (GqPCRs) by histamine potentiated stimulus-coupled secretion despite concurrently inhibiting Ca2+ influx through VGCCs. Histamine increased the size of the readily releasable pool of vesicles and in particular a subpool of fusion-competent vesicles localized in close proximity to VGCCs. Pharmacological characterization showed that potentiation of exocytosis depended on the activation of PLC but not protein kinase C. Overexpression of wild-type Munc13-1 by adenoviral infection had no effect on histamine-induced potentiation per se, whereas DAG-insensitive Munc13-1H567K completely abolished it.
This is the first endogenous mammalian GqPCR signaling pathway identified that engages Munc13-1 to increase stimulus-coupled secretion by recruiting vesicles to the immediately releasable pool. GqPCRs are therefore able to control exocytosis at the level of SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) complex formation to produce rapid, short-term potentiation of the secretory output of neurons and endocrine cells.
Keywords: Munc13-1, chromaffin, exocytosis, GPCR, phospholipase C, calcium channels
Introduction
G-protein-coupled receptors (GPCRs) modulate Ca2+-dependent exocytosis of neurotransmitters from synaptic vesicles as well as hormone and peptide release from large dense core vesicles (LDCVs) as part of a highly regulated information processing system. GPCRs that couple to Gα subunits of the Gq/11 type are known in particular for their potentiating effect on exocytosis, although the molecular mechanism is often unidentified (Majewski and Iannazzo, 1998; Gromada et al., 1999; Teschemacher and Seward, 2000). GqPCRs activate phospholipase C-β (PLCβ) and generate the second messenger inositol 1,4,5-trisphosphate (IP3) and 1,2-diacyl-sn-glycerol (DAG) by breakdown of phosphatidylinositol 4,5-bisphosphate (PIP2) (Smrcka et al., 1991). IP3 mobilizes Ca2+ from internal stores elevating resting cytoplasmic Ca2+ activity ([Ca2+]cy). Ca2+ together with DAG can then activate protein kinase C (PKC). PKC plays an important role in regulation of exocytosis (Majewski and Iannazzo, 1998). It is known to phosphorylate proteins involved in vesicle docking, priming, and fusion (Turner et al., 1999). Phorbol esters (PEs), which mimic the action of DAG, have been widely used to study the role of PKC in modulation of exocytosis.
These results are under debate since the recent discovery of Munc13 proteins. Initially, three homologs of the Caenorhabditis elegans unc-13 gene product, Munc13-1, -2, and -3, were found in the mammalian brain (Brose et al., 1995), and Munc13-1 was identified as a PE-binding protein that primes synaptic vesicles for exocytosis in neurons (Betz et al., 1998). A more general role for Munc13-1 in secretory cells is now emerging (Ashery et al., 2000; Sheu et al., 2003; Kang et al., 2006). Munc13-1 is thought to unfold and activate syntaxin by displacing Munc18, thereby promoting soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex formation and hence vesicle priming (Brose et al., 2004). Munc13-1, like PKC, contains the DAG/PE binding C1 domain, and a single point mutation in C1 (H567K) renders it DAG-insensitive (Betz et al., 1998). Expression of Munc13-1H567K nearly completely abolishes PE-dependent facilitation of synaptic transmission, which was originally attributed to PKC (Betz et al., 1998; Rhee et al., 2002). Moreover, mutant mice expressing DAG-insensitive Munc13-1H567K die shortly after birth, emphasizing the important role of Munc13-1 in DAG signaling in the nervous system. Unlike in C. elegans (Lackner et al., 1999), the endogenous mammalian neurotransmitter systems and receptor signaling pathways that use Munc13-1 to modulate exocytosis remain unknown. It is commonly believed, although never investigated directly, that like PKC, Munc13-1 may be regulated by GqPCRs. We have tested this hypothesis directly with the endogenous H1 histamine receptor in neurosecretory chromaffin cells, a well established model system for the study of GPCR-mediated modulation of exocytosis (von Ruden and Neher, 1993; Teschemacher and Seward, 2000; Marley, 2003; Chen et al., 2005). Voltage-clamp recordings were combined with membrane capacitance measurements to examine stimulus-coupled secretion under GqPCR modulation. We show that H1 histamine receptor activation potentiated exocytosis by increasing the size of the readily releasable pool (RRP) of vesicles and that this potentiation depended on the DAG sensitivity of Munc13-1. Munc13-1 therefore is an effective interface allowing PLC-coupled DAG-dependent signaling pathways to directly modulate secretory output.
Materials and Methods
DNA and adenoviral construction.
Full-length cDNA encoding Munc13-1 tagged with enhanced green fluorescent protein (EGFP) at the C terminus (Munc13-1–EGFP) (for details, see Ashery et al., 1999) was supplied by Nils Brose (MPI for Experimental Medicine, Goettingen, Germany). The diacylglycerol binding deficient Munc13-1H567K–EGFP was created according to Betz et al. (1998) using the Stratagene (La Jolla, CA) QuikChangeXL Site-Directed Mutagenesis kit (forward oligo, 5′-CTGCACCACGCCGAAGAACTTCGAGGTGTGG-3′, and reverse oligo, 3′-CCACACCTCGAAGTTCTTCGGCGTGGTGCAG-5′). Successful mutation was confirmed by DNA sequencing. Adenovirus encoding Munc13-1–EGFP and Munc13-1H567K–EGFP were constructed using BD Biosciences (Mountain View, CA) Adeno-X Expression System 2 per protocol. Viral titers were determined on HEK293 cells using the end point dilution assay. The titer for Munc13-1–EGFP was 1.5 × 108 pfu/ml and was 3 × 106 pfu/ml for Munc13-1H567K–EGFP.
Cell culture and infection.
Chromaffin cells were isolated from adult bovine adrenal glands as described previously (Teschemacher and Seward, 2000). Freshly isolated cells were cultured in DMEM (catalog #D6171; Sigma, St. Louis, MO) supplemented with 10% fetal calf serum (Invitrogen, San Diego, CA), 2 mm GlutaMax (Invitrogen), 0.1 mg/ml penicillin, 0.1 mg/ml streptomycin, 0.05 mg/ml gentamicin, 0.01 mm 5-fluoro-2′-deoxyuridine, 0.015 mm cytosine arabinoside (all from Sigma) while being plated on glass coverslips coated with BD Matrigel Matrix (VWR Scientific, West Chester, PA) at a density of 6 × 105 cells/cm2. One-half of the medium was changed after 18 h and cells were infected by addition of 1.5–50 μl of viral stock of Munc13-1–EGFP or Munc13-1H567K–EGFP to the culture medium. This resulted in a transfection efficiency of 30–50%. Possible toxic effects of adenoviral infection were assessed in control experiments using 1–4 μl of EGFP adenovirus (provided by T. Herbert, University of Leicester, Leicester, UK), CaMWT/EGFP or CaM1234/EGFP adenovirus (CaM sequence in EGFP-pIRES vector for coexpression; provided by D. Yue, Johns Hopkins University School of Medicine, Baltimore, MD). Those virus stocks had a titer of 6 × 1010 pfu/ml resulting in a transfection efficiency >70%. Infected cells were identified and classified by their EGFP fluorescence. Fluorescence was detected using 488 nm excitation provided by a monochromator (TILL Photonics, Gräfelfing, Germany). The intensity of EGFP emission from individual chromaffin cells was measured with a photomultiplier tube coupled to a variable aperture acquisition window (TILL Photonics). The fluorescence intensity was correlated to the cell size (brightness density, in volts per picofarad), and cells were classified as dim when brightness density was <0.05 V/pF.
Electrophysiology.
Electrophysiological data were acquired at room temperature by using a combination of voltage-clamp and membrane capacitance measurements as described previously (Powell et al., 2000). Cells were continuously superfused (1 ml/min) with external control solution containing the following (in mm): 140 NaCl, 2 KCl, 5 NaHCO3, 1 MgCl2, 2.5 CaCl2, 10 glucose, 10 HEPES, pH 7.25 (NaOH); osmolarity, ∼310 mOsm. Experiments were performed using the perforated patch-clamp configuration to minimize rundown of calcium currents and exocytosis, and to ensure a physiological regulation of [Ca2+]cy and Ca2+-dependent signaling pathways (Seward and Nowycky, 1996; Seward et al., 1996). For this, fire-polished borosilicate glass electrodes (Dow Corning, Midland, MI) coated with Sylgard 184 (Dow Corning) with a resistance of 1–2 MΩ were filled with internal solution containing the following (in mm): 140 Cs-glutamate (Calbiochem, La Jolla, CA; Merck Biosciences, Darmstadt, Germany), 10 HEPES, 8.5 NaCl, 0.3 BAPTA (Invitrogen), and 150–250 μg/ml gramicidin D (Sigma), pH 7.3 (CsOH; ICN Biomedicals, Aurora, OH); osmolarity, ∼300 mOsm. Experiments were started once the series resistance was <15 MΩ and compensated to 70% electronically using a patch-clamp amplifier (Axopatch 200B; Molecular Devices, Foster City, CA). Voltage protocol generation, data acquisition, and off-line data analysis were performed using custom-made software (kindly provided by Dr. A. P. Fox, University of Chicago, Chicago, IL). Changes in membrane capacitance (ΔC m) were measured using the software-based phase-tracking method relative to a 100 fF calibration signal (Seward et al., 1995). Capacitance measurements were interrupted to stimulate cells with depolarizing voltage steps from a holding potential of −80 mV to a test potential of +20 or +14 mV. No corrections were made for liquid junction potentials. I Ca was not leak subtracted and only cells with a leak current <15 pA were included in the analysis. Peak I Ca (in picoamperes) was detected and calcium entry was quantified by integration. The left integration limit was set at 3 ms into the voltage pulse to exclude Na+ current, the right limit was set to exclude tail current. Stimulus-evoked synchronous release was measured as ΔC m 40 ms after the end of the stimulus (Teschemacher and Seward, 2000); stimulus-independent asynchronous release 1.5 s later. Each cell was stimulated three to four times before addition of histamine. Basal [Ca2+]cy before application of histamine was not remarkably increased (3.5 ± 2.3%; n = 11) compared with [Ca2+]cy before repetitive stimulation. The exocytotic efficiency (ΔC m/∫Ca2+ influx) and the Ca2+ influx before application of histamine were averaged and normalized to 100%, respectively.
Double pulse experiments according to Gillis et al. (1996) and Voets et al. (1999) were performed to measure the RRP. Cells were stimulated every 35 s with two consecutive depolarizing pulses from −80 to +20 mV (first pulse) or +14 mV (second pulse) of 100 ms duration (interpulse interval, 100 ms). The depolarizing potentials were selected to give rise to quantitatively equivalent maximal I Ca counterbalancing calcium-dependent inactivation between the first and second stimulus. The RRP size was calculated from the evoked capacitance changes [ΔC m1 + ΔC m2/1 − R 2 (with R = ΔC m2/ΔC m1)]. Only cells with R < 0.7 were analyzed because pool size depletion is essential for accurate pool size estimation (Voets et al., 1999). Under control conditions, 50% of the tested cells (n = 10) showed pool size depletion (R = 0.57 ± 0.05; n = 5) and the R value was not significantly changed after addition of histamine (R = 0.41 ± 0.04; p = 0.089).
To further characterize the different pools of release-competent vesicles, cells were stimulated with a train of 6 × 10 ms depolarizations (390 ms interpulse interval) followed by 4 × 100 ms depolarizations (300 ms interpulse interval) according to Voets et al. (1999). The sum of ΔC m induced by the first six stimuli (ΔC mstim1–6) represents secretion from the immediately releasable vesicle pool (IRP), whereas the sum of ΔC m induced by the following 4 × 100 ms (ΔC mstim7–10) represents secretion of the remaining readily releasable plus part of the slowly releasable pool (SRP) of secretory vesicles (RRP/SRP).
All data are expressed as mean ± SEM and were collected in each case from at least three different batches of cultured cells. Statistical significance was determined with Student’s paired or unpaired t test (SigmaPlot).
Drug application.
All drugs were dissolved in H2O or DMSO to obtain ≥1000-fold stock solutions and stored as aliquots at +4 or −20°C. Drugs were diluted into the external solution immediately before the experiment. Calphostin C, 1-[6-((17β-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl]-1H-pyrrole-2,3-dione (U-73122), and 1-[6-((17β-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl]-2,5-pyrrolidine-dione (U-73443) were each applied for at least 5 min before addition of histamine. Bisindolylmaleimide I (Bis) was applied for 20–60 min before histamine. Histamine was purchased from Sigma; PMA, bisindolylmaleimide I hydrochloride, U-73122, U-73443, and calphostin C were from Calbiochem.
[Ca2+]cyt measurements.
Chromaffin cells were loaded at 37°C with 5 μm fura-2 AM (Invitrogen) for 30 min, and then washed for 15 min. Cytoplasmic Ca2+ activity ([Ca2+]cy) was ratiometrically measured as described by Teschemacher and Seward (2000) and displayed as the ratio of fluorescence intensity at 340/380 nm (R 340/380).
Results
Histamine potentiates stimulus-coupled exocytosis by increasing the size of the readily releasable pool of vesicles
Cells were voltage-clamped in the perforated-patch configuration and Ca2+-dependent exocytosis was evoked every 25 s with brief depolarizing pulses. Vesicle fusion was monitored from changes in membrane capacitance (ΔC m) and Ca2+ entry was measured by recording voltage-gated Ca2+ currents (I Ca). At the start of each experiment, the pulse duration was adjusted (25–100 ms) to elicit reproducible ΔC m of ∼100 fF. This low-frequency stimulus protocol resembles resting “breed and feed” conditions rather than the high-frequency stress stimulation associated with activation of the “fight or flight” response (Fulop et al., 2005) and avoids depletion of release competent vesicles and activity-dependent increases in vesicle priming caused by elevated basal [Ca2+]cy and PKC activation and changes in the exocytotic efficiency (Engisch et al., 1997; Smith et al., 1998) and is therefore suitable for studying the mechanism underlying GqPCR-mediated regulation of exocytosis (Teschemacher and Seward, 2000).
Activation of H1 receptors by superfusion with histamine inhibited peak I Ca (Fig. 1 A) and significantly reduced Ca2+ influx by 20% (Fig. 1 B). Histamine had no effect on the holding current used to clamp the cells at −80 mV (I Hold was −15 ± 1 pA before and −14 ± 2 pA in the presence of histamine; n = 9). The reduction of I Ca by histamine was attenuated by a short depolarizing prepulse to +100 mV (data not shown), a hallmark of G-protein modulation of voltage-gated calcium channels (VGCCs) (Dolphin, 2003). Despite the reduction in Ca2+ entry, histamine simultaneously caused a dramatic increase in depolarization-coupled ΔC m (Fig. 1 A), corresponding to a 400% increase in exocytotic efficiency (calculated ΔC m/∫Ca2+ influx) (Fig. 1 C). The effects of histamine on I Ca and secretion reversed during washout of the agonist.
Figure 1.

Histamine potentiates stimulus-coupled exocytosis in bovine chromaffin cells. A, Representative traces of I Ca (top) evoked by depolarizations before (ctrl) and during superfusion of histamine (100 μm). Corresponding membrane capacitance (ΔC m) changes (bottom) measured in the same cell in response to the Ca2+ entry evoked by the depolarizations. Capacitance detection was interrupted during depolarization (gap in ΔC m trace), and ΔC m was quantified relative to a 100 fF calibration step (indicated by ♦). B, C, Mean data ± SEM from n = 13 cells summarizing the effect of histamine (100 μm) on Ca2+ entry (B) and exocytotic efficiency (C). Data were normalized to control measurements made before application of histamine. The effects of histamine were completely reversed during washout of the agonist. **p < 0.01.
A potentiation in exocytotic efficiency could result either from an increase in the number of primed vesicles available for release or from a shift in the Ca2+ dependence of exocytosis. To distinguish between these possibilities, we adopted the double pulse protocol described by Gillis et al. (1996) and Voets et al. (1999) for measuring the size of the RRP in chromaffin cells. As shown in Figure 2 A, histamine caused a significant increase in the size of the RRP from 290 ± 85 to 805 ± 214 fF (p = 0.037) consistent with the view that activation of the receptor increased vesicle priming.
Figure 2.

Histamine increases the size of the RRP of vesicles. A, Sample traces of I Ca (top) evoked by the double pulse protocol illustrated used to measure the RRP (100 ms interpulse interval). Corresponding ΔC m1 and ΔC m2 (bottom) measured in the same cell before and during application of histamine (100 μm). B, Sample traces of ΔC m evoked by a train of 6 × 10 ms (390 ms interpulse interval) followed by 4 × 100 ms depolarizations (300 ms interpulse interval) measured in the same cell before and during application of histamine (100 μm). C, Mean ± SEM from n = 10 cells summarizing the effect of histamine on the exocytotic efficiency of a train of 6 × 10 ms followed by 4 × 100 ms depolarizations. The exocytotic efficiency was calculated as ΣΔC m/Σ∫Ca2+ for stimulus 1–6 and stimulus 7–10. **p < 0.01.
The RRP contains a subpool of vesicles that are released by short depolarizations and hence thought to be located close to VGCC (Voets et al., 1999). We assessed the contribution of this IRP to the effects of the agonist by stimulating cells with a train of six 10 ms depolarizations. Those stimuli were followed by four 100 ms depolarizations to elicit secretion of the remaining RRP and probably of a small fraction of the SRP (Voets et al., 1999, 2001). As illustrated in Figure 2 B, histamine caused a potentiation of depolarization-coupled ΔC m in response to the short stimuli (stim 1–6) but not the longer depolarizations (stim 7–10) and the exocytotic efficiency was significantly increased (Fig. 2 C) during stim 1–6. It would therefore appear that activation of the H1 receptor specifically and exclusively increases priming of vesicles localized close to VGCC to increase stimulus-coupled secretion.
Potentiation of exocytosis by histamine depends on activation of PLC but not PKC
We next examined the signaling pathway used by histamine to modulate exocytosis. Inhibition of PLC with U-73122 (1 μm) blocked histamine-induced potentiation of exocytotic efficiency (130 ± 20%; n = 5). The inactive analog U-73443 (1 μm) was without effect (exocytotic efficiency in presence of histamine plus U-73443 was 560 ± 70%; n = 4), suggesting that the inhibitor was acting specifically. Activated PLC generates the second messengers IP3 and DAG. IP3-induced Ca2+ release from stores is known to be required for histamine potentiation of secretion (von Ruden and Neher, 1993; Pan and Fox, 2000); the role of DAG-regulated proteins such as PKC is unclear (Zhang et al., 1995; Donald et al., 2002). The functionality of PKC and of alternative nonkinase DAG receptors is reported to be severely impaired by the C1-domain antagonist calphostin C (Brose and Rosenmund, 2002). We found that pretreatment of the cells with calphostin C blocked the histamine-induced increase in exocytotic efficiency (Fig. 3 A). In contrast, Bis, a specific inhibitor of PKC that acts at the ATP binding site, was completely ineffective (Fig. 3 A). VGCCs are known targets for PKC and activation of PKC by phorbol esters is reported to alter the size and kinetics of I Ca (Yang and Tsien, 1993; Zhu and Ikeda, 1994); we therefore checked whether the inhibition of PKC by Bis was effective in our cells by examining modulation of I Ca by PMA. In the same cells that Bis was ineffective at blocking histamine-induced potentiation of secretion, it was effective in preventing PMA-induced changes in I Ca (Fig. 3 B), demonstrating that the drug was active. Together, our pharmacology data indicate that histamine-induced potentiation of stimulus-coupled exocytosis depends on the activation of PLC and a downstream DAG receptor other than PKC. These results are in agreement with previous biochemical studies on histamine-evoked catecholamine release in unclamped chromaffin cells in which similar differences between the effectiveness of inhibitors that act at the kinase domain (Donald et al., 2002) versus DAG regulatory domain were also observed (Zhang et al., 1995).
Figure 3.
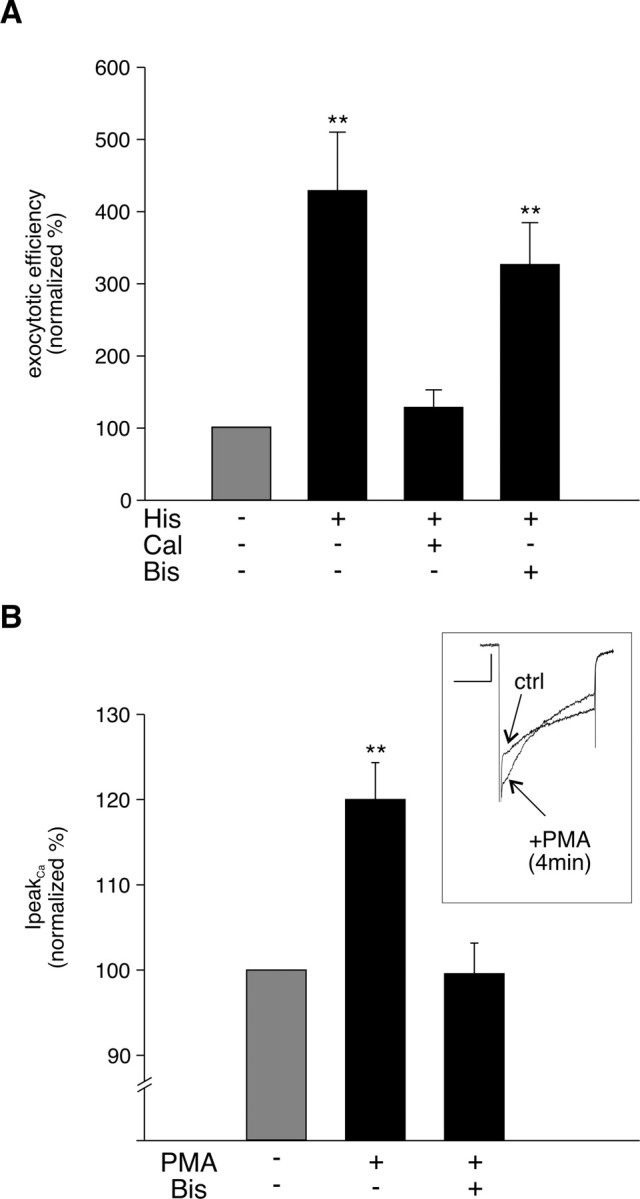
PKC is not involved in histamine potentiation. A, Mean data of normalized exocytotic efficiency measured in the presence of histamine (100 μm; n = 19), calphostin C (Cal) (100 nm) plus histamine (100 μm; n = 6), and Bis (0.5–1 μm) plus histamine (n = 7). B, Inset, Overlaid sample traces of I Ca before (ctrl) and 4 min into PMA (250 nm) application. Bar graph, Mean ± SEM percentage change in peak I Ca by PMA (250 nm) in untreated (n = 6) and Bis (0.5–1 μm)-treated cells (n = 7). Note that the experiment represented in B was performed on the same cells represented in A and confirms the effectiveness of the Bis pretreatment. **p < 0.01.
Adenoviral infection had no detrimental effect on stimulus-coupled exocytosis and H1 receptor signaling
To investigate a possible role of the DAG-regulated priming protein Munc13-1 in GqPCR modulation of exocytosis, we wanted to use a dominant-negative strategy that would specifically interfere with the activation of this protein by the receptor-coupled signaling pathway in otherwise uncompromised cells. For this, the DAG-insensitive mutant Munc13-1H567K was expressed in chromaffin cells using adenoviral infection. Experiments were done in parallel with EGFP-tagged wild-type Munc13-1 and EGFP. To assess possible negative effects of adenoviral infection itself on cell function, noninfected and culture-matched chromaffin cells expressing the reporter protein EGFP alone were stimulated with different depolarizing protocols and evoked stimulus-coupled secretion assessed (Fig. 4 A–C). High expression levels of EGFP are known to reduce I Ca in chromaffin cells (Thiagarajan et al., 2005), and therefore we restricted our experiments to cells with low EGFP fluorescence (see Materials and Methods). In contrast to observations made after Semliki Forest virus infection of chromaffin cells (Ashery et al., 1999; Duncan et al., 1999; Pan et al., 2002), adenoviral infection had no significant effect on stimulus-coupled secretion (Fig. 4 A) nor on poststimulus “asynchronous” release (Fig. 4 B). Figure 4 C shows that secretion from the different vesicle pools was also not affected by adenoviral infection. Ca2+ handling and H1 receptor signaling were also unaltered in adenoviral infected cells compared with noninfected cells (Fig. 4 D,E). Basal Ca2+ levels, peak I Ca, and histamine-induced Ca2+ release from stores were not significantly different in uninfected compared with adenoviral infected cells. Basal R 340/380 in noninfected cells (n = 4) was 1.7 ± 0.2 compared with 1.5 ± 0.2 (n = 13) in infected cells. R 340/380 increased after I Ca to 4.0 ± 0.4 in noninfected and to 3.9 ± 0.3 in infected cells and on His-induced Ca2+ release to 2.4 ± 0.3 in noninfected and to 2.8 ± 0.4 in infected cells. In uninfected cells, R 340/380 returned from its peak after store release to the basal level in 168 ± 18 s (n = 3) compared with 156 ± 1 s in infected cells (n = 8). Adenoviral infection did neither change the probability to observe histamine potentiation of stimulus-coupled exocytosis [85% in infected EGFP-expressing cells (n = 21) compared with 80% in uninfected control cells (n = 11)] nor the size of the response (Fig. 4 E). In summary, adenoviral infection proved to be a gentle and effective method for exogenous gene expression without affecting receptor signaling pathways and Ca2+ handling. By restricting our experiments to cells with low EGFP fluorescence, we not only avoided reduction in I Ca but also the reported increase in asynchronous release attributed to the expression of viral genes (Thiagarajan et al., 2005).
Figure 4.

Exogenous gene expression in bovine chromaffin cells using adenovirus. A, Mean ± SEM peak I Ca (top) and corresponding membrane capacitance changes (ΔC m) (bottom) evoked by a 200 ms depolarizing pulse from a holding potential of −80 to +20 mV in noninfected cells (n = 15) and culture-matched cells infected with adenovirus for EGFP expression (n = 11). B, Mean ± SEM stimulus-coupled synchronous (s) and poststimulus asynchronous (as) ΔC m in noninfected (black filled bar; n = 9) and culture-matched EGFP-expressing cells (gray filled bar; n = 12) evoked by a train of 10 depolarizations (550 ms interpulse interval). C, Mean ± SEM of ΔC m evoked by stimulus 1–6 (secretion from the IRP) and stimulus 7–10 (remaining RRP and part of SRP) in noninfected (black filled bar; n = 5) and culture-matched EGFP-expressing cells (gray filled bar; n = 5). Cells were stimulated by a train of 6 × 10 ms (390 ms interpulse interval) and 4 × 100 ms depolarizing pulses (300 ms interpulse interval). D, Sample traces of fura-2 measurements (R 340/380; top) and corresponding ΔC m (bottom) evoked by 50 ms depolarizing pulses from −80 to +20 mV (indicated by downward arrows) recorded in a cell expressing CaM1234/EGFP. The dashed line indicates basal Ca2+ levels. A 100 fF calibration step (indicated by ♦) is shown early in the C m trace. Histamine (100 μm) was added by superfusion as indicated. Increase in C m (open arrow) on Ca2+ release from internal stores (filled arrow) was rarely observed (3 of 23 noninfected and infected cells). E, Mean ± SEM percentage change in exocytotic efficiency induced by histamine in noninfected cells (n = 8) compared with adenoviral infected cells expressing EGFP (n = 11). **p < 0.01.
The ability of Munc13-1 to sense DAG is essential for histamine-induced potentiation of stimulus-coupled secretion
Bovine chromaffin cells express endogenous Munc13-1 and overexpression of wild-type or DAG-insensitive mutant Munc13-1H567K increases exocytosis (Ashery et al., 2000). In agreement with the work of Ashery et al., we found that cells overexpressing Munc13-1 or Munc13-1H567K after adenovirus infection also responded to a depolarizing stimulus (200 ms) with a significant increase in stimulus-coupled secretion and exocytotic efficiency compared with uninfected control cells (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). This confirms that overexpression of a functionally active Munc13-1 alone is sufficient to increase the size of the RRP of vesicles under basal conditions and that the DAG-sensing ability of Munc13-1 is important for regulating the protein rather than performing its priming function (Stevens et al. 2005).
As shown in Figure 5 A, cells overexpressing Munc13-1 responded to histamine with a decrease in I Ca and potentiation of ΔC m. In contrast, expression of Munc13-1H567K completely abolished histamine potentation of ΔC m but not the inhibition in I Ca (Fig. 5 B). In culture-matched noninfected and Munc13-1-overexpressing cells, histamine caused a significant increase of exocytotic efficiency that was completely blocked when Munc13-1H567K was expressed (Fig. 5 C). In all three groups of cells, I Ca was inhibited to a similar extent after activation of the GqPCR (Fig. 5 D). This indicates that G-protein activation and the initiation of the downstream signaling pathway was still functional in cells expressing Munc13-1H567K. Munc13-1H567K also had no effects on calcium release from internal stores or PMA regulation of I Ca (data not shown). Thus, expression of the DAG-insensitive Munc13-1 H567K selectively abolished the potentiation of exocytosis by histamine without affecting other aspects of receptor signaling.
Figure 5.

Histamine-induced potentiation of exocytosis is abolished in cells expressing DAG-insensitive Munc13-1H567K. A, Sample I Ca (top) and corresponding ΔC m (bottom) evoked in an adenovirus-infected chromaffin cell expressing Munc13-1–EGFP before (ctrl) and during superfusion with histamine (100 μm). B, Sample I Ca (top) and corresponding ΔC m (bottom) evoked in an adenovirus-infected chromaffin cell expressing Munc13-1H567K–EGFP before (ctrl) and during superfusion with histamine. C, Summary of effects of histamine on exocytotic efficiency normalized to (ctrl) for culture-matched, noninfected cells (n = 11), Munc13-1–EGFP-overexpressing (n = 10), and Munc13-1H567K–EGFP-expressing (n = 6) cells. D, Corresponding summary of effects of histamine on normalized Ca2+ entry for the same groups of cells. Error bars indicate SEM. **p < 0.01; *p < 0.05.
Discussion
We used membrane capacitance measurements combined with voltage-clamp recordings to assess directly the effect of agonist and signaling molecules on VGCC function and LDCV priming in chromaffin cells. We show that the DAG signals generated by endogenous H1 GqPCRs activate Munc13-1 to increase the pool of release-competent vesicles resulting in a short-term potentiation of exocytosis.
Histamine is a neurotransmitter as well as a neuromodulator in the nervous and neuroendocrine system (Schwartz et al., 1991). Its effects on catecholamine secretion, PIP2 turnover, and potassium channels in chromaffin cells are well documented (von Ruden and Neher, 1993; Zhang et al., 1995; Pan and Fox, 2000; Marley, 2003). Histamine activates H1 receptors that couple exclusively to the pertussis toxin-insensitive Gαq subunit (Hill et al., 1997). With receptor stimulation, Ca2+ influx is inhibited (Fig. 1) in a voltage-dependent way from a direct interaction of Gβγ subunits with VGCC (Currie and Fox, 2000). This inhibition is likely to involve the Gβ5 subunit, which couples almost exclusively to Gαq and is known to only modestly inhibit VGCC (Garcia et al., 1998; Arnot et al., 2000). Gβγ-mediated inhibition of I Ca is a widespread mechanism to inhibit Ca2+-dependent stimulus-coupled exocytosis (Miller, 1998; Powell et al., 2000). Activation of Gβγ from Gαi/o-coupled receptors may also inhibit exocytosis downstream of Ca2+ entry by affecting the activity of SNARE complexes (Blackmer et al., 2005; Chen et al., 2005; Gerachshenko et al., 2005; Photowala et al., 2006). Here, we find that activation of Gβγ from H1 receptors is predominantly associated with an increase in priming and exocytosis, indicating that any potential direct inhibitory effects of Gβγ on SNAREs are either prevented (Blackmer et al., 2005) or are Gβ isoform specific.
Up to now, receptor-mediated Munc13-1 activation in mammalian systems was widely assumed but still speculative (Brose et al., 2004). Our data are the first to show the longstanding hypothesis that PLCβ-coupled GqPCRs engage Munc13-1 to modulate vesicle priming and hence exocytosis in mammalian cells. At the neuromuscular junction of C. elegans, neurotransmitter release is enhanced by a presynaptic signaling pathway containing Gαq, PLCβ, and UNC-13 (Lackner et al., 1999). Mutation of the DAG-binding site in UNC-13 reduces facilitation, indicating that other DAG-binding proteins like PKC or DAG-activated Ca2+ channels may also contribute to the regulation. In contrast to C. elegans, DAG-insensitive Munc13-1H567K completely abolished the agonist-dependent potentiation of exocytosis in chromaffin cells (Fig. 5). Under our conditions, inhibition of PKC had no detectable effect (Fig. 3). It seems, therefore, that the well characterized effect of PKC on the RRP in chromaffin cells is more important for activity-dependent than for receptor-mediated potentiation of exocytosis (Smith, 1999; Park et al., 2006). We cannot dismiss a possible PKC-mediated modulation of the fusion pore, because this would not necessarily be apparent in C m measurements. A concerted regulation of vesicle priming and fusion by Munc13-1 and PKC may therefore occur under certain conditions. Experiments on unclamped chromaffin cells report that histamine induces phosphorylation of the syntaxin binding protein Munc18 by PKC and that this is correlated with a change in fusion pore kinetics (Barclay et al., 2003; Craig et al., 2003). A recent report suggests the role of PKC in agonist-dependent regulation of exocytosis may well depend on the presence of other mediators and activation of additional G-protein-coupled signaling pathways (Chen et al., 2005).
Ca2+ release from stores is also required for histamine potentiation of exocytosis (Pan and Fox, 2000); the molecular mechanism of this Ca2+ dependence is unknown but may involve the modulation of Munc13-1 activity by calmodulin (Junge et al., 2004) or DOC2 (Groffen et al., 2006). Interestingly, store-dependent Ca2+ influx at the plasma membrane does not appear to be needed for histamine-dependent potentiation of exocytosis because the holding current was not observed to change in the presence of agonist. This is in contrast to results obtained with angiotensin II and may be important in recruitment of additional signaling molecules, such as PKC by AT-1 receptors (Teschemacher and Seward, 2000).
In chromaffin cells, fusion-competent primed vesicles represent only a fraction of vesicles docked at the plasma membrane, suggesting that priming is a limiting process (Ashery et al., 2000). Because chromaffin cells constantly secrete at a low basal activity, limitation of priming may actually play an important physiological role and priming may be strictly regulated to avoid inappropriate and toxic overload of catecholamines in the bloodstream (Aunis and Langley, 1999). Bovine chromaffin cells express Munc13-1 and Munc13-3 (Ashery et al., 2000), and overexpression of Munc13-1 increases stimulus-coupled exocytosis by transferring vesicles from a pool of docked but unprimed vesicles to the RRP (Ashery et al., 2000). Low endogenous levels of Munc13-1 and normal secretion from adrenal slices of Munc13-1 knock-out mice led to the assumption that Munc13-1 is not the essential priming protein for LDCVs (Ashery et al., 2000; Stevens et al., 2005). Our data, however, clearly show that DAG-sensitive Munc13-1 is indispensable for the GqPCR-mediated short-term potentiation of LDCV secretion. The role of Munc13-3 in chromaffin cells is unclear, but one might speculate that, whereas Munc13-1 acts in receptor-mediated priming, Munc13-3 could cover basal priming. Short-term plasticity in hippocampal neurons depends on the predominant expressed Munc13 isoform (Rosenmund et al., 2002). Whereas Munc13-1-driven synapses show depression during high-frequency stimulation, Munc13-2-controlled synapses respond with augmentation. This augmentation is caused by [Ca2+]cy-dependent DAG production probably mediated by PLCδ (Rosenmund et al., 2002; Brose et al., 2004). It was speculated that the ability for depression or augmentation is an intrinsic feature of the respective Munc13 isoform (Rosenmund et al., 2002). We show here that Munc13-1 is necessary for agonist-potentiated exocytosis in chromaffin cells, indicating that a specific modulatory output may arise as a consequence of specific coupling of Munc13 isoforms with PLC isoforms in any given cellular background.
Unlike neurons, chromaffin cells lack molecular defined active zones where exocytosis occurs, but there is mounting evidence for sites of preferred docking and fusion (Schroeder et al., 1994; Oheim et al., 1999; Allersma et al., 2004). These microdomains of release can colocalize with Ca2+ microdomains created on opening of VGCC (Becherer et al., 2003). Not all vesicles within Ca2+ microdomains are released on stimulation, indicating that some vesicles are docked but not primed (Becherer et al., 2003). We previously suggested that during receptor-mediated potentiation of exocytosis, new release sites that had previously been silent become active (Teschemacher and Seward, 2000). It is tempting to speculate that GqPCR-mediated activation of Munc13-1 specifically recruits this pool of docked but unprimed vesicles in close proximity to VGCCs, hence the increase in the IRP observed in the presence of agonist (Fig. 2). Even a brief increase of [Ca2+]cy is sufficient to trigger exocytosis and therefore the size of this pool is an obvious target for plasticity.
There is increasing evidence that receptors, their signaling molecules, and their targets colocalize in signaling microdomains (Delmas et al., 2004; Levitan, 2006). The observed membrane-delimited modulation of the VGCCs by H1 stimulation, the Munc13-1-mediated DAG dependence of vesicle priming, and the specific priming of vesicles close to VGCCs indicates that GqPCRs, VGCCs, and LDCVs including their exocytotic machinery might form signaling microdomains dedicated to secretory plasticity. Under resting conditions, the DAG concentration in the plasma membrane is very low and PLC-mediated DAG production is tightly regulated in a spatial and temporal manner (Luo et al., 2004). Like UNC-13, which accumulates at release sites after receptor activation (Lackner et al., 1999), Munc13-1 could be recruited to signaling microdomains during DAG production, prime the residing vesicles and activate previously silent release sites. Because overexpression of wild-type Munc13-1 did not amplify histamine potentiation (Fig. 5), not Munc13-1 but the total number of signaling microdomains might limit the amount of plasticity that can be achieved in the presence of saturating concentrations of agonist. We propose a model in which Munc13-1 initially acts as a scaffold protein to generate and/or stabilize the signaling microdomains in the close proximity of GqPCR/PLCβ-generated DAG; it then promotes the open configuration of syntaxin molecules localized to these areas of release to trigger SNARE complex formation (Richmond et al., 2001) in these signaling microdomains, ultimately leading to priming of the vesicles docked in close proximity to the GqPCR/Munc13-1/VGCC complex. High-resolution imaging experiments of the signaling molecules and vesicle release sites are underway to test this model.
Footnotes
This work was supported by a project grant from the Wellcome Trust (E.P.S.). We gratefully acknowledge Drs. David Yue, Edward Levitan, and Terry Herbert for their generous gift of adenoviral constructs. We also thank Dr. Nils Brose for the Munc13-1–EGFP cDNA.
References
- Allersma MW, Wang L, Axelrod D, Holz RW. Visualization of regulated exocytosis with a granule-membrane probe using total internal reflection microscopy. Mol Biol Cell. 2004;15:4658–4668. doi: 10.1091/mbc.E04-02-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnot MI, Stotz SC, Jarvis SE, Zamponi GW. Differential modulation of N-type 1B and P/Q-type 1A calcium channels by different G protein subunit isoforms. J Physiol (Lond) 2000;527:203–212. doi: 10.1111/j.1469-7793.2000.00203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashery U, Betz A, Xu T, Brose N, Rettig J. An efficient method for infection of adrenal chromaffin cells using the Semliki Forest virus gene expression system. Eur J Cell Biol. 1999;78:525–532. doi: 10.1016/s0171-9335(99)80017-x. [DOI] [PubMed] [Google Scholar]
- Ashery U, Varoqueaux F, Voets T, Betz A, Thakur P, Koch H, Neher E, Brose N, Rettig J. Munc13-1 acts as a priming factor for large dense-core vesicles in bovine chromaffin cells. EMBO J. 2000;19:3586–3596. doi: 10.1093/emboj/19.14.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aunis D, Langley K. Physiological aspects of exocytosis in chromaffin cells of the adrenal medulla. Acta Physiol Scand. 1999;167:89–97. doi: 10.1046/j.1365-201x.1999.00580.x. [DOI] [PubMed] [Google Scholar]
- Barclay JW, Craig TJ, Fisher RJ, Ciufo LF, Evans GJ, Morgan A, Burgoyne RD. Phosphorylation of Munc18 by protein kinase C regulates the kinetics of exocytosis. J Biol Chem. 2003;278:10538–10545. doi: 10.1074/jbc.M211114200. [DOI] [PubMed] [Google Scholar]
- Becherer U, Moser T, Stuhmer W, Oheim M. Calcium regulates exocytosis at the level of single vesicles. Nat Neurosci. 2003;6:846–853. doi: 10.1038/nn1087. [DOI] [PubMed] [Google Scholar]
- Betz A, Ashery U, Rickmann M, Augustin I, Neher E, Sudhof TC, Rettig J, Brose N. Munc13-1 is a presynaptic phorbol ester receptor that enhances neurotransmitter release. Neuron. 1998;21:123–136. doi: 10.1016/s0896-6273(00)80520-6. [DOI] [PubMed] [Google Scholar]
- Blackmer T, Larsen EC, Bartleson C, Kowalchyk JA, Yoon EJ, Preininger AM, Alford S, Hamm HE, Martin TF. G protein betagamma directly regulates SNARE protein fusion machinery for secretory granule exocytosis. Nat Neurosci. 2005;8:421–425. doi: 10.1038/nn1423. [DOI] [PubMed] [Google Scholar]
- Brose N, Rosenmund C. Move over protein kinase C, you’ve got company: alternative cellular effectors of diacylglycerol and phorbol esters. J Cell Sci. 2002;115:4399–4411. doi: 10.1242/jcs.00122. [DOI] [PubMed] [Google Scholar]
- Brose N, Hofmann K, Hata Y, Sudhof TC. Mammalian homologues of Caenorhabditis elegans unc-13 gene define novel family of C2-domain proteins. J Biol Chem. 1995;270:25273–25280. doi: 10.1074/jbc.270.42.25273. [DOI] [PubMed] [Google Scholar]
- Brose N, Betz A, Wegmeyer H. Divergent and convergent signaling by the diacylglycerol second messenger pathway in mammals. Curr Opin Neurobiol. 2004;14:328–340. doi: 10.1016/j.conb.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Chen XK, Wang LC, Zhou Y, Cai Q, Prakriya M, Duan KL, Sheng ZH, Lingle C, Zhou Z. Activation of GPCRs modulates quantal size in chromaffin cells through Gβγ and PKC. Nat Neurosci. 2005;8:1160–1168. doi: 10.1038/nn1529. [DOI] [PubMed] [Google Scholar]
- Craig TJ, Evans GJ, Morgan A. Physiological regulation of Munc18/nSec1 phosphorylation on serine-313. J Neurochem. 2003;86:1450–1457. doi: 10.1046/j.1471-4159.2003.01955.x. [DOI] [PubMed] [Google Scholar]
- Currie KP, Fox AP. Voltage-dependent, pertussis toxin insensitive inhibition of calcium currents by histamine in bovine adrenal chromaffin cells. J Neurophysiol. 2000;83:1435–1442. doi: 10.1152/jn.2000.83.3.1435. [DOI] [PubMed] [Google Scholar]
- Delmas P, Crest M, Brown DA. Functional organization of PLC signaling microdomains in neurons. Trends Neurosci. 2004;27:41–47. doi: 10.1016/j.tins.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Dolphin AC. G protein modulation of voltage-gated calcium channels. Pharmacol Rev. 2003;55:607–627. doi: 10.1124/pr.55.4.3. [DOI] [PubMed] [Google Scholar]
- Donald AN, Wallace DJ, McKenzie S, Marley PD. Phospholipase C-mediated signalling is not required for histamine-induced catecholamine secretion from bovine chromaffin cells. J Neurochem. 2002;81:1116–1129. doi: 10.1046/j.1471-4159.2002.00915.x. [DOI] [PubMed] [Google Scholar]
- Duncan RR, Don-Wauchope AC, Tapechum S, Shipston MJ, Chow RH, Estibeiro P. High-efficiency Semliki Forest virus-mediated transduction in bovine adrenal chromaffin cells. Biochem J. 1999;342:497–501. [PMC free article] [PubMed] [Google Scholar]
- Engisch KL, Chernevskaya NI, Nowycky MC. Short-term changes in the Ca2+-exocytosis relationship during repetitive pulse protocols in bovine adrenal chromaffin cells. J Neurosci. 1997;17:9010–9025. doi: 10.1523/JNEUROSCI.17-23-09010.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulop T, Radabaugh S, Smith C. Activity-dependent differential transmitter release in mouse adrenal chromaffin cells. J Neurosci. 2005;25:7324–7332. doi: 10.1523/JNEUROSCI.2042-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia DE, Li B, Garcia-Ferreiro RE, Hernandez-Ochoa EO, Yan K, Gautam N, Catterall WA, Mackie K, Hille B. G-protein β-subunit specificity in the fast membrane-delimited inhibition of Ca2+ channels. J Neurosci. 1998;18:9163–9170. doi: 10.1523/JNEUROSCI.18-22-09163.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerachshenko T, Blackmer T, Yoon EJ, Bartleson C, Hamm HE, Alford S. Gβγ acts at the C terminus of SNAP-25 to mediate presynaptic inhibition. Nat Neurosci. 2005;8:597–605. doi: 10.1038/nn1439. [DOI] [PubMed] [Google Scholar]
- Gillis KD, Mossner R, Neher E. Protein kinase C enhances exocytosis from chromaffin cells by increasing the size of the readily releasable pool of secretory granules. Neuron. 1996;16:1209–1220. doi: 10.1016/s0896-6273(00)80147-6. [DOI] [PubMed] [Google Scholar]
- Groffen AJ, Friedrich R, Brian EC, Ashery U, Verhage M. DOC2A and DOC2B are sensors for neuronal activity with unique calcium-dependent and kinetic properties. J Neurochem. 2006;97:818–833. doi: 10.1111/j.1471-4159.2006.03755.x. [DOI] [PubMed] [Google Scholar]
- Gromada J, Hoy M, Renstrom E, Bokvist K, Eliasson L, Gopel S, Rorsman P. CaM kinase II-dependent mobilization of secretory granules underlies acetylcholine-induced stimulation of exocytosis in mouse pancreatic B-cells. J Physiol (Lond) 1999;518:745–759. doi: 10.1111/j.1469-7793.1999.0745p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill SJ, Ganellin CR, Timmerman H, Schwartz JC, Shankley NP, Young JM, Schunack W, Levi R, Haas HL. International Union of Pharmacology. XIII. Classification of histamine receptors. Pharmacol Rev. 1997;49:253–278. [PubMed] [Google Scholar]
- Junge HJ, Rhee JS, Jahn O, Varoqueaux F, Spiess J, Waxham MN, Rosenmund C, Brose N. Calmodulin and Munc13 form a Ca2+ sensor/effector complex that controls short-term synaptic plasticity. Cell. 2004;118:389–401. doi: 10.1016/j.cell.2004.06.029. [DOI] [PubMed] [Google Scholar]
- Kang L, He Z, Xu P, Fan J, Betz A, Brose N, Xu T. Munc13-1 is required for the sustained release of insulin from pancreatic beta cells. Cell Metab. 2006;3:1–6. doi: 10.1016/j.cmet.2006.04.012. [DOI] [PubMed] [Google Scholar]
- Lackner MR, Nurrish SJ, Kaplan JM. Facilitation of synaptic transmission by EGL-30 Gqalpha and EGL-8 PLCbeta: DAG binding to UNC-13 is required to stimulate acetylcholine release. Neuron. 1999;24:335–346. doi: 10.1016/s0896-6273(00)80848-x. [DOI] [PubMed] [Google Scholar]
- Levitan IB. Signaling protein complexes associated with neuronal ion channels. Nat Neurosci. 2006;9:305–310. doi: 10.1038/nn1647. [DOI] [PubMed] [Google Scholar]
- Luo B, Regier DS, Prescott SM, Topham MK. Diacylglycerol kinases. Cell Signal. 2004;16:983–989. doi: 10.1016/j.cellsig.2004.03.016. [DOI] [PubMed] [Google Scholar]
- Majewski H, Iannazzo L. Protein kinase C: a physiological mediator of enhanced transmitter output. Prog Neurobiol. 1998;55:463–475. doi: 10.1016/s0301-0082(98)00017-3. [DOI] [PubMed] [Google Scholar]
- Marley PD. Mechanisms in histamine-mediated secretion from adrenal chromaffin cells. Pharmacol Ther. 2003;98:1–34. doi: 10.1016/s0163-7258(03)00002-0. [DOI] [PubMed] [Google Scholar]
- Miller RJ. Presynaptic receptors. Annu Rev Pharmacol Toxicol. 1998;38:201–227. doi: 10.1146/annurev.pharmtox.38.1.201. [DOI] [PubMed] [Google Scholar]
- Oheim M, Loerke D, Stuhmer W, Chow RH. Multiple stimulation-dependent processes regulate the size of the releasable pool of vesicles. Eur Biophys J. 1999;28:91–101. doi: 10.1007/s002490050188. [DOI] [PubMed] [Google Scholar]
- Pan CY, Fox AP. Rundown of secretion after depletion of intracellular calcium stores in bovine adrenal chromaffin cells. J Neurochem. 2000;75:1132–1139. doi: 10.1046/j.1471-4159.2000.0751132.x. [DOI] [PubMed] [Google Scholar]
- Pan CY, Jeromin A, Lundstrom K, Yoo SH, Roder J, Fox AP. Alterations in exocytosis induced by neuronal Ca2+ sensor-1 in bovine chromaffin cells. J Neurosci. 2002;22:2427–2433. doi: 10.1523/JNEUROSCI.22-07-02427.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park YS, Hur EM, Choi BH, Kwak E, Jun DJ, Park SJ, Kim KT. Involvement of protein kinase C-ε in activity-dependent potentiation of large dense-core vesicle exocytosis in chromaffin cells. J Neurosci. 2006;26:8999–9005. doi: 10.1523/JNEUROSCI.2828-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Photowala H, Blackmer T, Schwartz E, Hamm HE, Alford S. G protein βγ-subunits activated by serotonin mediate presynaptic inhibition by regulating vesicle fusion properties. Proc Natl Acad Sci USA. 2006;103:4281–4286. doi: 10.1073/pnas.0600509103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell AD, Teschemacher AG, Seward EP. P2Y purinoceptors inhibit exocytosis in adrenal chromaffin cells via modulation of voltage-operated calcium channels. J Neurosci. 2000;20:606–616. doi: 10.1523/JNEUROSCI.20-02-00606.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee JS, Betz A, Pyott S, Reim K, Varoqueaux F, Augustin I, Hesse D, Sudhof TC, Takahashi M, Rosenmund C, Brose N. Beta phorbol ester- and diacylglycerol-induced augmentation of transmitter release is mediated by Munc13s and not by PKCs. Cell. 2002;108:121–133. doi: 10.1016/s0092-8674(01)00635-3. [DOI] [PubMed] [Google Scholar]
- Richmond JE, Weimer RM, Jorgensen EM. An open form of syntaxin bypasses the requirement for UNC-13 in vesicle priming. Nature. 2001;412:338–341. doi: 10.1038/35085583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Sigler A, Augustin I, Reim K, Brose N, Rhee JS. Differential control of vesicle priming and short-term plasticity by Munc13 isoforms. Neuron. 2002;33:411–424. doi: 10.1016/s0896-6273(02)00568-8. [DOI] [PubMed] [Google Scholar]
- Schroeder TJ, Jankowski JA, Senyshyn J, Holz RW, Wightman RM. Zones of exocytotic release on bovine adrenal medullary cells in culture. J Biol Chem. 1994;269:17215–17220. [PubMed] [Google Scholar]
- Schwartz JC, Arrang JM, Garbarg M, Pollard H, Ruat M. Histaminergic transmission in the mammalian brain. Physiol Rev. 1991;71:1–51. doi: 10.1152/physrev.1991.71.1.1. [DOI] [PubMed] [Google Scholar]
- Seward EP, Nowycky MC. Kinetics of stimulus-coupled secretion in dialyzed bovine chromaffin cells in response to trains of depolarizing pulses. J Neurosci. 1996;16:553–562. doi: 10.1523/JNEUROSCI.16-02-00553.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seward EP, Chernevskaya NI, Nowycky MC. Exocytosis in peptidergic nerve terminals exhibits two calcium-sensitive phases during pulsatile calcium entry. J Neurosci. 1995;15:3390–3399. doi: 10.1523/JNEUROSCI.15-05-03390.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seward EP, Chernevskaya NI, Nowycky MC. Ba2+ ions evoke two kinetically distinct patterns of exocytosis in chromaffin cells, but not neurohypophysial nerve terminals. J Neurosci. 1996;16:1370–1379. doi: 10.1523/JNEUROSCI.16-04-01370.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu L, Pasyk EA, Ji J, Huang X, Gao X, Varoqueaux F, Brose N, Gaisano HY. Regulation of insulin exocytosis by Munc13-1. J Biol Chem. 2003;278:27556–27563. doi: 10.1074/jbc.M303203200. [DOI] [PubMed] [Google Scholar]
- Smith C. A persistent activity-dependent facilitation in chromaffin cells is caused by Ca2+ activation of protein kinase C. J Neurosci. 1999;19:589–598. doi: 10.1523/JNEUROSCI.19-02-00589.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C, Moser T, Xu T, Neher E. Cytosolic Ca2+ acts by two separate pathways to modulate the supply of release-competent vesicles in chromaffin cells. Neuron. 1998;20:1243–1253. doi: 10.1016/s0896-6273(00)80504-8. [DOI] [PubMed] [Google Scholar]
- Smrcka AV, Hepler JR, Brown KO, Sternweis PC. Regulation of polyphosphoinositide-specific phospholipase C activity by purified Gq . Science. 1991;251:804–807. doi: 10.1126/science.1846707. [DOI] [PubMed] [Google Scholar]
- Stevens DR, Wu ZX, Matti U, Junge HJ, Schirra C, Becherer U, Wojcik SM, Brose N, Rettig J. Identification of the minimal protein domain required for priming activity of Munc13-1. Curr Biol. 2005;15:2243–2248. doi: 10.1016/j.cub.2005.10.055. [DOI] [PubMed] [Google Scholar]
- Teschemacher AG, Seward EP. Bidirectional modulation of exocytosis by Angiontensin II involves multiple G-protein-regulated transduction pathways in adrenal chromaffin cells. J Neurosci. 2000;20:4776–4785. doi: 10.1523/JNEUROSCI.20-13-04776.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagarajan R, Wilhelm J, Tewolde T, Li Y, Rich MM, Engisch KL. Enhancement of asynchronous and train-evoked exocytosis in bovine adrenal chromaffin cells infected with a replication deficient adenovirus. J Neurophysiol. 2005;94:3278–3291. doi: 10.1152/jn.00336.2005. [DOI] [PubMed] [Google Scholar]
- Turner KM, Burgoyne RD, Morgan A. Protein phosphorylation and the regulation of synaptic membrane traffic. Trends Neurosci. 1999;22:459–464. doi: 10.1016/s0166-2236(99)01436-8. [DOI] [PubMed] [Google Scholar]
- Voets T, Neher E, Moser T. Mechanisms underlying phasic and sustained secretion in chromaffin cells from mouse adrenal slices. Neuron. 1999;23:607–615. doi: 10.1016/s0896-6273(00)80812-0. [DOI] [PubMed] [Google Scholar]
- Voets T, Moser T, Lund PE, Chow RH, Geppert M, Sudhof TC, Neher E. Intracellular calcium dependence of large dense-core vesicle exocytosis in the absence of synaptotagmin I. Proc Natl Acad Sci USA. 2001;98:11680–11685. doi: 10.1073/pnas.201398798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Ruden L, Neher E. A Ca-dependent early step in the release of catecholamines from adrenal chromaffin cells. Science. 1993;262:1061–1065. doi: 10.1126/science.8235626. [DOI] [PubMed] [Google Scholar]
- Yang J, Tsien RW. Enhancement of N- and L-type calcium channel currents by protein kinase C in frog sympathetic neurons. Neuron. 1993;10:127–136. doi: 10.1016/0896-6273(93)90305-b. [DOI] [PubMed] [Google Scholar]
- Zhang L, Del Castillo AR, Trifaro JM. Histamine-evoked chromaffin cell scinderin redistribution, F-actin disassembly, and secretion: in the absence of cortical F-actin disassembly, an increase in intracellular Ca2+ fails to trigger exocytosis. J Neurochem. 1995;65:1297–1308. doi: 10.1046/j.1471-4159.1995.65031297.x. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Ikeda SR. Modulation of Ca2+-channel currents by protein kinase C in adult rat sympathetic neurons. J Neurophysiol. 1994;72:1549–1560. doi: 10.1152/jn.1994.72.4.1549. [DOI] [PubMed] [Google Scholar]