

Abstract
Prostaglandin D2 (PGD) is synthesized by hematopoietic PGD synthase (HPGDS) or lipocalin-type PGDS (L-PGDS), depending on the organ in which it is produced, and binds specifically to either DP1 or DP2 receptors. We investigated the role of PGD2 in the pathogenesis of hypoxic-ischemic encephalopathy (HIE) in neonatal mice at postnatal day 7. In wild-type mice, hypoxia-ischemia increased PGD2 production in the brain up to 90-fold compared with the level in sham-operated brains at 10 min after cessation of hypoxia. Whereas the size of the infarct was not changed in L-PGDS or DP2 knock-out mouse brains compared with that in the wild-type HIE brains, it was significantly increased in HPGDS–L-PGDS double knock-out or DP1 knock-out mice. The PGD2 level in L-PGDS, HPGDS, and HPGDS–L-PGDS knock-out mice at 10 min of reoxygenation was 46, 7, and 1%, respectively, of that in the wild-type ones, indicating the infarct size to be in inverse relation to the amount of PGD2 production. DP1 receptors were exclusively expressed in endothelial cells after 1 h of reoxygenation, and cerebral blood flow decreased more rapidly after the onset of hypoxia and did not return to the baseline level after reoxygenation in HPGDS–L-PGDS knock-out mice. Endothelial cells were severely damaged in HPGDS–L-PGDS and DP1 knock-out mice after 1 h of reoxygenation. In the human neonatal HIE brain, HPGDS-positive microglia were increased in number. In conclusion, it is probable that PGD2 protected the neonatal brain from hypoxic-ischemic injury mainly via DP1 receptors by preventing endothelial cell degeneration.
Keywords: hypoxic-ischemic encephalopathy, prostaglandin D2, hematopoietic PGD synthase, lipocalin-type PGD synthase, DP1 receptor, microangiopathy
Introduction
Neonatal hypoxic-ischemic encephalopathy (HIE) resulting from perinatal cerebral hypoxia-ischemia (HI) is one of the leading causes of acute mortality and chronic disability in infants and children (Vannucci and Hagberg, 2004; Miller et al., 2005). The incidence of systemic asphyxia is 2∼4/1,000 full-term infants (Vannucci and Vannucci, 2005), ∼10% of which cases are diagnosed as HIE (American College of Obstetricians and Gynecologists and American Academy of Pediatrics, 2003). The majority of infants with HIE die or exhibit permanent neuropsychological handicaps (Vannucci and Vannucci, 2005). Toward an understanding of the mechanism involved in neonatal HIE, a number of studies using a rat model for HIE (i.e., 7-d-old rats subjected to carotid ligation combined with hypoxia) were conducted previously (Rice et al., 1981; Vannucci et al., 1999). Because the brain of the 7-d-old rat is developmentally similar to that of the human fetus or newborn infant at the third trimester, this rat model (Grow and Barks, 2002) and similar mouse models (Sheldon et al., 1998) well represent human neonatal HIE. In the rat model, cerebral blood flow (CBF) in the hemisphere ipsilateral to the carotid ligation decreased during hypoxia, but was restored immediately after the cessation of the hypoxic insult (Grow and Barks, 2002). Investigations using these rodent models revealed that proinflammatory molecules including cytokines (Hedtjarn et al., 2005a,b) and prostaglandin (PG) (Nogawa et al., 1997; Manabe et al., 2004), along with activated microglia/macrophages (Kim et al., 2003; Hedtjarn et al., 2004), are involved in the pathogenesis of HIE. It is known that PGs are upregulated in the ischemic brain (Nogawa et al., 1997) and that inhibition of their production is neuroprotective (Nogawa et al., 1997; Nakayama et al., 1998; Manabe et al., 2004).
Prostaglandin D2 (PGD2) is a well known inflammatory mediator in peripheral tissues (Matsuoka et al., 2000; Honda et al., 2003; Kabashima and Narumiya, 2003), invoking airway inflammation (Matsuoka et al., 2000; Shiraishi et al., 2005), inhibiting platelet aggregation (Yun et al., 1991), and causing peripheral vasodilatation (Soter et al., 1983; Alving et al., 1991). PGD2 is synthesized by hematopoietic PGD synthase (HPGDS) and lipocalin-type PGDS (L-PGDS) (Urade and Hayaishi, 2000) and exerts its actions by binding to specific receptors, DP1 or DP2 (Kabashima and Narumiya, 2003; Nagata and Hirai, 2003). In the normal CNS, L-PGDS is expressed in oligodendroglia (Urade and Hayaishi, 2000), and HPGDS in microglia (Mohri et al., 2003). We found previously that both L-PGDS (Taniike et al., 2002) and HPGDS (Mohri et al., 2006) were upregulated together with specific elevation of PGD2 in the brains of twitcher mice, a model of chronic neuroinflammation. Suppression of HPGDS-PGD2-DP1 signaling resulted in the clinicopathological improvement in twitcher (Mohri et al., 2006).
In this study, we investigated the pathophysiological role of PGD2 in neonatal HIE by using wild-type HIE-model mice and those deficient in one PGDS or both or in DP receptors. Our results show that PGD2 alleviated microangiopathy and functioned as a protective molecule in neonatal HIE.
Materials and Methods
Animals and Surgical Procedure.
HPGDS knock-out (H-KO) (Mohri et al., 2006), L-PGDS knock-out (L-KO) (Eguchi et al., 1999), and DP1 knock-out (DP1-KO) (Matsuoka et al., 2000) mice were generated by standard gene targeting technology with embryonic stem cells derived from the 129 strain of mouse. H-KO, L-KO, and DP1-KO mice were backcrossed to the inbred C57BL/6J strain 15 times. DP2 knock-out mice (DP2-KO) (Satoh et al., 2006), which were on a BALB/cAJcl background, were kindly donated by Dr. Hirai (BML, Saitama, Japan) and Prof. Nakamura (Tokyo Medical and Dental University, Tokyo, Japan). Double knock-out mice for HPGDS and L-PGDS (HL-KO) were established by cross-mating H-KO and L-KO several times; HL-KO mice, confirmed by genome analysis, were selected for mating (Qu et al., 2006). Wild-type C57BL/6J and BALB/cAJcl were purchased from SLC (Shizuoka, Japan) and CLEA Japan (Tokyo, Japan), respectively. We checked the cerebrovascular anatomy of these mouse lines by the perfusion of dye and confirmed no developmental alterations in these genotypes (data not shown). These genetically manipulated mice showed no clinical signs and lived a normal life span.
All animal experiments conformed to the Japanese Law for the Protection of Experimental Animals and followed the protocols approved by the Institutional Animal Care and Use Committee at the Osaka Bioscience Institute, where the animal experiments were conducted. The mouse model for neonatal HIE was produced by following the method of Sheldon et al. (1998). Briefly, litters of postnatal day 7 (P7) pups were anesthetized with halothane and the left common carotid artery was permanently ligated. Thereafter, the pups were returned to their dam for a 1∼2 h recuperation period and then placed in a hypoxic chamber maintained at 37°C through which humidified 8% oxygen and balanced nitrogen flowed. After the pups had remained in the hypoxic environment for 30 min, they were returned to their dam until they were killed. In this model, CBF in the hemisphere ipsilateral to the carotid ligation did not decrease by the ligation alone because of the contralateral supply of blood via the circle of Willis, but it did decrease during hypoxia and was restored immediately after the cessation of the hypoxic insult, which we refer to as reoxygenation hereafter (Grow and Barks, 2002).
Quantification of PGs.
Ten min after reoxygenation had begun, HIE mice, together with those mice that had undergone a sham operation, were quickly decapitated and the amounts of PGs in their fresh-frozen brains were determined by enzyme immunoassays, as described previously (Ram et al., 1997).
Quantification of mRNA.
Quantitative PCR analysis of the contents of mRNAs for HPGDS, DP1, and glyceraldehyde 3-phosphate dehydrogenase (G3PDH) was performed with a LightCycler amplification and detection system (Roche Diagnostics, Indianapolis, IN), as described below. The animals were perfused with saline before the RNA isolation of RNA from their brain. Total RNA was extracted from mouse cerebrum by the guanidinium thiocyanate-phenol-chloroform method using ISOGEN (Nippon Gene, Tokyo, Japan). Two micrograms of total RNA was reverse transcribed into single-stranded cDNA for 30 min in a reaction mixture at 50°C containing 20 U of RNase inhibitor, 1× RNA PCR buffer, 1 mm dNTP mixture, 2.5 mm random primer, and 5 U avian myeloblastosis virus reverse transcriptase (Takara Biomedicals, Kyoto, Japan). The sequence-specific primers used were as follow: HPGDS forward primer, 5′-GAATAGAACAAGCTGACTGGC-3′; HPGDS reverse primer, 5′-AGCCAAATCTGTGTTTTTGG-3′; DP1 forward primer, 5′-TTTGGGAAGTTCGTGCAGTACT-3′; DP1 reverse primer, 5′-TGGCCTTCTTCAACAGCGT-3′; F4/80 forward primer, 5′-CGCTGCTGGTTGAATACAGAGA-3′; F4/80 reverse primer, 5′-GTCCAGGCAAGGAGGACAGA-3′; G3PDH forward primer, 5′-TGAACGGGAAGC-TCACTGG-3′; and G3PDH reverse primer, 5′-TCCACCACCCTG-TTGCT-3′. The constructs used to create a standard curve were made by cloning each amplified fragment into the HindIII site of a pGEM vector (Promega, Madison, WI). In each PCR experiment, the number of copies was estimated by referring to the standard curve prepared by using a dilution series of each fragment. The PCR mixture contained Taq polymerase, 1× LightCycler DNA master SYBR Green (Roche Diagnostics) reaction buffer, 3 mm MgCl2, and 12.5 pmol of each primer. After PCR had been performed with 40 cycles of denaturation (95°C for 1 s), annealing (57°C for 5 s), and enzymatic chain extension (72°C for 10 s), the products for HPGDS, DP1, and G3PDH were detected at 81, 80, and 87°C, respectively. All PCR products were visualized under UV light after electrophoresis in an agarose gel containing ethidium bromide.
Immunocytochemistry.
Under deep halothane anesthesia, the mice were perfused via the heart with physiological saline for 5 min, followed by 4% paraformaldehyde in 0.1 m sodium phosphate buffer (PB), pH 7.4, for 5 min. The brains were removed and postfixed in the same fixative overnight. Coronal sections of the brain were routinely processed into paraffin blocks. Several mice were perfused with physiological saline only and processed for frozen sections. Rabbit anti-mouse and anti-human HPGDS antibodies (Kanaoka et al., 2000) and guinea pig anti-mouse DP1 antibody (Mizoguchi et al., 2001) were raised in Osaka Bioscience Institute (Osaka, Japan). The other primary antibodies used in this study were as follows: anti-cow glial fibrillary acidic protein (GFAP) antibody (1:1000; DAKO, Glostrup, Denmark), monoclonal mouse anti-human von Willebrand factor (vWF) antibody (clone F8/86, 1:500; DAKO), and anti-human CD68 antibody (1:100; DAKO). Biotinylated Ricinus communis-agglutinin-1 (RCA-1), a marker for both microglia and endothelial cells (Mannoji et al., 1986), was purchased from Vector Laboratories (Burlingame, CA; 50 μg/ml).
HPGDS immunostaining was performed as described previously (Mohri et al., 2003). Rabbit anti-mouse HPGDS antibody was applied followed by biotinylated anti-rabbit IgG and avidin-biotin complex (ABC) from an ABC elite kit (Vector Laboratories). The immunoreactivity was visualized by using diaminobendizine hydrochloride (Dotite, Kumamoto, Japan) as the chromogen. The procedures for double labeling were described previously (Mohri et al., 2003). In the case of double labeling for HPGDS and biotinylated RCA-1, the paraffin-embedded sections were sequentially incubated with Texas Red-conjugated streptavidin (MP Biomedicals, Irvine, CA) and Alexa 488-conjugated anti-rabbit IgG (Invitrogen, Carlsbad, CA). In the case of DP1 and vWF double immunofluorescence, frozen sections were incubated with biotinylated anti-guinea pig IgG (Vector Laboratories) and Alexa 488-conjugated streptavidin (MP Biomedicals), followed by Cy3-labeled anti-mouse-IgG (Millipore, Temecula, CA). DP1 and GFAP double immunostaining was done in the same way as described above except that DP1 was labeled with Texas Red-conjugated streptavidin and vWF, with Alexa 488-conjugated anti-mouse IgG. The fluorescence was examined with a BX51 fluorescence microscope (Olympus, Tokyo, Japan).
Morphometric analysis of the infarct area.
At least 10 HIE model mice of each type (i.e., wild-type; either C57BL/6 or BALB/c) and H-KO, L-KO, HL-KO, DP1-KO, and DP2-KO at 24 h and 7 d after reoxygenation had begun were used in this study. Paraffin-embedded coronal sections (10 μm thickness) were cut at the level of the optic chiasm and infundibulum, identical to the bregma −1.8∼2.0 mm level of the adult mouse brain (Paxinos and Franklin, 2001) and were stained with hematoxylin and eosin (HE). The area of necrotic pallid lesions devoid of normal layered structures in the cerebral cortex and the basal ganglia on HE-stained sections was analyzed by investigators in a blind manner regarding the genotype (M. Taniike and H. Okabe-Arahori) using Macscope software (Mitani, Tokyo, Japan). The percentage of infarct area was calculated as the value of the ipsilateral (left) infarct area divided by the area of the contralateral (right) cerebral subdivisions (Cheng et al., 1997; Han and Holtzman, 2000). In the analysis of brains 7 d after reoxygenation, when brain damage had reached maturation, we calculated the infarct area plus lost area of the affected hemisphere.
Cerebral blood flow analysis.
CBF was analyzed by using a laser-Doppler flow meter (model TBF-LN1; Unique Medical, Tokyo, Japan) (Kitagawa et al., 2002). CBF, flow velocity, and flow volume were measured by a laser probe having a polyacrylamide sheath with an inner diameter of 0.8 mm attached with dental cement to the intact skull 3 mm posterior and 3 mm lateral to bregma. Body temperature was simultaneously monitored with a rectal thermometer. After confirming the stable flow wave of the ipsilateral hemisphere for at least 15 min, we subjected mice to hypoxia and then monitored the CBF until at least 15–20 min after the start of reoxygenation. By use of the software Unique Acquisition (Unique medical), the values of the above parameters were collected every 0.1 s. After any extraordinarily high value (>20 ml/min) caused by artifacts resulting from body movement of the mice had been omitted, the collected data were averaged every 1 min. The cerebral blood volume (CBV) was calculated as an integral of CBF.
Anatomical examination of cerebrovasculature.
Adult female wild-type, HL-KO, and DP1 KO (16–20 weeks old; n = 3) were deeply anesthetized before they were perfused with saline followed by India ink. Their brains were then carefully removed, positioned with the ventral side up, and photographed with a Nikon (Tokyo, Japan) D-200.
Electron microscopical analysis.
Cerebral hemispheres were coronally sectioned and fixed with 3% glutaraldehyde and 1% paraformaldehyde in 0.1 m PB, pH 7.2, followed by postfixation with 1% osmium tetrahydroxide, and routinely processed into Epon blocks. After observation of 1-μm-thick semithin sections stained with Toluidine blue, areas for preparation of additional ultrathin sections were selected, cut, stained with uranyl acetate and lead citrate, and examined with an H-7650 electron microscope (Hitachi High-Technologies, Tokyo, Japan) at 80 kV.
Evaluation of brain edema by tissue water content.
Water content of the brain hemisphere was measured to evaluate the extent of brain edema. HIE mice were killed by deep anesthesia and their cerebrums were removed and cut into hemispheres at 24 h after reoxygenation. Immediately thereafter, the wet weight was measured, and then the cerebral hemispheres were frozen at −30°C and kept frozen until they could be freeze-dried for 12 h. After the freeze-drying, the dry weight of each sample was measured. The water content in each hemisphere was calculated as %H2O = (wet weight − dry weight)/wet weight (Toung et al., 2002; Thiagarajah et al., 2005).
Human tissue source.
Paraffin sections of formalin-fixed human infant brains were obtained from Osaka Medical Center and the Research Institute for Maternal and Child Health (Osaka, Japan). Autopsy had been performed with written informed consent from the parents during the period of 1988∼2001. Twelve brains from infants who had died from HIE and eight from infants who had died because of non-neurological causes, such as stillbirth or cardiovascular disease without any apparent brain lesions, were examined. This study was approved by the institutional review boards of Osaka University Graduate School of Medicine, Osaka Medical Center, and Research Institute for Maternal and Child Health.
Statistical analysis.
Statistical comparisons were made by using Student's t test. Values of p < 0.05 were considered to be significant.
Results
Neuropathological characterization of model mice for HIE
Temporospatial progression of neuropathological changes was investigated at 10 min, 1 h, and 24 h after the hypoxic-ischemic insult, focusing on activation of microglia and astrocytes. No apparent neuropathological findings were observed on HE-stained sections at 10 min after reoxygenation had begun (Fig. 1A). Both RCA-1-positive microglia (Fig. 1G, arrow) and GFAP-positive astrocytes (Fig. 1M, arrow) possessed a morphology indicating rest or inactivation. Long, patent, and intact blood vessels having RCA-1-positive endothelial cells were also observed (Fig. 1G, arrowhead). Focal edema in the deep cortical layers (III–VI) was recognized in the ipsilateral hemisphere at 1 h after the insult (Fig. 1B, area delimited by the rectangle). One hour after the hypoxic insult, RCA-1-positive microglia increased in number (Fig. 1E) and changed their morphology to that of moderately activated microglia with swollen cytoplasm and short processes (Fig. 1H, arrow). Some RCA-1-positive blood vessels had contracted and their lumen had become narrower (Fig. 1H, arrowhead). GFAP-positive astrocytes still morphologically appeared slender and inactive at 1 h after reoxygenation (Fig. 1N, arrows). At 24 h of reoxygenation, a massive infarct was apparent with HE staining (Fig. 1C, asterisk). RCA-1-positive microglia (Fig. 1F) and GFAP-positive astrocytes (Fig. 1L) were increased in number in the hemisphere ipsilateral to the carotid ligation. Many amoeboid or activated microglia (Fig. 1I, arrow) were observed within the infarct lesions, and some of them had ingested dead cells. Hypertrophic or activated astrocytes (Fig. 1O, arrows) were also recognized at the rim of the infarct (Fig. 1L, wedged between the dotted lines). By 24 h of reoxygenation, almost all RCA-1-positive blood vessels had disappeared from the infarct (Fig. 1F, asterisk).
Figure 1.
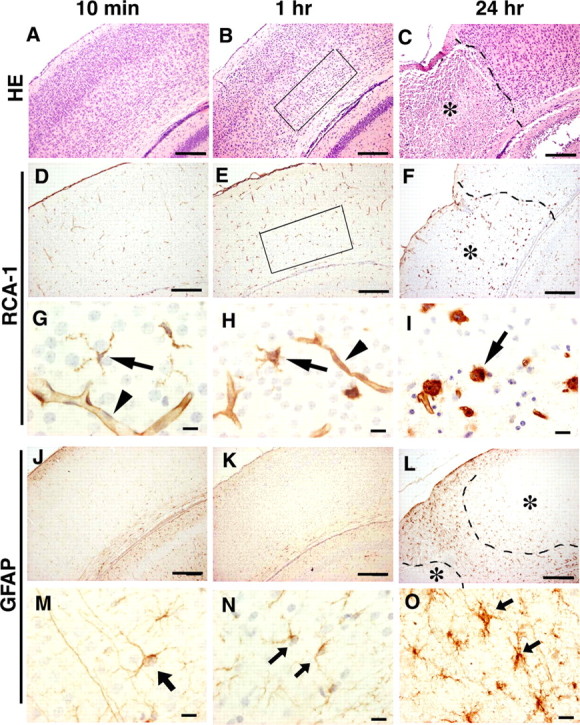
Neuropathology of the somatosensory cortex of a mouse model for HIE. A–C, HE stain. D–I, RCA-1 stain. J–O, GFAP immunostaining. Examination was done at 10 min (A, D, G, J, M), 1 h (B, E, H, K, N), and 24 h (C, F, I, L, O) after the start of reoxygenation. Arrowheads and arrows in G–I indicate RCA-1-positive blood vessels and microglia, respectively. Arrows in M–O indicate GFAP-positive astrocytes. Areas delimited by the rectangles in B and E indicate an edematous lesion in layers III–VI. The dotted lines in C, F, and L indicate the boundaries between infarct (asterisks) and apparently intact area. Scale bars: A–F, J–L, 200 μm; G–I, M–O, 5 μm.
PGD2 level was increased up to 90-fold together with upregulation of HPGDS and DP1 in HIE mouse brains
We quantified the level of three major PGs (i.e., PGD2, PGE2, and PGF2α) in the brain at 10 min after the start of reoxygenation. The level of PGD2 was increased up to 90-fold compared with that for the sham-operated brain. PGE2 was increased sixfold, and PGF2α, 45-fold (Fig. 2A). mRNA levels of HPGDS and DP1 in the ipsilateral hemispheres relative to those in the contralateral ones were unchanged at 1 h, but they increased up to 11-fold and threefold, respectively, at 24 h and then decreased at 48 h of reoxygenation (Fig. 2B).
Figure 2.

Temporal change in levels of PGD2 and PGD2-related molecules in the cerebral cortex of HIE mouse brains. A, Quantification of PGs in the whole brain at 10 min after the start of reoxygenation. Shaded boxes and black boxes indicate wild-type HIE mouse (WT HI) and wild-type sham controls (WT sham), respectively (n = 5). B, Expressional changes in mRNA for HPGDS and DP1 relative to those in G3PDH mRNA. The ordinate shows the ipsilateral/contralateral ratio (n = 4). Dotted lines indicate the level at which mRNA in the ipsilateral hemisphere equaled that in the contralateral hemisphere. The increases in mRNA expression of HPGDS and DP1 were not statistically significant. Error bars indicate mean ± SE. *p < 0.05; #p < 0.01, as indicated by the brackets.
Figure 3A shows the results of immunocytochemical analysis of HPGDS and DP1 in HIE mouse brains at 1 h of reoxygenation. HPGDS-positive cells reminiscent of microglia were present in both hemispheres as early as 1 h (Fig. 3A, left inset). DP1-positive cells were moderately increased in number in the ipsilateral hemisphere at 1 h after reoxygenation had begun (Fig. 3A). HPGDS-positive cells expressed RCA-1 at both 1 and 24 h (Fig. 3B, top), indicating that they were activated microglia, as reported previously (Mohri et al., 2003). In the brain parenchyma, DP1 was exclusively expressed in vWF-positive endothelial cells (Fig. 3B, bottom), at 1 h of reoxygenation. At 24 h after reoxygenation had commenced, DP1 was localized in GFAP-positive astrocytes (data not shown) as well as in vWF-positive endothelial cells (Fig. 3B, bottom)
Figure 3.

A, Immunocytochemical analysis of HPGDS and DP1 in the somatosensory cortex of HIE mouse brains. Insets show a high-magnification view of immunoreactive cells. The arrow and arrowheads indicate DP1-positive cells in parenchyma and meninges, respectively. B, Double fluorescence in ipsilateral hemisphere at 1 and 24 h after reoxygenation. Top, Double immunofluorescence for HPGDS and RCA-1; bottom, double immunofluorescence for DP1 and vWF; arrows and arrowheads indicate doubly and singly immunopositive cells, respectively. Scale bars: A, 100 μm; insets, 5 μm; B, 5 μm. cc, Corpus callosum.
Lack of PGD2-DP1 signaling augments HI lesion
We made HIE models by preparing mice deficient in PGDSs or DP receptors to investigate the pathophysiological role of the PGD2 increase in HIE. Representative HE-stained sections of HIE mouse brains at 24 h after reoxygenation in each genotype are shown in Figure 4A. The areas of infarct are clearly recognizable in this low-magnification view. Whereas the infarct area tended to be restricted to a part of the cerebral cortex in the wild-type HIE mice, it had expanded to almost the entire cortex and basal ganglia in the HL-KO HIE mice. Morphometric analysis was then performed, and the results are shown in Figure 4B. When compared with that for the wild-type HIE model, the infarct area in HL-KO mice was significantly enlarged, exceeding 50% of the cerebral cortex and 20% of the basal ganglia. The areas of infarct were enlarged less dramatically, but still significantly in DP1-KO and moderately in H-KO HIE mice, whereas this was not the case in L-KO mice. DP2-KO HIE mice on a BALB/c background showed a slightly reduced size of infarct compared with wild-type BALB/c mice.
Figure 4.
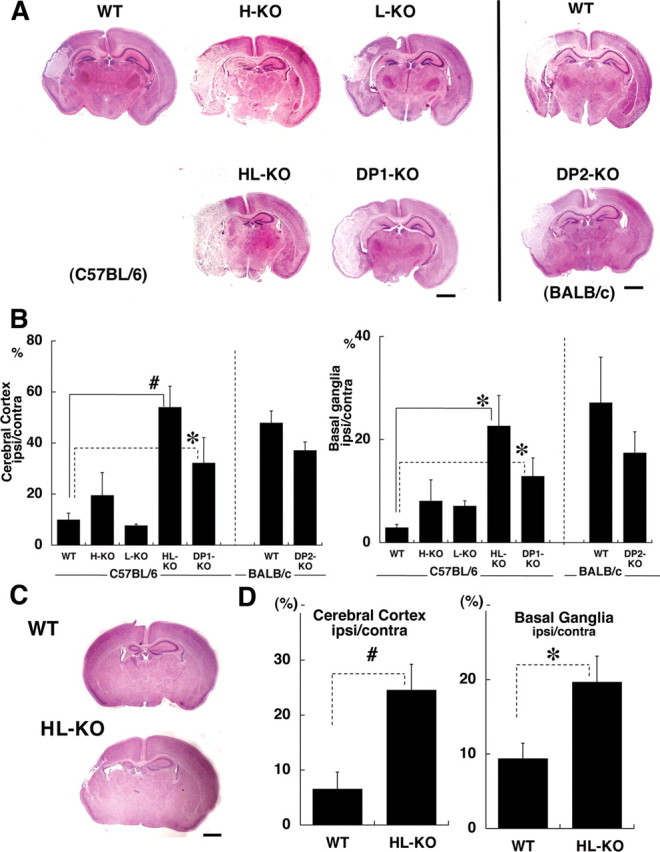
Morphometric comparison of the infarct area in different KO HIE mouse brains. A, C, Low-magnification view of HE-stained coronal sections of the HIE models at 24 h (A) and 7 d (C) after start of reoxygenation. Scale bars, 1 mm. B, D, Morphometric analysis of the infarct area in HIE brains at 24 h (B) and 7 d (D) after start of reoxygenation (n = 12). The results for wild-type C57BL/6 and BALB/c mice are shown separately. The differences between the HL-KO and the DP1-KO were not statistically significant. WT, Wild-type. Error bars indicate mean ± SE. *p < 0.05; #p < 0.01.
In addition, we evaluated the tissue loss in both wild-type and HL-KO mouse brain at 7 d after reoxygenation, when the brain damage had reached maturation. The edematous infarct had developed into tissue loss and atrophy. As shown in Figure 4, C and D, the cerebral tissue loss was significantly larger in HL-KO than in wild-type mice at 7 d after the hypoxic-ischemic insult, parallel to the evaluation at 24 h of reoxygenation. When PGD2 production in the brain at 10 min after reoxygenation was compared in the HIE mice on different genetic backgrounds, the highest was in the wild-type, and the production was reduced to 45.7 ± 0.7, 6.7 ± 0.2, and 1.2 ± 0.0% of the wild-type value in L-KO, H-KO, and HL-KO mice, respectively (Fig. 5). These results indicate that HPGDS widely contributed to PGD2 production and that the aggravation of neuropathology in HIE mice negatively correlated with PGD2 production. In view of our finding that DP2-KO mice did not show any aggravation of infarct, we confirmed that HPGDS-PGD2-DP1 signaling was the major pathway in the PGD2-mediated protection of the HIE brain.
Figure 5.

Quantification of PGD2 in the brains of wild-type sham, wild-type, H-KO (H-KO HI), L-KO (L-KO HI), and HL-KO HIE (HL-KO HI) mice at 10 min after the start of reoxygenation (n = 5). Error bars represent the mean ± SE. *p < 0.05, #p < 0.01, as indicated by the brackets.
Cerebral blood flow was decreased in HL- and DP1-KO HIE mice during hypoxia and failed to recover after reoxygenation in HL-KO HIE
Because DP1 expression was exclusively localized in blood vessels 1 h after reoxygenation had begun, we focused our investigation on the effect of PGD2 signaling on CBF during and after the hypoxia (Fig. 6). In wild-type mice, CBF gradually decreased during the hypoxic period, dropping to 60% of the baseline level (the average at 5 min before exposure to hypoxia) at 20 min of hypoxia and slowly recovered up to ∼80% of the baseline level within 15∼20 min after reoxygenation had been started (Fig. 6, open circles). However, CBF dropped to 60% of the baseline level as soon as 10 min after the start of hypoxia in HL- and DP1-KO HIE (Fig. 6, triangles and closed diamonds, respectively). CBV during hypoxia showed a 39.1 and 43.2% decrease in HL- and DP1-KO mice, respectively, whereas only a 29.6% CBV reduction was observed in the wild-type mice (data not shown). Moreover, CBF did not recover in the HL-KO mouse brains as it did in the wild-type ones. In HL-KO brains, the CBF level showed a transient increase during a few minutes after the start of reoxygenation, although its level decreased again to the lowest level seen during the hypoxia (Fig. 6, triangles). This low level of CBF lasted for >1 h after reoxygenation had begun in some of the HL-KO HIE animals (data not shown). However, DP1-KO HIE mice promptly recovered their normal CBF, reaching a higher level of CBF than that in the wild-type mice post hypoxia (Fig. 6, closed diamonds). We confirmed that there was no alteration of cerebrovascular anatomy among these three genotypes (data not shown).
Figure 6.

CBF analysis of neonatal HIE mice continuously monitored before, during, and after the start of reoxygenation. The averaged CBF during 5 min before hypoxia was defined as 100%. Symbols are defined in the graph. The time point 0 indicates the start of hypoxia, and the black bar at the bottom shows the duration of it. Values are indicated as the mean ± SE; *p < 0.05 wild-type versus DP1-KO; †p < 0.05 wild-type versus HL-KO.
These data suggest that CBF during the hypoxic period was reduced in HL-KO and DP1-KO mice more severely than in wild-type ones and failed to recover during the reoxygenation period in the HL-KO animals.
Absence of PGD2-DP1 signaling enhanced vascular endothelial cell degeneration and brain edema
As stated previously, RCA-1-positive blood vessels were already contracted at 1 h after the start of reoxygenation (Fig. 1E,H) and disappeared in the center of the infarct by 24 h in the wild-type HIE mice (Fig. 1F,I). We next investigated the early vascular pathology in detail. Figure 7A shows RCA-1 staining in a deep layer of the somatosensory cortex 1 h after reoxygenation had begun. In wild-type HIE mice, most blood vessels in the contralateral hemisphere showed smooth vascular walls, patent lumens, and intact nuclei (Fig. 7Aa,Ab), whereas some blood vessels had degenerated in the ipsilateral hemisphere (Fig. 7Ac,Ad). On the contrary, in the HL-KO HIE brains many degenerating blood vessels were recognized in the infarct area, with the endothelial cells showing shrunken cytoplasm and nuclear chromatin condensation, and often the vessels seemed to have disappeared (Fig. 7Ae,Af). In DP1-KO HIE, the vascular lumen had not narrowed and the number of vessels had not been reduced (Fig. 7Ag,Ah); however, the blood vessels were coiled, and the endothelial cells showed a darkened cytoplasm and condensed nuclear chromatin (Fig. 7Ah, inset).
Figure 7.
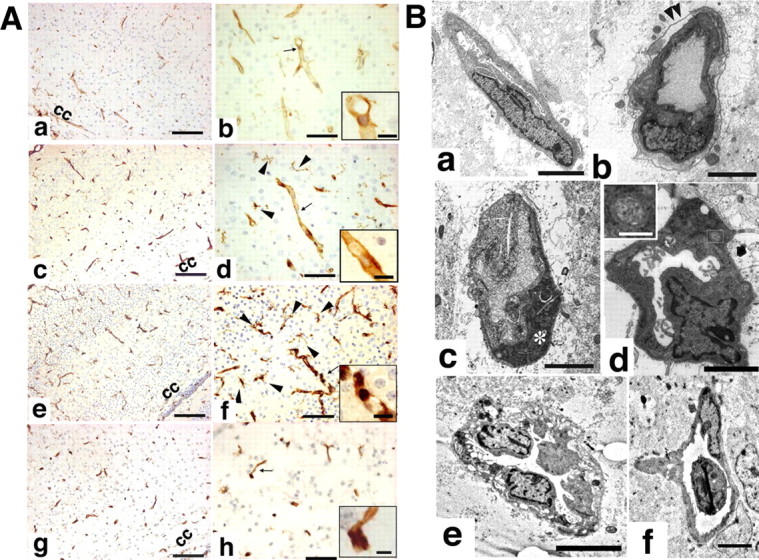
A, Representative pictures of RCA-1-stained HIE mouse brains from wild-type (contralateral and ipsilateral hemispheres, a, b and c, d, respectively), HL-KO (ipsilateral hemisphere, e, f), and DP1-KO HIE (ipsilateral hemisphere, g, h) mice 1 h after reoxygenation had begun. cc, Corpus callosum. B, Electron micrographs of small blood vessels in the ipsilateral hemisphere at 1 h after reoxygenation. a, Wild type; b–d, HL-KO; e, f, DP1-KO mice. Arrowheads in b indicate the basal lamina; the asterisk in c indicates a degenerating endothelial cell; the inset in d indicates the morphology of mitochondria. Scale bars: A, 50 μm; insets, 10 μm; B, 5 μm; insets, 0.5 μm.
By electron microscopic analysis, whereas perivascular edema was only partial, with a relatively intact basal lamina, in the wild-type HIE brain (Fig. 7Ba), a detached basal lamina (Fig. 7Bb, arrowheads) or degenerating endothelial cells (Fig. 7Bc, asterisk) with massive perivascular edema were frequently observed in the HL-KO mouse brain. In addition, we observed hyperlobulated nuclei with chromatin condensation and small spherical mitochondria with ambiguous cristae in HL-KO brains (Fig. 7Bd). These changes are compatible with the apoptotic process (Karbowski et al., 2004). In DP1-KO HIE mice, electron micrographs of vascular endothelial cells showed vacuolation of the cytoplasm, marked thickening of the vascular wall (Fig. 7Be), and newly formed branching blood vessels (Fig. 7Bf).
To evaluate the brain edema consequent to vascular injury (Fagan et al., 2004), we calculated the water content of the cerebral hemisphere as percent H2O in the cerebral hemispheres at 24 h after the start of reoxygenation. The water content in both hemispheres in wild-type, DP1-KO, and HL-KO HIE mice is given in Figure 8. It was increased significantly in the ipsilateral hemisphere in DP1-KO and HL-KO HIE, rising to 114.6 ± 13.6 and 125.9 ± 10.5%, respectively, of that in the contralateral hemisphere, whereas it was only increased to 100.7 ± 2.6% in the wild-type HIE. From these lines of evidence, we conclude that acute endothelial degeneration and subsequent perivascular edema in the absence of PGD2-DP1 signaling may account for failure to maintain a normal CBF and may have contributed to additional infarction-augmenting ischemia.
Figure 8.

Water content was determined at 24 h after the start of reoxygenation and calculated as (wet weight − dry weight)/wet weight (n = 6). Error bars indicate the mean ± SE. *p < 0.05, #p < 0.01, as indicated by the brackets.
HPGDS- immunopositive cells were also abundant in human HIE brain
To clarify the relevancy of our findings in this mouse model to the human disease, we investigated whether HPGDS was upregulated in human HIE brains. Figure 9A shows representative pictures of brain specimens from two HIE infants (Aa, Ab, Ae, Af) and a non-HIE control (Ac, Ad) immunostained for HPGDS. The two infants, who died from HIE a few days after birth were born at 40 weeks (Fig. 9Aa,Ab) and 37 weeks (Fig. 9Ae,Af) of gestation, respectively. The non-HIE control brain specimen was obtained from an infant who had died at 39 weeks of gestation. HPGDS-positive cells were much more abundant in the HIE brains (Fig. 9Aa,Ab) than in the control one (Fig. 9Ac,Ad). The specificity of HPGDS antibody was checked by omitting the first antibody (Fig. 9Ag). HPGDS immunoreactive cells were increased in number in HIE brains, especially in the cerebral cortex and hippocampus, which are the sites of predilection for the diffuse neuronal necrosis seen in neonatal HIE (Volpe, 2000). HPGDS-positive cells were either activated microglia or foamy macrophages as judged from their morphological characteristics (Fig. 9A). Double immunostaining confirmed that these HPGDS-positive cells in the human HIE brains were CD68-positive microglia/macrophages (Fig. 9B). These findings from human HIE samples were compatible with the results of our immunochemical study on HIE mouse brains.
Figure 9.

A, HPGDS immunocytochemistry in autopsied brains from human neonates with HIE (a, b, e, f) and a control human neonate (c, d). GM, Gray matter; WM, white matter. Arrowhead(s) in a and f and the arrow in e and f indicate HPGDS-immunopositive cells. Insets, Magnified views of the HPGDS-positive cells indicated by arrows. Scale bars: a–d, 50 μm; insets, e, f, 5 μm. g, Negative staining for HPGDS in a section adjacent to that of f. B, Double immunofluorescence of HPGDS and CD68, the latter being a microglial marker, in an HIE brain. Scale bars: 5 μm.
Discussion
PGD2, via DP1, protects neonatal brain from HI injury in a dose-dependent manner
PGD2 production was remarkably and specifically enhanced in HIE mouse brains at 10 min after the start of reoxygenation. It was remarkably inhibited in HL-KO and was partially suppressed in H-KO HIE (Fig. 5). The infarct area was the largest in HL-KO and DP1-KO, followed by H-KO HIE, at 24 h after the hypoxic-ischemic insult (Fig. 4A,B), and the infarct size showed an inverse relation to PGD2 production. The cerebral tissue loss at 7 d after the hypoxic-ischemic insult was also significantly larger in HL-KO than in the wild-type animals. These results indicate that PGD2 exerted its crucial protective role via DP1 in a dose-dependent manner. In addition, HPGDS-immunoreactive microglia increased in number in human neonatal HIE brains as in the mouse HIE brain (Fig. 9). Unfortunately, we could not perform DP1 staining on human brains; however, the similar distributional pattern of HPGDS-positive cells suggests that PGD2 also acts as a protective factor in the human brain.
Liang et al. (2005) reported that DP1 was present in neurons in brain-slice cultures and in cultures of neurons isolated from mouse brains and that PGD2 directly protected neurons from apoptosis. In the present study, we detected in vivo DP1 immunoreactivity in endothelial cells (Fig. 3B), suggesting that a different mechanism is involved in our neonatal HIE model.
Pathophysiology of PGD2-mediated brain protection in neonatal HIE
The role of PGD2 seems to be important in the extremely early phase, because PGD2 production had already increased to an extraordinarily high level as soon as 10 min after reoxygenation had been initiated (Fig. 2A). This abrupt increase in the PGD2 level may be achieved by HPGDS in microglia in the brain parenchyma; however, some PGD2 may have been produced by HPGDS in blood cells (Urade et al., 1990; Mahmud et al., 1997; Tanaka et al., 2000). Because decapitation and freezing the head should be done almost at the same time to avoid an artificial increase in PG levels, no time was allowed for washing out the blood with saline. However, PGD2, whether it came from inside the blood vessels or brain parenchyma, could have acted on DP1 expressed on endothelial cells.
Because DP1 was exclusively expressed on endothelial cells at 1 h after the start of reoxygenation, it is reasonable that PGD2 exerted its action on endothelial cells, possibly by modulating the CBF. The standard method for quantification of blood flow is the [14C]-iodoantipyrine method, but we chose to use the laser-Doppler flowmeter, which has been widely accepted in recent years (Aden et al., 2003; Ten et al., 2005; Gustavsson et al., 2007), because we had to assess the temporal change in blood flow in real time. HL-KO and DP1-KO mice showed a steep reduction in CBF during hypoxia and that CBF recovery post hypoxia was not achieved in the HL-KO mice (Fig. 6). The early endothelial cell damage was observed at 1 h after reoxygenation in the HL-KO and DP1-KO mouse brains. In the former, electron micrographs showed that endothelial cells were at the beginning of the apoptotic process and gave evidence of extensive perivascular edema (Fig. 7Bb–Bd). Mitochondria in these cells were small and spherical rather than the normal long oval shape. As a consequence of the early endothelial cell damage (Fig. 7), brain edema at 24 h of reoxygenation was greatly enhanced in the HL-KO and DP1-KO HIE animals (Fig. 8). Because up to now there has been no research on DP1 signaling in endothelial cells, the underlying molecular mechanism of PGD2-DP1-mediated endothelial cell protection needs to be elucidated; however, we speculate that PGD2 protects endothelial cells by increasing the intracellular cAMP level (Schildberg et al., 2005) through DP1, a Gs-coupled receptor. The intracellular cAMP acts via protein kinase A, which phosphorylates phospholamban (PLB), an intrinsic inhibitor of sarko/endoplasmic calcium ATPase (SERCA) at the endoplasmic reticulum (ER) (Sutliff et al., 1999). Hyperphosphorylation of PLB, however, relieves the inhibition of SERCA. Accordingly, an increased level of cAMP restores the action of calcium ATPase, which in turn stabilizes the Ca2+ levels in the ER and thus prevents apoptosis of the cells (Schildberg et al., 2005). Assuming that PGD2-mediated cytoprotection is caused by DP1 signaling, the question arises as to why the neuropathology and CBF recovery in the DP1-KO HIE mice was milder than those in HL-KO HIE. One possible clue to answering this question is the facilitation of the start of vascular repair in DP1-KO HIE. Although the blood vessels showed thickened vessel walls and vacuolation of endothelial cell cytoplasm in DP1-KO HIE brains, their lumens were not as narrow as those in HL-KO vessels, and vascular sprouting was often observed in DP1-KO HIE brains (Fig. 7Bf) (Yoshida et al., 1989). These unique features in vascular morphology imply the earlier start of vascular repair in DP1-KO HIE mice. We examined vascular endothelial growth factor (VEGF), a well established angiogenetic factor, in wild-type, DP1-KO, and HL-KO mouse brains (Ferrara et al., 2003). At 1 and 24 h after reoxygenation had begun, VEGF expressional pattern and its immunoreactivity were similar among the three genotypes mentioned above (data not shown). However, the RCA-1 staining revealed that the number of activated microglia increased at 1 h of reoxygenation in DP1-KO mice, but not in wild-type or HL-KO mice. In addition, the mRNA level of F4/80, a marker of activated microglia, was significantly greater in the DP1-KO than in the wild-type brain and slightly, although not significantly higher than that in the HL-KO HIE brain (data not shown). Because perivascular microglia were reported to increase in number in stroke (Hess et al., 2004) and to enhance vascular reconstruction after oxidative damage (Ritter et al., 2006), the more numerous microglia may have facilitated repair of the vascular components in the DP1-KO mice.
We also addressed the possibility that DP2 was induced in DP1-KO mice to compensate for the lack of the DP1 signal. We found that the level of DP2 mRNA was slightly, but not significantly greater in the DP1-KO than in the wild-type brain (data not shown). However, we suppose that a compensation mechanism by DP2 would not explain the discrepancy in the neuropathology for the following reasons: DP1 is a Gs-coupled receptor, and its signal increases the level of intracellular cyclic AMP, whereas DP2 is Gi-coupled, and stimulation of it decreases this level. In addition, Liang et al. (2005) documented in an in vitro study that DP1 receptor signaling was neuroprotective and that by the DP2 receptor it was neurotoxic. Because these two receptors were reported to have opposite functions, it is unlikely that DP2 would have compensated for the lack of DP1.
Another explanation for the pathophysiological difference between DP1-KO and HL-KO mice is the possible presence of a PGD2 signaling pathway not mediated via PGD2 receptors in DP1-KO HIE brains or, otherwise, there may be PGD2 receptors other than DP1 or DP2. Last, it may be possible that the extraordinarily high level of PGD2 may bind to other PG receptors that have low affinity for PGD2 (Narumiya and FitzGerald, 2001). These speculations need to be elucidated in the near future.
Clinical significance of PGD2-mediated cerebrovascular protection in neonatal HIE
Our findings that lack of PGD2-DP1 signaling enhanced endothelial degeneration (Fig. 1H, 7A,B), brain edema (Fig. 8), and severe pathology are consistent with a previous report that attributed severe HI injury to disrupted autoregulation of the CBF (Volpe, 2000). Volpe (2000) described that the decreased CBF seen in severely asphyxiated infants would cause impaired autoregulation of CBF. In another aspect, Riddle and Back (2006) attributed severe white-matter injury not only to the effect of decreased blood flow, but also to the immaturity of oligodendoglia lining the lesions (Riddle et al., 2006). Although our model differed from their model in that the major lesions were in the gray matter in our case and not in the white matter as in theirs, we cannot exclude the possibility of other determinants of the severity of tissue loss, aside from CBF, especially a difference in the maturation of the brain cells. However, our data showed that the extent of the brain lesion well correlated with the duration of the decreased CBF.
Therefore, in the clinical setting, every effort should be made to shorten the ischemic period and maintain CBF adequately in such infants. Furthermore, a proper targeting approach for vascular protection after HI injury is an important issue for the development of an effective neuroprotective strategy (Fagan et al., 2004). In this regard, our findings offer a new insight into the role of PGD2 in the cerebrovascular protection after HI brain injury. In conclusion, PGD2-DP1 signaling is probably one of the key components that attenuate cerebrovascular disruption and the subsequent brain damage in HIE model mice. Our future investigation includes developing a strategy for the effective delivery of DP1 agonists to the endothelial cells in the ischemic brain, which may possibly lead to a promising treatment for human neonatal HIE.
Footnotes
This work was supported by Grants-in-Aid for scientific research 17591085 (M.T.) and 18591218 (K.W.) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, and by the Mitsubishi Foundation (Y.U.), the Japan Foundation for Applied Enzymologist (Y.U.), the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovaiton (Y.U.), and Osaka City (Y.U.). We are grateful to Prof. Masataka Nakamura (Human Gene Sciences Center, Tokyo Medical and Dental University, Tokyo, Japan) and Dr. Hiroyuki Hirai (Department of Advanced Medicine and Development, Research and Development Center, BML, Saitama, Japan) for their generous gift of DP2-KO mice. We also thank Dr. Keiko Matsuoka (Osaka Medical Center and Research Institute for Maternal and Child Health, Osaka, Japan) for preparing autopsy samples, Dr. Kuriko Shimono (Osaka University Graduate School of Medicine, Osaka, Japan) for technical supervision, Dr. Naomi Eguchi (Osaka Bioscience Institute, Osaka, Japan) for providing us with anti-mouse DP1 antibody, and Prof. Kinuko Suzuki (University of North Carolina at Chapel Hill, Chapel Hill, NC) and Dr. Kazuo Kitagawa and Dr. Tsutomu Sasaki (Osaka University, Osaka, Japan) for their valuable comments.
References
- Aden U, Halldner L, Lagercrantz H, Dalmau I, Ledent C, Fredholm BB. Aggravated brain damage after hypoxic ischemia in immature adenosine A2A knock-out mice. Stroke. 2003;34:739–744. doi: 10.1161/01.STR.0000060204.67672.8B. [DOI] [PubMed] [Google Scholar]
- Alving K, Matran R, Lundberg JM. The possible role of prostaglandin D2 in the long-lasting airways vasodilatation induced by allergen in the sensitized pig. Acta Physiol Scand. 1991;143:93–103. doi: 10.1111/j.1748-1716.1991.tb09204.x. [DOI] [PubMed] [Google Scholar]
- American College of Obstetricians and Gynecologists, American Academy of Pediatrics. Washington, DC: American College of Obstetricians and Gynecologists; 2003. Neonatal encephalopathy and cerebral palsy: defining the pathogenesis and pathophysiology. [Google Scholar]
- Cheng Y, Gidday JM, Yan Q, Shah AR, Holtzman DM. Marked age-dependent neuroprotection by brain-derived neurotrophic factor against neonatal hypoxic-ischemic brain injury. Ann Neurol. 1997;41:521–529. doi: 10.1002/ana.410410416. [DOI] [PubMed] [Google Scholar]
- Eguchi N, Minami T, Shirafuji N, Kanaoka Y, Tanaka T, Nagata A, Yoshida N, Urade Y, Ito S, Hayaishi O. Lack of tactile pain (allodynia) in lipocalin-type prostaglandin D synthase-deficient mice. Proc Natl Acad Sci USA. 1999;96:726–730. doi: 10.1073/pnas.96.2.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan SC, Hess DC, Hohnadel EJ, Pollock DM, Ergul A. Targets for vascular protection after acute ischemic stroke. Stroke. 2004;35:2220–2225. doi: 10.1161/01.STR.0000138023.60272.9e. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- Grow J, Barks JD. Pathogenesis of hypoxic-ischemic cerebral injury in the term infant: current concepts. Clin Perinatol. 2002;29:585–602. doi: 10.1016/s0095-5108(02)00059-3. [DOI] [PubMed] [Google Scholar]
- Gustavsson M, Mallard C, Vannucci SJ, Wilson MA, Johnston MV, Hagberg H. Vascular response to hypoxic preconditioning in the immature brain. J Cereb Blood Flow Metab. 2007 doi: 10.1038/sj.jcbfm.9600408. in press. [DOI] [PubMed] [Google Scholar]
- Han BH, Holtzman DM. BDNF protects the neonatal brain from hypoxic-ischemic injury in vivo via the ERK pathway. J Neurosci. 2000;20:5775–5781. doi: 10.1523/JNEUROSCI.20-15-05775.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedtjarn M, Mallard C, Hagberg H. Inflammatory gene profiling in the developing mouse brain after hypoxia-ischemia. J Cereb Blood Flow Metab. 2004;24:1333–1351. doi: 10.1097/01.WCB.0000141559.17620.36. [DOI] [PubMed] [Google Scholar]
- Hedtjarn M, Mallard C, Arvidsson P, Hagberg H. White matter injury in the immature brain: role of interleukin-18. Neurosci Lett. 2005a;373:16–20. doi: 10.1016/j.neulet.2004.09.062. [DOI] [PubMed] [Google Scholar]
- Hedtjarn M, Mallard C, Iwakura Y, Hagberg H. Combined deficiency of IL-1beta18, but not IL-1alphabeta, reduces susceptibility to hypoxia-ischemia in the immature brain. Dev Neurosci. 2005b;27:143–148. doi: 10.1159/000085986. [DOI] [PubMed] [Google Scholar]
- Hess DC, Abe T, Hill WD, Studdard AM, Carothers J, Masuya M, Fleming PA, Drake CJ, Ogawa M. Hematopoietic origin of microglial and perivascular cells in brain. Exp Neurol. 2004;186:134–144. doi: 10.1016/j.expneurol.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Honda K, Arima M, Cheng G, Taki S, Hirata H, Eda F, Fukushima F, Yamaguchi B, Hatano M, Tokuhisa T, Fukuda T. Prostaglandin D2 reinforces Th2 type inflammatory responses of airways to low-dose antigen through bronchial expression of macrophage-derived chemokine. J Exp Med. 2003;198:533–543. doi: 10.1084/jem.20022218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashima K, Narumiya S. The DP receptor, allergic inflammation and asthma. Prostaglandins Leukot Essent Fatty Acids. 2003;69:187–194. doi: 10.1016/s0952-3278(03)00080-2. [DOI] [PubMed] [Google Scholar]
- Kanaoka Y, Fujimori K, Kikuno R, Sakaguchi Y, Urade Y, Hayaishi O. Structure and chromosomal localization of human and mouse genes for hematopoietic prostaglandin D synthase. Conservation of the ancestral genomic structure of sigma-class glutathione S-transferase. Eur J Biochem. 2000;267:3315–3322. doi: 10.1046/j.1432-1327.2000.01362.x. [DOI] [PubMed] [Google Scholar]
- Karbowski M, Arnoult D, Chen H, Chan DC, Smith CL, Youle RJ. Quantitation of mitochondrial dynamics by photolabeling of individual organelles shows that mitochondrial fusion is blocked during the Bax activation phase of apoptosis. J Cell Biol. 2004;164:493–499. doi: 10.1083/jcb.200309082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NG, Lee H, Son E, Kwon OY, Park JY, Park JH, Cho GJ, Choi WS, Suk K. Hypoxic induction of caspase-11/caspase-1/interleukin-1beta in brain microglia. Brain Res Mol Brain Res. 2003;114:107–114. doi: 10.1016/s0169-328x(03)00135-9. [DOI] [PubMed] [Google Scholar]
- Kitagawa K, Matsumoto M, Kuwabara K, Takasawa K, Tanaka S, Sasaki T, Matsushita K, Ohtsuki T, Yanagihara T, Hori M. Protective effect of apolipoprotein E against ischemic neuronal injury is mediated through antioxidant action. J Neurosci Res. 2002;68:226–232. doi: 10.1002/jnr.10209. [DOI] [PubMed] [Google Scholar]
- Liang X, Wu L, Hand T, Andreasson K. Prostaglandin D2 mediates neuronal protection via the DP1 receptor. J Neurochem. 2005;92:477–486. doi: 10.1111/j.1471-4159.2004.02870.x. [DOI] [PubMed] [Google Scholar]
- Mahmud I, Ueda N, Yamaguchi H, Yamashita R, Yamamoto S, Kanaoka Y, Urade Y, Hayaishi O. Prostaglandin D synthase in human megakaryoblastic cells. J Biol Chem. 1997;272:28263–28266. doi: 10.1074/jbc.272.45.28263. [DOI] [PubMed] [Google Scholar]
- Manabe Y, Anrather J, Kawano T, Niwa K, Zhou P, Ross ME, Iadecola C. Prostanoids, not reactive oxygen species, mediate COX-2-dependent neurotoxicity. Ann Neurol. 2004;55:668–675. doi: 10.1002/ana.20078. [DOI] [PubMed] [Google Scholar]
- Mannoji H, Yeger H, Becker LE. A specific histochemical marker (lectin Ricinus communis agglutinin-1) for normal human microglia, and application to routine histopathology. Acta Neuropathol (Berl) 1986;71:341–343. doi: 10.1007/BF00688060. [DOI] [PubMed] [Google Scholar]
- Matsuoka T, Hirata M, Tanaka H, Takahashi Y, Murata T, Kabashima K, Sugimoto Y, Kobayashi T, Ushikubi F, Aze Y, Eguchi N, Urade Y, Yoshida N, Kimura K, Mizoguchi A, Honda Y, Nagai H, Narumiya S. Prostaglandin D2 as a mediator of allergic asthma. Science. 2000;287:2013–2017. doi: 10.1126/science.287.5460.2013. [DOI] [PubMed] [Google Scholar]
- Miller SP, Ramaswamy V, Michelson D, Barkovich AJ, Holshouser B, Wycliffe N, Glidden DV, Deming D, Partridge JC, Wu YW, Ashwal S, Ferriero DM. Patterns of brain injury in term neonatal encephalopathy. J Pediatr. 2005;146:453–460. doi: 10.1016/j.jpeds.2004.12.026. [DOI] [PubMed] [Google Scholar]
- Mizoguchi A, Eguchi N, Kimura K, Kiyohara Y, Qu WM, Huang ZL, Mochizuki T, Lazarus M, Kobayashi T, Kaneko T, Narumiya S, Urade Y, Hayaishi O. Dominant localization of prostaglandin D receptors on arachnoid trabecular cells in mouse basal forebrain and their involvement in the regulation of non-rapid eye movement sleep. Proc Natl Acad Sci USA. 2001;98:11674–11679. doi: 10.1073/pnas.201398898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohri I, Eguchi N, Suzuki K, Urade Y, Taniike M. Hematopoietic prostaglandin D synthase is expressed in microglia in the developing postnatal mouse brain. Glia. 2003;42:263–274. doi: 10.1002/glia.10183. [DOI] [PubMed] [Google Scholar]
- Mohri I, Taniike M, Taniguchi H, Kanekiyo T, Aritake K, Inui T, Fukumoto N, Eguchi N, Kushi A, Sasai H, Kanaoka Y, Ozono K, Narumiya S, Suzuki K, Urade Y. Prostaglandin D2-mediated microglia/astrocyte interaction enhances astrogliosis and demyelination in twitcher. J Neurosci. 2006;26:4383–4393. doi: 10.1523/JNEUROSCI.4531-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata K, Hirai H. The second PGD(2) receptor CRTH2: structure, properties, and functions in leukocytes. Prostaglandins Leukot Essent Fatty Acids. 2003;69:169–177. doi: 10.1016/s0952-3278(03)00078-4. [DOI] [PubMed] [Google Scholar]
- Nakayama M, Uchimura K, Zhu RL, Nagayama T, Rose ME, Stetler RA, Isakson PC, Chen J, Graham SH. Cyclooxygenase-2 inhibition prevents delayed death of CA1 hippocampal neurons following global ischemia. Proc Natl Acad Sci USA. 1998;95:10954–10959. doi: 10.1073/pnas.95.18.10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narumiya S, FitzGerald GA. Genetic and pharmacological analysis of prostanoid receptor function. J Clin Invest. 2001;108:25–30. doi: 10.1172/JCI13455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogawa S, Zhang F, Ross ME, Iadecola C. Cyclo-oxygenase-2 gene expression in neurons contributes to ischemic brain damage. J Neurosci. 1997;17:2746–2755. doi: 10.1523/JNEUROSCI.17-08-02746.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. Ed 2. San Diego: Academic; 2001. [Google Scholar]
- Qu WM, Huang ZL, Xu XH, Aritake K, Eguchi N, Nambu F, Narumiya S, Urade Y, Hayaishi O. Lipocalin-type prostaglandin D synthase produces prostaglandin D2 involved in regulation of physiological sleep. Proc Natl Acad Sci USA. 2006;103:17949–17954. doi: 10.1073/pnas.0608581103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ram A, Pandey HP, Matsumura H, Kasahara-Orita K, Nakajima T, Takahata R, Satoh S, Terao A, Hayaishi O. CSF levels of prostaglandins, especially the level of prostaglandin D2, are correlated with increasing propensity towards sleep in rats. Brain Res. 1997;751:81–89. doi: 10.1016/s0006-8993(96)01401-1. [DOI] [PubMed] [Google Scholar]
- Rice JE, III, Vannucci RC, Brierley JB. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol. 1981;9:131–141. doi: 10.1002/ana.410090206. [DOI] [PubMed] [Google Scholar]
- Riddle A, Luo NL, Manese M, Beardsley DJ, Green L, Rorvik DA, Kelly KA, Barlow CH, Kelly JJ, Hohimer AR, Back SA. Spatial heterogeneity in oligodendrocyte lineage maturation and not cerebral blood flow predicts fetal ovine periventricular white matter injury. J Neurosci. 2006;26:3045–3055. doi: 10.1523/JNEUROSCI.5200-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter MR, Banin E, Moreno SK, Aguilar E, Dorrell MI, Friedlander M. Myeloid progenitors differentiate into microglia and promote vascular repair in a model of ischemic retinopathy. J Clin Invest. 2006;116:3266–3276. doi: 10.1172/JCI29683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T, Moroi R, Aritake K, Urade Y, Kanai Y, Sumi K, Yokozeki H, Hirai H, Nagata K, Hara T, Utsuyama M, Hirokawa K, Sugamura K, Nishioka K, Nakamura M. Prostaglandin D2 plays an essential role in chronic allergic inflammation of the skin via CRTH2 receptor. J Immunol. 2006;177:2621–2629. doi: 10.4049/jimmunol.177.4.2621. [DOI] [PubMed] [Google Scholar]
- Schildberg FA, Schulz S, Dombrowski F, Minor T. Cyclic AMP alleviates endoplasmic stress and programmed cell death induced by lipopolysaccharides in human endothelial cells. Cell Tissue Res. 2005;320:91–98. doi: 10.1007/s00441-004-1066-4. [DOI] [PubMed] [Google Scholar]
- Sheldon RA, Sedik C, Ferriero DM. Strain-related brain injury in neonatal mice subjected to hypoxia-ischemia. Brain Res. 1998;810:114–122. doi: 10.1016/s0006-8993(98)00892-0. [DOI] [PubMed] [Google Scholar]
- Shiraishi Y, Asano K, Nakajima T, Oguma T, Suzuki Y, Shiomi T, Sayama K, Niimi K, Wakaki M, Kagyo J, Ikeda E, Hirai H, Yamaguchi K, Ishizaka A. Prostaglandin D2-induced eosinophilic airway inflammation is mediated by CRTH2 receptor. J Pharmacol Exp Ther. 2005;312:954–960. doi: 10.1124/jpet.104.078212. [DOI] [PubMed] [Google Scholar]
- Soter NA, Lewis RA, Corey EJ, Austen KF. Local effects of synthetic leukotrienes (LTC4, LTD4, LTE4, and LTB4) in human skin. J Invest Dermatol. 1983;80:115–119. doi: 10.1111/1523-1747.ep12531738. [DOI] [PubMed] [Google Scholar]
- Sutliff RL, Hoying JB, Kadambi VJ, Kranias EG, Paul RJ. Phospholamban is present in endothelial cells and modulates endothelium-dependent relaxation. Evidence from phospholamban gene-ablated mice. Circ Res. 1999;84:360–364. doi: 10.1161/01.res.84.3.360. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Ogawa K, Sugamura K, Nakamura M, Takano S, Nagata K. Cutting edge: differential production of prostaglandin D2 by human helper T cell subsets. J Immunol. 2000;164:2277–2280. doi: 10.4049/jimmunol.164.5.2277. [DOI] [PubMed] [Google Scholar]
- Taniike M, Mohri I, Eguchi N, Beuckmann CT, Suzuki K, Urade Y. Perineuronal oligodendrocytes protect against neuronal apoptosis through the production of lipocalin-type prostaglandin D synthase in a genetic demyelinating model. J Neurosci. 2002;22:4885–4896. doi: 10.1523/JNEUROSCI.22-12-04885.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ten VS, Sosunov SA, Mazer SP, Stark RI, Caspersen C, Sughrue ME, Botto M, Connolly ES, Jr, Pinsky DJ. C1q-deficiency is neuroprotective against hypoxic-ischemic brain injury in neonatal mice. Stroke. 2005;36:2244–2250. doi: 10.1161/01.STR.0000182237.20807.d0. [DOI] [PubMed] [Google Scholar]
- Thiagarajah JR, Papadopoulos MC, Verkman AS. Noninvasive early detection of brain edema in mice by near-infrared light scattering. J Neurosci Res. 2005;80:293–299. doi: 10.1002/jnr.20439. [DOI] [PubMed] [Google Scholar]
- Toung TJ, Hurn PD, Traystman RJ, Bhardwaj A. Global brain water increases after experimental focal cerebral ischemia: effect of hypertonic saline. Crit Care Med. 2002;30:644–649. doi: 10.1097/00003246-200203000-00025. [DOI] [PubMed] [Google Scholar]
- Urade Y, Hayaishi O. Prostaglandin D synthase: structure and function. Vitam Horm. 2000;58:89–120. doi: 10.1016/s0083-6729(00)58022-4. [DOI] [PubMed] [Google Scholar]
- Urade Y, Ujihara M, Horiguchi Y, Igarashi M, Nagata A, Ikai K, Hayaishi O. Mast cells contain spleen-type prostaglandin D synthetase. J Biol Chem. 1990;265:371–375. [PubMed] [Google Scholar]
- Vannucci RC, Vannucci SJ. Perinatal hypoxic-ischemic brain damage: evolution of an animal model. Dev Neurosci. 2005;27:81–86. doi: 10.1159/000085978. [DOI] [PubMed] [Google Scholar]
- Vannucci RC, Connor JR, Mauger DT, Palmer C, Smith MB, Towfighi J, Vannucci SJ. Rat model of perinatal hypoxic-ischemic brain damage. J Neurosci Res. 1999;55:158–163. doi: 10.1002/(SICI)1097-4547(19990115)55:2<158::AID-JNR3>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Vannucci SJ, Hagberg H. Hypoxia-ischemia in the immature brain. J Exp Biol. 2004;207:3149–3154. doi: 10.1242/jeb.01064. [DOI] [PubMed] [Google Scholar]
- Volpe JJ. Neurology of the newborn. Ed 4. London: WB Saunders; 2000. [Google Scholar]
- Yoshida Y, Yamada M, Wakabayashi K, Ikuta F, Kumanishi T. Endothelial basement membrane and seamless-type endothelium in the repair process of cerebral infarction in rats. Virchows Arch A Pathol Anat Histopathol. 1989;414:385–392. doi: 10.1007/BF00718621. [DOI] [PubMed] [Google Scholar]
- Yun JC, Ohman KP, Gill JR, Jr, Keiser H. Effects of prostaglandins, cAMP, and changes in cytosolic calcium on platelet aggregation induced by a thromboxane A2 mimic (U46619) Can J Physiol Pharmacol. 1991;69:599–604. doi: 10.1139/y91-088. [DOI] [PubMed] [Google Scholar]