

Abstract
β-Arrestins bind to agonist-activated G-protein-coupled receptors regulating signaling events and initiating endocytosis. In β-arrestin2−/− (βarr2−/−) mice, a complex phenotype is observed that includes altered sensitivity to morphine. However, little is known of how β-arrestin2 affects μ receptor signaling. We investigated the coupling of μ receptors to voltage-gated Ca2+ channels (VGCCs) in βarr2+/+ and βarr2−/− dorsal root ganglion neurons. A lack of β-arrestin2 reduced the maximum inhibition of VGCCs by morphine and DAMGO (d-Ala2-N-Me-Phe4-glycol5-enkephalin) without affecting agonist potency, the onset of receptor desensitization, or the functional contribution of N-type VGCCs. The reduction in inhibition was accompanied by increased naltrexone-sensitive constitutive inhibitory coupling of μ receptors to VGCCs. Agonist-independent μ receptor inhibitory coupling was insensitive to CTAP (Cys-Tyr-d-Trp-Arg-Thr-Pen-Thr-NH2), a neutral antagonist that inhibited the inverse agonist action of naltrexone. These functional changes were accompanied by diminished constitutive recycling and increased cell-surface μ receptor expression in βarr2−/− compared with βarr2+/+ neurons. Such changes could not be explained by the classical role of β-arrestins in agonist-induced endocytosis. The localization of the nonreceptor tyrosine kinase c-Src appeared disrupted in βarr2−/− neurons, and there was reduced activation of c-Src by DAMGO. Using the Src inhibitor PP2 [4-amino-5-(4-chlorophenyl)-(t-butyl)pyrazolo[3,4-d]pyrimidine], we demonstrated that defective Src signaling mimics the βarr2−/− cellular phenotype of reduced μ agonist efficacy, increased constitutive μ receptor activity, and reduced constitutive recycling. We propose that β-arrestin2 is required to target c-Src to constitutively active μ receptors, resulting in their internalization, providing another dimension to the complex role of β-arrestin2 and c-Src in G-protein-coupled receptor function.
Keywords: opiate, analgesia, pain, desensitization, trafficking, internalization, tolerance, MOP receptor
Introduction
Agonist-bound μ opioid receptors activate inhibitory G-proteins, reducing neuronal excitability and neurotransmitter release through the combined inhibition of adenylyl cyclase, voltage-dependent Ca2+ channels (VGCCs), and activation of inwardly rectifying K+ channels (Williams et al., 2001). The acute actions of μ agonists in specific neuronal pathways underlie their rewarding and analgesic properties. Opioids also cause longer-term changes in neuronal function through more complex signal transduction cascades. These actions are of considerable interest because they may contribute to the tolerance associated with prolonged opioid use.
Studies of the prototypical G-protein-coupled receptor (GPCR), the β2 adrenergic receptor (β2AR), reveal that β-arrestins bind after receptor phosphorylation by GPCR kinases (GRKs). β-Arrestin-bound GPCRs undergo endocytosis and can either be degraded or recycled back to the cell membrane (Oakley et al., 2000; Lefkowitz and Shenoy, 2005). Agonist-induced receptor internalization and recycling promotes recovery from desensitization (Seachrist et al., 2000). β-Arrestins also orchestrate a complex pattern of β2AR signal transduction through intermediates such as mitogen-activated protein kinase (MAPK), Akt, phosphatidylinositol 3-kinase, nuclear factor κB, and the nonreceptor tyrosine kinase c-Src (Lefkowitz and Shenoy, 2005).
The role of β-arrestins in receptor internalization and signal transduction are somewhat receptor specific; therefore, generalization across the GPCR superfamily should be made with caution (Paing et al., 2002; Kohout and Lefkowitz, 2003). Even the highly related μ, δ, and κ receptors exhibit different requirements for binding to β-arrestin1 and β-arrestin2 (Cen et al., 2001) with different signaling outcomes (Oakley et al., 2000; Tohgo et al., 2003). Agonist-activated μ receptors recruit β-arrestin1 and β-arrestin2 (Bohn et al., 2004; Haberstock-Debic et al., 2005), although the efficacy of recruitment is agonist dependent (Groer et al., 2007). Furthermore, enhanced expression of recombinant β-arrestin2 and GRKs increases the tendency for morphine to desensitize and internalize μ receptors (Whistler and von Zastrow, 1998; Mace et al., 2005). Agonist-evoked internalization participates in the resensitization of μ receptors rendered tolerant to agonist (Whistler et al., 1999; Koch et al., 2005). Thus, a lack of β-arrestin2 would be anticipated to reduce internalization and thereby promote tolerance. In practice, however, βarr2−/− mice exhibit a complex phenotype of enhanced morphine analgesia and reward coupled with reduced morphine-induced tolerance, respiratory depression, and constipation (Bohn et al., 1999, 2000, 2002, 2004; Raehal et al., 2005). These findings underscore the need for a better understanding of the role of β-arrestin2 in μ receptor signaling.
We investigated the role of β-arrestin2 in μ receptor coupling to VGCCs in dorsal root ganglion (DRG) neurons. μ Agonists inhibited VGCCs with a lower efficacy in βarr2−/− compared with βarr2+/+ neurons, a phenomenon associated with increased agonist-independent μ receptor coupling to VGCCs. Increased constitutive activity was accompanied by reduced constitutive recycling and elevated surface μ receptor levels in βarr2−/− neurons. Furthermore, c-Src inhibition in βarr2+/+ neurons mimicked the cellular phenotype of βarr2−/− neurons. Importantly, c-Src was aberrantly targeted in βarr2−/− neurons and not activated by μ agonists. Together, our findings suggest that β-arrestin2 targets c-Src to constitutively active receptors, thereby initiating their deactivation and recycling.
Materials and Methods
Dorsal root ganglion neuron cultures.
Dorsal root ganglia were harvested from early postnatal mice [postnatal day 0 (P0) to P2], which contained both (βarr2+/+), one (βarr2+/−), or neither (βarr2−/−) of the β-arrestin2 alleles in the C57BL/6 background (Bohn et al., 1999). This line has been fully backcrossed to the C57BL/6 background, and the pups used were littermates or within two generations of β-arr2+/− mating. The DRGs were enzymatically and physically dissociated, and neurons were plated in different formats and densities for specific experiments. For electrophysiology and immunocytochemistry, 5 × 104 dissociated cells were plated on poly-d-lysine- (Sigma, St. Louis, MO) and laminin- (BD Biosciences, San Jose, CA) coated coverslips (10 mm diameter) in the center of 35 mm MatTek (Ashland, MA) dishes. For flow cytometry and quantitative PCR (qPCR) experiments, 1 × 106 cells were plated in similarly coated MatTek dishes, but the coverslips were larger (15 mm in diameter). Cells were cultured in Neurobasal A, B27 (2%), glutamax (0.5 mm), and antibiotic–antimycotic (12 U/ml penicillin, 12 U/ml streptomycin, and 30 ng/ml fungizone) media (Invitrogen, Carlsbad, CA) containing NGF (10 μg/ml; Roche, Indianapolis, IN) and kept for 2–3 d in vitro at 37°C and 5% CO2.
Electrophysiology.
The whole-cell patch-clamp technique was used to record VGCC activity from cultured DRG neurons (Axopatch 200A amplifier; Molecular Devices, Palo Alto, CA). Culture medium was replaced by an external solution that contained the following (in mm): 130 tetraethylammonium-Cl, 10 CaCl2, 5 HEPES, 25 d-glucose, and 0.25 tetrodotoxin, pH 7.2. Recording electrodes contained the following (in mm): 105 CsCl, 40 HEPES, 5 d-glucose, 2.5 MgCl2, 10 EGTA, 2 Mg2+-ATP, and 0.5 Na+-GTP, pH 7.2. The potential difference between the open electrode and the bath ground was zeroed before establishing a ≥1 GΩ resistance seal. No compensation was made for the cancellation of liquid junction potential. Ca2+ currents were activated by depolarizing neurons from −80 to 10 mV for 100 ms at 10 s intervals. In experiments examining constitutive inhibitory coupling to VGCCs, a two-pulse voltage protocol was used, and Ca2+ in the external solution was replaced by Ba2+ to prevent Ca2+-dependent inactivation. A depolarizing voltage step (80 ms duration) from −80 to 80 mV preceded (by 10 ms) a voltage-step from −80 to 10 mV (10 ms duration). The current amplitude evoked by the test pulse after an 80 mV prepulse (I+80) was expressed as a percentage of the current amplitude evoked by a similar test pulse in the absence of a depolarizing prepulse (I−80). In some instances, GTP-γ-S (300 μm) was included in the recording electrode to facilitate constitutive inhibitory coupling to VGCCs. In these experiments, recordings began >5 min after achieving the whole-cell configuration to allow time for GTP-γ-S to access the cell. Currents were low-pass filtered at 2 kHz and digitized (Digidata; Molecular Devices) at 10 kHz for storage on the hard drive of a Pentium personal computer. Leak currents were nulled using the P/4 subtraction method. DRG neurons were rapidly and continuously superfused (∼5 ml/min) with external solution in the chamber formed by the coverslip insert at the bottom of the 35-mm-diameter culture dish. The chamber had a volume of <500 μl, allowing rapid exchange of the bath solution. Opioid agonists, opioid antagonists, baclofen (all from Sigma), and ω-conotoxin GVIA (Calbiochem, La Jolla, CA) were diluted into external solution on the day of the experiment. Opioids and baclofen were rapidly applied through the perfusion system, and ω-conotoxin GVIA was applied locally by pressure ejection from a micropipette. Inhibition of Ca2+ currents by opioid agonists and ω-conotoxin GVIA reached maximum within 10 s of initial application. Experiments were performed at room temperature (22–24°C). Mean current amplitudes were measured (pClamp 9.0; Molecular Devices) between 5 and 10 ms after initiating the depolarizing step. Recordings that exhibited marked rundown were discarded. Stable recordings were fitted by a linear function to compare, by extrapolation, control current amplitude with the current amplitude recorded in the presence of opioid receptor agonists. Data are expressed as mean ± SEM and were compared using ANOVA with the post hoc Tukey's test.
qPCR.
Cultured DRG neurons from βarr2−/−, βarr2+/−, and βarr2+/+ P0–P2 pups were harvested in PBS/EDTA, spun (300 × g) for 5 min at 4°C, and lysed. mRNA was isolated (RNAqueous; Ambion, Austin, TX) and reverse transcribed (Superscript III; Invitrogen). Sequence-specific primer/probe sets (Invitrogen) for the mouse μ receptor (GenBank accession number NM_011013) and the control gene synaptophysin (GenBank accession number NM_009305) (Chen et al., 2001) were used to determine the relative transcript level, quantified by cycle number or count threshold (CT) at which the gene-specific fluorescence reached midlinear levels, using the Taqman 7700 (Applied Biosystems, Foster City, CA). The data are presented as the mean ± SEM of the ΔCT between the target and control genes for three separate experiments (supplemental data, available at www.jneurosci.org as supplemental material).
Flow cytometry.
After 3 d in vitro, cultured βarr2+/+ and βarr2−/− DRG neurons were harvested in ice-cold PBS/EDTA and spun at 300 × g for 5 min at 4°C. The cells were washed in ice-cold PBS containing 2% fetal bovine serum and 0.1% sodium azide (PBS/FBS/NaN3) and incubated with an anti-μ receptor antibody raised against the third extracellular loop, for 60 min at 4°C (1:100 dilution in PBS/FBS/NaN3; Millipore, Billerica, MA). Antibodies to this region of the μ receptor do not label neuronal tissue lacking the μ receptor (Guarna et al., 2003). Thereafter, the cells were washed and incubated in the secondary antibody [allophycocyanin (APC)-conjugated rabbit IgG; 1:100; BD Biosciences] for 60 min at room temperature. After a final wash, 5000 neurons per sample were acquired on a FACScalibur flow cytometer (BD Immunocytochemistry Systems, Mountain View, CA,) and analyzed using FCS express version 3.0 (De Novo Software, Thornhill, Ontario, Canada). Flow cytometry was also used to quantify the effect of DAMGO (d-Ala2-N-Me-Phe4-glycol5-enkephalin) on c-Src phosphorylation in cultured DRG neurons from βarr2+/+ and βarr2−/− mice. After 2–3 d in culture, control DRG neurons or neurons treated with DAMGO (1 μm for 20 min) were harvested and processed as described above but, in addition, were briefly fixed (5 min) in 2% paraformaldehyde, pH 7.4, rinsed, and permeabilized in 0.1% Tween 20 for 5 min before labeling with two primary antibodies: anti-c-Src (mouse monoclonal antibody raised against the C-terminal sequence; Santa Cruz Biotechnology, Santa Cruz, CA) and anti-phosphorylated Y416 c-Src (rabbit polyclonal antibody; Cell Signaling Technology, Danvers, MA). Thereafter, the samples were rinsed and labeled with the secondary antibodies: anti-mouse APC–IgG (1:100; BD Biosciences) and anti-rabbit IgG Alexa 488 (1:100; Invitrogen). Such analysis of protein levels by flow cytometry allows the neuronal population to be selected (Walwyn et al., 2004) and at least two epitopes, in this case c-Src and phospho-Y416-Src, to be monitored simultaneously.
Analysis of flow cytometry.
The acquired and analyzed parameters were size, (forward scatter, FSC-H), granularity (side scatter, SSC-H), and Alexa488 either alone or in combination with APC fluorescence, in channels FL1-H and FL4-H, respectively. Each sample within each experiment was analyzed using the same parameters of size, granularity, and FL1-H and FL4-H fluorescence. The neuronal population was first defined as region 1 (R1) by size and granularity and the Alexa488 or APC fluorescence of this population measured in FL1-H and/or FL4-H. Nonspecific fluorescence was subtracted, and the mean fluorescence intensity (FI) was obtained for each sample. Such mean FI values were normalized to the FI of the untreated βarr2+/+ or βarr2−/− sample for each experiment. Each experiment was repeated 6–10 times, and the data were analyzed by the unpaired or paired Student's t test, significance accepted at p < 0.05, and are presented as mean ± SEM.
Immunocytochemistry.
DRG neurons from βarr2+/+ or βarr2−/− were cultured in 35 mm MatTek dishes, fixed in 4% paraformaldehyde, pH 7.4 for 5 min, washed, permeabilized in 0.3% Triton X-100 for 10 min, and incubated in the C-terminal rabbit anti-μ receptor antibody (Walwyn et al., 2004) or C-terminal mouse monoclonal anti-c-Src antibody (1:200; Santa Cruz Biotechnology) overnight at 4°C in potassium PBS/1% normal goat serum. After an additional rinse, the secondary antibody Alexa555-conjugated anti-rabbit or anti-mouse IgG (1:1000; Invitrogen) was applied for 90 min at room temperature. The cells were finally washed, mounted in Slowfade (Invitrogen), and imaged using a Zeiss (Oberkochen, Germany) LSM 310 confocal laser-scanning microscope (CLSM).
Results
The absence of β-arrestin2 reduces the efficacy of μ agonist inhibition of VGCCs
The activation of opioid receptors in DRG neurons causes inhibition of VGCC activity through the mobilization of pertussis toxin-sensitive inhibitory G-proteins (Williams et al., 2001). We tested the ability of μ, δ, and κ opioid receptor agonists to inhibit Ca2+ currents activated by depolarizing βarr2+/+, βarr2+/−, and βarr2−/− neurons from −80 to 10 mV (Fig. 1). Cultured mouse DRG neurons are heterogeneous (Walwyn et al., 2005). To minimize heterogeneity and maximize the fidelity of voltage clamp, we recorded from neurons with a soma diameter of ∼15–30 μm, classified previously in culture as small- to medium-sized cells (Passmore, 2005). Agonists were rapidly applied through the perfusion system to the <500 μl recording chamber. As reported previously, all wild-type DRG neurons tested responded to DAMGO (1 μm), reaching maximal Ca2+ current inhibition within 10 s of DAMGO application (Walwyn et al., 2005). The inhibition evoked by DAMGO (1 μm) was significantly reduced from 33 ± 4% (n = 12) for βarr2+/+ neurons to 19 ± 4% (n = 10) and 14 ± 5% for βarr2+/− and βarr2−/− neurons, respectively (Fig. 1B). Morphine (1 μm) also caused a smaller inhibition of Ca2+ currents recorded from βarr2−/− compared with βarr2+/+ neurons (Fig. 1B). In contrast, reductions of the amplitudes of Ca2+ currents, elicited by the application of the κ receptor agonist U-50,488H [trans-(±)-3,4-dichloro-N-methyl-N-(2-[1-pyrrolidinyl]cyclohexyl) benzene-acetamide methanesulfonate] (1 μm) to βarr2+/+, βarr2+/−, and βarr2−/− DRG neurons, were not significantly different. Consistent with our previous findings, DPDPE ([d-Pen2,d-Pen5]-enkephalin) (1 μm) had no effect on the amplitude of Ca2+ currents recorded from wild-type DRG neurons (Walwyn et al., 2005). In contrast, a small inhibitory action of DPDPE occurred in βarr2−/− neurons (Fig. 1B).
Figure 1.
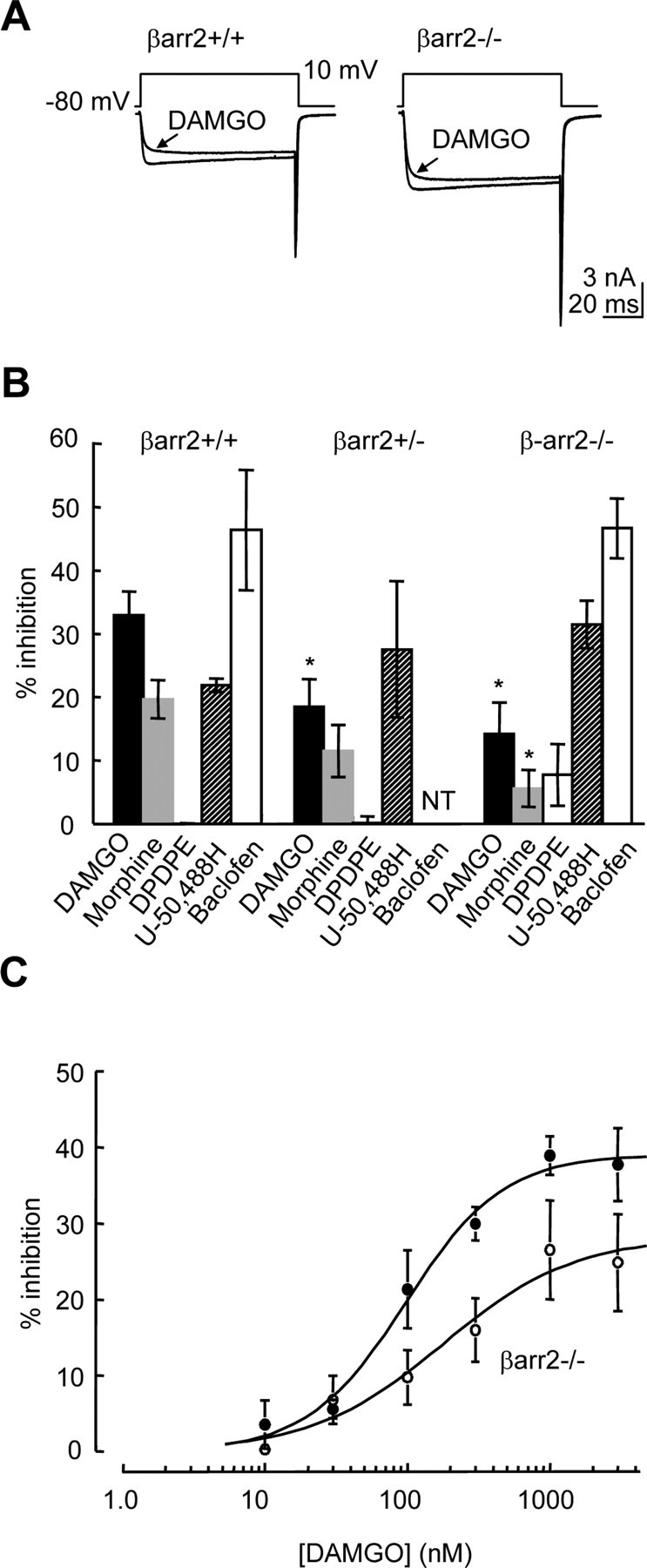
The absence of β-arrestin2 reduces the efficacy of μ receptor coupling to VGCCs. A, Whole-cell Ca2+ currents mediated by VGCCs recorded from βarr2+/+ and βarr2−/− DRG neurons in the absence and presence of DAMGO (1 μm). Neurons were depolarized from −80 to 10 mV. B, The bar graph illustrates the amplitudes of opioid and GABAB receptor-mediated inhibition of Ca2+ currents recorded from DRG neurons. Opioids were tested on βarr2+/+, βarr2+/−, and βarr2−/− neurons. Baclofen (50 μm) was tested on βarr2+/+ and βarr2−/− neurons but was not tested (NT) on βarr2+/− neurons. Opioids were applied at a concentration of 1 μm, and Ca2+ currents were recorded as indicated in A. Error bars represent ±SEM. Statistical significance was determined by ANOVA with post hoc Tukey's test, *p < 0.05 compared with inhibition of Ca2+ current amplitude by the agonist when applied to βarr2+/+ neurons. C, DAMGO caused a concentration-dependent inhibition of Ca2+ currents recorded from βarr2+/+ and βarr2−/− neurons. Data points are mean percentage inhibitions recorded from at least five cells. The logistic fits to the data were used to determine the IC50 value (βarr2+/+, 97 ± 17 nm), which was not significantly affected by the absence of β-arrestin2 (βarr2−/−, 177 ± 95 nm). In contrast, the maximum DAMGO-evoked inhibition of Ca2+ currents determined by the logistic fit to βarr2−/− data were significantly reduced (28 ± 5%) compared with the fit to βarr2+/+ data (39 ± 2%; p < 0.05, Student's t test). Error bars represent ±SEM.
We also tested the inhibitory response to baclofen (50 μm) of VGCC activity recorded from βarr2+/+ and βarr2−/− neurons. The GABAB receptor agonist inhibited Ca2+ currents by 46 ± 10 and 47 ± 5%, respectively (Fig. 1B). These data demonstrate that the deficit in inhibitory coupling to VGCCs in DRG neurons, caused by the absence of β-arrestin2, does not apply to all GPCRs.
We examined whether reduced inhibitory coupling between μ receptors and VGCCs in βarr2−/− DRG neurons results from a reduction in the potency and/or efficacy of DAMGO (Fig. 1C). DAMGO (0.01–3 μm) caused a concentration-dependent inhibition of Ca2+ currents in both βarr2+/+ and βarr2−/− DRG neurons. Logistic fits to the data revealed that DAMGO inhibited Ca2+ currents with IC50 values of 0.10 ± 0.02 and 0.18 ± 0.10 μm for βarr2+/+ and βarr2−/−, respectively, which were not significantly different (Fig. 1C). In contrast, there was a significant reduction in the maximum inhibition by DAMGO in βarr2+/+ compared with βarr2−/− neurons (Fig. 1C).
βarr2−/− DRG neurons have more cell-surface μ receptors than do βarr2+/+ DRG neurons
There are several factors that could reduce the efficacy of μ receptor coupling to VGCCs. The most obvious possibility is that there is a reduction in the number of μ receptors in the membranes of βarr2−/− DRG neurons. Initial analysis by CLSM showed no obvious difference in μ receptor immunolabeling (Fig. 2A). However, quantification of cell-surface μ receptor labeling of intact βarr2+/+ and βarr2−/− DRG neurons by flow cytometry (Fig. 2B,C) revealed a small increase (p < 0.05, Student's t test) in surface expression of μ receptors in βarr2−/− to 127 ± 11% of that in βarr2+/+ DRG neurons (n = 8; data not shown).
Figure 2.
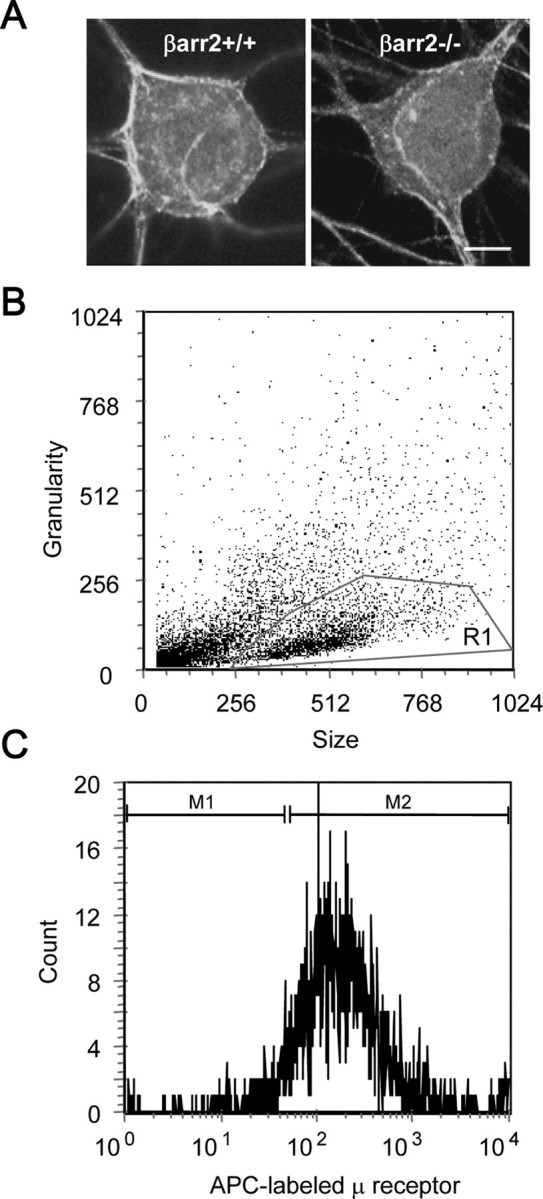
Confocal laser-scanning microscopy and flow cytometry to detect cell-surface μ receptors in DRG neurons. A, Confocal laser-scanning microscopy reveals no obvious difference in the cellular localization of the μ receptor in βarr2−/− and βarr2+/+ neurons. Scale bar, 10 μm. B, Flow cytometry was used to quantify cell-surface μ receptor levels in βarr2−/− and βarr2+/+ neurons. In all experiments, the neuronal population was first defined by the nonfluorescent parameters of size (FSC-H) and granularity (SSC-H) and labeled as R1. C, The mean fluorescence of the APC-labeled μ receptor of these R1 cells was then obtained (M2) after removing nonspecific background fluorescence (M1). Including neurons from βarr2+/+ and βarr2−/− neurons in the same experiment allowed a quantitative comparison of relative receptor levels across genotype revealing an increase in μ receptor levels in βarr2−/− neurons (see Results).
We used qPCR to examine whether the increased receptor surface expression of μ receptors in βarr2−/− compared with βarr2+/+ neurons was caused by increased gene expression. RNA was isolated from βarr2+/+, βarr2+/−, and βarr2−/− DRG neurons after 2 d in culture, and expression of the μ receptors was determined by qPCR using a sequence-specific primer probe set. The expression of synaptophysin was determined in each reaction as a control gene (Chen et al., 2001). The CT revealed that the expression of the μ receptor relative to synaptophysin was similar in βarr2+/+, βarr2+/−, and βarr2−/− DRG neurons (supplemental data, available at www.jneurosci.org as supplemental material). Therefore, increased μ receptor cell-surface expression in βarr2−/− neurons does not reflect increased gene expression.
Desensitization of μ receptor inhibitory coupling to VGCCs in βarr2+/+ and βarr2−/− neurons
Because there is no reduction in μ receptor number in βarr2−/− compared with βarr2+/+ DRG neurons, we looked for alternative explanations for reduced VGCC coupling efficacy in the former. Enhanced agonist-induced μ receptor desensitization in βarr2−/− neurons may reduce the apparent Ca2+ current inhibition by DAMGO and morphine. We measured the peak level of Ca2+ current inhibition by DAMGO (1 μm) or morphine (1 μm) and compared it with the level of inhibition after 200 s of exposure to the agonists to quantify acute μ receptor desensitization (Fig. 3A). After 200 s, the inhibition of Ca2+ current amplitude by DAMGO (10 μm) declined to 57 ± 12% (n = 5) and 44 ± 8% (n = 5) of the initial peak inhibitions in βarr2+/+ and βarr2−/− neurons, respectively (Fig. 3B). Likewise, there was no difference in the level of acute desensitization elicited by morphine (10 μm), 200 s exposure caused a decline in the inhibition to 37 ± 15% (n = 3) and 45 ± 7% (n = 4) of the initial peak inhibitions in βarr2+/+ and βarr2−/− neurons, respectively (Fig. 3B). Therefore, the absence of β-arrestin2 does not significantly alter the level of acute μ receptor desensitization.
Figure 3.
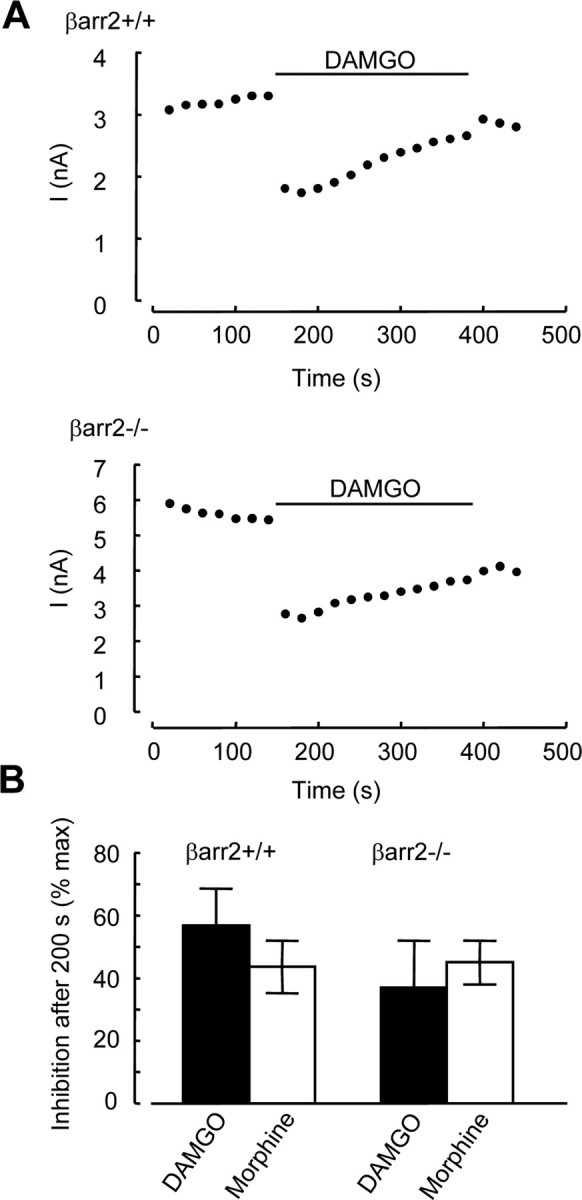
Desensitization of inhibitory coupling of μ receptors to VGCCs is similar in βarr2+/+ and βarr2−/− neurons. A, Time course of inhibition by DAMGO (10 μm) applied for 200 s to βarr2+/+ (top) and βarr2−/− (bottom) neurons. Ca2+ currents were activated by depolarizing from −80 to 10 mV using the voltage protocol illustrated in Figure 1A. Peak current amplitudes (pA) were measured and plotted against time. B, The inhibition of Ca2+ current amplitude measured after 200 s of exposure to either DAMGO (10 μm) or morphine (10 μm) was expressed as a percentage of the peak inhibition observed at the beginning of opioid application. Error bars represent ±SEM.
The absence of β-arrestin2 does not alter the current density of VGCCs or the proportion of N-type to non-N-type VGCCs in DRG neurons
It is possible that reduced inhibition of VGCCs by μ agonists could be caused by a deficit in G-protein-sensitive Ca2+ channels in βarr2−/− DRG neurons. Opioid receptors couple to high-voltage-activated Ca2+ channels (Williams et al., 2001). We compared the densities of Ca2+ current in βarr2+/+ and βarr2−/− DRG neurons over a range of voltages (Fig. 4). Currents activated in βarr2+/+ and βarr2−/− neurons, by depolarizing steps from −60 to 70 mV in 10 mV increments, appeared similar (Fig. 4A). Furthermore, when peak current amplitudes were normalized to the cell membrane capacitance (pA/pF), the resultant densities of current were similar at all voltages examined (Fig. 4B). These data suggest that the absence of β-arrestin2 does not cause a significant change in the expression of functional VGCCs.
Figure 4.
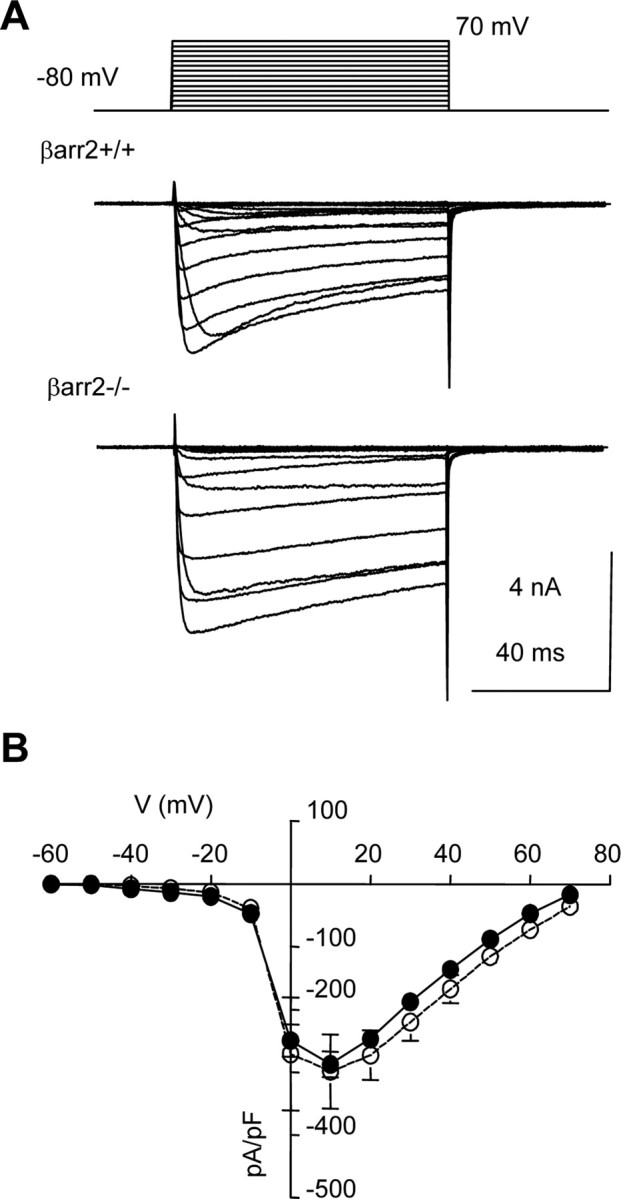
The densities of Ca2+ current mediated by VGCCs are similar in βarr2+/+ and βarr2−/− neurons. A, Superimposed Ca2+ currents recorded from βarr2+/+ (top) and βarr2−/− (bottom) DRG neurons in response to voltage steps from −80 mV to between −60 and 70 mV (10 mV increments every 15 s). B, The graph illustrates the densities of currents [determined by dividing peak current amplitudes (pA) by membrane capacitance (pF)] at all voltages tested in βarr2+/+ (filled circles) and βarr2−/− (open circles) neurons. Each data point represents the mean current density obtained from at least five cells.
In DRG neurons, opioid agonists predominantly inhibit the activity of N- and P/Q-type VGCCs (Rusin and Moises, 1995). Furthermore, β-arrestin1 has been implicated in the internalization of N-type channels (Puckerin et al., 2006). Therefore, we investigated the possibility that the absence of β-arrestin2 could also affect the contribution of N-type Ca2+ channels. Application of the selective N-type channel inhibitor ω-conotoxin GVIA (10 μm) from a local pressure pipette inhibited the amplitude of Ca2+ currents recorded from βarr2+/+ neurons by 55 ± 5% (n = 7) (Fig. 5A). The inhibition peaked within 10 s, was irreversible during a prolonged wash, and ranged from 43 to 73%. Application of ω-conotoxin GVIA caused a similar inhibition (51 ± 2%; n = 5) of Ca2+ currents recorded from βarr2−/− neurons (Fig. 5B). The inhibition by DAMGO (1 μm), after application of ω-conotoxin GVIA, was reduced to 21 ± 4% (n = 7) and 10 ± 4% (n = 4) in βarr2+/+ and βarr2−/− neurons, respectively (Fig. 5A,B). The reduction in the efficacy of DAMGO as an inhibitor of ω-conotoxin GVIA-insensitive current caused by a lack of β-arrestin2 was similar to the reduction seen in the absence of N-type channel inhibition. In the presence of ω-conotoxin GVIA, DAMGO inhibition of Ca2+ current in βarr2−/− neurons was ∼48% of that in βarr2+/+ neurons compared with ∼42% in the absence of ω-conotoxin GVIA. Furthermore, as mentioned above, baclofen, which also inhibits N- and P/Q-type channels, caused a similar level of inhibition in βarr2+/+ and βarr2−/− neurons (Fig. 1B), suggesting that there is no change in the contribution of these channels in the absence of β-arrestin2.
Figure 5.
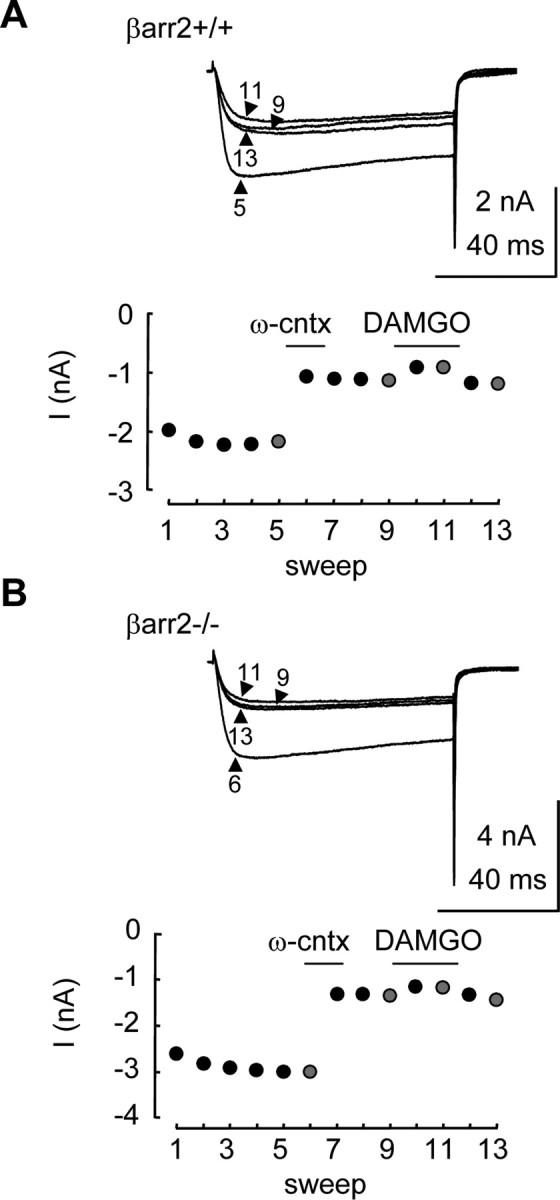
N-type VGCCs contribute equally to Ca2+ currents recorded from βarr2+/+ and βarr2−/− neurons. The application of ω-conotoxin GVIA (ω-cntx) inhibited currents recorded from βarr2+/+ and βarr2−/− neurons. A, Traces are Ca2+ currents recorded (using the protocol in Fig. 1A) in the absence and presence of ω-conotoxin GVIA (10 μm) and DAMGO (1 μm) at sweeps 5, 9, 11, and 13 in the plot of current amplitude versus time in the graph below. B, Traces are Ca2+ currents recorded at sweeps 6, 9, 11, and 13 in the plot of current amplitude versus time in the graph below.
Together, these data suggest that the reduction in the inhibition by DAMGO caused by the absence of β-arrestin2 was attributable to neither a change in the level of N-type VGCCs nor a shift in the proportion of the μ receptor inhibitory coupling from N-type to non-N-type VGCCs.
The absence of β-arrestin2 does not reduce G-protein coupling to VGCCs
Because in βarr2−/− DRG neurons there is (1) no reduction in the number of μ receptors, (2) unaltered desensitization after exposure to DAMGO, and (3) an apparently normal complement of functional VGCCs, we next examined the possibility of defective transduction between the μ receptor and effector.
Inhibitory coupling of GPCRs to N-type channels in DRG neurons has both voltage-dependent and voltage-independent components (Diverse-Pierluissi et al., 1995; Raingo et al., 2007). The direct interaction of the βγ subunits with the VGCCs is voltage dependent (Ikeda, 1996). Strong depolarization reverses this interaction and facilitates current in the presence of GPCR activation. In contrast, voltage-independent coupling remains in the presence of strong depolarization. Thus, a reduced efficacy of opioid receptor coupling to VGCCs in βarr2−/− neurons could be associated with a shift in the relative contribution of voltage-dependent and voltage-independent inhibitory components. To test this hypothesis, we compared the ability of a strong depolarization to reverse the inhibition of VGCCs by DAMGO and GTP-γ-S in βarr2+/+ and βarr2−/− neurons (Fig. 6). We used a two-pulse protocol to examine the effects of a prepulse (80 ms duration) to 80 mV delivered 10 ms before a test pulse (10 ms duration) to 10 mV (Fig. 6A). We used Ba2+ as the charge carrier in these experiments to avoid possible Ca2+-dependent inactivation of VGCCs. Control currents recorded from βarr2+/+ neurons with a preceding prepulse (+PP) to 80 mV were 97 ± 2% (n = 14) of the amplitude of currents recorded in the absence of a prepulse (−PP) (Fig. 6A). Interestingly, the prepulse caused a significantly (p < 0.05) larger increase (106 ± 3%; n = 9) in current amplitude in experiments performed on βarr2−/− neurons (Fig. 6D). After the application of DAMGO (1 μm), the depolarizing prepulse caused a marked facilitation of Ba2+ currents evoked by the test pulse compared with those recorded in the absence of a prepulse (Fig. 6B). The current amplitudes in the presence of DAMGO were enhanced by a depolarizing prepulse to 123 ± 8 and 134 ± 10% of those recorded in the absence of a prepulse from βarr2+/+ and βarr2−/− neurons, respectively (Fig. 6D). Because the inhibition by DAMGO is larger in βarr2+/+ neurons than βarr2−/− neurons (Fig. 1), a similar level of voltage-dependent facilitation in the presence of DAMGO could indicate a greater degree of voltage-dependent reversal of inhibition in βarr2−/− neurons. However, the depolarizing prepulse caused basal current facilitation in βarr2−/− neurons in the absence of DAMGO that was not seen in βarr2+/+ neurons (Fig. 6A). When the basal facilitation of βarr2−/− VGCC activity is taken into consideration, there is little difference in the efficacy of the 80 mV depolarizing prepulse to reverse the inhibitory effect of DAMGO on VGCC activity in βarr2−/− and βarr2+/+ neurons.
Figure 6.
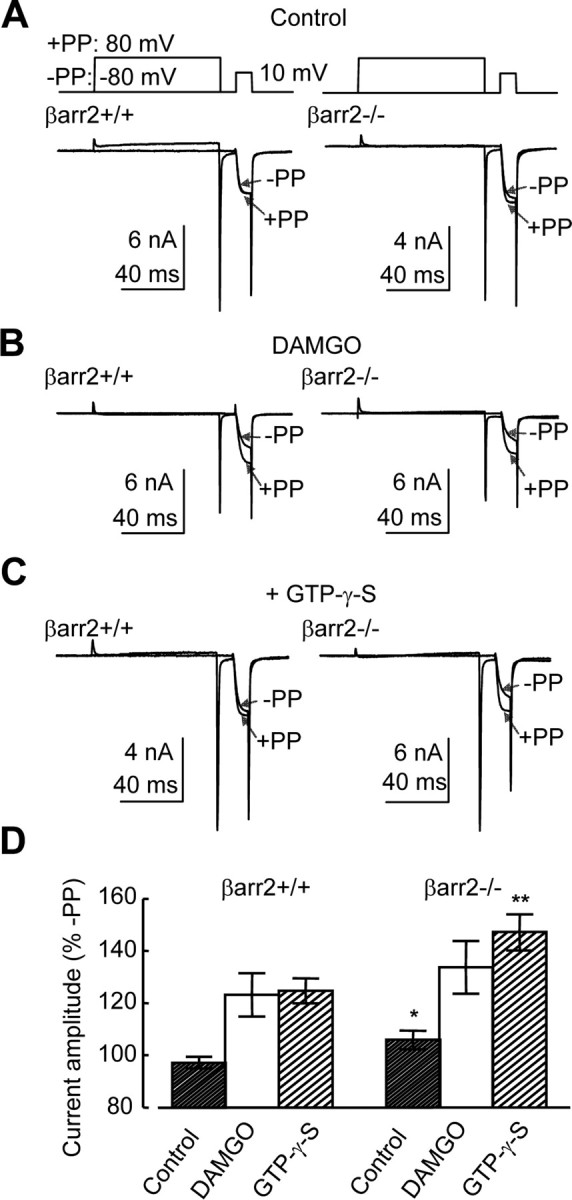
The absence of β-arrestin2 increases voltage-dependent facilitation of VGCCs recorded from DRG neurons. A, Typical control whole-cell Ba2+ currents recorded from βarr2+/+ (left) and βarr2−/− (right) neurons. Currents were evoked using the two-step voltage protocol illustrated above. The first sweep contains a single 10 ms test pulse to 10 mV from −80 mV. In the second sweep, the test pulse is preceded by a prepulse from −80 to 80 mV. Each trace depicts two current sweeps recorded from the same cell in the presence (+PP) and absence (−PP) of a depolarizing prepulse. Under control conditions, facilitation occurred in βarr2−/− but not βarr2+/+ neurons. B, The use of the two-step protocol caused a prepulse-evoked facilitation of Ba2+ currents recorded from βarr2+/+ (left) and βarr2−/− (right) neurons in the presence of DAMGO (1 μm). C, The depolarizing prepulse also caused facilitation of Ba2+ currents recorded from βarr2+/+ (left) and βarr2−/− (right) neurons evoked by the test pulse when GTP-γ-S (300 μm) was included in the electrode. D, Mean data from at least five βarr2+/+ and βarr2−/− neurons recorded in the presence and absence of either DAMGO or intracellular GTP-γ-S. The absence of β-arrestin2 caused a significant increase in the voltage-dependent facilitation in both the absence (*p < 0.05, ANOVA, post hoc Tukey's test) and presence (**p < 0.01) of GTP-γ-S. Error bars represent ±SEM.
We further explored the possibility of disrupted inhibitory G-protein coupling to VGCCs caused by the absence of β-arrestin2 by comparing the effect on VGCCs of GTP-γ-S (300 μm) applied through the recording electrode to the inside of βarr2−/− and βarr2+/+ neurons. The level of inhibition elicited by GTP-γ-S in the electrode solution was evaluated using the depolarizing prepulse approach (Fig. 6C). Inclusion of GTP-γ-S in the electrode increased facilitation in both neuronal populations (Fig. 6D). However, as seen under control conditions (Fig. 6A), voltage-dependent facilitation was significantly (p < 0.01) greater in βarr2−/− neurons (149 ± 7%; n = 10 of control) compared with βarr2+/+ neurons (125 ± 5%; n = 13) (Fig. 6D).
Together, these data demonstrate that reduced coupling between μ receptors and VGCCs in βarr2−/− DRG neurons is not caused by a disruption of inhibitory G-protein coupling.
Increased constitutive coupling of μ receptors to VGCCs in βarr2−/− neurons
The increased depolarization-evoked facilitation in βarr2−/− neurons observed under control conditions (Fig. 6A,D) may be caused by greater constitutive inhibition of VGCCs in βarr2−/− neurons compared with βarr2+/+ neurons (Fig. 6). Several GPCRs, including opioid receptors, exhibit constitutive coupling to cellular effectors (Costa and Herz, 1989; Wang et al., 1994; Sadee et al., 2005). Increased constitutive coupling of μ receptors to VGCCs in βarr2−/− neurons may also give rise to the reduced efficacy of μ agonists (Fig. 1), because receptors participating in constitutive coupling would not be available for mediating the effects of agonists such as morphine or DAMGO. Naltrexone has negative intrinsic activity and therefore acts as an inverse agonist able to inhibit constitutively active μ receptors (Sadee et al., 2005). We tested the effect of naltrexone on currents recorded from βarr2−/− and βarr2+/+ neurons (Fig. 7). We used the same protocol illustrated in Figure 6 to examine the effect of a prepulse to 80 mV on the current elicited by a test pulse to 10 mV recorded from βarr2−/− and βarr2+/+ neurons in the presence of naltrexone. GTP-γ-S (300 nm) was included in the electrode solution to increase the amplitude of facilitation (Fig. 6C,D). In the presence of GTP-γ-S, naltrexone had no significant effect on the level of facilitation in βarr2−/− neurons. Facilitation induced by the depolarizing prepulse was 139 ± 8 and 153 ± 7% (n = 6) in the presence and absence of naltrexone (1 μm), respectively (data not shown). GTP-γ-S is nonhydrolysable and therefore interacts irreversibly with activated G-protein α subunits. Thus, it is not possible to reverse such an interaction should it have taken place before administration of the inverse agonist naltrexone. There are several mechanisms that may facilitate the exchange of GDP, bound to Gα subunits, by intracellular GTP-γ-S: (1) endogenous agonist-mediated GPCR activity, (2) constitutive GPCR activity, and (3) intrinsic Gα subunit activity. Therefore, although it is possible that naltrexone slows the exchange process by inhibiting constitutive μ receptor activity in βarr2−/− neurons, this effect may be masked by the other mechanisms in play. Therefore, we performed the same experiment without inclusion of GTP-γ-S in the recording electrode (Fig. 7A). Naltrexone (1 μm) had no effect on the ratio of the current amplitude of the Ba2+ current recorded from βarr2+/+ neurons in the presence (I+80) and absence (I−80) of a prepulse (Fig. 7B). In contrast, naltrexone caused a significant (p < 0.01) inhibition of the I+80/I−80 ratio recorded from βarr2−/− neurons (Fig. 7B). The effects of inverse agonists such as naltrexone can be reversed by neutral competitive antagonists (Costa and Herz, 1989). We tested the effects of CTAP (Cys-Tyr-d-Trp-Arg-Thr-Pen-Thr-NH2) (1 μm), a μ receptor antagonist that has negligible negative efficacy (Wang et al., 1994), on the I+80/I−80 ratio recorded from both βarr2−/− and βarr2+/+ neurons (Fig. 7A,B). Consistent with the hypothesis that CTAP lacks negative efficacy, the antagonist had no effect on the I+80/I−80 ratio recorded from either βarr2−/− or βarr2+/+ neurons (Fig. 7A,B). However, CTAP did prevent the inhibitory effect of naltrexone on the I+80/I−80 ratio recorded from βarr2−/− neurons (Fig. 7B). These data support the hypothesis that naltrexone is acting as an inverse agonist at the μ receptor to reduce constitutive inhibitory coupling to VGCCs in βarr2−/− neurons but not βarr2+/+ neurons. The data suggest that constitutive coupling could account for the reduced efficacy of μ agonists as inhibitors of VGCCs in βarr2−/− compared with βarr2+/+ neurons.
Figure 7.

Constitutive inhibitory coupling of μ receptors to VGCCs in βarr2−/− neurons. A, Whole-cell Ba2+ currents recorded from βarr2+/+ (top traces) and βarr2−/− (bottom traces) DRG neurons. Currents were stimulated using a two-step voltage protocol illustrate in Figure 6. Each trace depicts two current sweeps recorded from a cell in the presence (+PP) and absence (−PP) of a depolarizing prepulse to 80 mV. Currents were recorded in the presence of the inverse agonist naltrexone (1 μm), the neutral antagonist CTAP (1 μm), or a combination of both drugs. Each pair of currents was recorded from a different cell. B, The bar graph of percentage facilitation by the 80 mV prepulse in the absence and presence of naltrexone and/or CTAP reveals that neither drug had a significant effect in βarr2+/+ neurons. In contrast, naltrexone inhibited the amplitude of the Ba2+ current activated in βarr2−/− neurons after the prepulse to 80 mV expressed as a percentage of that recorded in the absence of the prepulse (*p < 0.01, ANOVA, post hoc Tukey's test). Error bars represent ±SEM.
Constitutive recycling of μ receptors is impaired in βarr2−/− DRG neurons
Despite evidence for the constitutive activity of opioid receptors, there is little information regarding its functional role. Recent studies of other GPCRs suggest that constitutive activity drives constitutive recycling (Morris et al., 2004; Leterrier et al., 2006). Thus, it is possible that the impact of constitutively active GPCRs under normal circumstances is minimized by constitutive internalization. If this is the case, then increased constitutive activity in βarr2−/− neurons could be caused by a deficit of constitutive recycling, leading to an increase in spontaneous inhibitory coupling of μ receptors to VGCCs. We used flow cytometry (as described in Fig. 2B,C) to compare constitutive recycling in βarr2−/− and βarr2+/+ neurons. Exposure for 30 min to monensin (300 nm), an inhibitor of μ receptor recycling that blocks endosomal acidification causing receptor accumulation within the cytoplasm (Koch et al., 1998), decreased cell-surface APC-visualized μ receptor antibody labeling of βarr2+/+ DRG neurons. The FI of monensin-treated βarr2+/+ neurons was reduced to 86 ± 4% of that of untreated βarr2+/+ neurons (*p < 0.05, Student's t test; n = 6). In contrast, monensin had no effect on μ receptor levels in βarr2−/− neurons (FI of 101 ± 3% of untreated neurons; n = 10), indicating that constitutive recycling of the μ receptor is β-arrestin2 dependent. These data suggest that μ receptors are constitutively recycled in wild-type DRG neurons in a monensin-sensitive manner. Such GPCR recycling typically occurs within 200 nm of the plasma membrane and is therefore undetectable by traditional CLSM. This may account for the μ receptor labeling shown in Figure 2A, which appeared unaffected by β-arrestin2 deletion.
Aberrant distribution and phosphorylation of c-Src in βarr2−/− DRG neurons
A role for β-arrestin2 in agonist-induced internalization has been well established for several GPCRs, including the β2AR, α1a adrenergic receptors and angiotensin type 1 (AT1) receptors (Lefkowitz, 1998; Fessart et al., 2005; Pediani et al., 2005). Agonist stimulation of the β2AR causes β-arrestin-dependent formation of a complex between the receptor and the nonreceptor tyrosine kinase c-Src (Luttrell et al., 1999). In the case of the AT1 receptor and β2AR, c-Src is required for normal agonist-induced receptor internalization (Shumay et al., 2002; Fessart et al., 2005). We therefore investigated whether c-Src participates in β-arrestin2-dependent constitutive trafficking of μ receptors in DRG neurons.
Because β-arrestins are required for c-Src targeting to GPCRs (Miller et al., 2000), we first examined whether there is aberrant localization of c-Src in βarr2−/− neurons. We used high-resolution CLSM to compare the cellular distribution of c-Src in βarr2+/+ and βarr2−/− neurons (Fig. 8A). A dense “lattice-like” c-Src distribution is evident at the cell membrane in βarr2+/+ neurons, whereas βarr2−/− neurons exhibit a less structured c-Src distribution (Fig. 8A). This apparent qualitative difference in the distribution of c-Src was not evident in the processes of DRGs, perhaps attributable to the detection limits of CLSM. Altered c-Src distribution was hard to discern using immunocytochemistry; therefore, we used an antibody to the phosphorylated catalytic domain of c-Src, Y416 (Lefkowitz, 1998), to determine whether coupling between μ receptors and c-Src was disrupted by the absence of β-arrestin2. DAMGO (1 μm) exposure for 20 min caused a small but significant increase (p < 0.05 vs untreated neurons; n = 8) in phosphorylation and hence activation of c-Src in cultured βarr2+/+ DRG neurons as quantified by flow cytometry (Fig. 8B). In contrast, no increase in Y416 phospho-Src was observed after 20 min of βarr2−/− neuron stimulation by DAMGO. Thus, the appearance of aberrant targeting of c-Src in βarr2−/− neurons (Fig. 8A) was accompanied by an inability of DAMGO to induce phosphorylation of c-Src at Y416 (Fig. 8B).
Figure 8.
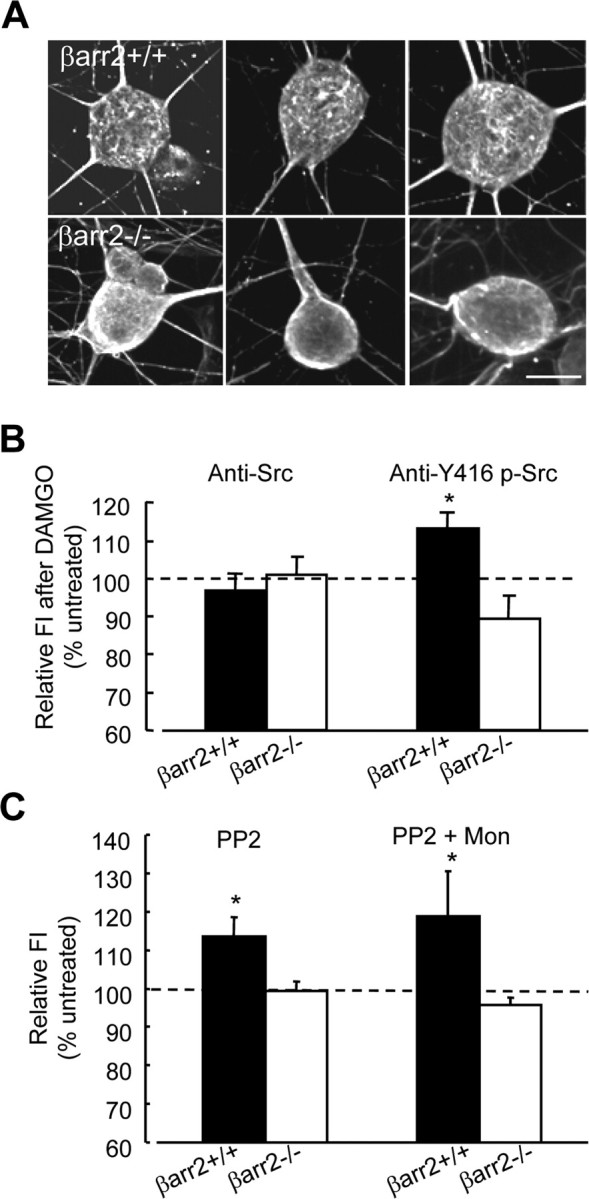
Constitutive trafficking of the μ receptor is β-arrestin2 and Src dependent. A, Confocal laser-scanning microscopy of c-Src-labeled DRG neurons showed a more dense and structured distribution of c-Src in the cell bodies of βarr2+/+ than βarr2−/− neurons. These cells were imaged by taking ∼30 serial x–y scans at 0.4–0.5 μm z-scale intervals through a 100× oil-immersion objective and merging the image stack into a single maximum-intensity projection of each cell. Scale bar, 5 μm. B, Flow cytometry was used to determine the effect of DAMGO, a μ agonist, on Src (using an APC-conjugated anti-Src antibody) and phospho-Y416 c-Src (using a fluorescein-conjugated anti-Y416 p-Src antibody) on the DRG neurons in R1 (see Fig. 2B,C). FI was expressed relative to that in cells that were not exposed to DAMGO. In βarr2+/+ neurons, DAMGO (20 min, 1 μm) caused an increase (*p < 0.05) in Y416 phosphorylation without affecting the levels of c-Src. There was no increase in Y416 phosphorylation in βarr2−/− neurons, indicating that μ agonist activation of c-Src is β-arrestin2 dependent. C, Treatment of βarr2+/+ neurons with the c-Src inhibitor PP2 (4 h, 10 μm) increased cell-surface μ receptor levels relative to untreated neurons (*p < 0.05; n = 9) determined by measurement of FI. In contrast to untreated neurons in which monensin reduced cell-surface μ receptor levels (see Results), monensin (300 nm) applied during the last 30 min of PP2 treatment had no effect. PP2 had no effect on cell-surface μ receptor levels in βarr2−/− neurons applied either alone or in combination with monensin. For designation of the neuronal population, see Figure 2, B and C. Statistical significance was determined by the Student's t test. Error bars represent ±SEM.
Inhibition of c-Src activity reduces constitutive recycling in DRG neurons
We examined whether defective c-Src signaling may be responsible for aberrant μ receptor trafficking and function in βarr2−/− neurons. Exposure of βarr2+/+ neurons to the selective Src family kinase inhibitor PP2 [4-amino-5-(4-chlorophenyl)-(t-butyl)pyrazolo[3,4-d]pyrimidine] (10 μm) for 4 h caused a small increase in cell-surface μ receptor antibody labeling (113 ± 4% of untreated βarr2+/+ neurons) determined by flow cytometry (Fig. 8C). The application of PP2 (10 μm) for 4 h with monensin (300 nm) during the final 30 min of PP2 exposure caused no significant increase in the level of cell-surface μ receptors compared with the increase observed when βarr2+/+ neurons were treated with PP2 alone (Fig. 8C). These data suggest that Src inhibition reduces the constitutive internalization of μ receptors, thereby ablating the effect of monensin on cell-surface receptor levels. PP2 had no effect on cell-surface μ receptor levels in βarr2−/− neurons when applied either alone or in combination with monensin (Fig. 8C). Together, these data are consistent with the hypothesis that defective c-Src signaling results in defective constitutive internalization and recycling of μ receptors in βarr2−/− DRG neurons.
Aberrant Src signaling reduces the inhibitory effect of μ agonists on VGCCs and increases constitutive activity of μ receptors in βarr2−/− DRG neurons
The absence of β-arrestin from DRG neurons disrupts both c-Src distribution and activation by a μ receptor agonist (Fig. 8). We investigated whether defective c-Src signaling may also give rise to the aberrant coupling of μ receptors to VGCCs in βarr2−/− DRG neurons. The effect of PP2 on inhibition of VGCCs by opioids in βarr2+/+ neurons (Fig. 9A) is similar to the effect of a lack of β-arrestin2 (Fig. 1B). Application of PP2 (10 μm, 5 h before recording) reduced the inhibition by DAMGO (1 μm) of VGCCs recorded from βarr2+/+ neurons (p < 0.01) (Fig. 9A). In contrast, PP2 had no significant effect on the inhibition by DAMGO of VGCCs recorded from βarr2−/− neurons (data not shown). The reduction of the inhibitory effect of morphine (1 μm) by PP2 in βarr2+/+ neurons was statistically insignificant. However, together these data support the hypothesis that the reduced efficacy of VGCC inhibition by μ agonists in βarr2−/− neurons is caused by aberrant c-Src signaling.
Figure 9.
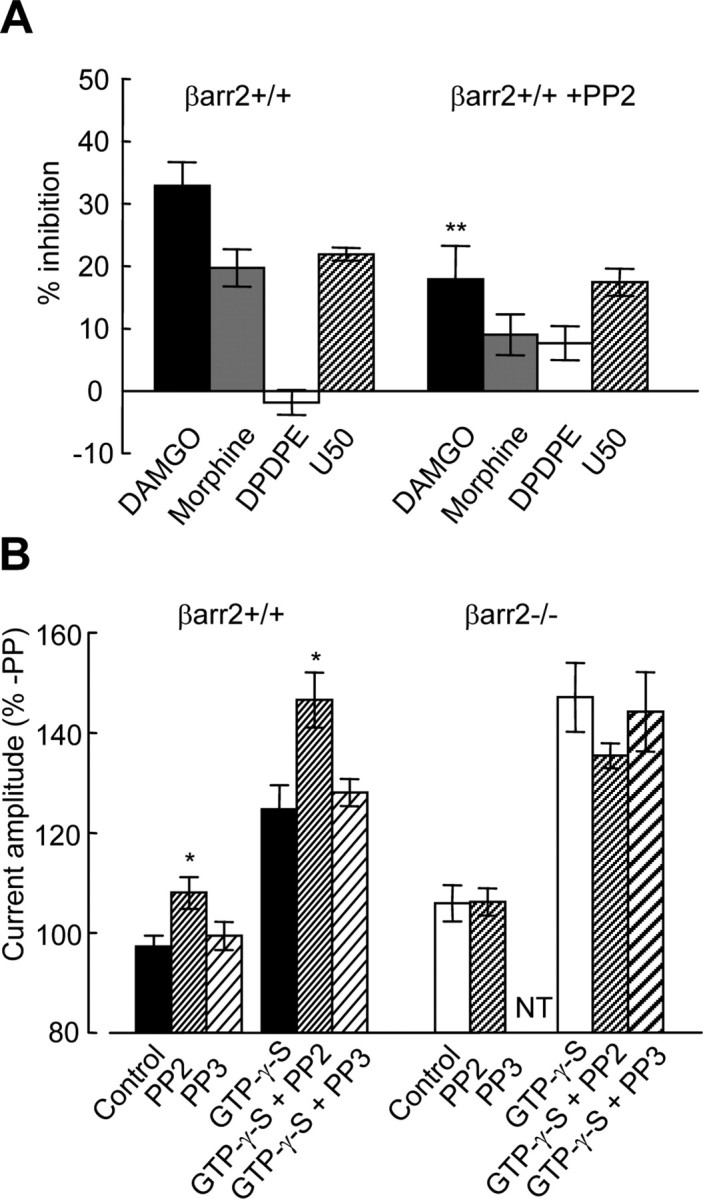
The Src inhibitor PP2 mimics the effects of the absence of β-arrestin2 on μ receptor coupling to VGCCs. A, The bar graph illustrates the amplitudes of opioid-mediated inhibition of Ca2+ currents recorded from βarr2+/+ DRG neurons in the absence or presence of the Src inhibitor PP2. Opioids were applied at a concentration of 1 μm, whereas Ca2+ currents were activated by depolarizing from −80 to 10 mV (as shown in Fig. 1A). Statistical significance (**p < 0.01) was determined using ANOVA with the post hoc Tukey's test. B, Voltage-dependent facilitation of Ba2+ currents recorded from βarr2+/+ and βarr2−/− neurons with and without GTP-γ-S (300 μm) in the recording electrode (voltage protocol illustrated in Fig. 5A). The Src inhibitor PP2 increased the level of facilitation (in both the presence and absence of GTP-γ-S) in βarr2+/+ but not βarr2−/− neurons (*p < 0.05). In contrast, PP3 had no effect on facilitation in either βarr2+/+ or βarr2−/− neurons. Statistical significance was determined using ANOVA with the post hoc Tukey's test. Error bars represent ±SEM.
In βarr2−/− neurons, reduced efficacy of μ agonists (Fig. 1) is associated with increased agonist-independent constitutive μ receptor inhibitory coupling to VGCCs (Fig. 7). We examined whether reduced inhibition by DAMGO and morphine caused by PP2 inhibition of Src is also associated with increased constitutive inhibition of VGCCs in βarr2+/+ DRG neurons (Fig. 9B). We tested the effect of PP2 application (10 μm, 5 h before recording) on prepulse-evoked facilitation recorded from both βarr2+/+ and βarr2−/− neurons (Fig. 9B). Consistent with the hypothesis that defective Src signaling gives rise to increased constitutive inhibition, PP2 caused an increase in the I+80/I−80 ratio recorded from βarr2+/+ neurons (p < 0.05) (Fig. 9B). This effect was evident either with or without GTP-γ-S (300 μm) in the recording electrode. In contrast, the inactive analog of PP2, PP3 (4-amino-7-phenylpyrazol[3,4-d]pyrimidine) (10 μm, applied 5 h before recording), had no effect on the I+80/I−80 ratio in recordings from βarr2+/+ DRG neurons. The increase in the I+80/I−80 ratio induced by PP2 (Fig. 9B) mimicked that caused by the absence of β-arrestin2 in βarr2−/− neurons (Fig. 6). Consistent with the hypothesis that defective Src signaling gives rise to increased constitutive μ receptor coupling to VGCCs in the absence of β-arrestin2, PP2 had no effect on the I80/I−80 ratio in βarr2−/− neurons.
Discussion
A lack of β-arrestin2 reduced the efficacy of μ receptor agonists in DRG neurons without significantly affecting VDCC inhibition by δ, κ, or GABAB receptor agonists. This deficit occurred without a change in the contribution of N-type channels. Furthermore, there was no change in the relative proportion of voltage-dependent versus voltage-independent inhibitory μ receptor coupling to VGCCs.
The reduction in the morphine- and DAMGO-mediated inhibition of VGCCs was accompanied by increased constitutive coupling of μ receptors to VGCCs in βarr2−/− neurons and an absence of monensin-sensitive constitutive μ receptor recycling. The absence of β-arrestin2 also prevented DAMGO from activating c-Src, a functional deficit associated with an aberrant localization of the kinase in βarr2−/− neurons. Accordingly, the phenotype of βarr2−/− neurons was mimicked in βarr2+/+ neurons treated with the Src inhibitor PP2. These findings suggest a novel role for the β-arrestin2/c-Src complex, recycling constitutively active μ receptors, thus limiting tonic coupling to cellular effectors.
Agonist activation of GPCRs recruits β-arrestins (Lefkowitz and Shenoy, 2005) which link the membrane-bound receptor with clathrin and α-adaptin-2 to initiate recycling and resensitization (Pippig et al., 1995; Goodman et al., 1996; Laporte et al., 1999, 2000). β-Arrestin would therefore be expected to accelerate μ receptor desensitization (Lowe et al., 2002) and enhance analgesic tolerance to morphine. However, βarr2−/− mice exhibit delayed tolerance and enhanced morphine-evoked analgesia and reward, whereas respiratory depression and constipation are diminished (Bohn et al., 1999, 2000, 2002, 2004; Raehal et al., 2005). This complex phenotype cannot be explained solely by the classical role of β-arrestins in promoting internalization of agonist-activated receptors.
Morphine is unusual among agonists; it causes little μ receptor internalization (Keith et al., 1996). Indeed, this property has been blamed for pronounced tolerance associated with morphine exposure (Whistler et al., 1999; Evans, 2000). The RAVE (for relative activity versus endocytosis) hypothesis postulates that non-internalizing μ agonists cause greater tolerance than do internalizing agonists. However, recent studies of μ receptor coupling to inwardly rectifying K+ channels demonstrate that neither the onset of nor the recovery from receptor desensitization (phenomena linked to tolerance) are ablated by inhibitors of internalization (Arttamangkul et al., 2006; Dang and Christie, 2006). In fact, recovery from desensitization occurs faster in the presence of inhibitors of internalization (Dang and Christie, 2006). Furthermore, the onset of desensitization, measured by μ receptor–VGCC coupling, is unrelated to internalization in DRG neurons (Walwyn et al., 2006). Consistent with these studies, we found no difference in the onset of desensitization of μ receptor coupling to VGCCs activated by morphine or DAMGO in βarr2+/+ and βarr2−/− neurons. Therefore, mechanisms other than classical agonist-mediated endocytosis must be at play to explain morphine-induced tolerance.
In addition to their role in agonist-induced internalization, β-arrestins participate in constitutive GPCR recycling (Pediani et al., 2005; Leterrier et al., 2006). In the absence of μ agonists, monensin, an inhibitor of GPCR recycling that accumulates receptors within the cytoplasm (Koch et al., 1998), reduced cell-surface μ receptor expression in βarr2+/+ neurons. These data support a role for ongoing constitutive recycling of neuronal μ receptors similar to that observed previously for recombinant receptors (Alvarez et al., 2002; Johnson et al., 2006, Walwyn et al., 2006). A lack of monensin-sensitive constitutive recycling in βarr2−/− neurons indicates a requirement for β-arrestin2 in keeping with other GPCRs that constitutively recycle in a β-arrestin-dependent manner (Pula et al., 2004; Pediani et al., 2005).
β-Arrestins target several proteins to μ receptors, including c-Src, which participates in μ receptor signaling (Kato et al., 2006). c-Src was refractory to activation by DAMGO in βarr2−/− neurons. The role of c-Src in facilitating ligand-induced internalization of the β2AR is well established (Shumay et al., 2002), affecting both the clathrin (Luttrell et al., 1999; Miller et al., 2000; van Koppen 2001; Fessart et al., 2005) and caveolin (Shajahan et al., 2004; Khan et al., 2006) internalization pathways. Inhibition of Src in βarr2+/+ neurons by PP2 increased μ receptor surface expression and abolished the monensin-induced downregulation of surface receptors. It is therefore consistent that c-Src inactivity, either through β-arrestin2 deletion or pharmacological inhibition, affected internalization and recycling, increasing cell-surface μ receptors. We therefore conclude that c-Src, recruited by β-arrestin2, is required for constitutive μ receptor recycling. Interestingly, p38 MAPK also initiates μ receptor constitutive internalization (Mace et al., 2005). Because MAPK and c-Src transduction pathways are orchestrated by β-arrestins (Lefkowitz and Shenoy, 2005), it will be interesting to establish the point at which their actions converge to regulate μ receptor recycling.
Inhibitory coupling of both μ and GABAB receptors to N-type VGCCs exhibits voltage-dependent and voltage-independent components (Richman et al., 2004; Raingo et al., 2007). The former requires the interaction of Src with a specific motif within a variant of the CaV2.2 α1 subunit that contains exon 37a (Raingo et al., 2007). Exons 37a and b are mutually exclusive in rat DRG neurons (Bell et al., 2004). A reduction in Src signaling caused by a lack of β-arrestin2 could reduce voltage-independent coupling of μ receptors to N-type channels. However, we observed no reduction in the voltage dependence of inhibition by DAMGO in βarr2−/− neurons. The whole-cell patch-clamp configuration may minimize the contribution of voltage-independent coupling to N-type channels (Raingo et al., 2007). Furthermore, the expression of exon 37a of the CaV2.2 α1 subunit is enriched is specific rat DRG neurons, particularly those containing low-threshold T-type VGCCs. Ca2+ current–voltage relationships revealed a lack of a low-threshold component; therefore, exon 37a may have been underrepresented in the cultured mouse neurons used in our study.
β-Arrestin1 participates in the internalization of N-type channels, raising the possibility that β-arrestin2 may play a similar role (Puckerin et al., 2006). However, we found that the functional contribution of N-type channels in DRG neurons was unaffected by the absence of β-arrestin2. Thus, β-arrestin1 may play a specific role in regulating N-type channel internalization/recycling. It remains to be determined whether this involves c-Src.
There is increasing evidence supporting a role for agonist-independent GPCR activity in triggering receptor recycling. Although neutral competitive antagonists are able to internalize with constitutively recycling GPCRs, the application of inverse agonists inhibits recycling, leading to cell-surface receptor upregulation (McCune et al., 2000; Miserey-Lenkei et al., 2002; Morris et al., 2004; Leterrier et al., 2006). Furthermore, mutations in the μ receptor, which enhance constitutive activity, also increase agonist-independent recycling in a manner that can be inhibited by inverse agonists (Li et al., 2001).
Many (perhaps all) wild-type GPCRs exhibit agonist-independent constitutive coupling to G-proteins (Costa and Herz, 1989; Costa and Cotecchia, 2005). Interestingly, the constitutive activity of μ receptors increases with morphine exposure and may participate in tolerance (Wang et al., 1994; Sadee et al., 2005). The inverse agonist naltrexone inhibits the constitutive activity of μ receptors, whereas the neutral antagonist CTAP lacks negative efficacy but can displace and thereby inhibit the action of naltrexone. We assayed μ receptor constitutive activity by exploiting the ability of strong depolarization to reverse inhibitory coupling to VGCCs. A strong depolarization drives Gβγ subunits off of inhibited N- and P/Q-type channels, causing current facilitation (Ikeda, 1996). Depolarizing prepulses were relatively ineffective at facilitating currents in βarr2+/+ neurons. In contrast, facilitation was significantly enhanced in βarr2−/− neurons and in βarr2+/+ neurons treated with the Src inhibitor PP2. The facilitation in βarr2−/− neurons was abolished by naltrexone but not by CTAP. CTAP did prevent the inhibitory effect of naltrexone on voltage-dependent facilitation. Therefore, a lack of c-Src activity caused by the absence of β-arrestin2 increased the constitutive μ receptor inhibition of VGCCs.
Together, our data suggest that c-Src-dependent μ receptor recycling limits the presence of constitutively active receptors in the cell membrane, thus minimizing constitutive inhibition of VGCCs. We hypothesize that constitutive μ receptor activity recruits β-arrestin2 and c-Src, leading to constitutive recycling, returning the μ receptor back to the cell membrane in a quiescent state. How might a deficit in this process, caused by the absence of β-arrestin2, give rise to reduced tolerance to morphine in βarr2−/− mice? In βarr2+/+ neurons, prolonged morphine exposure may increase μ receptor constitutive activity (Wang et al., 1994), triggering increased β-arrestin2/Src-dependent recycling. More receptors would therefore be located beneath the cell surface, perhaps deactivated by the action of Src. These events may contribute to morphine tolerance. In comparison, in the absence of β-arrestin2, there is a failure of internalization and recycling of constitutively active receptors. Therefore, more active receptors will be located in the membrane even after morphine exposure. This scenario may also contribute to the increased analgesic activity of morphine in βarr2−/− mice (Bohn et al., 2000).
Morphine preferentially recruits β-arrestin2 to the μ receptor (Bohn et al., 2004). We hypothesize that μ agonists, which are poor inducers of constitutive activity, less effectively recruit β-arrestin2 and cause less tolerance. Drug-induced downregulation of the β-arrestin2-mediated signaling process may ameliorate tolerance associated with analgesic opioids. Our data demonstrating that c-Src mediates many of the actions of β-arrestin2 on the μ receptor raise the possibility that this kinase may also be a suitable target for modulation of the therapeutic profiles of μ agonists.
Footnotes
This work was supported by National Institutes of Health/National Institute on Drug Abuse Grants DA05010 and DA00484. We thank Dr. R. Lefkowitz for providing β-arrestin2−/− mice. Flow cytometry was performed in the Jonsson Comprehensive Cancer Center and Center for AIDS Research Flow Cytometry Core Facility, University of California, Los Angeles.
References
- Alvarez VA, Arttamangkul S, Dang V, Salem A, Whistler JL, Von Zastrow M, Grandy DK, Williams JT. μ-Opioid receptors: Ligand-dependent activation of potassium conductance, desensitization, and internalization. J Neurosci. 2002;22:5769–5776. doi: 10.1523/JNEUROSCI.22-13-05769.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arttamangkul S, Torrecilla M, Kobayashi K, Okano H, Williams JT. Separation of μ-opioid receptor desensitization and internalization: endogenous receptors in primary neuronal cultures. J Neurosci. 2006;26:4118–4125. doi: 10.1523/JNEUROSCI.0303-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell TJ, Thaler C, Castiglioni AJ, Helton TD, Lipscombe D. Cell-specific alternative splicing increases Ca2+ channel current density in the pain pathway. Neuron. 2004;41:3–4. doi: 10.1016/s0896-6273(03)00801-8. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking β-arrestin 2. Science. 1999;286:2495–2498. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. μ-Opioid receptor desensitization by β-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000;408:720–723. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Caron MG. Differential mechanisms of morphine antinociceptive tolerance revealed in β-arrestin-2 knock-out mice. J Neurosci. 2002;22:10494–10500. doi: 10.1523/JNEUROSCI.22-23-10494.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Dykstra LA, Lefkowitz RJ, Caron MG, Barak LS. Relative opioid efficacy is determined by the complements of the G protein-coupled receptor desensitization machinery. Mol Pharmacol. 2004;66:106–112. doi: 10.1124/mol.66.1.106. [DOI] [PubMed] [Google Scholar]
- Cen B, Xiong Y, Ma L, Pei G. Direct and differential interaction of β-arrestins with the intracellular domains of different opioid receptors. Mol Pharmacol. 2001;59:758–764. doi: 10.1124/mol.59.4.758. [DOI] [PubMed] [Google Scholar]
- Chen J, Sochivko D, Beck H, Marechal D, Wiestler OD, Becker AJ. Activity-induced expression of common reference genes in individual CNS neurons. Lab Invest. 2001;81:913–916. doi: 10.1038/labinvest.3780300. [DOI] [PubMed] [Google Scholar]
- Costa T, Cotecchia S. Historical review: Negative efficacy and the constitutive activity of G-protein-coupled receptors. Trends Pharmacol Sci. 2005;26:618–624. doi: 10.1016/j.tips.2005.10.009. [DOI] [PubMed] [Google Scholar]
- Costa T, Herz A. Antagonists with negative intrinsic activity at δ opioid receptors coupled to GTP-binding proteins. Proc Natl Acad Sci USA. 1989;86:7321–7325. doi: 10.1073/pnas.86.19.7321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang V, Christie MJ. Beta-arrestin2 independent regulation of mu opioid receptor in locus coeruleus neurons. Soc Neurosci Abstr. 2006;32:426–11. [Google Scholar]
- Diverse-Pierluissi M, Goldsmith PK, Dunlap K. Transmitter-mediated inhibition of N-type calcium channels in sensory neurons involves multiple GTP-binding proteins and subunits. Neuron. 1995;14:191–200. doi: 10.1016/0896-6273(95)90254-6. [DOI] [PubMed] [Google Scholar]
- Evans CJ. Agonist selective μ-opioid receptor trafficking in rat central nervous system. Mol Psychiatry. 2000;5:121. doi: 10.1038/sj.mp.4000734. [DOI] [PubMed] [Google Scholar]
- Fessart D, Simaan M, Laporte SA. c-Src regulates clathrin adapter protein 2 interaction with β-arrestin and the angiotensin II type 1 receptor during clathrin-mediated internalization. Mol Endocrinol. 2005;19:491–503. doi: 10.1210/me.2004-0246. [DOI] [PubMed] [Google Scholar]
- Goodman OB, Jr, Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, Keen JH, Benovic JL. β-Arrestin acts as a clathrin adaptor in endocytosis of the β2-adrenergic receptor. Nature. 1996;383:447–450. doi: 10.1038/383447a0. [DOI] [PubMed] [Google Scholar]
- Groer CE, Tidgewell K, Moyer RA, Harding WW, Rothman RB, Prisinzano TE, Bohn LM. An opioid agonist that does not induce μ opioid receptor - arrestin interactions or receptor internalization. Mol Pharmacol. 2007;71:549–557. doi: 10.1124/mol.106.028258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarna M, Bartolini A, Ghelardini C, Galeotti N, Bracci L, Stefano GB, Bianchi E. Anti-μ opioid antiserum against the third external loop of the cloned μ-opioid receptor acts as a μ receptor neutral antagonist. Brain Res Mol Brain Res. 2003;119:100–110. doi: 10.1016/j.molbrainres.2003.08.019. [DOI] [PubMed] [Google Scholar]
- Haberstock-Debic H, Kim KA, Yu YJ, von Zastrow M. Morphine promotes rapid, arrestin-dependent endocytosis of μ-opioid receptors in striatal neurons. J Neurosci. 2005;25:7847–7857. doi: 10.1523/JNEUROSCI.5045-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G-protein βγ subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- Johnson EA, Oldfield S, Braksator E, Gonzalez-Cuello A, Couch D, Hall KJ, Mundell SJ, Bailey CP, Kelly E, Henderson G. Agonist-selective mechanisms of μ-opioid receptor desensitization in human embryonic kidney 293 cells. Mol Pharmacol. 2006;70:676–685. doi: 10.1124/mol.106.022376. [DOI] [PubMed] [Google Scholar]
- Kato H, Narita M, Miyoshi K, Narita M, Asato M, Hareyama N, Nozaki H, Takagi T, Suzuki M, Suzuki T. Implication of Src family kinase-dependent phosphorylation of NR2B subunit-containing NMDA receptor in the rewarding effect of morphine. Nihon Shinkei Seishin Yakurigaku Zasshi. 2006;26:119–124. [PubMed] [Google Scholar]
- Keith DE, Murray SR, Zaki PA, Chu PC, Lissin DV, Kang L, Evans CJ, von Zastrow M. Morphine activates opioid receptors without causing their rapid internalization. J Biol Chem. 1996;271:19021–19024. doi: 10.1074/jbc.271.32.19021. [DOI] [PubMed] [Google Scholar]
- Khan EM, Heidinger JM, Levy M, Lisanti MP, Ravid T, Goldkorn T. Epidermal growth factor receptor exposed to oxidative stress undergoes Src- and caveolin-1-dependent perinuclear trafficking. J Biol Chem. 2006;281:14486–14493. doi: 10.1074/jbc.M509332200. [DOI] [PubMed] [Google Scholar]
- Koch T, Schulz S, Schroder H, Wolf R, Raulf E, Hollt V. Carboxyl-terminal splicing of the rat μ opioid receptor modulates agonist-mediated internalization and receptor resensitization. J Biol Chem. 1998;273:13652–13657. doi: 10.1074/jbc.273.22.13652. [DOI] [PubMed] [Google Scholar]
- Koch T, Widera A, Bartzsch K, Schulz S, Brandenburg LO, Wundrack N, Beyer A, Grecksch G, Hollt V. Receptor endocytosis counteracts the development of opioid tolerance. Mol Pharmacol. 2005;67:280–287. doi: 10.1124/mol.104.004994. [DOI] [PubMed] [Google Scholar]
- Kohout TA, Lefkowitz RJ. Regulation of G protein-coupled receptor kinases and arrestins during receptor desensitization. Mol Pharmacol. 2003;63:9–18. doi: 10.1124/mol.63.1.9. [DOI] [PubMed] [Google Scholar]
- Laporte SA, Oakley RH, Zhang J, Holt JA, Ferguson SS, Caron MG, Barak LS. The β2-adrenergic receptor/βarrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc Natl Acad Sci USA. 1999;96:3712–3717. doi: 10.1073/pnas.96.7.3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laporte SA, Oakley RH, Holt JA, Barak LS, Caron MG. The interaction of β-arrestin with the AP-2 adaptor is required for the clustering of β2-adrenergic receptor into clathrin-coated pits. J Biol Chem. 2000;275:23120–23126. doi: 10.1074/jbc.M002581200. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ. G protein-coupled receptors. III. New roles for receptor kinases and β-arrestins in receptor signaling and desensitization. J Biol Chem. 1998;273:18677–18680. doi: 10.1074/jbc.273.30.18677. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by β-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- Leterrier C, Laine J, Darmon M, Boudin H, Rossier J, Lenkei Z. Constitutive activation drives compartment-selective endocytosis and axonal targeting of type 1 cannabinoid receptors. J Neurosci. 2006;26:3141–3153. doi: 10.1523/JNEUROSCI.5437-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Chen C, Huang P, Liu-Chen LY. Inverse agonist up-regulates the constitutively active D3.49(164)Q mutant of the rat μ-opioid receptor by stabilizing the structure and blocking constitutive internalization and down-regulation. Mol Pharmacol. 2001;60:1064–1075. doi: 10.1124/mol.60.5.1064. [DOI] [PubMed] [Google Scholar]
- Lowe JD, Celver JP, Gurevich VV, Chavkin C. μ-Opioid receptors desensitize less rapidly than δ-opioid receptors due to less efficient activation of arrestin. J Biol Chem. 2002;277:15729–15735. doi: 10.1074/jbc.M200612200. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin F, Kawakatsu H, Owada K, Luttrell DK, Caron MG, Lefkowitz RJ. β-Arrestin-dependent formation of β2 adrenergic receptor-Src protein kinase complexes. Science. 1999;283:655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- Mace G, Miaczynska M, Zerial M, Nebreda AR. Phosphorylation of EEA1 by p38 MAP kinase regulates μ opioid receptor endocytosis. EMBO J. 2005;24:3235–3246. doi: 10.1038/sj.emboj.7600799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCune DF, Edelmann SE, Olges JR, Post GR, Waldrop BA, Waugh DJ, Perez DM, Piascik MT. Regulation of the cellular localization and signaling properties of the α1B- and α1D-adrenoceptors by agonists and inverse agonists. Mol Pharmacol. 2000;57:659–666. doi: 10.1124/mol.57.4.659. [DOI] [PubMed] [Google Scholar]
- Miller WE, Maudsley S, Ahn S, Khan KD, Luttrell LM, Lefkowitz RJ. β-Arrestin1 interacts with the catalytic domain of the tyrosine kinase c-SRC. Role of β-arrestin1-dependent targeting of c-SRC in receptor endocytosis. J Biol Chem. 2000;275:11312–11319. doi: 10.1074/jbc.275.15.11312. [DOI] [PubMed] [Google Scholar]
- Miserey-Lenkei S, Parnot C, Bardin S, Corvol P, Clauser E. Constitutive internalization of constitutively active agiotensin II AT1A receptor mutants is blocked by inverse agonists. J Biol Chem. 2002;277:5891–5901. doi: 10.1074/jbc.M108398200. [DOI] [PubMed] [Google Scholar]
- Morris DP, Price RR, Smith MP, Lei B, Schwinn DA. Cellular trafficking of human α1a-adrenergic receptors is continuous and primarily agonist-independent. Mol Pharmacol. 2004;66:843–854. doi: 10.1124/mol.104.000430. [DOI] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS. Differential affinities of visual arrestin, β arrestin1, and β arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem. 2000;275:17201–17210. doi: 10.1074/jbc.M910348199. [DOI] [PubMed] [Google Scholar]
- Paing MM, Stutts AB, Kohout TA, Lefkowitz RJ, Trejo J. β-Arrestins regulate protease-activated receptor-1 desensitization but not internalization or down-regulation. J Biol Chem. 2002;277:1292–1300. doi: 10.1074/jbc.M109160200. [DOI] [PubMed] [Google Scholar]
- Passmore GM. Dorsal root ganglion neurones in culture: a model system for identifying novel analgesic targets? J Pharmacol Toxicol Methods. 2005;51:201–208. doi: 10.1016/j.vascn.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Pediani JD, Colston JF, Caldwell D, Milligan G, Daly CJ, McGrath JC. β-Arrestin-dependent spontaneous α1a-adrenoceptor endocytosis causes intracellular transportation of α-blockers via recycling compartments. Mol Pharmacol. 2005;67:992–1004. doi: 10.1124/mol.104.008417. [DOI] [PubMed] [Google Scholar]
- Pippig S, Andexinger S, Lohse MJ. Sequestration and recycling of β2-adrenergic receptors permit receptor resensitization. Mol Pharmacol. 1995;47:666–676. [PubMed] [Google Scholar]
- Puckerin A, Liu L, Permaul N, Carman P, Lee J, Diverse-Pierluissi MA. Arrestin is required for agonist-induced trafficking of voltage-dependent Ca2+ channels. J Biol Chem. 2006;281:31131–31141. doi: 10.1074/jbc.M605000200. [DOI] [PubMed] [Google Scholar]
- Pula G, Mundell SJ, Roberts PJ, Kelly E. Agonist-independent internalization of metabotropic glutamate receptor 1a is arrestin- and clathrin-dependent and is suppressed by receptor inverse agonists. J Neurochem. 2004;89:1009–1020. doi: 10.1111/j.1471-4159.2004.02387.x. [DOI] [PubMed] [Google Scholar]
- Raehal KM, Walker JK, Bohn LM. Morphine side effects in β-arrestin2 knockout mice. J Pharmacol Exp Ther. 2005;314:1195–1201. doi: 10.1124/jpet.105.087254. [DOI] [PubMed] [Google Scholar]
- Raingo J, Castiglioni AJ, Lipscombe D. Alternative splicing controls G protein-dependent inhibition of N-type Ca2+ channels in nociceptors. Nat Neurosci. 2007;10:285–292. doi: 10.1038/nn1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman RW, Tombler E, Lau KK, Anantharam A, Rodriguez J, O'Bryan JP, Diverse-Pierluissi MA. N-type Ca2+ channels as scaffold proteins in the assembly of signaling molecules for GABAB receptor effects. J Biol Chem. 2004;279:24649–24658. doi: 10.1074/jbc.M312182200. [DOI] [PubMed] [Google Scholar]
- Rusin KI, Moises HC. μ-Opioid receptor activation reduces multiple components of high-threshold calcium current in rat sensory neurons. J Neurosci. 1995;15:4315–4327. doi: 10.1523/JNEUROSCI.15-06-04315.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadee W, Wang D, Bilsky EJ. Basal opioid receptor activity, neutral antagonists, and therapeutic opportunities. Life Sci. 2005;76:1427–1437. doi: 10.1016/j.lfs.2004.10.024. [DOI] [PubMed] [Google Scholar]
- Seachrist JL, Anborgh PH, Ferguson SS. β2-Adrenergic receptor internalization, endosomal sorting, and plasma membrane recycling are regulated by rab GTPases. J Biol Chem. 2000;275:27221–27228. doi: 10.1074/jbc.M003657200. [DOI] [PubMed] [Google Scholar]
- Shajahan AN, Timblin BK, Sandoval R, Tiruppathi C, Malik AB, Minshall RD. Role of Src-induced dynamin-2 phosphorylation in caveolae-mediated endocytosis in endothelial cells. J Biol Chem. 2004;279:20392–20400. doi: 10.1074/jbc.M308710200. [DOI] [PubMed] [Google Scholar]
- Shumay E, Song X, Wang HY, Malbon CC. pp60Src mediates insulin-stimulated sequestration of the β2-adrenergic receptor: insulin stimulates pp60Src phosphorylation and activation. Mol Biol Cell. 2002;13:3943–3954. doi: 10.1091/mbc.E02-03-0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohgo A, Choy EW, Gesty-Palmer D, Pierce KL, Laporte S, Oakley RH, Caron MG, Lefkowitz RJ, Luttrell LM. The stability of the G protein-coupled receptor-β-arrestin interaction determines the mechanism and functional consequence of ERK activation. J Biol Chem. 2003;278:6258–6267. doi: 10.1074/jbc.M212231200. [DOI] [PubMed] [Google Scholar]
- van Koppen CJ. Multiple pathways for the dynamin-regulated internalization of muscarinic acetylcholine receptors. Biochem Soc Trans. 2001;29:505–508. doi: 10.1042/bst0290505. [DOI] [PubMed] [Google Scholar]
- Walwyn WM, Keith DE, Jr, Wei W, Tan AM, Xie CW, Evans CJ, Kieffer BL, Maidment NT. Functional coupling, desensitization and internalization of virally expressed μ opioid receptors in cultured dorsal root ganglion neurons from μ opioid receptor knockout mice. Neuroscience. 2004;123:111–121. doi: 10.1016/j.neuroscience.2003.08.060. [DOI] [PubMed] [Google Scholar]
- Walwyn W, Maidment NT, Sanders M, Evans CJ, Kieffer BL, Hales TG. Induction of δ opioid receptor function by up-regulation of membrane receptors in mouse primary afferent neurons. Mol Pharmacol. 2005;68:1688–1698. doi: 10.1124/mol.105.014829. [DOI] [PubMed] [Google Scholar]
- Walwyn WM, Wei W, Xie CW, Chiu K, Kieffer BL, Evans CJ, Maidment NT. μ Opioid receptor-effector coupling and trafficking in dorsal root ganglia neurons. Neuroscience. 2006;142:493–503. doi: 10.1016/j.neuroscience.2006.06.021. [DOI] [PubMed] [Google Scholar]
- Wang Z, Bilsky EJ, Porreca F, Sadee W. Constitutive μ opioid receptor activation as a regulatory mechanism underlying narcotic tolerance and dependence. Life Sci. 1994;54:PL339–PL350. doi: 10.1016/0024-3205(94)90022-1. [DOI] [PubMed] [Google Scholar]
- Whistler JL, von Zastrow M. Morphine-activated opioid receptors elude desensitization by β-arrestin. Proc Natl Acad Sci USA. 1998;95:9914–9919. doi: 10.1073/pnas.95.17.9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whistler JL, Chuang HH, Chu P, Jan LY, von Zastrow M. Functional dissociation of μ opioid receptor signaling and endocytosis: implications for the biology of opiate tolerance and addiction. Neuron. 1999;23:737–746. doi: 10.1016/s0896-6273(01)80032-5. [DOI] [PubMed] [Google Scholar]
- Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]