

Abstract
Prion protein (PrP) inhibits the activation of proapoptotic Bax in primary human neurons and MCF-7 cells. Because neuronal apoptosis occurs in human prion diseases, here we examine the anti-Bax function of familial PrP mutants. All Creutzfeldt-Jakob disease and fatal familial insomnia-associated prion protein mutations partially or completely lose the anti-Bax function in human neurons and, except for A117V and V203I, in MCF-7 cells. The ability of the mutants to protect against Bax-mediated cell death is divided into three groups: (1) group I, retention of anti-Bax function in both the Val129 and Met129 mutants; (2) group II, retention of anti-Bax function only in Val129 mutants; and (3) group III, reduction or no anti-Bax function in Val129 and Met129 mutants. The loss of anti-Bax function in these PrP mutants correlates completely with a significant decrease in the production of cytosolic PrP, a form of PrP shown previously to have anti-Bax function in human neurons. Cotransfection of the full-length PrP mutants with wild-type or mutant cytosolic PrP, but not with wild type full-length PrP, rescues the anti-Bax function of PrP. The results show that the failure of PrP mutants to produce cytosolic PrP is responsible for the loss of anti-Bax function and that the effect of the PrP mutants is dominant over wild-type PrP. Furthermore, these results imply that misfolded PrP that escapes retrotranslocation could accumulate at the cell surface and cause neuronal dysfunction.
Keywords: prion protein, apoptosis, Bax, familial prion protein mutations, cytosolic PrP, retrotranslocation
Introduction
Prion protein (PrP) is a glycoprotein that is synthesized through the secretory pathway and accumulates as a glycosyl phosphatidylinositol (GPI) anchored-protein at the cell surface (Caughey et al., 1989). In addition, PrP is retrotranslocated from the endoplasmic reticulum to the cytosol as cytosolic PrP (CyPrP) (Zanusso et al., 1999; Ma and Lindquist, 2001; Yedidia et al., 2001; Roucou et al., 2003). PrP is highly expressed in brain, and its sequence is well conserved among mammalian species (Kretzschmar et al., 1986; Wopfner et al., 1999), indicating an important function for the normal cellular PrP. PrP promotes cell survival (for review, see Roucou et al., 2004). Neuronal PrP knock-out cells are more susceptible to apoptosis than PrP-expressing cells (Kuwahara et al., 1999). PrP prevents tumor necrosis factor-α-induced cell death in the breast carcinoma MCF-7 cells (Diarra-Mehrpour et al., 2004). In primary cultures of human neurons and in MCF-7 cells, PrP prevents the Bax conformational change involved as the first step of Bax activation into a proapoptotic protein and subsequently Bax-mediated mitochondrial cytochrome c release and cell death (Bounhar et al., 2001; Roucou et al., 2003, 2005). Furthermore, PrP is able to overcome Bax-mediated cell death in yeast cells (Bounhar et al., 2006). Because Bax is a strong inducer of neuronal apoptosis (White et al., 1998), this could be a very important function for PrP.
Disease-specific mutations of the PrP gene (PRNP) correspond to 10–15% of human transmissible spongiform encephalopathies. More than 30 dominant single point mutations and insertions have been identified in PRNP; 11 single point mutations confer Creutzfeldt-Jacob disease (CJD) (Windl et al., 1999; Gambetti et al., 2003). In addition to the pathogenic mutations in PRNP, a polymorphism at codon 129 encodes either a methionine (Met129) or a valine (Val129). The codon 129 polymorphism affects the susceptibility to prion diseases, the phenotype of CJD, and dictates the manifestation of either CJD or fatal familial insomnia (FFI) associated with the D178N PrP mutation (Goldfarb et al., 1992; Wadsworth et al., 2004).
Because neuronal apoptosis occurs in CJD and FFI patients (Dorandeu et al., 1998; Gray et al., 1999; Kawashima et al., 2001; Ferrer, 2002), here we investigate the anti-Bax function of both the Met129 and Val129 alleles of 11 PrP CJD mutations, one FFI mutation, and one Gerstmann-Straussler-Scheinker disease (GSS) mutation. We show that all familial PrP mutations tested decrease or abolish the anti-Bax function in primary human neurons and, except for A117V and V203I, in MCF-7 cells. The loss of anti-Bax function in these PrP mutants correlates completely with a significant decrease in the production of CyPrP, a form of PrP shown previously to have anti-Bax function in human neurons. Furthermore, coexpression of normal or mutant cytosolic PrP, but not full-length PrP, rescues the loss of anti-Bax function in PrP mutants. These results suggest that CyPrP is essential to the anti-Bax function of PrP and that the defective retrotranslocation of PrP mutants is dominant.
Materials and Methods
Cell cultures
Human primary neurons, obtained from fetal brains with ethical approval from the McGill University Institutional Review Board, were cultured as described previously (LeBlanc et al., 1997). Breast carcinoma MCF-7 cells and N2a mouse neuroblastoma cells were obtained from American Type Culture Collection (Manassas, VA) and maintained in DMEM containing 10% fetal bovine serum.
Cloning, site-directed mutagenesis of human PrP, and sequencing of PrP mutants
Human wild-type (WT) PrP carrying a valine at the codon 129 (Bounhar et al., 2001) was subcloned in the NotI and XhoI sites of the pcDNA3.1(+) vector (Invitrogen, Burlington, Ontario, Canada). Single point mutations in PrP were produced from human WT PrP using the QuikChange Site-Directed Mutagenesis protocol (Stratagene, La Jolla, CA). Forward primers used to produce the different PrP mutations are listed below, and substituted nucleotides are indicated in bold: A117V, 5′-CTGGTGCTGCAGTAGCTGGGGCGGTGG-3′; V180I, 5′-CAACTTTGTCCACGACTGCATCAATATCACAATCAAGC-3′; T188A, 5′-CAATCAAGCAGCACGCGGTCACCACAACC-3′; E196K, 5′-ACAACCACCAAGGGGAAGAACTTCACCGAGAC-3′; R208H, 5′-CGTTAAGATGATGGAGCACGTGGTTGAGCAGATG-3′; V210I, 5-GATGATGGAGCGCGTGATTGAGCAGATGTGTATC-3′; M232R, 5′-GAGGATCGAGCAGGGTCCTCTTCTCC-3′; P238S, 5′-GTCCTCTTCTCCTCTTCACCTGTGATCCTCC-3′; 129M, 5-CCTTGGCGGCTACATGCTGGGAAGTGC-3′; and 129V, 5-CTTGGCGGCTACGTGCTGGGAAGTGC-3′. PrP D178N and PrP T183A had been obtained previously (Bounhar et al., 2001). PrP mutants E200K, V203I, and E211Q were a kind gift from Dr. Neena Singh (Case Western Reserve University, Cleveland, OH). All PrP mutations were confirmed by sequencing using the Sequenase Version 2.0 DNA Sequencing kit (USB, Cleveland, OH).
Enhanced green fluorescent protein (EGFP) and EGFP–Bax cDNAs were amplified by PCR amplification from pCep4β–EGFP and pCep4β–EGFP–Bax constructs (Roucou et al., 2003) with the upstream primer 5′-TTTGGTACCATGGTGAGCAAGGGCGAG-3′ and the downstream primer 5′-GGGAGATCTACTTGTACAGCTCGTCCAT-3′ for EGFP or the upstream primer 5′-TAAGCGGCCGCTATGGTGAGCAAGGGCGAGGA-3′ and the downstream primer 5′-CGCGGTACCTCAGCCCATCTTCTTCCACATC-3′ for EGFP–Bax. The PCR products were cloned into the pBudCE4.1 vector (designed for independent expression of two genes; Invitrogen) downstream of the elongation factor 1α (EF-1α) promoter using the KpnI and BglII sites for EGFP or the NotI and KpnI sites for EGFP–Bax, generating pBud–EGFP and pBud–EGFP–Bax, respectively. WT PrP and PrP mutant cDNAs were PCR amplified from pcDNA3.1(+)–PrP or PrP mutants with the upstream primer 5′-ATATGTCGACATGGCGAACCTTGGCTGCTGGAT-3′ and the downstream primer 5′-CGCGTCTAGATCATCCCACTATCAGGAAGAT-3′ and cloned into the SalI and XbaI sites under the cytomegalovirus (CMV) promoter in the pBud–EGFP or pBud–EGFP–Bax to generate pBud–EGFP/PrP or PrP mutants and pBud-EGFP–Bax/PrP or PrP mutants. Moreover, WT and mutant PrP cDNAs from pBud-EGFP/PrP or PrP mutants were subcloned into the BamH I and HindIII sites of the pCep4β vector to generate pCep4β–PrP or PrP mutants. CyPrP (amino acids 23–231) and CyPrP GPI (amino acids 23–253) from WT PrP and PrP mutants were produced by PCR amplification of pBud–EGFP/PrP or PrP mutants with the upstream primer 5′-GCAAGTCGACATGAAGAAGCGCCCGAAGCCTG-3′ and the downstream primer 5′-GCCAGTACTCAGCTCGATCCTCTCTGGTAATAGGC-3′ for CyPrP or 5′-CGCGTCTAGATCATCCCACTATCAGGAAGAT-3′ for CyPrP GPI and cloned into the SalI and ScaI sites or ScaI and XbaI sites, respectively, of pBud–EGFP. CyPrP and CyPrP GPI were then subcloned into the BamH I and HindIII sites of the pCep4β vector to generate pCep4β–CyPrP or pCep4β–CyPrP GPI.
Analysis of the PrP polymorphism at codon 129
The Met/Val129 polymorphism of PrP in MCF-7 cells and human neurons was detected by PCR amplification with the upstream 5′-GGAACAAGCCGAGTAAGCTAAAAACCAACATGAAGCAC-3′ primer and the downstream 5′-GGTTGTGGTGACCGCGTGCTGCTTGATTG-3′ primer. The PCR product was digested with NspI and separated on a 3% agarose gel. The Met129 corresponds to three fragments of 75, 90, and 116 bp, whereas the Val129 corresponds to two fragments of 116 and 165 bp. The MCF-7 cell endogenous PrP is heterozygous at codon 129. Endogenous PrP in human neurons was homozygous for Met, homozygous for Val, or heterozygous at codon 129.
Transfections
For cell death assays, human primary neurons were plated at a density of 3 × 106 cells/ml onto poly-d-lysine-coated (20 μg/ml; Sigma, Oakville, Ontario, Canada) plastic coverslips, and MCF-7 cells were plated at 2.5 × 105 cells/ml onto glass coverslips in 24-well plates. The human neurons and MCF-7 cells on coverslips were transfected using the Helios Gene Gun system (Bio-Rad, Mississauga, Ontario, Canada) at a shooting pressure of 100 and 220 psi, respectively, according to the protocol of the manufacturer. Transfection cartridges were prepared with 0.033 mg of DNA, 4.2 mg of gold microcarrier beads in 0.1 ml of 1 m calcium chloride, and 0.1 ml of 0.05 m spermidine, as described previously (Roucou et al., 2005). The microcarrier loading quantity of the transfection cartridges was 0.125 mg gold/shot, and the DNA loading ratio was 1 μg DNA/shot. When cells were transfected with two constructs, a 1:3 ratio of pBud–EGFP–Bax/PrP or PrP mutants to pCep4β–PrP, pCep4β–CyPrP, pCep4β–CyPrP mutant, pCep4β–CyPrP GPI, or pCep4β–CyPrP GPI mutant was used for the preparation of cartridges. For PrP expression, MCF-7 cells were plated in six-well plates at 1.5 × 106 cells/ml or in 24-well plates at 2.5 × 105 cells/ml and transfected with 4 or 0.8 μg DNA, respectively, at a 3:1 ratio with pCep4β–PrP:pCep4β–EGFP, using Lipofectamine 2000 (Invitrogen) as described previously (Roucou et al., 2005). The transfection efficiency was measured by counting EGFP-positive cells versus the total number of cells, using a Nikon (Mississauga, Ontario, Canada) eclipse TE2000-U microscope. The transfection efficiency in MCF-7 cells was constant at 20% for all mutants. N2a cells were plated in six-well plates at 0.8 × 106 cells/ml and transfected with 4 μg of pCep4β–PrP or PrP mutants, pCep4β–CyPrP or CyPrP mutants, or pCep4β–CyPrP GPI or CyPrP GPI mutants using Lipofectamine 2000. The transfection efficiency was 70%.
Cell death measurement
Twenty hours after transfection, human neurons and MCF-7 cells were washed with PBS and fixed for 20 min at room temperature in 4% paraformaldehyde and 4% sucrose in PBS (150 mm NaCl, 2.7 mm KCl, 1.3 mm KH2PO4, and 8.1 mm Na2HPO4, pH 7.4), and the chromatin was stained 20 min with 1 μg/ml Hoechst 33342 in PBS (Sigma). Cell death was measured by counting EGFP-positive cells displaying condensed chromatin versus the total number of EGFP-positive cells, using a Nikon eclipse TE2000-U microscope.
Detergent solubility assay
Forty-eight hours after transfection, MCF-7 cells in six-well plates were lysed with 0.2 ml of lysis buffer [150 mm NaCl, 2 mm EDTA, 0.5% Triton X-100 (v/v), 0.5% sodium deoxycholate (w/v), and 50 mm Tris-HCl, pH 7.5) for 20 min at 4°C. After a centrifugation at 11,500 × g for 10 min at 4°C, the supernatant was collected and the detergent insoluble pellet was resuspended in 0.2 ml of Laemli's sample buffer [2% SDS (w/v), 5% β-mercaptoethanol (v/v), 10% glycerol (v/v), 0.01% bromophenol blue (w/v), and 62.5 mm Tris-HCl, pH 6.8]. The supernatant proteins were precipitated with 4 vol of ice-cold methanol overnight at −20°C and solubilized in the Laemli's sample buffer.
Western blot analyses
The proteins were separated in a 15% SDS-PAGE and transferred to polyvinylidene fluoride membranes, and PrP was detected with 1:2500 3F4 antibody (anti-PrP109–112) (Kascsak et al., 1987). When indicated, 1:1000 polyclonal R155 antiserum (anti-PrP36–56; produced in our laboratory) or 1:1000 6H4 (anti-PrP144–156; Prionics, Schlieren, Switzerland) and 1:1000 8H4 (anti-PrP170–180; Dr. Man-Sun Sy, Case Western Reserve University, Cleveland, OH) were used for PrP detection. Antibodies specific for β-actin (1:1000, Clone AC-15; Sigma), GFP (1:500, B-2; Santa Cruz Biotechnology, Santa Cruz, CA), cytosolic heat shock protein 70 (Hsp70) (1:1000; Stressgen Biotechnologies, Victoria, British Columbia, Canada), mitochondrial Hsp70 (1:1000, Clone JG1; Affinity BioReagents, Golden, CO), cytochrome c (1:1000, Clone 7H8.2C12; BD Pharmingen, Mississauga, Ontario, Canada), and Tau (1:1000, Clone T-5530: Sigma) were also used for Western blot analyses. Immunoreactivity was detected with 1:5000 anti-mouse or rabbit IgG conjugated to horseradish peroxidase secondary antibodies (Jackson ImmunoResearch, West Grove, PA) using chemiluminescence (GE Healthcare, Baie d'Urfe, Quebec, Canada).
Metabolic labeling of PrP
Forty-eight hours after transfection, MCF-7 cells were starved for 1 h in methionine-free DMEM and metabolically labeled for 3 h with 100 μCi/ml [35S]methionine. Proteins were extracted and PrP was immunoprecipitated using the anti-PrP R155 antiserum, as described previously (Bounhar et al., 2001). The immune complexes were separated in a 15% SDS-PAGE and subjected to autoradiography.
PrP secretion in cell culture media
Forty-eight hours after transfection, MCF-7 cells in 24-well plates were washed once with serum-free DMEM, and fresh DMEM was added for 6 h. The cell culture media were then collected and centrifuged 5 min at 2000 × g. The cells were washed with cold PBS and lysed with lysis buffer. All of the proteins from the supernatant and 20% of the cell lysate were precipitated with 4 vol of ice-cold methanol overnight at −20°C, resuspended in Laemli's sample buffer, and submitted to Western blotting against 3F4. The intensity of the protein bands were analyzed with a Personal densitometer SI (Molecular Dynamics, Sunnyvale, CA), and the ratio of secreted PrP over cellular PrP was calculated.
Phosphatidylinositol–phospholipase C treatment
Phosphatidylinositol–phospholipase C treatment of protein extracts.
One hundred micrograms of protein extracts from transfected MCF-7 cells were treated with 0.0625 U of phosphatidylinositol–phospholipase C (PI-PLC) (Sigma) for 18 h at 37°C in the presence of complete protease inhibitor cocktail (Roche, Laval, Quebec, Canada).
PI-PLC treatment of MCF-7 cells.
Forty-eight hours after transfection, MCF-7 cells in 24-well plates were washed once with DMEM, and fresh DMEM containing 0.5 U/ml PI-PLC was added for 2 h. The cell culture media were collected and centrifuged 5 min at 2000 × g. The cells were washed with cold PBS and lysed with lysis buffer. All of the proteins from the supernatant and 20% of the cell lysate were precipitated with 4 vol of ice-cold methanol overnight at −20°C, resuspended in Laemli's sample buffer, and submitted to Western blotting against 3F4.
Proteinase K, peptide-N-glycosidase F, and endoglycosidase H digestion
Resistance to proteinase K (PK) was assessed by incubating 100 μg of proteins from MCF-7 cells extracted in lysis buffer for 20 min on ice with 2 μg/ml PK. For peptide-N-glycosidase F (PNGase F) and endoglycosidase H (Endo H) digestions, 100 μg of proteins from MCF-7 cells extracted in lysis buffer were adjusted to 0.5% SDS, boiled 10 min, and digested with 2 U of PNGase F (New England Biolabs, Pickering, Ontario, Canada) for 18 h at 37°C or with 25 U of Endo H (New England Biolabs) for 1 h at 37°C in the presence of protease inhibitors, as specified by the manufacturer. The reaction was terminated by the addition of Laemli's sample buffer and by boiling 2 min before submitting to Western blotting.
Subcellular fractionation
Subcellular fractionation was performed as described previously (Roucou et al., 2003) with some modifications. Twenty-four hours after transfection with pCep4β–PrP or PrP mutant, N2a cells were treated with 0.25 μm epoxomycin (BioMol, Plymouth Meeting, PA) and 5 μg/ml brefeldin A (BFA) (Sigma) for 18 h. Cells were washed with cold PBS and homogenized with 30 strokes in a Dounce homogenizer in homogenization buffer [8% sucrose (w/v), 20 mm HCl-tricine, pH 7.8, and 1 mm EDTA]. The homogenate was centrifuged three times 5 min at 2000 × g to eliminate unbroken cells and nuclei and then centrifuged at 100,000 × g for 30 min. The supernatant and the pellet represent the cytosolic and membrane fractions, respectively. The pellet was resuspended in lysis buffer, and proteins from the supernatant were precipitated with 4 vol of ice-cold methanol overnight at −20°C. The intensity of the proteins obtained from Western blot analyses was analyzed with a Personal densitometer SI (Molecular Dynamics), and the ratio of cytosolic PrP over membrane PrP was calculated.
Statistical analysis
The statistical significance of the results was analyzed by ANOVA followed by Scheffé's post hoc analysis, using StatView (SAS Institute, Cary, NC). A p value of <0.05 was taken as a significant difference. The correlation coefficient and its statistical significance were determined using a Fisher's r to z transformation (StatView).
Results
All PrP mutants, except A117V and V203I, partially or completely lose anti-Bax function of PrP in MCF-7 cells
To investigate the function of familial human PrP mutants against Bax-mediated cell death, we chose the breast carcinoma MCF-7 cell line as described previously (Roucou et al., 2005). MCF-7 cells do not express detectable endogenous levels of PrP and are transfected with relatively high efficiency, allowing us to study mutant PrP expression biochemically. The bigenic pBudCE4.1 vector was used for the study because we can express the Bax protein under the EF-1α promoter and PrP or PrP mutants under the CMV promoter. Furthermore, the CMV promoter is not highly expressed in MCF-7 cells and permits the study of the function of physiological levels of the mutant PrPs. MCF-7 cells are resistant to EGFP toxicity, but overexpression of Bax protein fused N-terminally with EGFP under the EF-1α promoter induces Bax-specific mediated cell death and allows visualization of the transfected cells (Roucou et al., 2005). All PrP mutations in this study are associated with CJD, except for the PrP mutations A117V and Met129D178N, which are associated with GSS and FFI, respectively (Fig. 1A). Each point mutation has been generated with a valine as well as with a methionine at codon 129. Eight of these PrP mutations are located at the C-terminal B or C α-helices of the protein. Only one mutation is between the B and C α-helices, two mutations are in the GPI signal peptide, and one is within the transmembrane domain.
Figure 1.

Anti-Bax function and toxicity of PrP mutants expressed in MCF-7 cells. A, Schematic diagram showing various features of human prion protein, the relative positions of familial PrP mutations, and the polymorphic codon 129. TM, CHO, and S-S indicate the transmembrane domain, the glycosylation sites, and the disulfide bond, respectively. The first and second β-sheet (β1 and β2) and the three α-helices (αA, αB, and αC) of PrP are indicated. The numbers represent the amino acids. B, Percentage of cell death measured in MCF-7 cells transfected with pBud–EGFP–Bax alone (solid line) or transfected with pBud–EGFP–Bax/PrP or PrP mutants carrying either a valine (Val129) or a methionine (Met129) at codon 129. C, Percentage of cell death in MCF-7 cells transfected with pBud–EGFP (solid line) or pBud–EGFP/PrP and PrP mutants carrying either a valine (Val129) or a methionine (Met129) at the codon 129. For B and C, data represent the mean ± SEM of three independent experiments. At least 600 cells were counted for each condition. *p < 0.05, statistically significant difference between pBud–EGFP and pBud–EGFP/PrP-transfected cells or pBud–EGFP–Bax- and pBud–EGFP–Bax/PrP-transfected cells. #p < 0.05, statistically significant difference between pBud–EGFP–Bax/PrP and pBud–EGFP–Bax/PrP mutants. D, Western blot analyses of PrP with 3F4 in codon Val129 pCep4β–PrP-, pCep4β–PrPM232R-, pCep4β–PrPP238S-, or pCep4β–PrPΔGPI-transfected MCF-7 cell protein extracts treated without (−) or with (+) PI-PLC and deglycosylated with PNGase F. The arrow indicates a mobility shift as expected after removal of the GPI anchor.
Expression of EGFP–Bax alone in MCF-7 cells induces 61% cell death (Fig. 1B). When coexpressed with EGFP-Bax, Val129 or Met129 WT PrP decreases cell death by 50%. This is lower than observed against non-EGFP-tagged Bax, which is less toxic than EGFP–Bax, but it is still sufficient to analyze the anti-Bax function of PrP (Bounhar et al., 2001; Roucou et al., 2003). Compared with WT PrP, 63% of Val/Met129 PrP mutants partially or completely lose their capacity to inhibit Bax-mediated cell death. Of these, 83% of Met129PrP mutants partially or completely lose their anti-Bax function compared with 42% of Val129 PrP mutants. The inhibition of Bax-induced cell death by PrP mutants can be separated into three groups (Fig. 1B, Table 1). In the first group, the Met/Val129 A117V and V203I anti-Bax function is normal. In the second group, the anti-Bax function is partially lost in Met129 V180I, E200K, or T188A and completely lost in Met129 E196K and R208H PrP mutants. In contrast, the Val129 mutants completely retain the anti-Bax function. In the third group, the Val129 and Met129 E211Q, D178N, M232R, V210I, and P238S partially or completely lose the anti-Bax function.
Table 1.
Identification of three groups of PrP mutants based on their protection against Bax-mediated cell death in MCF-7 cells
| Protection against Bax |
Amino acid |
Secondary structure | |||
|---|---|---|---|---|---|
| Val129 | Met129 | Polarity | Charge | ||
| Group I | |||||
| WT | Yes | Yes | − | − | β-Sheet 1 |
| A117V | Yes | Yes | − | − | TM |
| V203I | Yes | Yes | − | − | α-Helix C |
| Group II | |||||
| V180I | Yes | Partial | − | − | α-Helix B |
| E200K | Yes | Partial | − | + | α-Helix C |
| T188A | Yes | Partial | + | − | α-Helix B |
| E196K | Yes | No | − | + | Between α-helix B/C |
| R208H | Yes | No | − | − | α-Helix C |
| Group III | |||||
| E211Q | Partial | Partial | − | + | α-Helix C |
| D178N | Partial | No | − | + | α-Helix B |
| M232R | No | Partial | + | + | GPI anchor signal |
| V210I | No | No | − | − | α-Helix C |
| P238S | No | No | + | − | GPI anchor signal |
Change in the polarity or the charge of the amino acid in the PrP mutation are indicated, using + if there is a change and − if there is no change. The location of the substituted amino acid in the secondary structure of PrP is provided. TM, Transmembrane domain.
The amino acid substitution of the PrP mutants can change the amino acid charge or polarity. Interestingly, the two mutants that retain the anti-Bax function of PrP in MCF-7 cells have conserved mutation (Table 1). However, two other conserved substitutions in Met129V180I and Met/Val129V210I partially or completely lose the anti-Bax function, respectively. Therefore, simple nonconserved substitution of the amino acid cannot account for the loss of anti-Bax function. PrP mutations that alter the charge or the polarity of the substituted amino acid result in the loss of anti-Bax function but, in some cases, only if the codon 129 encodes methionine. Generally, the Met129 PrP mutants have less anti-Bax function than Val129 PrP mutants. Furthermore, the position of the PrP mutations in the secondary structure of the protein does not correlate with the loss of anti-Bax function. For example, neither the mutant with the Met129 or Val129 V210I conserved mutation in α-helix C protect against Bax-mediated cell death, whereas both mutants with the conserved Met129 and Val129 V203I mutation in α-helix C protect completely against Bax-mediated cell death. Together, these results indicate that there is no consensus change in either the polarity or charge of the substituted amino acids in PrP mutants that correlate with the loss of anti-Bax function.
The background levels of cell death in nontransfected and pBud–EGFP-transfected MCF-7 cells are 5.4 ± 0.8 and 5.2 ± 0.7%, respectively, indicating that EGFP is not toxic. Only the Val129E196K, Met/Val129V210I, and Met129P238S mutants are weakly cytotoxic (Fig. 1C). The cells look healthy and normal, indicating that the expression of the mutant PrPs is not inducing cell death. There is no correlation between PrP mutant cytotoxicity and loss of the anti-Bax function because the Val129E196K is slightly cytotoxic yet completely retains its anti-Bax function. Moreover, most of the PrP mutants that lose their anti-Bax function are not cytotoxic. These results show that the loss of anti-Bax function is not a direct consequence of mutant PrP-induced cytotoxicity.
The PrP M232R and PrP P238S mutations in the GPI anchor signal peptide allow cleavage of the C-terminal peptide and the addition of the GPI anchor
Normally, the C-terminal signal peptide of PrP is cleaved from amino acids 232–253 for the addition of the GPI anchor (Englund, 1993). Therefore, we investigated whether the mutations M232R and P238S result in proper GPI posttranslational processing because both lose the anti-Bax function despite the mutations being in a part of the PrP that should be removed in the mature protein. PI-PLC followed by deglycosylation treatments of protein extracts induces a mobility shift of WT PrP (Fig. 1D) as shown previously (Walmsley and Hooper, 2003). As expected, the control PrP lacking the GPI signal sequence (ΔGPI) does not shift with the PI-PLC treatment. However, both the M232R and P238S PrP mutants undergo the same mobility shift as WT PrP, indicating that they have acquired the GPI anchor. Furthermore, the molecular weight of the deglycosylated M232R and P238S PrP mutants is identical to that of WT PrP, indicating that the signal peptides have been removed adequately. These results indicate that loss of function of these two PrP mutations is not attributable to the improper addition of the GPI anchor.
Expression, limited PK resistance, or posttranslational modifications of the PrP mutants do not account for the loss of anti-Bax function
Steady-state PrP levels or detergent solubility does not account for the loss of anti-Bax function in the mutants (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). For example, Met129V203I and Val129E196K-transfected cells display lower levels of PrP but retain the anti-Bax function. Detergent insolubility is not variable among groups I, II, and III. PrP or PrP mutants in the first and second groups are very sensitive to limited PK digestion except for the Met/Val129 A117V and R208H mutants (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Both Val129 and Met129 PrP mutants in the third group are resistant to limited PK digestion. Therefore, some PrP mutants that retain or lose anti-Bax activity have limited PK resistance, whereas some PrP mutants with no anti-Bax activity are sensitive to PK. Therefore, the resistance or sensitivity of PrP mutants to limited PK digestion does not correlate with the anti-Bax function.
We then investigated whether posttranslational modifications could explain the loss of anti-Bax function in the PrP mutants. Glycosylation of PrP occurs at Asn181 and Asn197 (Endo et al., 1989). Unfortunately, whereas EGFP expression is easily detected by Western blotting of pBud–EGFP/PrP-transfected MCF-7 cells, PrP is not (Fig. 2A). Only very low levels of PrP can be seen by immunoprecipitation of metabolically labeled pBud–EGFP/PrP-transfected MCF-7 cells (Fig. 2B). However, the level of PrP is easily detected in MCF-7-transfected cells with the episomal pCep4β–PrP construct, which yields 50–100 copies of the cDNA per cell (Groger et al., 1989). To facilitate biochemical detection of the transfected mutant PrPs, we expressed the mutant PrPs under the CMV promoter of the pCep4β construct. PrP is absent in pCep4β-transfected MCF-7 cells, confirming that the immunoreactive PrP proteins are from the exogenous expression of PrP mutants (Fig. 2C). As a control, the ER-retained T183A PrP is almost completely sensitive to Endo H removal of high mannose sugars, as expected (Kiachopoulos et al., 2005). Val/Met129 WT PrP migrate mostly as mature glycosylated forms of PrP and are completely deglycosylated by PNGase F, resulting in 25 and 23 kDa proteins (Fig. 2D). The 25 kDa protein is consistent with the deglycosylated or immature form of PrP, whereas the 23 kDa likely represents a N-terminally cleaved product of deglycosylated PrP because the N-terminal-directed R155 PrP antibody does not recognize the 23 kDa protein (Fig. 2E). The immature high mannose glycosylated form of PrP represents only a small fraction of the total PrP, and this protein, migrating at ∼30 kDa, shifts to 25 kDa during treatment with Endo H (Fig. 2D). Similarly, all PrP mutants, except Met/Val129 V203I, Val129M232R, and Met129P238S, are sensitive to Endo H as indicated by the increase in the 25 kDa protein. All PrP mutants, with variations in the abundance of the glycosylated forms, generate the 23 and 25 kDa proteins with PNGase F treatment. This confirms previous observations with the V203I, V210I, and E211Q PrP mutants (Vetrugno et al., 1999; Mishra et al., 2003). However, in Met129A117V, Met129T188A, Met/Val129E196K, Met/Val129R208H, Val129M232R, and Met129P238S, the 25 and 23 kDa proteins migrate as doublets, indicating additional changes, possibly truncation of these PrP mutant proteins. These results indicate that, despite some slight differences, glycosylation of the PrP mutants cannot account for the loss of anti-Bax function.
Figure 2.

Expression and glycosylation status of PrP mutants in MCF-7 cells. A, Western blot analyses of PrP (3F4) and EGFP in MCF-7 cells transfected with pBudCE4.1 vector (pBud), pBud–EGFP, and pBud–EGFP/Val129WT PrP (pBud–EGFP/PrP). B, Autoradiogram of PrP immunoprecipitated from metabolically labeled MCF-7 cells transfected with pCep4β–Val129WT PrP, pBud–EGFP, and pBud–EGFP/PrP. C, Western blot of PrP with 3F4 of protein extracts treated without enzyme (−), with PNGase F (P), or with Endo H (E) from pCep4β (Control) and pCep4β–PrPT183A-transfected cells (right). D, Western blot of PrP with 3F4 of protein extracts treated without enzyme (−), with PNGase F (P), or with Endo H (E) from pCep4β–PrP (WT) or pCep4β–PrP mutant-transfected cells. E, Western blot of PrP in PNGase F-treated protein extracts from pCep4β–PrP Val129-transfected cells with R155, 6H4, 8H4, or 3F4 antibodies.
The levels of CyPrP generated by PrP mutants correlate with the anti-Bax function
CyPrP prevents Bax-mediated cell death in human neurons (Roucou et al., 2003); therefore, we investigated whether PrP mutations affect the levels of CyPrP (Fig. 3). pCep4β–PrP or PrP mutant-transfected MCF-7 cells were treated with BFA and epoxomycin to inhibit proteasomal degradation and were submitted to subcellular fractionations into membrane and cytosolic fractions, as done previously (Roucou et al., 2003). Unfortunately, BFA and epoxomycin treatment of MCF-7 cells results in the expression of endogenous human PrP (Fig. 3A). Therefore, we switched to mouse N2a cells because the endogenous mouse PrP is not recognized by the 3F4 antibody. The presence of mitochondrial Hsp70 and cytochrome c only in the membranes and the presence of Tau protein mainly in the cytosol confirm the purity of the subcellular fractions (Fig. 3B). In untreated cells, PrP is detected only in the membrane subcellular fraction. As shown previously, in the presence of epoxomycin and BFA, immature PrP is detected in the cytosol and membranes. The size of the CyPrP is consistent with retrotranslocated PrP lacking both the N- and C-terminal signal peptides because the deglycosylated CyPrP comigrates with recombinant human PrP23–231 (Fig. 3C). In group I, the ratio of CyPrP over membrane PrP is equivalent or tends to be slightly higher than that of WT PrP (Fig. 3D). Group II Val129PrP mutants that retain completely the anti-Bax function have the same level of CyPrP than WT PrP. In contrast, Met129PrP mutants that partially or completely lose the anti-Bax function have significantly decreased levels of CyPrP. Group III Val129 and Met129 PrP mutants that partially or completely lose anti-Bax function all have significantly lower levels of CyPrP. The loss of anti-Bax function for PrP mutants correlates well with the decrease of CyPrP (Fig. 3E). These results indicate that the reduction or loss of CyPrP from PrP mutants may be responsible for the deficit in anti-Bax function.
Figure 3.

Analysis of PrP mutants located in the cytosol. A, Western blot of PrP with 3F4 in protein extracts from pCep4β (−) or pCep4β-PrP Val129 (+)-transfected MCF-7 cells treated without or with epoxomycin (Epoxo) and BFA. B, Western blot of PrP with 3F4, mitochondrial Hsp70 (mHsp70), cytochrome c (Cyt c), or Tau in proteins from membrane and cytosolic fractions of pCep4β–PrP Val129-transfected N2a cells treated or untreated (control) with epoxomycin and BFA. Equal amounts of membrane and cytosolic fractions were loaded on the gel except for the PrP detection in which cytosol and membrane were loaded as a ratio of 10:1, respectively. C, Western blot with 3F4 of recombinant human PrP (rPrP, amino acid 23–231) and untreated (−) or PNGase F deglycosylated (+) cytosolic protein extracts from pCep4β–PrP Val129-transfected N2a cells treated with epoxomycin and BFA. D, Quantification of the Western blot analyses for CyPrP in N2a cells transfected with pCep4β–PrP or pCep4β–PrP mutants carrying either a valine (Val129) or a methionine (Met129) at codon 129. The data represent the mean ± SEM of three independent experiments. *p < 0.05 indicates a statistically significant difference between pCep4β–PrP and pCep4β–PrP mutant-transfected cells. E, Correlation between the anti-Bax function of PrP mutants in MCF-7 cells and CyPrP level. The correlation is statistically significant (p < 0.0001).
The inability of PrP mutants to generate normal levels of CyPrP is not accompanied with abnormal cell surface GPI-anchored PrP levels
To examine whether there are other trafficking problems with the mutant PrPs, we looked at cell surface PrP. As observed previously (Winklhofer et al., 2003; Kiachopoulos et al., 2005), PI-PLC treatment of the pCep4β–PrP-transfected MCF-7 cells releases significant amounts of the WT or mutant PrPs (Fig. 4A). There is no consistent difference between group I, II, or III or between Val129 and Met129 PrP mutants, which indicates that the defect in making CyPrP is not accompanied by a problem with trafficking to the cell surface.
Figure 4.

PrP mutants are GPI anchored. A, Western blot of PI-PLC-treated pCep4β- (Control), pCep4β–PrPVal129- (V), or Met129 (M) and pCep4β–PrP mutant-transfected MCF-7 cellular and released PrP with 3F4. B, Western blot of secreted and cellular PrP from PrPΔGPI, empty vector (Control), Val129 (V), or Met129 (M) WT and mutant PrP-transfected MCF-7 cells. Ratio corresponds to the ratio of secreted PrP over cellular PrP.
Similarly, secretion of PrP is not affected. In primary human neuron cultures, PrP is secreted into the media (Bounhar et al., 2001) as is WT or mutant PrPs from pCep4β-transfected MCF-7 cells (Fig. 4B). Both the 25 kDa immature and 36–40 kDa mature glycosylated forms of PrP are secreted. However, compared with the secretion of the positive control PrP lacking the GPI signal sequence (ΔGPI), PrP and PrP mutants are weakly secreted. The ratios of secreted PrP over cellular PrP indicates that Met129 T188A, E196K, and R208H, and the Val/Met129 E211Q and D178N mutants are more abundantly secreted relative to WT PrP. Whether PrP is released by an endogenous lipase or is actually secreted without attaching to the cell surface is not clear. However, the level of PrP secretion does not correlate with the anti-Bax function because ratios vary from 0.01 to 46 in group III mutants, in which all have partially or completely lost anti-Bax function. Together, these results indicate that the loss of anti-Bax function in PrP mutants is not explained by the loss of cell surface GPI-anchored PrP or by altered secretion of PrP.
Cotransfection of PrP mutants with wild-type or mutant CyPrP, but not with wild-type full-length PrP, rescues the loss of anti-Bax function in PrP mutants
To determine whether the decrease of CyPrP explains the loss of anti-Bax function in PrP mutants, we coexpressed CyPrP with all PrP mutants that have partially or completely lost the anti-Bax function. For this, we cotransfected MCF-7 cells with pBud–EGFP–Bax/PrP mutants and pCep4β–CyPrP (Fig. 5). CyPrP confers anti-Bax function to all pBud–EGFP–Bax/PrP mutant-transfected cells. To determine whether the PrP mutation also affect the anti-Bax function, we repeated the rescue experiment with pCep4β–CyPrP mutants carrying the same mutation as that of the pBud–EGFP–Bax/PrP mutant. CyPrP mutant expression was confirmed (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Two of the CyPrP mutants (M232R and P238S) were expressed with the GPI signal peptide because the mutations are present in this domain. Again, all mutant CyPrPs rescue the anti-Bax activity (Fig. 5).
Figure 5.

CyPrP and CyPrP mutants, but not full-length WT PrP, rescue the loss of anti-Bax function of full-length PrP mutants. Percentage of cell death in MCF-7 cells transfected with two constructs with a ratio of 1:3 of pBud–EGFP–Bax:pCep4β (solid line), pBud–EGFP–Bax/PrP Val129 (WT) or pBud–EGFP–Bax/PrP mutants (as indicated in x-axis):pCep4β–PrP Val129 (PrP, black bar), pBud-EGFP-Bax/PrP Val129 or pBud–EGFP–Bax/PrP mutants:pCep4β–CyPrP Val129 (CyPrP, gray bar), pBud–EGFP–Bax/PrP Val129 or pBud–EGFP–Bax/PrP mutants: pCep4β–CyPrP mutant Val129 (CyPrP mutant, white bar), pBud–EGFP–Bax/PrP Val129 or pBud–EGFP–Bax/PrP mutants:pCep4β–CyPrP GPI, or pBud–EGFP–Bax/PrP Val129 or pBud–EGFP–Bax/PrP mutants:pCep4β–CyPrP GPI mutants. Mutations in pCep4β–CyPrP and pCep4β–CyPrP GPI are identical to that tested in the pBud construct (indicated in x-axis). PrP mutants carry either a valine (V) or a methionine (M) at codon 129. The WT PrP shown here has a Val129 codon, and the results were similar in PrP Met129. Data represent the mean ± SEM of three independent experiments. At least 400 cells were counted for each condition. *p < 0.05, statistically significant difference between pBud–EGFP–Bax- and pBud–EGFP–Bax/PrP- or PrP mutant-transfected cells. #p < 0.05, statistically significant difference between pBud–EGFP–Bax/PrP and pBud–EGFP–Bax/PrP mutant-transfected cells.
Finally, we attempted to rescue the loss of anti-Bax function in PrP mutants by cotransfecting with pCep4β–PrP. Unexpectedly, wild-type PrP expression does not rescue the loss of anti-Bax function in any of the pBud–EGFP–Bax/PrP mutant-transfected MCF-7 cells.
Together, these results indicate that the loss of CyPrP is responsible for the loss of anti-Bax function in PrP mutants. Furthermore, the inability of full-length WT PrP to rescue suggests that the PrP mutants have a dominant effect on the ability of wild-type PrP to retrotranslocate into the cytosol.
The loss of anti-Bax function in PrP mutants is more prominent in human neurons than in MCF-7 cells
We next investigated whether the mutant PrPs retain the anti-Bax function in primary cultures of human neurons. EGFP–Bax expression induces cell death within 24 h (Fig. 6A). As seen in MCF-7 cells, WT PrP inhibits EGFP–Bax-mediated cell death by 50%. The anti-Bax function of PrP is lost in 83% of PrP mutants. All Met129 PrP mutants and 67% of Val129 PrP mutants partially or completely lose their anti-Bax function. The codon 129 of endogenously expressed PrP in human neurons does not affect the anti-Bax function of PrP mutants (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). In group I, only Met/Val129 WT PrP protects against Bax. In group II, Val129 A117V, V203I, T188A, and E200K mutants retain protection against Bax, whereas the Met129 counterparts lose this protection. In group III, both Val129 and Met129 D178N, R208H, E211Q, V180I, E196K, V210I, M232R, and P238S PrP mutants lose anti-Bax function. Only three mutants partially retain function: the Met129D178N, the Val129R208H, and the Val129E211Q. The loss of anti-Bax function by PrP mutants is not associated with a change in either the polarity or charge of the substituted amino acids (Table 2). Furthermore, the position of the PrP mutations in the secondary structure of the protein does not correlate with the loss of anti-Bax function.
Figure 6.
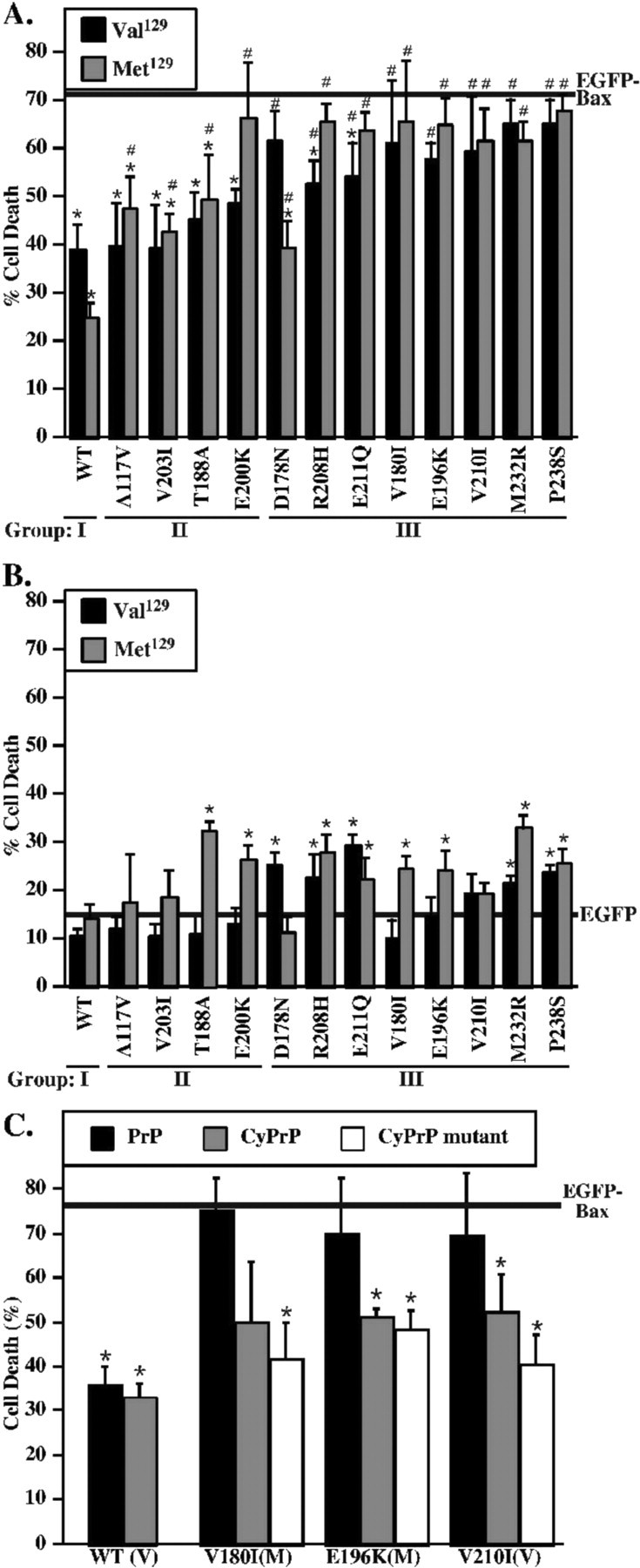
Anti-Bax function, toxicity, and rescue of anti-Bax function in human neurons transfected with PrP mutants. A, Percentage of cell death in human neurons transfected with pBud–EGFP–Bax alone (solid line) or transfected with pBud–EGFP–Bax/PrP or PrP mutants carrying either a valine (Val129) or a methionine (Met129) at codon 129. Data represent the mean ± SEM of three independent experiments. At least 150 cells were counted for each condition. *p < 0.05, statistically significant difference between pBud–EGFP–Bax and pBud–EGFP–Bax/PrP. #p < 0.05, statistically significant difference between pBud–EGFP–Bax/PrP- and pBud–EGFP–Bax/PrP mutant-transfected cells. B, Percentage of cell death in human neurons induced by EGFP (solid line) or EGFP expressed with WT or mutant PrP carrying either a valine (Val129) or a methionine (Met129) at codon 129. Data represent the mean ± SEM of three independent experiments. At least 150 cells were counted for each condition. *p < 0.05, statistically significant difference between pBud–EGFP- and pBud–EGFP/PrP- or PrP mutant-transfected cells. The codon 129 of endogenous PrP in human neurons is shown at supplemental Figure 4. C, Percentage of cell death in human neurons transfected with pBud–EGFP–Bax: pCep4β (solid line), pBud–EGFP–Bax/PrP Val129 (WT PrP) or pBud–EGFP–Bax/PrP mutant (indicated in x-axis):pCep4β–PrP Val129 (PrP, black bar), pCep4β–CyPrP Val129 (gray bar), or pCep4β–CyPrP mutant (white bar). PrP mutants carry either a valine (V) or a methionine (M) at codon 129. Data represent the mean ± SEM of three independent experiments. At least 50 cells were counted for each condition. *p < 0.05, statistically significant difference between pBud–EGFP–Bax- and pBud–EGFP–Bax/PrP- or PrP mutant-transfected cells.
Table 2.
Identification of three groups of PrP mutants based on their protection against Bax-mediated cell death in human neurons
| Protection against Bax |
Amino acid |
Secondary structure | |||
|---|---|---|---|---|---|
| Val129 | Met129 | Polarity | Charge | ||
| Group I | |||||
| WT | Yes | Yes | − | − | β-Sheet 1 |
| Group II | |||||
| A117V | Yes | Partial | − | − | TM |
| V203I | Yes | Partial | − | − | α-Helix C |
| T188A | Yes | Partial | + | − | α-Helix B |
| E200K | Yes | No | − | + | α-Helix C |
| Group III | |||||
| D178N | No | Partial | − | + | α-Helix B |
| R208H | Partial | No | − | − | α-Helix C |
| E211Q | Partial | No | − | + | α-Helix C |
| V180I | No | No | − | − | α-Helix B |
| E196K | No | No | − | + | Between α-helix B/C |
| V210I | No | No | − | − | α-Helix C |
| M232R | No | No | + | + | GPI anchor signal |
| P238S | No | No | + | − | GPI anchor signal |
Change in the polarity or the charge of the amino acid by PrP mutation are indicated, using + if there is a change and − if there is no change. The location of the substituted amino acid in the secondary structure of PrP is provided. TM, Transmembrane domain.
In contrast to the MCF-7 cells, human neurons were more susceptible to the mutant PrP expressions. The background level of cell death in nontransfected and EGFP-transfected human neurons is 15 ± 3 and 16 ± 3%, respectively. Therefore, EGFP is not toxic to these neurons. The Met129 T188A, E200K, R208H, E211Q, V180I, E196K, M232R, and P238S and the Val129 D178N, R208H, E211Q, M232R, and P238S mutants exhibit low but statistically significant cytotoxicity (Fig. 6B). However, as observed in MCF-7 cells, cytotoxicity does not correlate with the loss of the anti-Bax function.
Because it is not possible to transfect the human neurons with high efficiency, these studies are restricted to single-cell analyses. However, we did perform rescue experiments in three PrP mutants of the third group that completely lose the anti-Bax function. pCep4β–CyPrP reduces cell death in pBud–EGFP–Bax/PrP V180I Met129-, E196K Met129-, and V210I Val129-transfected human neurons, but the difference does not reach statistical significance for pBud–EGFP–Bax/PrP V180I Met129 (Fig. 6C). As seen in MCF-7 cells, all CyPrP mutants rescue against the loss of anti-Bax function, but wild-type full-length PrP cannot rescue the loss of anti-Bax function in these PrP mutants. These results show that, as in MCF-7 cells, CyPrP or CyPrP mutants rescue human neurons from the loss of anti-Bax function of PrP mutants, and the effect is dominant. This likely explains why the PrP mutants lose their anti-Bax function despite high expression of endogenously expressed PrP in these cells.
Discussion
We show here that most of the familial PrP mutants exhibit a significant loss of the PrP anti-Bax function in human neurons and MCF-7 cells. The ability of mutants to protect against Bax-mediated cell death is divided into three groups: (1) group I, retention of anti-Bax function in both Val129 and Met129 mutants; (2) group II, retention of anti-Bax function only in Val129 mutants; and (3) group III, reduction or loss of anti-Bax function in both Val129 and Met129 mutants. The loss of anti-Bax function in PrP mutants correlates entirely with a significant decrease in the production of CyPrP. CyPrP arises from incomplete translocation into the endoplasmic reticulum or retrotranslocation from the endoplasmic reticulum (Ma and Lindquist, 2001; Roucou et al., 2003; Rane et al., 2004). Incomplete translocation results in a PrP that lacks the N-terminal signal peptide but retains the C-terminal GPI anchor signal peptide. Here, we find that the CyPrP does not contain the N-terminal or GPI signal peptides and thus is generated through retrotranslocation. The CyPrP has been shown to be cytotoxic in mouse neuroblastoma N2a cells and in cerebellar granular neurons, possibly through aberrant interaction with membrane lipids (Ma and Lindquist, 2001, 2002; Wang et al., 2006). The decrease in retrotranslocated CyPrP is unexpected because familial PrP mutants are associated with neuronal degeneration and cell death in human disease, a condition that could have been initiated by CyPrP cytotoxicity. However, CyPrP is not toxic and protects human neurons in primary cultures (Roucou et al., 2003) and MCF-7 cells (D. T. S. Lin and A. C. LeBlanc, unpublished observations) against Bax-mediated cell death. In addition, CyPrP is not toxic to a number of other human neuroblastoma cells (Roucou et al., 2003). Here, we show that wild-type CyPrP rescues the loss of anti-Bax function in PrP mutants. Therefore, CyPrP is essential to PrP anti-Bax function. Furthermore, the mutant CyPrP also rescue against the loss of anti-Bax function in PrP mutants, indicating that the mutations in PrP do not alter the structural entity responsible for the anti-Bax function. Therefore, we conclude that these PrP mutants lose the ability to prevent Bax-mediated cell death because they cannot generate enough CyPrP.
Exactly how all of these PrP mutations alter retrotranslocation of PrP is not clear. The change in the charge, the polarity, or the position of the substituted amino acids in mutant PrPs do not correlate with the loss of anti-Bax function or the production of CyPrP. It is generally assumed that PrP mutations alter the conformation of the PrP. D178N, V180I, R208H, and V210I amino acid substitutions thermodynamically destabilize the PrP (Riek et al., 1998; Liemann and Glockshuber, 1999; Apetri et al., 2004). However, not all PrP mutants are affected in this way. Moreover, mutant PrPs, such as D178N, possess misfolded protein characteristics (Lehmann and Harris, 1996). Interestingly, even the M232R and P238S mutations, which are posttranslationally removed during GPI anchor addition, have reduced CyPrP levels. These results indicate that the C terminus of PrP regulates retrotranslocation of PrP before PrP is completely processed in the endoplasmic reticulum. Furthermore, the codon 129 polymorphism regulates retro-translocation because in group II, Val129 containing mutants have normal levels of CyPrP and retain their anti-Bax function, whereas Met129 containing mutants have reduced CyPrP levels and partially or completely lose the anti-Bax function. Relative to Val129, the Met129 increases the ability of PrP to fold with a higher β-sheet content and to oligomerize (Tahiri-Alaoui et al., 2004). Therefore, it is likely that codon 129 and familial mutations slightly alter the conformation of PrP, thus resulting in altered posttranslational trafficking.
We also find that the mutant PrPs do not accumulate into the endoplasmic reticulum, are normally glycosylated, and accumulate at the cell surface as GPI-anchored proteins. These results suggest that the mutant PrPs escape the normal regulatory checkpoint for misfolded proteins, which constitutes 10% of newly synthesized WT PrP (Yedidia et al., 2001). Because many PrP mutants resist limited PK digestion, the mutants are likely misfolded. Cell surface PrP has been shown to induce several beneficial signaling transduction pathways (for review, see Roucou et al., 2004; Roucou and LeBlanc, 2005). The accumulation of these misfolded proteins could be detrimental to the normal function of PrP at the cell surface of the neurons.
Whereas wild-type or mutant CyPrP can rescue the anti-Bax function, the full-length wild-type PrP cannot rescue. This indicates that the mutant PrPs have a dominant effect with regards to retrotranslocation of PrP into the cytosol. The results explain why there is a loss of PrP anti-Bax function in PrP mutant-transfected primary cultures of human neurons, which express relatively high levels of endogenous normal PrP. The results are also consistent with the autosomal dominant nature of familial PrP mutations.
The retention of protection with the Val129 allele in group II is consistent with observations that show a protective effect of Val129 against scrapie infections (Wadsworth et al., 2004). Our results in human neurons showing abrogation of anti-Bax function in all Met129 PrP mutants are also consistent with the observations that individuals homozygous for methionine are more susceptible to develop a prion disease (Kovacs et al., 2005). Interestingly, all CJD human familial mutations encode a Met129 on the mutant allele. In addition, D178N and E200K mutations are also found with a Val129 mutant allele (Hoque et al., 1996; Windl et al., 1999; Peoc'h et al., 2000; Kovacs et al., 2005). Codon 129 strongly regulates the phenotypic manifestation of CJD (Val129) and FFI (Met129) associated with the D178N mutation and the prion disease linked to the E200K mutation (Goldfarb et al., 1992; Hainfellner et al., 1999). Moreover, codon 129 polymorphism affects the type of pathological PrP in genetic prion diseases and, in some cases, the age of onset (Baker et al., 1991; Monari et al., 1994; Gambetti et al., 2003; Kovacs et al., 2005). Our observations show the additional influence of codon 129 polymorphism on the retrotranslocation and anti-Bax function of PrP mutants.
We confirm that the loss of anti-Bax function also occurs in human neurons. However, the loss is more prominent in human neurons than in MCF-7 cells. The Met/Val129 A117V and V203I group I mutations that retain protection against Bax in MCF-7 cells shift to group II in human neurons. Moreover, group II mutations Val129 V180I, E196K, and R208H that retain function in MCF-7 cells shift to group III in human neurons. One possible explanation for this difference is that the mutant PrPs are more cytotoxic to human neurons than MCF-7 cells. Unfortunately, it is impossible to study in these human neurons the level of CyPrP to determine whether it is lower in all of the mutants because primary cultures of human neurons are highly resistant to infections or transfections and ballistic transfection into the human neurons occurs at very low transfection efficiency and limits us to single-cell analyses. However, we show by cotransfection experiments that CyPrP and CyPrP mutants, but not WT full-length PrP, also rescue the anti-Bax function of PrP mutants in human neurons. These results indicate that CyPrP deficits are also responsible for the loss of anti-Bax function in human neurons.
Neuronal apoptosis is observed in FFI and familial CJD (Gray et al., 1999; Ferrer, 2002). Because clinical symptoms are manifested during aging in familial cases, it is possible that the loss of anti-Bax prion function contributes to age-dependent Bax-mediated apoptosis. Several age-dependent insults, such as oxidative stress and endoplasmic reticulum stress, activate Bax (Savory et al., 1999; Keller et al., 2002; Phaneuf and Leeuwenburgh, 2002). Interestingly, the universal anti-Bax inhibitor protein Bcl-2 decreases in the aging CNS (Merry et al., 1994). Because Bcl-2 has wide-ranging effects on many proapoptotic proteins, it is possible that PrP, which is quite specific against Bax (Roucou et al., 2005), replaces the broader Bcl-2 protein for a more focused effect against Bax-mediated cell death in the aging CNS. Therefore, it is possible that the loss of anti-Bax function in PrP mutants contributes to the loss of neurons in prion diseases.
In summary, we find that PrP mutants associated with familial prion diseases lose their anti-Bax function and that this loss of function is attributable to a deficit in generating CyPrP. The loss of the anti-Bax function in mutant PrPs may result in a higher susceptibility of neurons to age-dependent Bax-mediated neuronal cell death in familial prion diseases. In addition, these results suggest that the mutant PrPs escape the retrotranslocation pathway normally responsible for removing misfolded protein. In these conditions, the accumulation of misfolded PrP could adversely affect neuronal function.
Footnotes
This work was supported by National Institutes for Health Grant 1RO1 NS-40431, Canadian Institutes of Health Research Grant MOP49594, and the Fonds de Recherche en Santé du Québec. We are grateful to Dr. Neena Singh (Case Western Reserve University, Cleveland, OH) for providing the E200K, V203I, and E211Q PrP mutants, Dr. Man-Sun Sy (Case Western Reserve University, Cleveland, OH) for providing the 8H4 antibody, the Birth Defects Research Laboratory (University of Washington, Seattle, WA) for providing conceptal tissue for research (National Institutes of Health Grant HD 000836), Jennifer Hammond for the culture of the human neurons, and Iwona Link, Myriam Pilon, Thi Huong Tra Truong, and Paresa Giannopoulos for the purification of DNA.
References
- Apetri AC, Surewicz K, Surewicz WK. The effect of disease-associated mutations on the folding pathway of human prion protein. J Biol Chem. 2004;279:18008–18014. doi: 10.1074/jbc.M313581200. [DOI] [PubMed] [Google Scholar]
- Baker HE, Poulter M, Crow TJ, Frith CD, Lofthouse R, Ridley RM. Aminoacid polymorphism in human prion protein and age at death in inherited prion disease. Lancet. 1991;337:1286. doi: 10.1016/0140-6736(91)92953-y. [DOI] [PubMed] [Google Scholar]
- Bounhar Y, Zhang Y, Goodyer CG, LeBlanc A. Prion protein protects human neurons against Bax-mediated apoptosis. J Biol Chem. 2001;276:39145–39149. doi: 10.1074/jbc.C100443200. [DOI] [PubMed] [Google Scholar]
- Bounhar Y, Mann KK, Roucou X, LeBlanc AC. Prion protein prevents Bax-mediated cell death in the absence of other Bcl-2 family members in Saccharomyces cerevisiae. FEMS Yeast Res. 2006;6:1204–1212. doi: 10.1111/j.1567-1364.2006.00122.x. [DOI] [PubMed] [Google Scholar]
- Caughey B, Race RE, Ernst D, Buchmeier MJ, Chesebro B. Prion protein biosynthesis in scrapie-infected and uninfected neuroblastoma cells. J Virol. 1989;63:175–181. doi: 10.1128/jvi.63.1.175-181.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diarra-Mehrpour M, Arrabal S, Jalil A, Pinson X, Gaudin C, Pietu G, Pitaval A, Ripoche H, Eloit M, Dormont D, Chouaib S. Prion protein prevents human breast carcinoma cell line from tumor necrosis factor alpha-induced cell death. Cancer Res. 2004;64:719–727. doi: 10.1158/0008-5472.can-03-1735. [DOI] [PubMed] [Google Scholar]
- Dorandeu A, Wingertsmann L, Chretien F, Delisle MB, Vital C, Parchi P, Montagna P, Lugaresi E, Ironside JW, Budka H, Gambetti P, Gray F. Neuronal apoptosis in fatal familial insomnia. Brain Pathol. 1998;8:531–537. doi: 10.1111/j.1750-3639.1998.tb00175.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo T, Groth D, Prusiner SB, Kobata A. Diversity of oligosaccharide structures linked to asparagines of the scrapie prion protein. Biochemistry. 1989;28:8380–8388. doi: 10.1021/bi00447a017. [DOI] [PubMed] [Google Scholar]
- Englund PT. The structure and biosynthesis of glycosyl phosphatidylinositol protein anchors. Annu Rev Biochem. 1993;62:121–138. doi: 10.1146/annurev.bi.62.070193.001005. [DOI] [PubMed] [Google Scholar]
- Ferrer I. Synaptic pathology and cell death in the cerebellum in Creutzfeldt-Jakob disease. Cerebellum. 2002;1:213–222. doi: 10.1080/14734220260418448. [DOI] [PubMed] [Google Scholar]
- Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. Sporadic and familial CJD: classification and characterisation. Br Med Bull. 2003;66:213–239. doi: 10.1093/bmb/66.1.213. [DOI] [PubMed] [Google Scholar]
- Goldfarb LG, Petersen RB, Tabaton M, Brown P, LeBlanc AC, Montagna P, Cortelli P, Julien J, Vital C, Pendelbury WW, Haltia M, Wills PR, Hauw JJ, McKeever PE, Morani L, Schrank B, Swergold GD, Autilio-Gambetti L, Gajdusek DC, Lugaresi E, Gambetti P. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: disease phenotype determined by a DNA polymorphism. Science. 1992;258:806–808. doi: 10.1126/science.1439789. [DOI] [PubMed] [Google Scholar]
- Gray F, Chretien F, Adle-Biassette H, Dorandeu A, Ereau T, Delisle MB, Kopp N, Ironside JW, Vital C. Neuronal apoptosis in Creutzfeldt-Jakob disease. J Neuropathol Exp Neurol. 1999;58:321–328. doi: 10.1097/00005072-199904000-00002. [DOI] [PubMed] [Google Scholar]
- Groger R, Morrow D, Tyckocinski M. Directional antisense and sense cloning using Epstein-Barr virus episomal expression vectors. Gene. 1989;81:285–294. doi: 10.1016/0378-1119(89)90189-3. [DOI] [PubMed] [Google Scholar]
- Hainfellner JA, Parchi P, Kitamoto T, Jarius C, Gambetti P, Budka H. A novel phenotype in familial Creutzfeldt-Jakob disease: prion protein gene E200K mutation coupled with valine at codon 129 and type 2 protease-resistant prion protein. Ann Neurol. 1999;45:812–816. [PubMed] [Google Scholar]
- Hoque MZ, Kitamoto T, Furukawa H, Muramoto T, Tateishi J. Mutation in the prion protein gene at codon 232 in Japanese patients with Creutzfeldt-Jakob disease: a clinicopathological, immunohistochemical and transmission study. Acta Neuropathol. 1996;92:441–446. doi: 10.1007/s004010050544. [DOI] [PubMed] [Google Scholar]
- Kascsak RJ, Rubenstein R, Merz PA, Tonna-DeMasi M, Fersko R, Carp RI, Wisniewski HM, Diringer H. Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J Virol. 1987;61:3688–3693. doi: 10.1128/jvi.61.12.3688-3693.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima T, Doh-ura K, Ogomori K, Iwaki T. Apoptotic bodies in the cerebellum of Japanese patients with Creutzfeldt-Jakob disease. Pathol Int. 2001;51:140–144. doi: 10.1046/j.1440-1827.2001.01181.x. [DOI] [PubMed] [Google Scholar]
- Keller JN, Gee J, Ding Q. The proteasome in brain aging. Ageing Res Rev. 2002;1:279–293. doi: 10.1016/s1568-1637(01)00006-x. [DOI] [PubMed] [Google Scholar]
- Kiachopoulos S, Bracher A, Winklhofer KF, Tatzelt J. Pathogenic mutations located in the hydrophobic core of the prion protein interfere with folding and attachment of the glycosylphosphatidylinositol anchor. J Biol Chem. 2005;280:9320–9329. doi: 10.1074/jbc.M412525200. [DOI] [PubMed] [Google Scholar]
- Kovacs GG, Puopolo M, Ladogana A, Pocchiari M, Budka H, van Duijn C, Collins SJ, Boyd A, Giulivi A, Coulthart M, Delasnerie-Laupretre N, Brandel JP, Zerr I, Kretzschmar HA, de Pedro-Cuesta J, Calero-Lara M, Glatzel M, Aguzzi A, Bishop M, Knight R, Belay G, Will R, Mitrova E. Genetic prion disease: the EUROCJD experience. Hum Genet. 2005;118:166–174. doi: 10.1007/s00439-005-0020-1. [DOI] [PubMed] [Google Scholar]
- Kretzschmar HA, Prusiner SB, Stowring LE, DeArmond SJ. Scrapie prion proteins are synthesized in neurons. Am J Pathol. 1986;122:1–5. [PMC free article] [PubMed] [Google Scholar]
- Kuwahara C, Takeuchi AM, Nishimura T, Haraguchi K, Kubosaki A, Matsumoto Y, Saeki K, Yokoyama T, Itohara S, Onodera T. Prions prevent neuronal cell-line death. Nature. 1999;400:225–226. doi: 10.1038/22241. [DOI] [PubMed] [Google Scholar]
- LeBlanc AC, Papadopoulos M, Belair C, Chu W, Crosato M, Powell J, Goodyer CG. Processing of amyloid precursor protein in human primary neuron and astrocyte cultures. J Neurochem. 1997;68:1183–1190. doi: 10.1046/j.1471-4159.1997.68031183.x. [DOI] [PubMed] [Google Scholar]
- Lehmann S, Harris DA. Mutant and infectious prion proteins display common biochemical properties in cultured cells. J Biol Chem. 1996;271:1633–1637. doi: 10.1074/jbc.271.3.1633. [DOI] [PubMed] [Google Scholar]
- Liemann S, Glockshuber R. Influence of amino acid substitutions related to inherited human prion diseases on the thermodynamic stability of the cellular prion protein. Biochemistry. 1999;38:3258–3267. doi: 10.1021/bi982714g. [DOI] [PubMed] [Google Scholar]
- Ma J, Lindquist S. Wild-type PrP and a mutant associated with prion disease are subject to retrograde transport and proteasome degradation. Proc Natl Acad Sci USA. 2001;98:14955–14960. doi: 10.1073/pnas.011578098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Lindquist S. Conversion of PrP to a self-perpetuating PrPSc-like conformation in the cytosol. Science. 2002;298:1785–1788. doi: 10.1126/science.1073619. [DOI] [PubMed] [Google Scholar]
- Merry DE, Veis DJ, Hickey WF, Korsmeyer SJ. bcl-2 protein expression is widespread in the developing nervous system and retained in the adult PNS. Development. 1994;120:301–311. doi: 10.1242/dev.120.2.301. [DOI] [PubMed] [Google Scholar]
- Mishra RS, Bose S, Gu Y, Li R, Singh N. Aggresome formation by mutant prion proteins: the unfolding role of proteasomes in familial prion disorders. J Alzheimers Dis. 2003;5:15–23. doi: 10.3233/jad-2003-5103. [DOI] [PubMed] [Google Scholar]
- Monari L, Chen SG, Brown P, Parchi P, Petersen RB, Mikol J, Gray F, Cortelli P, Montagna P, Ghetti B, Goldfarb LG, Gajdusek DC, Lugaresi E, Gambetti P, Autilio-Gambetti L. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: different prion proteins determined by a DNA polymorphism. Proc Natl Acad Sci USA. 1994;91:2839–2842. doi: 10.1073/pnas.91.7.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peoc'h K, Manivet P, Beaudry P, Attane F, Besson G, Hannequin D, Delasnerie-Laupretre N, Laplanche JL. Identification of three novel mutations (E196K, V203I, E211Q) in the prion protein gene (PRNP) in inherited prion diseases with Creutzfeldt-Jakob disease phenotype. Hum Mutat. 2000;15:482. doi: 10.1002/(SICI)1098-1004(200005)15:5<482::AID-HUMU16>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Phaneuf S, Leeuwenburgh C. Cytochrome c release from mitochondria in the aging heart: a possible mechanism for apoptosis with age. Am J Physiol Regul Integr Comp Physiol. 2002;282:R423–R430. doi: 10.1152/ajpregu.00296.2001. [DOI] [PubMed] [Google Scholar]
- Rane NS, Yonkovich JL, Hegde RS. Protection from cytosolic prion protein toxicity by modulation of protein translocation. EMBO J. 2004;23:4550–4559. doi: 10.1038/sj.emboj.7600462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riek R, Wider G, Billeter M, Hornemann S, Glockshuber R, Wuthrich K. Prion protein NMR structure and familial human spongiform encephalopathies. Proc Natl Acad Sci USA. 1998;95:11667–11672. doi: 10.1073/pnas.95.20.11667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roucou X, LeBlanc AC. Cellular prion protein neuroprotective function: implications in prion diseases. J Mol Med. 2005;83:3–11. doi: 10.1007/s00109-004-0605-5. [DOI] [PubMed] [Google Scholar]
- Roucou X, Guo Q, Zhang Y, Goodyer CG, LeBlanc AC. Cytosolic prion protein is not toxic and protects against Bax-mediated cell death in human primary neurons. J Biol Chem. 2003;278:40877–40881. doi: 10.1074/jbc.M306177200. [DOI] [PubMed] [Google Scholar]
- Roucou X, Gains M, LeBlanc AC. Neuroprotective functions of prion protein. J Neurosci Res. 2004;75:153–161. doi: 10.1002/jnr.10864. [DOI] [PubMed] [Google Scholar]
- Roucou X, Giannopoulos PN, Zhang Y, Jodoin J, Goodyer CG, LeBlanc A. Cellular prion protein inhibits proapoptotic Bax conformational change in human neurons and in breast carcinoma MCF-7 cells. Cell Death Differ. 2005;12:783–795. doi: 10.1038/sj.cdd.4401629. [DOI] [PubMed] [Google Scholar]
- Savory J, Rao JK, Huang Y, Letada PR, Herman MM. Age-related hippocampal changes in Bcl-2:Bax ratio, oxidative stress, redox-active iron and apoptosis associated with aluminum-induced neurodegeneration: increased susceptibility with aging. Neurotoxicology. 1999;20:805–817. [PubMed] [Google Scholar]
- Tahiri-Alaoui A, Gill AC, Disterer P, James W. Methionine 129 variant of human prion protein oligomerizes more rapidly than the valine 129 variant: implications for disease susceptibility to Creutzfeldt-Jakob disease. J Biol Chem. 2004;279:31390–31397. doi: 10.1074/jbc.M401754200. [DOI] [PubMed] [Google Scholar]
- Vetrugno V, Malchow M, Liu Q, Marziali G, Battistini A, Pocchiari M. Expression of wild-type and V210I mutant prion protein in human neuroblastoma cells. Neurosci Lett. 1999;270:41–44. doi: 10.1016/s0304-3940(99)00460-7. [DOI] [PubMed] [Google Scholar]
- Wadsworth JD, Asante EA, Desbruslais M, Linehan JM, Joiner S, Gowland I, Welch J, Stone L, Lloyd SE, Hill AF, Brandner S, Collinge J. Human prion protein with valine 129 prevents expression of variant CJD phenotype. Science. 2004;306:1793–1796. doi: 10.1126/science.1103932. [DOI] [PubMed] [Google Scholar]
- Walmsley AR, Hooper NM. Distance of sequons to the C-terminus influences the cellular N-glycosylation of the prion protein. Biochem J. 2003;370:351–355. doi: 10.1042/BJ20021303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang F, Arterburn L, Wollmann R, Ma J. The interaction between cytoplasmic prion protein and the hydrophobic lipid core of membrane correlates with neurotoxicity. J Biol Chem. 2006;281:13559–13565. doi: 10.1074/jbc.M512306200. [DOI] [PubMed] [Google Scholar]
- White FA, Keller-Peck CR, Knudson CM, Korsmeyer SJ, Snider WD. Widespread elimination of naturally occurring neuronal death in Bax-deficient mice. J Neurosci. 1998;18:1428–1439. doi: 10.1523/JNEUROSCI.18-04-01428.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windl O, Giese A, Schulz-Schaeffer W, Zerr I, Skworc K, Arendt S, Oberdieck C, Bodemer M, Poser S, Kretzschmar HA. Molecular genetics of human prion diseases in Germany. Hum Genet. 1999;105:244–252. doi: 10.1007/s004399900124. [DOI] [PubMed] [Google Scholar]
- Winklhofer KF, Heske J, Heller U, Reintjes A, Muranyi W, Moarefi I, Tatzelt J. Determinants of the in vivo folding of the prion protein. A bipartite function of helix 1 in folding and aggregation. J Biol Chem. 2003;278:14961–14970. doi: 10.1074/jbc.M209942200. [DOI] [PubMed] [Google Scholar]
- Wopfner F, Weidenhofer G, Schneider R, von Brunn A, Gilch S, Schwarz TF, Werner T, Schatzl HM. Analysis of 27 mammalian and 9 avian PrPs reveals high conservation of flexible regions of the prion protein. J Mol Biol. 1999;289:1163–1178. doi: 10.1006/jmbi.1999.2831. [DOI] [PubMed] [Google Scholar]
- Yedidia Y, Horonchik L, Tzaban S, Yanai A, Taraboulos A. Proteasomes and ubiquitin are involved in the turnover of the wild-type prion protein. EMBO J. 2001;20:5383–5391. doi: 10.1093/emboj/20.19.5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanusso G, Petersen RB, Taocong J, Jing Y, Kanoush R, Ferrari S, Gambetti P, Singh N. Proteosomal degradation and N-terminal protease resistance of the codon 145 mutant prion protein. J Biol Chem. 1999;274:23396–23404. doi: 10.1074/jbc.274.33.23396. [DOI] [PubMed] [Google Scholar]