

Abstract
Neurofibrillary tangles (NFTs) are a pathological hallmark of Alzheimer's disease and other tauopathies, but recent studies in a conditional mouse model of tauopathy (rTg4510) have suggested that NFT formation can be dissociated from memory loss and neurodegeneration. This suggests that NFTs are not the major neurotoxic tau species, at least during the early stages of pathogenesis. To identify other neurotoxic tau protein species, we performed biochemical analyses on brain tissues from the rTg4510 mouse model and then correlated the levels of these tau proteins with memory loss. We describe the identification and characterization of two forms of tau multimers (140 and 170 kDa), whose molecular weight suggests an oligomeric aggregate, that accumulate early in the pathogenic cascade in this mouse model. Similar tau multimers were detected in a second mouse model of tauopathy (JNPL3) and in tissue from patients with Alzheimer's disease and FTDP-17 (frontotemporal dementia and parkinsonism linked to chromosome 17). Moreover, levels of the tau multimers correlated consistently with memory loss at various ages in the rTg4510 mouse model. Our findings suggest that accumulation of early-stage aggregated tau species, before the formation of NFT, is associated with the development of functional deficits during the pathogenic progression of tauopathy.
Keywords: tau, transgenic, neurodegeneration, Alzheimer's disease, FTDP-17, toxicity
Introduction
Tauopathies encompass more than 20 different disorders, including Alzheimer's disease (AD), progressive supranuclear palsy, and frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17), which are characterized by intracellular accumulations of filamentous tau (Lee et al., 2001). The identification of tau mutations in FTDP-17 patients established that tau dysfunction alone can lead to neurodegeneration (Hutton et al., 1998; Poorkaj et al., 1998; Spillantini et al., 1998). The causal role of these mutations is supported by experiments in animal models, in which expression of mutant tau leads to neurodegeneration and the development of functional deficits (Lewis et al., 2000; Wittmann et al., 2001; Santacruz et al., 2005).
Neurofibrillary tangles (NFTs) are a common pathological hallmark of the tauopathies (Lee et al., 2001) and were previously implicated in mediating neurodegeneration and dementia in Alzheimer's disease (Arriagada et al., 1992; Gotz et al., 2001; Lewis et al., 2001; Guillozet et al., 2003; Bennett et al., 2004). However, recent studies in a novel conditional mouse model of tauopathy (rTg4510), expressing human mutant P301L tau, suggested that NFT formation could be dissociated from neuronal dysfunction (Santacruz et al., 2005). This in turn implied that NFTs are unlikely to be the major toxic tau species, at least in the early stages of tauopathy.
The apparent dissociation between behavioral dysfunction/tau toxicity and NFT is supported by additional observations. Inhibition of tau hyperphosphorylation in a second mouse model of tauopathy (JNPL3), expressing mutant human P301L tau, led to delayed development of motor dysfunction without affecting NFT numbers (Le Corre et al., 2006). Neuronal cell death also occurred independently of histologically observed tangles in mice that express all six isoforms of wild-type human tau (Andorfer et al., 2005). Finally, overexpression of wild-type and mutant R406W tau in Drosophila led to neurodegeneration without formation of neurofibrillary tangles (Wittmann et al., 2001).
These findings raised the possibility that specific tau aggregation/hyperphosphorylation intermediates, formed before the development of NFT, could be neurotoxic. The role of oligomeric aggregation intermediates has recently received considerable attention in several neurodegenerative diseases. SDS-stable Aβ oligomers, α-synuclein oligomers, and prion oligomers have all been linked to toxicity (Conway et al., 2000; Lashuel et al., 2002; Walsh et al., 2002; Sharon et al., 2003; Cleary et al., 2005; Lesne et al., 2006; Novitskaya et al., 2006).
This led us to explore the formation of similar early tau aggregated species during pathogenic progression in the conditional rTg4510 model. In addition, we determined the relationship of these early aggregates and other pathological tau species with the development of memory deficits in rTg4510.
Materials and Methods
Transgenic mice.
rTg4510 mice were bred by crossing mice expressing the responder mutant tau P301L transgene (FVB/NCr strain; Charles River Laboratories, Wilmington, MA) with mice expressing the tetracycline-dependent transcription activator (tTa) activator transgene (129S6 strain; Taconic, Germantown, NY) as described previously (Santacruz et al., 2005). 129/FVB F1 littermates were used in all experiments. Briefly, the responder transgene consists of a tetracycline operon-responsive element placed upstream of a cDNA encoding human tau with four microtubule binding repeats (4R tau) and the P301L mutation. The activator transgene contains the tet-off open reading frame placed downstream of Ca2+/calmodulin kinase II promoter elements.
To suppress transgene expression, doxycycline (dox) (200 ppm) in chow was administered to mice ad libitum during the course of the study. In addition, doxycycline (1.5 mg/ml) was administered for the first 2 d in drinking water in 4% sucrose (controls were given 4% sucrose only). JNPL3 mice were bred as described previously (Lewis et al., 2000) and maintained on an outbred Swiss Webster Background (Taconic). All animals were housed and tested according to standards established by the Mayo Clinic Institutional Animal Care and Use Committee. Mice were killed using cervical dislocation or under deep anesthesia.
Behavioral protocols.
The reference memory (place discrimination) Morris water maze test (MWM) test was preceded by 3 d cued (visible platform) version of the test. During this test, the water maze tank was surrounded by a curtain, and the platform location and point of release of a mouse were changed semirandomly each trial. The reference memory MWM test was run for 9 d with four training trials per day. An escape platform submerged under water was always positioned in the center of one quadrant (target quadrant). During the training, four 60 s probe trials on days 1, 3, 6, 9, and 10 of training were administered to evaluate spatial memory. The spatial memory for the platform location during probe trials was evaluated by computing the annulus crossing index, referred to as memory index. The memory index represents the number of crosses over the platform site in the target quadrant, adjusted for crosses over corresponding sites in other quadrants, and, because this transforms the raw data, the total number of platform crosses in all four quadrants of the pool was also analyzed. The mean memory index representing the average from the last two probe trials at the end of the 9 d training period (days 9 and 10) was used. Complete details of the water maze apparatus, procedure, and data analysis are provided in the supplemental methods (available at www.jneurosci.org as supplemental material).
Preparation of brain/spinal cord extracts and tau biochemistry.
Tissue (brain or spinal cord) was homogenized in 6× volume of the following (in mm): 50 Tris base, pH 8.0, 274 NaCl, 5 KCl, 2 EGTA, 2 EDTA, protease inhibitor cocktail (Sigma, St. Louis, MO), phosphatase inhibitor cocktail I and II (Sigma), and 1 PMSF, except for experiments testing the presence of disulfide bonds (supplemental methods, available at www.jneurosci.org as supplemental material).
The extract (homogenate) was spun for 15 min at 13,000 × g, and supernatant was used as a total fraction (NFTs and cell debris were removed in the pellet). Purification of sarkosyl-insoluble tau (P3 fraction) was based on previously published procedures (Greenberg and Davies, 1990). The amount of starting material was adjusted for protein concentration to obtain comparable results. The supernatant was then further centrifuged at 150,000 × g for 15 min to separate proteins into soluble (S1, supernatant) and insoluble (pellet) fraction. Pellet was re-extracted in 10 mm Tris, pH 7.4, 0.8 m NaCl, 10% sucrose, 1 mm EGTA, 1 mm PMSF, protease inhibitor cocktail (Sigma), and phosphatase inhibitor cocktail I and II (Sigma) and centrifuged at 150,000 × g for 15 min. The pellet was discarded, and the supernatant was incubated with 1% sarkosyl at 37°C for 1 h and centrifuged at 150,000 × g for 30 min, briefly washed with the same buffer, and centrifuged again. The pellet (solubilized in Tris-EDTA buffer) contained sarkosyl-insoluble tau (P3 fraction). Typically 150 μl of total extract was used to obtain 50 μl of P3 fraction. We estimated that P3 fraction obtained using this protocol contained <5% of the 64 kDa tau in the total brain extracts.
Western blotting.
A total of 10 μg of proteins for the total fraction, 15 μg for the S1 fraction, and 10 μl for the P3 fraction were mixed with equivalent volume of 2× SDS-sample buffer (Invitrogen, Carlsbad, CA) (62.5 mm Tris-HCl, pH 6.8, 2% SDS, 25% glycerol, and 0.1% bromophenol blue) and 5% β-mercapthoethanol (unless mentioned otherwise), boiled for 5 min, and loaded on 10% polyacrylamide Tris-glycine gels [Invitrogen or Bio-Rad (Hercules, CA)]. The gels were resolved at 15 mA for ∼2 h and then transferred onto polyvinylidene difluoride membrane using wet transfer system at 200 mA for 2 h. The membrane was briefly washed in TBST (1% Triton X-100, 10 mm Tris, and 140 mm NaCl), blocked in 5% milk, and incubated in primary antibody overnight. Membrane was then washed three times for 10 min with TBST and incubated in secondary antibody for 0.5–2 h, washed three times for 10 min with TBST, and incubated with Western lightning Chemiluminescence reagent plus (PerkinElmer, Wellesley, MA) and exposed on Eastman Kodak (Rochester, NY) BioMax Light Film. Bands on the films were scanned, and band intensities were quantified using Scion (Frederick, MD) Image Alfa 4.0.3.2. The intensity of the protein band of interest was divided by the intensity of the band representing a loading control [glyceraldehyde-3-phosphate dehydrogenase (GAPDH)] to calculate the relative amount. The intensity of the bands was compared on the same membrane when possible (sample size permitting), or the intensity was adjusted according to a standard run on both gels.
We used the following antibodies: E1 (1:2000), CP13 (1:1000), PHF1 (1:1000), AT8 (1:1000; Pierce, Rockford, IL), PS422 (1:1000; Biosource International, Camarillo, CA), GAPDH (1:10,000; Biodesign International, Kennebunk, ME), tau 12 (1:200,000), TauC3 (1:1000), and anti-rabbit and anti-mouse (1:2000 to 1:10,000; Jackson ImmunoResearch, West Grove, PA). Total tau 64 kDa was always quantified using AT8 antibody because this provided better separation between 64 and 55 kDa tau.
Tau dephosphorylation.
Low-speed supernatant, 60 μl, was mixed with 400 μl of 7 m guanidium hydrochloride and 5% β-mercaptoethanol and dialyzed overnight against water. The next day, samples were lyophilized and then incubated with 50 μl of hydrofluoric acid overnight. On the following day, samples were again lyophilized and resuspended in 0.5 m Tris, pH 8.6.
Size exclusion chromatography.
Column preparation and general methodology were described previously (Nichols et al., 2002). In brief, sample (one brain or hemibrain in total volume of 2 ml) was loaded on to a HiPrep 16/60 Sephacryl S-300HR column (Amersham Biosciences, Arlington Heights, IL) attached to a Pharmacia LKB (Gaithersburg, MD) system. The column was pre-equilibrated in the buffer in which the brain was homogenized (see above) at 25°C and run at a flow rate of 0.8 ml/min. Two milliliter fractions were collected and concentrated to 50–200 μ and 5–10 μl were used for Western blot analysis, using the E1 antibody. The size exclusion chromatography (SEC) profile was constructed from Western blot data, using fractions 19–32, containing monomeric and multimeric tau (on Western blots). Tau ∼55 and 64 kDa, were also found in fractions corresponding to particles of smaller size (data not shown) but not multimers. The absence of the curve (see Fig. 2D,E) in a particular fraction indicates the absence of the species. Blue dextran was used to determine the void volume and other standards (thyroglobulin, ∼670 kDa; apoferritin, ∼450 kDa; and BSA, ∼70 kDa) were used to determine the approximate molecular weights corresponding to the fractions of the inclusion volume.
Figure 2.
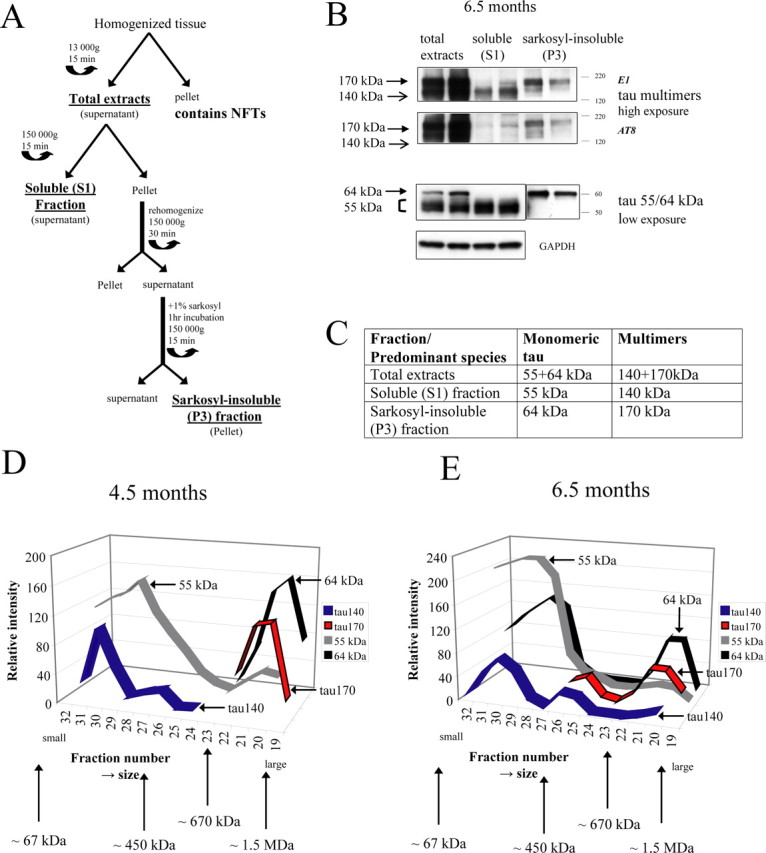
Biochemical properties of tau species. A, Schematic diagram of the tau extraction procedure that is based on previously published methology (Greenberg and Davies, 1990). For details, see Material and Methods. B, Brain extracts from 6.5-month-old rTg4510 were processed as described in A. Tau ∼55 kDa and tau 64 kDa are present in total extracts at 6.5 months of age (assessed by E1 antibody). Tau ∼55 kDa is found in soluble (S1) fraction, whereas hyperphosphorylated tau 64 kDa is found in the sarkosyl-insoluble (P3) fraction. Tau170 and tau140 are present in total lysate from 6.5-month-old rTg4510. Tau140 is mostly found in soluble (S1) fraction, whereas tau170 is mostly in the sarkosyl-insoluble (P3) fraction, as assessed by E1 antibody. Tau170 kDa, present in the sarkosyl-insoluble (P3) fraction, is strongly immunoreactive with AT8, whereas tau140, present in soluble (S1) fraction, exhibits very little immunoreactivity with AT8 antibody. C, Summary of how the various tau species are extracted into the different fractions. D, Analysis of the size of the different tau species in vivo. Size-exclusion chromatography was performed with total extracts from 4.5-month-old rTg4510. Tau140 and tau 55 kDa elute at a similar size and appear relatively small, whereas tau170 and tau 64 kDa are part of larger aggregates. Tau140 and tau170 have distinct sizes that do not overlap at this time point. Molecular weight scale corresponds to standards (see Materials and Methods) eluting at respective fractions shown on the graph (∼67 kDa eluted in fraction 34). E, Size-exclusion chromatography analysis using extracts of older 6.5-month-old rTg4510. Hyperphosphorylated species (64 kDa, tau170) are characterized by two distinct elution peaks, suggesting they are derived from two distinct populations of small and large aggregated species. Tau140 and tau170 have overlapping size ranges at 6.5 months of age.
Neurofibrillary tangle counts in rTg4510 mouse brains.
Brains were dissected, and one hemisphere of the brains was drop fixed in 10% Formalin for 1 week. After fixation, brains were embedded in paraffin, sections were cut at 5 μm, and Gallyas silver staining was performed (supplemental methods, available at www.jneurosci.org as supplemental material). Stained sections were viewed using an Olympus Optical (Center Valley, PA) 40i microscope and imaged using a Retiga EXi (QImaging, Burnaby, British Columbia, Canada) and MetaMorph imaging software (Molecular Devices, Downington, PA). Total number of neurofibrillary tangles in each section was counted in the insular, motor, and somatosensory cortices as defined by the atlas of Paxinos and Franklin (2001).
All mice in the 5.5-month-old cohort evaluated for biochemical tau species were included in the NFT counts. In the 8-month-old cohort, two brains were excluded from the analysis of NFT counts (compared with biochemistry analysis) because of a substantial damage of the cortical region that prevented us from obtaining accurate NFT count.
Tissue from human subjects.
For initial evaluation of the presence of multimers in human tissue, frozen brain tissue was obtained from 10 subjects with pathologically confirmed Alzheimer's disease (six women and four men; average age at death, 81 ± 8 years; average Braak neurofibrillary tangle stage, 5.6 ± 0.5; average postmortem delay, 11 ± 6 h) and two subjects from the pallido-ponto-nigral degeneration family (Wszolek et al., 1992; Reed et al., 1998), who have N279K mutations in microtubule-associated protein tau (MAPT) (two women; average age at death, 51 ± 4 years; average Braak neurofibrillary tangle stage, 0.8 ± 0.4; average postmortem delay, 10 ± 1 h). Alzheimer's diagnosis was made with established criteria according to previously reported methods (Uchikado et al., 2006).
Results
Tau multimers in a conditional model of tauopathy, rTg4510
To determine whether tau was able to form oligomers (multimers) similar to those described for other proteins implicated in neurodegenerative disorders (Conway et al., 2000; Lashuel et al., 2002; Walsh et al., 2002; Sharon et al., 2003; Cleary et al., 2005; Lesne et al., 2006), we analyzed Western blots prepared from brain extracts from the rTg4510 conditional tau mouse model. We used rTg4510 mice because they exhibit robust neurofibrillary pathology, cell loss, and memory impairment (Santacruz et al., 2005). Initial studies used 3.5-month-old mice, a stage at which the rTg4510 line shows memory impairments but has not yet developed mature NFTs or detectable hippocampal neuronal cell loss (Santacruz et al., 2005).
Standard Western blot analysis was first performed on total brain extracts that represent the supernatant obtained after centrifuging the brain homogenate at low speed (13,000 × g). At this age, the human-specific tau antibody E1 (residues 19–33) (Crowe et al., 1991) detected multiple tau species migrating at ∼55 kDa (Fig. 1A). These bands correspond to normal soluble tau species with different phosphorylation states, as reported previously (Sahara et al., 2002; Santacruz et al., 2005). In contrast, there was no evidence of the pathologically hyperphosphorylated tau migrating at 64 kDa (Fig. 1A) that accumulates in rTg4510 mice at older ages (Santacruz et al., 2005).
Figure 1.

Tau multimers in rTg4510 mice. A, Western blot of total extracts from 3.5-month-old rTg4510 and age-matched tTa animals (without mutant P301L tau transgene). Tau migrating at ∼55 kDa can be detected with the human-specific E1 antibody in rTg4510 but not in tTa animals. Tau multimers migrating at ∼170 (called tau170) and ∼140 kDa (called tau140) can be seen in rTg4510 but not in age-matched tTa animals, when film is exposed for a longer time. B, Schematic location of epitopes on tau that are recognized by phosphorylation-independent antibodies tau12, E1, TauC3, and tau46. Tau12 and E1 are N-terminal, whereas tau46 is C-terminal. TauC3 (which does not detect tau multimers) is selective for tau cleaved at Asp421. C, Tau multimers are detected in total brain extracts from 3.5-month-old rTg4510 with a variety of tau antibodies (E1, CP13, PHF1, PS422, AT8, and T46). Age-matched tTa animals (without mutant P301L tau transgene) were used as a control. Tau170 and tau140 are seen by E1, CP13, PHF1, and T46, whereas PS422 and AT8 selectively detects tau170. D, Dephosphorylation leads to disappearance of tau170, although tau140 is still present.
Oligomeric tau species was not observed when the film was exposed for the time necessary to detect ∼55 kDa tau. However, after prolonged exposure, we observed what appeared to be oligomeric tau species (termed tau “multimers”) migrating as two major bands with apparent molecular weights of ∼140 kDa (termed tau140) and ∼170 kDa (tau170) (Fig. 1A) (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). These species were detected in rTg4510 tau animals (with both tau and tTa transgenes) but not tTa control animals (with only the activator tTa transgene) (Fig. 1A). The apparent molecular weight of these species is sufficient to suggest that they are multimeric structures (i.e., species with two or more tau molecules).
To further characterize the tau multimers, we used different antibodies recognizing epitopes located in N- and C-terminal region of tau, which are independent of tau phosphorylation status (Fig. 1B). Tau multimers (tau140 and tau170) were detected with the E1 antibody (residues 19–33 of human tau) (Crowe et al., 1991) (Fig. 1A,C) and with the tau12 antibody (data not shown) recognizing residues 9–18 of human tau (Horowitz et al., 2004). Moreover, tau multimers were also detected with the tau46 antibody (Fig. 1C) that recognizes an epitope near the C terminus (428–441 amino acids) (Fig. 1B,C). In contrast, the tau multimers were not detected using the TauC3 antibody (data not shown) that recognizes tau truncated at Asp421 (Gamblin et al., 2003), although we cannot exclude the possibility that this negative result may be attributable to insufficient sensitivity or that other truncated tau species may be present. Nonetheless, our results are consistent with the multimers containing full-length tau species. In addition, the tau multimers were also not detected by antibodies that recognize MAP1, MAP2, or mouse tau, which were previously suggested to affect tau aggregation (Alonso et al., 1997; Andorfer et al., 2003).
Both tau multimer species were SDS-stable, even after boiling in SDS for 30 min (data not shown). Moreover, levels of the multimers were not altered by prolonged incubation in 0.1% SDS, which mimics the conditions during SDS-PAGE (data not shown), demonstrating that the tau multimers are not an artifact of SDS-PAGE.
We next determined the phosphorylation state of the tau170 and tau140 multimers, using antibodies that recognize different tau phospho-epitopes. Both tau170 and tau140 were detected with CP13 and PHF1 antibodies that also recognize phospho-epitopes on normal tau, migrating at ∼55 kDa (Fig. 1C). However, antibodies that selectively recognize pathological phospho-epitopes, in both human AD patients and rTg4510 mice (AT8 and PS422), preferentially detected the tau170 multimers (Fig. 1C), despite higher abundance of the tau140 species, in 3.5-month-old rTg4510 mice. Dephosphorylation of the protein extracts, through treatment with hydrofluoric acid, resulted in disappearance of the tau170 species, suggesting that tau170 exists in a hyperphosphorylated state (Fig. 1D). In summary, tau140 and tau170 exhibit different levels of phosphorylation.
Our results clearly show that the tau multimers described above are detected in the presence of reducing agents. However, species reminiscent of tau multimers were previously shown to form in vitro under nonreducing conditions through the formation of intermolecular disulfide bonds (Schweers et al., 1994, 1995; Friedhoff et al., 1998). We were therefore interested to determine whether such “disulfide-bond-dependent” multimers might also exist in vivo in rTg4510 because these species would have been lost in our standard SDS-PAGE Western blot analysis attributable to the reducing conditions used in this technique. To test this hypothesis without in vitro artifacts, we performed rTg4510 brain extractions under conditions that enabled us to capture the state of sulfhydryl groups and disulfide bonds in vivo (see Material and Methods) and then performed Western blot analysis of tau multimer species under reducing and nonreducing conditions. However, these studies showed no evidence of additional disulphide-bond-dependent multimers in rTg4510 mice (supplemental Fig. S1, available at www.jneurosci.org as supplemental material).
Properties of tau multimers in rTg4510 mice
Tau multimers were initially detected in total brain extracts (Fig. 1). To characterize the extent of aggregation of tau multimers in vivo, we initially used a fractionation procedure (Greenberg and Davies, 1990), frequently used to separate aggregated tau species (see schematic in Fig. 2A). Total extracts were first centrifuged at high speed (150,000 × g), leading to accumulation of larger insoluble tau aggregates in the pellet, whereas monomeric or oligomeric tau species remain in the supernatant (Fig. 2A). The supernatant is the soluble (S1) fraction, whereas the pellet is further extracted with the detergent sarkosyl to obtain the sarkosyl-insoluble (P3) fraction.
The soluble (S1) fraction contains the normal human tau that migrates as a series of diffuse bands at ∼55 kDa on SDS-PAGE. In contrast, the sarkosyl-insoluble (P3) fraction (from the extracts of rTg4510 mice >4 months) contains pathological hyperphosphorylated tau species that migrates as 64 kDa species (Fig. 2B). The material in sarkosyl-insoluble (P3) fraction also ultrastructurally resembles NFTs. However, the P3 fraction does not contain mature NFTs because these large structures are removed by low-speed centrifugation during the initial preparation of the total brain extracts (see schematic in Fig. 2A) (Lewis et al., 2000, 2001; Sahara et al., 2002; Santacruz et al., 2005).
In fractionated brain extracts from rTg4510 mice, tau multimers were detected in both soluble (S1) and sarkosyl-insoluble (P3) fractions (Fig. 2B). However, in the soluble fraction, tau140 multimers were the major species, whereas tau170 was essentially absent. In contrast, the tau170 species were most abundant in the sarkosyl-insoluble (P3) fraction (Fig. 2B). Significantly, tau170 multimers present in the P3 fraction were strongly immunoreactive with the AT8 antibody, known to selectively recognize pathological hyperphosphorylated tau, including the 64 kDa species. However, the tau140 multimers present in the soluble (S1) fraction showed little AT8 immunoreactivity. These data suggest that tau140 exists mostly as relatively small aggregates in vivo, which are not hyperphosphorylated, whereas in contrast tau170 is derived from larger aggregated structures that are also hyperphosphorylated.
Although generally similar results were obtained in rTg4510 at all ages examined (see Figs. 3, 4, 6), it is important to emphasize that, in young mice (up to 5.5 months) (see Figs. 3, 4), only the tau140 multimers and the 55 kDa tau species were detected in the soluble (S1) fraction. In contrast, the hyperphosphorylated tau170 multimers and 64 kDa tau species were not detected in the soluble (S1) fraction. However, in older mice (>6.5 months), a relatively small quantity of tau170 was observed in the soluble (S1) fraction (see Figs. 2B, 6B). Similarly, in 6.5-month-old rTg4510, 64 kDa tau could be detected in soluble (S1) fraction with the phospho-specific AT8 antibody (data not shown). These results suggest that hyperphosphorylated tau is only present in large aggregates early in the disease cascade.
Figure 3.

Changes in the levels of tau species between 1 and 4.5 months. A, Tau species present in total extracts in 1- and 4.5-month-old rTg4510. Abundant ∼55 kDa tau is detected at both time points, and the levels of this species appear to increase over this time period. Hyperphosphorylated tau 64 kDa can be detected at 4.5 months with the AT8 antibody (arrow, the top band in the doublet). Levels of the tau140 multimers are increased at 4.5 months, relative to 1 month, and tau170, also detected by the AT8 antibody, is detectable only at 4.5 months. B, Tau species present in the soluble (S1) fraction in 1- and 4.5-month-old rTg4510. Only tau140 is present in the soluble (S1) fraction, whereas tau170 is absent. Tau140 levels increase between 1 and 4.5 months similar to that observed in total fraction. C, Tau ∼55 kDa positively correlates with the levels of the multimers (tau140) in the soluble (S1) fraction. Data from 1-month-old animals are depicted in squares, whereas data from 4.5-month-old animals are in triangles; the same applies to D. D, Levels of tau140 multimers positively correlate with levels of tau170 in total extracts.
Figure 4.

Analysis of tau species present at 5.5-month-old rTg4510. A, Tau species present in total extracts in 5.5-month-old rTg4510. The abundant ∼55 kDa tau is observed with the E1 antibody without obvious variability between mice. Hyperphosphorylated 64 kDa tau is detected both by E1 and the phospho-epitope-specific AT8 antibody. Both tau170 and tau140 are seen with the E1 antibody, whereas only tau170 is recognized specifically by AT8. Levels of the tau multimers (tau170 and tau140) appear to be variable between different animals. B, Soluble (S1) fraction contains tau ∼55 kDa and tau140, but tau 64 kDa and tau170 are absent. C, Levels of multimers in total extracts exhibit a higher extent of variability across the group (SD of 0.33; 48% of the mean) compared with the levels of multimers in the soluble (S1) fraction (SD of 0.06; 21% of the mean).
Figure 6.
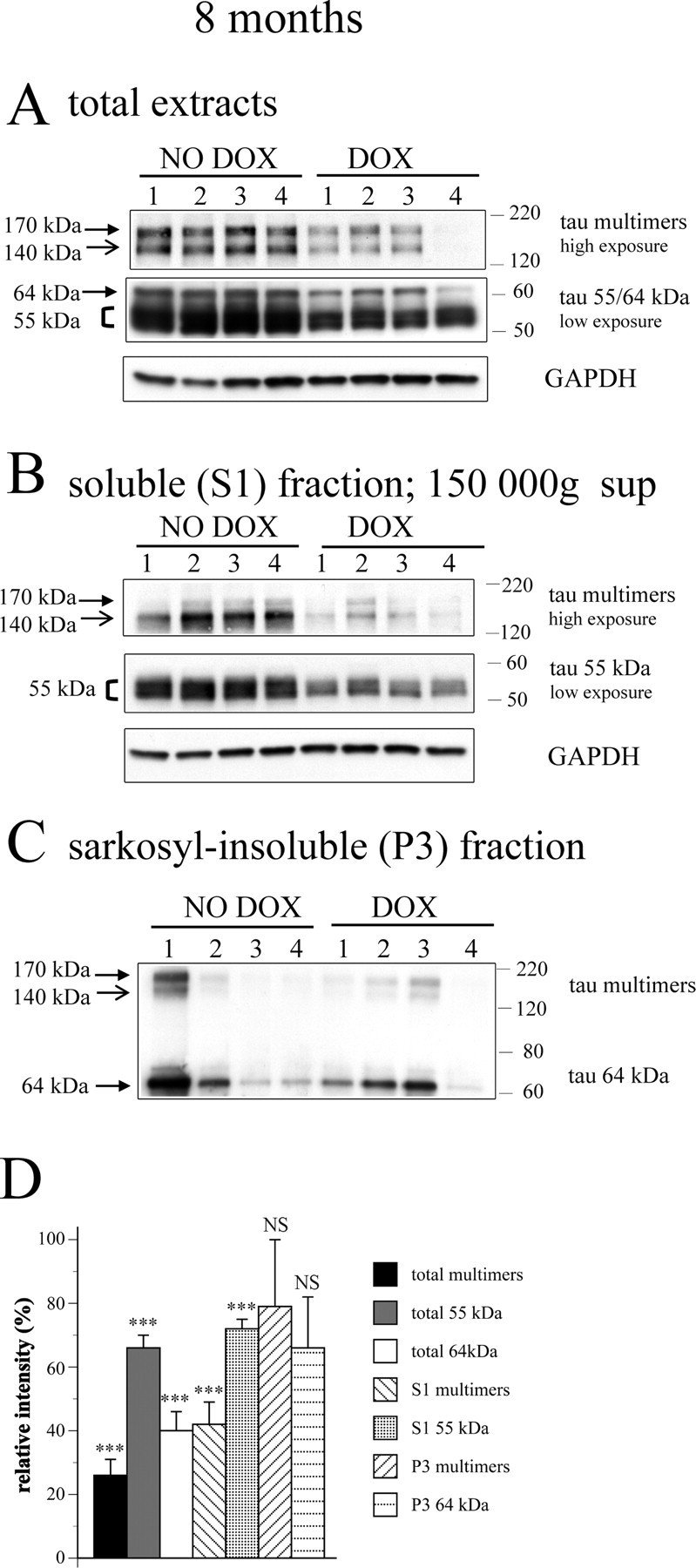
Changes in tau species after suppression of transgene expression with doxycycline between 6.5 and 8 months. A, Transgene suppression with doxycycline led to decreased levels of ∼55 kDa tau, 64 kDa tau, and tau multimers (tau170 and tau140) in total extracts. B, Transgene suppression with doxycycline also led to decreased levels of tau species in the soluble (S1) fraction. C, Levels of sarkosyl-insoluble 64 kDa tau and tau multimers in the P3 fraction were variable and did not show a consistent decrease with transgene suppression. D, Quantification of changes of different tau species from experiment in Figure 6A–C. The values of tau species in animals not treated with doxycycline was set as 100% (n = 10 for animals without doxycycline; n = 11 for animals treated with doxycycline). ***p < 0.001; NS p > 0.05. Two-sample two-tailed t test was used.
To confirm the apparent differences in the relative size of the various tau species, we next performed SEC on total extracts from rTg4510 at 4.5 and 6.5 months of age. We used these two ages because hyperphosphorylated tau species (tau170 and 64 kDa tau) can be detected only in insoluble fractions at 4.5 months, although they appear in both soluble (S1) and insoluble (P3) fractions at 6.5 months.
As expected, SEC on brain extracts from 4.5-month-old rTg4510 showed a clear distinction between the relative sizes of the tau140 and tau170 species with no overlap in their elution profile (Fig. 2D). Tau140 was primarily detected in fractions 30–32 that correspond to a molecular weight just above ∼70 kDa, raising the possibility that this species may exist as a single 140 kDa unit. A shoulder on the tau140 elution profile (fractions 26–28) also suggests that a small proportion of this species is present in larger aggregates. In contrast, tau170 was only found in fractions that correspond to molecular weights between ∼670 kDa and ∼1.5 MDa, suggesting that tau170 is exclusively a component of larger aggregated structures at 4.5 months of age. The 64 kDa species also appeared only in fractions with high molecular weight, at 4.5 months, indicating that it is also found in large aggregates, similar to tau170. Interestingly, the ∼55 kDa tau species exhibited a broad elution profile, suggesting that some normally phosphorylated tau (that migrates as ∼55 kDa tau on Western blots) is part of larger aggregates (fractions 20–22).
In older 6.5-month-old rTg4510 mice, SEC again showed that the majority of tau140 elutes with smaller size than tau170, similar to results at 4.5 months (Fig. 2E). However, in the older mice (6.5 months), tau140 and tau170 also give overlapping elution profiles with both present in detectable amounts in fractions 24–26 (Fig. 2E). This indicates that, in older mice, unlike at 4.5 months, the two tau multimer species can be present in aggregates of similar intermediate size. Interestingly, 64 kDa tau also gave two distinct SEC elution peaks in 6.5-month-old mice, indicating that this species is not only present in large aggregates (as at 4.5 months) but is also derived from smaller aggregates and possibly hyperphosphorylated tau monomers.
Age-dependent changes in tau species in rTg4510
To determine how levels of the tau multimers and other tau species change with age in rTg4510, we analyzed in parallel brain extracts of 1-month-old and 4.5-month-old mice. In the period from 1 to 4.5 months, rTg4510 mice develop large numbers of pretangle lesions and also mature silver-positive NFTs (Santacruz et al., 2005). We analyzed total extracts and soluble (S1) fractions.
Hyperphosphorylated tau migrating at 64 kDa was observed in total extracts of 4.5-month-old rTg4510, although this species was not detected at 1 month of age (Fig. 3A). The 64 kDa species was absent in the S1 fraction at both ages (Fig. 3B), consistent with previous observations that the 64 kDa tau is not detected in the S1 fraction in young (<6.5 month old) mice. Tau ∼55 kDa was relatively abundant (compared with 64 kDa tau) in total extracts both at 1 and 4.5 months, as assessed with the E1 antibody. The levels of ∼55 kDa tau in total extracts significantly increased from 1 to 4.5 months (1.4-fold; p = 0.006, two-tailed t test; n = 16). Similarly, ∼55 kDa tau detected in the soluble (S1) fraction also significantly increased during this period (1.5-fold; p = 0.007, two-tailed t test; n = 16). This increase in ∼55 kDa tau might reflect an increase in transgene-driven human tau expression during this period or alternatively it may be attributable to the formation of tau aggregates containing the ∼55 kDa species as suggested by the SEC studies (Fig. 2E).
Tau140 multimers were detected in total extracts even at 1 month of age, whereas tau170 multimers were absent. At 4.5 months, both tau140 and tau170 were present in total extracts (Fig. 3A). In the soluble (S1) fractions, from both ages, only tau140 was present with a threefold increase in levels (p < 0.001, two-tailed t test; n = 16) observed from 1 to 4.5 months (Fig. 3B). Similarly, tau140 multimers in total extracts increased significantly during this period (threefold; p < 0.001, two-tailed t test; n = 16). Levels of ∼55 kDa tau correlated positively with tau140 in the soluble (S1) fractions (R2 = 0.75; p < 0.001) (Fig. 3C); moreover, levels of tau140 exhibited a significant positive correlation with levels of tau170 in total extracts (R2 = 0.30; p < 0.05) (Fig. 3D).
Relationship between tau species and memory deficits in 5.5-month-old rTg4510
To test the relationship of tau species and memory deficits in rTg4510 mice, we analyzed the spatial memory of a cohort of 12 mice at 5.5 months, using the reference memory version of the Morris water maze test (Morris, 1981). The evaluation of spatial memory was performed by computing an annulus crossing index, referred to as the “memory index” throughout, during the probe trials (see Material and Methods) (Janus, 2004). The time point of 5.5 months was chosen because partial recovery of memory function was observed in rTg4510 after transgene suppression at 5.5 months, although rTg4510 exhibits significant memory impairment and neuronal loss in the hippocampus at this age (Santacruz et al., 2005).
Western blot analysis of this cohort of 5.5-month-old rTg4510 mice, with the E1 antibody (Fig. 4A), detected both the ∼55 kDa and hyperphosphorylated 64 kDa tau species in total brain extracts. The hyperphosphorylated 64 kDa tau was again selectively recognized by the AT8 antibody (Fig. 4A). In addition, in total extracts, the tau170 and tau140 multimers were also detected with the E1 antibody and tau170 was recognized by AT8 (but not tau140) (Fig. 4A). In contrast, in the soluble (S1) fraction, only the ∼55 kDa tau and tau140 multimers were observed (Fig. 4B).
The levels of both tau multimer species (tau170 and tau140) in total extracts displayed marked variability between individual mice within the group (SD of 0.33; 48% of the mean) (Fig. 4C). In contrast, levels of tau140 present in the soluble (S1) fraction appeared less variable (SD of 0.06; 21% of the mean) (Fig. 4C).
The combined levels of the two tau multimer species (140 and 170 kDa) in total extracts correlated negatively with the memory index (R2 = 0.57; p < 0.01) (Fig. 5A). In addition, levels of individual 140 and 170 kDa multimer species (determined using the E1 antibody) also correlated negatively with memory index to a similar extent (tau140, R2 = 0.52, p < 0.01; tau170, R2 = 0.57, p < 0.01; data not shown). Levels of tau170 analyzed with the AT8 antibody also showed a similar correlation (R2 = 0.56; p < 0.01; data not shown). In contrast, levels of the soluble multimers (S1 fraction) did not show any correlation with memory index (R2 = 0.008; p = 0.80) (Fig. 5A).
Figure 5.

Relationship of tau species and memory impairment (memory index) in 5.5-month-old rTg4510. A, Levels of multimers (tau170 and tau140) in total extracts showed a significant negative correlation with memory index, whereas multimers in the soluble (S1) fraction did not. B, Levels of ∼55 kDa tau in total extracts or in the soluble (S1) fraction did not significantly correlate with memory index. C, Levels of hyperphosphorylated tau ∼64 kDa in total extracts did not exhibit significant correlation with memory index. However, the subpopulation of this species that was extracted into the sarkosyl-insoluble (P3) fraction correlated significantly and negatively with memory index.
Next we examined the correlation of other tau species with memory index in the cohort of 5.5-month-old rTg4510 mice. The ∼55 kDa tau present in total extracts did not exhibit significant correlation with memory index (R2 = 0.12; p = 0.26) (Fig. 5B). Similarly, ∼55 kDa tau in soluble (S1) fraction did not show a significant correlation with memory index (R2 = 0.14; p = 0.47) (Fig. 5B). Levels of hyperphosphorylated 64 kDa tau in total extracts also did not show a significant correlation with memory index (R2 = 0.22; p = 0.13) (Fig. 5C). However, sarkosyl-insoluble 64 kDa tau species, present in the P3 fraction, negatively correlated with memory index (R2 = 0.35; p < 0.05) (Fig. 5C).
Single transgenic littermates with the tau responder transgene alone did not develop tau multimers or the 64 kDa tau species (data not shown). Consistent with this lack of pathological tau species, the single transgenic mice displayed normal spatial memory performance that was indistinguishable from nontransgenic littermates (data not shown).
In summary, pathological, aggregated tau species, tau multimers in the total extracts, and the 64 kDa in the sarkosyl-insoluble (P3) fraction exhibited a significant negative correlation with the memory index in individual 5.5-month-old rTg4510 mice. In contrast, levels of soluble tau species (tau140 and ∼55 kDa) did not correlate with the memory index.
Relationship between tau species and memory deficits after suppression of transgene expression
Suppression of transgene expression in rTg4510 mice at 2.5 months and 5.5 months of age was associated with recovery of spatial reference memory (Santacruz et al., 2005). This paradigm allows us to determine whether levels of tau multimers or other species are associated with not only the development of memory deficits but also subsequent recovery. We therefore suppressed tau expression in a cohort of rTg4510 mice from 6.5 months until 8 months of age, using dox-supplemented mouse chow and compared them with age-matched rTg4510 mice on control chow (no dox; for details, see Material and Methods).
Transgene suppression in this cohort resulted in reduced levels of normal ∼55 kDa tau in both total brain extracts and in the soluble (S1) fraction (33 and 28% reduction, respectively; p < 0.001) (Fig. 6A,B,D), which was consistent with our previous findings (Santacruz et al., 2005). In addition, levels of hyperphosphorylated 64 kDa tau species in the total extracts were also significantly reduced (51% reduction; p < 0.001) (Fig. 6A,D). Levels of sarkosyl-insoluble (P3) fraction 64 kDa tau species were variable (Fig. 6C) and did not show a significant reduction relative to 8-month-old no-dox mice (p = 0.17) (Fig. 6d), which is again consistent with our previous studies (Santacruz et al., 2005).
Levels of tau multimers (tau170 and tau140) in total extracts were dramatically decreased after transgene suppression in this cohort (74% reduction; p < 0.001) (Fig. 6A,D), and a similar reduction was observed when tau140 and tau170 were assessed individually in total extracts (74 and 73% reduction, respectively; p < 0.001). In the soluble (S1) fraction, tau multimers (mostly tau140) were reduced by 58% (p < 0.001) (Fig. 6B,D).
Transgene suppression from 6.5–8 months resulted in improved memory function. The memory index was significantly higher in the dox rTg4510 group compared with animals kept on control chow (mean ± SEM memory index, 0.02 ± 0.37 mice without dox; 1.00 ± 0.30 mice with dox; p < 0.05, two-sample one-tailed t test).
Next we examined the relationship between the levels of different tau species and spatial memory in individual rTg4510 mice, after transgene suppression. The memory index negatively correlated with the levels of combined multimers (tau140 and tau170 species) in total extracts (R2 = 0.22; p < 0.02) (Fig. 7A). A similar correlation was observed when tau140 and tau170 multimers in total extracts were assessed individually (R2 = 0.28, p < 0.01 and R2 = 0.27, p < 0.01, respectively; data not shown). However, multimers in the soluble (S1) fraction (mostly tau140) again did not correlate significantly with memory index (R2 = 0.08; p = 0.09) (Fig. 7A).
Figure 7.

Relationship of tau species and memory index during transgene suppression by doxycycline between 6.5–8 months. A, Levels of multimers in total extracts correlated significantly and negatively with memory index, whereas levels of multimers in the soluble (S1) fraction do not. B, Levels of ∼55 kDa tau in total extracts or in soluble (S1) fraction did not exhibit significant correlation with memory index. C, Levels of hyperphosphorylated tau 64 kDa in total extracts did not significantly correlate with memory, whereas sarkosyl-insoluble 64 kDa tau in P3 fraction does exhibit significant and negative correlation with memory index.
Levels of the ∼55 kDa tau in total extracts and the soluble S1 fractions did not significantly correlate with memory index [R2 = 0.10, p = 0.09 (Fig. 7B) and R2 = 0.05, p = 0.17 (Fig. 7B), respectively]. Levels of hyperphosphorylated 64 kDa tau in total extracts did not show a significant correlation with memory index in this cohort (R2 = 0.08; p = 0.11) (Fig. 7C). However, interestingly, sarkosyl-insoluble 64 kDa tau (P3) did show a significant negative correlation with memory index (R2 = 0.22; p < 0.02) (Fig. 7C). This result is somewhat surprising given that sarkosyl-insoluble (P3) 64 kDa tau did not show a significant reduction after doxycycline treatment in our study but illustrates the importance of assessing tau species in individual mice rather than simply mean values from groups of animals. In summary, levels of pathological aggregated tau species, multimers in total extracts, and 64 kDa tau, in the sarkosyl-insoluble (P3) fraction, again gave significant negative correlations with memory index, in transgene suppressed mice, whereas levels of soluble tau species did not show such association.
The relationship of tau multimers and memory deficits in 3-month-old rTg4510
To test whether tau multimers correlate with memory in the absence of sarkosyl-insoluble (P3) 64 kDa tau, we studied a cohort of 3-month-old rTg4510. At this age, the rTg4510 mice developed significant memory impairment (t(24) = 2.7; p < 0.02, two-tail t test, with an outlier removed; see below) compared with nontransgenic littermates. Only tau140 multimers were present in both total extracts and soluble (S1) fraction (data not shown). Hyperphosphorylated tau170 multimers were not detected in total extracts at this age.
Levels of tau140 multimers in total extracts were not significantly correlated with memory indices in 3-month-old rTg4510 mice (R2 = 0.15; p = 0.10) (Fig. 8A). However, the inspection of the memory scores of these mice revealed one extreme score of 8.5 (Fig. 8A). Normalizing the scores using z-score transformation revealed that this score of 8.5 was an outlier deviating 2.62 SD from the mean with the probability of occurrence of p < 0.01. After removing this score from the analysis, the negative correlation between the tau140 multimers in the total extracts and memory reached a significance at α = 0.05 (R2 = 0.45; p < 0.02). In contrast, we again did not observe a correlation between levels of tau140 multimers in soluble (S1) fractions and the memory index (R2 = 0.15; p = 0.41) (Fig. 8B), and this result was not affected by excluding the single outlier (R2 = 0.11; p = 0.16). Similar analysis performed on the cohorts of rTg4510 at 5.5 and 8 months of age (Figs. 5, 7) did not alter the level of statistical significance of these correlations (i.e., all correlations above α = 0.05 remained above this level and vice versa).
Figure 8.

Tau multimers in 3-month-old rTg4510. A, Levels of multimers in total extracts did not correlate significantly with memory index at 3.5 months. However, removal of an outlier animal with memory index of 8.5 (square) resulted in a significant and negative correlation between levels of tau140 multimers in total extracts and memory index (R2 = 0.45; p = 0.01). B, Levels of multimers in soluble (S1) fraction did not correlate significantly with memory index at 3.5 months, and this analysis is not affected by removing the outlier animal (R2 = 0.11; p = 0.16).
Relationship of different tau protein species
Our studies demonstrated that both the tau140 and tau170 multimers in total extracts and the sarkosyl-insoluble 64 kDa tau consistently correlated with memory index in two different cohorts of rTg4510 mice at different ages. Given these observations, we next explored the relationship between these pathological tau species. We observed a strong positive correlation between tau140 and tau170 multimers in total extracts (5.5 months, R2 = 0.88, p < 0.001; 8 months, R2 = 0.88, p < 0.001) (supplemental Fig. S3A, available at www.jneurosci.org as supplemental material). This observation is consistent with our previous results that show that levels of the tau140 and tau170 multimers correlate with memory index to a similar extent. Moreover, we also saw a positive correlation between levels of the multimers (tau170 and tau140) in total extracts and sarkosyl-insoluble 64 kDa tau in the P3 fraction (5.5 months, R2 = 0.36, p < 0.05; 8 months, R2 = 0.16, p < 0.05) (supplemental Fig. S3B, available at www.jneurosci.org as supplemental material). This indicates a possible relationship between the formation of the tau140, tau170 multimers, and tau 64 kDa. Interestingly, this positive correlation was even stronger when we analyzed the tau multimers and tau 64 kDa, present in the sarkosyl-insoluble (P3) fraction (5.5 months, R2 = 0.69, p < 0.001; 8 months, R2 = 0.91, p < 0.001) (supplemental Fig. S3C, available at www.jneurosci.org as supplemental material). This likely reflects the fact that the P3 fraction contains almost exclusively tau170 multimers that, like 64 kDa tau, are hyperphosphorylated and appear to be derived from large aggregated species (Fig. 2D).
Relationship of NFT counts in rTg4510 mice to tau multimers and memory deficits
To compare the biochemical results described above with the extent of pathology in individual mice, we determined NFT counts in the same series of 5.5-month-old rTg4510 mice. We observed a significant correlation between NFT numbers and levels of the tau multimers (R2 = 0.50; p < 0.001; data not shown) in this cohort of mice consistent with both the multimers and end-stage NFT being part of a continuum of pathological tau aggregation events. In addition, the NFT counts in individual mice displayed a borderline negative correlation with memory index (R2 = 0.33; p = 0.05) (Fig. 9A). In contrast, multimer levels gave a highly significant negative correlation with memory index in the same cohort of mice (Fig. 5A), suggesting that levels of multimers are more predictive of memory function, at least using with the methodology used in this study.
Figure 9.

NFT counts and memory index in 5.5- and 8-month-old rTg4510. A, The number of neurofibrillary tangles in individual rTg4510 mice and memory index displayed a borderline significant negative correlation (R2 = 0.33; p = 0.05) in 5.5-month-old rTg4510. B, In 8-month-old rTg4510 mice, the number of neurofibrillary tangles correlated negatively with memory index only in the no-dox cohort (R2 = 0.47; p = 0.02), whereas this correlation was completely lost after transgene suppression in the dox cohort (R2 = 0.03; p = 0.33). Dox treatment was for 6 weeks from 6.5 to 8 months.
In a similar manner, we also examined the relationship of NFT counts to memory index in 8-month-old rTg4510 mice treated with dox for 6 weeks to suppress transgene expression. Although the levels of the NFTs were not significantly different between the dox and no-dox groups (p = 0.33), this analysis revealed that, although NFT numbers again showed a significant negative correlation with memory in the no-dox group (R2 = 0.47; p = 0.02), this relationship was completely lost after transgene suppression with dox (R2 = 0.03; p = 0.33) (Fig. 9B). This result is in a stark contrast to previous analysis of the correlation between memory and levels of tau multimers or P3 64 kDa tau (Fig. 7A–C), in which similar negative correlations for these species was observed in both dox and no-dox groups.
The association of NFT counts with memory index in rTg4510 mice constitutively expressing mutant tau at 5.5 months and 8 months of age (Fig. 9) is reminiscent of the consistent correlation observed between NFT pathology and memory deficits in human AD patients (Arriagada et al., 1992; Guillozet et al., 2003). Moreover, the loss of the correlation between NFT and memory index in individual 8-month-old mice, after transgene suppression, extends our previous observation that NFT formation can be dissociated from memory loss in rTg4510 (Santacruz et al., 2005).
Tau species in JNPL3 mice
We also evaluated the presence of different tau species, including tau multimers, in a second mouse model of tauopathy, the JNPL3 transgenic mouse line that expresses the same mutant tau isoform (0N4R P301L isoform) as rTg4510. Average levels of human tau expression, in JNPL3 mice, are similar to levels of endogenous mouse tau. JNPL3 mice exhibit motor deficits with variable onset age, starting as early as 6.5 months, associated with the development of neurodegenerative changes in the spinal cord (Lewis et al., 2000).
We observed tau of ∼55 kDa and tau140 multimer species in total spinal cord extracts from 2-month-old JNPL3 mice (Fig. 10A), demonstrating that tau140 multimers are formed early in the pathogenic progression in this model. At this age, sarkosyl-insoluble 64 kDa tau (in P3 fraction) was not detected (Lewis et al., 2000).
Figure 10.

Relationship of tau species to motor deficits in JNPL3 mice. A, Multimers were detected in spinal cord extracts from 2-month-old JNPL3 mice (1–3) but were absent in nontransgenic littermates (NT). Asterisk marks nonspecific band also present in nontransgenic littermates, which is attributable to nonspecific binding of the secondary antibody. B, Spinal cord and brain extracts from JNPL3 female mice at the age of 12 (1) and 14 months (2). Tau multimers (tau170 and tau140) and 64 kDa tau species are present in spinal cord but are not clearly visible in brain. C, Tau multimers (tau170 and tau140) and hyperphosphorylated 64 kDa are detected in JNPL3 mice severely affected with motor dysfunction, but neither of these species is seen in unaffected JNPL3 mice of similar age. D, Tau multimers are not present in mice overexpressing wild-type human tau. wt tau, Spinal cord extracts from mice overexpressing wild-type human tau (JN25 line) at 14-months of age; JNPL3, spinal cord extracts from 14-month-old JNPL3 mice. Note that there is no evidence of tau multimers in mice expressing human wild-type tau, despite significantly higher levels of total tau being loaded onto the blot, compared with JNPL3 mice. The apparent low level of tau 55/64 kDa in the JNPL3 mice seen on this Western blot is only attributable to this adjustment (∼3 times more protein was loaded for wild-type tau).
We then compared tau species present in total extracts of spinal cord and brain from animals with functional (motor) deficits at 12–14 months of age. Both tau multimer species (tau170 and tau140) and hyperphosphorylated 64 kDa tau were present in total extracts in aged JNPL3 mice (Fig. 10B). Moreover, higher levels of these species were present in spinal cord compared with brain extracts (Fig. 10B), consistent with the distribution of NFT pathology in this model (Lewis et al., 2000). We also observed higher levels of multimers (tau140 and tau170) and 64 kDa tau in 12- to 15.5-month-old JNPL3 mice with functional (motor) deficits compared with unaffected mice of similar age, although these two groups of mice displayed similar levels of normal ∼55 kDa tau species (Fig. 10C). In contrast, no tau140 or tau170 multimers were detected in spinal cord extracts from 14-month-old animals expressing wild-type human tau (Fig. 10D) that do not develop NFT pathology or motor deficits (Lewis et al., 2000). The same mouse prion promoter was used in the lines expressing wild-type or mutant tau P301L (Lewis et al., 2000). These results demonstrate that, similar to rTg4510 mice, the accumulation of tau multimers and hyperphosphorylated 64 kDa tau is associated with the development of functional deficits in a second mouse model of tauopathy, the JNPL3 mice.
Tau species with similar characteristics to tau multimers accumulate in human tauopathies
To test whether similar multimer species form during the development of human tauopathy, we examined brain extracts from two patients with the N279K MAPT mutation (Wszolek et al., 1992; Reed et al., 1998; Arvanitakis et al., 2006). We observed tau-immunoreactive bands at molecular weight ∼170 kDa in extracts from temporal cortices (a region affected by tau pathology) from these individuals (Fig. 11A, lanes D1, D2), whereas these species were absent in extracts from the cerebellum (a region without tau pathology in these patients) and were also absent in extracts from both regions of nondemented controls. These bands appeared relatively well defined and migrated at identical molecular weight to the tau170 species that we described in tau mice. The levels of the multimers in tissue from the FTDP-17 patients appeared relatively low (compared with AD brains, see below) (Fig. 11B). This is consistent with the low Braak stage in the FTDP-17 patients with the N279K mutation (0.8 ± 0.4) (Wszolek et al., 2005). In summary, these data suggest that species similar to tau multimers are present in human tauopathy associated with an MAPT mutation.
Figure 11.

Species similar to tau multimers are found in extracts from patients with AD and FTDP-17. A, Species similar to tau multimers are present in total extracts from the temporal cortex of FTDP-17 patients (D1, D2) with the N279K mutation but not in extracts from the cerebellum. Note that the low-intensity band present in control individuals (C) or in cerebellum is attributable to nonspecific binding of the secondary antibody (data not shown). B, Comparison of the molecular weight of the multimers in total extracts from the spinal cords of JNPL3 mice (affected, 12 and 14 months of age), brain extracts from 6.5-month-old rTg4510, an AD patient (Braak stage 5 and 6), and two FTDP-17 cases with the N279K mutation. In both panels, the human-specific E1 antibody was used for detection of tau, and GAPDH was used as a loading control.
Previous studies reported the presence of high molecular tau species in brain extracts from patients with Alzheimer's disease (Morishima-Kawashima et al., 1993; Weaver et al., 2000; Maeda et al., 2006), although their possible role in disease was not addressed. We analyzed these species to compare them with the tau multimers in the tau mice. In AD brain, a more complex mixture of species was observed that migrated as a compact smear with a similar molecular weight range to the tau multimers in mice (∼140–170 kDa) (Fig. 11B) (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). The increased complexity of the multimer-like species in AD patients may well reflect the fact that, in AD, all six tau isoforms are incorporated into the pathological tau aggregates, whereas in FTDP-17 patients with the N279K mutation, it is just the four-repeat tau isoforms. The multimer-like species in AD were detected in brain regions with NFT pathology (frontal cortices) but were absent in unaffected regions (cerebellum) (data not shown).
Discussion
Characterization of pathological tau species in mouse models of tauopathy
Previous results in the rTg4510 mouse model suggested the existence of toxic tau species that accumulate before the formation of significant numbers of mature NFT (Santacruz et al., 2005). We therefore undertook a series of biochemical investigations to identify candidate neurotoxic tau species. These studies identified tau multimers both in rTg4510 and a second mouse model of tauopathy (JNPL3) that expresses mutant human tau P301L.
Tau multimers migrate on SDS-PAGE with apparent molecular weights of 140 and 170 kDa (tau140 and tau170). We speculate that these species may represent tau dimers, with tau140 consisting of two “55 kDa” tau molecules and tau170 containing two “64 kDa” tau molecules; the apparent discrepancy in molecular weight (140 vs 55 × 2 = 110) may be accounted for by their SDS-stable conformation or by unequal binding of SDS to this species. However, we cannot exclude the possibility that tau multimers may be composed of tau in complex with another protein(s).
Regardless of the exact composition, the two multimer species represent tau aggregation states that are stable during exposure to SDS. This is in contrast to previously reported aggregated tau species that are sensitive to SDS and that are broken down to individual monomers during incubation with SDS. On Western blots, these SDS-sensitive aggregated species are observed as the hyperphosphorylated 64 kDa tau species. SEC, performed on total brain extracts from young (4.5 month) rTg4510 mice, demonstrate that both the 64 kDa and the tau170 species are subunits of much larger tau aggregates. It is, however, unknown whether these two tau species are derived from same or different aggregates that elute with similar size on SEC.
In contrast, similar SEC analysis indicates that tau140 multimers predominantly exist in smaller structures, possibly even as a single 140 kDa unit. The tau140 multimers are the earliest pathological tau species detected in the rTg4510 mice. In contrast, the tau170 and 64 kDa species (derived from larger aggregates) appear later in the pathological progression. Importantly, the tau170 and 64 kDa species are hyperphosphorylated, whereas tau140 exhibits a phosphorylation state similar to normal (55 kDa) tau species. This suggests that pathological tau hyperphosphorylation is an event that likely occurs after initial tau aggregation has taken place, at least during the initial stages of disease development. An alternative explanation is that hyperphosphorylated monomeric tau species normally have an extremely short half-life before they are degraded or incorporated into large aggregated structures, with the result that they are undetectable in soluble extracts during early stages of disease in rTg4510 mice. However, this seems less likely because small tau aggregates with a normal pattern of phosphorylation (e.g., tau140) are observed at this stage, and, as a result, initial tau aggregation events in vivo are clearly not absolutely dependent on previous tau hyperphosphorylation. This is a critical point because the majority of current hypotheses regarding the role of tau hyperphosphorylation have assumed that pathological hyperphosphorylation of tau is required for the release of tau from microtubules and for subsequent tau aggregation (Lee et al., 2001).
Interestingly, both fractionation through centrifugation and SEC elution profiles show that small hyperphosphorylated tau aggregates and possibly hyperphosphorylated tau monomers are present in older rTg4510 mice (6.5 months) (Fig. 2E and data not shown). Whether this reflects the breakdown of larger hyperphosphorylated aggregates (perhaps as a consequence of widespread neuronal death) or a shift to the early hyperphosphorylation of monomeric tau in older animals is unknown. Significantly, hyperphosphorylated soluble tau species were observed previously in extracts from the brains of AD patients, usually at a relatively late disease stage. Our data, in the rTg4510 mice, suggest that, in human AD, there may also be an initial stage during which hyperphosphorylated tau occurs only within large tau aggregates. Regardless, the SEC data obtained from the rTg4510 model of tauopathy suggest that a fundamental revision of the role of tau hyperphosphorylation in AD pathogenesis may be necessary.
Tau species and functional deficits in mouse models of tauopathy
Levels of tau multimers (tau140 and tau170) in total extracts and 64 kDa tau in the sarkosyl-insoluble fraction displayed a significant negative correlation with memory index in individual rTg4510 mice at 5.5 and 8 months. We also observed an association between the onset of motor impairment in the JNPL3 mouse model of tauopathy and elevated levels of both the tau multimers and the 64 kDa species. Importantly, we also observed a correlation between tau140 multimers, in total extracts, and memory index in rTg4510 at 3 months, when sarkosyl-insoluble 64 kDa tau was not detectable. This indicates that formation of sarkosyl-insoluble 64 kDa tau occurs after the initial accumulation of the tau140 multimers and also that formation of this insoluble 64 kDa species does not appear to be necessary for the development of initial memory impairment in rTg4510, consistent with our previous observations (Santacruz et al., 2005). Low levels of tau140 were also observed at 1 month, before the onset of memory deficits (Fig. 3), suggesting that tau multimers and/or other aggregated species, if they are toxic, must accumulate to a critical level before affecting neuronal function.
Although we consistently observed a significant correlation between memory index and levels of tau multimers in total extracts, we did not observe a similar correlation with the levels of tau multimers in the soluble (S1) fraction. Thus, we conclude that it is the accumulation of larger (more aggregated) tau species that predominantly correlated with memory index. However, the dissociation of NFT counts from the memory index after transgene suppression in rTg4510 mice (Fig. 9B) (Santacruz et al., 2005) suggests that the aggregates associated with memory dysfunction are of intermediate size and smaller than NFTs. Based on these observations, it is tempting to speculate that the early accumulation of intermediate aggregated tau species, composed at least in part of tau multimers, is necessary to induce neurotoxicity in these transgenic mice. However, these observations do not exclude the possibility that other, yet unidentified small soluble tau species also contribute to neurodegeneration and memory loss in the rTg4510 model. If such species do exist, however, their formation is likely to be closely linked to the process of tau aggregation.
Relationships between tau species
Our data argue against the possibility that formation of the tau140 and tau170 species is simply attributable to overexpression of tau in transgenic mouse models. First, overexpression of wild-type human tau in transgenic mouse models is not sufficient to cause formation of similar multimer species. Second, species similar to tau multimers are observed in cases of human tauopathy (FTDP-17) without tau overexpression. Therefore, tau overexpression alone is neither necessary nor sufficient to cause formation of tau multimers. In addition, tau140 and tau170 show a close correlation, making it unlikely that they are formed by two unrelated mechanisms. A much more obvious possibility is that the tau170 represents a hyperphosphorylated version of tau140 that has been incorporated into bigger aggregates. The difference in the elution profiles of these two species in SEC studies is clearly consistent with this hypothesis.
Tau multimer-like species in AD and FTD
We also detected species similar to the multimers in brain extracts from FTDP-17 patients. These species are well defined and migrate at the same molecular weigh as tau170. We also observed similarities between tau multimers and reported previously high-molecular-weight tau species observed in brain extracts from AD patients (Morishima-Kawashima et al., 1993). However, they also exhibit significant differences from the multimers, notably the greater complexity, which is reflected in their migration as a smear on Western blots rather than discrete bands as in the tau mice and FTDP-17 patients. This greater complexity likely in part reflects the fact that all six tau isoforms are incorporated into the aggregates in the AD patients compared with three 4R isoforms in the N279K FTDP-17 patients and one mutant 4R isoform present in the transgenic mouse models. In addition, the interpretation of the these species in human AD is complicated by the likely presence of polyubiquitinated and other modified forms of tau (Morishima-Kawashima et al., 1993) that migrate in a similar size range.
Conclusion
Our results identify multimeric tau species that accumulate during pathogenesis in two mouse models of tauopathy and that correlate closely with functional deficits. This correlation is predominantly with tau multimers derived from larger aggregated structures. Consistent with this observation, the only other tau species that correlated with functional deficits in our mouse models is the previously described sarkosyl-insoluble 64 kDa tau species. The appearance of multimers before the formation of the sarkosyl-insoluble 64 kDa tau and the presence of multimers in both soluble and insoluble fractions imply that they reflect an early aggregation event and should be further examined for their potential role in tau-associated neuronal toxicity and degeneration. In addition, our data clearly imply that the formation of aggregated tau species is either necessary for neuronal dysfunction and memory loss in the rTg4510 mouse model or that tau aggregation is tightly linked to the mechanism of tau toxicity.
Footnotes
This work was supported by an EMBO long-term fellowship (Z.B.), National Institutes of Health Grants P01-AG017216 (M.H.), R01-NS4635 (J.L.), and P01-AG014449 and P30 AG10161 (D.D.), John Douglas French Alzheimer's Foundation (C.A.), Alzheimer's Disease Research Center Pilot Project and American Federation for Aging Research grants (C.J.), and Morris K. Udall Parkinson's Disease Research Center of Excellence Grant P50-NS40256-06 (Z.W., D.D.). We are grateful to Dr. S. H. Yen for helpful discussion, Dr. Peter Davies for providing CP13 and PHF1 tau antibodies, and Dr. Lester Binder for tau12 and TauC3 antibodies. We are grateful to Judith Wilson for technical assistance, Zeshan Ahmed and John Gonzalez for help with samples from human tauopathies, and Dr. Jason Eriksen, Dr. Cynthia Zehr, and Linda Rousseau for assistance with imaging and NFT counts.
References
- Alonso AD, Grundke-Iqbal I, Barra HS, Iqbal K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc Natl Acad Sci USA. 1997;94:298–303. doi: 10.1073/pnas.94.1.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andorfer C, Kress Y, Espinoza M, de Silva R, Tucker KL, Barde YA, Duff K, Davies P. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem. 2003;86:582–590. doi: 10.1046/j.1471-4159.2003.01879.x. [DOI] [PubMed] [Google Scholar]
- Andorfer C, Acker CM, Kress Y, Hof PR, Duff K, Davies P. Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci. 2005;25:5446–5454. doi: 10.1523/JNEUROSCI.4637-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- Arvanitakis Z, Witte RJ, Dickson DW, Tsuboi Y, Uitti RJ, Slowinski J, Hutton ML, Lin SC, Boeve BF, Cheshire WP, Pooley RA, Liss JM, Caviness JN, Strongosky AJ, Wszolek ZK. Clinical-pathologic study of biomarkers in FTDP-17 (PPND family with N279K tau mutation) Parkinsonism Relat Disord. 2006 doi: 10.1016/j.parkreldis.2006.10.007. in press. [DOI] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE. Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch Neurol. 2004;61:378–384. doi: 10.1001/archneur.61.3.378. [DOI] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT., Jr Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson's disease: implications for pathogenesis and therapy. Proc Natl Acad Sci USA. 2000;97:571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe A, Ksiezak-Reding H, Liu WK, Dickson DW, Yen SH. The N terminal region of human tau is present in Alzheimer's disease protein A68 and is incorporated into paired helical filaments. Am J Pathol. 1991;139:1463–1470. [PMC free article] [PubMed] [Google Scholar]
- Friedhoff P, von Bergen M, Mandelkow EM, Davies P, Mandelkow E. A nucleated assembly mechanism of Alzheimer paired helical filaments. Proc Natl Acad Sci USA. 1998;95:15712–15717. doi: 10.1073/pnas.95.26.15712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL, Lu M, Fu Y, Garcia-Sierra F, LaPointe N, Miller R, Berry RW, Binder LI, Cryns VL. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease. Proc Natl Acad Sci USA. 2003;100:10032–10037. doi: 10.1073/pnas.1630428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293:1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- Greenberg SG, Davies P. A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci USA. 1990;87:5827–5831. doi: 10.1073/pnas.87.15.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillozet AL, Weintraub S, Mash DC, Mesulam MM. Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Arch Neurol. 2003;60:729–736. doi: 10.1001/archneur.60.5.729. [DOI] [PubMed] [Google Scholar]
- Horowitz PM, Patterson KR, Guillozet-Bongaarts AL, Reynolds MR, Carroll CA, Weintraub ST, Bennett DA, Cryns VL, Berry RW, Binder LI. Early N-terminal changes and caspase-6 cleavage of tau in Alzheimer's disease. J Neurosci. 2004;24:7895–7902. doi: 10.1523/JNEUROSCI.1988-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- Janus C. Search strategies used by APP transgenic mice during spatial navigation in the Morris water maze. Learn Mem. 2004;11:337–346. doi: 10.1101/lm.70104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashuel HA, Petre BM, Wall J, Simon M, Nowak RJ, Walz T, Lansbury PT., Jr Alpha-synuclein, especially the Parkinson's disease-associated mutants, forms pore-like annular and tubular protofibrils. J Mol Biol. 2002;322:1089–1102. doi: 10.1016/s0022-2836(02)00735-0. [DOI] [PubMed] [Google Scholar]
- Le Corre S, Klafki HW, Plesnila N, Hubinger G, Obermeier A, Sahagun H, Monse B, Seneci P, Lewis J, Eriksen J, Zehr C, Yue M, McGowan E, Dickson DW, Hutton M, Roder HM. An inhibitor of tau hyperphosphorylation prevents severe motor impairments in tau transgenic mice. Proc Natl Acad Sci USA. 2006;103:9673–9678. doi: 10.1073/pnas.0602913103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- Maeda S, Sahara N, Saito Y, Murayama S, Ikai A, Takashima A. Increased levels of granular tau oligomers: an early sign of brain aging and Alzheimer's disease. Neurosci Res. 2006;54:197–201. doi: 10.1016/j.neures.2005.11.009. [DOI] [PubMed] [Google Scholar]
- Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Titani K, Ihara Y. Ubiquitin is conjugated with amino-terminally processed tau in paired helical filaments. Neuron. 1993;10:1151–1160. doi: 10.1016/0896-6273(93)90063-w. [DOI] [PubMed] [Google Scholar]
- Morris RGM. Spatial localization does not require the presence of local cues. Learn Motiv. 1981;12:239–260. [Google Scholar]
- Nichols MR, Moss MA, Reed DK, Lin WL, Mukhopadhyay R, Hoh JH, Rosenberry TL. Growth of beta-amyloid(1–s;40) protofibrils by monomer elongation and lateral association. Characterization of distinct products by light scattering and atomic force microscopy. Biochemistry. 2002;41:6115–6127. doi: 10.1021/bi015985r. [DOI] [PubMed] [Google Scholar]
- Novitskaya V, Bocharova OV, Bronstein I, Baskakov IV. Amyloid fibrils of mammalian prion protein are highly toxic to cultured cells and primary neurons. J Biol Chem. 2006;281:13828–13836. doi: 10.1074/jbc.M511174200. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin K. The mouse brain in stereotaxic coordinates, Ed 2. San Diego: Elsevier; 2001. [Google Scholar]
- Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–825. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- Reed LA, Schmidt ML, Wszolek ZK, Balin BJ, Soontornniyomkij V, Lee VM, Trojanowski JQ, Schelper RL. The neuropathology of a chromosome 17-linked autosomal dominant parkinsonism and dementia (“pallido-ponto-nigral degeneration”) J Neuropathol Exp Neurol. 1998;57:588–601. doi: 10.1097/00005072-199806000-00006. [DOI] [PubMed] [Google Scholar]
- Sahara N, Lewis J, DeTure M, McGowan E, Dickson DW, Hutton M, Yen SH. Assembly of tau in transgenic animals expressing P301L tau: alteration of phosphorylation and solubility. J Neurochem. 2002;83:1498–1508. doi: 10.1046/j.1471-4159.2002.01241.x. [DOI] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweers O, Schonbrunn-Hanebeck E, Marx A, Mandelkow E. Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J Biol Chem. 1994;269:24290–24297. [PubMed] [Google Scholar]
- Schweers O, Mandelkow EM, Biernat J, Mandelkow E. Oxidation of cysteine-322 in the repeat domain of microtubule-associated protein tau controls the in vitro assembly of paired helical filaments. Proc Natl Acad Sci USA. 1995;92:8463–8467. doi: 10.1073/pnas.92.18.8463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon R, Bar-Joseph I, Frosch MP, Walsh DM, Hamilton JA, Selkoe DJ. The formation of highly soluble oligomers of alpha-synuclein is regulated by fatty acids and enhanced in Parkinson's disease. Neuron. 2003;37:583–595. doi: 10.1016/s0896-6273(03)00024-2. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA. 1998;95:7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchikado H, Lin WL, DeLucia MW, Dickson DW. Alzheimer disease with amygdala Lewy bodies: a distinct form of alpha-synucleinopathy. J Neuropathol Exp Neurol. 2006;65:685–697. doi: 10.1097/01.jnen.0000225908.90052.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Weaver CL, Espinoza M, Kress Y, Davies P. Conformational change as one of the earliest alterations of tau in Alzheimerf's disease. Neurobiol Aging. 2000;21:719–727. doi: 10.1016/s0197-4580(00)00157-3. [DOI] [PubMed] [Google Scholar]
- Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. 2001;293:711–714. doi: 10.1126/science.1062382. [DOI] [PubMed] [Google Scholar]
- Wszolek ZK, Slowinski J, Golan M, Dickson DW. Frontotemporal dementia and parkinsonism linked to chromosome 17. Folia Neuropathol. 2005;43:258–270. [PubMed] [Google Scholar]
- Wszolek ZK, Pfeiffer RF, Bhatt MH, Schelper RL, Cordes M, Snow BJ, Rodnitzky RL, Wolters EC, Arwert F, Calne DB. Rapidly progressive autosomal dominant parkinsonism and dementia with pallido-ponto-nigral degeneration. Ann Neurol. 1992;32:312–320. doi: 10.1002/ana.410320303. [DOI] [PubMed] [Google Scholar]