

Abstract
Several types of neurons are able to regulate their synaptic inputs via releasing retrograde signal molecules, such as endocannabinoids or nitric oxide (NO). Here we show that, during activation of cholinergic receptors, retrograde signaling by NO controls CB1 cannabinoid receptor (CB1R)-dependent depolarization-induced suppression of inhibition (DSI). Spontaneously occurring IPSCs were recorded in CA1 pyramidal neurons in the presence of carbachol, and DSI was induced by a 1-s-long depolarization step. We found that, in addition to the inhibition of CB1Rs, blocking the NO signaling pathway at various points also disrupted DSI. Inhibitors of NO synthase (NOS) or NO-sensitive guanylyl cyclase (NO-sGC) diminished DSI, whereas a cGMP analog or an NO donor inhibited IPSCs and partially occluded DSI in a CB1R-dependent manner. Furthermore, an NO scavenger applied extracellularly or postsynaptically also decreased DSI, whereas l-arginine, the precursor for NO, prolonged it. DSI of electrically evoked IPSCs was also blocked by an inhibitor of NOS in the presence, but not in the absence, of carbachol. In line with our electrophysiological data, double immunohistochemical staining revealed an NO-donor-induced cGMP accumulation in CB1R-positive axon terminals. Using electron microscopy, we demonstrated the postsynaptic localization of neuronal NOS at symmetrical synapses formed by CB1R-positive axon terminals on pyramidal cell bodies, whereas NO-sGC was found in the presynaptic terminals. These electrophysiological and anatomical results in the hippocampus suggest that NO is involved in depolarization-induced CB1R-mediated suppression of IPSCs as a retrograde signal molecule and that operation of this cascade is conditional on cholinergic receptor activation.
Keywords: synaptic plasticity, hippocampus, GABA, CB1 receptor, cGMP, retrograde
Introduction
Retrograde signaling in synaptic communication regulates information flow in neuronal networks by altering the neurotransmitter release from presynaptic axon terminals in response to activation of a postsynaptic neuron. Several chemically distinct molecules mediating retrograde signaling have been proposed, for instance, classical neurotransmitters (e.g., glutamate and GABA), peptides (e.g., BDNF and dynorphin), gaseous molecules [e.g., nitric oxide (NO) and carbon monoxide], or lipids (e.g., arachidonic acid and endocannabinoids) (Tao and Poo, 2001; Alger, 2002). One of the best studied forms of retrograde signaling is the short-term depression of GABA release induced by depolarization of the postsynaptic neuron. This phenomenon, called depolarization-induced suppression of inhibition (DSI) was first described in the cerebellum (Llano et al., 1991) and the hippocampus (Pitler and Alger, 1992) and was later discovered in other brain areas (for review, see Chevaleyre et al., 2006). In DSI, the postsynaptic rise of Ca2+ concentration and the subsequent activation of presynaptically localized CB1 cannabinoid receptors (CB1R) are essential (Di Marzo et al., 1998; Lenz and Alger, 1999; Freund et al., 2003; Isokawa and Alger, 2006). According to the currently accepted model, depolarization of the postsynaptic cell is thought to lead to the release of endocannabinoids, which activates presynaptic CB1Rs, which, in turn, suppresses GABA release from those axon terminals (Kreitzer and Regehr, 2001; Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001).
However, a rise of Ca2+ concentration during depolarization may influence several signaling cascades in addition to the endocannabinoid system. One example is NO, a well known intercellular signaling molecule (Medina and Izquierdo, 1995; Prast and Philippu, 2001). NO can be synthesized by neurons in a Ca2+-dependent manner (Prast and Philippu, 2001) from l-arginine by NO synthases (NOS), which enzymes are present in hippocampal pyramidal cells (Wendland et al., 1994; Burette et al., 2002), and their activities could be significantly enhanced during the activation of acetylcholine receptors (Arnal et al., 1999; Christopoulos and El-Fakahany, 1999). Stimulation of NO-sensitive guanylyl cyclase (NO-sGC) is one of the main effector targets of NO, resulting in elevation of cGMP concentration. Because NO is a gaseous molecule that freely crosses membranes, it has been implicated as a retrograde messenger, e.g., in long-term potentiation in the hippocampus (O'Dell et al., 1991; Medina and Izquierdo, 1995; Prast and Philippu, 2001) (but see Cummings et al., 1994).
The aim of the present study was to investigate the contribution of NO signaling to DSI. We recorded action potential-dependent IPSCs in CA1 pyramidal cells while synaptic activity was enhanced by the cholinergic receptor agonist carbachol. Pharmacological manipulation of the NO signaling pathway at various points was used to examine its interference with DSI. We also tested the involvement of NO signaling in DSI of electrically evoked IPSCs. Moreover, immunohistochemical double staining was performed to investigate the NO donor-induced elevation of cGMP in CB1R-expressing boutons and the localization of neuronal NOS and NO-sGC at symmetrical synapses formed by CB1R-immunopositive axon terminals.
Materials and Methods
Slice preparation and electrophysiology.
Experiments were performed according to the guidelines of the institutional ethical code and the Hungarian Act of Animal Care and Experimentation (1998; XXVIII, section 243/1998.). Animals were deeply anesthetized with isoflurane and decapitated, and the brain was removed and placed in ice-cold cutting solution containing the following (in mm): 252 sucrose, 2.5 KCl, 1.25 NaH2PO4, 5 MgCl2, 0.5 CaCl2, 26 NaHCO3, and 10 glucose. The cutting solution had been bubbled with 95% O2/5% CO2 (carbogen gas). Horizontal slices of the hippocampus (350–400 μm thick) were prepared from male Wistar rats (14–19 d old) or 300-μm-thick sections from the hippocampus of littermate wild-type (CD1) or CB1R knock-out mice [22–41 d old of both sexes (Ledent et al., 1999)] using a Leica (Nussloch, Germany) VT1000S vibratome. Slices were incubated for 1 h in artificial CSF (ACSF) (in mm: 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2 MgCl2, 2 CaCl2, 26 NaHCO3, and 10 glucose) at room temperature in interface conditions and then transferred to a submerged type of recording chamber that was optimized for laminar flow. The flow rate was 3–4 ml/min, ensuring better oxygenization and more stable recordings (N. Hájos, unpublished observations). Whole-cell recordings were performed at 24–26°C under visual guidance using a Zeiss (Jena, Germany) Axioskop. Patch electrodes had resistances of 3–6 MΩ when filled with the intrapipette solution containing the following (in mm): 80 CsCl, 60 Cs-gluconate, 1 MgCl2, 2 Mg-ATP, 3 NaCl, 10 HEPES, and 5 QX-314 [2(triethylamino)-N-(2,6-dimethylphenyl) acetamine], pH 7.3 (290 mOsm). The pipette solution contained 0.1–0.3% biocytin in a subset of the experiments, and post hoc morphological identification of recorded cells using immunoperoxidase reaction confirmed that these were all pyramidal cells (n = 73). All experiments were performed on at least two independent preparations.
Spontaneous IPSCs (sIPSCs) were measured in ACSF containing the following (in mm): 126 NaCl, 2.5 KCl, 26 NaHCO3, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 10 glucose, 2 kynurenic acid, and 0.005 carbachol (equilibrated with 5% CO2–95% O2 gas). Access resistance (between 5 and 20 MΩ, compensated 65–75%) was frequently monitored and did not change substantially (±25%) during the analyzed period. Signals were recorded with an Axopatch 200B or Multiclamp 700A amplifier (Molecular Devices, Foster City, CA), filtered at 2 kHz, digitized at 6–10 kHz with a PCI-6024E board (National Instruments, Austin, TX), and off-line analyzed using the EVAN software (http://www.thotec.com/evan). Cells were held at −60 mV, and DSI was elicited usually every 100–130 s by step depolarizations to 0 mV for 1 s. After automatic event detection, integrated charge transfer of sIPSCs was determined. For calculation of DSI, the average charge of sIPSCs was measured during 10 s before depolarization (chargectr) and during 5 s after depolarization (chargedepol), discarding the first second after the voltage step, which includes the latency period of DSI (Wilson et al., 2001). The magnitude of DSI is expressed as DSI = 100 × (1 − chargedepol/chargectr), i.e., the percentage reduction of charge transfer. DSI was calculated from the averaged results of two to four DSIs under control and two to three DSIs under drug-treated conditions. In experiments in which drug effects were measured in the same cells, we analyzed cells only when DSI in the control period was at least 30% (±15% maximum SD) on average and exhibited at least >3 pA (pC for 1 s) basal charge transfer. In experiments in which differently treated groups were compared (see Figs. 1E,F and 4, and experiments with intracellular inhibitors), all measured cells receiving sufficient action potential-dependent inhibitory input (>3 pA) were included in the analysis regardless of their ability to express DSI. In these cases, control and drug-treated slices from the same brains were selected randomly. Basal charge transfer was usually measured for 10 s preceding depolarization except for Figure 4A–D, in which charge was measured for 30 s. There was no correlation between control basal charge and DSI (p > 0.5) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The recovery time course of DSI was fitted with single-exponential decay. When different intrapipette solutions were compared, we routinely applied weak positive pressure after breaking into whole-cell configuration to improve equilibrium of the pipette solution in the cytoplasm, and DSIs were measured between 5 and 15 min after break-in.
Figure 1.

Postsynaptically produced NO is involved in DSI. Spontaneous IPSCs (downward deflections from the baseline) showed transient reduction after a depolarizing step (arrow) in control (ctr) conditions (left; DSI); this alteration was compared after drug applications (middle). A–D, Left and Middle, Representative recordings of pyramidal cells before (left) and during (middle) application of 2 μm AM251 (CB1R antagonist; A), 100 μm l-NAME (NOS antagonist; B), 100 μm d-NAME (inactive analog of l-NAME; C), and 100 μm 7-NI (NOS inhibitor; D) for 5–10 min. Right, Magnitude and kinetics of DSI under control conditions (filled squares) and during application (open squares) of 2 μm AM251 or SR141716A (pooled, n = 6) (A), 100 μm l-NAME (n = 8) (B), 100 μm d-NAME (n = 4) (C), or 100 μm 7-NI (n = 7) (D). Charge was normalized to the control period before depolarization. E, DSI in two representative cells recorded using intrapipette solution containing only vehicle (0.1% DMSO; left) or 1 mm CPTIO (NO scavenger; middle). Right, Summary of DSI measured with pipettes containing DMSO (filled squares; n = 7) or CPTIO (open squares; n = 7). F, DSI in two cells pretreated for 30–40 min with 0.1% DMSO (left) or 500 μm extracellular CPTIO (middle). Right, Summary of DSI measured in DMSO (filled squares; n = 15) or in CPTIO (open squares; n = 9). G, Left and Middle, Representative recordings of pyramidal cells before (left) and during (middle) application of 0.2–1 mm l-arginine (NO precursor). Right, Magnitude and kinetics of DSI under control conditions (filled squares) and during application of l-arginine (n = 6). Except d-NAME, all drugs significantly altered DSI. Calibration: 50 pA, 5 s.
Figure 4.

CB1R function is a prerequisite for modulation of DSI by Br-cGMP and ODQ. A, B, Effect of 100 μm 8-Br-cGMP (A) and 10 μm ODQ (B) on sIPSCs in control (ctr) cells. C, D, Effect of 100 μm 8-Br-cGMP (C) and 10 μm ODQ (D) on sIPSCs in cells pretreated with 2 μm AM251 for >15 min. E, Summary of the effect of 100 μm 8-Br-cGMP and 10 μm ODQ on sIPSC charge, in control and in AM251-pretreated cells (n = 7 and 7 for 8-Br-cGMP; n = 8 and 8 for ODQ). Values are expressed as percentage of control before perfusion of the drugs. Note that changes in sIPSC charge may reflect both amplitude and frequency alteration. Calibration: 100 pA, 500 ms. **p < 0.01.
Drugs were applied for 5–15 min. We noticed that bath application of l-arginine for >10–15 min significantly reduced sIPSC charge; therefore, in these experiments, those periods were compared with the control, in which the charge was not significantly decreased.
Miniature IPSCs (mIPSCs) were measured after the addition of 0.5 μm tetrodotoxin (TTX) to the extracellular solution used for measurement of sIPSCs. Only those cells were tested for mIPSCs that expressed DSI of spontaneous IPSCs before application of TTX.
Evoked IPSCs were triggered every 3 s (0.33 Hz) with silver wires inserted in an ACSF-containing theta electrode, which was placed to the border of strata radiatum and pyramidale (within ∼50–100 μm from the recorded cell). Current impulses of 0.1 ms and 10–50 μA were used in all experiments, aiming to activate a single presynaptic axon. In this case, the mean amplitude of IPSCs showed a clear step without additional changes with increasing stimulus intensity (Hájos et al., 2000). The extracellular solution was the same as for sIPSCs, except for carbachol concentration, which was either 0 or 5 μm. DSI was calculated as DSI = 100 × (1 − amplitudedepol/amplitudectr), where amplitudectr is the average of the last 10 events before depolarization, and amplitudedepol is the average of the first three events after depolarization. Note that the value of DSI can be positive or negative (representing a decrease or increase, respectively, of amplitude after depolarization). To give a measure of DSI, two to four DSIs of evoked IPSCs were averaged under each conditions. Because the variance in the magnitude of DSI was smaller than for DSI of sIPSCs, we analyzed cells in which DSI in the control period was at least 20% on average.
For measurement of Ca2+ currents, the ACSF described above was complemented with 30 mm tetraethylammonium (TEA)-Cl (instead of 30 mm NaCl) to reduce K+ currents and 50 μm picrotoxin to inhibit all synaptic activity. Pipette solution contained the following (in mm): 110 CsCl, 30 TEA-Cl, 1 CaCl2, 10 EGTA, 1 MgCl2, 4 ATP, 3 NaCl, and 10 HEPES, pH 7.3 (300 mOsm). Voltage-gated Ca2+ currents were evoked every 15 s by a 500-ms-long depolarizing step from −60 to 0 mV. Currents were normalized as Cd2+-sensitive current component (measured at the end of the experiments by adding 200 μm Cd2+) and corrected for rundown.
In the experiments with the NO donor, slices prepared from 14- to 32-d-old rats were first preincubated with a broad-spectrum inhibitor of phosphodiesterases 3-isobuthyl-1-methylxanthine (IBMX) (1 mm) for 15–30 min, followed by the application of the NO donor sodium nitroprusside (SNP) for 10 min. For immunohistochemical studies, incubations were performed both at room temperature as well as at 34–35°C.
Immunohistochemistry.
After incubation, rat hippocampal slices were fixed for 2 h in freshly prepared ice-cold 4% paraformaldehyde dissolved in 0.1 m phosphate buffer (PB). During this step and throughout the staining procedure, the sections were continuously rinsed in a free-floating manner. The slices were then washed in PB for 10 min, pasted onto wax plates, and resectioned into 50 μm thin sections using a Leica VT1000S vibratome. The sections were first washed four times for 15 min in PB and then twice for 20 min in 0.05 m Tris-buffered saline (TBS) containing 0.3% Triton X-100 (TBST; Sigma, St. Louis, MO). Blocking was performed in 10% normal donkey serum (Vector Laboratories, Burlingame, CA) dissolved in TBST for 45 min. Incubation with primary antibodies was performed at 4°C for 48 h. As primary antibody, sheep anti-cGMP (1:4000) was used in combination with either rabbit anti-CB1R (1:2000) or rabbit anti-parvalbumin (PV) (1:2000; Sigma), all dissolved in TBS. The specificity and an estimate of the detection limit of the anti-formaldehyde-fixed cGMP antisera have been described previously (Tanaka et al., 1997). The specificity of the anti-CB1R antibody was proved in CB1R knock-out animals (Hájos et al., 2000). After primary antibody incubation, the sections were washed extensively in TBS and then incubated for 2 h in a mixture of the following secondary antisera: Alexa Fluor 488 donkey anti-sheep and Alexa Fluor 594 donkey anti-rabbit (Invitrogen, Carlsbad, CA), both diluted 1:200 in TBS. Afterward, the sections were washed three times for 15 min in TBS, then mounted onto glass slides, covered in Vectashield (Vector Laboratories), and sealed the coverslips with nail polish. Immunofluorescence staining was analyzed with an Olympus Optical (Tokyo, Japan) FluoView300 confocal laser scanning microscope using a sequential scanning mode. The ratio of labeled terminals was not different in slices treated at room temperature or 34–35°C; therefore, data were pooled.
Quantification of immunostaining.
Altogether, 1800 axon terminals were analyzed to establish the presence or absence of NO donor-induced or NO-independent cGMP immunoreactivity in either CB1R-containing or PV-positive axon terminals in three animals. Quantification of cGMP immunostaining was performed by an observer blind to the type of pretreatment. Images were taken from three slices from each animal from each the four experimental paradigms. Randomly selected areas in the CA1 pyramidal layer were photographed at 60× magnification separately, for either CB1R/cGMP or PV/cGMP immunoreactivity. Images were then analyzed using Adobe Photoshop 8.0 (Adobe Systems, San Jose, CA). First, 50 CB1R- or PV-immunoreactive axon terminals were selected and numbered in the image randomly, the corresponding image with the cGMP immunoreactivity was copied onto the first image, and the presence or absence of immunocytochemically detectable amount of cGMP was determined for each numbered bouton. The quantity of NO donor-induced increase in cGMP immunoreactivity may depend on several factors, such as the specificity of the inhibitor for cGMP-degrading phosphodiesterases, the concentration of NO donor (van Staveren et al., 2005), its penetration into the 400-μm-thick slices, and the success of immediate fixation, whereas penetration of primary antibodies for CB1R or PV may also vary. Therefore, in our analysis, we compared the ratio of cGMP-positive CB1R- and PV-immunoreactive axon terminals between NO donor-treated and untreated slices, which were derived from the same animal and were processed in parallel. Note also that absence of cGMP immunoreactivity does not rule out the presence of cGMP in immunohistochemically undetectable amounts in some other terminals. Thus, data obtained using this method always represent the minimal estimation of axon terminals, in which NO donor-dependent cGMP induction occurs.
Preembedding immunogold and immunoperoxidase reactions.
Six adult male Wistar rats (Charles River, Budapest, Hungary) were anesthetized with an intraperitoneal injection of an anesthetic mixture (containing 2.5% ketamine, 0.5% xylazine hydrochloride, 0.25% promethazine hydrochloride, 0.0025% benzetonium chloride, and 0.002% hydrochinone) used at 0.2 ml/100 g body weight. Animals were perfused through the heart with 0.9% NaCl saline. Then, in the experiments localizing neuronal NOS (nNOS), it was followed by fixative perfusion containing 1% paraformaldehyde in 0.1 m PB, pH 7.4, for 60 min. In the experiments localizing NO-sGC at the electron microscopic level, rats were perfused first for 2 min with a fixative containing 4% paraformaldehyde in Na-acetate buffer, pH 6.0, and then with a fixative containing 4% paraformaldehyde in Borax buffer, pH 8.5, for 40 min. Fixative perfusions were followed by perfusion with 0.1 m PB for 10 min, and then the brains were removed from the skulls. Blocks from the dorsal hippocampus were dissected and sectioned on a vibratome at 60 μm thickness, followed by washing in 0.1 m PB, and then they were incubated in 10 and 30% sucrose for cryoprotection and freeze thawed over liquid nitrogen several times. After repeated washes in 0.1 m PB, the sections were processed for immunostaining.
First, sections were incubated in 1% human serum albumin (HSA) (Sigma) diluted in TBS. In case of the experiments with nNOS, this was followed by a 48 h incubation in solutions of primary antibodies raised against neuronal NOS (rabbit polyclonal antibody, 1:500; Zymed Laboratories, San Francisco, CA), with or without CB1R antibody (goat polyclonal antibody, 1:2000). The specificity of this antibody developed against CB1R was proven in CB1R knock-out animals. In the experiments with NO-sGC, sections were incubated in solutions of primary antibodies raised against α1 subunit of NO-sGC (rabbit polyclonal antibody, 1:1000; Sigma), with the CB1R antibody. After repeated washes in TBS, sections were treated with blocking solution (Gel-BS) containing 0.5% cold water fish skin gelatin (GE Healthcare, Little Chalfont, UK) and 0.5% HSA in TBS for 1 h. In the experiments with nNOS, this was followed by 24 h incubation with 0.8 nm gold-conjugated anti-rabbit antibody (donkey polyclonal antibody, 1:80; Aurion, Wageningem, The Netherlands), whereas in the experiments with NO-sGC α1 subunit, this was followed by incubation with 0.8 nm gold-conjugated anti-goat antibody (donkey polyclonal antibody, 1:80; Aurion) to recognize CB1R, and it was diluted in Gel-BS. After intensive washes in TBS, the sections were treated with 2% glutaraldehyde in PB for 15 min, to fix the gold particles into the tissue. This was followed by washes in PB and in enhancement conditioning solution (ECS) (Aurion). After this, sections were incubated in silver enhancement solution for electron microscopy (SE-EM; Aurion) for 60 min at room temperature. This was followed by wash in ECS and repeated washes in PB. Then sections in the experiments with nNOS were incubated in biotinylated anti-goat (horse polyclonal antibody, 1:200; Vector Laboratories) for 24 h, whereas sections in the experiments with NO-sGC were incubated in biotinylated anti-rabbit (biotin-SP-conjugated AffiniPure donkey polyclonal antibody, 1:200; Jackson ImmunoResearch, West Grove, PA), followed by incubation in Elite ABC (1:300; Vector Laboratories) diluted in TBS for 3 h. The immunoperoxidase reaction was developed using 3,3 diaminobenzidine (Sigma) as chromogen. All sections were treated with OsO4 in PB on ice followed by dehydration in ascending alcohol series and propylene oxide and were embedded in Durcupan. During dehydration, the sections were treated with 1% uranyl acetate in 70% ethanol for 30 min. Finally, sections were reembedded and investigated by electron microscope.
Materials and data analysis.
Nω-Nitro-l-arginine methyl ester (l-NAME), Nω-nitro-d-arginine methyl ester (d-NAME), l-arginine, SNP, 8-Br-cGMP, and tetrodotoxin (Alomone Labs, Jerusalem, Israel) were dissolved in distilled water. 7-Nitroindazole (7-NI), 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (CPTIO), AF-DX116 (11-[[2-[(diethylamino)methyl]-1-piperidinyl]acetyl]-5, 11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one) (Tocris Bioscience, Bristol, UK), IBMX, SR141716A [N-piperidino-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-3-pyrazole carboxamide] (National Institute on Drug Abuse drug supply service), and AM251 [N-1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-1-piperidinyl-1H-pyrazole-3-carboxamide] (Tocris Bioscience) were dissolved in DMSO. WIN 55,212-2 [R-(+)-(2,3-dihydro-5-methyl-3-[(4-morpholinyl)methyl]pyrol[1,2,3-de]-1,4-benzoxazin-6-yl)-(1-naphthalenyl) methanone monomethanesulfonate] was dissolved in 0.1N HCl. Drugs were prepared as stock solutions and diluted at least by 1:1000. All drugs were obtained from Sigma-Aldrich (St. Louis, MO), unless otherwise stated.
For statistical analysis of electrophysiological data, the Wilcoxon's matched pairs test (for dependent samples), the Mann–Whitney U test (for independent samples), or the Kolmogorov–Smirnov test (for mIPSC distributions) was used. Fluorescent immunohistochemical results were analyzed using two-way repeated-measures ANOVA with Tukey's post hoc test (Statistica 6.0; StatSoft, Tulsa, OK). p < 0.05 was considered significant. Data are presented as mean ± SEM, unless otherwise indicated.
Results
We recorded sIPSCs enhanced by bath application of 5 μm carbachol in CA1 pyramidal cells, using whole-cell patch-clamp technique. Under these conditions, synaptic currents had fast rise times (10–90% rise time, 1.21 ± 0.13 ms; calculated from events measured in 10 randomly selected cells), suggesting that they primarily originated from the perisomatic region (Maccaferri et al., 2000). As shown previously, a form of retrograde signaling triggered by depolarization of the postsynaptic neuron (a phenomenon called depolarization-induced suppression of inhibition, or DSI) can be reliably studied, when cholinergic receptors are activated (Pitler and Alger, 1992). Consistent with previous data (Pitler and Alger, 1992; Wilson and Nicoll, 2001), depolarization of CA1 pyramidal cells from −60 to 0 mV for 1 s temporarily reduced charge transfer by sIPSCs (Fig. 1). The magnitude of DSI was independent of basal IPSC charge (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). In line with previous descriptions (Wilson and Nicoll, 2001), DSI of sIPSCs was dependent on the activation of CB1Rs, because the CB1R antagonists SR141716A or AM251 (2 μm for both) significantly reduced DSI from 77.7 ± 9.2 to 18.1 ± 4.3% (pooled, n = 6; p < 0.05) (Fig. 1A), and it was absent in CB1R knock-out mice (knock-out, 7.9 ± 2.6%, n = 6; wild-type littermates, 52.3 ± 8.6%, n = 6; p < 0.01).
Blockers of nitric oxide synthesis or application of NO scavengers inhibit DSI
To test the involvement of NO in DSI, we first applied inhibitors of the NOS enzymes. Bath application of the nonspecific NOS inhibitor l-NAME (100 μm) for 5–10 min reduced the magnitude of DSI from 64.7 ± 5.5 to 24.0 ± 4.7% (n = 8; p < 0.05) (Fig. 1B) (see also a complete representative recording in supplemental Fig. 2, available at www.jneurosci.org as supplemental material), whereas the inactive analog d-NAME (100 μm) did not affect DSI (control, 75.6 ± 7.8%; d-NAME, 71.2 ± 12.3%; n = 4; p > 0.6) (Fig. 1C). Similar to l-NAME, another NOS blocker, 7-NI (100 μm), also reduced DSI from 56.1 ± 4.6 to 15.1 ± 6.4% (n = 7; p < 0.05) (Fig. 1D). Thus, under our recording conditions, blockers of NO synthase inhibited DSI as efficiently as blockers of CB1Rs.
To determine whether the source of NO was the postsynaptic pyramidal cell, we added l-NAME (200 μm) to the intrapipette solution and compared DSI of cells from the same preparations using either control (vehicle-containing) or l-NAME-containing pipettes. Although DSI was detected in all control cells (64.1 ± 6.0%; n = 9), DSI was variable but on average significantly reduced in l-NAME-treated cells (34.0 ± 10.5%; n = 8; p < 0.05). However, l-NAME is membrane permeable; therefore, it cannot be ruled out that it exerted its effect not only in the pyramidal cell but also in the neighboring cellular compartments. Therefore, we also tested the NO scavenger CPTIO, which is considered to be membrane impermeable (Ko and Kelly, 1999). Although control cells (measured with pipette solution containing the vehicle DMSO only) exhibited robust DSI (80.2 ± 3.1%; n = 7), DSI was significantly reduced in cells treated with 1 mm intracellular CPTIO (27.5 ± 9.9%; n = 7; p < 0.01) (Fig. 1E). These results indicate that the postsynaptic presence of NO is necessary for DSI. To assess whether NO may act as an intercellular messenger in DSI, we tested whether extracellular application of CPTIO also affects the phenomenon. In cells pretreated for 30–40 min with 500 μm extracellular CPTIO, DSI was inhibited compared with control cells that were pretreated with vehicle only (DSI in 0.1% DMSO, 54.3 ± 6.1%, n = 15; DSI in 500 μm CPTIO, 15.4 ± 9.7%, n = 9; p < 0.01) (Fig. 1F). NOS enzymes use l-arginine as a substrate for NO production (Palmer et al., 1988). Thus, exogenously applied l-arginine could potentiate NO production by NOS, which might enhance DSI. Indeed, after 5-min-long application of 0.2–1 mm l-arginine, the decay time of DSI was significantly prolonged to 140.3 ± 18.9% of control (control τ, 13.3 ± 2.2 s vs l-arginine τ, 17.2 ± 1.7 s; n = 6; p < 0.05), and its magnitude was also enhanced (control, 45.8 ± 6.2%; l-arginine, 58.9 ± 3.3%; n = 6; p < 0.05) (Fig. 1G).
In conclusion, these results suggest that, during DSI of sIPSCs in the presence of carbachol, NO could be released postsynaptically and diffuse through the extracellular space to reach its (presumably) presynaptic target.
NO affects DSI via GC–cGMP pathways
The main effector molecule of NO is NO-sGC, which converts GTP to cGMP. To examine whether activation of NO-sGC mediates the effect of NO on DSI, we tested a specific NO-sGC blocker, ODQ. Similar to NOS inhibitors, bath application of ODQ (10 μm) for 5–10 min reduced DSI from 72.0 ± 8.2 to 18.8 ± 10.9% (n = 7; p < 0.05) (Fig. 2A,B). Moreover, ODQ significantly increased basal charge transfer to 174.7 ± 17.7% of control (p < 0.05) (Fig. 2B) (see Fig. 4E), suggesting that tonic activation of NO-sGC suppresses sIPSCs under control conditions. In contrast, when ODQ was included in the intrapipette solution, the magnitude of DSI (DSI in 0.05% DMSO, 47.3 ± 5.1%, n = 10; DSI in 10 μm ODQ, 40.3 ± 5.1, n = 11; p > 0.3) or the charge transfer (control in 0.05% DMSO, 14.4 ± 2.1 pA, n = 10; in 10 μm ODQ, 17.6 ± 2.1 pA, n = 11; p > 0.2) did not differ significantly even after 10–15 min of recordings. Subsequent bath application of ODQ in the same cells for 5–7 min could still inhibit DSI (DSI with intrapipette ODQ, 37.4 ± 4.8%; DSI after bath application of ODQ, 14.4 ± 9.2%, n = 4; p < 0.05). These data imply that, under these conditions, NO activates NO-sGC in the presynaptic axon terminals rather than in the postsynaptic pyramidal cells.
Figure 2.

cGMP production by NO-sGC is involved in DSI. A, Representative experiment of DSI in a cell before (left) and during (right) application of the NO-sGC inhibitor ODQ (10 μm). B, Left, Mean current and DSI under control conditions (filled squares) and in ODQ (open squares). Mean current was calculated as the sum of sIPSCs charge in every second. Right, sIPSC charge transfer before (c) and after (d) depolarization under control conditions and after addition of ODQ (n = 7). Charge was normalized to the average value before depolarization under control conditions. C, Representative DSI before (left) and during (right) application of 100 μm 8-Br-cGMP. D, Effect of 8-Br-cGMP on DSI and charge transfer (n = 8; see B). Calibration: 100 pA, 5 s. ctr, Control.
Additional evidence for the involvement of NO-sGC activation in DSI was provided by bath application of the membrane-permeable cGMP analog 8-Br-cGMP (100 μm) for 5–10 min, which decreased sIPSC charge transfer to 48.2 ± 7.7% of control (n = 8; p < 0.05) (Fig. 2C,D) (see also Fig. 4E) and partially occluded DSI (control DSI, 63.2 ± 4.8%; in 8-Br-cGMP, 42.5 ± 8.5%; n = 8; p < 0.05) (Fig. 2C,D).
Application of ODQ significantly increased basal charge transfer, suggesting that a basal activity of NO-sGC might influence sIPSCs under our conditions. In addition to ODQ, l-NAME also caused a smaller but significant increase in charge transfer (136.7 ± 16.9%; p < 0.05). Other drugs, although some of them also tended to increase charge transfer, did not cause significant alteration in sIPSC charge (AM251 or SR141716A, 170.8 ± 75.3, p > 0.7; 7-NI, 162.9 ± 34.8%, p > 0.05). Theoretically, enhanced GABA release from other, CB1R-lacking axon terminals might mask the reduction of sIPSCs from DSI-sensitive boutons after depolarization, and, therefore, inhibition of DSI with drugs that enhance charge transfer might be indirect. To test this possibility, we used a muscarinic acetylcholine receptor type 2 (M2R) antagonist to specifically enhance GABA release from axon terminals expressing M2R that innervate the perisomatic region of pyramidal cells and are distinct from CB1R bearing axon endings (Hájos et al., 1998; Freund et al., 2003; Fukudome et al., 2004). Because we recorded sIPSCs in the presence of carbachol, a general agonist of acetylcholine receptors, M2Rs were likely to be constitutively activated under our recording conditions. Thus, application of a selective antagonist of M2Rs should specifically augment GABA release from those axon terminals that do not express CB1Rs. In 6 of 10 experiments, bath application of 0.1–0.2 μm AF-DX116, an M2R antagonist, enhanced charge transfer. The increase of basal IPSC charge in these six cells was 164.9 ± 8.9% of control (p < 0.05), yet the magnitude of DSI only slightly changed (control, 55.5 ± 4.9%; AF-DX116, 43.9 ± 3.8; n = 6; p > 0.1) (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). These data show that IPSC enhancement from cannabinoid-insensitive axon terminals cannot explain the significant reduction of DSI, when charge transfer increases after application of various drugs.
To exclude the possibility that drugs used to inhibit NO signaling influenced postsynaptic GABAA receptors directly, we measured miniature IPSCs in the presence of 0.5 μm tetrodotoxin. Amplitude and frequency of mIPSCs were not affected by l-NAME or ODQ (Fig. 3), implying that mainly action potential-dependent GABA release and not postsynaptic GABA sensitivity was affected by these drugs. Furthermore, we also confirmed that the inhibitory effect of NOS and NO-sGC blockers on DSI was not the result of postsynaptic reduction of Ca2+ influx, because charge transfer carried by voltage-gated Ca2+ channels was not influenced by 100 μm l-NAME (103.3 ± 4.1% of control; n = 4; p > 0.7) or 10 μm ODQ (94.1 ± 6.7% of control; n = 4; p > 0.2).
Figure 3.

Effect of NO signaling modulators on miniature IPSCs. After cells were positively tested for DSI of spontaneous IPSCs, 0.5 μm TTX was applied to inhibit sIPSCs and uncover mIPSCs. After obtaining mIPSC baseline, l-NAME (100 μm, n = 6; A) or ODQ (10 μm, n = 6; B) was applied for 5–8 min. Left panels show representative recordings from individual cells (3 traces consecutive in time) before and after drug application. Amplitude (middle panels) and interevent interval (right panels) cumulative probabilities were compared on 60 randomly selected mIPSCs from every cell both under control (ctr) conditions and after drug application, using Kolmogorov–Smirnov statistics showing no significant effect. Calibration: 20 pA, 0.5 s.
CB1R activation is downstream of NO signaling in DSI
A plausible explanation of the above results is that, in response to the depolarization-evoked Ca2+ signal, NO is released postsynaptically, diffuses to the presynaptic terminal, and increases cGMP synthesis via NO-sGC activation, which leads to suppression of GABA release by a mechanism that involves CB1Rs. To test the involvement of CB1Rs in the effects of the NO signaling pathway, we first compared the effect of 100 μm 8-Br-cGMP on spontaneous IPSCs between cells from control slices and from slices pretreated with AM251 (2 μm) for 15–20 min. Because AM251 eliminated DSI (5.1 ± 2.9%, n = 15; DSI in control slices, 55.2 ± 5.9%, n = 15; p < 0.001), we analyzed the effect of 8-Br-cGMP on basal charge transfer. Whereas in control cells 8-Br-cGMP strongly decreased charge transfer to 50.3 ± 8.5% of control (n = 7) (Fig. 4A,E), in AM251-pretreated cells, the charge transfer measured after application of 8-Br-cGMP was not changed on average (101.3 ± 22.0%; n = 7) (Fig. 4C,E). The effect of 8-Br-cGMP differed significantly between control and AM251-pretreated cells (p < 0.01). In additional experiments, we tested the effect of inhibition of NO-sGC on spontaneous IPSCs in the absence and presence of AM251. Application of ODQ (10 μm) increased sIPSC charge to 157.8 ± 17.1% of control (n = 8) (Fig. 4B,E), whereas in cells from slices pretreated with AM251, the charge transfer remained unchanged by application of ODQ (103.6 ± 3.8%; n = 8) (Fig. 4D,E), although in some cases increased appearance of small-amplitude IPSCs, which did not affect total charge substantially, could still be observed. Again, the effect of ODQ was significantly different between control and AM251-pretreated cells (p < 0.01). In summary, these data suggest that the NO signaling cascade acts either upstream of CB1Rs or at a target that is dependent on CB1R function.
To further test this hypothesis, we asked whether, on the contrary, impairment of NO signaling would influence the effect of cannabinoid agonists on sIPSCs and DSI. We compared the effect of the CB1R agonist WIN 55,212-2 between control cells and cells that were pretreated with the NO-sGC inhibitor ODQ (10 μm) for >10 min. As described before (Fig. 2), ODQ treatment strongly inhibited DSI (9.0 ± 5.8%; n = 12) compared with control cells (51.8 ± 6.5%; n = 13; p < 0.001). However, application of WIN 55,212-2 (2 μm) for 10 min decreased sIPSC charge similarly in the two cell groups (control, to 74.5 ± 10.1%, n = 7; ODQ-pretreated, to 68.2 ± 3.7%, n = 6; p > 0.3). Thus, results of this experiment exclude the possibility that inhibition of NO-sGC signaling decreased DSI by impairing the effect of CB1R activation on GABA release.
Contribution of NO signaling to DSI of electrically evoked IPSCs
All data presented so far clearly show that, in the presence of carbachol, NO plays an important role in DSI of spontaneously occurring IPSCs. However, any change in the DSI of sIPSCs on interfering NO signaling could be attributable to the alteration in GABA release at the presynaptic site, to the modification of firing properties of inhibitory neurons, or both. To investigate whether NO could indeed control GABA release from the axon terminals, we evoked IPSCs by minimal electrical stimulation of fibers in the close vicinity of cell bodies (Hájos et al., 2000) and tested their DSI sensitivity. In the presence of carbachol, 10 of 45 evoked IPSCs showed clearly detectable DSI (i.e., larger than a 20% reduction in the amplitude after 1-s-long depolarization in a given cell). After recording a control period, the effect of l-NAME (100 μm) was successfully tested in seven cells. Without changing the basal amplitude (103.4 ± 20.4% of control; p > 0.5; n = 7), the NOS inhibitor abolished DSI (control DSI, 43.2 ± 2.9%; in l-NAME, −5.7 ± 6.3%; p < 0.05; n = 7) (Fig. 5A,C). These results undoubtedly indicate that DSI of evoked IPSCs is also dependent on NO when cholinergic receptors are activated and also suggest that the NO-cGMP signaling cascade exerts its effect directly on the axons of CB1R-positive interneurons.
Figure 5.

Involvement of NO in DSI of evoked IPSCs with (A, C) or without (B, D) carbachol (CCh). A, B, Left and Middle, Representative recordings of pyramidal cells before (left) and during (middle) application of 100 μm l-NAME (NOS inhibitor). IPSCs (black) evoked by minimal stimulation of fibers in the close vicinity of cell bodies showed transient reduction after a depolarizing step (gray) under control conditions (ctr) (left; DSI). DSI was absent after drug application (A, middle) in the presence of carbachol, but it was unaltered (B, middle) in recordings without carbachol. Traces are averages of three and three consecutive events before and after depolarization, respectively. Stimulus artifacts were removed. Calibration: 50 pA, 5 s. Right, Magnitude and kinetics of DSI under control conditions (filled squares) and during application (open squares) of 100 μm l-NAME in the presence (C; n = 7) or in the absence (D; n = 5) of carbachol. Amplitude was normalized to the control period before depolarization.
Because DSI of evoked IPSCs is routinely studied in the absence of carbachol, we investigated whether NO is involved under these circumstances as well. In 5 of 21 cells, a substantial DSI of evoked IPSCs was observed (control, 33.4 ± 3.8%; n = 5). In contrast to the results measured in the presence of carbachol, bath application of 100 μm l-NAME did not alter DSI in the absence of the cholinergic agonist (control, 33.4 ± 3.8%; in l-NAME, 30.9 ± 4.4%; n = 5; p > 0.5) (Fig. 5B,D). These data suggest that, without activation of cholinergic receptors, retrograde signaling at GABAergic synapses does not involve NO.
NO donor-induced cGMP production in CB1R-expressing boutons
In cortical networks, at least two major GABAergic interneuron populations with separate physiological functions control the output of pyramidal neurons (Freund, 2003). Axon terminals of these interneuron populations can be visualized by immunostaining for either CB1Rs [and cholecystokinin (CCK)] or parvalbumin (Katona et al., 1999; Hájos et al., 2000). It has been established that only GABAergic synapses formed by CB1R-containing axon terminals can undergo DSI (Ohno-Shosaku et al., 2001; Wilson et al., 2001). Thus, we hypothesized that NO signaling should preferentially induce cGMP production in axon terminals expressing CB1Rs. Indeed, after incubation of hippocampal slices in the presence of the NO donor SNP (0.1–0.2 mm) and the phosphodiesterase blocker IBMX (1 mm) in standard carbachol-containing ACSF, cGMP immunoreactivity appeared in 67.0 ± 1.0% of CB1R-containing axon terminals (Fig. 6A–C), which represents a ninefold increase relative to basal conditions in the presence of IBMX only (7.3 ± 1.2%) (Fig. 6G) (supplemental Fig. 4A–C, available at www.jneurosci.org as supplemental material). In contrast, SNP treatment did not increase the level of cGMP immunoreactivity in parvalbumin-positive boutons (basal, 19.3 ± 4.8%; SNP-treated, 18.0 ± 3.8%) (Fig. 6D–F). Thus, cGMP formation on NO donor-induced sGC activity is proved to be present in axon terminals, which contain CB1Rs. In line with the anatomical results, application of SNP (100 μm) in IBMX-pretreated slices partially occluded DSI of sIPSCs (DSI in IBMX only, 42.7 ± 7.1%; DSI in SNP, 21.7 ± 11.8%; n = 9; p < 0.05) (Fig. 6H). It is also important to note that no enhancement or appearance of cGMP immunoreactivity during application of SNP has been observed in the somata of pyramidal cells (Fig. 6B,E).
Figure 6.
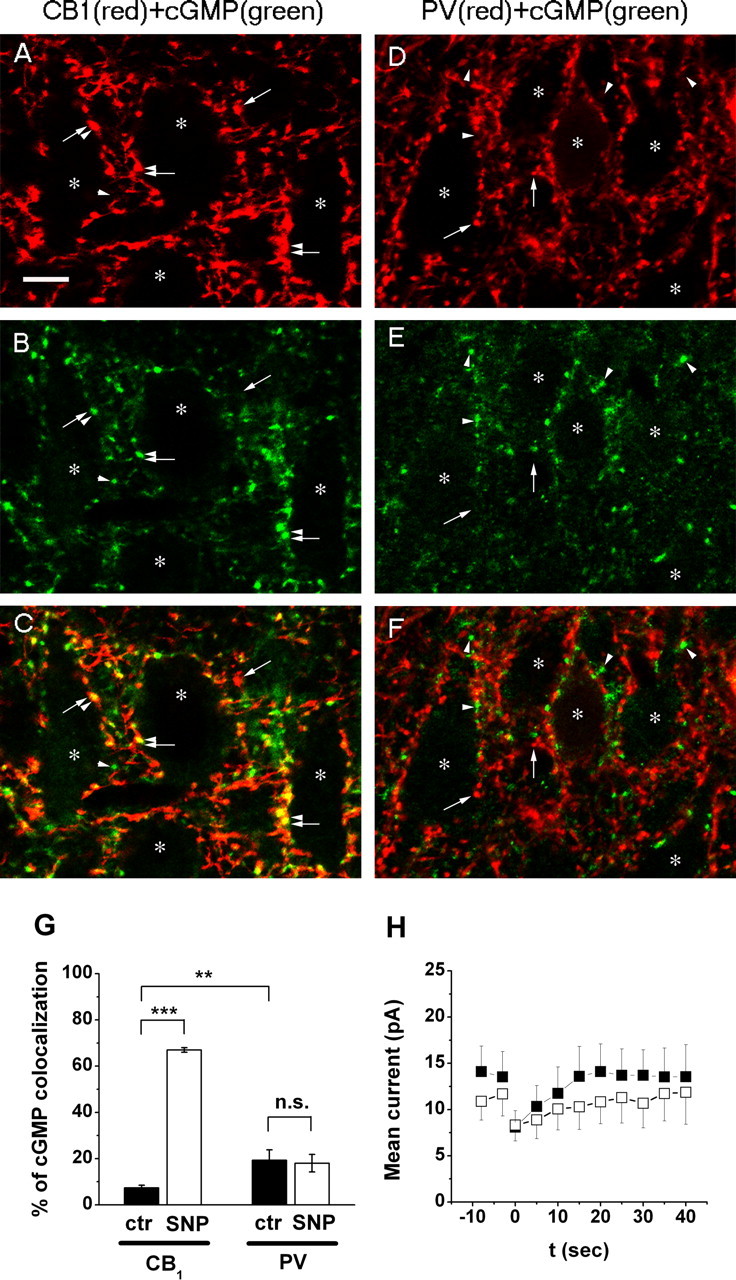
NO donor-induced cGMP immunoreactivity is enhanced in CB1 cannabinoid receptor-containing axon terminals after IBMX treatment. A, B, Immunostaining for CB1R (A) and cGMP (B) visualizes a dense meshwork of axons in the stratum pyramidale of the CA1 subfield, which forms characteristic pericellular arrays of boutons around immunonegative cell bodies of pyramidal neurons (depicted by asterisks). C, Overlay of the two images reveals that the majority of these axon terminals contain both CB1R and cGMP. D, E, In contrast, boutons of the other basket cell population visualized by immunostaining for PV (D) do not contain NO donor (SNP)-induced cGMP immunoreactivity (E). F, In the overlaid image, red parvalbumin-positive boutons and green cGMP-positive axon terminals are located in a primarily complementary position. Double-positive boutons are labeled by the combination of arrow and arrowhead throughout the images, whereas single-positive CB1R- and PV- or cGMP-immunoreactive boutons are marked by single arrows or arrowheads, respectively. Asterisks label double-immunonegative pyramidal cell bodies. Scale bar, 10 μm. G, Summary of the ratio of cGMP-containing boutons of all perisomatic axon terminals positive for either CB1R or PV. cGMP immunoreactivity appears in 67% of CB1R-positive axon terminals after activation of NO-sGC by SNP. In contrast, SNP treatment does not change the ratio of cGMP immunostaining in PV-positive boutons. **p < 0.01, ***p < 0.001; ctr, Control; n.s., not significant. H, Mean current and DSI in the presence of 1 mm IBMX only (control, filled squares) and after application of 100 μm SNP (open squares; n = 9).
Molecular and anatomical basis of NO signaling at synapses formed by CB1R-immunopositive axon terminals
Finally, we asked whether nNOS, which has been shown to be present in the postsynaptic densities at glutamatergic synapses (Burette et al., 2002), could also be located at GABAergic synapses. Using immunogold labeling in mildly fixed hippocampal tissue, nNOS labeling could be found not only in dendritic spines at excitatory synapses in the CA1 region (data not shown) but also at symmetrical synapses on pyramidal cell somata (Fig. 7A–C). The labeling was specifically associated with the symmetrical synaptic active zones on pyramidal cell bodies and avoided extrasynaptic membranes. Some labeling was also found in the cytoplasm of the pyramidal cells around the endoplasmic reticulum (Fig. 7C/b). Next, we specifically tested whether nNOS is present at those GABAergic synapses that express CB1Rs. Experiments using immunogold–immunoperoxidase double staining revealed that nNOS labeling was indeed localized to the postsynaptic membranes of symmetrical synapses formed by CB1R-positive axon terminals (Fig. 7D–F). If NO is a retrograde signaling molecule in DSI, then its receptor, NO-sGC, should be present in these presynaptic terminals. In fact, using double immunostainings, α1 subunits of NO-sGC were unequivocally found in CB1R-expressing axon endings (Fig. 7G,H). These anatomical data provide the structural basis of NO-mediated retrograde signaling in CB1R-dependent DSI.
Figure 7.
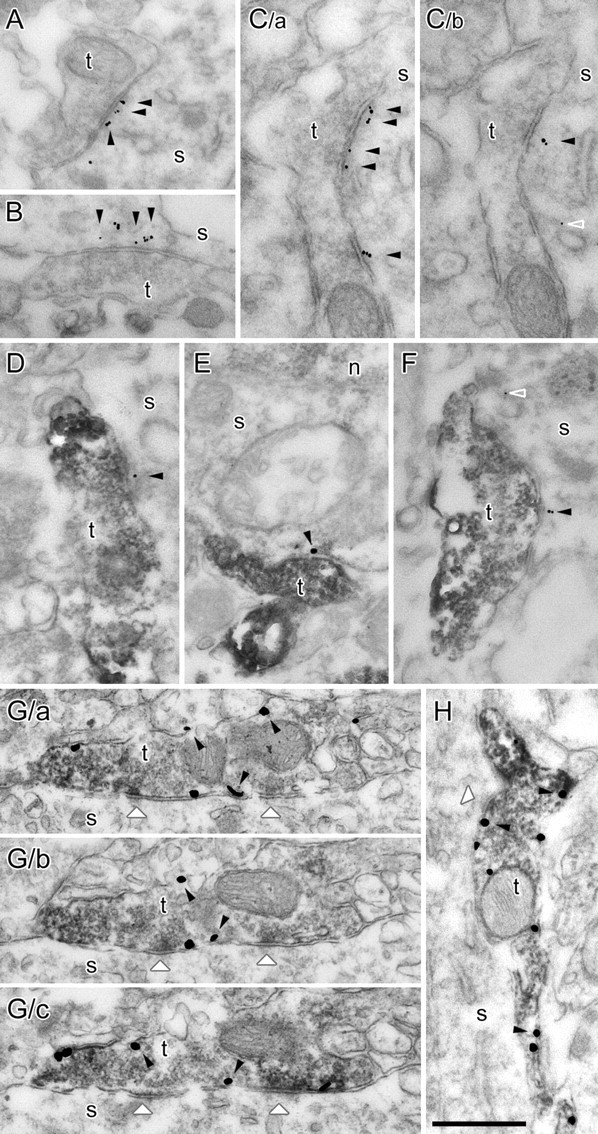
Neuronal NOS labeling is present postsynaptically at CB1R-positive axon terminals, which terminals also show immunopositivity for NO-sGC. A–C, Electron micrographs from immunogold labeling experiments in hippocampal CA1 area of the rat brain. Gold particles show that postsynaptic nNOS labeling (arrowheads) is closely associated with the synapses established by three terminals (t) on pyramidal cell somata (C/a and C/b are serial sections of the same terminal). D–F, Electron micrographs from immunogold–immunoperoxidase double-labeling experiments. CB1R labeling is shown with dark DAB precipitation in the terminals (t) that establish symmetrical synapses on pyramidal cell somata (s). Gold particles show nNOS labeling postsynaptically (arrowheads) or occasionally in the cytoplasm around endoplasmic reticulum membranes (open arrowhead in C/b and F). The smaller numbers of gold particles is attributable to technical limitations of the double-labeling method. G, H, Electron micrographs show the colocalization of CB1R and NO-sGC in axon terminals contacting CA1 pyramidal cells (G/a–G/c are serial sections of the same terminal). NO-sGC labeling is shown with dark DAB precipitation in the terminals (t) that establish symmetrical synapses (white arrowheads) on pyramidal cell somata (s). CB1Rs are labeled with silver-intensified gold particles (some of them are labeled with black arrowheads). n, Nucleus. Scale bar, 500 nm.
Discussion
In this study, we investigated the role of nitric oxide in short-term retrograde regulation of GABAergic synaptic transmission. We found that inhibition of NOS and NO-sGC enzymes diminished DSI at cholinergically enhanced spontaneously active inhibitory inputs of CA1 pyramidal cells, whereas a cGMP analog mimicked and partially occluded DSI in a CB1R-dependent manner. Moreover, increasing NO levels by l-arginine enhanced DSI. In the presence, but not in the absence, of carbachol, DSI of evoked IPSCs was eliminated by an NOS inhibitor. We also found that an NO donor, which partially occluded DSI, increased the level of cGMP preferentially in CB1R-positive axon terminals, providing an anatomical link between the two signaling systems. In addition, electron microscopic investigation revealed that neuronal NOS was postsynaptically located at symmetrical synapses formed by CB1R-immunopositive axon terminals. Importantly, in these presynaptic terminals, the presence of NO-sGC immunopositivity was also demonstrated. We propose that, in the presence of cholinergic receptor agonists, activation of NO signaling by depolarization-induced Ca2+ transients is involved in a form of CB1R-dependent DSI in the presence of cholinergic receptor agonists.
Several lines of evidence support the involvement of CB1Rs and the retrograde action of endocannabinoids in hippocampal DSI (Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001). Cannabinoid analogs reduce GABA release measured with either electrophysiological (Hájos et al., 2000; Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001) or biochemical (Katona et al., 1999) techniques and partially occlude DSI (Wilson and Nicoll, 2001), whereas pharmacological suppression or genetic deletion of CB1Rs abolishes DSI (Ohno-Shosaku et al., 2001; Varma et al., 2001; Wilson et al., 2001). Endogenous cannabinoids can be produced by neurons in a Ca2+-dependent manner (Di Marzo et al., 1998; Freund et al., 2003). Although the enzyme responsible for endocannabinoid production at inhibitory synapses has not yet been identified, CB1Rs are expressed at a very high density on presynaptic terminals of CCK-positive interneurons (Nyiri et al., 2005). Finally, inhibition of endocannabinoid degradation by cyclooxygenase-2 (Kim and Alger, 2004) or by monoacylglycerol lipase inhibitors (Makara et al., 2005; Hashimotodani et al., 2007) prolongs DSI.
Although endocannabinoids are currently the most widely accepted retrograde signal molecules, other modulators of synaptic activity have also been considered previously (Medina and Izquierdo, 1995; Alger, 2002). However, according to the literature, only some of these molecules, e.g., adenosine or glutamate (Pitler and Alger, 1994; Morishita et al., 1998), have been investigated in hippocampal DSI. The rapidly diffusing NO is ideally suited for intercellular signaling and has been implicated as retrograde messenger in hippocampal long-term potentiation (O'Dell et al., 1991; Prast and Philippu, 2001). Several arguments, presented in this study or reported previously, support the idea that NO may also modulate DSI as a retrograde messenger, generated in the postsynaptic cell and acting at the presynaptic terminal. The nNOS enzyme is activated by Ca2+ (Schmidt et al., 1992), and it is reasonable to assume that the large postsynaptic Ca2+ signal evoked by the DSI paradigm (Isokawa and Alger, 2006) is sufficient to stimulate NO production. In support of NO as a retrograde signal molecule, previous data showed that nNOS could be found at the postsynaptic densities of glutamatergic synapses (Wendland et al., 1994; Burette et al., 2002), and presynaptic localization of NO-sGC in glutamatergic afferents terminating on CA1 pyramidal cells has also been described (Burette et al., 2002). We provide anatomical evidence that postsynaptic nNOS and presynaptic NO-sGC are expressed also at inhibitory synapses and that axon terminals forming such inhibitory synapses are equipped with CB1Rs. Moreover, we found that, after inhibiting the activity of phosphodiesterases with IBMX, an NO donor could evoke cGMP production in CB1R-positive axon terminals. Beside the anatomical evidence, we also show that inhibition of NOS or scavenging NO intracellularly in pyramidal cells decreases DSI, suggesting that NO is generated by the postsynaptic pyramidal cells. Conversely, blocking NO-sGC inhibits DSI only when applied extracellularly but not postsynaptically. Supporting the idea that NO should diffuse in the extracellular space to reach its target, extracellular application of a membrane-impermeable NO scavenger substantially reduced DSI. This is concordant with our immunocytochemical results, which show cGMP accumulation in CB1R-expressing axon terminals but not in the postsynaptic pyramidal cells. These observations suggest that NO could trigger cGMP synthesis on the presynaptic rather than the postsynaptic side of synapses formed by cannabinoid-sensitive axon terminals and strongly support a role for NO in modulating the function of GABAergic synapses as a retrograde messenger. Importantly, the effect of ODQ and 8-Br-cGMP on sIPSCs was inhibited by pretreatment with the CB1R antagonist AM251, whereas the effect of the CB1R agonist WIN 55,212-2 was not influenced by pretreatment with the NO-sGC antagonist ODQ. These experiments point to a functional interaction between the two cascades instead of a simple parallel effect on GABA release and suggest that cGMP exerted its effect either upstream of CB1R activation or at a target that is crucially dependent on CB1Rs. Although the above results strongly support the idea that the observed effects of NO on DSI were mediated at the presynaptic terminals, a postsynaptic site of effect, e.g., an enhancement of endocannabinoid production by NO, cannot be ruled out. The α2/β1 subunit composition of NO-sGC is expressed by pyramidal cells, but the protein has been reported to be predominantly presynaptic in glutamatergic synapses (Burette et al., 2002), whereas the α1 subunit of NO-sGC (either protein or mRNA) is not detectable in postsynaptic pyramidal cells (Szabadits et al., 2007). Although low levels of sGC may be present also in pyramidal cell bodies, their activation by NO donors did not result in a detectable increase in cGMP levels. The highly diffusible nature of NO and the lack of membrane-impermeable inhibitors of NOS and sGC also complicate the interpretation of pharmacological experiments, conducted in the tightly packed structure of presynaptic and postsynaptic compartments in the slice. For example, although the extracellular NO scavenger inhibited DSI, this effect might have been the result of “sinking” fast diffusible NO molecules from the postsynaptic cell. It should be noted that a putative postsynaptic action of NO does not rule out a simultaneous presynaptic effect, which is favored by the evidence we presented. Additional experiments are required to pinpoint the precise site of action of NO as a signaling molecule in DSI.
Importantly, we have shown that NO represents a crucial component of the signaling machinery in DSI only when cholinergic receptors are activated. Activation of acetylcholine receptors is critical for the detection of DSI of spontaneous, action potential-dependent IPSCs, because DSI of these currents is rarely observed in the absence of a cholinergic agonist (Martin and Alger, 1999). Carbachol increases firing of DSI-susceptible interneurons (Martin and Alger, 1999; Martin et al., 2001) and favors GABA release from CB1R-positive axon terminals but inhibits release from PV-containing boutons likely via activation of presynaptically located M2Rs (Hájos et al., 1998; Fukudome et al., 2004; present study). Moreover, postsynaptic activation of type 1 and 3 muscarinic acetylcholine receptors (Martin and Alger, 1999; Kim et al., 2002; Ohno-Shosaku et al., 2003; Edwards et al., 2006) were shown to enhance DSI in slices, although the explanation for their precise mode of action is still lacking (Edwards et al., 2006). Based on our data with evoked IPSCs, in which l-NAME inhibited DSI only in the presence of carbachol, we propose that activation of cholinergic receptors is essential for the induction of the NO signaling cascade. This induction mechanism is well established in the vascular system, in which activation of muscarinic acetylcholine receptors has long been known to enhance the activity of endothelial NOS (Furchgott and Vanhoutte, 1989; Arnal et al., 1999). Neuronal NOS can also be activated by muscarinic receptors, either by Ca2+ mobilization or via Ca2+-independent pathways (Christopoulos and El-Fakahany, 1999), and stimulatory effect of cholinergic agonists on NO and cGMP production in the brain has been observed (de Vente, 2004). Such cholinergic augmentation of DSI by NO could take place in vivo under conditions when tissue concentration of acetylcholine is high, e.g., during exploratory activity (Marrosu et al., 1995).
In summary, the physiological and anatomical results presented in this study strongly suggest that NO as a retrograde signal molecule cooperates with endocannabinoids to control GABA release from CB1R-expressing axon terminals in an activity-dependent manner, when cholinergic receptors are activated. In vivo, CB1R-expressing interneurons could play a role in the temporal coordination of pyramidal cell ensembles necessary for normal hippocampal function (Klausberger et al., 2005; Robbe et al., 2006). Convergence of distinct, state-dependent physiological stimuli simultaneously activating NO and endocannabinoid signaling in a select population of pyramidal neurons may change their entrainment by inhibition as well as the precise timing of their action potentials that is critical for temporal coding. Thus, a fast, activity-dependent and target-selective retrograde signaling achieved by the cooperation of NO and endocannabinoids may shed new light on the regulation of neuronal activity patterns associated with information coding and transfer (Freund et al., 2003), as well as with pathophysiological states such as anxiety (Haller et al., 2002) and epilepsy (Chen et al., 2003).
Footnotes
This work was supported by the Howard Hughes Medical Institute, European Union Grant LSHM-CT-2004-005166, National Research and Development Programs Grant 1A/002/2004, Hungarian Scientific Research Fund Grants T46820 and F046407, Hungarian Medical Research Council Grant 474/2003, and National Institutes of Health (NIH) Grant NS30549. N.H. is the recipient of a Wellcome Trust International Senior Research Fellowship. I.K. is a grantee of the Bolyai János Scholarship. We thank Eszter Szabadits and Csaba Cserép for their essential contribution to the anatomical experiments and observations. We thank Drs. György Buzsáki, István Mody, Roger Nicoll, Péter Somogyi, Thomas Klausberger, and Zoltán Nusser for advice and comments on a previous version of this manuscript and Dr. Zsolt Borhegyi for help with confocal microscopy. We also thank Dr. Ken Mackie (supported by NIH Grant DA11322) for the rabbit anti-CB1R antibody. The excellent technical assistance of Katalin Lengyel, Emőke Simon, Katalin Iványi, Gabriella Urbán, and Győző Goda is also gratefully acknowledged.
References
- Alger BE. Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Prog Neurobiol. 2002;68:247–286. doi: 10.1016/s0301-0082(02)00080-1. [DOI] [PubMed] [Google Scholar]
- Arnal JF, Dinh-Xuan AT, Pueyo M, Darblade B, Rami J. Endothelium-derived nitric oxide and vascular physiology and pathology. Cell Mol Life Sci. 1999;55:1078–1087. doi: 10.1007/s000180050358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burette A, Zabel U, Weinberg RJ, Schmidt HH, Valtschanoff JG. Synaptic localization of nitric oxide synthase and soluble guanylyl cyclase in the hippocampus. J Neurosci. 2002;22:8961–8970. doi: 10.1523/JNEUROSCI.22-20-08961.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Ratzliff A, Hilgenberg L, Gulyas A, Freund TF, Smith M, Dinh TP, Piomelli D, Mackie K, Soltesz I. Long-term plasticity of endocannabinoid signaling induced by developmental febrile seizures. Neuron. 2003;39:599–611. doi: 10.1016/s0896-6273(03)00499-9. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Takahashi KA, Castillo PE. Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci. 2006;29:37–75. doi: 10.1146/annurev.neuro.29.051605.112834. [DOI] [PubMed] [Google Scholar]
- Christopoulos A, El-Fakahany EE. The generation of nitric oxide by G protein-coupled receptors. Life Sci. 1999;64:1–15. doi: 10.1016/s0024-3205(98)00348-8. [DOI] [PubMed] [Google Scholar]
- Cummings JA, Nicola SM, Malenka RC. Induction in the rat hippocampus of long-term potentiation (LTP) and long-term depression (LTD) in the presence of a nitric oxide synthase inhibitor. Neurosci Lett. 1994;176:110–114. doi: 10.1016/0304-3940(94)90883-4. [DOI] [PubMed] [Google Scholar]
- de Vente J. cGMP: a second messenger for acetylcholine in the brain? Neurochem Int. 2004;45:799–812. doi: 10.1016/j.neuint.2004.03.010. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Melck D, Bisogno T, De Petrocellis L. Endocannabinoids: endogenous cannabinoid receptor ligands with neuromodulatory action. Trends Neurosci. 1998;21:521–528. doi: 10.1016/s0166-2236(98)01283-1. [DOI] [PubMed] [Google Scholar]
- Edwards DA, Kim J, Alger BE. Multiple mechanisms of endocannabinoid response initiation in hippocampus. J Neurophysiol. 2006;95:67–75. doi: 10.1152/jn.00813.2005. [DOI] [PubMed] [Google Scholar]
- Freund TF. Interneuron diversity series: rhythm and mood in perisomatic inhibition. Trends Neurosci. 2003;26:489–495. doi: 10.1016/S0166-2236(03)00227-3. [DOI] [PubMed] [Google Scholar]
- Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev. 2003;83:1017–1066. doi: 10.1152/physrev.00004.2003. [DOI] [PubMed] [Google Scholar]
- Fukudome Y, Ohno-Shosaku T, Matsui M, Omori Y, Fukaya M, Tsubokawa H, Taketo MM, Watanabe M., Manabe T, Kano M. Two distinct classes of muscarinic action on hippocampal inhibitory synapses: M2-mediated direct suppression and M1/M3-mediated indirect suppression through endocannabinoid signaling. Eur J Neurosci. 2004;19:2682–2692. doi: 10.1111/j.0953-816X.2004.03384.x. [DOI] [PubMed] [Google Scholar]
- Furchgott RF, Vanhoutte PM. Endothelium-derived relaxing and contracting factors. FASEB J. 1989;3:2007–2018. [PubMed] [Google Scholar]
- Hájos N, Papp EC, Acsady L, Levey AI, Freund TF. Distinct interneuron types express m2 muscarinic receptor immunoreactivity on their dendrites or axon terminals in the hippocampus. Neuroscience. 1998;82:355–376. doi: 10.1016/s0306-4522(97)00300-x. [DOI] [PubMed] [Google Scholar]
- Hájos N, Katona I, Naiem SS, Mackie K, Ledent C, Mody I, Freund TF. Cannabinoids inhibit hippocampal GABAergic transmission and network oscillations. Eur J Neurosci. 2000;12:3239–3249. doi: 10.1046/j.1460-9568.2000.00217.x. [DOI] [PubMed] [Google Scholar]
- Haller J, Bakos N, Szirmay M, Ledent C, Freund TF. The effects of genetic and pharmacological blockade of the CB1 cannabinoid receptor on anxiety. Eur J Neurosci. 2002;16:1395–1398. doi: 10.1046/j.1460-9568.2002.02192.x. [DOI] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Kano M. Presynaptic monoacylglycerol lipase activity determines basal endocannabinoid tone and terminates retrograde endocannabinoid signaling in the hippocampus. J Neurosci. 2007;27:1211–1219. doi: 10.1523/JNEUROSCI.4159-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isokawa M, Alger BE. Ryanodine receptor regulates endogenous cannabinoid mobilization in the hippocampus. J Neurophysiol. 2006;95:3001–3011. doi: 10.1152/jn.00975.2005. [DOI] [PubMed] [Google Scholar]
- Katona I, Sperlagh B, Sik A, Kofalvi A, Vizi ES, Mackie K, Freund TF. Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci. 1999;19:4544–4558. doi: 10.1523/JNEUROSCI.19-11-04544.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Alger BE. Inhibition of cyclooxygenase-2 potentiates retrograde endocannabinoid effects in hippocampus. Nat Neurosci. 2004;7:697–698. doi: 10.1038/nn1262. [DOI] [PubMed] [Google Scholar]
- Kim J, Isokawa M, Ledent C, Alger BE. Activation of muscarinic acetylcholine receptors enhances the release of endogenous cannabinoids in the hippocampus. J Neurosci. 2002;22:10182–10191. doi: 10.1523/JNEUROSCI.22-23-10182.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T, Marton LF, O'Neill J, Huck JH, Dalezios Y, Fuentealba P, Suen WY, Papp E, Kaneko T, Watanabe M, Csicsvari J, Somogyi P. Complementary roles of cholecystokinin- and parvalbumin-expressing GABAergic neurons in hippocampal network oscillations. J Neurosci. 2005;25:9782–9793. doi: 10.1523/JNEUROSCI.3269-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko GY, Kelly PT. Nitric oxide acts as a postsynaptic signaling molecule in calcium/calmodulin-induced synaptic potentiation in hippocampal CA1 pyramidal neurons. J Neurosci. 1999;19:6784–6794. doi: 10.1523/JNEUROSCI.19-16-06784.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Cerebellar depolarization-induced suppression of inhibition is mediated by endogenous cannabinoids. J Neurosci. 2001;21(RC174):1–5. doi: 10.1523/JNEUROSCI.21-20-j0005.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledent C, Valverde O, Cossu G, Petitet F, Aubert JF, Beslot F, Bohme GA, Imperato A, Pedrazzini T, Roques BP, Vassart G, Fratta W, Parmentier M. Unresponsiveness to cannabinoids and reduced addictive effects of opiates in CB1 receptor knockout mice. Science. 1999;283:401–404. doi: 10.1126/science.283.5400.401. [DOI] [PubMed] [Google Scholar]
- Lenz RA, Alger BE. Calcium dependence of depolarization-induced suppression of inhibition in rat hippocampal CA1 pyramidal neurons. J Physiol (Lond) 1999;521:147–157. doi: 10.1111/j.1469-7793.1999.00147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llano I, Leresche N, Marty A. Calcium entry increases the sensitivity of cerebellar Purkinje cells to applied GABA and decreases inhibitory synaptic currents. Neuron. 1991;6:565–574. doi: 10.1016/0896-6273(91)90059-9. [DOI] [PubMed] [Google Scholar]
- Maccaferri G, Roberts JD, Szucs P, Cottingham CA, Somogyi P. Cell surface domain specific postsynaptic currents evoked by identified GABAergic neurones in rat hippocampus in vitro. J Physiol (Lond) 2000;524:91–116. doi: 10.1111/j.1469-7793.2000.t01-3-00091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makara JK, Mor M, Fegley D, Szabo SI, Kathuria S, Astarita G, Duranti A, Tontini A, Tarzia G, Rivara S, Freund TF, Piomelli D. Selective inhibition of 2-AG hydrolysis enhances endocannabinoid signaling in hippocampus. Nat Neurosci. 2005;8:1139–1141. doi: 10.1038/nn1521. [DOI] [PubMed] [Google Scholar]
- Marrosu F, Portas C, Mascia MS, Casu MA, Fa M, Giagheddu M, Imperato A, Gessa GL. Microdialysis measurement of cortical and hippocampal acetylcholine release during sleep-wake cycle in freely moving cats. Brain Res. 1995;671:329–332. doi: 10.1016/0006-8993(94)01399-3. [DOI] [PubMed] [Google Scholar]
- Martin LA, Alger BE. Muscarinic facilitation of the occurrence of depolarization-induced suppression of inhibition in rat hippocampus. Neuroscience. 1999;92:61–71. doi: 10.1016/s0306-4522(98)00745-3. [DOI] [PubMed] [Google Scholar]
- Martin LA, Wei DS, Alger BE. Heterogeneous susceptibility of GABA(A) receptor-mediated IPSCs to depolarization-induced suppression of inhibition in rat hippocampus. J Physiol (Lond) 2001;532:685–700. doi: 10.1111/j.1469-7793.2001.0685e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina JH, Izquierdo I. Retrograde messengers, long-term potentiation and memory. Brain Res Brain Res Rev. 1995;21:185–194. doi: 10.1016/0165-0173(95)00013-5. [DOI] [PubMed] [Google Scholar]
- Morishita W, Kirov SA, Alger BE. Evidence for metabotropic glutamate receptor activation in the induction of depolarization-induced suppression of inhibition in hippocampal CA1. J Neurosci. 1998;18:4870–4882. doi: 10.1523/JNEUROSCI.18-13-04870.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyiri G, Cserep C, Szabadits E, Mackie K, Freund TF. CB1 cannabinoid receptors are enriched in the perisynaptic annulus and on preterminal segments of hippocampal GABAergic axons. Neuroscience. 2005;136:811–822. doi: 10.1016/j.neuroscience.2005.01.026. [DOI] [PubMed] [Google Scholar]
- O'Dell TJ, Hawkins RD, Kandel ER, Arancio O. Tests of the roles of two diffusible substances in long-term potentiation: evidence for nitric oxide as a possible early retrograde messenger. Proc Natl Acad Sci USA. 1991;88:11285–11289. doi: 10.1073/pnas.88.24.11285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Maejima T, Kano M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron. 2001;29:729–738. doi: 10.1016/s0896-6273(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Matsui M, Fukudome Y, Shosaku J, Tsubokawa H, Taketo MM, Manabe T, Kano M. Postsynaptic M1 and M3 receptors are responsible for the muscarinic enhancement of retrograde endocannabinoid signalling in the hippocampus. Eur J Neurosci. 2003;18:109–116. doi: 10.1046/j.1460-9568.2003.02732.x. [DOI] [PubMed] [Google Scholar]
- Palmer RM, Ashton DS, Moncada S. Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature. 1988;333:664–666. doi: 10.1038/333664a0. [DOI] [PubMed] [Google Scholar]
- Pitler TA, Alger BE. Postsynaptic spike firing reduces synaptic GABAA responses in hippocampal pyramidal cells. J Neurosci. 1992;12:4122–4132. doi: 10.1523/JNEUROSCI.12-10-04122.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitler TA, Alger BE. Depolarization-induced suppression of GABAergic inhibition in rat hippocampal pyramidal cells: G protein involvement in a presynaptic mechanism. Neuron. 1994;13:1447–1455. doi: 10.1016/0896-6273(94)90430-8. [DOI] [PubMed] [Google Scholar]
- Prast H, Philippu A. Nitric oxide as modulator of neuronal function. Prog Neurobiol. 2001;64:51–68. doi: 10.1016/s0301-0082(00)00044-7. [DOI] [PubMed] [Google Scholar]
- Robbe D, Montgomery SM, Thome A, Rueda-Orozco PE, McNaughton BL, Buzsaki G. Cannabinoids reveal importance of spike timing coordination in hippocampal function. Nat Neurosci. 2006;9:1526–1533. doi: 10.1038/nn1801. [DOI] [PubMed] [Google Scholar]
- Schmidt HH, Pollock JS, Nakane M, Forstermann U, Murad F. Ca2+/calmodulin-regulated nitric oxide synthases. Cell Calcium. 1992;13:427–434. doi: 10.1016/0143-4160(92)90055-w. [DOI] [PubMed] [Google Scholar]
- Szabadits E, Cserep C, Ludanyi A, Katona I, Gracia-Llanes J, Freund TF, Nyiri G. Hippocampal GABAergic synapses possess the molecular machinery for retrograde nitric oxide signaling. J Neurosci. 2007;27:8101–8111. doi: 10.1523/JNEUROSCI.1912-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka J, Markerink-van Ittersum M, Steinbusch HW, de Vente J. Nitric oxide-mediated cGMP synthesis in oligodendrocytes in the developing rat brain. Glia. 1997;19:286–297. [PubMed] [Google Scholar]
- Tao HW, Poo M. Retrograde signaling at central synapses. Proc Natl Acad Sci USA. 2001;98:11009–11015. doi: 10.1073/pnas.191351698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Staveren WC, Markerink-van Ittersum M, Steinbusch HW, Behrends S, de Vente J. Localization and characterization of cGMP-immunoreactive structures in rat brain slices after NO-dependent and NO-independent stimulation of soluble guanylyl cyclase. Brain Res. 2005;1036:77–89. doi: 10.1016/j.brainres.2004.12.040. [DOI] [PubMed] [Google Scholar]
- Varma N, Carlson GC, Ledent C, Alger BE. Metabotropic glutamate receptors drive the endocannabinoid system in hippocampus. J Neurosci. 2001;21(RC188):1–5. doi: 10.1523/JNEUROSCI.21-24-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendland B, Schweizer FE, Ryan TA, Nakane M, Murad F, Scheller RH, Tsien RW. Existence of nitric oxide synthase in rat hippocampal pyramidal cells. Proc Natl Acad Sci USA. 1994;91:2151–2155. doi: 10.1073/pnas.91.6.2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Kunos G, Nicoll RA. Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron. 2001;31:453–462. doi: 10.1016/s0896-6273(01)00372-5. [DOI] [PubMed] [Google Scholar]