

Abstract
Fast inhibitory synaptic transmission is predominantly mediated by GABAA receptor (GABAAR) in the CNS. Although several types of neuronal activity-dependent plasticity at GABAergic synapses have been reported, the detailed mechanism is elusive. Here we show that binding of structurally altered GABAAR-associated protein (GABARAP) to GABAAR γ2 subunit and to tubulin is critical for long-term potentiation [called rebound potentiation (RP)] at inhibitory synapses on a cerebellar Purkinje neuron (PN). Either inhibition of GABARAP association with GABAARγ2 or deletion of tubulin binding region of GABARAP impaired RP. Inhibition of tubulin polymerization also suppressed RP. Thus, precise regulation of GABAARγ2–GABARAP–microtubule interaction is critical for RP. Furthermore, competitive inhibition of GABARAP binding to GABAARγ2 after the RP establishment attenuated the potentiated response, suggesting that GABARAP is critical not only for the induction but also for the maintenance of RP. Fluorescence resonance energy transfer analysis revealed that GABARAP underwent sustained structural alteration after brief depolarization of a PN depending on the activity of Ca2+/calmodulin-dependent protein kinase II (CaMKII), which is required for the RP induction. The susceptibility of GABARAP to undergo structural alteration was abolished by an amino acid replacement in GABARAP. Furthermore, RP was impaired by expression of the mutant GABARAP with the replacement. Together, we conclude that GABAAR association with structurally altered GABARAP downstream of CaMKII activation is essential for RP.
Keywords: synaptic plasticity, GABAAR, GABARAP, FRET, microtubule, Purkinje neuron
Introduction
Precise regulation of inhibitory synaptic transmission is critical for the proper function of CNS as well as that of excitatory synaptic transmission. Neuronal activity-dependent plasticity of synaptic transmission has been regarded as a cellular basis for learning and memory (Bailey et al., 2000; Hansel et al., 2001; Kandel, 2001). Various types of synaptic plasticity have been reported at both excitatory and inhibitory synapses in the CNS (Kano et al., 1992; Komatsu, 1996; Nusser et al., 1998; Oda et al., 1998; Bailey et al., 2000; Hansel et al., 2001; Kandel, 2001; Gaiarsa et al., 2002; Diana and Bregestovski, 2005). Extensive studies have uncovered the regulatory molecular mechanism of excitatory synaptic plasticity. Phosphorylation of glutamate receptors modulates their properties such as channel conductance and the affinity to scaffold proteins changing the number of receptors at the postsynaptic density (Hayashi et al., 2000; Lee et al., 2000; Sheng and Kim, 2002). Conversely, little is known about the molecular mechanism of plasticity at inhibitory synapses. Some forms of inhibitory synaptic plasticity are mediated by the change in postsynaptic responsiveness of GABAA receptor (GABAAR), major receptors at inhibitory synapses (Kano et al., 1992; Nusser et al., 1998). However, details of how GABAAR changes its properties are unclear.
At GABAergic synapses on a cerebellar Purkinje neuron (PN), postsynaptic depolarization caused by excitatory inputs, such as those from a climbing fiber, potentiates the inhibitory synaptic transmission mediated by GABAAR for a long time, which is called rebound potentiation (RP) (Kano et al., 1992). The increase in intracellular Ca2+ concentration and resultant activation of Ca2+/calmodulin-dependent protein kinase II (CaMKII) are required for the induction of RP (Kano et al., 1996; Kawaguchi and Hirano, 2002). However, how CaMKII activation augments the GABAAR function during the RP induction remains to be clarified. In the synaptic plasticity at excitatory synapses, proteins that directly bind to glutamate receptors, such as PICK1 (protein interacting with C kinase), GRIP (glutamate receptor interacting protein), and TARP (transmembrane AMPA receptor regulatory protein), play critical roles by regulating intracellular trafficking of glutamate receptors (Hirbec et al., 2003; Rouach et al., 2005; Tomita et al., 2005). We considered that binding proteins for GABAAR might be involved in RP. Recently, several GABAAR binding proteins have been identified (Wang et al., 1999; Bedford et al., 2001; Kanematsu et al., 2002; Chen and Olsen, 2007). They are GABARAP (GABAAR-associated protein), PRIP (phospholipase C-related, catalytically inactive protein), Plic-1, GODZ (Golgi-specific DHHC zinc finger protein), etc. Among them, we attended to GABARAP, a linker protein between GABAAR and tubulin (Wang et al., 1999). The number and/or function of surface GABAAR can be regulated by GABARAP in both heterologous expression systems and neurons (Chen et al., 2000, 2005; Nymann-Andersen et al., 2002; Everitt et al., 2004; Leil et al., 2004; Luu et al., 2006). However, implication of GABARAP in synaptic plasticity has not been explored. Here, we examined involvement of GABARAP in RP using cultured PNs. Our results indicate that precise regulation of microtubule–GABARAP–GABAAR association is essential for the induction and maintenance of RP.
Materials and Methods
Culture.
The method for preparing primary culture of cerebellar neurons was similar to a previous study (Kawaguchi and Hirano, 2006). Experiments including whole-cell patch-clamp recordings, immunocytochemistry, and fluorescent imagings were performed 3–4 weeks after preparation of culture. Experimental procedures were performed in accordance with the guideline regarding care and use of animals for experimental procedures of National Institutes of Health (United States) and Kyoto University and approved by the local committee for handling experimental animals in the Graduate School of Science, Kyoto University.
Electrophysiology.
Methods of electrophysiological experiments were similar to previous studies (Kawaguchi and Hirano, 2000, 2002, 2006). Briefly, whole-cell patch-clamp recording from a cerebellar PN grown in culture for 3–4 weeks was performed with an amplifier (EPC9; HEKA Elektronik, Lambrecht/Pfalz, Germany) in the solution containing the following (in mm): 145 NaCl, 5 KOH, 2 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose, pH 7.3, at room temperature (20–24°C), unless otherwise stated. The solution contained 6-cyano-7-nitroquinoxaline-2, 3-dione disodium (10 μm; Tocris Bioscience, Bristol, UK), tetrodotoxin (TTX; 1 μm; Wako, Osaka, Japan), and SCH50911 [(2S)(+)5,5-dimethyl-2-morpholineacetic acid; 10 μm; Tocris Bioscience] to inhibit glutamatergic EPSCs, action potentials, and GABABR activation, respectively, unless otherwise stated. A PN was visually identified by a large cell body and thick dendrites. A patch pipette used to record from a PN was filled with an internal solution (pH 7.3) containing the following (in mm): 155 CsCl, 10.5 CsOH, 0.5 EGTA, 10 HEPES, 2 Mg-ATP (Sigma, St. Louis, MO), and 0.2 Na-GTP (Sigma). Mg-ATP and Na-GTP were used to minimize rundown of GABAAR. For recordings from a presynaptic inhibitory interneuron, the following internal solution (pH 7.3) was used (in mm): 147 KCl, 15 KOH, 5 EGTA, 10 HEPES, 2 Mg-ATP, and 0.2 Na-GTP. The membrane potential of a PN was corrected for the liquid junction potential (−3 mV) and was held at −70 mV. When GABA responses, miniature IPSCs (mIPSCs), and evoked IPSCs were recorded for a long duration from a PN, only recordings with an input resistance of >100 MΩ and series resistance of <25 MΩ were accepted. To minimize the voltage-clamp error, the amplitude of GABA response at the beginning of experiments was set to ∼200 pA. Series resistance and input resistance were monitored every 2 min, and experiments were terminated when a change of >20% was detected. The Ba2+ currents through voltage-gated Ca2+ channels were measured by applying a 50 ms depolarizing pulse to −20 mV in the external solution containing 2 mm Ba2+ instead of Ca2+, 1 μm TTX, 1 mm 4-aminopyridine, and 10 mm tetraethylammonium (the latter two are blockers for K+ channels). In the Ba2+ current recording, the series resistance was kept between 7 and 12 MΩ and compensated (40%, 10 μs lag).
In the mIPSCs analysis, events larger than 6 pA with appropriate time course were selected visually, and the amplitudes were measured with the Mini Analysis software (Synaptsoft, Fort Lee, NJ). The mean amplitude was calculated from 200–400 events. Time courses (10–90% rise time and half-height width) were obtained by averaging 20 mIPSCs in each cell. The method for iontophoretic application of GABA was similar to previous studies (Kawaguchi and Hirano, 2000, 2002, 2006). A glass pipette containing 10 mm GABA was aimed at a proximal dendrite, and 20 ms positive voltage pulses were applied every 20 s. In some experiments, the temperature was regulated by Thermo Plate (Tokai Hit, Shizuoka, Japan). The 18 amino acids peptide of GABAAR γ2 subunit (γ2 peptide) or the scrambled control peptide (100 μm, purity >95%; Invitrogen, Carlsbad, CA) was applied into a PN through a patch pipette. Data are presented as mean ± SEM, unless otherwise stated. Statistical significance was assessed by unpaired Student's t test or by one-way ANOVA when multiple groups were compared at the same time.
Immunocytochemistry.
Cultured neurons were fixed with 4% paraformaldehyde, permeabilized in PBS containing 0.5% Tween 20 and 2% skim milk, and labeled with primary antibodies followed by treatment with secondary antibodies. Surface staining of GABAARα1 was performed after fixation under a nonpermeabilized condition without Tween 20, followed by wash and subsequent staining of enhanced green fluorescent protein (EGFP) or calbindin in a permeabilized condition with Tween 20. Primary and secondary antibodies used were as follows: a mouse monoclonal antibody (mAb) against calbindin D28 (1:1000; Swant, Bellinzona, Switzerland), an mAb against α-tubulin (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA), a rabbit polyclonal antibody (pAb) against GABAAR α1 subunit (1:1000; Alomone Labs, Jerusalem, Israel), a rabbit pAb against GABAAR γ2 subunit (1:300; Alomone Labs), an mAb against GABAAR β2/3 subunit (1:500; Upstate, Charlottesville, VA), a rabbit pAb against calbindin (1:1000; Chemicon, Temecula, CA), a chick pAb against GFP (1:1000; Chemicon), and Alexa 568-conjugated pAb against rabbit or mouse IgG and Alexa 488-conjugated pAb against chick, rabbit, or mouse IgG (Invitrogen, Carlsbad, CA). Fluorescent images were recorded with a confocal laser microscope (CSU 10; Yokokawa Electric, Tokyo, Japan) equipped to an upright microscope (Eclipse E800; Nikon, Tokyo, Japan) and analyzed using IP Lab software (Solution Systems, Chiba, Japan). High K+-containing conditioning treatment solution was made by replacing 50 mm Na+ with K+ in the normal external solution. The averaged fluorescent signal for surface GABAAR divided by the area positive for EGFP or calbindin was compared.
DNA construction.
Human GABARAP cDNA was a generous gift from Drs. Kanematsu and Hirata (Kyushu University, Fukuoka, Japan). The amino acid sequence of human GABARAP is identical to that of rat GABARAP. The mutant GABARAP, such as Δ27, V33E, and P37A, was produced by PCR-mediated introduction of each mutation. cDNA of wild-type or mutant GABARAP were inserted into pCAGplay expression vector (Kawaguchi and Hirano, 2006) at BglII/PstI site. Venus cDNA was produced by PCR-mediated mutagenesis from enhanced yellow fluorescent protein cDNA (Invitrogen). Coding regions of enhanced cyan fluorescent protein (ECFP) and Venus were inserted into pCAGplay at EcoRI/BglII site and at PstI/XhoI site, respectively. To avoid the possible cleavage at GABARAP C-terminal L by Atg-family protease (Tanida et al., 2004), two amino acids G116 and L117 were removed from CGV probe. Coding region for GABARAP residues 1–115 was inserted in-frame into the BglII/PstI site between coding regions for ECFP and Venus in pCAGplay vector. Expression plasmid encoding hemagglutinin (HA)–GABARAP was constructed by in-frame insertion of double-strand DNA primers encoding HA tag at EcoRI/BglII site of pCAGplay vector containing full-length GABARAP cDNA between BglII and PstI sites. The expression plasmid (50 ng/μl) encoding wild-type or mutant GABARAP or CGV probe was injected into a nucleus of PN through a sharp glass pipette. The electrophysiological, fluorescence resonance energy transfer (FRET), or immunocytochemical experiments were performed 1–3 d after the injection.
FRET imaging.
FRET images were obtained from a PN transfected with CGV probe using an upright microscope IX71 equipped with FV1000 fluorescence imaging system (Olympus, Tokyo, Japan). CGV probe was excited at 440 nm, and the emission between 460 and 500 nm (for CFP) and that between 515 and 615 nm (for Venus) were recorded. The fluorescence intensities of ECFP and Venus in a thick shaft of proximal dendrite were measured. Then, the fluorescence ratio (Venus/ECFP) was calculated.
Results
RP impairment by competitive inhibition of GABARAP binding to GABAARγ2
In a PN, most GABAARs are composed of α1, β2/3, and γ2 subunits (Laurie et al., 1992). This was confirmed by immunocytochemistry showing that fluorescent signal for GABAAR α1 subunit and that for γ2 subunit almost completely overlapped with that for β2/3 subunit (data not shown). GABARAP binds to both GABAARγ2 subunits and tubulin (Wang et al., 1999). The binding site of γ2 subunits to GABARAP lies at a region consisting of 18 amino acids RTGAWRHGRIHIRIAKMD in the intracellular loop between third and fourth transmembrane segments (Wang et al., 1999; Coyle et al., 2002; Nymann-Andersen et al., 2002). We first examined whether GABARAP is involved in RP by competitively inhibiting association of GABARAP with γ2 subunit using a fusion protein containing the 18 amino acids and Venus, a variant of yellow fluorescent protein (Nagai et al., 2002).
An expression plasmid encoding the fusion protein was constructed (γ2 peptide–Venus) and intranuclearly injected into a PN. GABAAR responsiveness was monitored with the current response to GABA applied iontophoretically to the proximal dendrites of a PN (Kawaguchi and Hirano, 2000, 2002). In a control PN transfected with EGFP, conditioning depolarization (0 mV for 500 ms, 5 times at 0.5 Hz) potentiated the amplitude of GABA response within 2 min after the conditioning (245 ± 13% at 2 min, mean ± SEM), and the potentiation lasted for >30 min (172 ± 11% at 30 min) (Fig. 1A,B). Thus, RP is rapidly induced after depolarization of a PN and lasts for a long time. Conversely, expression of γ2 peptide–Venus in a PN suppressed the potentiation of GABA response at 30 min (100 ± 3%, p < 0.005, Student's t test), although the reduced potentiation at 2–10 min after the conditioning remained (183 ± 21%, p < 0.05, 2 min) (Fig. 1A,B). Thus, inhibition of GABARAP binding to GABAAR γ2 subunit completely suppressed the sustained phase (at 10–30 min) of RP, although the suppressive effect on the early phase (at 2–10 min) was limited.
Figure 1.
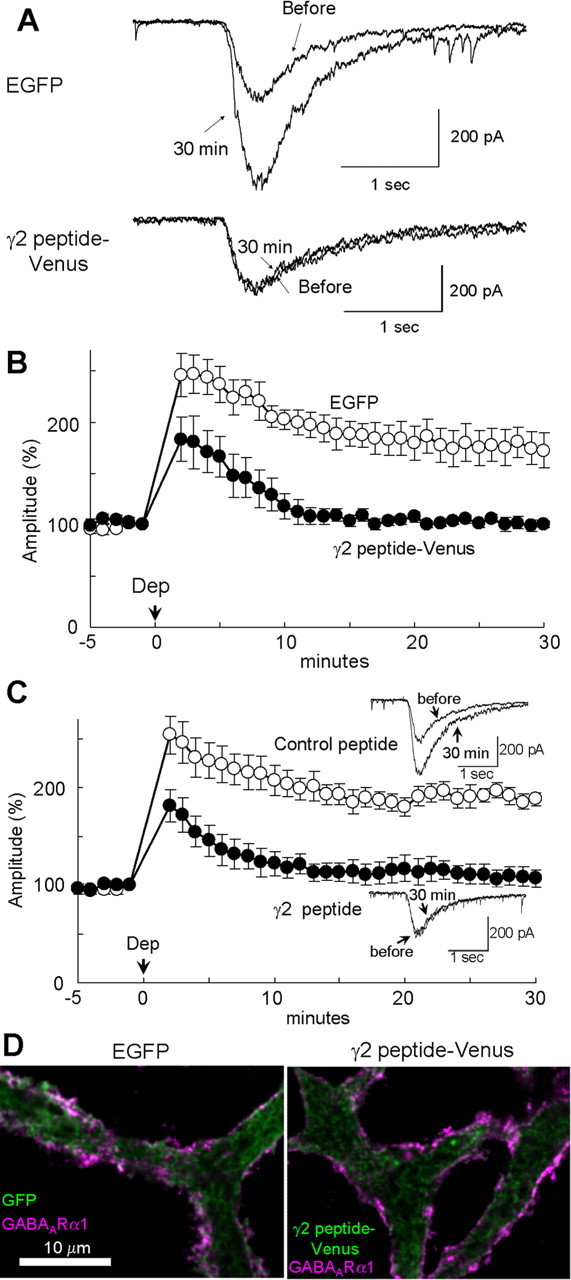
Involvement of GABARAP in RP. A, B, Representative traces (A) and time courses of amplitudes (B) of current responses to GABA before and after the conditioning depolarization (0 mV for 500 ms, 5 times at 0.5 Hz) recorded from a PN transfected with EGFP or fusion protein of 18 amino acids peptide of GABAAR γ2 subunit and Venus. n = 5 for each. C, Representative traces and time courses of amplitudes of GABA responses before and after the conditioning depolarization with γ2 or control peptide applied through a patch pipette. n = 5 for each. D, Immunocytochemical staining of GABAAR α1 subunit and EGFP transfected into a PN. Dendrites of PNs are shown.
To confirm the conclusion, we next applied the 18 amino acids peptide (γ2 peptide, RTGAWRHGRIHIRIAKMD) directly into a PN through a patch pipette. The γ2 peptide reduced the extent of RP in the early phase (182 ± 17%, 2 min) and almost completely suppressed the late phase of RP (107 ± 9%, 30 min), whereas a control peptide with randomized amino acids sequence (AAKGHRIMWGRIRTDHI) did not (2 min, 255 ± 15%, p < 0.05; 30 min, 188 ± 14%, p < 0.005) (Fig. 1C). These results suggest that association of GABARAP with GABAARγ2 subunit is critical for RP. We then examined whether the γ2 peptide suppresses the RP induction at a near-physiological temperature. At 32–33°C, the early phase of RP attenuated faster than at room temperature (199 ± 16% at 2 min), followed by the gradual increase of GABAAR responsiveness (supplemental Fig. 1A, Control peptide, available at www.jneurosci.org as supplemental material). The γ2 peptide impaired the late phase of RP but not the early phase (2 min, 230 ± 7%, p > 0.12; 20 min, 96 ± 9%, p < 0.05). These results suggest that association of GABARAP to GABAAR γ2 subunit is critical for long-term induction of RP.
GABARAP plays a role in trafficking of GABAAR toward the postsynaptic membrane of dendritic inhibitory synapses and in regulation of the GABAAR number on the plasma membrane in hippocampal neurons (Leil et al., 2004). Thus, we examined whether inhibition of GABARAP and GABAARγ2 association affects the subcellular localization of GABAAR in a PN. Immunocytochemical analysis showed that both GABAAR α1 and γ2 subunits were located along the plasma membrane and that expression of γ2 peptide–Venus did not apparently affect the localization pattern in a PN (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). To further examine the effect of γ2 peptide–Venus on trafficking of GABAAR toward the plasma membrane, we stained the surface GABAARα1 on the plasma membrane in a nonpermeabilized condition, followed by staining of intracellular EGFP or γ2 peptide–Venus after permeabilization. Expression of γ2 peptide–Venus did not affect the localization pattern and amount of GABAARα1 on the plasma membrane (Fig. 1D). The surface GABAAR signal in a PN transfected with γ2 peptide–Venus was 115 ± 14% (n = 12) of that in a PN transfected with EGFP (n = 25; p > 0.35). Thus, it is suggested that competitive inhibition of GABARAP and GABAARγ2 binding impairs RP without clear alteration in subcellular localization of GABAAR in a PN.
Next, we examined whether GABARAP localization changes in response to depolarization in a PN. Because no good anti-GABARAP antibody that can stain endogenous GABARAP was available, we constructed an expression vector encoding HA-tagged GABARAP. HA–GABARAP was diffusely distributed in the basal condition, and hardly colocalized with GABAAR detected by an antibody against β2/3 subunit (supplemental Fig. 2C, available at www.jneurosci.org as supplemental material). The depolarizing conditioning treatment (for 30 s) with the external solution containing 50 mm K+ seemed to change the GABARAP distribution. The fluorescent signal for HA–GABARAP tended to become more conspicuous near the plasma membrane 10–30 min after the conditioning, and colocalization of HA–GABARAP and GABAAR signals was sometimes observed (supplemental Fig. 2C, available at www.jneurosci.org as supplemental material). These results suggest that depolarization of a PN tends to translocate GABARAP toward the plasma membrane and that the surface GABAAR might become to be bound with GABARAP.
We next examined whether binding of GABARAP to GABAARγ2 subunit is required for RP at synapses. Conditioning depolarization potentiated the amplitude of mIPSCs in the presence of control peptide in a PN (2 min, 192 ± 18%; 30 min, 168 ± 16%) (Fig. 2A–C). In contrast, the conditioning failed to establish the sustained potentiation in the presence of γ2 peptide (2 min, 147 ± 13%, p > 0.07; 30 min, 96 ± 16%, p < 0.05) (Fig. 2A–C). Thus, synaptic GABAAR needs to be bound by GABARAP for long-term expression of RP. We also noticed the increase of mIPSC frequency, presumably reflecting the increased probability of synaptic vesicle release after the conditioning depolarization (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). This phenomenon might correspond to the depolarization-induced potentiation of inhibition (DPI) (Duguid and Smart, 2004). The extent of changes in mIPSCs frequency by the depolarization was not affected by the γ2 peptide in a postsynaptic PN (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material), supporting the idea that presynaptic DPI is distinct from postsynaptic RP.
Figure 2.

Requirement of GABARAP association with GABAARγ2 for RP at synapses. A, Representative traces of mIPSCs before and 30 min after the conditioning depolarization in the presence of control peptide or γ2 peptide. B, Cumulative probability histogram of amplitudes of mIPSCs (>400 events for each) before and 30 min after the conditioning depolarization in the presence of control or γ2 peptide. C, Time courses of amplitudes of mIPSCs with γ2 or control peptide applied through a patch pipette. n = 5 for each. Dep, Depolarization. D, Representative traces of presynaptic action potentials and postsynaptic IPSCs before and 20 min after the conditioning depolarization in the presence of control or γ2 peptide. IN, Inhibitory interneuron. Each trace was obtained by averaging 15 events. E, Time courses of amplitudes of evoked IPSCs in the presence of control or γ2 peptide.
We next examined whether the γ2 peptide suppresses the RP of evoked IPSCs. Paired recordings from a presynaptic inhibitory interneuron and a postsynaptic PN were performed without TTX. A presynaptic inhibitory interneuron was maintained in a whole-cell voltage-clamp or current-clamp condition, and Na+ current or an action potential was evoked by a depolarization pulse or by the positive current injection. The evoked IPSC was recorded from a whole-cell voltage-clamped PN. The amplitude of evoked IPSC was potentiated for >30 min after the conditioning depolarization with the control peptide (2 min, 153 ± 23%; 20 min, 264 ± 51%) (Fig. 2D,E). In contrast, the late phase of RP was impaired with the γ2 peptide (2 min, 132 ± 21%, p > 0.52; 20 min, 90 ± 3%, p < 0.05) (Fig. 2D,E). The potentiation of evoked IPSC at 2 min after the depolarization was somewhat weak compared with RP of GABA responses and mIPSCs, presumably attributable to the depolarization-induced potentiation of suppression (DSI). DSI is the transient (∼2 min) decrease of presynaptic transmitter release mediated by endocannabinoid released from a depolarized PN (Kreitzer and Regehr, 2001; Diana et al., 2002).
Neither transfection of EGFP or γ2 peptide–Venus, nor application of the γ2 or the control peptide affected the basal mIPSC amplitude, time course, frequency, or the currents through voltage-gated Ca2+ channels (VGCCs) (supplemental Table 1, available at www.jneurosci.org as supplemental material). Together, we suggest that RP at inhibitory synapses on a PN depends on association of GABAARγ2 and GABARAP. Hereafter, we focus on RP, the postsynaptic alteration of GABAAR responsiveness induced by the conditioning depolarization reflected with the amplitudes of mIPSCs or GABA responses.
GABARAP binding to tubulin is required for RP
In addition to binding to GABAARγ2, GABARAP also associates with tubulin through the N-terminal 27 amino acids region (Fig. 3A) (Wang et al., 1999; Coyle et al., 2002). A mutant GABARAP in which N-terminal 27 amino acids were deleted (Δ27) retains the ability to associate with GABAARγ2 subunit but is unable to bind to tubulin. Transfection of this mutant GABARAP impaired RP induction with slight reduction in the early phase and almost complete suppression of the sustained phase (2 min, 172 ± 10%; 30 min, 99 ± 5%), whereas expression of the full-length GABARAP did not affect RP (2 min, 226 ± 10%, p < 0.01; 30 min, 171 ± 17%, p < 0.05) (Fig. 3B). These results suggest that association of GABAARγ2 and GABARAP lacking binding site to microtubule suppresses RP. Thus, the ability of GABARAP to bind to tubulin is indispensable for RP. RP of mIPSC was also impaired by transfection of γD27 GABARAP (2 min, 133 ± 7%; 30 min, 114 ± 5%), whereas it was not affected by the full-length GABARAP transfection (2 min, 174 ± 17%, p > 0.06; 30 min, 171 ± 14%, p < 0.05) (Fig. 3C). Thus, RP of synaptic transmission also depends on the binding ability of GABARAP to tubulin. Transfection of full-length or γD27 GABARAP did not affect the basal mIPSC amplitude, time course, frequency, or VGCC current (supplemental Table 1, available at www.jneurosci.org as supplemental material).
Figure 3.
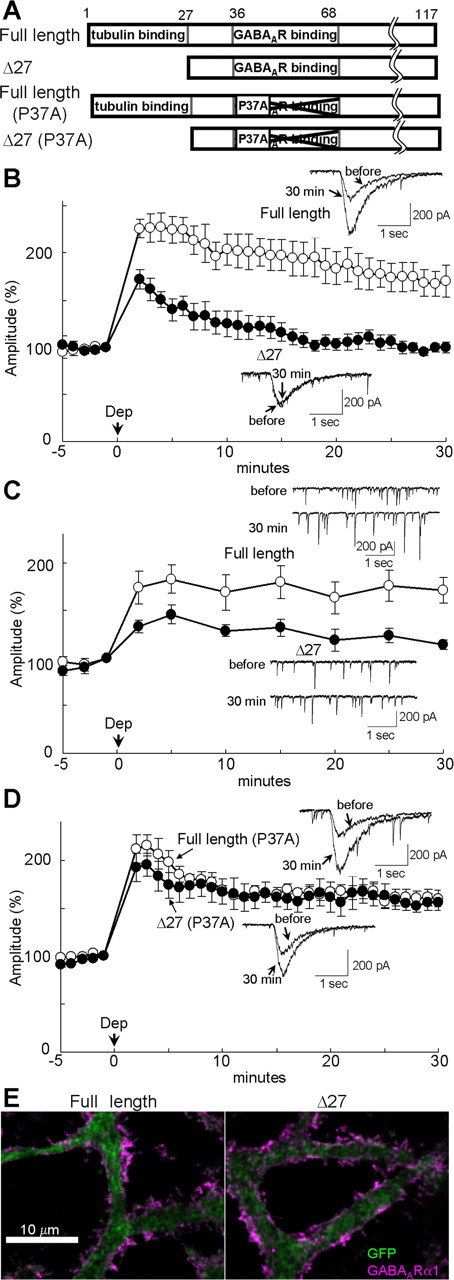
Binding of GABARAP to tubulin is critical for RP. A, The wild-type and mutant GABARAP proteins. Deletion of N-terminal 27 amino acids abolishes tubulin binding. P37A point mutation reduces the affinity to GABAARγ2 subunit. B, Representative traces and time courses of amplitudes of GABA responses in a PN transfected with either full-length or Δ27 GABARAP. n = 5 for each. C, Representative traces and time courses of amplitudes of mIPSCs in a PN transfected with either full-length (n = 5) or Δ27 GABARAP (n = 8). D, Representative traces and time courses of amplitudes of GABA responses in a PN transfected with either full-length (n = 6) or Δ27 GABARAP (n = 5) containing P37A point mutation. E, Immunocytochemical staining of EGFP and surface GABAAR α1 subunit in a PN transfected with either full-length or Δ27 GABARAP together with EGFP.
Next, using a point mutation P37A, which is reported to primarily decrease the affinity of GABARAP to GABAARγ2 subunit (Fig. 3A) (Leil et al., 2004), we examined whether the suppressive effect of γD27 GABARAP on RP is mediated by direct binding of the mutant GABARAP to GABAARγ2 subunit. As shown in Figure 3D, the suppressive effect of γD27 GABARAP on RP was abolished by the point mutation P37A (2 min, 193 ± 15%, p > 0.25; 30 min, 156 ± 8%, p < 0.001) compared with γD27, suggesting that impairment of RP was attributable to direct association of GABAARγ2 and the mutant GABARAP lacking the tubulin binding region. Conversely, P37A mutation in the full-length GABARAP did not affect the RP induction (2 min, 212 ± 15%, p > 0.4; 30 min, 160 ± 9%; p > 0.7) compared with the wild-type GABARAP (Fig. 3D), suggesting that the mutant GABARAP lacking the ability to associate with GABAARγ2 subunit does not disturb the function of endogenous GABARAP in RP. Competition of the P37A mutant with the endogenous GABARAP for binding to tubulin might be limited because of the large amount of tubulin in a PN.
Taking all above results together, we suggest that precise regulation of microtubule–GABARAP–GABAARγ2 association is essential for RP. The amount of GABAAR γa1 subunit on the plasma membrane and the localization pattern of GABAAR γa1 and γ2 subunits in a PN were not affected by expression of γD27 GABARAP (Fig. 3E) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The surface GABAARγa1 signal in PNs transfected with the full-length GABARAP or γD27 GABARAP were 102 ± 7% (n = 16) and 108 ± 7% (n = 16) of that in EGFP-transfected PNs (p > 0.53).
Sustained GABARAP structural alteration by depolarization
To obtain additional insight into the role of GABARAP in RP, we developed a FRET-based probe protein that could be used for sensing the conformational change of GABARAP in a living cell. GABARAP was fused with ECFP and Venus at the N and C terminals, respectively (named CGV probe). We expected that the FRET efficiency of CGV probe might be changed by the structural alteration of GABARAP in a similar manner to other FRET-based probe proteins used for the analysis on synaptic plasticity at excitatory synapses (Okamoto et al., 2004; Takao et al., 2005). CGV probe was transfected into a PN, in which simultaneous FRET imaging and a whole-cell patch-clamp recording were performed.
When CGV probe in a PN was illuminated by 440 nm laser, which preferentially excites ECFP, it emitted fluorescence with the intensity peaks for both ECFP (∼470 nm) and Venus (∼520 nm), indicating that FRET occurred between ECFP and Venus fused to each end of GABARAP (Fig. 4C). Thus, the fluorescence ratio of the Venus signal (515–615 nm) divided by the ECFP signal (460–500 nm) decreased (Fig. 4B). These results suggest that the distance between ECFP and Venus in the CGV probe was increased or the angle between them was altered by the conditioning depolarization of a PN, implying occurrence of some conformational change in GABARAP. The decrease of FRET efficiency in CGV probe caused by the depolarization reached the peak within 30 s and lasted for >15 min (depolarization, 85 ± 3% at 15 min; no depolarization, 99 ± 2%; p < 0.001) (Fig. 5A,B). Thus, it seems that GABARAP conformation change induced by the transient depolarization of a PN lasts for a long time.
Figure 4.

FRET decrease of CGV probe in response to depolarization of a PN. A, Representative fluorescence images of ECFP and Venus and the ratio images of Venus/ECFP before and immediately after the conditioning depolarization (Dep). B, Time courses of fluorescence intensity of ECFP (blue) and Venus (green) and that of the fluorescence ratio (Venus/ECFP, black). The conditioning depolarization was applied at 0 s. A.U., Arbitrary unit. C, Fluorescence emission spectrum of CGV probe excited by 440 nm laser. Data obtained before (black) and 10 min after the conditioning depolarization (red) are shown.
Figure 5.
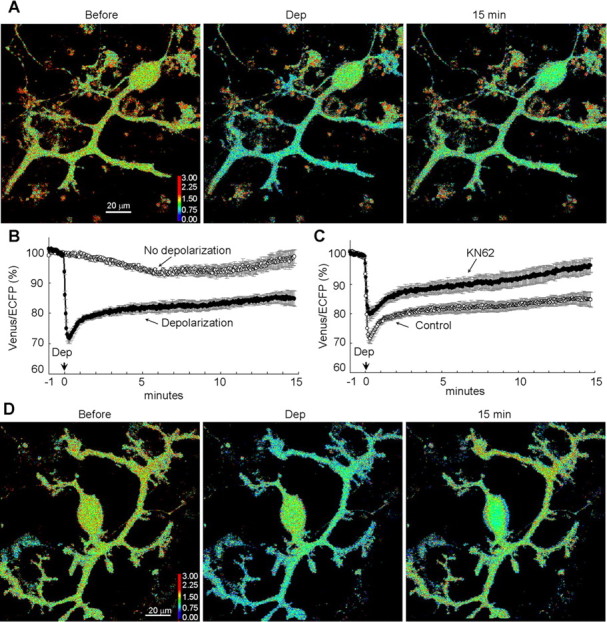
Long-lasting GABARAP structural alteration detected with FRET. A, Representative pseudocolor fluorescence ratio images (Venus/ECFP) before, during, and 15 min after the conditioning depolarization (Dep) in a PN. B, Time courses of the normalized fluorescence ratio in a proximal dendrite of PNs. The conditioning depolarization was applied at 0 min. n = 13 (Depolarization) and n = 7 (No depolarization). C, Time courses of the normalized fluorescence ratio in the absence or presence of KN62 (n = 8). The control data are same as the depolarization data in B. D, Representative images of the fluorescence ratio of CGV probe before, during, and 15 min after the conditioning depolarization in the presence of KN62.
We next attempted to clarify how the decrease in FRET efficiency of CGV probe is brought about. Because CaMKII is critical for the RP induction, we examined whether it is also involved in the FRET decrease of CGV probe. Application of KN62 (1-[N,O-bis(5-isoquinolinesulphonyl)-N-methyl-l-tyrosyl]-4-phenyl-piperazine) (5 μm), an inhibitor of CaMKII, suppressed the sustained FRET decrease, although a transient decrease was observed (96 ± 2%; p < 0.005) (Fig. 5C,D). Thus, CaMKII activity seems to be necessary for the sustained conformation change of CGV probe. These results suggest that the sustained GABARAP structural alteration might be closely related to the RP induction.
Role of sustained GABARAP conformation change in RP
To obtain a molecular tool for analyzing the relation of GABARAP conformation change and the RP induction, we searched for a mutation in GABARAP that suppresses the FRET decrease. We also intended to confirm that the FRET decrease of CGV probe was attributable to structural alteration in GABARAP. When V33 of GABARAP was replaced by E in the CGV probe, the conditioning depolarization failed to induce the sustained decrease of FRET efficiency (depolarization, 95 ± 2%; no depolarization, 98 ± 1%; p > 0.3) (Fig. 6A,B), suggesting that V33 is important for the structural alteration of GABARAP. The fact that a point mutation of GABARAP abolished the sustained FRET decrease of CGV probe confirms that the FRET decrease of CGV probe was attributable to the structural alteration of GABARAP region but not to the change in linker regions or in fluorophores.
Figure 6.
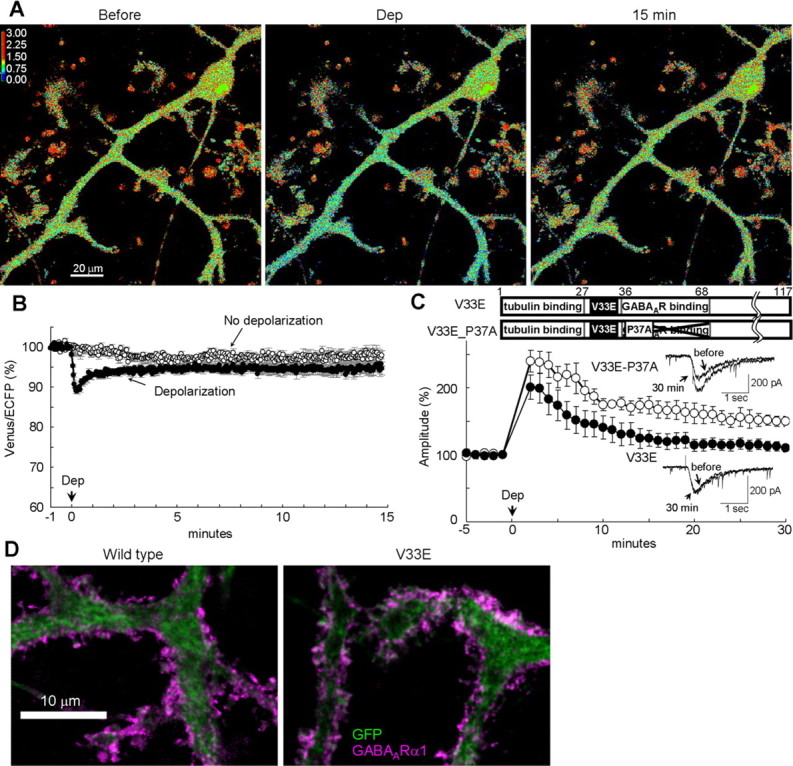
A critical role of GABARAP conformation change in RP. A, Representative ratio images (Venus/ECFP) before, during, and 15 min after the conditioning depolarization (Dep) in a PN transfected with the CGV probe containing V33E mutation. B, Time courses of normalized fluorescent ratio (Venus/ECFP) in a PN transfected with the CGV probe containing V33E mutation. n = 9 (Depolarization) and n = 6 (No depolarization). C, Representative traces and time courses of amplitudes of GABA response before and after the conditioning depolarization in PNs transfected either with the full-length GABARAP containing V33E mutation alone (n = 6) or with both V33E and P37A mutations (n = 5). D, Immunocytochemical images for staining of EGFP and surface GABAAR α1 subunit on the plasma membrane of PN dendrites. Either the full-length GABARAP (Wild type) or V33E GABARAP (V33E) together with EGFP was transfected into a PN.
We next examined whether the V33E GABARAP affects RP. Transfection of V33E GABARAP in a PN impaired RP with slight reduction in the early phase and almost complete suppression in the sustained phase (2 min, 201 ± 18%, p > 0.25; 30 min, 110 ± 6%, p < 0.05) compared with the wild-type GABARAP transfection. This effect was cancelled by an additional point mutation P37A reducing the affinity to GABAARγ2 subunit (2 min, 240 ± 16%, p > 0.1; 30 min, 150 ± 7%, p < 0.01) (Fig. 6C), suggesting that direct binding of structurally unchangeable GABARAP to GABAARγ2 impaired RP. V33E GABARAP did not affect the basal mIPSC amplitude, time course, frequency, or VGCC current (supplemental Table 1, available at www.jneurosci.org as supplemental material). Together, structurally unchangeable GABARAP suppressed RP through direct binding to GABAARγ2 subunit, indicating an essential role of structural alteration of GABARAP in RP. Δ27 GABARAP with V33E mutation also suppressed RP (2 min, 186 ± 15%, p > 0.05; 30 min, 100 ± 2%, p < 0.05 compared with full-length wild-type GABARAP) (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material), similar to Δ27 GABARAP without V33E mutation. The suppressive effect of Δ27 GABARAP with V33E mutation was also abolished by an additional point mutation P37A reducing the affinity to GABAARγ2 (2 min, 224 ± 14%, p > 0.12; 30 min, 154 ± 7%, p < 0.001) (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material). We also examined whether V33E mutated GABARAP affects the localization of GABAAR in PNs. As shown in Figure 6D and supplemental Figure 2 (available at www.jneurosci.org as supplemental material), V33E GABARAP did not affect the amount of surface GABAARα1 on the plasma membrane (116 ± 9%, n = 15, p > 0.15 compared with wild-type GABARAP) and the overall localization pattern of GABAAR α1 and γ2 subunits in PN dendrites. Taking above results together, we suggest that structural alteration of GABARAP is critical for RP.
Binding of structurally altered GABARAP to GABAARγ2 directly mediates RP expression
How does binding of structurally altered GABARAP to GABAARγ2 subunit contribute to expression of RP? To address this issue, we attempted to inhibit binding of GABAARγ2 subunit to structurally altered GABARAP after the induction of RP and examined the effect on established RP. To this end, two patch pipettes were prepared: one contained the normal internal solution for whole-cell recording, and the other contained the γ2 peptide (100 μm) to competitively inhibit GABARAP binding to γ2 subunit. Both pipettes were placed on a soma of PN, and gigaseals were formed. The membrane under the former pipette was ruptured for whole-cell recording. The GABA response was monitored, and RP was induced by the conditioning depolarization (247 ± 12% at 2 min) (Fig. 7B). Five minutes after the depolarization, the membrane under the latter pipette was ruptured, allowing entry of the γ2 peptide into the PN (Fig. 7A). As shown in Figure 7B, the potentiated GABAAR responsiveness started to decline after rupture of the membrane with the second pipette containing the γ2 peptide but not of that containing the control peptide. The γ2 peptide finally attenuated the amplitude of GABAAR response to the basal level at 30 min (γ2 peptide, 112 ± 10%; control peptide, 161 ± 11%; p < 0.05). These results suggest that the once established RP was cancelled by inhibition of binding between structurally altered GABARAP and GABAAR γ2 subunit. Thus, it seems that the potentiation of GABAAR responsiveness is maintained by direct binding of structurally altered GABARAP to GABAARγ2.
Figure 7.
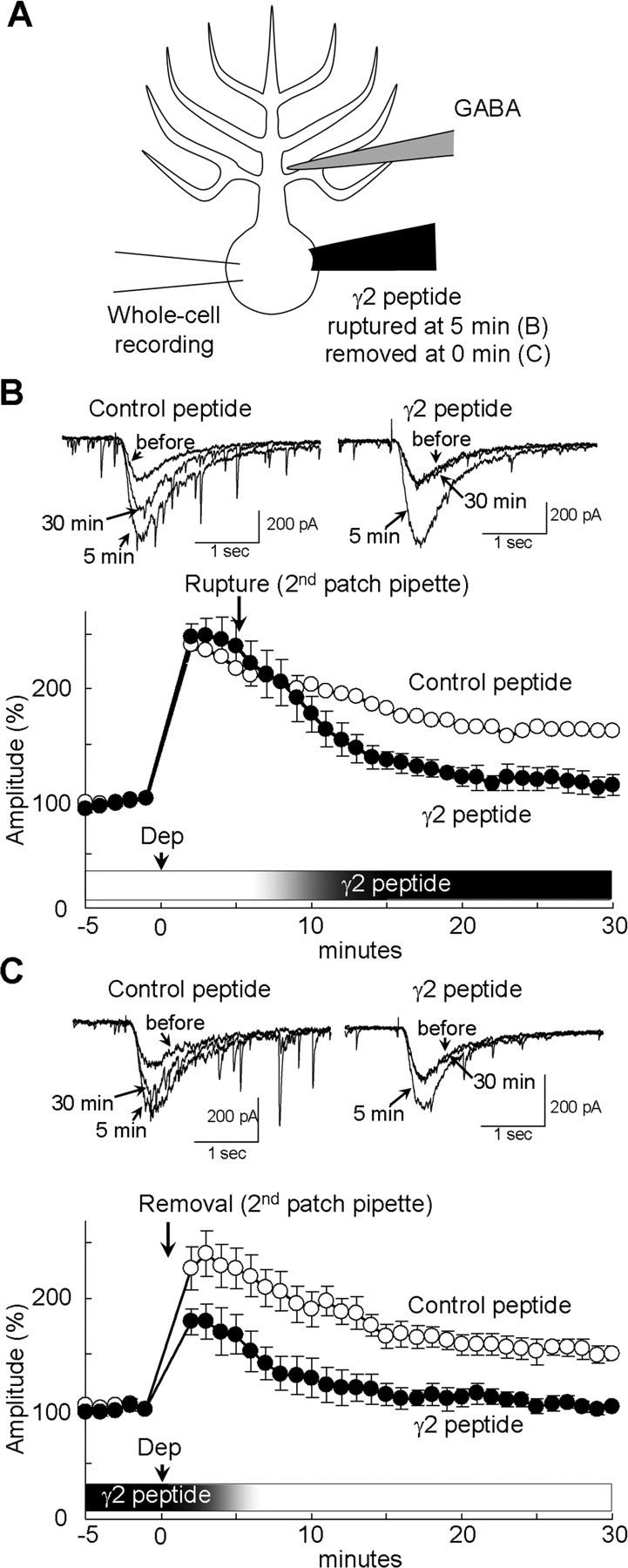
Binding of GABARAP and GABAARγ2 subunit is required for the maintenance of RP. A, Schematic diagram to show the experimental design. GABARAP binding to GABAARγ2 subunit was inhibited by the γ2 peptide (100 μm) from 5 min after the depolarization (B) or until the end of conditioning depolarization (C). B, C, Representative traces and time courses of amplitudes of GABA responses before and after the conditioning depolarization (Dep) with either the γ2 peptide or the control peptide introduced into a PN from 5 min (B) or until 0.5 min (C) through the second patch pipette. n = 5 for each.
We next tried to examine whether association of GABARAP to GABAARγ2 is important only in the maintenance of RP. The first pipette containing only the normal internal solution and the second pipette containing also the γ2 peptide were kept in a whole-cell recording condition from the start of experiment. Then the second pipette was removed immediately after the conditioning depolarization, expecting that the γ2 peptide would be washed out from the PN through the first pipette. The results (Fig. 7C) were similar to those without washout of the γ2 peptide (Fig. 1C). The amplitude of GABA response did not increase after removal of the γ2 peptide-containing pipette (γ2 peptide, 101 ± 5%; control peptide, 150 ± 8% at 30 min; p < 0.005). These results might suggest that the GABARAP association with GABAAR γ2 subunit during the induction phase is critical for establishment of RP, although the possibility that the γ2 peptide was not washed out efficiently and remained bound with GABARAP in a PN cannot be excluded.
Involvement of microtubule in RP
To evaluate the relevance of microtubule–GABARAP–GABAARγ2 interaction to RP, we next examined the effect of destruction of microtubule on RP. When tubulin polymerization was inhibited by a pharmacological agent vincristine (5 μm) for >2 h, microtubule structure was altered in a PN (Fig. 8A). However, the localization pattern of GABAAR α1 subunit was not apparently changed by the treatment in accordance with a previous report (Fig. 8B) (Allison et al., 2000). In such a PN, the conditioning depolarization failed to induce sustained potentiation of GABA responses (30 min, 105 ± 4%, p < 0.005), whereas the early phase of RP was not affected (2 min, 229 ± 13%, p > 0.7) (Fig. 8C). Similarly, acute inhibition of tubulin polymerization by vincristine (2 μm) applied through a patch pipette suppressed the sustained phase of RP in both GABA responses and mIPSCs (GABA responses, 105 ± 4%, p < 0.0005; mIPSCs, 115 ± 10%, p < 0.001 at 30 min) but not the early phase (GABA responses, 216 ± 9%, p > 0.16; mIPSCs, 164 ± 11%, p > 0.29 at 2 min) (Fig. 8D,E). Thus, microtubule is involved in the sustained phase but not in the early phase of RP. Vincristine application through a patch pipette did not affect the basal mIPSC amplitude, time course, frequency, or VGCC current (supplemental Table 1, available at www.jneurosci.org as supplemental material).
Figure 8.
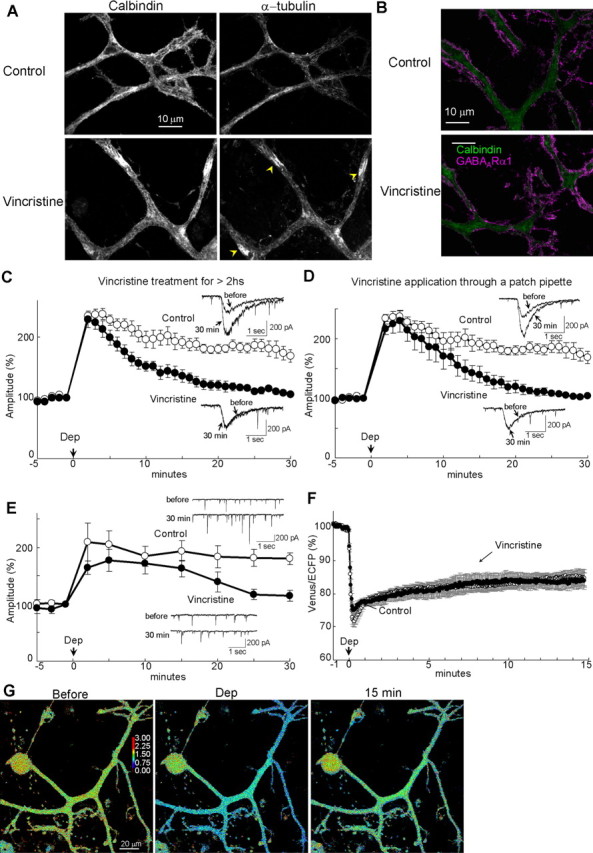
Involvement of microtubule in RP. A, B, Immunocytochemical staining of calbindin and α-tubulin (A) or GABAARα1 (B) in dendrites of PNs with or without (control) vincristine treatment. Distribution of α-tubulin was changed by the vincristine treatment from filamentous distribution along dendritic shafts to irregular distribution consisting of sparse and accumulated regions (yellow arrowheads). Despite microtubule alteration, GABAAR distribution was not clearly affected by the treatment. C, D, Representative traces and time courses of amplitudes of GABA responses before and after the conditioning depolarization (Dep) in a PN. C, Neurons were treated with vincristine for longer than 2 h. n = 6 (Vincristine) and n = 5 (Control). D, Vincristine was applied through a patch pipette. E, Representative traces and time courses of amplitudes of mIPSCs in a PN to which vincristine was applied through a patch pipette. n = 5 (Vincristine) and n = 6 (Control). F, Time courses of the normalized fluorescence ratio (Venus/ECFP) in the presence (Vincristine, n = 7) or absence (Control, n = 13) of vincristine applied through a patch pipette. G, Fluorescence intensity ratio images (Venus/ECFP) before, during, and 15 min after the conditioning depolarization in the presence of vincristine in the patch pipette.
Next, we examined whether tubulin polymerization is also involved in the sustained conformation change of GABARAP monitored with the FRET decrease of CGV probe. Inhibition of tubulin polymerization by vincristine applied through a patch pipette did not affect the FRET decrease caused by the conditioning depolarization (84 ± 2%, p > 0.8) (Fig. 8F,G), suggesting that the sustained structural alteration of GABARAP is independent of microtubule. Microtubule might contribute to RP by working as a scaffold for structurally altered GABARAP to stably bind to GABAAR γ2 subunit.
Discussion
We have shown that GABARAP is a critical mediator of synaptic plasticity at inhibitory synapses on a PN. GABARAP underwent sustained structural alteration depending on the CaMKII activity. RP expression depended on binding of the structurally altered GABARAP to GABAARγ2 subunit. Binding of GABARAP and tubulin was also required for RP. Together, it is suggested that regulation of microtubule–GABARAP–GABAAR association downstream of CaMKII activation is essential for potentiation of GABAAR responsiveness in a PN.
Role of GABARAP in RP
There are two possibilities how binding of the structurally altered GABARAP to GABAARγ2 subunit potentiates GABAAR function (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). GABARAP might increase the number of surface GABAAR by facilitating transportation of intracellular GABAAR to the plasma membrane. Alternatively, GABARAP binding to GABAARγ2 might change the channel property such as single-channel conductance. Numerous studies reported that GABARAP plays roles in trafficking of GABAAR to the plasma membrane (Kneussel et al., 2000; Kittler et al., 2001; Moss and Smart, 2001; Kneussel, 2002; Nymann-Andersen et al., 2002; Leil et al., 2004; Luscher and Keller, 2004; Chen and Olsen, 2007). However, we showed that neither transfection of γ2 peptide–Venus nor that of wild-type or mutant GABARAP affected the localization pattern or the amount of surface GABAAR in a PN. GABAAR trafficking might be mainly mediated by molecules other than GABARAP in a PN. Preliminary data suggest that the amount of surface GABAAR does not increase after depolarization of a PN (data not shown). Together, GABARAP might contribute to RP not through facilitated transportation of intracellular GABAAR toward the plasma membrane, although this possibility cannot be excluded. We speculate that functional modulation of GABAARs by binding of structurally altered GABARAP is responsible for the RP expression. When GABARAP is expressed with GABAAR composed of α1, β2/3, and γ2 subunits in L929 fibroblasts, both single-channel conductance and mean open time of GABAAR are increased (Everitt et al., 2004; Luu et al., 2006). Considering that the majority of GABAAR subunits expressed in a PN are α1, β2/3, and γ2, interaction of GABARAP and GABAARγ2 might upregulate functional properties of GABAAR in a PN. We noticed GABARAP translocation from the intracellular compartment to the periplasma membrane regions and colocalization of GABARAP with GABAAR after depolarization of a PN. It is noteworthy that CaMKII directly phosphorylates GABAAR β and γ subunits (Moss and Smart, 2001; Kneussel, 2002; Luscher and Keller, 2004). CaMKII-mediated phosphorylation of GABAAR and binding of structurally altered GABARAP to γ2 subunit might synergistically bring about potentiation of GABAAR function.
We demonstrated that neuronal activity altered GABARAP conformation stably through the CaMKII activity. Thus, GABARAP seems to have at least two structural states, and the transition is controlled by the neuronal activity. Previous studies using crystallographic analysis and nuclear magnetic resonance analysis reported that GABARAP takes two conformations in the N-terminal region (Coyle et al., 2002; Stangler et al., 2002), which might correspond to the different conformations we reported here. It remains to be elucidated how GABARAP structure is altered by depolarization depending on the CaMKII activity. GABARAP seems not to contain potential phosphorylation sites by CaMKII. Thus, GABARAP conformation change might be stabilized indirectly by the CaMKII activity through phosphorylation of other molecules.
We showed that the GABARAP conformation change caused by the conditioning depolarization was sustained for >15 min and that inhibition of GABARAP association with GABAARγ2 after the induction cancelled once established RP. Thus, long-term maintenance of RP might depend on the retention of GABARAP conformation change. In addition, transient application of the γ2 peptide before and during the conditioning depolarization was sufficient to suppress the RP induction. Thus, the binding of GABAARγ2 and GABARAP may be critical for both the induction and maintenance of RP, although the possibility that the γ2 peptide was not effectively washed out in the transient application experiment cannot be excluded. Inhibition of GABARAP binding to GABAARγ2 subunit or expression of GABARAP mutant unable to associate with tubulin or to undergo conformational alteration not only completely suppressed the sustained phases of RP but also depressed the early phase. These results support the idea that GABARAP is essential for both the expression and maintenance of RP. Conversely, destruction of microtubule impaired only the sustained phase of RP but not the early phase. Thus, microtubule might work as a scaffold for sustained association of GABARAP with GABAARγ2 contributing to maintenance of RP.
We want to note that all of the present results were obtained using cultured cerebellar neurons, which is clearly advantageous for combined application of multiple techniques such as patch-clamp recording, immunocytochemistry, gene expression, and fluorescent protein imaging, including FRET measurement. However, it is not confirmed presently that the RP regulation mechanism revealed here using the culture preparation operates in vivo. It should be also noted that GABARAP is a member of microtubule-associated protein family. Another member GEC1 (GABARAP L1), which has 94% similarity in amino acids sequence to GABARAP and binds to both GABAARγ2 and tubulin, is also expressed in the cerebellum (Mansuy-Schlick et al., 2006). The γ2 peptide and expression of mutant GABARAP used in this study were likely to disturb functions of not only GABARAP but also GEC1. Therefore, GEC1 might also play roles in RP synergistically with GABARAP in a PN. Thus, knock-out of only GABARAP might not affect RP. Indeed, it was reported that GABARAP knock-out mice showed no apparent abnormality in GABAAR distribution at synapses in cortical neurons (O'Sullivan et al., 2005), despite that there are many reports demonstrating implication of GABARAP in GABAAR trafficking to the plasma membrane.
RP and its regulation
RP is heterosynaptically induced by postsynaptic depolarization caused by excitatory synaptic inputs such as those from a climbing fiber to a PN (Kano et al., 1992). We reported previously that presynaptic activation in conjunction with the postsynaptic depolarization suppresses the RP induction through postsynaptic metabotropic GABAB receptor activation (Kawaguchi and Hirano, 2000). GABAB receptor activation negatively regulates the RP induction through reduction of PKA activity. The reduced PKA activity allows calcineurin to effectively dephosphorylate DARPP-32 (dopamine and cAMP-regulated phosphoprotein-32), resulting in release of protein phosphatase-1 (PP-1) from inhibition. PP-1 attenuates and counteracts the CaMKII activity (Kawaguchi and Hirano, 2000, 2002). In this way, the induction of RP is regulated by the activities of both presynaptic and postsynaptic neurons through interaction of intracellular signaling cascades. Balancing excitatory and inhibitory inputs is critical for proper function of the nervous system. Disturbance of the balance in cerebellar circuits results in ataxia (Watanabe et al., 1998). One role of RP might be to keep the adequate balance by regulating GABAAR function, sensing frequency and amplitudes of excitatory and inhibitory synaptic inputs. It has also been reported that RP is negatively regulated by the activity of cell-adhesion molecule integrin α3β1 through Src tyrosine kinase (Kawaguchi and Hirano, 2006).
Molecules involved in RP, such as GABAARγ2 subunit, GABARAP, tubulin, and CaMKII, are widely expressed in the CNS. Thus, a similar regulatory mechanism might work at other inhibitory synapses in the CNS. Previous studies reported that Ca2+/calmodulin signaling augments the GABAergic synaptic transmission depending on the cytoskeleton in hippocampal CA1 pyramidal neurons (Wei et al., 2004). Recently, Houston and Smart (2006) reported that CaMKII augments function of GABAAR composed of α1β3γ2 subunits in cerebellar granule cells and neuroblastoma NG108-15 cells. Without γ2 subunit expression, the extent of augmentation was limited, suggesting an important role of γ2 subunits in the CaMKII-mediated upregulation of GABAAR function. The upregulation might be mediated by GABARAP association with γ2 subunit.
Synaptic plasticity at inhibitory synapses on a PN
Two forms of synaptic plasticity other than RP occur at the GABAergic synapses on a PN. One is DSI lasting for 1–2 min, which is presynaptically expressed with the decreased transmitter release probability caused by endocannabinoid release from a postsynaptic PN (Kreitzer and Regehr, 2001; Diana et al., 2002). The other is DPI expressed as the increased release probability of transmitter after the DSI (Duguid and Smart, 2004). DPI is induced by activation of NMDA type of glutamate receptors on the presynaptic terminals by retrograde release of glutamate from a PN in a slice preparation. We also observed a DPI-like phenomenon, an increase in the mIPSC frequency after the conditioning depolarization, although it lasted longer than the previous report. The difference might be ascribed to the different neuronal condition in the culture and in the slice or to the different conditioning depolarization. Compared with RP of GABA response or mIPSC amplitude, the extent of potentiation of evoked-IPSC amplitude, which reflects all of RP, DSI, and DPI, was limited at 2 min and clear at 20 min after the conditioning depolarization, presumably attributable to DSI and DPI. Multiple synaptic regulation mechanisms work at inhibitory synapses on a PN.
Footnotes
This work was supported by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (S.K., T.H.). We thank Drs. Y. Tagawa, S. Yawata, and T. Yoshida for critical reading of this manuscript and for helpful comments.
References
- Allison DW, Chervin AS, Gelfand VI, Craig AM. Postsynaptic scaffolds of excitatory and inhibitory synapses in hippocampal neurons: maintenance of core components independent of actin filaments and microtubules. J Neurosci. 2000;20:4545–4554. doi: 10.1523/JNEUROSCI.20-12-04545.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CH, Giustetto M, Huang YY, Hawkins RD, Kandel ER. Is heterosynaptic modulation essential for stabilizing Hebbian plasticity and memory? Nat Rev Neurosci. 2000;1:11–20. doi: 10.1038/35036191. [DOI] [PubMed] [Google Scholar]
- Bedford FK, Kittler JT, Muller E, Thomas P, Uren JM, Merlo D, Wisden W, Triller A, Smart TG, Moss SJ. GABA(A) receptor cell surface number and subunit stability are regulated by the ubiquitin-like protein Plic-1. Nat Neurosci. 2001;4:908–916. doi: 10.1038/nn0901-908. [DOI] [PubMed] [Google Scholar]
- Chen L, Wang H, Vicini S, Olsen RW. The gamma-aminobutyric acid type A (GABAA) receptor-associated protein (GABARAP) promotes GABAA receptor clustering and modulates the channel kinetics. Proc Natl Acad Sci USA. 2000;97:11557–11562. doi: 10.1073/pnas.190133497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Olsen RW. GABAA receptor associated proteins: a key factor regulating GABAA receptor function. J Neurochem. 2007;100:279–294. doi: 10.1111/j.1471-4159.2006.04206.x. [DOI] [PubMed] [Google Scholar]
- Chen ZW, Chang CS, Leil TA, Olcese R, Olsen RW. GABAA receptor-associated protein regulates GABAA receptor cell-surface number in Xenopus laevis oocytes. Mol Pharmacol. 2005;68:152–159. doi: 10.1124/mol.104.009878. [DOI] [PubMed] [Google Scholar]
- Coyle JE, Qamar S, Rajashankar KR, Nikolov DB. Structure of GABARAP in two conformations: implications for GABA(A) receptor localization and tubulin binding. Neuron. 2002;33:63–74. doi: 10.1016/s0896-6273(01)00558-x. [DOI] [PubMed] [Google Scholar]
- Diana MA, Bregestovski P. Calcium and endocannabinoids in the modulation of inhibitory synaptic transmission. Cell Calcium. 2005;37:497–505. doi: 10.1016/j.ceca.2005.01.020. [DOI] [PubMed] [Google Scholar]
- Diana MA, Levenes C, Mackie K, Marty A. Short-term retrograde inhibition of GABAergic synaptic currents in rat Purkinje cells is mediated by endogenous cannabinoids. J Neurosci. 2002;22:200–208. doi: 10.1523/JNEUROSCI.22-01-00200.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duguid IC, Smart TG. Retrograde activation of presynaptic NMDA receptors enhances GABA release at cerebellar interneuron-Purkinje cell synapses. Nat Neurosci. 2004;7:525–533. doi: 10.1038/nn1227. [DOI] [PubMed] [Google Scholar]
- Everitt AB, Luu T, Cromer B, Tierney ML, Birnir B, Olsen RW, Gage PW. Conductance of recombinant GABA(A) channels is increased in cells co-expressing GABA(A) receptor-associated protein. J Biol Chem. 2004;279:21701–21706. doi: 10.1074/jbc.M312806200. [DOI] [PubMed] [Google Scholar]
- Gaiarsa JL, Caillard O, Ben-Ari Y. Long-term plasticity at GABAergic and glycinergic synapses: mechanisms and functional significance. Trends Neurosci. 2002;25:564–570. doi: 10.1016/s0166-2236(02)02269-5. [DOI] [PubMed] [Google Scholar]
- Hansel C, Linden DJ, D'Angelo E. Beyond parallel fiber LTD: the diversity of synaptic and non-synaptic plasticity in the cerebellum. Nat Neurosci. 2001;4:467–475. doi: 10.1038/87419. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- Hirbec H, Francis JC, Lauri SE, Braithwaite SP, Coussen F, Mulle C, Dev KK, Coutinho V, Meyer G, Isaac JT, Collingridge GL, Henley JM. Rapid and differential regulation of AMPA and kainate receptors at hippocampal mossy fibre synapses by PICK1 and GRIP. Neuron. 2003;37:625–638. doi: 10.1016/s0896-6273(02)01191-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houston CM, Smart TG. CaMK-II modulation of GABA(A) receptors expressed in HEK293, NG108–15 and rat cerebellar granule neurons. Eur J Neurosci. 2006;24:2504–2514. doi: 10.1111/j.1460-9568.2006.05145.x. [DOI] [PubMed] [Google Scholar]
- Kandel ER. The molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001;294:1030–1038. doi: 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- Kanematsu T, Jang IS, Yamaguchi T, Nagahama H, Yoshimura K, Hidaka K, Matsuda M, Takeuchi H, Misumi Y, Nakayama K, Yamamoto T, Akaike N, Hirata M, Nakayama K. Role of the PLC-related, catalytically inactive protein p130 in GABA(A) receptor function. EMBO J. 2002;21:1004–1011. doi: 10.1093/emboj/21.5.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Rexhausen U, Dreessen J, Konnerth A. Synaptic excitation produces a long-lasting rebound potentiation of inhibitory synaptic signals in cerebellar Purkinje cells. Nature. 1992;356:601–604. doi: 10.1038/356601a0. [DOI] [PubMed] [Google Scholar]
- Kano M, Kano M, Fukunaga K, Konnerth A. Ca2+-induced rebound potentiation of γ-aminobutyric acid-mediated currents requires activation of Ca2+/calmodulin-dependent kinase II. Proc Natl Acad Sci USA. 1996;93:13351–13356. doi: 10.1073/pnas.93.23.13351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi S, Hirano T. Suppression of inhibitory synaptic potentiation by presynaptic activity through postsynaptic GABAB receptors in a Purkinje neuron. Neuron. 2000;27:339–347. doi: 10.1016/s0896-6273(00)00041-6. [DOI] [PubMed] [Google Scholar]
- Kawaguchi S, Hirano T. Signaling cascade regulating long-term potentiation of GABAA receptor responsiveness in cerebellar Purkinje neurons. J Neurosci. 2002;22:3969–3976. doi: 10.1523/JNEUROSCI.22-10-03969.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi S, Hirano T. Integrin α3β1 suppresses long-term potentiation at inhibitory synapses on the cerebellar Purkinje neuron. Mol Cell Neurosci. 2006;31:416–426. doi: 10.1016/j.mcn.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Kittler JT, Rostaing P, Schiavo G, Fritschy JM, Olsen R, Triller A, Moss SJ. The subcellular distribution of GABARAP and its ability to interact with NSF suggest a role for this protein in the intracellular transport of GABA(A) receptors. Mol Cell Neurosci. 2001;18:13–25. doi: 10.1006/mcne.2001.1005. [DOI] [PubMed] [Google Scholar]
- Kneussel M. Dynamic regulation of GABA(A) receptors at synaptic sites. Brain Res Brain Res Rev. 2002;39:74–83. doi: 10.1016/s0165-0173(02)00159-5. [DOI] [PubMed] [Google Scholar]
- Kneussel M, Haverkamp S, Fuhrmann JC, Wang H, Wassle H, Olsen RW, Betz H. The gamma-aminobutyric acid type A receptor (GABAAR)- associated protein GABARAP interacts with gephyrin but is not involved in receptor anchoring at the synapse. Proc Natl Acad Sci USA. 2000;97:8594–8599. doi: 10.1073/pnas.97.15.8594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu Y. GABAB receptors, monoamine receptors, and postsynaptic inositol trisphosphate-induced Ca2+ release are involved in the induction of long-term potentiation at visual cortical inhibitory synapses. J Neurosci. 1996;16:6342–6352. doi: 10.1523/JNEUROSCI.16-20-06342.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Cerebellar depolarization-induced suppression of inhibition is mediated by endogenous cannabinoids. J Neurosci. 2001;21(RC174):1–5. doi: 10.1523/JNEUROSCI.21-20-j0005.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurie DJ, Seeburg PH, Wisden W. The distribution of 13 GABAA receptor subunit mRNAs in the rat brain. II. Olfactory bulb and cerebellum. J Neurosci. 1992;12:1063–1076. doi: 10.1523/JNEUROSCI.12-03-01063.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. 2000;405:955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- Leil TA, Chen ZW, Chang CS, Olsen RW. GABAA receptor- associated protein traffics GABAA receptors to the plasma membrane in neurons. J Neurosci. 2004;24:11429–11438. doi: 10.1523/JNEUROSCI.3355-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher B, Keller CA. Regulation of GABAA receptor trafficking, channel activity, and functional plasticity of inhibitory synapses. Pharmacol Ther. 2004;102:195–221. doi: 10.1016/j.pharmthera.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Luu T, Gage PW, Tierney ML. GABA increases both the conductance and mean open time of recombinant GABAA channels co-expressed with GABARAP. J Biol Chem. 2006;281:35699–35708. doi: 10.1074/jbc.M605590200. [DOI] [PubMed] [Google Scholar]
- Mansuy-Schlick V, Tolle F, Delage-Mourroux R, Fraichard A, Risold PY, Jouvenot M. Specific distribution of gabarap, gec1/gabarap Like 1, gate16/gabarap Like 2, lc3 messenger RNAs in rat brain areas by quantitative real-time PCR. Brain Res. 2006;1073–1074:83–87. doi: 10.1016/j.brainres.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Moss SJ, Smart TG. Constructing inhibitory synapses. Nat Rev Neurosci. 2001;2:240–250. doi: 10.1038/35067500. [DOI] [PubMed] [Google Scholar]
- Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol. 2002;20:87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Hajos N, Somogyi P, Mody I. Increased number of synaptic GABA(A) receptors underlies potentiation at hippocampal inhibitory synapses. Nature. 1998;395:172–177. doi: 10.1038/25999. [DOI] [PubMed] [Google Scholar]
- Nymann-Andersen J, Sawyer GW, Olsen RW. Subunit specificity and interaction domain between GABA(A) receptor-associated protein (GABARAP) and GABA(A) receptors. J Neurochem. 2002;80:815–823. doi: 10.1046/j.0022-3042.2002.00762.x. [DOI] [PubMed] [Google Scholar]
- Oda Y, Kawasaki K, Morita M, Korn H, Matsui H. Inhibitory long-term potentiation underlies auditory conditioning of goldfish escape behavior. Nature. 1998;394:182–185. doi: 10.1038/28172. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci. 2004;7:1104–1112. doi: 10.1038/nn1311. [DOI] [PubMed] [Google Scholar]
- O'Sullivan GA, Kneussel M, Elazar Z, Betz H. GABARAP is not essential for GABA receptor targeting to the synapse. Eur J Neurosci. 2005;22:2644–2648. doi: 10.1111/j.1460-9568.2005.04448.x. [DOI] [PubMed] [Google Scholar]
- Rouach N, Pebay A, Meme W, Cordier J, Ezan P, Etienne E, Giaume C, Tence M. TARP gamma-8 controls hippocampal AMPA receptor number, distribution and synaptic plasticity. Nat Neurosci. 2005;8:1525–1533. doi: 10.1038/nn1551. [DOI] [PubMed] [Google Scholar]
- Sheng M, Kim MJ. Postsynaptic signaling and plasticity mechanisms. Science. 2002;298:776–780. doi: 10.1126/science.1075333. [DOI] [PubMed] [Google Scholar]
- Stangler T, Mayr LM, Willbold D. Solution structure of human GABA(A) receptor-associated protein GABARAP: implications for biological function and its regulation. J Biol Chem. 2002;277:13363–13366. doi: 10.1074/jbc.C200050200. [DOI] [PubMed] [Google Scholar]
- Takao K, Okamoto K, Nakagawa T, Neve RL, Nagai T, Miyawaki A, Hashikawa T, Kobayashi S, Hayashi Y. Visualization of synaptic Ca2+/calmodulin-dependent protein kinase II activity in living neurons. J Neurosci. 2005;25:3107–3112. doi: 10.1523/JNEUROSCI.0085-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I, Sou YS, Ezaki J, Minematsu-Ikeguchi N, Ueno T, Kominami E. HsAtg4B/HsApg4B/autophagin-1 cleaves the carboxyl termini of three human Atg8 homologues and delipidates microtubule-associated protein light chain 3- and GABAA receptor-associated protein-phospholipid conjugates. J Biol Chem. 2004;279:36268–36276. doi: 10.1074/jbc.M401461200. [DOI] [PubMed] [Google Scholar]
- Tomita S, Stein V, Stocker TJ, Nicoll RA, Bredt DS. Bidirectional synaptic plasticity regulated by phosphorylation of stargazin-like TARPs. Neuron. 2005;45:269–277. doi: 10.1016/j.neuron.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Wang H, Bedford FK, Brandon NJ, Moss SJ, Olsen RW. GABA(A)-receptor-associated protein links GABA(A) receptors and the cytoskeleton. Nature. 1999;397:69–72. doi: 10.1038/16264. [DOI] [PubMed] [Google Scholar]
- Watanabe D, Inokawa H, Hashimoto K, Suzuki N, Kano M, Shigemoto R, Hirano T, Toyama K, Kaneko S, Yokoi M, Moriyoshi K, Suzuki M, Kobayashi K, Nagatsu T, Kreitman RJ, Pastan I, Nakanishi S. Ablation of cerebellar Golgi cells disrupts synaptic integration involving GABA inhibition and NMDA receptor activation in motor coordination. Cell. 1998;95:17–27. doi: 10.1016/s0092-8674(00)81779-1. [DOI] [PubMed] [Google Scholar]
- Wei J, Zhang M, Zhu Y, Wang JH. Ca2+-calmodulin signalling pathway up-regulates GABA synaptic transmission through cytoskeleton-mediated mechanisms. Neuroscience. 2004;127:637–647. doi: 10.1016/j.neuroscience.2004.05.056. [DOI] [PubMed] [Google Scholar]