

Abstract
Evidence has been accumulated that glioblastoma cells release and exploit glutamate for proliferation and migration by autocrine or paracrine loops through Ca2+-permeable AMPA-type glutamate receptors. Here, we show that Ca2+ signaling mediated by AMPA receptor regulates the growth and motility of glioblastoma cells via activation of Akt. Ca2+ supplied through Ca2+-permeable AMPA receptor phosphorylated Akt at Ser-473, thereby facilitating proliferation and mobility. A dominant-negative form of Akt inhibited cell proliferation and migration accelerated by overexpression of Ca2+-permeable AMPA receptor. In contrast, introduction of a constitutively active form of Akt rescued tumor cells from apoptosis induced by the conversion of Ca2+-permeable AMPA receptor to Ca2+-impermeable receptors by the delivery of GluR2 cDNA. Therefore, Akt functions as downstream effectors for Ca2+-signaling mediated by AMPA receptor in glioblastoma cells. The activation of the glutamate-AMPA receptor-Akt pathway may contribute to the high degree of anaplasia and invasive growth of human glioblastoma. This novel pathway might give an alternative therapeutic target.
Keywords: brain tumor, glioblastoma, AMPA receptors, Akt, invasion, calcium signaling
Introduction
Glioblastoma (GBM) is the most common and the most malignant tumor occurring in the CNS and is notorious for its highly infiltrative and invasive behavior, which makes surgical intervention ineffective (Kleihues et al., 2000; Maher et al., 2001). The prognosis for intermediate grade glioma patients has been improved by the combination of modern therapies such as surgery, radiation, and chemotherapy, but the overall survival has not increased in patients with glioblastoma (Bailey and Cushing, 1926; Maher et al., 2001). Establishment of a curative treatment for glioblastoma will require better understanding of the molecular mechanisms underlying the proliferation, migration, and invasion of the tumor cells.
Several lines of evidence have been accumulated regarding signal transduction pathways in glioma cells. Amplification of the epidermal growth factor receptor (EGFR) gene occurs in 40–50% of glioblastomas, and the tumor cells usually show overexpression of EGFR (Smith et al., 2001). Stimulation by a growth factor causes the activation of phosphatidylinositol-3-OH kinase (PI3K), which catalyzes the conversion of phosphatidylinositol (4,5)-biphosphate (PIP2) into phosphatidylinositol (3,4,5)-triphosphate (PIP3) (Maehama and Dixon, 1998). Membrane-associated PIP3 attracts and activates the protein serine-threonine kinase Akt [also called protein kinase B (PKB)] (Cantley and Neel, 1999). Loss of a tumor suppressor gene called phosphatase/tensin homolog on chromosome 10 (PTEN) also is detected frequently in glioblastoma as well as breast, prostate, and endometrial carcinomas and melanoma (Li et al., 1997). PTEN is a lipid phosphatase, which blocks the activation of Akt by converting PIP3 to PIP2, thereby promoting cell cycle arrest and apoptosis. Loss of the PTEN function further accelerates the hyperactivation of Akt and facilitates the growth and anti-apoptotic survival of glioblastoma cells (Di Cristofano et al., 1998). Therefore, the PI3K-Akt pathway may play a key role in the anti-apoptotic survival of glioblastoma cells as well as other cancer cells (Li et al., 1997; Tamura et al., 1998; Choe et al., 2003).
The Ca2+ influx mediated by NMDA-type glutamate receptors or voltage-dependent Ca2+ channels activates Akt independently of the PI3K pathway, which in turn phosphorylates the bclII-associated death promoter and protects cells from apoptosis (Koike et al., 1989; Datta et al., 1997; Yano et al., 1998). We have shown previously that activation of Ca2+-permeable AMPA-type glutamate receptors facilitates the migration and proliferation of human glioblastoma cells (Ishiuchi et al., 2002). AMPA receptors mediate fast neurotransmission in most excitatory synapses in the CNS (Seeburg, 1993; Hollmann and Heinemann, 1994; Ozawa et al., 1998) and consist of subunits taken from a set of four proteins, GluR1–4 (Seeburg, 1993; Hollmann and Heinemann, 1994). The Ca2+ permeability of AMPA receptors depends on the subunit composition: receptors with the GluR2 subunit exhibit little permeability, whereas those without GluR2 exhibit high permeability. Abundance of the GluR2 subunit has been shown to decrease Ca2+ permeability (Burnashev et al., 1992; Bochet et al., 1994; Geiger et al., 1995). The unique properties of GluR2 can be traced to a single amino acid residue in the second hydrophobic segment. This residue is arginine (R) in GluR2 and glutamine (Q) in the other subunits. Replacement of the arginine with glutamine at this critical site (Q/R site) causes the homomeric receptors assembled from the mutant GluR2Q to show high Ca2+ permeability (Seeburg, 1993; Hollmann and Heinemann, 1994). Interestingly, a glutamine codon (CAG) is encoded at the Q/R position in genomic sequences for the GluR1 through GluR4 subunits, whereas an arginine codon (CGG) is found only in the cDNA sequence of the GluR2 subunit. This adenosine-to-guanosine change resulting from an RNA editing process is developmentally controlled. Although GluR2 exists exclusively in the edited form in the adult brain, the unedited and edited forms coexist in the fetal brain (Burnashev et al., 1992) and glioblastoma tissues and cell lines (Maas et al., 2001; Yoshida et al., 2006). We showed previously that glioblastoma cells express Ca2+-permeable AMPA receptors assembled mainly from the GluR1 and/or GluR4 subunits, although some primary cultured cells coexpress GluR2 (Ishiuchi et al., 2002). Both GluR2-lacking, and GluR2Q-assembling, forms of the AMPA receptor are Ca2+ permeable, and the Ca2+ influx through these receptors may contribute to the invasive and aggressive growth behavior of glioblastomas (Maas et al., 2001; Ishiuchi et al., 2002). However, no link has been established between Ca2+-triggered signaling through glutamate receptors and activation of Akt.
The present study investigated whether Akt functions as a downstream effector molecule of Ca2+-permeable AMPA receptors to promote cell growth and mobility in human glioblastoma cells.
Materials and Methods
Surgical specimens and cell cultures.
The surgical specimens examined in this study were histologically identified as glioblastoma multiforme, anaplastic astrocytoma (AA), and diffuse astrocytoma according to the World Health Organization classification (Kleihues et al., 2000). Cell cultures were prepared as described previously (Ishiuchi et al., 1998). Cells were cultivated in Eagle’s minimal essential medium (Nissui, Tokyo, Japan) supplemented with 10% FBS and 2 mm glutamine. The concentration of glutamate in this medium, which was referred to thereafter as control media, was determined by enzymatic cycling as 107 ± 5 μm (n = 4) (Sakurai and Okada, 1992). Glutamate-free media were constituted with DMEM (low glucose) (Invitrogen, San Diego, CA) supplemented with 10% dialyzed FBS. The concentration of glutamate in this medium was <5 μm (n = 4).
Amino acid analysis.
Glutamate concentrations in tissue culture media (1 ml) in which cultured glial cells (∼0.5 × 105 cells, or 0.5 mg per culture) were incubated for 48 h under control media, glutamate-free media, media containing 100 μm glutamate, and media containing 100 μm glutamate together with 100 μm GYKI-52466, a noncompetitive AMPA receptor antagonist. Samples were stored at 60°C until use. For determination of glutamate levels, 300 μl of each culture medium was mixed with 450 μl of 3.89% sulfosalicylic acid in a vortex-mixer for deproteination and was stirred and centrifuged (9000 × g) at 4°C for 10 min. The clear supernatant (30 μl) was applied to L-8500 amino acid analyzer (Hitachi, Japan, Tokyo). By using lithium citrate buffers with ninhydrin as derivatization agent and fluorescence detection (emission at 440 nm for proline and hydroxyproline, and emission at 570 nm for other amino acids), excellent separation of 41 amino acids was obtained in the lower range (∼1.0–9.0 nmol/ml). Samples were analyzed in triplicate.
Gene transfer using adenoviral vectors.
We constructed the following recombinant adenoviruses: (1) AxCALNLGluR1, AxCALNLGluR2, and AxCALNLGluR2Q, expression-switching units for expression of the rat GluR1, GluR2, and GluR2Q subunits, respectively, consisting of the CAG promoter, a loxP sequence, a stuffer gene (neo-resistance gene), a poly (A) signal, a second loxP site, the GluR1, GluR2, or GluR2Q coding sequence, and another poly (A) signal [rat GluR1 and GluR2 cDNAs were generous gifts from Drs. Stephan F. Heinemann (Salk Institute, La Jolla, CA) and M. Hollmann (Ruhr University, Bochum, Germany)]; (2) AxCANCre and AxCAGFP, recombinant adenoviruses for the expression of Cre recombinase and green fluorescent protein (GFP) (Clontech, Cambridge, UK), respectively; and (3) AxCALNLmyr-Akt-HA and AxCALNLmyr-Akt-AA, the cDNA fragments comprising the entire coding region for human Akt1 were isolated from human embryonic kidney 293 cDNA by PCR. Constitutively active Akt, which lacks the pleckstrin homology domain (amino acid 4–129) but has the src-myristoylation signal sequence (MGSSKSKPKDPSQRR) (Resh, 1994) fused to its N-terminal end and a hemagglutininepitope (HA) tag at its C-terminal end (Kohn et al., 1996a) was prepared by PCR. Akt-AA (rat Akt1 T308A/S473A), which also contains HA tag in its N terminal and works as a dominant-negative mutant of Akt (Kitamura et al., 1998), was kindly provided by Drs. M. Kasuga and W. Ogawa (Kobe University, Kobe, Japan). The recombinant adenoviral vectors were constructed by the COS-terminal protein complex method (Miyake et al., 1996). Cells were infected at a multiplicity of infection of five with the recombinant adenoviruses 2–5 d before experiments except for AxCALNLmyr-Akt-AA, which was used at a multiplicity of infection of 20.
Reverse transcription-PCR.
Aliquots of 100 ng of RNA prepared from samples were reverse-transcribed with random haxamers and the reverse transcription (RT) product was subjected to 35 PCR cycles (94°C, 20 s; 50°C, 20 s; 72°C, 20 s) with primers that simultaneously amplified GluR1–4 (Bochet et al., 1994) (sense primer, CCTTTGGCCTATGAGATCTGGATGTG; antisense primer, TCGTACCACCATTTGTTTTTCA). A second PCR (35 cycles) for subunit-specific amplification was performed with the sense primers specific for GluR1 (AAGAGGGACGAGACCAGACAAC at 1712–1733; position 1 being the first nucleotide of the coding sequence), GluR2 (GAAGATGGAAGAGAAACA-CAAAGT at 1732–1755), GluR3 (GGAAGACAACAATGAAGAACCTC at 1749–1771), or GluR4 (GAAGGACCCAGCGACCAGCC at 1747–1766) and the common antisense primer that was used for the first amplification. The products of this second amplification (637 bp for GluR1, 638 bp for GluR2, 657 bp for GluR3, and 626 bp for GluR4) were digested by restriction enzymes specific for GluR1 (BglI), GluR2 (Bsp1286 I), GluR3 (AvaI), and GluR4 (PvuII).
Analysis of RNA editing.
The PCR was performed in 20 μl containing 1 μl of first-strand cDNA. The reaction was performed for 20 cycles (94°C, 30 s; 49°C, 2 min) with GluR2-specific primer and AMPA receptor-common lower primer. PCR products were digested with Q/R site-specific restriction enzymes, BbvI, and separated by agarose gel electrophoresis (Yoshida et al., 2006).
Western blot analysis.
Samples (∼100 mg) were homogenized in 200 μl of whole-cell lysis buffer (0.1% SDS, 1% deoxycholic acid, 30 mm Tris, pH 7.6, 1% Triton X-100, 1 mm phenylmethyl-sulfonyl-fluoride, and 1 mm EDTA) mixed with 20 μl of protease inhibitor mixture (Boehringer Mannheim, Indianapolis, IN), and then lysed on ice. Protein samples (20 μg) were subjected to 10% SDS-PAGE and transferred to poly-vinylidene difluoride membrane. Primary antibodies for phospho-phosphoinositide-dependent kinase-1 (PDK-1) (Ser-241), PDK-1, Akt, Phospho-Akt (Ser-473), and Phospho-Akt (Thr-308) (Cell Signaling Technology, Beverly, MA) were used at 1:1000 dilutions. Secondary peroxidase-conjugated antibody (Dako, High Wycombe, UK) was used at 1:2000, and the GE Healthcare (Arlington Heights, IL) ECL system was used for visualizing the antibody-bound protein.
Histology and immunofluorescence.
Indirect immunofluorescence staining was performed as described previously (Ishiuchi et al., 2002). Selective antibodies against GluR1, GluR2, GluR2/GluR3, GluR4 (Chemicon, Temecula, CA), and vimentin (V9; Dako) were used. For dual immunofluorescence, FITC-, rhodamine-, Alexa 594-labeled, and Texas red secondary antibodies (Invitrogen, Eugene, OR) were used to visualize the bound antibodies. The stained cells were viewed with a laser-scanning confocal microscope (MRC-1024E, Bio-Rad, Hercules, CA; Pascal LSM5, Zeiss, Thornwood, NY). DNA counterstaining was performed with 10 μg/ml propidium iodide (PI) or 5 μg/ml 4,6-diamidino-2-phenylindole (DAPI). For detection of fluorescence intensity of pAkt immunostaining, 11 cross sections taken at 0.45 μm spacing were collected, and images were analyzed by Pascal Quick operation version 3.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling, proliferation, and migration assays.
The terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) assay was performed using an In Situ Cell Death Detection kit (MBL, Tokyo, Japan), and the cell proliferation assay was performed using anti-Ki-67 monoclonal antibody (Dako) staining indices as described previously (Ishiuchi et al., 2002). The migration assay was performed as described previously (Ishiuchi et al., 2002). YM872 was kindly provided by Astellas Pharmaceutical (Tokyo, Japan).
Animal models.
CGNH-89 cells were suspended at 107/100 μl in culture medium. Cell suspensions (1 × 107/100 μl) were injected subcutaneously into the flank of 5- to 6-week-old nude mice (body weight, 18–20 g). Adenoviruses, either AxCALNLmyr-Akt-HA plus AxCANCre or AxCALNLAkt-AA plus AxCANCre, and either AxCALNLGluR2 plus AxCANCre or AxCALNLGluR2Q plus AxCANCre (1 × 107 pfu; diluted in a total of 100 μl of PBS), were administered intratumorally once 5 d after tumor inoculation with a 27-gauge needle. AxCALNLmyr-Akt without AxCANCre or AxCALNLGluR2 without AxCANCre was administered as a control. Tumor volume was calculated according to the formula (length × width2)/2. At the end of each experiment, tumor tissues were subjected to histological analysis. The Animal Care and Experimentation Committee of Gunma University, Showa Campus, approved all animal experiments. The Ethical Committee of Gunma University, Showa Campus, approved experiments using human tissues derived from glioma patients.
Data analysis.
Data are expressed as the mean ± SEM. Statistical comparisons were performed with an unpaired t test or one-way ANOVA (Scheffe’s test for post hoc comparison).
Results
Glutamate stimulates cell proliferation and migration
Glutamate, a principal excitatory neurotransmitter in the CNS, stimulates proliferation and migration of neuronal progenitors and immature neurons in the developing brain (Ikonomidou et al., 1999), and the biology of proliferative glioblastoma cells is reminiscent of cell division, migration, and proliferation of neural stem cells (Sanai et al., 2005). Therefore, we examined the influence of glutamate on tumor proliferation using a human glioblastoma cell line (CGNH-89), which expresses Ca2+-permeable AMPA receptors as well as other cell lines and primary cultures (Ishiuchi et al., 2002).
To examine how glutamate influences tumor growth, we maintained glioblastoma cells in the control medium for 12 h after plating and then cultured the cells in glutamate- and glutamine-free medium supplemented with 10% dialyzed FBS (referred to as the glutamate-free medium) for 2 d with or without 100 μm glutamate. The culture medium was changed daily in each case. We estimated the proliferative activity of CGNH-89 cells using the MIB-1 (Ki-67) staining index and estimated the apoptotic index using the TUNEL assay. Deprivation of glutamate from the culture medium markedly suppressed the proliferative activity of tumor cells and prompted apoptosis (Fig. 1A). The Ki-67 staining and apoptotic indices were 16.0 ± 6.0% (n = 3) and 3.0 ± 0.5% (n = 3), respectively, in the control medium and <0.1% (n = 3; p < 0.001) and 18.0 ± 2.5% (n = 3; p < 0.001), respectively, in the glutamate-free medium. The addition of 100 μm glutamate to the glutamate-free medium restored the Ki-67 staining index to 18.5 ± 3.0% (n = 3; p < 0.001) and reduced the apoptotic index to 3.0 ± 2.0% (n = 3; p < 0.001) (Fig. 1A). The further addition of 20 μm 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo (F)-quinoxaline (NBQX), an antagonist of AMPA receptors, markedly inhibited the effect of glutamate on proliferation, reduced the Ki-67 staining index to 2.0 ± 1.0% (n = 3; p < 0.001), and increased the apoptotic index to 13.0 ± 2.0% (n = 3; p < 0.05) (Fig. 1A). In the presence of NBQX, most tumor cells exhibited flat morphologies under the phase-contrast microscope (Fig. 1A). Another AMPA antagonist, [2,3-dioxo-7-(1H-imidazol-1-yl)-6-nitro-1,2,3,4-tetrahydroquinoxalin-1-yl] acetic acid monohydrate (YM872) (Kohara et al., 1998), also inhibited the effect of glutamate on cell proliferation. In the presence of both 100 μm YM872 and 100 μm glutamate, the Ki-67 staining index was 4.0 ± 1.0% (n = 3; p < 0.001), and the apoptotic index was 20.0 ± 2.0% (n = 3; p < 0.05).
Figure 1.
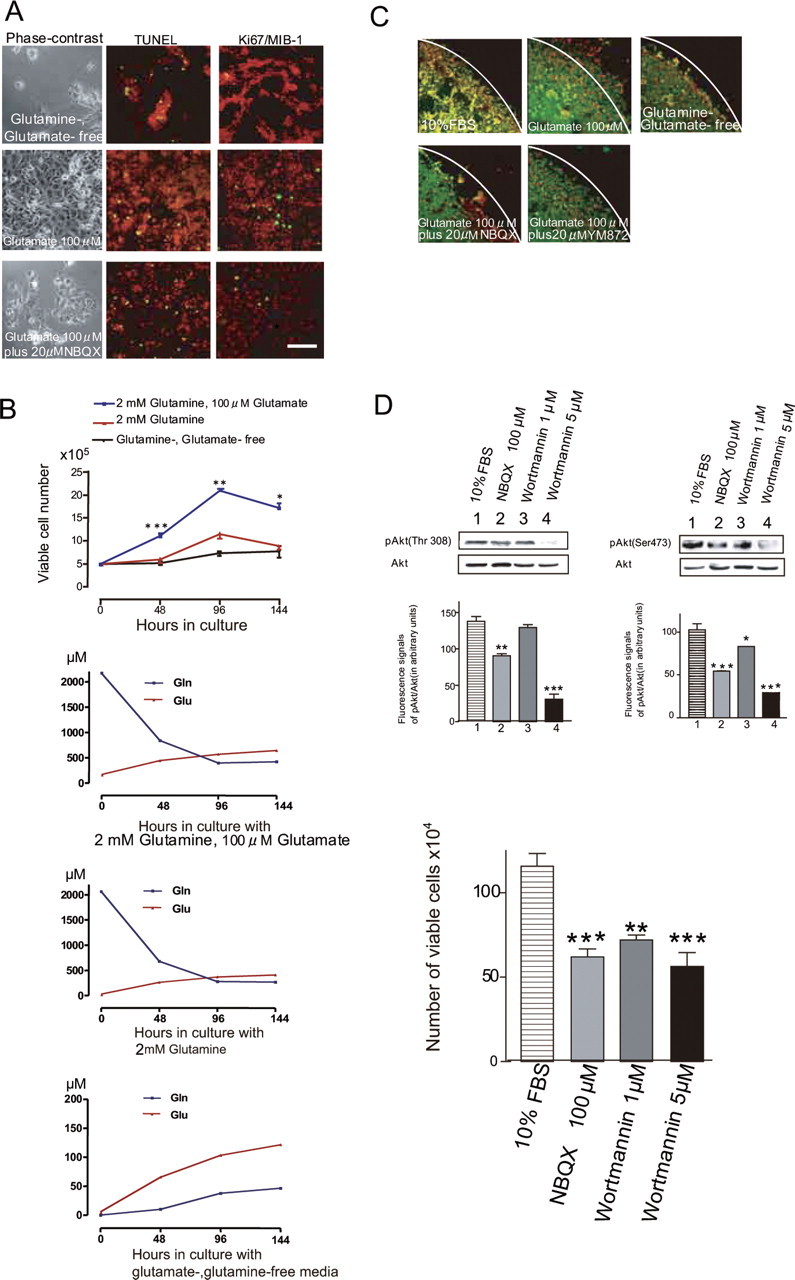
Effects of glutamate on tumor cell proliferation and migration. A, Human glioblastoma cells (CGNH-89) cultured in glutamine-, glutamate-free medium (top row), with 100 μm glutamate (middle row), or 100 μm glutamate plus 20 μm NBQX (bottom row) for 2 d. Phase-contrast micrographs (left column) and results of TUNEL staining (FITC, green; center column) and Ki-67/MIB-1 staining (FITC, green; right column) are shown. The DNA was counterstained with PI (red). Scale bar (in A): A, 50 μm; C, 100 μm. B, Growth curve of viable cells cultured in glutamine-, glutamate-free medium (black line), with 2 mm glutamine (red line), or 100 μm glutamate plus 2 mm glutamine (blue line). Cells (5 × 105) plated in the medium containing 10% FBS were incubated for 12 h, and then the medium was changed to either glutamine-, glutamate-free medium or medium containing 2 mm glutamine or 100 μm glutamate plus 2 mm glutamine for 6 d, respectively. Viable cells were counted after Trypan blue staining. Glutamate significantly promoted tumor cell proliferation (n = 3; *p < 0.05, **p < 0.01, ***p < 0.001). The bottom three graphs showed that determination of extracellular glutamate (glu) and glutamine (gln) in control medium, medium containing 2 mm glutamine, or glutamine-, glutamate-free medium maintained without medium change for 6 d (n = 3). C, Motility assays using a cloning ring. The culture immediately following the removal of the cloning ring after incubation in the medium containing 10% FBS for 24 h (top left) and migratory cells incubated in glutamine-, glutamate-free medium, with or without 100 μm glutamate, or 100 μm glutamate plus 20 μm NBQX or 20 μm YM872 at 24 h after the removal of the cloning ring are shown. Cells were stained with monoclonal antibody for vimentin (FITC, green), and nuclei were counterstained with PI (red). White lines indicate the border of the ring. D, Immunoblotting with anti-phospho-Akt (Thr-308), anti-phospho-Akt (Ser-473), and anti-Akt antibodies for glioblastoma cells, which shows the effects of 100 μm NBQX, 1 μm wortmannin, or 5 μm wortmannin on phosphorylation of Akt. The relative fluorescent signal intensity of pAktThr308 to Akt, and that of pAktSer473 to Akt (in arbitrary units) for each treated cell (n = 3 each) is shown in the graphs below the blots. The amount of Akt phosphorylated at Thr-308 was significantly reduced in cells treated with 5 μm wortmannin (***p < 0.001) compared with the control. The amount of Akt phosphorylated at Ser-473 was significantly reduced in cells treated with 100 μm NBQX (***p < 0.001), 1 μm wortmannin (*p < 0.05), and 5 μm wortmannin (***p < 0.001) compared with the control. E, Effects of NBQX and wortmannin on cell growth. Cells (5 × 105) plated in the medium containing 10% FBS were incubated for 12 h, and then the medium was changed and 100 μm NBQX, 1 μm wortmannin, or 5 μm wortmannin was added for 2 d. Viable cells were counted after Trypan blue staining. A significant decrease in growth was found in cells treated with 100 μm NBQX (n = 3; ***p < 0.001), 1 μm wortmannin (n = 3; **p < 0.01), and 5 μm wortmannin (n = 3; ***p < 0.001) compared with the control.
To confirm the effect of glutamate on cell proliferation, we counted the number of viable cells using Trypan blue staining. Cells were incubated in the glutamate-free medium for 6 d, and the effects of the addition of 2 mm glutamine or 100 μm glutamate plus 2 mm glutamine were tested. A substantial increase in cell number was found in the medium containing 100 μm glutamate. In contrast, the addition of only 2 mm glutamine had little effect on cell proliferation (Fig. 1B). These results indicate that glutamate facilitated the proliferation of tumor cells and inhibited apoptosis by activating AMPA receptors. We next examined glutamate accumulation in media of cultured cells by HPLC. The presence of glutamate significantly influences cell growth at the start of growth assays. Even in the glutamate- and glutamine-free condition, glioma cells release a significant amount of glutamate (>100 μm; 6 d in vitro) as well as a glutamate precursor glutamine, and that released glutamate stimulates gradual cell proliferation in autocrine and paracrine loops (Fig. 1B).
We next examined the effect of glutamate on the mobility of tumor cells using a cloning ring (7 mm in diameter) in the center of the culture dish. Cells were cultured in the control medium inside the ring, which was removed 24 h after plating. The cells were then exposed to glutamate-free medium during the next 24 h, and the number of cells that crossed the border of the cloning ring was counted. The addition of 100 μm glutamate markedly increased the number of cells that crossed the border from 33.2 ± 5.3 (n = 5) to 331.2 ± 123.3 (n = 5; p < 0.001) (Fig. 1C). The further addition of either 100 μm NBQX or 100 μm YM872 reduced the number of cells crossing to 38.2 ± 5.3 (n = 5) and 40.2 ± 9.3 (n = 5; p < 0.001), respectively. These results indicate that glutamate regulated cell migration by activating AMPA receptors.
AMPA antagonists inhibit phosphorylation of Akt in glioblastoma cells
Akt is a well-established survival factor exerting anti-apoptotic activity, and its dysregulation is implicated in tumorigenesis (Feng et al., 2004). Glioblastoma cells grow vigorously in medium with 10% FBS containing ∼100 μm glutamate as well as cytokines and growth factors (Sasaki et al., 1998). Multiple pathways stimulated by these factors concomitantly activate Akt (Datta et al., 1997; Cantley and Neel, 1999). The activation of Akt requires phosphorylation at Thr-308 and Ser-473 (Alessi and Cohen, 1998; Williams et al., 2000).
We first examined the extent of phosphorylation at Thr-308 of Akt in glioblastoma cells using an antibody against anti-phosphorylated Akt, which detects Akt only if phosphorylated at this site. Cells cultured in the 10% FBS-containing medium had both phosphorylated and unphosphorylated Akt at Thr-308. The extent of phosphorylation was estimated by calculating the fluorescence intensity of the phosphorylated Akt (pAktThr308) stained with the specific antibody relative to that of total Akt. Wortmannin (5 μm), a specific inhibitor of PI3K, significantly reduced the relative amount of pAktThr308/Akt (in arbitrary units) from 136.3 ± 12.4 (n = 3) to 31.0 ± 13.8 (n = 3; p < 0.001), although 1 μm wortmannin had no significant effect (129.3 ± 12.5; n = 4) (Fig. 1D). The addition of 100 μm NBQX significantly reduced this amount (98.6 ± 11.2; n = 3; p < 0.01).
We next examined the extent of phosphorylation at Ser-473 using an antibody against anti-phosphorylated Akt, which detects phosphorylated Akt at this site. Wortmannin at 5 μm reduced markedly the relative amount of pAktSer473/Akt (in arbitrary units) from 102.3 ± 14.0 (n = 3) to 28.0 ± 5.0 (n = 3; p < 0.001) (Fig. 1D). Wortmannin at 1 μm also had a significant effect (83.0 ± 2.0; n = 3; p < 0.05). Addition of 100 μm NBQX caused a significant decrease in this amount to 54.5 ± 4.0 (n = 3; p < 0.001). LY294002, another specific inhibitor for PI3K, suppressed the activation of Akt at Ser-473 in a similar manner to wortmannin. The fluorescent intensities in arbitrary units of pAktSer473/Akt in cells treated with 20 μm LY294002, 100 μm LY294002, and control cells were 110.0 ± 8.0, 38.0 ± 8.5, and 126.0 ± 5.0, respectively (n = 3). Therefore, high doses of the PI3K inhibitors (100 μm LY294002 and 5 μm wortmannin) significantly suppressed the phosphorylation of Akt at Ser-473 (p < 0.001).
To test the anti-proliferative activity of NBQX or wortmannin, we measured the number of viable cells using Trypan blue staining in cell cultures treated with each agent for 2 d. Treatment with 100 μm NBQX (n = 3; p < 0.001), 1 μm wortmannin (n = 3; p < 0.01), or 5 μm wortmannin (n = 3; p < 0.001) significantly reduced the number of viable cells (Fig. 1E), indicating that the suppression of Akt at Ser-473 was inversely correlated with the rate of tumor growth. The TUNEL assay revealed that 100 μm NBQX and 5 μm wortmannin increased the apoptotic indices from 3.0 ± 1.5 to 20.0 ± 5.0% (n = 3; p < 0.001) and 17.0 ± 8.0% (n = 3; p < 0.001), respectively.
GluR2 inhibits but GluR2Q stimulates activation of Akt at Ser-473
To test the effect of Ca2+ influx through AMPA receptors mediated by activation of Akt, we attempted to convert Ca2+-permeable AMPA receptors into Ca2+-impermeable receptors by adenoviral-mediated transfer of GluR2 cDNA (Iino et al., 2001; Ishiuchi et al., 2001). Two recombinant adenoviruses were constructed, one for expression of Cre recombinase (AxCANCre) (Kanegae et al., 1996) and another with a switching unit for expression of GluR2 (AxCALNLGluR2). We also constructed a recombinant adenovirus with GFP cDNA (AxCAGFP) for marking adenovirus-infected cells (Fig. 2A).
Figure 2.

Effects of expression of GluR2 and GluR2Q on cell morphology and phosphorylation of Akt. A, Cultured cells (CGNH-89) infected with AxCAGFP (top row) for 4 d. The DNA was stained with PI. GFP fluorescence was detected in almost all cells. At 4 d postinfection with AxCANCre alone, cells were stained with anti-vimentin antibody (FITC, green) and anti-phospho Akt antibody (Ser-473) (Texas red; red, bottom row). Merged images are also provided (right column). B, Cultured cells (CGNH-89) infected with AxCALNLGluR2 and AxCANCre for expression of GluR2. Cells were stained with anti-GluR2 antibody (FITC, green) and anti-phospho Akt antibody (Ser-473) (Texas red; red, top row), and anti-vimentin antibody (FITC, green) and anti-phospho Akt antibody (Ser-473) (Texas red; red, bottom row). Merged images are also provided (right column). C, Cultured cells (CGNH-89) infected with AxCALNLGluR2Q and AxCANCre for expression of GluR2Q. Cells were stained with anti-GluR2 antibody (FITC, green) and anti-phospho Akt antibody (Ser-473) (Texas red; red, top row), and anti-vimentin antibody (FITC; green) and anti-phospho Akt antibody (Ser-473) (Texas red; red, bottom row). Merged images are also provided (right column). Scale bar: A (top), 200 μm; A (bottom), 100 μm; B, C, 50 μm. D, Immunoblotting with anti-phospho-Akt (Thr-308), anti-phospho-Akt (Ser-473), and anti-Akt antibodies for glioblastoma cells, showing the effects of the adenoviral-mediated expressions of GluR2, GluR2Q, and GluR1 with or without the addition of wortmannin on phosphorylation of Akt. For the control, cells cultured in the medium containing 10% FBS were infected with AxCALNLGluR2Q without AxCANCre (vector). Cells were infected with AxCALNLGluR2, AxCALNLGluR2Q, and AxCALNLGluR1, respectively, together with AxCANCre to express GluR2, GluR2Q, and GluR1. Immunoblotting was performed at 4 d postinfection. Effects of wortmannin were tested by adding at either 1 or 5 μm during these periods. The relative fluorescent signal intensity of pAktSer473 to Akt and that of pAktThr308 to Akt (in arbitrary units) for each treated cell (n = 3 each) are provided (right column). The amount of Akt phosphorylated at Ser-473 was significantly reduced by expression of GluR2 or 5 μm wortmannin compared with the control but increased by expression of GluR2Q regardless of the presence of wortmannin (***p < 0.001). The amount of Akt phosphorylated at Thr-308 was significantly reduced only if the cells were treated with 5 μm wortmannin (***p < 0.001), and this effect of wortmannin was reversed by expression of GluR2Q (n = 3; *p < 0.05, **p < 0.01, ***p < 0.001).
Human glioblastoma cells have high affinity for recombinant adenoviruses, and almost all cells were infected at a multiplicity of infection of five. The adenoviral-mediated delivery of GluR2 cDNA induced flattening of the cell shape and cytoskeletal disorganization as revealed by immunofluorescence microscopy using an antibody for vimentin (Fig. 2B). Immunofluorescence analysis using anti-phospho-Akt (Ser-473) and anti-GluR2 antibodies showed that GluR2 was localized in the perinuclear cytoplasm, and the phosphorylation of Akt was inhibited almost completely, consistent with the result of Western blotting (Fig. 2D). These results indicate that the suppression of the Ca2+ influx through AMPA receptors inhibited the phosphorylation of Akt.
To confirm the involvement of Ca2+-permeable AMPA receptors in the activation of Akt in glioblastoma cells, we transferred GluR2Q cDNA into glioblastoma cells using the recombinant adenoviruses AxCANCre and AxCALNLGluR2Q. Cells with transferred GluR2Q cDNA became ubiquitously immunopositive for an antibody to pAkt (Ser-473) (Fig. 2C). Cell processes and cell bodies were stained more markedly as revealed by immunofluorescence analysis for pAkt (Ser-473) compared with those of control cells (Fig. 2A,C). Western blotting showed that the introduction of GluR2Q cDNA into cells increased the phosphorylation of Akt (Ser-473) compared with the control (146.3 ± 7.5 vs 84.3 ± 4.4, fluorescent intensity in arbitrary units; n = 3; p < 0.001) (Fig. 2D). The transfer of GluR1 cDNA increased the phosphorylation of Akt (Ser-473) (128.0 ± 11.0; n = 3; p < 0.001), whereas the transfer of GluR2 cDNA reduced it significantly (54.5 ± 3.1; n = 3; p < 0.001) (Fig. 2D). Interestingly, the phosphorylation of Akt (Ser-473) induced by overexpression of GluR2Q was not inhibited by wortmannin even at 5 μm, a concentration that normally markedly inhibited phosphorylation of Akt (53.5 ± 5.0 vs 146.3 ± 11 U; n = 3; p < 0.001) (Fig. 2D), indicating that the activation of Akt at Ser-473 caused by Ca2+ influx through AMPA receptors was independent of PI3K.
The adenoviral-mediated transfer of GluR2 cDNA, GluR2Q cDNA, and GluR1 cDNA had no significant effects on the phosphorylation of Akt at Thr-308 (81.0 ± 23.4, 79.8 ± 17.4, and 87.0 ± 2.5 U, respectively; n = 3), although 5 μm wortmannin caused significant reduction compared with the control (71.5 ± 5.7 vs 28.0 ± 3.2 U; n = 3; p < 0.001) (Fig. 2D). In the presence of 5 μm wortmannin, the delivery of GluR2Q restored the phosphorylation of Akt at Thr-308 to the control level (69.0 ± 24.0 U; n = 3) (Fig. 2D). These results indicated that the Ca2+ influx through AMPA receptors restored the phosphorylation of Akt at Thr-308 to the basal level even when PI3K was inhibited by wortmannin.
Akt stimulates the extension of cell processes
The expression of AMPA receptors causes a marked change in the morphology of both normal astrocytes and neoplastic glial cells (Ishiuchi et al., 2001, 2002). To clarify whether the phosphorylation of Akt is involved in the AMPA receptor-mediated morphological changes in glioblastoma cells, we tested the effect of the expression of a dominant-negative form of Akt (Akt-AA) (Takata et al., 1999; Namikawa et al., 2000) on the cell shape of the tumor cells. From days 5–7 postinfection with AxCANCre and AxCALNLmyr-Akt-AA for the delivery of Akt-AA cDNA, the cells gradually exhibited polygonal or round morphology. Dual immunofluorescence labeling for vimentin and pAkt revealed disrupted vimentin filaments and sparsely distributed pAkt in the Akt-AA-expressing cells (Fig. 3A). Similar changes were observed after the transfer of GluR2 cDNA (Fig. 2A). To estimate the morphological change quantitatively, we calculated the percentage of cells with processes longer than the diameter of the long axes of the cell bodies in randomly selected 10 high-power fields (200×) and defined the mean value as the morphological index. This index was 53.5 ± 7.5% (n = 6) in control cells and was markedly reduced to 10.5 ± 5.5% (n = 6; p < 0.001) in Akt-AA-expressing cells. In contrast, the adenoviral-mediated expression of the constitutively active form of Akt (Akt-HA) (Takata et al., 1999; Namikawa et al., 2000) caused elongation of the cellular processes (Fig. 3A), similar to that induced by the overexpression of GluR2Q. The morphological index defined in cells at 5 d postinfection with AxCANCre and AxCALNLmyr-Akt-HA for the delivery of Akt-HA cDNA was 85.6 ± 1.3% (n = 6), significantly greater than that in control cells (p < 0.01).
Figure 3.

Effects of expression of constitutively active and dominant-negative forms of Akt on cell morphology, proliferation, and migration. A, Cultured cells (CGNH-89) infected with AxCALNLmyr-Akt without AxCANCre (vector for control; top row), AxCALNLmyr-Akt-AA and AxCANCre (for dominant-negative form of Akt; Akt-AA; middle row), or AxCALNLmyr-Akt-HA and AxCANCre (for constitutively active form of Akt; Akt-HA; bottom row). Cells were stained with anti-vimentin antibody (FITC, green) and anti-pAktSer473 antibody (Texas red; red). Merged images are also provided (right column). B, Triple immunofluorescence for anti-pAktSer473 antibody (Texas red; red), anti-HA-tag antibody (FITC, green), and DAPI (blue) for nuclear staining in cells after adenoviral-mediated transfer of only the vector, Akt-HA cDNA, and Akt-AA cDNA, respectively. Control cells were stained positively for pAkt with moderate intensity and exhibited healthy nuclear staining. Cells infected with AxCALNLmyr-Akt-AA and AxCANCre had no phosphorylated Akt and exhibited nuclear blebbing and apoptotic bodies with HA-tag staining. Akt-HA cDNA-transferred cells showed dense staining for pAkt together with HA-tag. C, Effects of expression of Akt-HA and Akt-AA on cell motility. Cells (5 × 105) plated with 100 μl of 10% FBS in a cloning ring were incubated for 12 h and then infected with AxCALNLmyr-Akt-HA without AxCANCre (vector for control), AxCALNLmyr-Akt-AA with AxCANCre (expression of Akt-AA), or AxCALNLmyr-Akt-HA with AxCANCre (expression of Akt-HA) for 3 d. At 48 h after removal of the cloning ring, cells were stained with DAPI. White lines indicate the border of the ring. Scale bar (in C): A, 100 μm; B, 50 μm; C, 150 μm.
Akt-HA stimulates tumor cell proliferation, and Akt-AA induces apoptosis
In the NG108 neuroblastoma cell line, the Ca2+ influx through the NMDA receptors activates Ca2+/calmodulin-dependent protein kinase kinase, which in turn phosphorylates Akt (Yano et al., 1998). The activation of Akt is known to release various anti-apoptotic signals, thereby facilitating cell survival (Datta et al., 1997). Thus, we examined the biological effect of Akt-HA cDNA or Akt-AA cDNA on the proliferation of glioma cells by estimating mitotic indices in a high-power field (200×) after PI nuclear staining. The mitotic indices, calculated as the number of mitotic cells per field for only vector (control), Akt-AA, and Akt-HA were 6.0 ± 1.2, 2.3 ± 1.3, and 12.5 ± 1.7, respectively. We estimated the proliferative potential with a monoclonal antibody for Ki-67. The Ki-67 indices were 17.1 ± 7.3, 5.3 ± 2.1, and 28.0 ± 5.4% in cells expressing GFP, iAkt-AA, and Akt-HA, respectively. These results indicate that activation of Akt stimulated tumor cell proliferation (p < 0.001), whereas inactivation inhibited proliferation (p < 0.001). We also examined the anti-apoptotic effect of Akt using DAPI staining (Fig. 3B). The numbers of apoptotic cells per field (200×) were 6.3 ± 1.0, 17.0 ± 5.1, and 0.3 ± 0.5 (n = 3 each) in the cells expressing GFP, Akt-AA, and Akt-HA, respectively. Therefore, the activation of Akt significantly affected the number of apoptotic tumor cells (p < 0.001).
Akt activation promotes tumor cell migration
Elongation of the cellular processes is considered to be the first step in cell migration, followed by translocation of the cell soma and retraction of the rear processes (Mitchison and Cramer, 1996). Because the activation of Akt induced cytoskeletal reorganization, we examined the effects of adenoviral-mediated expression of Akt-AA and Akt-HA on the motile activity of the tumor cells using the cloning ring. Cells were cultured inside the ring, which was then removed 72 h after viral infection. The number of cells that crossed the border of the cloning ring during the next 48 h was counted. The mean number of cells in three experiments was 355 ± 121, 120 ± 51, and 1320 ± 341 for cells expressing GFP, Akt-AA, and Akt-HA, respectively (Fig. 3C). Therefore, the expression of Akt-AA significantly suppressed (p < 0.001), whereas the expression of Akt-HA promoted, cell mobility (p < 0.001).
Akt functions as a downstream molecule of AMPA receptors
The above results suggest that Akt functions as a downstream effector for Ca2+ signaling mediated by AMPA receptors in glioblastoma cells. To test this idea, we examined whether the retraction of cellular processes induced by the adenoviral-mediated delivery of GluR2 was overcome by the simultaneous expression of the constitutive active form of Akt-HA. In contrast to the flat shape with retracted processes in cells with only GluR2 cDNA, the protrusion of cellular processes was seen in most cells with both Akt-HA and GluR2 cDNA (Fig. 4A). The morphological index was 11.3 ± 6.3% (n = 4) in cells expressing GluR2, but 87.3 ± 3.1% in cells expressing both GluR2 and Akt-HA (n = 4; p < 0.001). Therefore, Akt-HA overcame the effects of GluR2 on the shapes of the tumor cells.
Figure 4.

Reversal of effects of GluR2 and GluR2Q by expression of Akt-HA and Akt-AA, respectively. A, Reversal of effects of GluR2 by expression of Akt-HA. Cultured cells (CGNH-89) were infected with either AxCALNLGluR2 and AxCANCre (top row) or AxCALNLmyr-Akt-HA, AxCALNLGluR2, and AxCANCre (bottom row). Cells were stained with anti-GluR2 antibody (FITC; green) and anti-pAktSer473 (Texas red; red). Scale bar, 50 μm. B, Reversal of effects of GluR2Q by expression of Akt-AA. Cultured cells (CGNH-89) were infected with either AxCALNLGluR2Q and AxCANCre (top row) or AxCALNLmyr-Akt-AA, AxCALNLGluR2Q, and AxCANCre (bottom row). Cells were stained with anti-GluR2 antibody (FITC; green) and anti-pAktSer473 antibody (Texas red; red). Scale bar, 50 μm. C, Promotion of the motility of cells expressing GluR2 by the Akt-HA. Migration assays were conducted for cells expressing GFP (left), GluR2 (center), and GluR2 together with Akt-HA (right). Cells were doubly stained by anti-pAktSer473 antibody (Texas red; red) and anti-GluR2 antibody (FITC; green). Nuclei were stained by DAPI. Scale bars: C–E, 75 μm. D, Inhibition of the motility of cells expressing GluR2Q by the Akt-AA. Migration assays were conducted for cells expressing GFP (left), GluR2Q (center), and GluR2Q together with Akt-AA (right). Cells were stained by anti-vimentin monoclonal antibody (Texas red; red, left) and doubly stained by anti-vimentin monoclonal antibody (Texas red; red) and anti-GluR2 polyclonal antibody (FITC; green, center and right). Nuclei were stained by DAPI. E, No effect of wortmannin on GluR2Q-mediated promotion of cell motility. Migration assays were conducted for cells expressing GFP (left), GFP in the presence of 5 μm wortmannin (center), and GluR2Q in the presence of 5 μm wortmannin (right). Cells were stained with anti-vimentin monoclonal antibody (Texas red; red, left and center), and doubly stained by anti-vimentin monoclonal antibody (Texas red; red) and anti-GluR2 polyclonal antibody (FITC; green, right). Nuclei were stained by DAPI.
We next examined whether the introduction of Akt-AA to these cells inhibits the morphological changes induced by GluR2Q. Cells were infected either with only GluR2Q or both Akt-AA and GluR2Q cDNA. At day 5 postinfection, most of the latter cells exhibited flat shapes with retracted cell processes (Fig. 4B). The morphological index was 85.0 ± 4.0% (n = 6) in cells expressing GluR2Q but 10.0 ± 4.0% in cells expressing both GluR2Q and Akt-AA (n = 6; p < 0.001). This result further supports the idea that Akt acts downstream of the AMPA receptors in the maintenance of cell shapes.
We also tested whether the above idea is applicable to cell migration and proliferation. The introduction of Akt-HA cDNA into GluR2-expressing cells significantly activated cell migration. The mean number of migrated cells 24 h after the removal of the ring and 72 h after viral infection was 125 ± 24, 15 ± 5, and 535 ± 55 for cells expressing GFP, GluR2, and GluR2 plus Akt-HA, respectively (n = 3; p < 0.001) (Fig. 4C). The mitotic indices, calculated as the number of mitotic cells per field (200×), were 5.0 ± 1.0, 0.5 ± 0.6, and 16.3 ± 5.0 for GFP-expressing cells, GluR2-expressing cells, and GluR2 plus Akt-HA-expressing cells, respectively (n = 4; p < 0.001). The apoptotic cells per field (200×) were 5.0 ± 1.7, 18.5 ± 3.0, and 2.3 ± 1.3 in cells expressing GFP, GluR2, and GluR2 plus Akt-HA, respectively (n = 4; p < 0.001). Therefore, the introduction of Akt-HA rescued tumor cells from apoptosis induced by the adenovirus-mediated transfer of the GluR2 cDNA.
We also introduced Akt-AA into cells expressing GluR2Q and examined the effect on cell migration and proliferation. The mean number of migrated cells 24 h after removal of the ring and 72 h after viral infection was 235 ± 57 for cells expressing GluR2Q, and 15 ± 5 for cells expressing GluR2Q plus Akt-AA (n = 4; p < 0.001) (Fig. 4D), indicating that the introduction of Akt-AA cDNA into GluR2Q-expressing cells significantly inhibited migration. The apoptotic index was also increased in cells expressing GluR2Q plus Akt-AA (15.0 ± 2.3; n = 4) compared with cells expressing only GluR2Q (4.5 ± 0.6; n = 4; p < 0.001). Therefore, the Akt-AA inhibited migration and induced apoptosis in cells expressing GluR2Q. Together, these results indicate that Akt functions as a downstream effector molecule in AMPA receptor-mediated Ca2+ signaling.
GluR2Q promotes motility of cells treated with wortmannin
Akt controls cell migration as a part of the PI3K signaling pathway (Kim et al., 2001; Lefranc et al., 2005). Expression of GluR2Q stimulates Akt activation, so we examined whether GluR2Q influences the inhibition of cell migration caused by PI3K in-hibitors. Cells were cultured inside the ring, which was then removed 48 h after viral infection by either AxCAGFP or AxCALNLGluR2Q plus AxCANCre. The cells were incubated for an additional 24 h in the presence or absence of 5 μm wortmannin, and the number of cells that crossed the border of the cloning ring was counted. The introduction of GluR2Q cDNA increased cell motility as reported previously (Ishiuchi et al., 2002). Treatment with 5 μm wortmannin inhibited cell migration, and the number of flat polygonal cells was increased. The mean number of migrated cells 24 h after the removal of the ring in three experiments was 174 ± 39, 56 ± 17, and 325 ± 57 for GFP-expressing cells, GFP-expressing cells treated with 5 μm wortmannin, and GluR2Q-expressing cells treated with 5 μm wortmannin, respectively (Fig. 4E). These results indicate that the treatment of wortmannin suppressed cell migration (p < 0.001), and cells expressing GluR2Q facilitated cell migration regardless of the presence of wortmannin (p < 0.001). We next examined the anti-apoptotic effect of GluR2Q on wortmannin-treated cells. The number of apoptotic cells per field (200×) was 3.5 ± 1.3 (n = 6), 18.3 ± 6.4 (n = 6), and 4.3 ± 1.0 (n = 6) in GFP-expressing cells, GFP-expressing cells treated with 5 μm wortmannin, and GluR2Q-expressing cells with 5 μm wortmannin, respectively, indicating that GluR2Q rescued wortmannin-treated cells from apoptosis (n = 6; p < 0.01).
Glutamate-AMPA receptor-Akt signaling in other cell lines and primary cultures
To examine the molecular heterogeneity of AMPA subunit expression in GBM, AA, low-grade astrocytoma (LGA), and normal astrocytes, we conducted immunohistochemical staining with selected antibodies against GluR1, GluR2, GluR2/3, and GluR4 on 18 surgical samples (17 GBMs, one AA), 12 cell cultures (four GBMs, four AAs, two LGAs, and two normal astrocytes), and five glioblastoma cell lines. We also performed RT-PCR studies with primers specific for GluR1–4 followed by PCR amplification on five surgical samples (four GBMs, one AA), 12 primary cultures (four GBMs, four AAs, two LGAs, and two normal astrocytes), and five glioblastoma cell lines. The amplified products were then investigated by restriction analysis with enzymes specific for each of the GluR1–4 fragments. Immunohistochemically, all tumor cells expressed AMPA receptor subunits, regardless of tumor grade. Above all, samples derived from high-grade astrocytomas (GBM and AA) had a tendency of abundant expression of GluR1 and/or GluR4, and that of GluR2 was moderate or weak. Expression of AMPA subunits by RT-PCR analysis is completely consistent with immunohistochemical results and summarized in Table 1. Analysis of RNA editing of GluR2 mRNA using Q/R site-specific reaction enzymes revealed that five surgical specimens expressed GluR2 mRNA, of which one GBM and one AA sample coexpressed GluR2Q and GluR2R. One AA and one GBM cell line (U87MG) expressed both GluR2R and GluR2Q mRNA in 10 cell cultures and two cell lines examined. Figure 5A shows a representative result obtained in a primary culture of glioblastoma cells (GNS- 3314). In this case, expression of GluR1 mRNA was dominant, but all GluR1–4 mRNAs were detected. In control brain tissues adjacent to the tumor, all GluR1–4 mRNAs were also detected (Fig. 5B). Whether glutamate-AMPA receptor-Akt signaling observed in CGNH-89 cells was adopted in low-grade astrocytoma and anaplastic astrocytoma cells, we next examined the influence of glutamate on phosphorylated-Akt (Ser473) expression by modulating extracellular glutamate concentrations using primary culture cells (two AAs, two LGAs). Figure 6, A (GNS-3901, diffuse astrocytoma) and D (GNS-6330, anaplastic astrocytoma), shows a representative AMPA subunit expression by immunofluorescence for GluR1, GluR2, GluR2/3, or GluR4. By using these cultures, we performed bioassays of cell proliferation in changing culture condition with control media, media containing 100 μm glutamate, glutamate-free media, or media containing 100 μm glutamate plus 100 μm GYKI-52466. We found that glutamate enhanced not only Akt activation but also stimulated tumor cell proliferation in both low-grade astrocytomas (Fig. 6B,C) and anaplastic astrocytomas (Fig. 6E,F). Glutamate release in extracellular space was detected even in a glutamate-free condition in cell cultures derived from AAs and LGAs. We next examined the influence of glutamate on Akt phosphorylation, cell proliferation, and cell migration in a glioblastoma cell line (U87-MG) and four primary glioblastoma cell cultures. Figure 7 showed a representative image of Akt phosphorylation and cell migration in cultured cells incubated with control media, glutamate-free medium, media containing 100 μm glutamate, or media containing 100 μm glutamate plus 100 μm GYKI-52466 in a GBM cell line (U87-MG) (Fig. 7A–C) and a primary culture of GBM (GNS-3314) (Fig. 7D–F). Glutamate influences Akt phosphorylation and cell migration as well as cell proliferation. Moreover, glutamate accumulation in glutamate-free media was observed in GBM cell cultures, both in cell lines and primary cultures.
Table 1.
Expression of AMPA receptor subunits in human glioma
| Immunohistochemistry | RT-PCR | E | Histology | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | R2/R3 | R4 | R1 | R2 | R3 | R4 | |||
| Surgical specimen | ||||||||||
| GNS-3000 | +++ | + | + | ++ | GBM | |||||
| GNS-3114 | ++ | ++ | ++ | +++ | + | + | + | + | Q/R | GBM |
| GNS-3148 | ++ | +/− | +/− | ++ | GBM | |||||
| GNS-3186 | ++ | + | + | ++ | GBM | |||||
| GNS-3195 | ++ | + | + | ++ | GBM | |||||
| GNS-3199 | ++ | + | + | ++ | GBM | |||||
| GNS-3210 | +++ | + | + | +++ | GBM | |||||
| GNS-3226 | ++ | + | + | ++ | GBM | |||||
| GNS-3243 | ++ | ++ | ++ | +++ | GBM | |||||
| GNS-3245 | ++ | + | + | ++ | + | + | + | + | R | GBM |
| GNS-3262 | ++ | + | + | ++ | GBM | |||||
| GNS-3275 | ++ | + | + | ++ | GBM | |||||
| GNS-3296 | ++ | + | + | ++ | GBM | |||||
| GNS-3314 | +++ | + | + | +++ | + | + | + | + | R | GBM |
| G-22 | + | − | − | + | GBM | |||||
| G-87535 | ++ | − | − | + | GBM | |||||
| GNS-4167 | +++ | ++ | ++ | ++ | + | + | + | + | R | GBM |
| GNS-828 | ++ | ++ | + | ++ | + | + | + | + | Q/R | AA |
| Cell culture | ||||||||||
| GNS-3245 | +++ | − | + | + | + | − | + | + | GBM | |
| GNS-3296 | ++ | + | + | ++ | + | + | + | + | R | GBM |
| GNS-3302 | +++ | + | + | +++ | + | + | + | + | R | GBM |
| GNS-3314 | +++ | ++ | ++ | +++ | + | + | + | + | R | GBM |
| G-98 | +++ | + | + | + | + | + | + | + | R | All |
| GNS-3901 | ++ | + | ++ | ++ | + | + | + | + | R | All |
| GNS-6330 | ++ | + | ++ | ++ | + | + | + | + | R | AA |
| GNS-828 | ++ | ++ | + | ++ | + | + | + | + | Q/R | AA |
| G-151 | +++ | + | + | + | + | + | + | + | R | AA |
| GNS-2093 | ++ | ++ | + | ++ | + | + | + | + | R | AA |
| Cerebellar astrocytes | + | − | − | + | + | − | − | + | a | |
| Cerebral astrocytes | − | + | + | − | − | + | + | − | R | b |
| Cell line | ||||||||||
| CGNH-89 | ++ | − | − | ++ | + | − | − | + | GBM | |
| CGNH-NM | ++ | − | + | − | + | − | + | − | GBM | |
| CGNH-PM | +++ | − | − | + | + | − | − | + | GBM | |
| U87-MG | + | + | ++ | − | + | + | + | − | Q/Rc | GBM |
| C6 | ++ | ++ | + | ++ | + | + | + | + | R | GBM |
In immunohistochemistry, +++, ++, +, +/−, and − indicate high, moderate, low, faint, and no expression, respectively. In RT-PCR, + and − indicate that the digested bands were detected and not detected, respectively. Editing status (E) of GluR2 mRNA is shown as R (edited form) or Q/R (unedited and edited form). GBM, AA, and All indicate glioblastoma, astrocytoma, and astrocytoma grade II, respectively.
aRat cerebellar fusiform Bergmann glia-like cells (Ishiuchi et al., 2001).
bRat cerebral flat type 1-like astrocytes (McCarthy and de Vellis, 1980).
cReported by Yoshida et al. (2006).
Figure 5.

RT-PCR analysis with specific primers for GluR1–4 in tumor cells in a primary culture (GNS-3314) (A) and in normal brain tissue adjacent to the tumor before resection (B). In both A and B, the top and bottom four lanes (R1–R4) show electrophoresis of PCR products before and after digestion by restriction enzymes specific for each GluR1–4 DNA, respectively. The size of digested fragments on the right lanes is as follows: 189 and 448 bp for GluR1 cut by BglI, 368 and 270 bp for GluR2 cut by Bsp1286 I, 420 and 237 bp for GluR3 cut by AvaI, and 379 and 247 bp for GluR4 cut by PvuII. Lane in the center shows DNA size marker with the dense 500 bp band.
Figure 6.
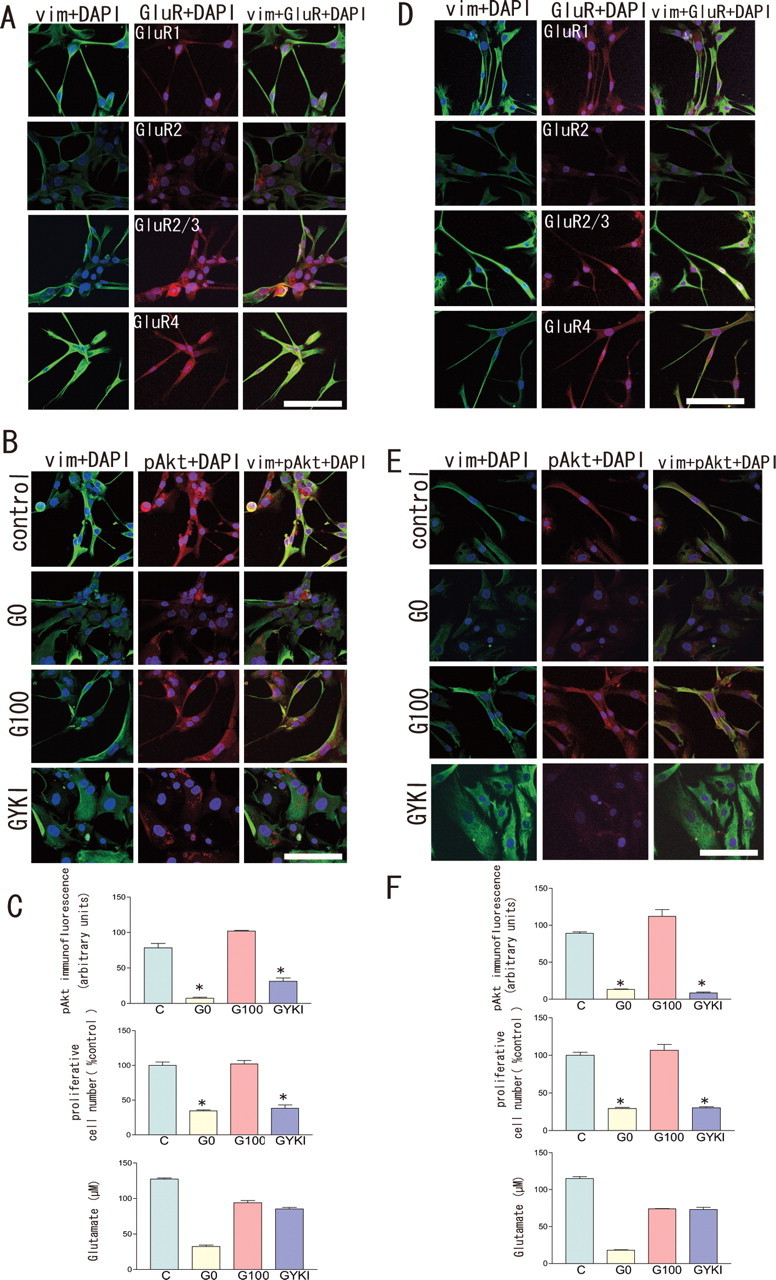
Expression of AMPA receptor subunits and effect of glutamate on Akt activation and cell proliferation in low-grade astrocytoma and anaplastic astrocytoma cell cultures. A, D, GNS-3901 (diffuse astrocytoma) (A) and GNS-6330 (anaplastic astrocytoma) (D) showing a representative expression of dual immunofluorescence for GluR1, GluR2, GluR2/3, or GluR4 (Alexa 594; red) together with vimentin (FITC; green), respectively. Scale bar, 100 μm. B, E, GNS-3901 (diffuse astrocytoma) (B) and GNS-6330 (anaplastic astrocytoma) (E) showing a representative image of Akt phosphorylation in cultured cells incubated with control media or glutamine-, glutamate-free medium (G0), medium containing 100 μm glutamate (G100), or medium containing 100 μm glutamate together with 100 μm GYKI-52466 (GYKI) for 2 d, immunostained with anti-vimentin (FITC; green), anti-phospho Akt (Ser-473) antibody (Texas red; red) together with DAPI (blue) for nuclear staining, respectively. Scale bar, 50 μm. GNS-3901 (diffuse astrocytoma) (C) and GNS-6330 (anaplastic astrocytoma) (F) showing intensity of Akt immunofluorescence, cell number, and extracellular glutamate concentrations under conditions with control media (C) or glutamine-, glutamate-free medium (G0), medium containing 100 μm glutamate (G100), or medium containing 100 μm glutamate plus 100 μm GYKI-52466 (GYKI) for 2 d, respectively (n = 3; *p < 0.05).
Figure 7.
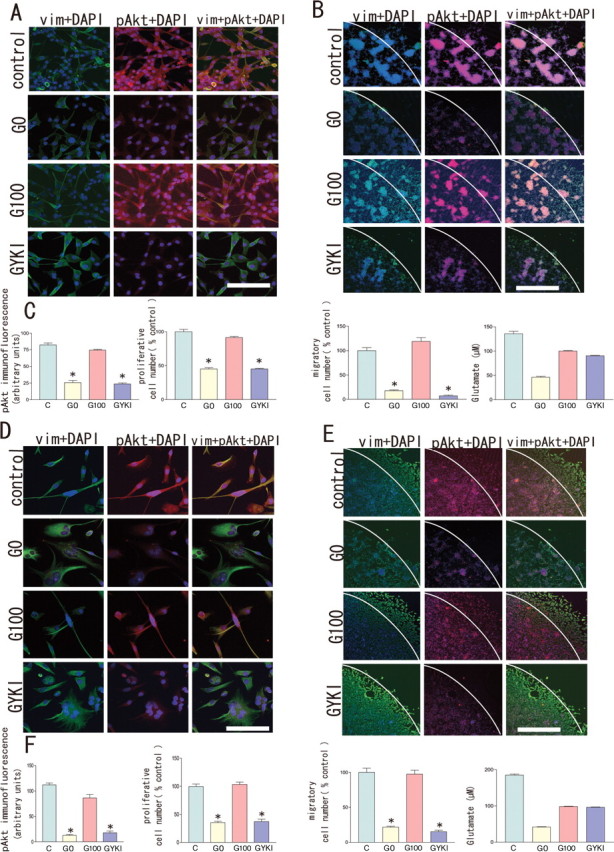
Effect of glutamate on Akt activation, cell proliferation, and cell migration in glioblastoma cell culture. A, D, U87-MG (a human glioblastoma cell line) (A) and GNS-3314 (a primary culture of GBM) (D) showing a representative image of Akt phosphorylation in cultured cells incubated with control medium, glutamine-, glutamate-free medium (G0), medium containing 100 μm glutamate (G100), or medium containing 100 μm glutamate plus 100 μm GYKI-52466 (GYKI), immunostained with anti-vimentin (FITC; green), anti-phospho Akt (Ser-473) antibody (Texas red; red) together with DAPI (blue) for nuclear staining, respectively. Scale bar, 50 μm. B, E, U-87MG (B) and GNS-3314 (E) showing cell motility assays. Cultured cells were incubated with control media, glutamine-, glutamate-free medium (G0), medium containing 100 μm glutamate (G100), or medium containing 100 μm glutamate plus 100 μm GYKI-52466 (GYKI). At 48 h after removal of the cloning ring, cells were stained with anti-vimentin (FITC; green), anti-phospho Akt (Ser-473) antibody (Texas red; red) together with DAPI (blue) for nuclear staining, respectively. Scale bar, 200 μm. C, F, U87-MG (C) and GNS-3314 (F) showing intensity of Akt immunofluorescence, cell number compared with control, number of migratory cells outside the ring, and extracellular glutamate concentrations under conditions with control media (C), glutamine-, glutamate-free medium (G0), medium containing 100 μm glutamate (G100), or medium containing 100 μm glutamate plus 100 μm GYKI-52466 (GYKI) for 2 d, respectively (n = 3; *p < 0.05).
These results indicate that the glutamate-AMPA receptor-Akt pathway plays an important role for tumor growth in all glioblastomas, anaplastic astrocytomas, and low-grade astrocytomas. These results obtained by primary cultures and other cell lines were consistent with those of the CGNH-89 cell line.
Autophosphorylation of PDK-1 in glioblastoma cells
The phosphorylation of Akt is critically regulated at Thr-308 in a kinase domain and Ser-473 in a C-terminal regulatory alignment (Datta et al., 1999). PDK-1 has been demonstrated to be the upstream kinase for Thr-308 of the Akt molecule. PDK-2, a not yet fully identified Ser-473 kinase, eventually causes the activation of Akt (Vanhaesebroeck and Alessi, 2000). Full activation of this molecule is considered to require the phosphorylation of both Thr-308 and Ser-473 sites, and the phosphorylation of Thr-308 is a crucial step for the full activation of Akt (Toker and Newton, 2000). Because PDK-1 is a pivotal upstream kinase that stimulates activation at Thr-308, we examined how PDK-1 activity is regulated when human glioblastoma cells are treated with GluR2 and/or Akt-HA cDNA, or GluR2Q and/or Akt-AA cDNA. LY294002 (20 or 100 μm) is a potent inhibitor of PI3K and had a similar anti-proliferative effect to wortmannin (1 or 5 μm). Ubiquitous PDK-1 phosphorylation was detected in all cells except those treated with LY294002, which significantly reduced the activation of PDK-1 at 20 or 100 μm (77.8 ± 21.0 and 35.3 ± 13.0, fluorescent intensity of pPDK-1/PDK-1 in arbitrary units; n = 3; p < 0.01 and p < 0.001, respectively) compared with the control (104.4 ± 11.0 U; n = 3) (Fig. 8). These findings indicate that the activation of Akt is not regulated exclusively by PDK-1 in human glioblastoma cells.
Figure 8.

Effects of manipulating the activities of AMPA receptors, Akt, and PI3K on phosphorylation of PDK-1. Immunoblotting with anti-phospho-PDK-1 (Ser-241) and anti-PDK-1 antibody in cells after the adenoviral transfer of GluR2 cDNA, GluR2 cDNA plus Akt-HA cDNA, GluR2Q cDNA, GluR2Q cDNA plus Akt-AA cDNA, Akt-HA cDNA, or Akt-AA cDNA. AxCALNLmyr-AKT-HA without AxCANCre was used for the control (vector). The effects of LA294002, a potent inhibitor of PI3K, at 20 and 100 μm were also tested. The relative fluorescent signal intensity of pPDK-1 to PDK-1 (in arbitrary units) for each treated cell is provided in the graph below. Phosphorylated PDK-1 was seen in all cells examined and was affected only by the inhibitor of PI3K (n = 3; **p < 0.01, ***p < 0.001).
Constitutively active Akt stimulates tumor growth, whereas dominant-negative Akt reduces tumor size in vivo
Finally, we estimated the effect of Akt on tumor growth in vivo by quantitative assessment of whether the manipulation of Akt-HA and Akt-AA alters tumor growth by grafting CGNH-89 cells into the subcutaneous tissue of nude mice (Fig. 9). The adenoviral-mediated expression of Akt-AA significantly reduced the rate of tumor growth as well as the size of the tumor at 22 d after grafting, compared with the control (p < 0.001). The expression of Akt-HA accelerated tumor growth and increased the size of the tumor at 22 d after grafting (p < 0.001) (Fig. 9A). Macroscopically, the introduction of Akt-HA increased the incidence of intratumoral hemorrhage (10 of 12) compared with the control (5 of 12) and Akt-AA (0 of 12) groups (Fig. 9B). Histological analysis revealed the presence of a larger number of mitotic cells in 10 randomly selected high-power fields (200×) in the Akt-HA group (50 ± 5) (p < 0.001) compared with tumor tissues treated with only the vehicle (20 ± 3) or the Akt-AA group (10 ± 7) (Fig. 9B). A high degree of tumor anaplasia, including nuclear and cytoplasmic pleomorphism, mitosis, and multinucleated giant cells, was evident in Akt-HA-expressing cells (Fig. 9B). These cells were markedly immunopositive for pAktSer473, whereas cells overexpressing Akt-AA were immunonegative for pAktSer473 (Fig. 9C).
Figure 9.
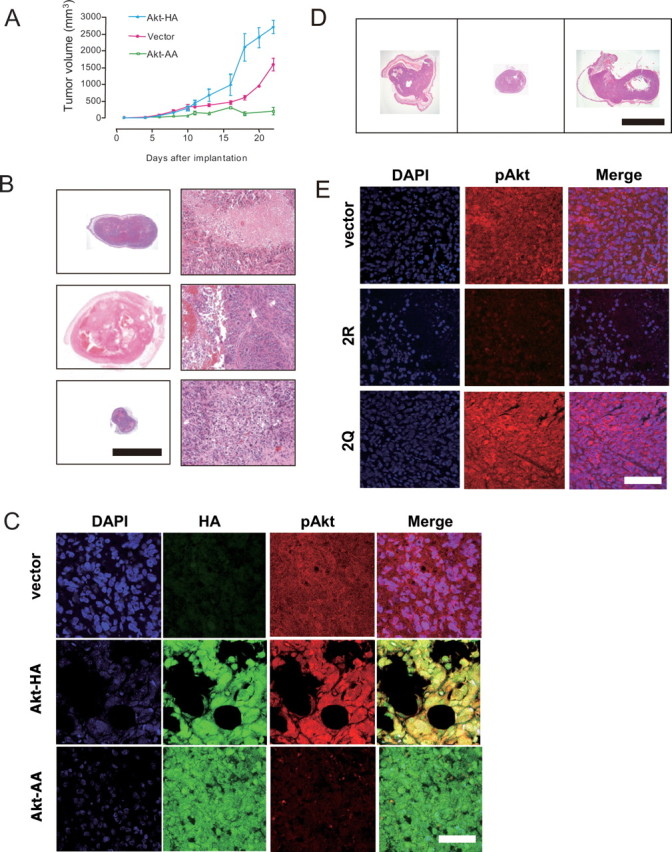
Effects of manipulating Akt activities on tumor growth in vivo. A, Effects of adenoviral transfer of Akt-HA cDNA and Akt-AA cDNA on growth rates of tumors grafted into the subcutaneous tissue of nude mice: injection of AxCALNLmyr-Akt-HA without AxCANCre (vector for control; red line); AxCALNLmyr-Akt-HA with AxCANCre (expression of Akt-HA; blue line); and AxCALNLmyr-Akt-AA with AxCANCre (expression of Akt-AA, green line). For each treatment, 12 animals were used. Each plot represents the mean ± SEM (n = 12) of the tumor volume. B, Histology of tumor tissues treated with only the vector (top row), Akt-HA cDNA (middle row), or Akt-AA cDNA (bottom row). Macroscopic (left column) and microscopic (right column) views of HE staining are shown. Akt-HA-expressing tumors exhibited a high degree of anaplasia with several large, thrombosed tumor vessels. Scale bars: left column, 1 cm; right column, 100 μm. C, Immunostaining with anti-HA-tag antibody (left column, green; FITC) and anti-pAktSer473 antibody (center column, red; Texas red) for tumor tissues treated with only the vector (top row), Akt-HA cDNA (middle row), and Akt-AA cDNA (bottom row). Merged images are also provided (right column). The Akt-HA-expressing tumor (middle row) exhibited bizarre giant cells and showed a high degree of Akt activation. The Akt-AA-expressing tumor (bottom row) exhibited diminished Akt activation. Scale bar, 50 μm. D, Macroscopic images of tumor tissues treated with only the vector (left row), GluR2 cDNA (middle row), or GluR2Q cDNA (right row). HE staining is shown. Scale bar, 1 cm. E, Immunostaining with an anti-pAktSer473 antibody (center column, red; Texas red) for tumor tissues treated with only the vector (top row), GluR2R cDNA (middle row), and GluR2Q cDNA (bottom row), together with nuclear staining with DAPI (left column, blue). Merged images are also provided (right column). Scale bar, 50 μm.
In this nude mice model, we also confirmed that the phosphorylation of Akt at Ser-473 was enhanced in cells with transferred GluR2Q cDNA (n = 6), whereas it was diminished in cells with transferred GluR2 cDNA (n = 6) (Fig. 9D,E). Histological analysis revealed the decline of a number of mitotic cells in 10 randomly selected high-power fields (200×) in the GluR2 cDNA transferred group (7.0 ± 3.0; p < 0.01) compared with tumor tissues treated with only the vehicle (65 ± 13) or the GluR2Q cDNA transferred group (75 ± 7). Apoptotic indices of the GluR2 cDNA transferred group (33.5 ± 7.5%; n = 6) was significantly increased compared with those of control (15.5 ± 3.5%; n = 6; p < 0.05) or the GluR2Q cDNA transferred group (7.0 ± 2.0%; n = 6; p < 0.001).
Discussion
The identification of Akt as a key regulator of cellular survival has significant implications for current glioma biology (Datta et al., 1999; Choe et al., 2003). Combined activation of Ras and Akt in neural progenitors induced glioblastoma formation in mouse glioma models (Holland et al., 2000). Elevated Ras activity and the phosphorylated form of Akt, as well as the deletion of PTEN, which downregulates Akt signaling, has been demonstrated in surgical specimens derived from human gliomas (Guha et al., 1997; Stambolic et al., 1998; Wu et al., 1998). Therefore, deletion of active PTEN and overexpression of active Ras, combined with the overexpression of active PI3K, renders cancer cells resistant to apoptosis by blocking adaptive cellular apoptosis through the hyperactivation of Akt. Investigation of human glioblastoma models is necessary to clarify the full involvement of the cascade of Akt signaling in tumor growth and invasion. The novel cell line, CGNH-89, used in this study is a valuable tool, because it exhibits all histological hallmarks of human glioblastomas (Ishiuchi et al., 2002). Using CGNH-89 cells and primary human glioblastoma cultures, both of which possess Ca2+-permeable AMPA receptors, we examined the potential role of the AMPA receptor as an effector molecule for the regulation of Akt activity. Both NBQX (100 μm), an AMPA antagonist, and wortmannin (5 μm), an inhibitor of PI3K, dephosphorylated Akt at both Thr-308 and Ser-473. Wortmannin at 5 μm significantly abolished the phosphorylation of Akt at Ser-473 as well as Thr-308. However, adenoviral-mediated transfer of GluR2Q cDNA followed by treatment with 5 μm wortmannin resulted in a twofold increase in Akt phosphorylation at Ser-473 compared with the control. Moreover, there was no significant difference in the activation of Akt between cells with GluR2Q cDNA in the presence and absence of 5 μm wortmannin, indicating that the phosphorylation of Akt at Ser-473 mediated by Ca2+-permeable AMPA receptors occurs independently of PI3K. Similarly, overexpression of GluR1 activated Akt. In contrast, transfer of GluR2 cDNA resulted in the inactivation of Akt at Ser-473. Moreover, there was no difference in the activation of Akt at Thr-308 between the cells with GluR2 cDNA and GuR2Q cDNA. Consistent with these findings, the phosphorylation of PDK-1 was similar between cells with GluR2 cDNA and GluR2Q cDNA. These findings strongly suggest that Ca2+-permeable AMPA receptors induce the activation of Akt directly at Ser-473, and blockade of these receptors causes Akt inactivation, resulting in induction of cell apoptosis and inhibition of cell migration (Fig. 10).
Figure 10.
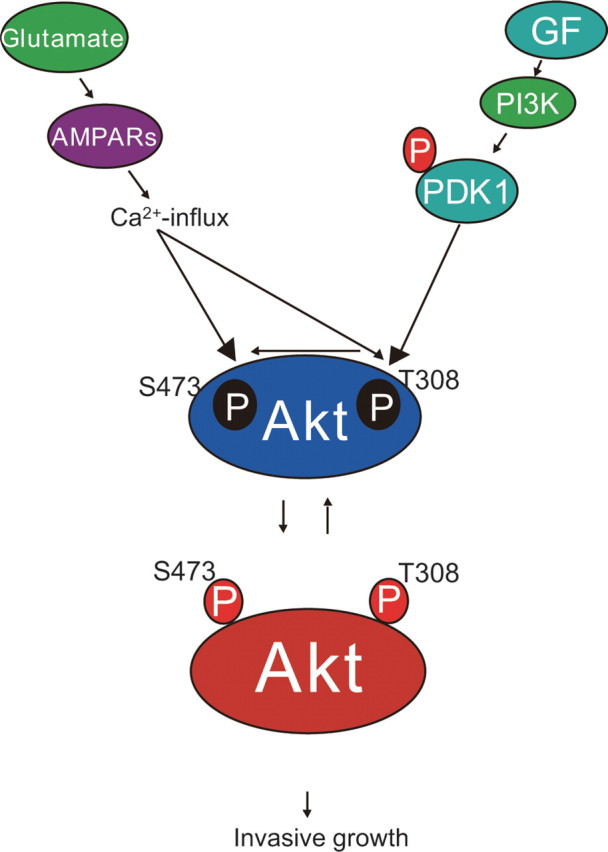
Model depicting the mechanism by which phosphorylation of Akt contributes to the invasive growth of glioblastoma. Tumor cells can pervert both growth factors (GF) and neurotransmitter glutamate secreted in an autocrine or paracrine manner. Independent of the PI3K-PDK-1-Akt pathway, in which PDK-1 is autophosphorylated, glutamate stimulates the phosphorylation of Akt at the site of Ser-473 via Ca2+ entry through Ca2+-permeable AMPA receptors. This novel pathway contributes to the high degree of anaplasia and invasive growth of human glioblastomas. This pathway also phosphorylates Akt at Thr-308 when the PI3K-PDK-1-Akt pathway is downregulated. Tumor cells can take advantage of their survival using at least these two pathways.
PDK-1 is a kinase known to be a potential upstream molecule, which efficiently phosphorylates Akt at Thr-308 in the PI3K-Akt pathway (Toker and Newton, 2000). In this study, PDK-1 had a high level of basal activity for autophosphorylation independent of Ca2+ entry through AMPA receptors in glioblastoma cells.
Stimulation of NMDA-type glutamate receptors in neuroblastoma cells resulted in a threefold or fourfold increase in the activation of endogenous PKB/Akt (Yano et al., 1998). Ca2+ through NMDA-type glutamate receptor is believed to bind to calmodulin, and the Ca2+/calmodulin complex activates the calmodulin-dependent kinase kinase, which directly induces activation of Akt at Thr-308. In this study, the Ca2+ influx through Ca2+-permeable AMPA receptors enhanced the phosphorylation of Akt at Ser-473, presumably by acting on a yet unidentified PDK-2. This pathway seemed to activate Akt at Thr-308 only if the PI3K-Akt pathway was tightly inhibited (Fig. 2D). Although the intracellular Ca2+ rise induced by cAMP, a protein kinase A agonist, also stimulates Akt unaffected by wortmannin (Sable et al., 1997; Filippa et al., 1999), the mechanism by which Akt is activated at Ser-473 remains to be clarified (Toker and Newton, 2000; Vanhaesebroeck and Alessi, 2000).
We found that phosphorylation at Ser-473 is a more important mediator of tumor cell proliferation and migration than phosphorylation at Thr-308. Therefore, the identification of the kinase responsible for the phosphorylation of Ser-473 is an urgent task for glioma biology. DNA protein kinase is a candidate for PDK-2, and glioma cells deficient of this kinase still retain constitutively active Akt phosphorylated at Ser-473, indicating that multiple pathways can phosphorylate this hydrophobic motif site (Feng et al., 2004). Recently, the mammalian target of rapamycin (mTOR) kinase and its associated protein rictor were found to be necessary for the phosphorylation of Akt at Ser-473, and that reduced expression of rictor or mTOR in human cancer cells inhibited an Akt/PKB effector (Sarbassov et al., 2005). The relationship between AMPA receptor-mediated signaling and the rictor-mTOR complex in the PI3K-Akt pathway is important to clarify, because both factors are attractive targets in cancer therapy.
Anaplastic features, including nuclear or cytoplasmic pleomorphism, the appearance of giant cells, brisk mitotic figures, microvascular proliferation, and necrosis with pseudopalisading, are all histological hallmarks of human glioblastomas. Although the PI3K-Akt pathway has been considered to be the main cascade of glioblastoma cell proliferation, the function and role of Akt in glioblastoma biology has not been elucidated fully in human disease models. Using CGNH-89 cells possessing the hallmarks of human glioblastomas after grafting into the subcutaneous tissue of nude mice as a model system, we found that introduction of a constitutively active form of Akt (Akt-HA) enhanced tumor anaplasia and increased the incidence of intratumoral hemorrhage. In contrast, a dominant-negative form of Akt (Akt-AA) reduced the incidence of intratumoral bleeding. Neuropathological analysis revealed that tumors expressing Akt-HA had large thrombosed vessels or increased vascularity. Glioblastomas are known for a high frequency of tumor bleeding (Kleihues et al., 2000). Akt may be responsible for this malignant behavior. In addition, the introduction of Akt-HA enhanced the growth rate, whereas introduction of Akt-AA reduced the growth rate, compared with the control group. Histological analysis further confirmed that Akt-HA stimulated tumor cell proliferation, prompted microvascular proliferation, and inhibited tumor cell apoptosis, whereas Akt-AA inhibited cell proliferation, reduced vascularity, and induced apoptosis.
Molecular heterogeneity is a challenge for clinical targeted therapy of glioblastoma. Growth factors acting through receptors, and tyrosine kinases and their signaling pathways are now primarily the focus of therapeutic research (Osaki et al., 2004). Pathway inhibitors, including kinase inhibitors and monoclonal antibodies, have been developed (Shawver et al., 2002). Primary glioblastoma multiforme commonly overexpresses EGFR and its ligand-independent mutant EGFRvIII (Mellinghoff et al., 2005). Platelet-derived growth factor receptors (PDGFRs) are frequently overexpressed and involved in the same pathway as EGFR and EGFRvIII. Signaling for vascular endothelial growth factors and their receptors is involved in the angiogenesis of glioblastoma and also involves the same pathway as EGFR, EGFRvIII, and PDGFR (Lokker et al., 2002). These growth factors finally activate the major downstream molecule PKB. A constitutively active mutant variant of EGFR, EGFRvIII, preferentially activates PI3K-Akt signaling, so the patients expressing EGFRvIII have been found to respond to EGFR inhibitors. PTEN acts as a tumor suppressor, and cells lacking PTEN show increased Akt activity. Loss of PTEN function markedly diminished the responsiveness of EGFR inhibitors. Thus, coexpression of EGFRvIII and PTEN is associated with clinical responsiveness to EGFR inhibitors (Mellinghoff et al., 2005). C-terminal modulator protein (CTMP), a protein partner for Akt that binds specifically to the C-terminal regulatory domain of Akt at the plasma membrane, also negatively regulates PKB activity (Maira et al., 2001). A protein phosphatase, PH domain leucine-rich repeat protein phosphatase (PHLPP) dephosphorylates the hydrophobic motif of Akt at Ser473 and suppresses tumor growth (Gao et al., 2005).
Like PTEN function, decreased cellular level of CTMP protein and the expression of PHLPP inhibits proliferation and abolishes tumorigenesis in nude mice. We found that glutamate is also involved in Akt activity through Ca2+-permeable AMPA receptors as shown in Figure 10. This novel pathway is not specific for the human glioblastoma cell line used in this study but is also adapted to the growth mechanism of many glioblastoma cell lines and primary cultures. Cell cultures derived from operative specimens regardless tumor grade possessed Ca2+-permeable AMPA receptors. These cells showed ubiquitously phosphorylated Akt at Ser-473, and this phosphorylation was abolished by the treatment of AMPA antagonists, which simultaneously reduced proliferation and migration, indicating that glutamate-AMPA receptor-Akt signaling is commonly observed in glioblastoma cells and that this pathway is essential for tumor survival.
Molecular heterogeneity was observed in glioblastoma cells, that is, PTEN status and the expression level of CTMP, but almost all glioma cells, including glioblastoma, anaplastic astrocytoma, and low-grade astrocytoma, possessed Ca2+-permeable AMPA receptors, and this pathway may function as the collateral flow to activate Akt if the cell is starved of the growth factors. This back-up pathway efficiently works in the early phase of plating before environmental cues were not fully established (Fig. 1B). Indeed, cells in glutamate-containing medium proliferated more rapidly than cells in glutamate- free medium. This was the most significant at 48 h after plating. Previously, we reported that the effect of glutamate in U87-MG cells was detected only in 0.5% but not 10% FBS-containing media. Plating of high cell density and the prolonged cultivation (>78 h after plating) in the previous experiment would hinder the detection of glutamate-mediated signaling (Yoshida et al., 2006).
In vivo, cells of the invasion front or metastatic cells isolated from the tumor bulk may use glutamate as the energy supply before establishing the autocrine and paracrine loops of various growth factors. Because the CNS is rich in glutamine and glutamate, tumor cells can use glutamate to expand for invasion to destroy surrounding tissues, as well as for proliferation and cell movement. This pathway may become active if constitutively active growth factor receptors are inhibited. Unfortunately, only 10–20% of patients are responsive to EGFR inhibitor, and this response acquires resistance. The glutamate-AMPA receptor-Akt pathway may give an alternative therapeutic target. Moreover, this novel pathway may impact on neuroscience field in two respects. First, excess glutamate released by glioma cells identified in our experiments in primary cultures causes excitotoxicity of glutamatergenic synapses surrounding neurons, which plays a role of pathophysiology in brain edema and epilepsy of brain tumor patients. Second, glioblastoma cells are not only fascinating as a disease but also a model system, because the biology displayed by proliferative glioblastoma cells is reminiscent of cell division, migration, and proliferation of normal brain progenitor cells during development of the nervous system. Hence, research on the biology of neoplastic glial cells has begun to cross-fertilize with developmental biology of glial cells from which glioma cells are derived.
In summary, the present study demonstrated that Ca2+ influx through Ca2+-permeable AMPA receptors activated Akt, and this effect was independent of PI3K. Several lines of evidence indicated that the Ca2+-permeable AMPA receptors were an upstream effector molecule of Akt. Exogenous GluR2Q or GluR1 phosphorylated Akt at Ser-473 and stimulated the growth of glioma, whereas GluR2 dephosphorylated Akt and inhibited tumor cell proliferation. Furthermore, a constitutively active form of Akt stimulated tumor growth, whereas a dominant-negative form of Akt reduced tumor volume. Finally, the introduction of a dominant-negative form of Akt into cells expressing GluR2Q reduced glioma cell proliferation, whereas the transfer of a constitutively active form of Akt into cells expressing GluR2 prevented apoptosis. Therefore, we conclude that Ca2+ entry through Ca2+-permeable AMPA receptors promotes glioblastoma cell proliferation via the activation of Akt, and that this Ca2+ signaling is a novel pathway independent of PI3K.
The discovery and development of a novel Ca2+ signaling pathway via Ca2+-permeable AMPA receptors will have important implications in cancer research (Rzeski et al., 2001). This receptor as well as the Akt molecule may serve as targets for the treatment of glioblastoma. Furthermore, glioma cells pervert the neurotransmitter glutamate as well as a variety of cytokines and growth factors (Labrakakis et al., 1998; Sasaki et al., 2001; Takano et al., 2001; Sontheimer, 2003), and thus a combination of AMPA antagonists and growth factor receptor antagonists may be useful for treating malignant gliomas (Fig. 10). Whether this novel cascade is adopted in other human malignancies remains to be investigated.
Footnotes
This work was supported in part by a Grant-in-Aid for Scientific Research (B) (15390429) from the Japanese Ministry of Education, Culture, Sports, Science, and Technology (S.I.). We thank Drs. Izumu Saito and Yumi Kanegae for providing materials for constructing recombinant adenoviruses and Mitsue Maniwa for technical assistance. We also thank Drs. Kazuhiko Namikawa and Hiroshi Kiyama for providing us with AxCALNLmyr-Akt-HA and AxCALNLmyr-Akt-AA.
References
- Alessi DR, Cohen P. Mechanism of activation and function of protein kinase B. Curr Opin Genet Dev. 1998;8:55–62. doi: 10.1016/s0959-437x(98)80062-2. [DOI] [PubMed] [Google Scholar]
- Bailey P, Cushing H. Philadelphia: Lippincott; 1926. A classification of tumors of the glioma group on a histogenetic basis with a correlation study of prognosis. [Google Scholar]
- Bochet P, Audinat E, Lambolez B, Crépel F, Rossier J, Iino M, Tsuzuki K, Ozawa S. Subunit composition at the single-cell level explains functional properties of a glutamate-gated channel. Neuron. 1994;12:383–388. doi: 10.1016/0896-6273(94)90279-8. [DOI] [PubMed] [Google Scholar]
- Burnashev N, Monyer H, Seeburg PH, Sakmann B. Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron. 1992;8:189–198. doi: 10.1016/0896-6273(92)90120-3. [DOI] [PubMed] [Google Scholar]
- Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci USA. 1999;96:4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe G, Horvath S, Cloughesy TF, Crosby K, Seligson D, Palotie A, Inge L, Smith BL, Sawyers CL, Mischel PS. Analysis of the phosphatidylinositol 3′-kinase signaling pathway in glioblastoma patients in vivo. Cancer Res. 2003;63:2742–2746. [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19:348–355. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- Feng J, Park J, Cron P, Hess D, Hemmings BA. Identification of a PKB/Akt hydrophobic motif Ser-473 kinase as DNA-dependent protein kinase. J Biol Chem. 2004;279:41189–41196. doi: 10.1074/jbc.M406731200. [DOI] [PubMed] [Google Scholar]
- Filippa N, Sable CL, Filloux C, Hemmings BA, Van Obberghen E. Mechanism of protein kinase B activation by cyclic AMP-dependent protein kinase. Mol Cell Biol. 1999;19:4989–5000. doi: 10.1128/mcb.19.7.4989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell. 2005;18:13–24. doi: 10.1016/j.molcel.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Geiger JRP, Melcher T, Koh DS, Sackmann B., Seeburg PH, Jonas P, Monyer H. Relative abundance of subunit mRNAs determining gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron. 1995;15:193–204. doi: 10.1016/0896-6273(95)90076-4. [DOI] [PubMed] [Google Scholar]
- Guha A, Feldkamp MM, Lau N, Boss G, Pawson A. Proliferation of human malignant astrocytomas is dependent on Ras activation. Oncogene. 1997;15:2755–2765. doi: 10.1038/sj.onc.1201455. [DOI] [PubMed] [Google Scholar]
- Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet. 2000;25:55–57. doi: 10.1038/75596. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- Iino M, Goto K, Kakegawa W, Okado H, Sudo M, Ishiuchi S, Miwa A, Takayasu Y, Saito I, Tsuzuki K, Ozawa S. Glia-synapse interaction through Ca2+-permeable AMPA receptors in Bergmann glia. Science. 2001;292:926–929. doi: 10.1126/science.1058827. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vöckler J, Dikranian K, Tenkova T, Stefovska V, Turski L, Olneny JW. Blokade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–74. doi: 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- Ishiuchi S, Nakazato Y, Iino M, Ozawa S, Tamura M, Ohye C. In vitro neuronal and glial production and differentiation of human central neurocytoma cells. J Neurosci Res. 1998;51:526–535. doi: 10.1002/(SICI)1097-4547(19980215)51:4<526::AID-JNR12>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Ishiuchi S, Tsuzuki K, Yamada N, Okado H, Miwa A, Kuromi H, Yokoo H, Nakazato Y, Sasaki T, Ozawa S. Extension of glial processes by activation of Ca2+-permeable AMPA receptor channels. NeuroReport. 2001;12:745–748. doi: 10.1097/00001756-200103260-00026. [DOI] [PubMed] [Google Scholar]
- Ishiuchi S, Tsuzuki K, Yoshida Y, Yamada N, Hagimura N, Okado H, Miwa A, Kurihara H, Nakazato Y, Tamura M, Sasaki T, Ozawa S. Blockage of Ca2+-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat Med. 2002;8:971–978. doi: 10.1038/nm746. [DOI] [PubMed] [Google Scholar]
- Kanegae Y, Takamori K, Sato Y, Lee G, Nakai M, Saito I. Efficient gene activation system on mammalian cell chromosomes using recombinant adenovirus producing Cre recombinase. Gene. 1996;181:207–212. doi: 10.1016/s0378-1119(96)00516-1. [DOI] [PubMed] [Google Scholar]
- Kim D, Kim S, Koh H, Yoon SO, Chung AS, Cho KS, Chung J. Akt /PKB promotes cancer cell invasion via increased motility and metalloproteinase production. FASEB J. 2001;15:1953–1962. doi: 10.1096/fj.01-0198com. [DOI] [PubMed] [Google Scholar]
- Kitamura T, Ogawa W, Sakaue H, Hino Y, Kuroda S, Takata M, Matsumoto M, Maeda T, Konishi H, Kikkawa U, Ogawa W, Kasuga M. Requirement for activation of the serine-threonine kinase Akt (protein kinase B) in insulin stimulation of protein synthesis but not of glucose transport. Mol Cell Biol. 1998;18:3708–3717. doi: 10.1128/mcb.18.7.3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleihues P, Burger PC, Collins VP, Newcombb EW, Ohgaki H, Cavenee WK. Glioblastoma. In: Kleihues P, Cavenee WK, editors. World Health Organization classification of tumours. Pathology and genetics of tumours of the nervous system. Lyon, France: ARC; 2000. pp. 29–39. [Google Scholar]
- Kohara A, Okada M, Tsutsumi R, Ohno K, Takahashi M, Shimizu-Sasamata M, Shishikura J, Inami H, Sakamoto S, Yamaguchi T. In vitro characterization of YM872, a selective, potent and highly water-soluble α-amino-3-hydroxy-5-methylisoxazole-4-propionate receptor antagonist. J Pharm Pharmacol. 1998;50:795–801. doi: 10.1111/j.2042-7158.1998.tb07142.x. [DOI] [PubMed] [Google Scholar]
- Kohn AD, Takeuchi F, Roth RA. Akt, apleckstrin homology domain containing kinase is activated primarily by phosphorylation. J Biol Chem. 1996a;271:21920–21926. doi: 10.1074/jbc.271.36.21920. [DOI] [PubMed] [Google Scholar]
- Koike T, Martin DP, Johnson EM., Jr Role of Ca2+ channels in the ability of membrane depolarization to prevent neuronal death induced by trophic factor deprivation: evidence that levels of internal Ca2+ determine nerve growth factor dependence of sympathetic ganglion cells. Proc Natl Acad Sci USA. 1989;86:6421–6425. doi: 10.1073/pnas.86.16.6421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrakakis C, Patt S, Hartmann J, Kettenmann H. Glutamate receptor activation can trigger electrical activity in human glioma cells. Eur J Neurosci. 1998;10:2153–2162. doi: 10.1046/j.1460-9568.1998.00226.x. [DOI] [PubMed] [Google Scholar]
- Lefranc F, Britchi J, Kiss R. Possible future issues in the treatment of glioblastomas: special emphasis on cell migration and the resistance of migrating glioblastoma cells to apoptosis. J Clin Oncol. 2005;23:2411–2422. doi: 10.1200/JCO.2005.03.089. [DOI] [PubMed] [Google Scholar]
- Li J, Yen C, Liaw D, Podsypanina K, Bose S, Want SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bignaer SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wiqler MH, Parsons R. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- Lokker NA, Sullivan CM, Hollenbach SJ, Israel MA, Giese NA. Platelet-derived growth factor (PDGF) autocrine signaling regulates survival and mitogenic pathways in glioblastoma cells: evidence that the novel PDGF-C and PDGF-D ligands may play a role in the development of brain tumors. Cancer Res. 2002;62:3729–3735. [PubMed] [Google Scholar]
- Maas S, Patt S, Schrey M, Rich A. Underediting of glutamate receptor GluR-B mRNA in malignant gliomas. Proc Natl Acad Sci USA. 2001;98:14687–14692. doi: 10.1073/pnas.251531398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- Maher EA, Furnari FB, Bachoo RM, Rowitch DH, Louis DN, Cavenee WK, DePinho RA. Malignant glioma: genetics and biology of a grave matter. Genes Dev. 2001;15:1311–1333. doi: 10.1101/gad.891601. [DOI] [PubMed] [Google Scholar]
- Maira SM, Galetic I, Brazil DP, Kaech S, Ingley E, Thelen M, Hemmings BA. Carboxyl-terminal modulator protein (CTMP), a negative regulator of PKB/Akt and v-Akt at the plasma membrane. Science. 2001;294:374–380. doi: 10.1126/science.1062030. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellinghoff IK, Wang MD, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, Lu KV, Yoshimoto K, Huang JHY, Chute DJ, Riggs BL, Horvath S, Liau LM, Cavenee WK, Rao PN, Beroukhim R, Peck TC, Lee JC, Sellers WR, Stokoe D, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353:2012–2024. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- Mitchison TJ, Cramer LP. Actin-based cell motility and cell locomotion. Cell. 1996;84:371–379. doi: 10.1016/s0092-8674(00)81281-7. [DOI] [PubMed] [Google Scholar]
- Miyake S, Makimura M, Kanegae Y, Harada S, Sato Y, Takamori K, Tokuda C, Saito I. Efficient generation of recombinant adenoviruses using DNA-terminal protein complex and a cosmid bearing the full-length virus genome. Proc Natl Acad Sci USA. 1996;93:1320–1324. doi: 10.1073/pnas.93.3.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namikawa K, Honma M, Abe K, Takeda M, Mansur K, Obata T, Miwa A, Okado H, Kiyama H. Akt/protein kinase B prevents injury-induced motoneuron death and accelerates axonal regeneration. J Neurosci. 2000;20:2875–2886. doi: 10.1523/JNEUROSCI.20-08-02875.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osaki M, Oshimura M, Ito H. PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis. 2004;9:667–676. doi: 10.1023/B:APPT.0000045801.15585.dd. [DOI] [PubMed] [Google Scholar]
- Ozawa S, Kamiya H, Tsuzuki K. Glutamate receptors in the mammalian central nervous system. Prog Neurobiol. 1998;54:581–618. doi: 10.1016/s0301-0082(97)00085-3. [DOI] [PubMed] [Google Scholar]
- Resh MD. Myristylation and palmitylation of Src family members: the fasts of the matter. Cell. 1994;76:411–413. doi: 10.1016/0092-8674(94)90104-x. [DOI] [PubMed] [Google Scholar]
- Rzeski W, Turski L, Ikonomidou C. Glutamate antagonists limit tumor growth. Proc Natl Acad Sci USA. 2001;98:6372–6377. doi: 10.1073/pnas.091113598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sable CL, Filippa N, Hemmings B, Van Obberghen E. cAMP stimulates protein kinase B in a wortmannin-insensitive manner. FEBS Lett. 1997;409:253–257. doi: 10.1016/s0014-5793(97)00518-8. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Okada Y. Selective reduction of glutamate in the rat superior colliculus and dorsal lateral geniculate nucleus after contralateral enucleation. Brain Res. 1992;573:197–203. doi: 10.1016/0006-8993(92)90763-y. [DOI] [PubMed] [Google Scholar]
- Sanai N, Alvarez-Buylla A, Berger MS. Neural stem cells and origin of gliomas. N Engl J Med. 2005;353:811–822. doi: 10.1056/NEJMra043666. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by Rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Sasaki A, Tamura M, Hasegawa M, Ishiuchi S, Hirato J, Nakazato Y. Expression of interleukin-1beta mRNA and protein in human gliomas assessed by RT-PCR and immunohistochemistry. J Neuropathol Exp Neurol. 1998;57:653–663. doi: 10.1097/00005072-199807000-00002. [DOI] [PubMed] [Google Scholar]
- Sasaki A, Ishiuchi S, Kanda T, Hasegawa M, Nakazato Y. Analysis of interleukin-6 gene expression in primary human gliomas, glioblastoma xenografts and glioblastoma cell lines. Brain Tumor Pathol. 2001;18:13–21. doi: 10.1007/BF02478920. [DOI] [PubMed] [Google Scholar]
- Seeburg PH. The molecular biology of mammalian glutamate receptor channels. Trends Neurosci. 1993;16:359–365. doi: 10.1016/0166-2236(93)90093-2. [DOI] [PubMed] [Google Scholar]
- Shawver LK, Salmon D, Ullrich HM. Smart drugs: tyrosine kinase inhibitors in cancer therapy. Cancer Cell. 2002;1:117–123. doi: 10.1016/s1535-6108(02)00039-9. [DOI] [PubMed] [Google Scholar]
- Smith JS, Tachibana I, Passe SM, Huntley BK, Borell TJ, Iturria N, O’Fallon JR, Schaefer PL, Scheithauer BW, James CD, Buckner JC, Jenkins RB. PTEN mutation, EGFR amplification, and outcome in patients with anaplastic astrocytoma and glioblastoma multiforme. J Natl Cancer Inst. 2001;93:1246–1256. doi: 10.1093/jnci/93.16.1246. [DOI] [PubMed] [Google Scholar]
- Sontheimer H. Malignant gliomas: perverting glutamate and ion homeostasis for selective advantage. Trends Neurosci. 2003;26:543–549. doi: 10.1016/j.tins.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- Takano T, Lin JH, Arcuino G, Gao Q, Yang J, Nedergaard M. Glutamate release promotes growth of malignant gliomas. Nat Med. 2001;7:1010–1015. doi: 10.1038/nm0901-1010. [DOI] [PubMed] [Google Scholar]
- Takata M, Ogawa W, Kitamura T, Hino Y, Kuroda S, Kotani K, Klip A, Gingras AC, Sonenberg N, Kasuga M. Requirement for Akt (protein kinase B) in insulin-induced activation of glycogen synthase and phosphorylation of 4E-BP1 (PHAS-1) J Biol Chem. 1999;274:20611–20618. doi: 10.1074/jbc.274.29.20611. [DOI] [PubMed] [Google Scholar]
- Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science. 1998;280:1614–1617. doi: 10.1126/science.280.5369.1614. [DOI] [PubMed] [Google Scholar]
- Toker A, Newton AC. Akt/protein kinase B is regulated by autophosphorylation at the hypothetical PDK-2 site. J Biol Chem. 2000;275:8271–8274. doi: 10.1074/jbc.275.12.8271. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Alessi DR. The PI3K-PDK1 connection: more than just a road to PKB. Biochem J. 2000;346:561–576. [PMC free article] [PubMed] [Google Scholar]
- Williams MR, Arthur JS, Balendran A, van der Kaay J, Poli V, Choen P, Alessi DR. The role of 3-phoshoinositide-dependent protein kinase 1 in activating AGC kinases defined in embryonic stem cells. Curr Biol. 2000;10:439–448. doi: 10.1016/s0960-9822(00)00441-3. [DOI] [PubMed] [Google Scholar]
- Wu X, Senechal K, Neshat MS, Whang YE, Sawyers CL. The PTEN/MMAC1 tumor suppressor phosphatase functions as a negative regulator of the phosphoinositide 3-kinase/Akt pathway. Proc Natl Acad Sci USA. 1998;95:15587–15591. doi: 10.1073/pnas.95.26.15587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano S, Tokumitsu H, Soderling TR. Calcium promotes cell survival through CaM-K kinase activation of the protein-kinase-B pathway. Nature. 1998;396:584–587. doi: 10.1038/25147. [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Tsuzuki K, Ishiuchi S, Ozawa S. Serum-dependence AMPA receptor mediated proliferation in glioma cells. Pathol Int. 2006;56:262–271. doi: 10.1111/j.1440-1827.2006.01954.x. [DOI] [PubMed] [Google Scholar]