

Abstract
Dorsal root injury (DRI) disrupts the flow of sensory information to the spinal cord. Although primary afferents do not regenerate to their original targets, spontaneous recovery can, by unknown mechanisms, occur after DRI. Here, we show that brain-derived neurotrophic factor (BDNF) and neurotrophin-3 (NT-3), but not nerve growth factor or neurotrophin-4, are upregulated in the spinal gray matter after DRI. Because endognous BDNF and NT-3 have well established roles in synaptic and axonal plasticity, we hypothesized that they contributed to spontaneous recovery after DRI. We first developed a model of DRI-induced mechanosensory dysfunction: rat C7/8 DRI produced a deficit in low-threshold cutaneous mechanosensation that spontaneously improved within 10 d but did not recover completely. To determine the effects of endogenous BDNF and NT-3, we administered TrkB-Fc or TrkC-Fc fusion proteins throughout the recovery period. To our surprise, TrkB-Fc stimulated complete recovery of mechanosensation by 6 d after DRI. It also stimulated mechanosensory axon sprouting but prevented deafferentation-induced serotonergic sprouting. TrkC-Fc had no effect on low-threshold mechanosensory behavior or axonal plasticity. There was no mechanosensory improvement with single-bolus TrkB-Fc infusions at 10 d after DRI (despite significantly reducing rhizotomy-induced cold pain), indicating that neuromodulatory effects of BDNF did not underlie mechanosensory recovery. Continuous infusion of the pan-neurotrophin antagonist K252a also stimulated behavioral and anatomical plasticity, indicating that these effects of TrkB-Fc treatment occurred independent of signaling by other neurotrophins. These results illustrate a novel, plasticity-suppressing effect of endogenous TrkB ligands on mechanosensation and mechanosensory primary afferent axons after spinal deafferentation.
Keywords: mechanosensation, plasticity, spinal cord, vesicular glutamate transporter, neurotrophin-3, brain-derived neurotrophic factor
Introduction
It is widely recognized that spontaneous recovery of function after spinal cord injury (SCI) depends on synaptic and/or axonal plasticity of spared neurons. Although mechanisms remain primarily undefined, such plasticity is evident in limited functional recovery after incomplete SCI (Goldberger and Murray, 1974; Murray and Goldberger, 1974; Kaegi et al., 2002). Dorsal root injury (DRI), which disconnects sensory axons from the spinal cord, is a useful model in which to investigate injury-induced structural and functional changes in spared spinal axons: it is sensory specific, precisely titratable in the sense that the extent of deafferentation can be controlled by varying the number of roots cut, and like incomplete SCI, some recovery occurs when lesions are limited. For example, postural reflexes partially return in the rhizotomized cat (Goldberger and Murray, 1974), and restricted DRI leads to a transient loss of manual dexterity in monkeys (Darian-Smith and Ciferri, 2005). Such recovery implicates improvements in mechanosensation.
DRI also results in the upregulation of brain-derived neurotrophic factor (BDNF) and neurotrophin-3 (NT-3) in spinal gray matter (Johnson et al., 2000), where they have been implicated in enhancing synaptic plasticity in respiratory circuitry (Kinkead et al., 1998; Baker-Herman et al., 2004). Several convergent lines of evidence suggest that BDNF and/or NT-3 may also mediate spontaneous mechanosensory recovery after DRI and that this may occur through their modulatory effects on synaptic transmission and/or via promoting morphological plasticity (sprouting) of relevant axonal populations. For example, both BDNF and NT-3 are known to facilitate glutamatergic synaptic efficacy between primary afferent axons and second-order neurons in the spinal cord (Kerr et al., 1999; Arvanian et al., 2003; Ji et al., 2003). In addition, the normally inhibitory effect of GABA on spinal projection neurons becomes excitatory in the presence of elevated BDNF (Coull et al., 2005), increasing excitability and enhancing sensory transmission. BDNF and NT-3 also play essential roles in establishing and maintaining mechanosensory circuitry (Airaksinen et al., 1996; Carroll et al., 1998), and NT-3 promotes functional mechanosensory axon regeneration into and within the spinal cord after both SCI and DRI (Bradbury et al., 1999; Ramer et al., 2000, 2002). Given that mechanosensory recovery and spinal primary afferent sprouting have been shown to occur after dorsal rhizotomy (Darian-Smith, 2004) and that mechanosensory axons express TrkB and TrkC but not TrkA (McMahon et al., 1994), it is also important to ask whether DRI-induced expression of these neurotrophins effects sprouting of their terminal arbors.
Here, we establish a model of mechanosensory dysfunction after partial forepaw deafferentation in the rat. In this injury model, mechanosensory behavior partially recovers spontaneously within 10 d of injury, in the absence of spinal primary afferent sprouting. However, mechanosensation does not recover to preoperative levels, and mechanosensory deficits in the deafferented forepaw persist for months after injury. We hypothesized that spinally upregulated neurotrophins might underpin the early partial functional restitution. To our surprise, sequestering endogenous BDNF/neurotrophin-4 (NT-4), but not NT-3, resulted in more complete behavioral recovery and stimulated sprouting of mechanosensory primary afferent axons in the spinal cord.
Materials and Methods
Surgery and animal care
All procedures conformed to the Canadian Council on Animal Care guidelines on the use of experimental animals and were approved by the University of British Columbia (UBC) animal care committee. Adult male Sprague Dawley rats (UBC Animal Care Facility) were housed in groups of three to five, on a 12 h light/dark cycle, and had ad libitum access to standard rodent chow and water. After preoperative behavioral testing (see below), rats were anesthetized with an intraperitoneal injection of ketamine HCl/medetomidine HCl (75 mg/kg and 0.5 mg/kg, respectively). After laminectomy and durotomy, the exposed seventh and eighth cervical dorsal roots were transected with microscissors or left intact (sham).
For continuous intrathecal infusions, osmotic minipumps (models 2001 and 2002; Alzet, Cupertino, CA) and intrathecal cannulas (0.64 mm outer diameter, 0.3 mm inner diameter) were filled with a solution of PBS, whole human IgG (Sigma-Aldrich Canada, Oakville, Ontario, Canada) in PBS, TrkB-Fc (Sigma, St. Louis, MO) in PBS, TrkC-Fc (R & D Systems, Minneapolis, MN) in PBS, 10% dimethylsulfoxide (DMSO) in PBS, or the pan-Trk antagonist K252a in 10% DMSO in PBS. Cannulas were inserted into the intrathecal space via the atlanto-occipital membrane such that the tip rested above the C6 spinal cord. Minipumps were inserted into a subcutaneous pocket just below the scapulae. Based on previous experiments demonstrating efficacy of these and similar drugs in vivo (Kerr et al., 1999; Sung et al., 2003; Yajima et al., 2005; McPhail et al., 2007), IgG and Trk-Fc chimeras were delivered at a rate of 3 μg/d for 10 or 20 d; K252a was delivered at a rate of 2 μg/d. In the 20 d animals, the 2002 (2 week) model pump was exchanged for a 2001 (1 week) model pump on day 15. Individuals performing the surgeries coded the animals for blind behavioral analysis (see below). At the end of the behavioral study, rats were reanesthetized, and their ipsilateral median nerve was exposed in the upper forelimb. Using a pulled glass micropipette fitted to a Hamilton syringe, the nerve was injected with 0.5 μl of a 1% solution of the B fragment of cholera toxin (CTB; Cedarlane, Hornby, Ontario, Canada) to transganglionically label the spinal terminal fields of myelinated primary afferent axons.
For bolus infusions of IgG or TrkB-Fc, saline-filled intrathecal cannulas were inserted into ketamine/medetomidine-anesthetized rats on the 7th postoperative day. These were externalized over the skull and sealed with cyanoacrylate gel. On days 9 and 10, rats received either a bolus injection of IgG or TrkB-Fc (0.2 mg/ml, 10 μl injections followed by 10 μl saline flush). Beginning 30 min after infusions and continuing for no more than 1 h, rats underwent blind behavioral assessment (see below).
Mechanosensory testing
All behavioral experiments were performed by observers who were blind with respect to treatment. Rats underwent training for habituation to the testing environment on two occasions before preoperative behavioral testing. Low-threshold cutaneous mechanosensation across the palmar surface of the forepaw was assessed using an adhesive removal test (Thallmair et al., 1998; Bradbury et al., 2002; Onifer et al., 2005; Starkey et al., 2005; Moreno-Flores et al., 2006). Circular stickers (6.4 mm in diameter) were applied to the palm of either forepaw, and the rat was placed in a cage for observation. The time required for the rat to sense the sticker, indicated by a brisk paw shake and/or bringing the paw to the mouth, was measured for each paw to a maximum time of 150 s. This maximum was imposed to distinguish sticker sensation in the palm from accidental sticker discovery during grooming, behavior that typically begins after 2–3 min of test cage exploration. Trials in which the sticker was discovered during grooming before the 150 s maximum were excluded and repeated. To characterize the injury-induced mechanosensory deficit, rats that received sham injury or C7/8 DRI only were tested over 90 postoperative days. To examine the effects of neurotrophin signaling antagonism, rats that received C7/8 DRI plus IgG, Trk-Fc, K252a, or K252a vehicle (10% DMSO in PBS) were tested over 20 postoperative days. In all cases, daily test scores represent averages of two trials, and trials were separated by at least 1 h. Between trials, rats underwent a mock trial in which the experimenter picked up the rat and applied pressure to the forepaws to mimic sticker application.
The Dynamic Plantar Aesthesiometer (Ugo Basile, Comerio, Italy) was used to test for mechanical allodynia. Rats were positioned in a raised cage with a wire mesh floor over the stimulator unit. The filament was applied to the center of the palmar surface of the forepaw, and upward force was increased from 1 to 50 g over 7 s. Force at withdrawal was recorded for both forepaws.
Cold sensitivity was examined by measuring the duration of response to acetone (10 μl) applied to the palmar forepaw, as we have done previously (Ramer et al., 2004). Rats were placed in a raised cage with a wire mesh bottom, and acetone was applied from below. A response was defined as withdrawal, biting, licking, or shaking of the paw. A brief withdrawal without further attention to the paw was assigned a score of 1 s. Response duration was recorded for both forepaws.
Electrophysiology
In a separate group of rats that did not undergo behavioral testing, we recorded dorsal horn activity using a 16-electrode matrix (2 × 8) microarray inserted into the superficial dorsal horn between the seventh and eighth cervical segments (Borisoff et al., 2006). Neural activity in urethane-anesthetized (1.5 g/kg, i.p.), paralyzed (gallamine triethiodide, 60 mg/kg, intra-arterially), and ventilated rats was amplified and recorded using a multichannel Pentusa neurophysiology workstation (Tucker-Davis Technologies, Alachua, FL). Sensory receptive fields were electrically stimulated in left forepaw digits using steel pin electrodes (1 ms pulse width, 4 mA current amplitude, 0.5 Hz). Postsynaptic responses from 100 trials were recorded from each electrode in the array and stored for off-line analysis. At the end of the experiment, rats were killed with an intra-arterial injection of chloral hydrate (1 g/kg).
We have described in detail a quantitative cluster analysis-based method to characterize electrophysiological activity in the dorsal horn arising from electrical stimulation of individual digits (Borisoff et al., 2006). This method is based on the discrimination accuracy of specific digit stimulation sites, solely determined from an automatic analysis of recorded neural activity in the spinal cord. Briefly, signal features based on spike rate data were calculated for each stimulation event. The features consisted of the number of threshold-detected fast (Aβ fiber-evoked) spikes in each of 12 consecutive bins of 4 ms in length, calculated for each of the 16 channels. Thus 192 features were generated for each event from the all but 2.5 ms of the first 48 ms of recorded data after stimulation onset. Data from the very first 2.5 ms after stimulus were not included in the analysis to eliminate the influence of stimulus artifact.
A classification scheme using a k-means-derived codebook, calculated from the training feature sets, was used. The feature set dimensionality was first reduced to the top three principal components using principal component analysis (PCA). The PCA transform matrix was calculated during codebook generation and later used during classification of the test features. During codebook generation, each class of the three-dimensional training features was reduced to k = 2 codebook vectors using k-means clustering. During testing, one-Nearest Neighbors classification was used to discriminate test features, using the previously calculated two-vector classes as a codebook.
Tissue processing and image analysis
At the end of the behavioral experiments, animals were killed with an overdose of chloral hydrate (1 g/kg, i.p.; Sigma) and perfused with PBS, followed by 4% paraformaldehyde in 0.1 m phosphate buffer (PB). Cords and cannulas were inspected for damage and/or blockage, and those animals in which evidence for either was overt were removed from all behavioral and anatomical analyses. Final sample sizes for each group are presented in Table 1. The cervical spinal cords (segments C6–T1) were harvested, postfixed overnight, cryoprotected in 20% sucrose in 0.1 m PB, frozen over liquid nitrogen, and sectioned on a cryostat at 16 μm. Sections were thaw mounted onto glass slides and stored at −80°C. Slides were blocked in a 10% solution of normal donkey serum in PBS plus Triton X-100 (0.1%) for 20 min, followed by primary antibody solutions overnight (Table 2). After three 15 min washes in PBS, secondary antibodies raised in donkey and conjugated to Cy3 (Jackson ImmunoResearch, West Grove, PA), Alexa 488 (Invitrogen, Eugene, OR), or AMCA (7-amino-4-methylcoumarin-3-acetic acid; Jackson ImmunoResearch) were applied (1:200) for 2 h. Coverslipped slides were inspected using a Zeiss (Jena, Germany) Axioplan II microscope, and digital images were captured using Northern Eclipse software (Empix Imaging, Mississauga, Ontario, Canada) via a digital camera (QImaging, Burnaby, British Columbia, Canada). Exposure, gain, and offset settings were set to optimize signal-to-noise ratios for each antigen.
Table 1.
Sample sizes
| Outcome measure | Treatment | Sample size |
|||
|---|---|---|---|---|---|
| Total included | 10 d | 20 d | 90 d | ||
| Behavioral pilot | Sham | 7 | 7 | 7 | 7 |
| C7/8 DRI | 7 | 7 | 7 | 7 | |
| Electrophysiology | 19a | 6 | |||
| Behavioral study | |||||
| Continuous treatment (all C7/8 DRI) | IgG | 13 | 13 | 8 | |
| TrkB-Fc | 11 | 11 | 5 | ||
| TrkC-Fc | 9 | 9 | 4 | ||
| 10% DMSO | 15 | 15 | 6 | ||
| K252a | 15 | 15 | 6 | ||
| Acute treatment (all C7/8 DRI) | IgG/TrkB-Fcb | 10 | 10 | ||
| Anatomical study | Uninjured | 7 | |||
| C7/8 DRIc | 15 | 5 | 5 | ||
| IgGd,e | 10 | 5 | 5 | ||
| TrkB-Fcd,e | 10 | 5 | 5 | ||
| TrkC-Fcd,e | 9 | 5 | 4 | ||
| 10% DMSOd | 11 | 5 | 6 | ||
| K252ad | 11 | 5 | 6 | ||
aThirteen intact animals underwent terminal electrophysiological experiments. Of these, six also received acute C7/C8 rhizotomies to assess the contribution of the C7 and C8 roots to digit discrimination.
bTen animals with C7/8 DRI received an intrathecal bolus of TrkB-Fc or IgG 9 and 10 d after injury.
cAnimals with 3, 10, and 20 d C7/8 DRI used to map the spinal deafferentation gap and assess mechanosensory sprouting therein.
dTissue processed from behaviorally tested animals.
eTissue from the 20 d group used as control material in assessing Trk-Fc and IgG penetration.
Table 2.
Primary antibodies used
| Host | Antigen | Target tissue/structures | Dilution | Source |
|---|---|---|---|---|
| Sheep | NGF | 1:1000 | Neuromics (Medina, MN) | |
| Chicken | BDNF | 1:1000 | Neuromics | |
| Goat | NT-3 | 1:100 | Neuromics | |
| Rabbit | NT-4 | 1:1000 | Chemicon (Temecula, CA) | |
| Goat | Human Fc | 1:100 | Sigma-Aldrich Canada | |
| Mouse | Cd11b (Ox-42) | Microglia | 1:500 | Sigma-Aldrich Canada |
| Rabbit | GFAP | Astrocytes | 1:1000 | Dako Canada (Mississauga, Ontario, Canada) |
| Mouse | RECA-1 | Blood vessels | 1:1000 | Serotec (Raleigh, NC) |
| Guinea pig | VGLUT1 | Mechanosensory/ corticospinal terminals | 1:4000 | Chemicon |
| Goat | CTB | Mechanosensory terminals | 1:2000 | Cedarlane |
| Rabbit | SERT | Serotonergic axons | 1:1200 | Immunostar (Hudson, WI) |
GFAP, Glial fibrillary acidic protein; RECA-1, rat endothelial cell antigen 1; SERT, serotonin transporter.
Quantitative analyses of terminal density in the dorsal horn were performed by measuring terminal density as a function of depth in the dorsal horn, as we have done previously (Ramer et al., 2001, 2004; MacDermid et al., 2004; Scott et al., 2005). Densitometric analyses of vesicular glutamate transporter 1 (VGLUT1)- and CTB-positive terminals were performed within the consistent deafferentation gap apparent in C7 after C7/8 DRI. Images of five randomly selected sections from the C7 spinal segment were captured for each animal listed in Table 1. To measure density, images were first passed through a Laplacian omnidirectional edge-detection filter (SigmaScan Pro version 5.0), which optimizes the signal-to-noise ratio and corrects for small variations in background staining across images. Terminal profiles in the filtered images were selected with a threshold overlay, to give all immunopositive pixels equal weight regardless of their brightness in the original image. The density of terminals was measured from the overlay in laminas II or III/IV of the deafferentation gap. To control for variability in tracing efficacy, the density of CTB-positive terminals within the gap was normalized to that of laterally adjacent spared terminals, directly beneath the dorsal horn apex (see Fig. 1C, arrow). Behavioral and densitometric results are presented as means ± SEM.
Figure 1.
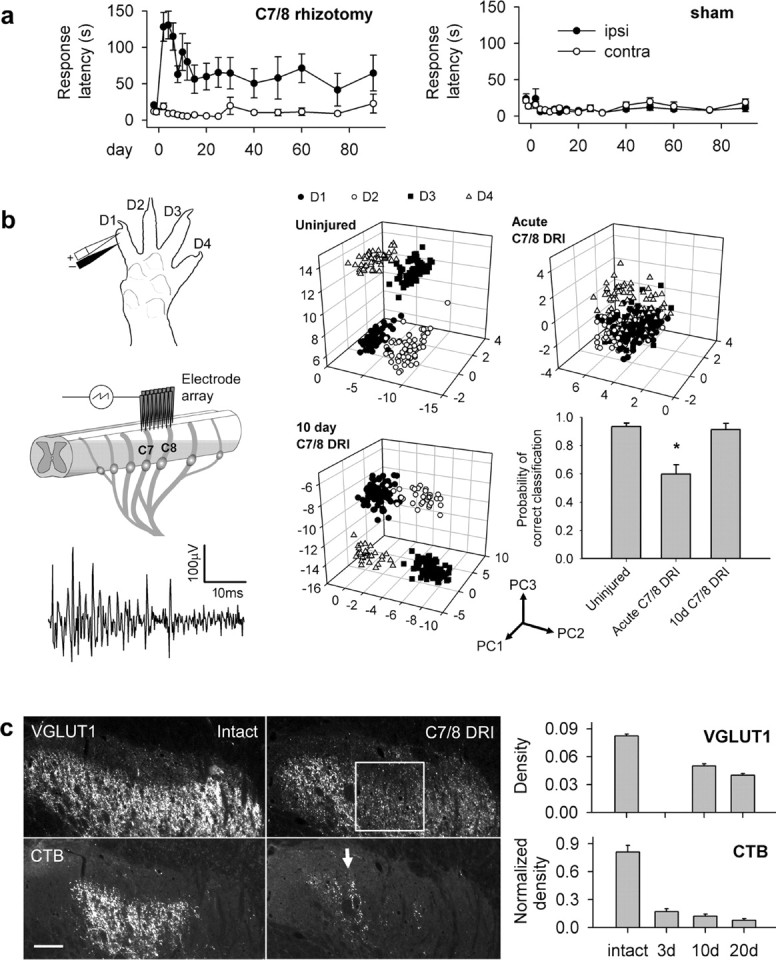
Behavioral and physiological deficits after C7/8 DRI partially recover in the absence of mechanosensory axon sprouting. a, Time to detect a sticker on the palm in sham-operated and C7/8 DRI rats. In rhizotomized animals, all postoperative ipsilateral (ipsi)–contralateral (contra) latencies differed significantly. b, Electrophysiological assessment of functional deficits and recovery. Left, Experimental setup and a sample trace from a single channel of the electrode array. Right, Feature space plots in various experimental conditions (Borisoff et al., 2006). Acute C7/8 rhizotomy decreased the discrimination of digit simulation clusters. Ten days after C7/8 DRI, all clusters were distinct, and the probability of correctly classifying evoked responses as being from separate digits was restored. Principal component (PC) axes are indicated. Asterisk indicates significant reduction in probability of correct digit classification. c, C7/8 DRI produces a prominent deafferentation gap in VGLUT1- and CTB-positive terminals in the C7 dorsal horn (box); density did not change therein for 20 d after injury. The arrow points to spared C6/T1 axons. Scale bar, 100 μm.
We also quantified the size distribution of VGLUT1-positive terminals in deafferented regions of the dorsal horn. To do so, images were captured at high power (63×, oil immersion), and the diameter of all structures distinguishable as individual boutons was measured in each field using SigmaScan Pro version 5.0. Because the distribution of profiles can lead to overestimation of the proportion of small-diameter boutons (attributable to end-cuts of larger-diameter ones), recursive translation (Rose and Rohrlich, 1988) was used to convert the distribution of bouton profiles to the distribution of whole boutons.
Statistics
A list of sample sizes for all experiments is presented in Table 1. All quantitative behavioral, histological, and electrophysiological data are expressed as means ± SEMs. For all statistical outcomes, significance was set at p < 0.05.
Behavior.
For the sticker, dynamic plantar anaesthesiometer and acetone tests, differences between ipsilateral and contralateral latencies were normally distributed and compared using Student's t test. For the sticker test, mean latency difference scores (ipsilateral minus contralateral) were averaged over plateau phases (days 10–20) and compared using a one-way ANOVA followed by the Holm-Sidak test for pairwise differences. To detect differences between IgG and TrkB-Fc bolus infusions in the same animals, we used a paired t test.
Electrophysiology.
The single trial digit-discrimination accuracy for each animal was calculated using five separate sequential (80% training and 20% test) feature sets and averaging the results (fivefold cross-validation). A parametric one-way ANOVA was used to detect differences in discrimination accuracy.
Histology.
Terminal densities were subjected to one-way ANOVAs followed by Holm-Sidak post hoc tests. Because DRI can result in contralateral effects (Ramer et al., 2004), ipsilateral densities after rhizotomy were compared with those in uninjured animals. Densities were all normally distributed. Statistical differences in size distributions of VGLUT1-positive terminals were detected using the Kolmogorov–Smirnov goodness-of-fit test.
Results
Partial mechanosensory recovery after C7/8 DRI
We first characterized changes in low-threshold mechanosensation after unilateral transection of the seventh and eighth cervical dorsal roots (C7/8 DRI). In experimental and sham-operated rats, we measured the time to attend to a sticker applied to the palm (Fig. 1a) (Bradbury et al., 2002). Normally, rats sensed the sticker within 11 ± 7 s of its application. Two days after C7/8 DRI, response latencies increased significantly (p < 0.001), to 128 ± 20 s. By 2 weeks after injury, detection latency had decreased to 56 ± 19 s, but was still significantly different from the contralateral side (p = 0.026), and remained so thereafter. Response latencies were unaffected in sham-operated rats, assessed concurrently in a blinded manner. These results demonstrate a rapid and spontaneous but incomplete return of mechanosensory function after C7/8 DRI.
It is conceivable that an increased detection rate may have been the result of supraspinal learning [i.e., an association between handling/deposition into the test environment (a potential conditioning stimulus) and the presence of a sticker (the test stimulus)]. Such associative learning was ruled out through mock behavioral trials in which the rats were handled and deposited into test cages in exactly the same way but did not have stickers applied. In these mock trials, rats did not attend to their forepaws as though stickers were present.
An electrophysiological correlate of behavioral recovery
Although partial spontaneous behavioral recovery implied emergent connectivity between decentralized dermatomes and spinal neurons and although mock behavioral trials (without stickers) did not elicit responses, we wanted to also verify that more rapid attention to the sticker was not purely a result of associative learning (Onifer et al., 2005). If primary afferent-driven activity at the level of the spinal cord did not return in parallel with behavioral recovery, this may indicate that cues other than the sticker elicit faster response times in behavioral experiments. We therefore tracked recovery electrophysiologically in the spinal cord (Borisoff et al., 2006) (Fig. 1b). Using an electrode microarray inserted into the dorsal horn, we sought to discriminate specific locations of peripheral electrical digit stimulation from postsynaptic neural activity. Evoked activity from each of four digits was subjected to machine learning classification using PCA and k-means clustering (Borisoff et al., 2006). Such classification generated identifiable clusters in three-dimensional feature spaces and reliably discriminated between the digits stimulated in intact animals. Acute C7/8 DRI significantly reduced the probability of digit discrimination (p < 0.001). However, 10 d after C7/8 DRI, discernable sites of digit stimulation reemerged, and classification accuracy was restored to that of the uninjured state. Together with mock sticker trials, these data indicate that more rapid sticker detection does not occur through associative learning, but is mediated by changes in the spinal cord. They also show that although changes in connectivity occur at the level of the dorsal horn to such an extent that digit discrimination probability is fully restored, they are insufficient to completely restore normal behavior.
C7/8 DRI produces a stable deafferentation gap in the dorsal horn
We next investigated changes in primary afferent innervation of the dorsal horn. Two markers were used: VGLUT1 and the axon tracer CTB, both of which label mechanosensory afferents in the rat dorsal horn (Todd et al., 2003; Alvarez et al., 2004; Persson et al., 2006) (Fig. 1c). VGLUT1 is a particularly useful marker because it only labels terminal boutons and not presynaptic growth cones; we found that CTB-labeled axotomized C7 and C8 axons halted at the dorsal root entry zone do not contain VGLUT1 (data not shown). C7/8 DRI produced a consistent deafferentation gap in laminas III–IV of the C7 dorsal horn. Densitometric analyses of VGLUT1- and CTB-positive terminals were performed within the deafferentation gap, by measuring terminal density as a function of depth in the dorsal horn (Ramer et al., 2001, 2004; MacDermid et al., 2004; Scott et al., 2005). After C7/8 DRI, the density of both VGLUT1- and CTB-labeled terminals was decreased but remained unchanged for at least 20 d postoperatively. VGLUT1 immunohistochemistry was not performed at the 3 d time point because of the confounding influence of slow clearance of degenerating primary afferents (George and Griffin, 1994). Spontaneous partial recovery therefore occurs in the absence of spinal mechanosensory axon sprouting.
C7/8 DRI induces BDNF and NT-3 expression in the dorsal horn
Because BDNF and NT-3 are upregulated in the spinal ventral horn after DRI (Johnson et al., 2000), we asked whether the same was true in the dorsal horn (Fig. 2). In uninjured animals, BDNF was restricted to superficial primary afferent terminals (Fig. 2a). C7/8 DRI induced BDNF expression in Ox-42-positive microglia/macrophages throughout the ipsilateral dorsal gray and white matter (Fig. 2e). Although the NT-3 antibody used efficiently labeled cerebellar Purkinje cells and their processes (Fig. 2f, inset), NT-3 immunoreactivity was undetectable in uninjured spinal cord (Fig. 2f). However, C7/8 DRI induced NT-3 expression in the ipsilateral gray and white matter (Fig. 2g–i). This was localized to blood vessel-associated astrocytic cell bodies and their processes (Fig. 2h,j). Both neurotrophins were upregulated by 3 d after injury and persisted for at least 20 d after DRI. Although nerve growth factor (NGF) and NT-4 immunoreactivities were obvious in positive control tissues (injured dorsal roots and cerebellar Purkinje cells, respectively), they were undetectable in uninjured or deafferented cords at all timepoints (data not shown).
Figure 2.
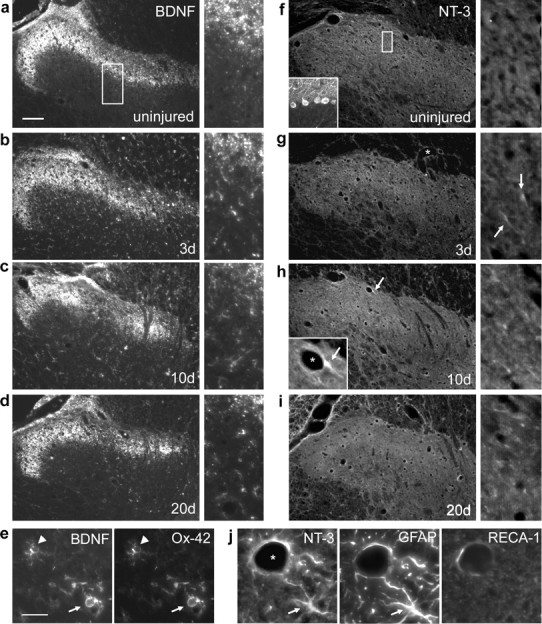
DRI induces BDNF and NT-3 expression in the dorsal horn. a, In the intact spinal cord, BDNF immunoreactivity was restricted to the terminals of primary afferent axons. Higher-power images from the same sections are shown on the right (boxes in lower-power images indicate their approximate origins). b–d, BDNF was upregulated in glial cells by 3 d after C7/8 DRI and remained elevated for at least 20 d. e, Ox-42 immunohistochemistry revealed BDNF expression by microglia in early (arrowhead) and intermediate (arrow) stages of reactivity. f–h, NT-3 immunoreactivity was intense in cerebellar Purkinje cells (inset), absent from the uninjured dorsal horn, but present in blood vessel-associated (asterisk) cellular processes as early as 3 d after DRI (g, h, arrows). h, i, DRI-induced NT-3 expression persisted for at least 20 d. j, NT-3 was expressed in glial fibrillary acidic protein (GFAP)-positive astrocytes around rat endothelial cell antigen 1 (RECA-1)-positive vasculature (a white-matter astrocyte from the degenerating dorsal columns is shown). Scale bars: a, 100 μm; e, i, 25 μm.
Continuous TrkB-Fc treatment improves mechanosensory recovery
To test the hypothesis that upregulated neurotrophins mediate partial recovery, we administered Trk-Fc “receptor bodies” intrathecally to sequester spinal BDNF/NT-4 and NT-3 (Cabelli et al., 1997; Seebach et al., 1999; Chan et al., 2001). An antibody specific for the human Fc moiety was used to assess spinal Trk-Fc penetration (Fig. 3a). Prominent staining was evident in dorsal white matter, and more so in dorsolateral white and gray matter, as we have demonstrated previously with a similar fusion protein (MacDermid et al., 2004). Behaviorally, IgG treatment resulted in a pattern of behavior similar to untreated rats in which mechanosensory ability reached a plateau that remained significantly different from the contralateral side (Fig. 3b). Surprisingly, intrathecal TrkC-Fc was without effect, and TrkB-Fc treatment actually enhanced recovery: ipsilateral and contralateral detection latencies were statistically equivalent (p = 0.08) by the 6th postoperative day and remained so thereafter. To directly determine whether the extent of recovery differed between groups, we compared response latency differences (ipsilateral minus contralateral response times) over the plateau phase (averaged data from days 10–20). There was a significant difference between IgG-treated (41 ± 9 s) and TrkB-Fc treated (8 ± 3 s; p = 0.016) rats, but not between IgG-treated and TrkC-Fc-treated rats (46 ± 16 s; p = 0.711).
Figure 3.

Sequestration of injury-induced BDNF promotes complete mechanosensory recovery after C7/8 DRI. a, Fc immunohistochemistry demonstrating spinal penetration of intrathecally delivered proteins. Scale bar, 100 μm. b, Only continuous TrkB-Fc treatment improved mechanosensation. c, Mechanical allodynia did not emerge in IgG- or TrkB-Fc-treated rats, and withdrawal thresholds were increased with TrkC-Fc treatment. Asterisks in b and c indicate significant ipsilateral (ipsi)–contralateral (contra) differences.
Possible mechanical allodynia after C7/8 DRI was examined using the Dynamic Plantar Aesthesiometer. These experiments were done to determine whether more rapid sticker detection was related to evolution of mechanical allodynia [i.e., did any pain (potentially associated with the presence of the sticker) motivate more rapid detection?] When increasing punctate force was applied to the center of the palmar forepaw, no differences in force at withdrawal were detected between forepaws in IgG-treated rats and TrkB-Fc-treated rats (Fig. 3c). In rats that received TrkC-Fc, withdrawal thresholds were increased significantly at 5 d (p = 0.005), 10 d (p = 0.003), and 15 d (p = 0.02) after DRI, possibly as a result of decreased synaptic efficacy between deep-terminating mechanosensory axons and motoneurons (Arvanian et al., 2003). These results indicate that mechanical allodynia did not incite more rapid sticker withdrawal in TrkB-Fc-treated rats.
Because of the unexpected improvement in mechanosensation afforded by TrkB-Fc treatment, we did not pursue electrophysiological assessment further: even in untreated rats, digit discrimination probability recovered to close to 100% by 10 d after DRI, rendering additional electrophysiological experiments superfluous.
TrkB-Fc treatment enhances mechanosensory recovery independent of effects on synaptic transmission
Of the two TrkB ligands, only BDNF has acute effects on neurotransmission in the spinal cord. Unlike BDNF, NT-4 has no effect on activity-dependent synaptic plasticity or neuropathic pain (Heppenstall and Lewin, 2001; Yajima et al., 2002). To determine whether potential neuromodulatory effects of BDNF contributed to mechanosensory recovery, we gave rats bolus intrathecal injections of either IgG or TrkB-Fc (2 μg of protein) on days 9 and 10 after DRI. We chose this time point based on the statistically complete recovery in rats treated continuously with TrkB-Fc. Mechanosensory deficits were unaffected by acute BDNF antagonism (Fig. 4c), indicating that recovery was not attributable to acute effects of BDNF on synaptic transmission.
Figure 4.

Acute BDNF antagonism does not affect mechanosensation 10 d after rhizotomy. a, Time course of mechanosensory recovery over the first 10 d after DRI. b, Time course of forepaw cold-pain development (increased response duration to an acetone squirt) in the same animals. Arrows indicate days on which intrathecal boli of IgG or TrkB-Fc were administered. c, Intrathecal boli of TrkB-Fc did not alter mechanosensory response latencies 30–90 min after administration. d, Acute BDNF antagonism reduced response durations to acetone in the same animals. Asterisks in a and b indicate significant ipsilateral (ipsi)–contralateral (contra) differences. Asterisks in c and d indicate significant differences between IgG andTrkB-Fc treatments.
To verify efficacy of bolus TrkB-Fc infusions, we measured rhizotomy-induced cold hypersensitivity in the same animals (Ramer et al., 2004).
We found that a 2 μg intrathecal bolus of TrkB-Fc reduced ipsilateral response durations to forepaw acetone stimulation from 9.2 ± 1.1 to 5.8 ± 0.8 s (p = 0.03) (Fig. 4d). Contralateral responses were unaffected. These results indicate a neuromodulatory role for endogenous spinal BDNF after DRI that contributes to cold pain and provide additional support for an absence of the acute synaptic involvement of BDNF in mediating improved mechanosensation.
TrkB-Fc treatment induces spinal mechanosensory axon sprouting
Could the unexpected improvement conferred by TrkB-Fc treatment be mediated by mechanosensory axon sprouting? VGLUT1-positive terminal density in the deafferentation gap was significantly increased by TrkB-Fc (Fig. 5a,c,d) at both 10 d (p = 0.008) and 20 d (p = 0.01) after C7/8 DRI, correlating with behavioral improvement. Because it is also expressed in corticospinal axons (Persson et al., 2006), we measured VGLUT1 density within lamina II, where corticospinal but not myelinated sensory axons terminate. No increases in density occurred in lamina II in any treatment group. TrkB-Fc treatment, but not IgG or TrkC-Fc-treatment, also increased CTB-positive terminal density in laminas III–IV (Fig. 5b,e) at 10 d (p = 0.02) and 20 d (p < 0.001) after DRI; CTB-positive terminals were absent from lamina II.
Figure 5.
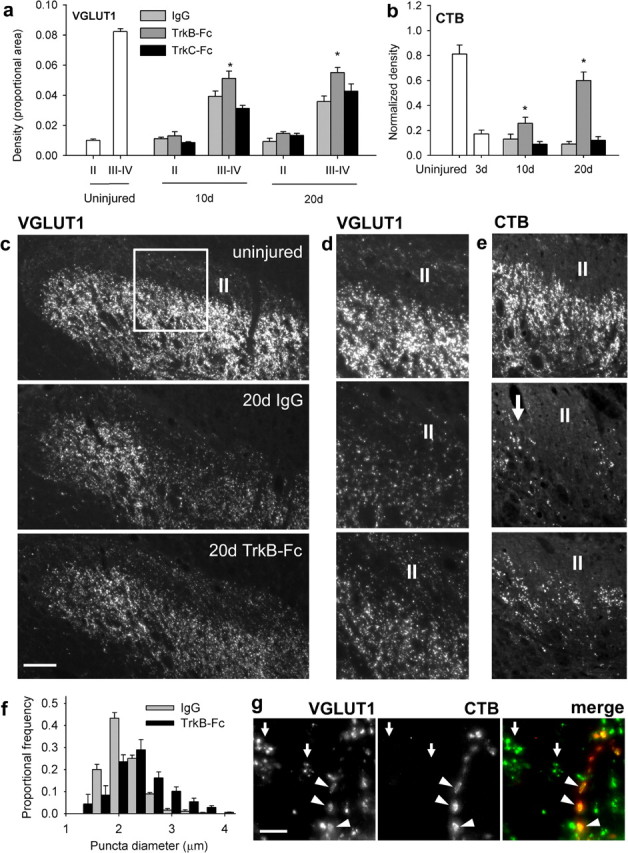
TrkB-Fc promotes mechanosensory axon sprouting after C7/8 DRI. a, b, VGLUT1-positive (a) and CTB-positive (b) terminal density in superficial (II) and deep (III–IV) spinal laminas in uninjured animals. DRI reduced density in deeper laminas, which was partially reversed by intrathecal TrkB-Fc. Asterisks indicate significant differences between IgG- and Trk-Fc-treated rats. c, Low-power micrographs of VGLUT1 immunoreactivity (lamina II indicated). d, e, Higher-power micrographs of VGLUT1 (d) and CTB (e) staining from regions similar to that indicated in c (arrow indicates spared C6/T1 axons). f, The size distribution of VGLUT1-positive terminals was significantly right-shifted in TrkB-Fc-treated rats. g, Large VGLUT1-positive terminals were also CTB filled (arrowheads). Clusters of small VGLUT1-positive terminals are indicated by arrows. Scale bars: c, 100 μm; g, 20 μm.
Because VGLUT1-containing mechanosensory terminals are larger than corticospinal boutons (Valtschanoff et al., 1993), we quantified the size distribution of VGLUT1-positive puncta in the deafferentation gap of 20 d IgG-treated (n = 1111 puncta from five rats) and TrkB-Fc-treated (n = 1270 puncta from five rats) animals. This was performed to determine whether the increase in density could have been attributable to corticospinal axons. Terminals were significantly larger (p < 0.05) in TrkB-Fc-treated rats, indicating that mechanosensory axon terminals were present in higher proportions in TrkB-Fc-treated rats than IgG-treated controls (Fig. 5f). Larger terminals were also CTB filled (Fig. 5g, arrowheads). Thus, endogenous TrkB ligands suppress mechanosensory axon sprouting after DRI. These data also validate VGLUT1 as a reliable marker of mechanosensory axons.
C7/8 DRI induces TrkB-dependent serotonergic sprouting in the dorsal horn
To determine whether endogenous DRI-induced neurotrophins had a generalized suppressive effect on axonal plasticity, we analyzed changes in spinal serotonergic axon density. Serotonergic sprouting is a well recognized consequence of spinal deafferentation (Polistina et al., 1990; Wang et al., 1991; Zhang et al., 1993; Kinkead et al., 1998; MacDermid et al., 2004; Ramer et al., 2004; Scott et al., 2005), although the underlying mechanism has remained unknown. Because descending serotonergic axons express TrkB and TrkC (King et al., 1999), we asked whether the responsible stimulus was BDNF/NT-4 and/or NT-3. Serotonergic axon density had increased significantly in outer lamina II (IIo; p = 0.004), inner lamina II (IIi; p = 0.01), and laminas III/IV (p < 0.001) by 20 d after DRI in rats treated with IgG (Fig. 6a–c). The same was true for TrkC-Fc-treated rats (IIo, p < 0.001; IIi, p = 0.005; III/IV, p < 0.001). TrkB-Fc treatment completely prevented serotonergic sprouting, illustrating that for some axonal populations, BDNF/NT-4 does indeed enhance plasticity. These results support the notion of hierarchical plasticity in the spinal cord after spinal deafferentation (Polistina et al., 1990): processes that favor sprouting of some axonal populations (serotonergic) also suppress sprouting in others (mechanosensory primary afferent).
Figure 6.
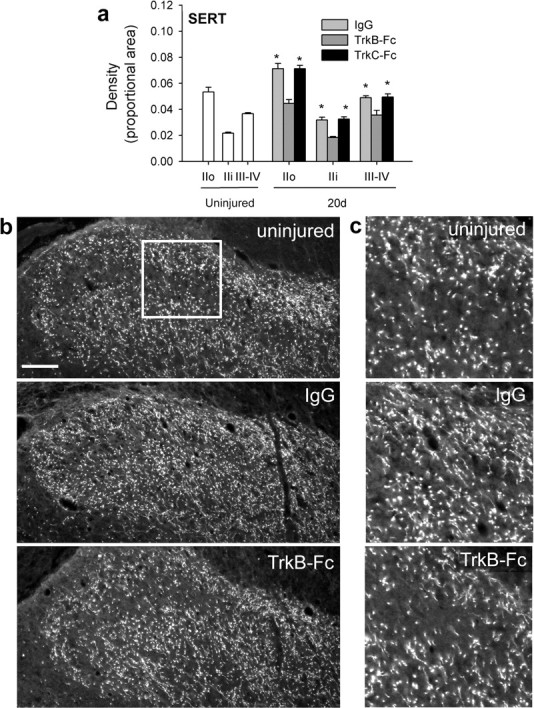
Endogenous TrkB ligands stimulate serotonergic sprouting after C7/8 DRI. a, Serotonergic axon density measurements from superficial (IIo and IIi) and deeper (III/IV) spinal laminas: TrkB-Fc treatment prevented DRI-induced sprouting. Asterisks indicate significant differences from intact rats. b, Low-power micrographs of serotonin transporter (SERT)-expressing axons in the spinal dorsal horn. c, Higher-power micrographs of SERT immunohistochemistry from regions similar to that indicated in b. Scale bar, 100 μm.
TrkB antagonsim promotes functional plasticity independent of NT-3
We next asked what role spinally upregulated NT-3/TrkC signaling might play in functional recovery and plasticity in the absence of BDNF/NT-4/TrkB signaling. We continuously administered the pan-Trk antagonist K252a (Knusel and Hefti, 1992) intrathecally in animals with C7/8 DRI, reasoning that if NT-3 was responsible for functional improvements and enhanced plasticity in the absence of TrkB signaling, additionally blocking TrkC signaling would abolish both effects. Like TrkB-Fc, intrathecal K252a significantly improved the behavioral performance of rhizotomized animals (Fig. 7a): ipsilateral and contralateral detection latencies were equivalent by the 8th postoperative day (p = 0.08) and remained so thereafter. Plateau latency difference scores were significantly higher in vehicle-treated rats than in K252a-treated rats (56 ± 16 s vs 18 ± 6 s; p = 0.04).
Figure 7.

K252a improves mechanosensation and enhances mechanosensory sprouting after C7/8 DRI. a, Ipsilateral-contralateral latencies remained significantly different in vehicle-treated rats; differences disappeared by 8 d after DRI in K252a-treated rats. Asterisks indicate significant ipsilateral (ipsi)–contralateral (contra) differences. b, VGLUT1 density (pooled 10 and 20 d animals) was elevated after K252a treatment. Asterisks indicate significant difference from deafferented control rats. c, VGLUT1 immunohistochemistry. Scale bar, 100 μm.
K252a treatment also significantly increased the density of VGLUT1-positive terminals in the deafferentation gap in laminas III–IV (Fig. 7b,c) (p < 0.001). Elevated NT-3 therefore does not contribute to enhanced recovery and plasticity when TrkB is antagonized.
Discussion
We set about this work to test the hypothesis that spinally upregulated BDNF and/or NT-3 mediate partial spontaneous mechanosensory recovery after limited spinal deafferentation, a rationale supported not only by the known neuromodulatory effects of these neurotrophins (particularly those of BDNF) (Kerr et al., 1999; Arvanian et al., 2003; Ji et al., 2003; Baker-Herman et al., 2004; Coull et al., 2005; Vaynman and Gomez-Pinilla, 2005), but also by their roles in mechanosensory development (Airaksinen et al., 1996; Carroll et al., 1998) and regeneration (Bradbury et al., 1999; Ramer et al., 2000, 2002). We find that spontaneous recovery during the early postoperative period occurs in the absence of primary afferent sprouting and is, in fact, neurotrophin independent: recovery was identical between Trk antagonizing treatments and their respective controls. The more surprising outcome is that beyond the spontaneous recovery period, endogenous TrkB ligands have a suppressive effect on mechanosensory plasticity: TrkB antagonism results in complete recovery of mechanosensation. Acute neuromodulatory effects of BDNF do not underpin recovery because bolus injections of TrkB-Fc were without effect. Finally, we show that, in addition to promoting behavioral recovery, TrkB antangonism stimulates mechanosensory axon sprouting in the deafferented spinal cord. Endogenous BDNF/NT-4 do not have a general, plasticity-limiting influence on spinal axons, because TrkB-Fc treatment prevented rhizotomy-induced sprouting of serotonergic axons in the dorsal horn.
Potential mechanisms of spontaneous recovery after DRI
The reactivation of dorsal horn neurons by electrical stimulation of decentralized digits not only provides an electrophysiological correlate of behavioral improvement but also illustrates the spinal nature of spontaneous recovery. It is conceivable, for example, that faster response times could have been caused solely by learning-associated changes at supraspinal loci and may not have manifested as increased discrimination probability at the level of the spinal cord. However, the reappearance of primary afferent-driven activity occurred in the absence of training (a prerequisite for associative learning). The same holds for experiments in which cats underwent similar electrophysiological studies in the absence of behavioral training (Basbaum and Wall, 1976). These findings argue very strongly against supraspinal associative learning as underlying neurophysiological recovery at the level of the spinal cord.
The present results implicate a recovery mechanism operating locally, within the partially deafferented cord. Synaptic unmasking and/or strengthening is the most plausible mechanism. A physiological process that may underlie partial recovery is the GABAergic presynaptic control [via primary afferent depolarization (PAD)] of impulse transmission along branches of individual mechanosensory axons (Wall, 1995): although some collaterals of myelinated axons fail to propagate action potentials in intact animals, this conduction block is reversed with time after rhizotomy of caudal segments. Successful conduction emerges by 5–7 d after DRI, correlating remarkably well with the partial behavioral recovery reported here.
In the rat, many C6 and T1 sensory neurons also innervate C7 and C8 dermatomes (Ramer et al., 2004). Therefore, the most likely mechanism underlying spontaneous recovery involves the (neurotrophin-independent) unmasking and/or strengthening of synapses between C6/T1 primary afferents and postsynaptic targets. Although others have suggested that this functional connectivity emerges subsequent to deafferentation-induced sprouting (Sengelaub et al., 1997; Darian-Smith and Brown, 2000; Darian-Smith, 2004), such axonal plasticity occurs too slowly to account for rapid partial spontaneous recovery after C7/8 DRI.
TrkB ligands arrest full spontaneous mechanosensory recovery independent of synaptic neuromodulation
BDNF (but not NT-4) has been shown to both enhance and suppress synaptic transmission in the dorsal horn (Pezet and McMahon, 2006). Our original hypothesis was based in part on the previously demonstrated GABAergic suppression of impulse transmission along branches of mechanosensory axons (Wall and McMahon, 1994; Wall, 1995) and on the known effects of BDNF on this type of inhibitory control. Because exogenous BDNF enhances GABA release in the spinal cord (Pezet et al., 2002), one might have expected DRI-induced BDNF to augment GABA-mediated PAD. Strengthened PAD would lead to successful action potential propagation in previously blocked Aβ fiber collaterals and activation of second-order neurons, a mechanism that has been proposed to underlie increased activation of pain-responsive second-order neurons in nerve injury and inflammation (Willis, 1999). BDNF is also known to render postsynaptic GABAergic inhibition of nociceptive projection neurons excitatory (Coull et al., 2005). The potential applicability of this mechanism to mechanosensory second-order neurons, or to wide dynamic-range neurons, had initially led us to predict that the effects of BDNF on GABAergic transmission participated in partial spontaneous recovery.
On the other hand, BDNF has been shown to inhibit glutamatergic signaling in the nucleus of the solitatry tract (Balkowiec et al., 2000) and in lateral motor nuclei in the spinal cord (Seebach et al., 1999; Arvanian and Mendell, 2001). It might therefore have been argued that endogenous BDNF prevents complete recovery by inhibiting glutamatergic transmission in the dorsal horn. However, because recovery occurred at its usual rate over the first several days after DRI even in the presence of TrkB-Fc (and K252a), we conclude that it is attributable neither to the effects of BDNF on GABAergic transmission nor on glutamatergic transmission during this period. Furthermore, acute treatment with a bolus injection of TrkB-Fc had no effect on mechanosensory ability during the plateau phase after DRI, despite significantly reducing cold pain.
In neonatal rats, chronic TrkB-Fc treatment has been shown to strengthen the connection between Ia afferents and motoneurons, evident as an increase in the size of EPSPs (Seebach et al., 1999). It might therefore be argued that TrkB-Fc treatment results in larger EPSPs in dorsal horn neurons subsequent to peripheral stimulation of cutaneous mechanosensory axons. Although this is an interesting question, its relevance to behavioral recovery becomes questionable when we consider that because of sprouting and synapse formation (the latter implied by the increased density of VGLUT1-positive terminals), more dorsal horn neurons are being driven by the same number of dorsal root axons. That is, the behavioral improvement that occurs with TrkB-Fc treatment may simply be the result of the larger population of neurons activated by C6/T1 primary afferents.
How does TrkB-Fc treatment promote primary afferent sprouting?
It is important to define putative direct and indirect processes by which BDNF (and possibly NT-4) suppresses plasticity. Evidence for a direct mechanism comes from work on dissociated adult sensory neurons (Gavazzi et al., 1999) where addition of BDNF had a marked inhibitory effect on spontaneous neurite outgrowth from large-diameter cells. Such inhibition may involve the p75 neurotrophin receptor, which in DRG neurons is coexpressed with Trk receptors (Wright and Snider, 1995): activating p75 with BDNF in TrkB-negative cells inhibits outgrowth mediated by NGF/TrkA signaling (MacPhee and Barker, 1997; Kimpinski et al., 1999; Kohn et al., 1999), suggesting that in the present experiments, spinally upregulated BDNF (or NT-4) may have been similarly interfering with NT-3/TrkC signaling. However, TrkB-Fc and K252a data were remarkably similar, indicating that whether or not p75 is involved, improved function and anatomical changes occurring in TrkB-Fc-treated animals were NT-3 independent. In combination with the absence of TrkA from low-threshold mechanosensory axons (McMahon et al., 1994; Wright and Snider, 1995), and no evidence for NGF expression in the spinal cord or upregulation after DRI, the K252a data also argue against a role for NGF-TrkA signaling in mechanosensory improvement.
TrkB activation is more likely to indirectly suppress mechanosensory axon sprouting by selectively enhancing plasticity in other spinally projecting systems. Both BDNF and NT-4, whether delivered via osmotic minipumps or genetically modified cell grafts, can induce growth of serotonergic fibers in the injured spinal cord (Bregman et al., 1997; Tobias et al., 2003; Blesch et al., 2004). TrkB-expressing descending serotonergic fibers (Bradbury et al., 1998; King et al., 1999) sprout earlier and more profusely after DRI than primary afferent and descending noradrenergic axons (Zhang et al., 1993), and the present data show for the first time that removing the influence of endogenous TrkB ligands prevents rhizotomy-induced serotonergic sprouting. Although exogenous or virally overexpressed NT-3 has also been shown to promote serotonergic axon growth after CNS lesions (Bregman et al., 1997; Grider et al., 2005), the present results demonstrate that endogenous NT-3 is not necessary for DRI-induced sprouting.
It is now clear that endogenous TrkB ligands have an important negative impact on mechanosensory plasticity after DRI. Although the relative contribution of NT-4 to these results has yet to be formally defined, it is likely to be minor because its mRNA is expressed at very low levels compared with other neurotrophins (Timmusk et al., 1993; Ibanez, 1996; Widenfalk et al., 2001) and its protein is undetectable in the intact and deafferented cord (data not shown). Because optimizing synaptic and axonal plasticity of CNS-projecting axons remains a necessary objective in the search for a viable therapy for neural trauma (Scott et al., 2006), clarifying the positive and negative effects of injury-induced molecules such as BDNF represents a vital intermediate step.
Footnotes
This work was supported by grants from the Canadian Institutes of Health Research (CIHR) and the International Spinal Research Trust. L.M.R. is a Michael Smith Foundation for Health Research (MSFHR) and a Natural Science and Engineering Council of Canada (NSERC) senior graduate trainee. L.T.M. and J.F.B. are MSFHR and NSERC-supported postdoctoral fellows. T.K.Y.K., J.W.T.S., and L.P.R.H. were supported by NSERC summer studentships. M.S.R. is a MSFHR Senior Scholar and a CIHR New Investigator.
References
- Airaksinen MS, Koltzenburg M, Lewin GR, Masu Y, Helbig C, Wolf E, Brem G, Toyka KV, Thoenen H, Meyer M. Specific subtypes of cutaneous mechanoreceptors require neurotrophin-3 following peripheral target innervation. Neuron. 1996;16:287–295. doi: 10.1016/s0896-6273(00)80047-1. [DOI] [PubMed] [Google Scholar]
- Alvarez FJ, Villalba RM, Zerda R, Schneider SP. Vesicular glutamate transporters in the spinal cord, with special reference to sensory primary afferent synapses. J Comp Neurol. 2004;472:257–280. doi: 10.1002/cne.20012. [DOI] [PubMed] [Google Scholar]
- Arvanian VL, Mendell LM. Acute modulation of synaptic transmission to motoneurons by BDNF in the neonatal rat spinal cord. Eur J Neurosci. 2001;14:1800–1808. doi: 10.1046/j.0953-816x.2001.01811.x. [DOI] [PubMed] [Google Scholar]
- Arvanian VL, Horner PJ, Gage FH, Mendell LM. Chronic neurotrophin-3 strengthens synaptic connections to motoneurons in the neonatal rat. J Neurosci. 2003;23:8706–8712. doi: 10.1523/JNEUROSCI.23-25-08706.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker-Herman TL, Fuller DD, Bavis RW, Zabka AG, Golder FJ, Doperalski NJ, Johnson RA, Watters JJ, Mitchell GS. BDNF is necessary and sufficient for spinal respiratory plasticity following intermittent hypoxia. Nat Neurosci. 2004;7:48–55. doi: 10.1038/nn1166. [DOI] [PubMed] [Google Scholar]
- Balkowiec A, Kunze DL, Katz DM. Brain-derived neurotrophic factor acutely inhibits AMPA-mediated currents in developing sensory relay neurons. J Neurosci. 2000;20:1904–1911. doi: 10.1523/JNEUROSCI.20-05-01904.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbaum AI, Wall PD. Chronic changes in the response of cells in adult cat dorsal horn following partial deafferentation: the appearance of responding cells in a previously non-responsive region. Brain Res. 1976;116:181–204. doi: 10.1016/0006-8993(76)90899-4. [DOI] [PubMed] [Google Scholar]
- Blesch A, Yang H, Weidner N, Hoang A, Otero D. Axonal responses to cellularly delivered NT-4/5 after spinal cord injury. Mol Cell Neurosci. 2004;27:190–201. doi: 10.1016/j.mcn.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Borisoff JF, McPhail LT, Saunders JWT, Birch GE, Ramer MS. Detection and classification of sensory information from acute spinal cord recordings. IEEE Trans Biomed Eng. 2006;53:1715–1719. doi: 10.1109/TBME.2006.877800. [DOI] [PubMed] [Google Scholar]
- Bradbury EJ, King VR, Simmons LJ, Priestley JV, McMahon SB. NT-3, but not BDNF, prevents atrophy and death of axotomized spinal cord projection neurons. Eur J Neurosci. 1998;10:3058–3068. doi: 10.1046/j.1460-9568.1998.00307.x. [DOI] [PubMed] [Google Scholar]
- Bradbury EJ, Khemani S, Von R, King, Priestley JV, McMahon SB. NT-3 promotes growth of lesioned adult rat sensory axons ascending in the dorsal columns of the spinal cord. Eur J Neurosci. 1999;11:3873–3883. doi: 10.1046/j.1460-9568.1999.00809.x. [DOI] [PubMed] [Google Scholar]
- Bradbury EJ, Moon LD, Popat RJ, King VR, Bennett GS, Patel PN, Fawcett JW, McMahon SB. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature. 2002;416:636–640. doi: 10.1038/416636a. [DOI] [PubMed] [Google Scholar]
- Bregman BS, McAtee M, Dai HN, Kuhn PL. Neurotrophic factors increase axonal growth after spinal cord injury and transplantation in the adult rat. Exp Neurol. 1997;148:475–494. doi: 10.1006/exnr.1997.6705. [DOI] [PubMed] [Google Scholar]
- Cabelli RJ, Shelton DL, Segal RA, Shatz CJ. Blockade of endogenous ligands of trkB inhibits formation of ocular dominance columns. Neuron. 1997;19:63–76. doi: 10.1016/s0896-6273(00)80348-7. [DOI] [PubMed] [Google Scholar]
- Carroll P, Lewin GR, Koltzenburg M, Toyka KV, Thoenen H. A role for BDNF in mechanosensation. Nat Neurosci. 1998;1:42–46. doi: 10.1038/242. [DOI] [PubMed] [Google Scholar]
- Chan JR, Cosgaya JM, Wu YJ, Shooter EM. Neurotrophins are key mediators of the myelination program in the peripheral nervous system. Proc Natl Acad Sci USA. 2001;98:14661–14668. doi: 10.1073/pnas.251543398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- Darian-Smith C. Primary afferent terminal sprouting after a cervical dorsal rootlet section in the macaque monkey. J Comp Neurol. 2004;470:134–150. doi: 10.1002/cne.11030. [DOI] [PubMed] [Google Scholar]
- Darian-Smith C, Brown S. Functional changes at periphery and cortex following dorsal root lesions in adult monkeys. Nat Neurosci. 2000;3:476–481. doi: 10.1038/74852. [DOI] [PubMed] [Google Scholar]
- Darian-Smith C, Ciferri MM. Loss and recovery of voluntary hand movements in the macaque following a cervical dorsal rhizotomy. J Comp Neurol. 2005;491:27–45. doi: 10.1002/cne.20686. [DOI] [PubMed] [Google Scholar]
- Gavazzi I, Kumar RD, McMahon SB, Cohen J. Growth responses of different subpopulations of adult sensory neurons to neurotrophic factors in vitro. Eur J Neurosci. 1999;11:3405–3414. doi: 10.1046/j.1460-9568.1999.00756.x. [DOI] [PubMed] [Google Scholar]
- George R, Griffin JW. Delayed macrophage responses and myelin clearance during Wallerian degeneration in the central nervous system: the dorsal radiculotomy model. Exp Neurol. 1994;129:225–236. doi: 10.1006/exnr.1994.1164. [DOI] [PubMed] [Google Scholar]
- Goldberger ME, Murray M. Restitution of function and collateral sprouting in the cat spinal cord: the deafferented animal. J Comp Neurol. 1974;158:37–53. doi: 10.1002/cne.901580104. [DOI] [PubMed] [Google Scholar]
- Grider MH, Mamounas LA, Le W, Shine HD. In situ expression of brain-derived neurotrophic factor or neurotrophin-3 promotes sprouting of cortical serotonergic axons following a neurotoxic lesion. J Neurosci Res. 2005;82:404–412. doi: 10.1002/jnr.20635. [DOI] [PubMed] [Google Scholar]
- Heppenstall PA, Lewin GR. BDNF but not NT-4 is required for normal flexion reflex plasticity and function. Proc Natl Acad Sci USA. 2001;98:8107–8112. doi: 10.1073/pnas.141015098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibanez CF. Neurotrophin-4: the odd one out in the neurotrophin family. Neurochem Res. 1996;21:787–793. doi: 10.1007/BF02532301. [DOI] [PubMed] [Google Scholar]
- Ji RR, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci. 2003;26:696–705. doi: 10.1016/j.tins.2003.09.017. [DOI] [PubMed] [Google Scholar]
- Johnson RA, Okragly AJ, Haak-Frendscho M, Mitchell GS. Cervical dorsal rhizotomy increases brain-derived neurotrophic factor and neurotrophin-3 expression in the ventral spinal cord. J Neurosci. 2000;20(RC77):1–5. doi: 10.1523/JNEUROSCI.20-10-j0005.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaegi S, Schwab ME, Dietz V, Fouad K. Electromyographic activity associated with spontaneous functional recovery after spinal cord injury in rats. Eur J Neurosci. 2002;16:249–258. doi: 10.1046/j.1460-9568.2002.02076.x. [DOI] [PubMed] [Google Scholar]
- Kerr BJ, Bradbury EJ, Bennett DL, Trivedi PM, Dassan P, French J, Shelton DB, McMahon SB, Thompson SW. Brain-derived neurotrophic factor modulates nociceptive sensory inputs and NMDA-evoked responses in the rat spinal cord. J Neurosci. 1999;19:5138–5148. doi: 10.1523/JNEUROSCI.19-12-05138.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimpinski K, Jelinski S, Mearow K. The anti-p75 antibody, MC192, and brain-derived neurotrophic factor inhibit nerve growth factor-dependent neurite growth from adult sensory neurons. Neuroscience. 1999;93:253–263. doi: 10.1016/s0306-4522(99)00156-6. [DOI] [PubMed] [Google Scholar]
- King VR, Michael GJ, Joshi RK, Priestley JV. trkA, trkB, and trkC messenger RNA expression by bulbospinal cells of the rat. Neuroscience. 1999;92:935–944. doi: 10.1016/s0306-4522(99)00072-x. [DOI] [PubMed] [Google Scholar]
- Kinkead R, Zhan WZ, Prakash YS, Bach KB, Sieck GC, Mitchell GS. Cervical dorsal rhizotomy enhances serotonergic innervation of phrenic motoneurons and serotonin-dependent long-term facilitation of respiratory motor output in rats. J Neurosci. 1998;18:8436–8443. doi: 10.1523/JNEUROSCI.18-20-08436.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knusel B, Hefti F. K-252 compounds: modulators of neurotrophin signal transduction. J Neurochem. 1992;59:1987–1996. doi: 10.1111/j.1471-4159.1992.tb10085.x. [DOI] [PubMed] [Google Scholar]
- Kohn J, Aloyz RS, Toma JG, Haak-Frendscho M, Miller FD. Functionally antagonistic interactions between the TrkA and p75 neurotrophin receptors regulate sympathetic neuron growth and target innervation. J Neurosci. 1999;19:5393–5408. doi: 10.1523/JNEUROSCI.19-13-05393.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDermid VE, McPhail LT, Tsang B, Rosenthal A, Davies A, Ramer MS. A soluble Nogo receptor differentially affects plasticity of spinally projecting axons. Eur J Neurosci. 2004;20:2567–2579. doi: 10.1111/j.1460-9568.2004.03715.x. [DOI] [PubMed] [Google Scholar]
- MacPhee IJ, Barker PA. Brain-derived neurotrophic factor binding to the p75 neurotrophin receptor reduces TrkA signaling while increasing serine phosphorylation in the TrkA intracellular domain. J Biol Chem. 1997;272:23547–23551. doi: 10.1074/jbc.272.38.23547. [DOI] [PubMed] [Google Scholar]
- McMahon SB, Armanini MP, Ling LH, Phillips HS. Expression and coexpression of Trk receptors in subpopulations of adult primary sensory neurons projecting to identified peripheral targets. Neuron. 1994;12:1161–1171. doi: 10.1016/0896-6273(94)90323-9. [DOI] [PubMed] [Google Scholar]
- McPhail LT, Borisoff JF, Tsang B, Hwi LP, Kwiecien JM, Ramer MS. Protracted myelin clearance hinders central primary afferent regeneration following dorsal rhizotomy and delayed neurotrophin-3 treatment. Neurosci Lett. 2007;411:206–211. doi: 10.1016/j.neulet.2006.09.076. [DOI] [PubMed] [Google Scholar]
- Moreno-Flores MT, Bradbury EJ, Martin-Bermejo MJ, Agudo M, Lim F, Pastrana E, Avila J, Diaz-Nido J, McMahon SB, Wandosell F. A clonal cell line from immortalized olfactory ensheathing glia promotes functional recovery in the injured spinal cord. Mol Ther. 2006;13:598–608. doi: 10.1016/j.ymthe.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Murray M, Goldberger ME. Restitution of function and collateral sprouting in the cat spinal cord: the partially hemisected animal. J Comp Neurol. 1974;158:19–36. doi: 10.1002/cne.901580103. [DOI] [PubMed] [Google Scholar]
- Onifer SM, Zhang YP, Burke DA, Brooks DL, Decker JA, McClure NJ, Floyd AR, Hall J, Proffitt BL, Shields CB, Magnuson DS. Adult rat forelimb dysfunction after dorsal cervical spinal cord injury. Exp Neurol. 2005;192:25–38. doi: 10.1016/j.expneurol.2004.10.016. [DOI] [PubMed] [Google Scholar]
- Persson S, Boulland JL, Aspling M, Larsson M, Fremeau RT, Jr, Edwards RH, Storm-Mathisen J, Chaudhry FA, Broman J. Distribution of vesicular glutamate transporters 1 and 2 in the rat spinal cord, with a note on the spinocervical tract. J Comp Neurol. 2006;497:683–701. doi: 10.1002/cne.20987. [DOI] [PubMed] [Google Scholar]
- Pezet S, McMahon SB. Neurotrophins: mediators and modulators of pain. Annu Rev Neurosci. 2006;29:507–538. doi: 10.1146/annurev.neuro.29.051605.112929. [DOI] [PubMed] [Google Scholar]
- Pezet S, Cunningham J, Patel J, Grist J, Gavazzi I, Lever IJ, Malcangio M. BDNF modulates sensory neuron synaptic activity by a facilitation of GABA transmission in the dorsal horn. Mol Cell Neurosci. 2002;21:51–62. doi: 10.1006/mcne.2002.1166. [DOI] [PubMed] [Google Scholar]
- Polistina DC, Murray M, Goldberger ME. Plasticity of dorsal root and descending serotoninergic projections after partial deafferentation of the adult rat spinal cord. J Comp Neurol. 1990;299:349–363. doi: 10.1002/cne.902990307. [DOI] [PubMed] [Google Scholar]
- Ramer LM, Borisoff JF, Ramer MS. Rho-kinase inhibition enhances axonal plasticity and attenuates cold hyperalgesia after dorsal rhizotomy. J Neurosci. 2004;24:10796–10805. doi: 10.1523/JNEUROSCI.3337-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramer MS, Priestley JV, McMahon SB. Functional regeneration of sensory axons into the adult spinal cord. Nature. 2000;403:312–316. doi: 10.1038/35002084. [DOI] [PubMed] [Google Scholar]
- Ramer MS, Bradbury EJ, McMahon SB. Nerve growth factor induces P2X(3) expression in sensory neurons. J Neurochem. 2001;77:864–875. doi: 10.1046/j.1471-4159.2001.00288.x. [DOI] [PubMed] [Google Scholar]
- Ramer MS, Bishop T, Dockery P, Mobarak MS, O'Leary D, Fraher JP, Priestley JV, McMahon SB. Neurotrophin-3-mediated regeneration and recovery of proprioception following dorsal rhizotomy. Mol Cell Neurosci. 2002;19:239–249. doi: 10.1006/mcne.2001.1067. [DOI] [PubMed] [Google Scholar]
- Rose RD, Rohrlich D. Counting sectioned cells via mathematical reconstruction. J Comp Neurol. 1988;272:365–386. [PubMed] [Google Scholar]
- Scott AL, Borisoff JF, Ramer MS. Deafferentation and neurotrophin-mediated intraspinal sprouting: a central role for the p75 neurotrophin receptor. Eur J Neurosci. 2005;21:81–92. doi: 10.1111/j.1460-9568.2004.03838.x. [DOI] [PubMed] [Google Scholar]
- Scott AL, Ramer LM, Soril LJ, Ramer MS. Targeting myelin to optimize plasticity of spared spinal axons. Mol Neurobiol. 2006;33:91–111. doi: 10.1385/MN:33:2:91. [DOI] [PubMed] [Google Scholar]
- Seebach BS, Arvanov V, Mendell LM. Effects of BDNF and NT-3 on development of Ia/motoneuron functional connectivity in neonatal rats. J Neurophysiol. 1999;81:2398–2405. doi: 10.1152/jn.1999.81.5.2398. [DOI] [PubMed] [Google Scholar]
- Sengelaub DR, Muja N, Mills AC, Myers WA, Churchill JD, Garraghty PE. Denervation-induced sprouting of intact peripheral afferents into the cuneate nucleus of adult rats. Brain Res. 1997;769:256–262. doi: 10.1016/s0006-8993(97)00708-7. [DOI] [PubMed] [Google Scholar]
- Starkey ML, Barritt AW, Yip PK, Davies M, Hamers FP, McMahon SB, Bradbury EJ. Assessing behavioral function following a pyramidotomy lesion of the corticospinal tract in adult mice. Exp Neurol. 2005;195:524–539. doi: 10.1016/j.expneurol.2005.06.017. [DOI] [PubMed] [Google Scholar]
- Sung B, Lim G, Mao J. Altered expression and uptake activity of spinal glutamate transporters after nerve injury contribute to the pathogenesis of neuropathic pain in rats. J Neurosci. 2003;23:2899–2910. doi: 10.1523/JNEUROSCI.23-07-02899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thallmair M, Metz GA, Z'Graggen WJ, Raineteau O, Kartje GL, Schwab ME. Neurite growth inhibitors restrict plasticity and functional recovery following corticospinal tract lesions. Nat Neurosci. 1998;1:124–131. doi: 10.1038/373. [DOI] [PubMed] [Google Scholar]
- Timmusk T, Belluardo N, Metsis M, Persson H. Widespread and developmentally regulated expression of neurotrophin-4 mRNA in rat brain and peripheral tissues. Eur J Neurosci. 1993;5:605–613. doi: 10.1111/j.1460-9568.1993.tb00526.x. [DOI] [PubMed] [Google Scholar]
- Tobias CA, Shumsky JS, Shibata M, Tuszynski MH, Fischer I, Tessler A, Murray M. Delayed grafting of BDNF and NT-3 producing fibroblasts into the injured spinal cord stimulates sprouting, partially rescues axotomized red nucleus neurons from loss and atrophy, and provides limited regeneration. Exp Neurol. 2003;184:97–113. doi: 10.1016/s0014-4886(03)00394-7. [DOI] [PubMed] [Google Scholar]
- Todd AJ, Hughes DI, Polgar E, Nagy GG, Mackie M, Ottersen OP, Maxwell DJ. The expression of vesicular glutamate transporters VGLUT1 and VGLUT2 in neurochemically defined axonal populations in the rat spinal cord with emphasis on the dorsal horn. Eur J Neurosci. 2003;17:13–27. doi: 10.1046/j.1460-9568.2003.02406.x. [DOI] [PubMed] [Google Scholar]
- Valtschanoff JG, Weinberg RJ, Rustioni A. Amino acid immunoreactivity in corticospinal terminals. Exp Brain Res. 1993;93:95–103. doi: 10.1007/BF00227784. [DOI] [PubMed] [Google Scholar]
- Vaynman S, Gomez-Pinilla F. License to run: exercise impacts functional plasticity in the intact and injured central nervous system by using neurotrophins. Neurorehabil Neural Repair. 2005;19:283–295. doi: 10.1177/1545968305280753. [DOI] [PubMed] [Google Scholar]
- Wall PD. Do nerve impulses penetrate terminal arborizations? A pre-presynaptic control mechanism. Trends Neurosci. 1995;18:99–103. doi: 10.1016/0166-2236(95)93883-y. [DOI] [PubMed] [Google Scholar]
- Wall PD, McMahon SB. Long range afferents in rat spinal cord. III. Failure of impulse transmission in axons and relief of the failure after rhizotomy of dorsal roots. Philos Trans R Soc Lond B Biol Sci. 1994;343:211–223. doi: 10.1098/rstb.1994.0022. [DOI] [PubMed] [Google Scholar]
- Wang SD, Goldberger ME, Murray M. Plasticity of spinal systems after unilateral lumbosacral dorsal rhizotomy in the adult rat. J Comp Neurol. 1991;304:555–568. doi: 10.1002/cne.903040405. [DOI] [PubMed] [Google Scholar]
- Widenfalk J, Lundstromer K, Jubran M, Brene S, Olson L. Neurotrophic factors and receptors in the immature and adult spinal cord after mechanical injury or kainic acid. J Neurosci. 2001;21:3457–3475. doi: 10.1523/JNEUROSCI.21-10-03457.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis WD., Jr Dorsal root potentials and dorsal root reflexes: a double-edged sword. Exp Brain Res. 1999;124:395–421. doi: 10.1007/s002210050637. [DOI] [PubMed] [Google Scholar]
- Wright DE, Snider WD. Neurotrophin receptor mRNA expression defines distinct populations of neurons in rat dorsal root ganglia. J Comp Neurol. 1995;351:329–338. doi: 10.1002/cne.903510302. [DOI] [PubMed] [Google Scholar]
- Yajima Y, Narita M, Narita M, Matsumoto N, Suzuki T. Involvement of a spinal brain-derived neurotrophic factor/full-length TrkB pathway in the development of nerve injury-induced thermal hyperalgesia in mice. Brain Res. 2002;958:338–346. doi: 10.1016/s0006-8993(02)03666-1. [DOI] [PubMed] [Google Scholar]
- Yajima Y, Narita M, Usui A, Kaneko C, Miyatake M, Narita M, Yamaguchi T, Tamaki H, Wachi H, Seyama Y, Suzuki T. Direct evidence for the involvement of brain-derived neurotrophic factor in the development of a neuropathic pain-like state in mice. J Neurochem. 2005;93:584–594. doi: 10.1111/j.1471-4159.2005.03045.x. [DOI] [PubMed] [Google Scholar]
- Zhang B, Goldberger ME, Murray M. Proliferation of SP- and 5HT-containing terminals in lamina II of rat spinal cord following dorsal rhizotomy: quantitative EM-immunocytochemical studies. Exp Neurol. 1993;123:51–63. doi: 10.1006/exnr.1993.1139. [DOI] [PubMed] [Google Scholar]