

Abstract
Loss of mitochondrial membrane integrity and release of apoptogenic factors are a key step in the signaling cascade leading to neuronal cell death in various neurological disorders, including ischemic injury. Emerging evidence has suggested that the intramitochondrial protein apoptosis-inducing factor (AIF) translocates to the nucleus and promotes caspase-independent cell death induced by glutamate toxicity, oxidative stress, hypoxia, or ischemia. However, the mechanism by which AIF is released from mitochondria after neuronal injury is not fully understood. In this study, we identified calpain I as a direct activator of AIF release in neuronal cultures challenged with oxygen–glucose deprivation and in the rat model of transient global ischemia. Normally residing in both neuronal cytosol and mitochondrial intermembrane space, calpain I was found to be activated in neurons after ischemia and to cleave intramitochondrial AIF near its N terminus. The truncation of AIF by calpain activity appeared to be essential for its translocation from mitochondria to the nucleus, because neuronal transfection of the mutant AIF resistant to calpain cleavage was not released after oxygen–glucose deprivation. Adeno-associated virus-mediated overexpression of calpastatin, a specific calpain-inhibitory protein, or small interfering RNA-mediated knockdown of calpain I expression in neurons prevented ischemia-induced AIF translocation. Moreover, overexpression of calpastatin or knockdown of AIF expression conferred neuroprotection against cell death in neuronal cultures and in hippocampal CA1 neurons after transient global ischemia. Together, these results define calpain I-dependent AIF release as a novel signaling pathway that mediates neuronal cell death after cerebral ischemia.
Keywords: ischemia, apoptosis, mitochondria, caspase, neuroprotection, neuronal death
Introduction
Release of apoptogenic factors from mitochondria plays a critical role in mediating neuronal cell death after cerebral ischemia (Fujimura et al., 1999; Sugawara et al., 1999). Activation of the mitochondrial-signaling cascade can activate both caspase-dependent and caspase-independent cell death-execution pathways (Graham and Chen, 2001). In the caspase-independent mechanism, loss of mitochondrial membrane integrity leads to the release of apoptosis-inducing factor (AIF), which kills cells by degrading the nuclear genome without requiring caspase activity (Susin et al., 1999; Widlak et al., 2001). Recent studies have demonstrated that AIF is translocated from mitochondria to the nucleus after neuronal insults, including excitotoxicity, oxidative stress, hypoxia, and cerebral ischemia (Cregan et al., 2002; Yu et al., 2002; Zhang et al., 2002; Cao et al., 2003; Zhu et al., 2003). This caspase-independent mechanism appears to be particularly relevant to adult brain injury, because AIF is expressed abundantly in adult brain (Cao et al., 2003), whereas the expression of caspases and the caspase dependence of ischemic neuronal death markedly decline with brain maturation (H. Chen et al., 2001; Yakovlev et al., 2001; Cao et al., 2002; Tanaka et al., 2004). Indeed, several studies have shown neuroprotective effects against neuronal cell death by either neutralizing intracellular AIF or genetically reducing the expression of AIF (Yu et al., 2002; Cheung et al., 2005; Culmsee et al., 2005). Hence, the AIF-dependent death pathway is emerging as a legitimate potential therapeutic target for ischemic neuronal injury.
The recent study by Cheung et al. (2006) has provided important evidence that AIF-induced cell death is dependent on the translocation of AIF to the nucleus and not attributable to the loss of AIF from mitochondria per se. It is thought that, through direct interaction with genomic DNA, AIF degrades the nucleus, leading to large-scale DNA fragmentation, chromatin condensation, and chromatinolysis (Cande et al., 2002; Ye et al., 2002). The physiological role of AIF in mitochondria is not fully understood; however, several studies suggest that AIF possesses important nicotinamide adenine dinucleotide oxidase activity and that deletion of AIF could result in reduced mitochondrial complex I activity, impaired oxidative phosphorylation (Vahsen et al., 2004), and reduced neuronal survival during brain development (Cheung et al., 2006). Thus, from the perspective of therapeutic intervention in the brain, it will not be feasible to alleviate the cell-killing effect of AIF by permanently lowering its expression without impairing the normal function of mitochondria. Strategically, an alternative approach would be to inhibit the release of AIF from mitochondria after neuronal injury.
Several intracellular signaling events have been mechanistically linked to AIF release. These include poly(ADP-ribose) polymerase-1 (PARP-1) activation (Yu et al., 2002; Du et al., 2003; Alano et al., 2004), activation of p53 and its downstream proteins Bax (Bcl-2-associated X protein) (Cregan et al., 2002), and mitochondrial translocation of Bid (BH3-interacting domain death agonist) (van Loo et al., 2002; Culmsee et al., 2005). Despite these discoveries, however, the precise mechanism responsible for AIF release in ischemic neuronal injury remains elusive. Therefore, in the present study, the signaling mechanism for AIF release was investigated in both in vitro and in vivo models of ischemic neuronal injury. The results presented here suggest that calpain I activation plays a critical role in triggering AIF release from mitochondria in ischemic neurons.
Materials and Methods
cDNA isolation, site-directed mutagenesis, and generation of recombinant proteins.
The rat and human full-length AIF cDNAs were obtained using PCR from a rat brain cDNA library and a human kidney cDNA library, respectively. The rat calpastatin cDNA was amplified from the rat brain cDNA library using the primers 5′-atg att tct gct ggt gga gag agt g-3′ and 5′-ttc act agt gat tct agt cat ctt ttg-3′. The mutant AIF (containing double mutations L101G/L103G) was generated by site-directed mutagenesis using the Gene Editor system (Promega, Madison, WI). The sequences of all cDNAs were verified at the University of Pittsburgh Gene Service Facility by sequencing reactions on both strands.
Recombinant proteins of full-length, truncated, and mutant AIF, and calpastatin were generated using the standard method (Chen et al., 2000; Cao et al., 2001). In brief, the cDNA was fused into the glutathione S-transferase (GST) gene in PGEX-2T vector (GE Healthcare, Little Chalfont, UK). The GST fusion protein was expressed in Escherichia coli BL21 cells, absorbed to a glutathione-Sepharose 4B column, and then cleaved by thrombin to remove GST.
Construction of small interfering RNA expression vectors.
Mammalian expression plasmids directing the transcription of short hairpin (sh) small interfering RNAs (siRNAs) against AIF, calpain I, or calpain II were constructed according to the principles described previously (Brummelkamp et al., 2002) and using our published protocols (Chu et al., 2005). This expression plasmid uses the pcDNA3.1 (+) backbone, and, in its SpeI/EcoRV cloning sites, it contains the histone H1 (H1)–RNA promoter followed by the gene-specific targeting sequence (Table 1). For each of the targeted genes, the scramble sequence, which contains the same nucleotide composition as the targeted sequence but in randomized order, was also constructed and served as the control. During construction, all inserts in the expression plasmids were verified by sequencing reaction on both strands.
Table 1.
Sequences of short hairpin RNA for gene knockdown
| Construct | Sequences |
|---|---|
| AIFt | Targeting sequence 1, 5′-GAGAAACAGAGAAGAGCCA-3′ |
| Targeting sequence 2, 5′-GTCACGTCCCTTTCCTGCT-3′ | |
| AIFs | Scramble sequence, 5′-AACAGGAACAGACGGAGAA-3′ |
| GFPt | Targeting sequence, 5′-AACAGCTGCTAGGATTACACA-3′ |
| calp-It | Targeting sequence 1, 5′-CAGTTTCTCATAGCTTGGGCTTCAA-3′ |
| Targeting sequence 2, 5′-ATCAAGTGGAAGCGTCCTACGGAA-3′ | |
| calp-Is | Scramble sequence, 5′-TTAAGCTTACCTTGGATAGTGCCT-3′ |
| calp-IIt | Targeting sequence 1, 5′-AAAGCTCAGGAAGCCTGCAGAAATT-3′ |
| Targeting sequence 2, 5′-CAAATACCTTCTGGATGAACCCTCA-3′ | |
| calp-IIs | Scramble sequence, 5′-TTTGAAGAACAAGCAGACAAGGCCT-3′ |
calp-IIt, Calpain II-targeting sequence; calp-IIs, calpain II scramble sequence.
Production of adeno-associated virus vectors.
Adeno-associated virus (AAV) vectors carrying either the short hairpin siRNA AIF targeting sequence (AAV–AIFt), its scramble control sequence (AAV–AIFs), the calpastatin cDNA (AAV–Cps), or rat or human AIF cDNA (AAV–AIF) were constructed. Compared with several other types of viral vectors, AAV shows higher efficiency for neuronal or brain gene transfection with minimal cytotoxicity or inflammation inducibility. In this study, the expressing vector plasmid, which contains an simian virus 40 splicing site and a polyadenylation signal, was flanked by the AAV inverted terminal repeats. The pH1–AIF or its scramble control sequence was isolated by EcoRV and NotI from its parental plasmid and then inserted into the AAV expressing vector plasmid. Large-scale production of the AAV vector was performed using the adenovirus-free triple-plasmid cotransfection method described previously (Xiao et al., 1998). A total of 50 μg of plasmid mixture was cotransfected into human embryonic kidney HEK293 cells with the assistance of 0.25 m CaCl2, and the cells were grown in DMEM containing 10% fetal bovine serum (Invitrogen, Carlsbad, CA) for 48 h and then harvested. AAV vectors were purified using an fast protein liquid chromatography system in conjunction with HiTrap Heparin columns (GE Healthcare). The titers of the virus particles were determined by the DNA dot blot method.
Primary neuronal culture and in vitro model of ischemia.
Primary cultures of hippocampal/cortical neurons were prepared from 17 d Sprague Dawley rat embryos as described previously (Cao et al., 2001). Experiments were conducted at 12 d in vitro (DIV), when cultures consisted primarily of neurons (∼97%) as determined using cell phenotype-specific immunocytochemistry (Cao et al., 2001).
To model ischemia-like conditions in vitro, primary cultures were exposed to transient oxygen and glucose deprivation (OGD) (Cao et al., 2001, 2003). In brief, two-thirds of the culture medium was replaced four times with serum- and glucose-free medium, resulting in a final glucose concentration of <1 mm. The glucose-deprived cultures were then placed in a Billups-Rothenberg (Del Mar, CA) modular incubator chamber, which was flushed for 15 min with 95% argon and 5% CO2 and then sealed. The chamber was placed in a water-jacketed incubator (Thermo Electron Corporation, Cambridge, UK) at 37°C for 60 min and then returned to 95% air, 5% CO2 and glucose-containing medium for the period of time indicated in each experiment. Control glucose-containing cultures were incubated for the same periods of time at 37°C in humidified 95% air and 5% CO2.
Fluorescence of Alamar blue (Accumed International, Westlake, OH), an indicator that changes from blue to red and fluoresces when reduced by cellular metabolic activity, was used to measure the viability of the cultured neuron at 24–72 h after OGD (Nagayama et al., 1999; Cao et al., 2004). One-half of the culture medium was replaced with MEM-Pak containing 10% (v/v) Alamar blue, and cultures were incubated for 1.5 h at 37°C in humidified 95% air and 5% CO2. Fluorescence was determined in a Millipore (Billerica, MA) CytoFluor 2300 automated plate-reading fluorometer, with excitation at 530 nm and emission at 590 nm. As demonstrated previously, Alamar blue fluorescence in these cultures varies linearly with cell number, decreases with exposure to hypoxia or OGD, and correlates with the extent of cellular injury determined by lactate dehydrogenase (White et al., 1996).
OGD-induced cell death was quantified by measuring lactate dehydrogenase (LDH) release from damaged cells into the culture medium (Cao et al., 2003). In brief, 10 μl aliquots of medium taken from the cell culture wells were added to 200 μl of LDH reagent (Sigma, St. Louis, MO). Using a spectrophotometer plate reader (Molecular Devices, Sunnyvale, CA), the emission was measured at 340 nm, which is proportional to the amount of LDH in the medium. The percentage of cell death was calculated as follows: the difference between the amounts of LDH measured in the media of OGD-challenged and control nontreated cells was divided by the difference between the amounts of LDH measured in the media of totally lysed cells and control nontreated cells. Total cell lysis was achieved by incubating the cells in 1% Triton X-100 solution for 10 min at 37°C.
In selective experiments, cell death was evaluated after OGD using Hoechst 33258 nuclear staining, propidium iodide uptake, or Klenow-mediated DNA nick-end labeling (Nagayama et al., 1999). The percentages of cells showing chromatin condensation or DNA damage were quantified by counting at least 3000 cells under each experimental condition (three randomly selected fields per well, four to six wells per condition per experiment, and three independent experiments).
Gene transfection in primary neurons by AAV vectors and nucleofector.
The neuronal cultures were infected with the AAV–AIFt, AAV–AIFs, or AAV–Cps vector or the control vector [AAV–green fluorescent protein (GFP) or empty AAV] at the particle/cell ratio of 1 × 105/1iter for 6 h in serum-free media and then incubated in vector-free normal media for 72 h (Cao et al., 2004; Chu et al., 2005). The overexpression of calpastatin in neurons was confirmed by immunocytochemistry and Western blot, respectively, using the anti-hemagglutinin (HA) antibody. The AIF knockdown effect by AAV–AIFt in neurons was examined using Northern blot analysis, Western blot (with the anti-AIF antibody), and double-label immunofluorescence [AIF/cytochrome c oxidase IV (COX IV)] and confocal microscopy.
Knockdown of calpain I or calpain II was achieved in neurons by transfection of the shRNA plasmids using the Nucleofector device (Amaxa, Gaithersburg, MD) according to the protocol of the manufacturer. The calpain I- or calpain II-targeting plasmid or their respective scramble control plasmids were transfected immediately before cell plating. Using the rat neuron Nucleofector kit, each transfection reaction was done using 4 × 106 neurons, which were plated at the densities of 5 × 104 for the 96-well plate, 3 × 105 for the 24-well plate, or 4 × 106 for the six-well plate. Neurons were used for OGD experiments at 12 DIV.
Calpain activity assay.
Calpain activity assay was performed using a fluorescent calpain I substrate as described previously (Cuerrier et al., 2005). In brief, whole-cell or mitochondrial proteins at the indicated amounts were incubated with calpain reaction buffer [20 mm HEPES, pH 7.6, 1 mm EDTA, 50 mm NaCl, and 0.1% (v/v) 2-mercaptoethanol] containing 10 μm calpain I fluorescent substrate H-E(EDANS)PLF∼AERK(DABCYL)-OH (Calbiochem, La Jolla, CA). The reaction was initiated by addition of CaCl2 (final concentration of 5 μm) and incubated at 37°C for 30 min. The activity of calpain was measured by detecting the increase in fluorescence using excitation/emission wavelengths of 335/500 nm. Calpain activity was calculated quantitatively based on the standard curve generated using recombinant calpain I (Calbiochem) and expressed as units per milligram protein.
Isolation of mitochondria.
Nonsynaptic brain mitochondria were isolated from mice according to previously described methods with modifications (Berman and Hastings, 1999). In brief, brain tissues were homogenized in mitochondria isolation buffer (MIB) containing 225 mm mannitol, 75 mm sucrose, 5 mm HEPES, pH 7.4, 1 mm EGTA, and 1 mg/ml bovine serum albumin and centrifuged at 1200 × g for 10 min. The mitochondria were collected by centrifugation at 12,000 × g for 10 min, resuspended in 10 ml of MIB containing 0.02% digitonin, and centrifuged at 12,000 × g for 10 min. The mitochondrial pellets were then resuspended in MIB and further purified by a sucrose step gradient consisting of 2 ml each of 1.2 and 1.6 m sucrose by centrifugation at 40,000 × g for 1 h at 4°C. The brownish-colored mitochondria-containing band, which was located at the interface of 1.2 and 1.6 m sucrose, was recovered, washed once with MIB, and resuspended in MIB without BSA and EGTA. For mitochondrial isolation from primary neuron cultures, cells were suspended in MIB and placed on ice for 15 min. The cells were homogenized and mitochondria were resuspended in MIB and further purified by a sucrose step gradient as described above.
To generate mitochondrial subfractions, a protocol using a combination of lower concentrations of digitonin and sonication was used as described previously with minor modifications (Kim et al., 2004). Mitochondria were resuspended at a concentration of 3 μg/μl and treated with digitonin at a final ratio of 0.4% (w/w, digitonin/mitochondrial protein). We found that, at this concentration, digitonin can break the outer membrane (OM) of brain mitochondria [allowing the separation of inner membrane (IM) from mitochondria] but does not significantly dissolve membrane proteins. The mixtures were placed at 4°C with gentle shaking for 15 min and then centrifuged at 12,000 × g for 15 min. The resulting supernatants were further centrifuged at 100,000 × g for 1 h to collect the intermembrane space (IMS) (supernatant) and outer membrane (pellet) fraction. The resulting pellets, which contain both outer and inner membrane, were resuspended in hypotonic buffer containing 20 mm HEPES-KOH, pH 7.4, for 20 min at 0°C and sonicated for three times for 10 s at 50% amplitude in a cell disruptor equipped with a microtip (three times for 10 s at 50% amplitude or 8 W output). Residual intact mitochondria and large fragments were removed by centrifugation at 16,000 × g for 20 min at 4°C. Submitochondrial membrane in the supernatant was layered onto linear sucrose gradients consisting of 0.8 ml each of 1.0, 1.2, 1.4, and 1.6 m sucrose (in 10 mm KCI and 5 mm HEPES-KOH, pH 7.4) and centrifuged at 140,000 × g for 16 h at 4°C. Fractions were sequentially collected from the bottom, and each contained ∼200 μl of sample. After protein concentration determinations, the purity of these fractions was analyzed by immunoblot with antibodies against mitochondrial outer membrane [voltage-dependent anion channel (VDAC)], inner membrane (COX IV), and intermembrane space [adenylate kinase 2 (AK2)] markers.
Protein binding assay.
Protein–protein binding between AIF and heat shock protein 10 (HSP10) was studied using in vitro translation products using the cell-free assay as described previously (Cao et al., 2004). In brief, [35S]AIF (1.0 μl) was incubated with 0.5 or 1.0 μl of Flag–HSP10 (in the absence or presence of activated calpain I) at 30°C for 1 h in the protein binding buffer containing 150 mm NaCl, 5 mm MgCl2, 1 mm DTT, and 10 μg/ml each of pepstatin, leupeptin, and PMSF. The mixture was then immunoprecipitated with 2 μg of anti-Flag polyclonal antibody (Sigma), and the immunoprecipitates were then subjected to autoradiography for the detection of [35S]AIF.
Intracerebral infusion of AAV and transient cerebral ischemia.
AAV vectors were infused into the CA1 sector in the dorsal hippocampus (two injection spots; coordinates: anteroposterior, −3.5 mm and lateral, 2.0/3.5 mm from bregma) using the convection enhancement technology. Briefly, male Sprague Dawley rats weighing 250–275 g were anesthetized using isoflurane. The AAV vectors (AAV–Cps, AAV–AIFt, AAV–AIFs, or control vectors, including AAV–enhanced GFP and empty AAV vector) dissolved in physiologic saline [2 × 1010 DNase-resistant particles/μl] were infused at the rate of 0.1 μl/min over 50 min via a custom-made convection-enhanced microinfusion system linked to a UMP2 microsyringe pump (World Precision Instruments, Sarasota, FL). The animals were allowed to recover for up to 14 d to enable sufficient gene expression. Expression of calpastatin in the hippocampal CA1 was examined by immunohistochemistry and Western blot using the anti-HA antibody. The effect of AAV–AIFt on AIF expression in the hippocampal CA1 was examined by Western blot using the anti-AIF antibody and by double-label immunofluorescence for GFP and AIF.
At 14 d after AAV infusion, global ischemia was induced for 15 min in isoflurane-anesthetized rats using the four-vessel occlusion method (Chen et al., 1998). Blood pressure, blood gases, and blood glucose concentration were monitored and maintained in the normal range throughout the experiments. Rectal temperature was continually monitored and kept at 37–37.5°C using a heating pad and a temperature-regulated heating lamp. Brain temperature was monitored using a 29 gauge thermocouple implanted in the right caudate–putamen and was kept at 35.8 ± 0.2°C during ischemia and at 37–37.5°C thereafter. Electroencephalogram was monitored in all animals to ensure isoelectricity within 10 s after the induction of ischemia. Sham operations were performed in additional animals using the same anesthesia and surgical exposure procedures except that the arteries were not occluded. Rats were killed at prescheduled time points after ischemia: for assessments by Western blot or calpain activity assays, each group consisted of six rats; for immunohistochemistry, each group consisted of four to five rats; and for histological assessments (7 d after ischemia), each group consisted of nine rats.
For the hippocampal cell death assessments, coronal sections (40 μm thick) were cut throughout the dorsal hippocampal formation at the coronal levels between −2.5 and −4.5 mm from bregma. Every fourth section was stained for cresyl violet, and the immediate adjacent section was stained for DNA fragmentation using Klenow-mediated DNA nick-end labeling (Nagayama et al., 1999). Viable CA1 neurons and neurons containing DNA damage were quantified using stereology by an investigator who was blinded to the experimental conditions.
Stereological cell counting.
The stereology principles were described previously in detail (Gundersen, 1986; Schmitz, 1997). In the present study, the Bioquant Image Analysis program (Bioquant, Nashville, TN) was used to count CA1 neurons. The Bioquant software is interfaced with a stage encoder to interpret x-, y-, and z-axis movement of the microscope stage. The entire CA1 subfield from a given section was captured with a color CCD camera, and grid squares of 50 × 50 μm were generated over the region of interest. The optical dissector method requires that 100–150 cells be counted in a given structure to estimate the total number. The number of points needed to be assessed within the outlined area was empirically determined. Previous study has shown that a 25 × 25 μm counting frame and a focus depth of 25 μm in a 40 μm section with 7.5 μm guard regions above and below should be used. The 25 × 25 × 25 μm box in which the neurons were counted was defined as a dissector. An average of nine dissectors per section yields the desired number of counted neurons throughout the CA1 at the levels of the dorsal hippocampus. The number of dissectors in the CA1 of a given section was maintained at a constant ratio from section to section. The rostrocaudal length of the CA1 was 2 mm, obtained by multiplying section thickness by the number of sections (50 sections). Every fourth section was counted. The total number of neurons was calculated using the optical dissector, equal to the quotient of the total number of neurons counted and the product of the fractions for sampling section frequency (SSF) (fraction of sections counted), area section frequency (ASF) (sampling area/area between dissectors), and thickness section frequency (TSF) (dissector depth/section thickness), or N = Σ neurons counted × 1/SSF × 1/ASF × 1/TSF). For our study, SSF = 1/4 sections, ASF = 25 × 25 μm/50 × 50 μm, and TSF = 25 μm/40 μm.
Western blot analysis.
Western blot was performed using the standard method and the enhanced chemiluminescence detection reagents (GE Healthcare). The following antibodies were used. The rabbit monoclonal anti-AIF antibody (clone E20, 1:1000) was purchased from Epitomics (Burlingame, CA). Two rabbit polyclonal antibodies recognizing the internal peptides of rat AIF (NSVLVLIVGLSTIGA and LGLSPEEKQRRAIAS) were custom-made (Bio-Synthesis, Lewisville, TX) and used at dilutions of 1:1500. The mouse monoclonal anti-cytochrome c oxidase IV antibody (1:1000) was from Invitrogen (Carlsbad, CA). The rabbit polyclonal antibodies against HSP10 (1:500), HSP60 (1:1000), AK2 (1:500), or histone-1 (1:500) were from Santa Cruz Biotechnology (Santa Cruz, CA). The rabbit polyclonal antibodies against calpain I (1:1000), calpain II (1:500), or calpastatin (1:1000) were from Chemicon (Temecula, CA).
Pulse-field gel electrophoresis for large-scale DNA fragmentation.
Pulse-field gel electrophoresis (PFGE) was performed to detect large-scale (50 or 10 kbp) DNA fragmentation, a hallmark of AIF-induced genomic DNA degradation, in brain tissues as described previously (Cao et al., 2003). Frozen tissues dissected from CA1 (30 mg) were used to prepare agarose plugs. High-molecular-weight DNA was separated using a PFGE system (CHEF Mapper systems' Bio-Rad, Hercules, CA). A slide of 0.02 ml of prepared cast agarose plug was loaded onto a well in 0.8% agarose gel in Tris-borate EDTA (TBE) electrophoresis buffer and sealed in place with 1% low-melting-point agarose. The gel was subsequently run on a Q-life (Kingston, Ontario, Canada) autobase PFG system with software-assisted ROM card 3 (resolution, 8–500 kbp; Q-Life) in TBE buffer at 14°C. After staining with ethidium bromide, the gel was distained and photographed under UV transillumination.
Statistical analysis.
Results are reported as mean ± SE values. The significance of difference between means was assessed by Student's t test (single comparisons) or by ANOVA and post hoc Bonferroni's/Dunn's tests, with p < 0.05 considered statistically significant.
Results
AIF knockdown reduces cell death induced by oxygen–glucose deprivation
The first objective of the present study was to determine whether AIF plays a critical role in neuronal cell death induced by OGD, an in vitro model that simulates the major pathologic components of ischemic injury. To achieve this, the RNA interference approach was taken. We designed a panel of six shRNA sequences targeting various segments of rat AIF mRNA. Subsequent tests in a rat cell line identified two sequences that were highly effective in knocking down AIF. To test the effect of AIF knockdown in primary neurons, recombinant adeno-associated virus vectors carrying the two AIF-targeting sequences (AAV–AIFt) were made (Table 1), together with control AAV carrying the scramble sequence (AAV–AIFs). Infection with AAV–AIFt (Fig. 1A–C), but not AAV–AIFs, of cultured cortical neurons resulted in time-dependent decreases in AIF expression, as confirmed using Northern blot (data not shown), Western blot, and immunofluorescent staining. Semiquantitatively, AAV–AIFt reduced AIF protein expression by ∼80% at 72 h and ∼90% at 96 h. AAV–AIFt infection did not change the expression levels of the mitochondrial proteins COX IV, VDAC, and HSP60.
Figure 1.
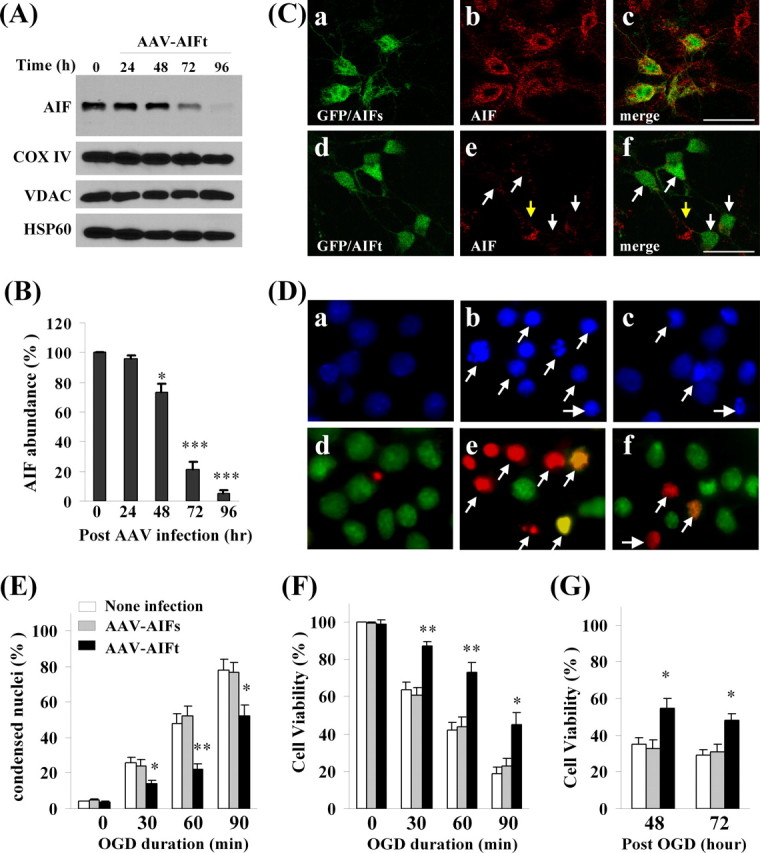
Neuroprotective effect of AIF knockdown against oxygen–glucose deprivation injury in primary hippocampal/cortical neurons. A, Western blots show that AIF expression was decreased time dependently in neurons infected with AAV expressing the AIF targeting sequence (AAV–AIFt) (Table 1). The expression levels of COX IV, VDAC, and HSP60 were unchanged. B, Semiquantitative data of A, based on four independent experiments. *p < 0.05, ***p < 0.001 versus empty AAV-infected cells. C, Confocal images showing AIF expression (red) in neurons coinfected with AAV–AIFs (scramble control sequence) and AAV–GFP (a–c) or AAV–AIFt and AAV–GFP (d–f). The arrows in e and f point to infected neurons showing decreased AIF expression. D, Hoechst 33258 (top) and dual TUNEL/Cyto11 green fluorescent nuclear staining (bottom) in normal unstressed neurons (a, d) and neurons at 24 h after 60 min of OGD (b, c, e, f). After OGD, many neurons showed condensed nuclei (b) and TUNEL-positive nuclei (yellow–red, e); condensed nuclei and TUNEL-positive nuclei were present less frequently in cells infected with AAV–AIFt (c, f). E, Percentages of condensed nuclei were quantified at 24 h after OGD of different duration in neurons either nontransfected or infected with AAV–AIFs or AAV–AIFt. F, Cell viability was measured based on Alamar blue fluorescence at 24 h after OGD of different duration. G, Cell viability was measured at 48 and 72 h after OGD (60 min). *p < 0.05, **p < 0.01 versus nontransfected neurons. Data are mean ± SE; n = 12 per experimental condition from three independent transfection experiments.
To determine whether AIF knockdown per se had adverse effects on neurons, cell viability was evaluated using Alamar blue fluorescence at 72, 96, and 120 h after AAV infection (AAV–AIFt, AAV–AIFs, and AAV–GFP). AAV–AIFt did not significantly decrease cell survival over control vectors (data not shown). The effect of AIF knockdown on selective respiratory complex activities was also examined in neurons at 96 h after AAV infection. Infection of AAV–AIFt resulted in insignificant reductions in mitochondrial activities (11% decrease in complex I activity, 8% decrease in complex II activity, 12.3% decrease in complex II–III activity) (supplemental data 1, available at www.jneurosci.org as supplemental material).
Neurons infected with AAV–AIFt, but not control vectors, were significantly protected against OGD-induced cell death (Fig. 1D–G). The neuroprotective effect by AAV–AIFt was detected using three different methods, including Alamar blue fluorescence, nuclear staining [Hoechst and terminal deoxynucleotidyl transferase-mediated biotinylated dUTP nick end labeling (TUNEL)], and LDH release (data not shown) after different levels of insult (OGD for 30, 60, or 90 min). To determine whether the neuroprotective effect of AIF knockdown manifested simply as a delay in cell death, cell viability was also measured at 48 and 72 h after 60 min of OGD. At both time points, AAV–AIFt-infected neurons showed significantly increased cell viability compared with controls.
To further ensure that the prosurvival effect of AAV–AIFt was specifically attributable to AIF knockdown, two additional controls were tested. First, neurons were coinfected with AAV–AIFt and AAV–hAIF; the latter delivered the human AIF gene that was completely resistant to the rat AIFt [the targeting sequence shares 50% homology between rat (GenBank accession number AF375656, targeting sequence 318–336) and human (GenBank accession number AF100928) AIF]. Compared with AAV–AIFt infection alone, AAV–hAIF/AAV–AIFt coinfection significantly rescued the OGD-induced cell death phenotype (supplemental data 2A–C, available at www.jneurosci.org as supplemental material). Second, we tested the effect of an AIF-unrelated targeting sequence, i.e., AAV–GFPt. Overexpression of GFPt in neurons effectively depleted the expression of GFP delivered by AAV–GFP. AAV–GFPt had no significant effect on ischemic cell death, determined at 24 h after 60 or 90 min of OGD (supplemental data 2D–F, available at www.jneurosci.org as supplemental material).
Translocation of a truncated AIF in neurons after OGD
The next objective of this study was to elucidate the mechanism by which AIF is released from mitochondria after OGD. As the first step, we characterized the temporal profile of AIF release in neurons subjected to 60 min of OGD using Western blot. Increased AIF was readily detectable in the nuclear and cytosolic fractions at 2 h after OGD and throughout the experiment (24 h); a corresponding decrease in mitochondrial AIF content was detected at 6–24 h after OGD (Fig. 2B).
Figure 2.

Translocation of a truncated AIF in neurons after OGD. A, Western blots show that endogenous AIF in brain or cultures of primary neurons or astrocytes has a molecular size of ∼62 kDa. Recombinant proteins AIF67 (proform AIF), AIF62 (lacking the first 50 amino acids), and AIF57 (lacking the first 100 amino acids) served as size controls. B, Western blots after subcellular fractionation show that a truncated AIF at the size of 57 kDa was translocated to both nucleus and cytosol after OGD (60 min), whereas the levels of mitochondrial AIF (62 kDa) were decreased after OGD. Recombinant proteins AIF62 and AIF57 served as size controls. The graph at the bottom illustrates the time-dependent changes of AIF in three fractions after OGD. Data are based on four independent experiments. *p < 0.05 versus non-OGD neurons. C, Western blots based on total-cell extracts show that AIF was truncated to yield the 57 kDa fragment in neurons at 2, 6, and 12 h after OGD (60 min). D, Western blots show that both antibodies raised against amino acids 105–118 (AIF105–118) and amino acids 65–80 (AIF65–80) of pro-AIF were able to detect endogenous AIF in brain extracts and cultured neurons. Recombinant proteins AIF67, AIF62, and AIF57 served as positive controls. E, AIF that translocated into nucleus at 2, 6, and 16 h after OGD (60 min) was detectable using the AIF105–118 antibody but not the AIF65–80 antibody. All blots are representative of at least three sets of independent data with similar results.
Initially, for AIF immunoblots, two recombinant AIF proteins with high purity were used as positive controls, i.e., the proform AIF (67 kDa, before it enters mitochondria) and the mature AIF (57 kDa, the reported size for mitochondrial AIF) lacking the first 100 amino acids of the proform (Susin et al., 1999). To our surprise, we consistently observed size discrepancies between the positive controls and the endogenous AIF in extracts from brain or cultured primary neurons or astrocytes. In particular, the endogenous mitochondrial AIF (∼62 kDa) was found to be larger than the 57 kDa AIF (Fig. 2A), whereas the nuclear or cytosolic AIF (released) was 57 kDa (Fig. 2B). The results suggest that the size of mature AIF in mitochondria is larger than originally reported (Susin et al., 1999) but is reduced during release from mitochondria. Indeed, the mitochondrial AIF showed the same size as the 62 kDa AIF recombinant protein (Fig. 2B), which lacks the first 50 amino acids of the proform. Moreover, the AIF immunoblots of whole-cell extracts from OGD-treated neurons detected an additional band consistent with the size of 57 kDa that was absent in nontreated neurons (Fig. 2C), indicating that AIF was partially truncated after OGD.
To test the predication that the mature mitochondrial AIF is ∼40–50 amino acids larger than the released AIF and that the truncation of AIF in OGD neurons likely occurs at the N terminus (because the antibody against the C-terminal sequence detected both mitochondrial and released AIF), we raised two polyclonal antibodies based on the peptide sequences corresponding to amino acids 65–80 (AIF65–80) and 105–118 (AIF105–118) of proform AIF, respectively. On Western blots, both antibodies immunoreacted with AIF in brain extracts and neuron cultures (Fig. 2D), but only the AIF105–118 antibody immunoreacted with the nuclear AIF in OGD neurons (Fig. 2E), indicating that the nuclear-translocated AIF is a truncated (at the N terminus) form of mitochondrial AIF. Thus, mitochondrial AIF was subjected to proteolytic cleavage to generate the 57 kDa AIF fragment that was translocated into the nucleus after OGD.
Calpain I is capable of truncating AIF and releasing AIF from mitochondria in cell-free assays
A number of cysteine proteases, including caspases, lysosomal enzymes, and calpain, may be activated in neurons after ischemic injury (Graham and Chen, 2001). To determine which specific protease is able to cleave AIF and specifically generate the 57 kDa fragment, we performed an in vitro cell-free assay in which the mitochondrial AIF (62 kDa) was incubated with various purified cysteine proteases. None of the active caspases tested, including caspase-3, caspase-9, and caspase-8, was capable of cleaving AIF. The lysosomal enzymes cathepsin B and cathepsin L caused nonspecific cleavage of AIF, generating fragments with sizes <40 kDa. In contrast, incubation of recombinant AIF with active calpain I, but not calpain II, was shown to specifically produce the 57 kDa fragment (supplemental data 3A, available at www.jneurosci.org as supplemental material). Subsequently, the calpain I-generated AIF fragment was purified and subjected to N-terminal sequencing using Edman degradation chemistry (Bio-Synthesis). The results revealed that calpain I truncated AIF between L103 and S104. To produce a calpain I-resistant AIF, several single or double mutations were made in the stretch of amino acids 100–103 using site-directed mutagenesis, and then each of the mutant proteins was tested for its cleavage by calpain I. The AIF protein containing a double mutation (L101G/L103G) showed complete resistance to calpain I activity (supplemental data 3B, available at www.jneurosci.org as supplemental material).
We and others reported recently that activated calpain I can directly trigger the release of AIF from isolated mitochondria (Liou et al., 2005; Polster et al., 2005). However, the biochemical basis of this action by calpain I is not fully understood. In the present study, we further investigated the effect of calpain I on isolated mitochondria, determining whether calpain I causes truncation of AIF in mitochondria, whether calpain-induced AIF release is blocked by mitochondrial permeability transition (MPT) pore inhibitors and whether calpain-induced AIF release depends on the presence of the proapoptotic protein Bax or Bid.
In the calpain I/mitochondria assay, the Ca2+ ion concentration required for both calpain I activation and AIF release was found to be between 2 and 5 μm in the absence of the divalent ion chelator EGTA in the reaction buffer. At the above concentrations, Ca2+ alone neither induced AIF release from mitochondria nor caused mitochondrial swelling. With the presence of activated calpain I, the truncated AIF with the size of 57 kDa was released from mitochondria (Fig. 3A). Although the 57 kDa AIF fragment was only a small fraction of total AIF in the pellet (mitochondrial fraction), this was the only size of AIF present in the supernatant (released from mitochondria), suggesting that AIF was truncated by calpain I before its release from mitochondria. Calpain I-induced AIF truncation and release could be inhibited by recombinant calpastatin (Fig. 3B). Calpain I-induced AIF release does not appear to depend on MPT (Fig. 3C), because the MPT inhibitors cyclosporin A and bongkrekic acid at concentrations (1 μm) sufficient to block calcium-induced mitochondrial swelling (Fig. 3D) or cytochrome c release (Fig. 3E) failed to inhibit AIF release induced by calpain I. Moreover, calpain I at the concentration capable of inducing AIF release failed to induce mitochondrial swelling (Fig. 3D).
Figure 3.

Calpain I induces truncation and release of AIF from mitochondria in cell-free assays. A, Western blot shows that active calpain I (Calp-I) (Ca2+ at 5 μm) was capable of truncating AIF and inducing its release from isolated brain mitochondria. Immunoblotting was performed using both pellets (mitochondria, 35 μg) and supernatant from the cell-free assays. The recombinant proteins (Rp) AIF62 and AIF57 served as positive control. B, The addition of recombinant calpastatin (micrograms per milliliter) in the assay inhibited calpain I (0.3 U/5 μm Ca2+)-induced AIF release from isolated brain mitochondria. Blot for VDAC from total mitochondria served as loading control. C, The addition of the MPT inhibitor cyclosporin A (CSA, 1 μm) or bongkrekic acid (BA, 1 μm) in the cell-free assay failed to inhibit calpain I (0.3 U/5 μm Ca2+)-induced AIF release from isolated brain mitochondria. D, Left, Mitochondrial swelling assays show that calpain I at the concentration (1 U/5 μm calcium) that induces AIF release did not induce mitochondrial swelling. Right, CSA or BA at the concentration (1 μm) that did not inhibit calpain I-induced AIF release was sufficient to block calcium (500 μm)-induced mitochondrial swelling. E, CSA or BA at the concentration (1 μm) that did not inhibit calpain I-induced AIF release was sufficient to block calcium (500 μm)-induced cytochrome c release. Blot for VDAC from total mitochondria served as loading control. F, Cell-free assays were performed using isolated brain mitochondria (35 μg) from Bid-deficient (−/−), Bid/Bax-double deficient, or wild-type (Wt) mice. Blot for VDAC from total mitochondria served as loading control. G, The synergism between calpain I and Bid to induce AIF release. Mitochondria (35 μg) were incubated with recombinant tBid (3 or 30 ng) or Bax (30 or 300 ng) in the absence or presence of calpain I (0.1 or 1 U) for 60 min, and AIF and cytochrome c release was detected by Western blot. The relative density of AIF or cytochrome c band under each experimental condition is indicated at the bottom. Note that tBid enhanced AIF release induced by calpain I. All data are representative of at least three independent experiments with similar results.
It is controversial whether Bid or Bax is involved in the regulation of AIF release from mitochondria (Arnoult et al., 2002; van Loo et al., 2002). Nevertheless, there is considerable evidence suggesting that activated calpain I can cleave Bid or Bax into active fragments that could subsequently damage mitochondria. Thus, we intended to determine whether calpain I-induced AIF release includes any effect from Bid or Bax. The assays were first performed using isolated brain mitochondria from Bid knock-out mice, Bid/Bax double-knock-out mice, or wild-type littermates. As shown (Fig. 3F), calpain I induced AIF release from mitochondria regardless of the genetic phenotypes of the mice, indicating that calpain I could induce AIF release independent of Bid and Bax. Next, we wanted to determine whether recombinant truncated Bid (tBid) or Bax protein could induce AIF release in isolated mitochondria as calpain I does. The results revealed that, although the recombinant Bax or tBid potently induced the release of cytochrome c, neither of them was capable of inducing AIF release at the concentrations tested (Fig. 3G). However, the presence of tBid increased calpain I-induced AIF release from mitochondria (Fig. 3G), suggesting that tBid can enhance AIF release in the presence of calpain I.
Calpain I is obligatory for OGD-induced AIF release in neurons
Based on the results from the in vitro cell-free assays, we hypothesized that calpain I may mediate the truncation and release of AIF from mitochondria in neurons under ischemic conditions. We tested this hypothesis using primary neuron cultures, addressing the following specific questions. (1) Does inhibition of calpain activation prevent AIF truncation and release in neurons after OGD? (2) Does knockdown of calpain I expression in neurons prevent AIF truncation and release after OGD? (3) How does calpain I gain access to the mitochondrial AIF after OGD?
To inhibit calpain activity with the ultimate specificity, we overexpressed calpastatin, a well characterized calpain-inhibitory protein, in neurons using AAV vectors (AAV–Cps). The control vectors included empty AAV vector and AAV–GFP. As determined using immunocytochemistry against the triple-HA tag, infection of AAV–Cps but not the control vector in cultures for 72 h resulted in overexpression of calpastatin in 86.6 ± 3.1% of neurons (mean ± SEM, 12 wells from three independent experiments). This was reflected in Western blot as 8- to 10-fold increases over the endogenous calpastatin levels in neurons (Fig. 4A). Overexpression of calpastatin effectively attenuated OGD-induced calpain activation, showing reduced cleavage of α-Fodrin to its 140 kDa fragments after OGD (Fig. 4B) and decreased calpain substrate-cleavage activity in cell extracts (Fig. 4C). Calpain inhibition by AAV–Cps had profound inhibitory effects on OGD-induced release of AIF in neurons, as determined at the cellular level by triple-label (AIF/mitochondria/nuclei) immunofluorescence (Fig. 4D,E). Western blots further confirmed that the truncation and nuclear translocation of AIF was inhibited by calpastatin expression after OGD (Fig. 4F). Consistent with the neuroprotective effect of calpastatin overexpression, OGD-induced cell death was significantly attenuated in AAV–Cps-infected cultures (Fig. 4G–I).
Figure 4.
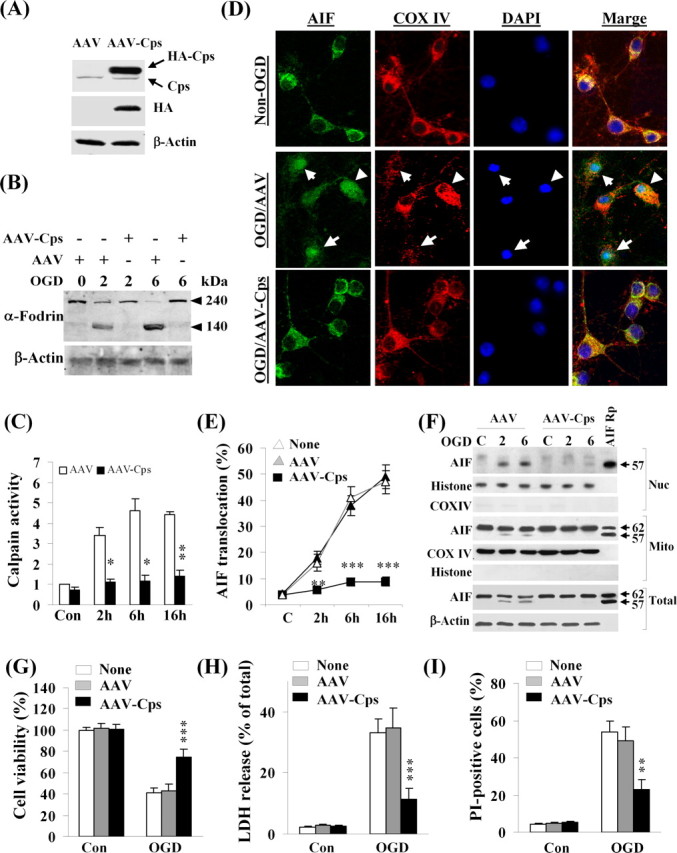
Calpain inhibition prevents OGD-induced AIF release in neurons. A, Primary neurons were infected for 3 d with empty AAV or AAV carrying the calpastatin cDNA (AAV–Cps), and the expression of Cps (tagged with triple HA) was confirmed by Western blot using antibodies against Cps and HA, respectively. B, Neurons infected with empty AAV or AAV–Cps were subjected to OGD (60 min), and Western blot was performed to detect α-fodrin cleavage, a marker for calpain activation, at 2 and 6 h after OGD. C, Neurons infected with empty AAV or AAV–Cps were subjected to OGD (60 min), and calpain activity was measured using substrate-based assays at 2, 6, and 16 h after OGD, and the data are expressed as fold increase over non-OGD controls (Con). *p < 0.05, **p < 0.01 versus empty AAV-infected neurons, from three independent experiments. D, Representative confocal images show the inhibitory effect of Cps overexpression on OGD-induced AIF release in neurons. Triple-label immunofluorescent staining for AIF (green), COX IV (red), and 4′-6′-diamidino-2-phenylindole (DAPI; blue) was performed at 6 h after OGD (60 min) in neurons infected with empty AAV or AAV–Cps. The arrows in the middle point to neurons that showed nuclear translocation of AIF after OGD. E, Percentages of neurons showing nuclear translocation of AIF at 2, 6, and 16 h after OGD (60 min). **p < 0.01, ***p < 0.001 versus empty AAV-infected neurons, from four independent experiments. F, Western blots for AIF using total cell extracts or nuclear (Nuc) and mitochondrial (Mito) fractions from non-OGD control neurons (C) or 2 and 6 h after OGD in neurons infected with empty AAV or AAV–Cps. The recombinant proteins AIF62 and/or AIF57 served as positive controls. The blots are representatives of three independent experiments. G–I, Cps overexpression attenuated OGD-induced neuronal cell death. Neurons were infected for 3 d with empty AAV or AAV–Cps and then subjected to OGD for 60 min. Cell death was measured at 24 h after OGD using Alamar blue fluorescence (G), LDH release (H), and phosphatidylinositol (PI) uptake (I). Data are mean ± SE; n = 12 per experimental condition from three independent experiments. **p < 0.01, ***p < 0.001 versus empty AAV-infected neurons.
Because calpastatin has no preference for calpain I over other calpain subtypes, we wanted to further address the role of calpain I using shRNA. Therefore, shRNA vectors specifically targeting calpain I and calpain II, the two most abundant neuronal calpain subtypes, were constructed, respectively, together with the scramble-sequence controls (Table 1). All targeting sequences for calpain I or calpain II shown in Table 1 were selected from five to six sequences for each targeted gene based on their effectiveness in a rat cell line. Gene transfection of neurons was performed using the Nucleofector device with the rat cortical neuron transfection kit (Amaxa). Using this methodology, transfection efficiency of ∼60% was achieved. As demonstrated in Western blots, transfection of shRNAs specifically reduced the neuronal levels of calpain I (Fig. 5A) and calpain II (data not shown), respectively, whereas the scramble controls had no effect. After OGD challenge (60 min), the calpain I-deficient (Fig. 5B), but not calpain II-deficient (data not shown), neurons showed decreased truncation and nuclear translocation of AIF compared with the control neurons. Moreover, only the calpain I-deficient neurons showed significantly increased cell survival after OGD (Fig. 5C,D).
Figure 5.

Calpain I activation contributes to OGD-induced AIF release in neurons. A, Left, Neurons were transfected with empty plasmid (lane 1), calpain I scramble shRNA sequence (Calp-Is; lanes 2, 3), or calpain I targeting shRNA sequence (Calp-It) for 12 d and then immunoblotted against calpain I. **p < 0.01 versus plasmid controls; n = 3. Right, Neurons were transfected with GFP expression plasmid alone, both GFP and Calp-Is, or both GFP and Calp-It for 12 d. Transfected neurons were collected by GFP cell sorting and then immunoblotted against calpain I and GFP. ***p < 0.001 versus GFP alone; n = 3. B, Transfected neurons were subjected to OGD for 60 min, and, at 6 h after OGD, nuclear (Nuc), mitochondrial (Mito), or total-cell extracts were immunoblotted against AIF. The graph at the bottom illustrates the semiquantitative results of nuclear AIF from three experiments. *p < 0.05 versus non-OGD controls; #p < 0.05 versus empty plasmid-transfected OGD neurons. C, D, Knockdown of calpain I attenuated OGD-induced neuronal cell death. Cell viability and cell death were measured at 24 h after OGD (60 min) using Alamar blue fluorescence (C) and LDH release (D), respectively. Data are mean ± SE; n = 12 per experimental condition from three independent experiments. *p < 0.05 versus empty plasmid-transfected neurons. E, Western blot detects endogenous calpain I in brain mitochondria. Purified brain mitochondria were subfractionated in the presence of digitonin at either 0.4% (outer membrane broken but no membrane protein dissolving) or 1.0% (resulting in membrane protein dissolving) and then immunoblotted against calpain I and AIF and fraction marker proteins COX IV (IM), VDAC (OM), and AK2 (IMS). Note that, under 0.4% digitonin, calpain I is present exclusively in the IMS, whereas AIF is associated with the IM; under 1.0% digitonin, both AIF and VDAC are dissolved from membranes and appear in all fractions, whereas calpain I remains in the IMS. For calpain I and AIF, the recombinant proteins (Rp) served as markers. The blots are representatives of three experiments. F, Translocation of calpain I and AIF within mitochondria after OGD. Neurons were subjected to OGD (60 min); 2 h later, mitochondria were purified and subfractionated under 0.4% digitonin. Note that calpain I is present in IM, IMS, and, to a lesser extent, in OM fractions after OGD; AIF appears in the IMS fraction at the size of 57 kDa after OGD. G, Semiquantitative analysis of calpain I and AIF levels in total or subfractions of mitochondria after OGD, based on three independent experiments described in F. Levels are expressed as fold increase over non-OGD neurons. *p < 0.05, **p < 0.01 versus non-OGD controls. H, Calpain activity measured in isolated whole mitochondria (30 μg protein/reaction) before or 2 h after OGD from neurons transfected with empty plasmid, Calp-Is, or Calp-It. #p < 0.05 versus non-OGD controls (con); *p < 0.05 versus empty plasmid-transfected neurons, from three experiments. I, Calpain activity measured in subfractions (IMS, 30 μg protein/reaction; OM or IM, 20 μg protein/reaction) of isolated mitochondria before or 2 h after OGD. *p < 0.05 versus non-OGD controls, from three experiments. J, Active calpain I is capable of entering mitochondria. Isolated brain mitochondria were incubated for 20 min with either inactive calpain I (1 U) or active calpain I (1 U) [preincubated with calcium (10 μm)], and subfractions (0.4% digitonin) were immunoblotted against calpain I and AIF. Note that calcium-activated calpain I translocates to IM and OM fractions, whereas the IM-bound AIF is truncated (57 kDa) and released to the IMS. The blots are representatives of three experiments.
The recent study by Garcia et al. (2005) demonstrated the expression of endogenous calpain I protein in purified rat brain mitochondria, suggesting that calpain I may be in close proximity to its mitochondrial substrates. To further understand how calpain I may gain access to the inner-membrane-bound AIF after ischemic injury, we examined the expression and activity of calpain I in purified mitochondria and mitochondrial subfractions. Using an antibody specific for domain III of the large subunit, we readily detected calpain I in purified mitochondria from mouse brain (Fig. 5E) or cultured neurons (Fig. 5F). Immunoblots of subfractions showed that calpain I was present exclusively in the IMS in normal tissues, whereas AIF was normally associated with the IM. Interestingly, in neurons challenged with OGD, calpain I was also detected in the IM and, to lesser extent, in the OM; AIF (the 57 kDa form) was also detected in the IMS after OGD (Fig. 5F,G). These results suggest that, after OGD, calpain I translocated to the IM and AIF is subject to truncation and translocation from IM to IMS.
To determine whether calpain I translocation within mitochondria is associated with alterations of calpain activity, we measured calpain-specific activity using mitochondrial protein extracts. Using whole-mitochondrial extracts from neurons, we detected a 1.8-fold increase in calpain activity in OGD neurons compared with control non-OGD neurons (Fig. 5H). In neurons transfected with the calpain I shRNA construct (calp-It), calpain activity was significantly decreased compared with empty plasmid or scramble sequence (calp-Is)-transfected neurons (Fig. 5H), indicating that calpain I was obligatory to mitochondrial calpain activity. We then examined calpain activity in mitochondrial subfractions, and, consistent with the immunoblot data, calpain activity was detected almost exclusively in the IMS of control non-OGD neurons. In contrast, calpain activity was significantly increased in all three fractions in OGD neurons (Fig. 5I).
Given that the total amount of mitochondrial calpain I appeared to be increased after OGD (Fig. 5F,G) and that an exogenous calpain I can gain access to AIF in isolated mitochondria (Liou et al., 2005; Polster et al., 2005), we wanted to determine whether exogenous calpain I can indeed move into mitochondria. Therefore, the cell-free assay (described in Fig. 3) was performed using purified brain mitochondria and active calpain I. Indeed, calcium-activated calpain I, but not the inactive calpain I, was detected in both IM and OM of mitochondria (Fig. 5J).
Calpain activation is sufficient to induce AIF release in neurons
Because calpain I was found to be obligatory for AIF release after OGD, it is important to determine whether other types of neuronal insults that activate calpain can also induce AIF release in neurons. We tested two non-OGD insults well known for their ability to activate calpain, the calcium ionophore A23187 (5-methylamino-2-[[(2S,3R,5R,8S,9S)-3,5,9-trimethyl-2-[1-oxo-1(1H-pyrrol-2-yl)propan-2-yl]-1,7-dioxaspiro[5.5]undecan-8-yl]methyl]benzoxazole-4-carboxylic acid) and the excitatory neurotoxin NMDA. Determined at 2 and 6 h after A23187 (10 μm for 30 min) challenge or 6 and 16 h after NMDA (100 μm for 15 min) challenge, both insults resulted in truncation (supplemental data 4A,C, available at www.jneurosci.org as supplemental material) and nuclear translocation (supplemental data 4B,D, available at www.jneurosci.org as supplemental material) of AIF. As predicted, overexpression of calpastatin attenuated nuclear translocation of AIF induced by either A23187 or NMDA.
Truncation of AIF is essential for calpain-mediated AIF release in OGD neurons
It has been reported previously that either the full-length (proform) AIF or its 57 kDa truncated form can degrade isolated nuclei with equal potency (Susin et al., 1999; Cao et al., 2003), suggesting that the truncation of AIF may not be important for its cell death-execution activity in the nucleus. However, a critical issue that remains to be addressed is whether the truncation of AIF to the 57 kDa fragment is essential for its release from mitochondria. To address this question, we constructed AAV vectors carrying either the wild-type full-length AIF or the calpain-resistant L101G/L103G mutant AIF, both with a GFP fusion protein at their C terminus. Neurons infected with the vectors carrying the wild-type or mutant AIF showed mitochondrial location of the GFP fusion protein as demonstrated by confocal microscopy (Fig. 6A). After OGD challenge, the number of neurons showing a nuclear localization of AIF–GFP fusion protein was markedly increased in cultures transfected with the wild-type AIF construct, whereas the number was not significantly increased in cultures transfected with the mutant AIF (Fig. 6A,B). Western blot confirmed that the mutant AIF fusion protein in mitochondria was resistant to OGD-induced truncation or nuclear translocation (Fig. 6C).
Figure 6.
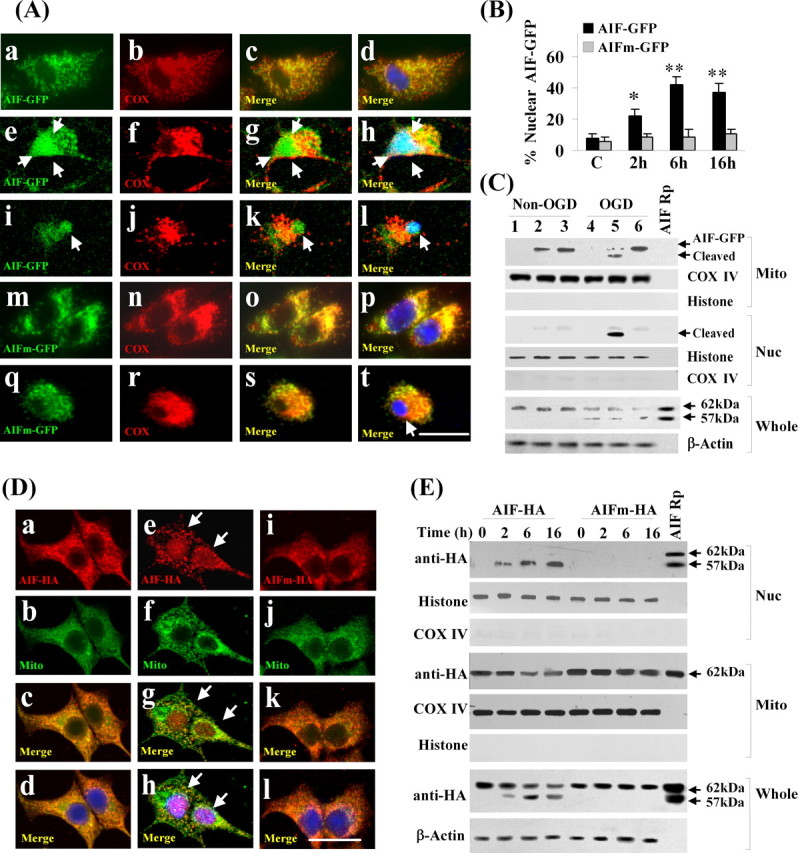
Calpain-dependent truncation of AIF is required for its release after OGD. A, Neurons were infected for 3 d with AAV carrying either the AIF-GFP fusion construct (a–l) or the calpain-resistant mutant (L101/103G) AIFm-GFP construct (m–t) and then subjected to OGD for 60 min. Confocal images were taken on triple-label immunofluorescent neurons (GFP, green; COX IV, red; Hoechst 33258, blue) in non-OGD cultures (a–d, m–p) or at 2 h (e–h, q–t) and 6 h (i–l) after OGD. In non-OGD neurons, AIF–GFP and AIFm–GFP are localized in mitochondria; after OGD, AIF–GFP is translocated into the nucleus (e–l), whereas AIFm–GFP remains in the mitochondria (q–t). B, Percentages of transfected neurons showing nuclear localization of AIF–GFP or AIFm–GFP at 2, 6, and 16 h after 60 min OGD. *p < 0.05, **p < 0.01 versus non-OGD neurons, from four independent experiments. C, Western blots of the mitochondrial and nuclear fractions using anti-GFP antibody in non-OGD and OGD neurons (lanes 1, 4, empty AAV; lanes 2, 5, AIF–GFP transfected; lanes 3, 6, AIFm–GFP transfected). Note that AIF–GFP, but not AIFm–GFP, is cleaved and translocated into the nucleus after OGD. The whole-cell extracts were immunoblotted with the anti-AIF antibody, showing the truncation of endogenous AIF after OGD. All blots are representative of three independent experiments. D, Neurons were infected with AAV carrying either C-terminal HA-tagged AIF construct (a–h) or the calpain-resistant mutant (L101/103G) AIFm–HA (i–l) and then subjected to OGD for 60 min. Confocal images were taken on triple-label immunofluorescent neurons (HA, red; COX IV, green; Hoechst 33258, blue) in non-OGD cultures (a–d) or at 2 h (e–h) after OGD. AIF–HA is translocated into the nucleus after OGD (e–h, arrows), whereas AIFm–HA remains in the mitochondria (i–l). E, Western blots using anti-HA antibody on whole-cell extracts or nuclear and mitochondrial fractions of transfected neurons at 2, 6, and 16 h after OGD. AIF–HA, but not AIFm–HA, is subjected to truncation and nuclear translocation after OGD. Blots are representative of three independent experiments.
To determine whether the resistance of L101G/L103G mutant AIF to OGD-induced nuclear translocation was an artifact attributable to GFP fusion, we tested additional constructs in which the wild-type and mutant AIF were tagged with an HA sequence instead of GFP. Results from both confocal microscopy and Western blot confirmed that the mutant AIF was resistant to OGD-induced nuclear translocation (Fig. 6D,E).
Truncation of AIF enables its disassociation from HSP10 in the mitochondria
One potential mechanism by which AIF releases after its truncation is that truncation of AIF may enable its disassociation from a binding partner within the mitochondria. In an effort to identify potential AIF binding partners, we performed coimmunoprecipitation with AIF against 12 different mitochondrial proteins and found that AIF bound to the mitochondrial chaperone HSP10 (Fig. 7A). The ability of AIF to bind to HSP10 was subsequently confirmed in a cell-free assay using isotope-labeled proteins generated through the in vitro transcription/translation system (Fig. 7B). Results from the domain-deletion experiments suggested that the N-terminal sequence of the 62 kDa form of AIF is required for AIF to bind to HSP10, because the 57 kDa AIF protein could not bind to HSP10 (Fig. 7C). Furthermore, the binding between 62 kDa AIF and HSP10 was decreased in the presence of calpain I in the assay, whereas the binding between the calpain-resistant mutant AIF and HSP10 was not affected by calpain I (Fig. 7C).
Figure 7.

Truncation of AIF enables its disassociation with HSP10 in the mitochondria. A, HSP10 is an endogenous binding protein for AIF in neurons. Left, Proteins from cultured neurons were first immunoprecipitated with anti-HSP60 or anti-HSP10 antibody and then immunoblotted with anti-AIF, anti-HSP60, and anti-HSP10 antibodies. Right, Proteins were first immunoprecipitated with anti-AIF or anti-HSP60 antibody and then immunoblotted with anti-AIF, anti-HSP60, and anti-HSP10 antibodies. B, Autoradiograph shows direct protein–protein interaction between AIF and HSP10. The protein-binding assay was performed using in vitro translated proteins (the Flag–HSP10 and [35S]AIF). The reaction products were immunoprecipitated using the anti-Flag antibody, electrophoresed, and then autoradiographed. C, Autoradiograph for [35S]AIF shows that Flag–HSP10 can directly bind to AIF62 but not to AIF57 and that calpain I, which cleaves AIF, inhibited AIF62/HSP10 binding but not the binding between HSP10 and the calpain-resistant AIFm. The protein-binding assay was performed as described in B. D, Western blots show decreased AIF/HSP10 binding after OGD in empty AAV-infected neurons. Cell extracts were immunoprecipitated using the anti-HSP10 antibody and then immunoblotted with the anti-AIF antibody. The AIF/HSP10 binding was restored in neurons transfected with AAV–Cps. The graph at the bottom illustrates the relative changes of three independent experiments. *p < 0.05 versus non-OGD controls. E, Neurons were infected with AAV–AIF–HA or AAV–AIFm–HA (calpain-resistant mutant) and then subjected to OGD (60 min). Immunoprecipitation (IP) was performed using the anti-HA antibody and then immunoblotted with HSP10 and HA antibodies. Note that the binding of HSP10/AIF, but not HSP10/AIFm, was decreased after OGD. Blots are representative of three experiments.
To determine whether the binding profile between AIF and HSP10 was altered in neurons after ischemic insults, coimmunoprecipitation was performed using neurons challenged with OGD for 60 min. Although the total amount of endogenous HSP10 or AIF was not changed after OGD, the binding between HSP10 and AIF was decreased in OGD neurons (Fig. 7D). This change in HSP10/AIF binding profile was also replicated in AIF–HA (wild-type AIF)-transfected neurons (Fig. 7E), whereas the binding between the mutant AIF–HA and HSP10 was not reduced after OGD.
The wild-type AIF but not calpain-resistant mutant AIF restores OGD-induced cell death of AIF-deficient neurons
Because the data suggested that calpain I-mediated truncation of AIF is critical for OGD-induced release of AIF from mitochondria, we wanted to test the hypothesis that overexpression of the calpain-resistant AIF in neurons lacking endogenous AIF would not restore the levels of OGD-induced cell death, but the wild-type AIF would. Thus, we performed gene transfection of AIF and the L101G/L103G mutant AIF (Fig. 8C), respectively, in neuronal cultures derived from Harlequin (Hq) mice that express ∼20% of normal AIF levels in the brain (Vahsen et al., 2004). Compared with the wild-type neurons, Hq neurons displayed 27–34% decreases in cell death at 24 h after 60 min of OGD, as measured using two independent methods (Fig. 8A,B). Transfection of the wild-type AIF, but not the calpain-resistant mutant AIF, significantly restored the levels of OGD-induced cell death in Hq neurons (Fig. 8A,B).
Figure 8.

The wild-type AIF, but not the calpain-resistant mutant AIF, restores OGD-induced cell death of AIF-deficient neurons. A, B, Neurons from Hq mice or wild-type (Wt) mice were infected with AAV–AIF, AAV–AIFm, or empty AAV for 3 d and then subjected to OGD for 60 min. Cell viability (A) and cell death (B) were measured at 24 and 48 h after OGD using Alamar blue fluorescence and phosphatidylinositol/Hoechst 33258 staining, respectively. *p < 0.05 versus wild-type neurons after OGD; #p < 0.05 versus Hq neurons infected with AAV–AIFm. n = 12, from three independent experiments. C, Western blots (anti-AIF and anti-HA) show the expression levels of AIF or AIFm in Hq neurons 3 d after AAV infection. AIF recombinant proteins (AIF Rp) served as size markers. D, E, AAV-mediated overexpression of calpastatin (Cps) in Hq neurons resulted in additive prosurvival effect against OGD. Neurons from Hq mice were infected with AAV–Cps or empty AAV for 3 d and then subjected to OGD for 60 min. Cell viability (D) and cell death (E) were measured at 48 h after OGD using Alamar blue and phosphatidylinositol/Hoechst 33258 staining, respectively. *p < 0.05 versus noninfected Hq neurons, n = 14 per condition, from three independent experiments.
To determine whether the increased calpain activity after OGD relies exclusively on AIF release to produce cytotoxicity, additional AAV–Cps transfection experiments were performed using Hq neurons. Transfection of calpastatin into Hq neurons resulted in significantly greater increase in cell survival (12% net increases) over untransfected Hq neurons (Fig. 8D,E). This result suggests that, despite the key role AIF plays in mediating calpain activity-dependent cell death, there are other calpain substrates that also contribute to ischemic cell death.
Calpastatin overexpression inhibits AIF translocation in CA1 neurons after global ischemia
Our next objective was to determine whether calpain activation is the underlying mechanism for nuclear translocation of AIF in an in vivo model of cerebral ischemia. In a previous study, we investigated the spatial distribution and temporal profile of AIF release in the rat transient global ischemia model, observing a highly reproducible pattern of AIF release in CA1 neurons at 24–72 h after ischemia (Cao et al., 2003). Moreover, calpain activation in CA1 after transient global cerebral ischemia has been previously well documented (Roberts-Lewis et al., 1994; Neumar et al., 2001). Therefore, in the present study, we investigated the effect of calpastatin overexpression in CA1 neurons on ischemia-induced AIF release. AAV vectors were infused into the rat hippocampus using convection-enhanced gene delivery technology (Bankiewicz et al., 2000; Sanftner et al., 2005). This technique resulted in widespread gene transfection in the CA1 sector of dorsal hippocampus as examined 14 d after microinfusion (Fig. 9A). Transient global ischemia was then induced 14 d after AAV infusion, and AIF release in the CA1 sector was evaluated using Western blot (n = 6 per group) at 36 and 72 h after ischemia. Compared with controls, AAV–Cps-infected CA1 showed an attenuation of calpain activation (Fig. 9B,C) and significantly reduced nuclear translocation of AIF after ischemia (Fig. 9D–G). Diminished AIF release by calpastatin overexpression was confirmed at the cellular level (n = 5 per group) using double-label immunofluorescence (Fig. 9D). Finally, CA1 cell death/survival was examined using stereological cell counting at 7 d after global ischemia (n = 9 per group). Calpastatin overexpression significantly enhanced CA1 neuronal survival (Fig. 9H) (supplemental data 5A, available at www.jneurosci.org as supplemental material) and significantly decreased the number of CA1 neurons containing DNA fragmentation after ischemia (Fig. 9I).
Figure 9.
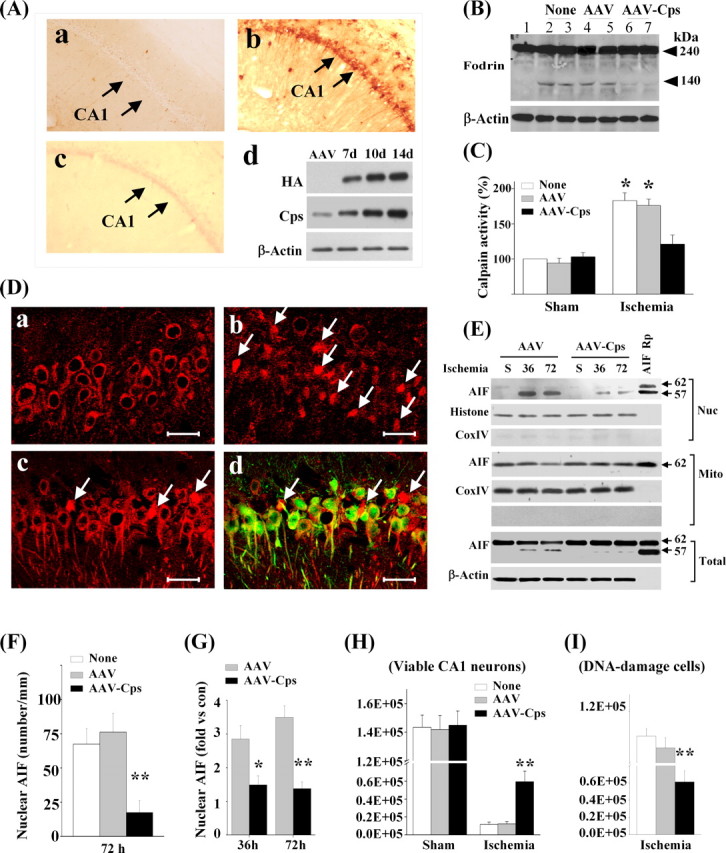
Calpastatin overexpression inhibits AIF translocation in CA1 neurons after global ischemia. A, Overexpression of calpastatin (Cps) after AAV infection in the rat brain. AAV was infused into the hippocampus using convection methodology to achieve widespread expression in the CA1 sector. Images show HA immunoreactivity (HA–Cps) at 14 d after AAV–Cps infection (b); preabsorption of the antibody with Cps protein abolished the signals (c). No Cps immunoreactivity is seen in CA1 after empty vector infection (a). Western blots (anti-HA or anti-Cps) confirm the expression of Cps protein in the CA1 sector at 7–14 d after AAV–Cps infection; n = 5 per group. B, C, AAV–Cps infection inhibited global ischemia-induced calpain activation in CA1. Western blot (B) was performed at 36 h after ischemia to detect α-fodrin cleavage, which was inhibited by AAV–Cps (lanes 6, 7) but not by empty AAV (lanes 4, 5). Substrate-based activity assays (C) show that ischemia-induced increases in calpain activity were inhibited by AAV–Cps. *p < 0.05 versus non-ischemic controls; n = 6 per group. D, Representative immunofluorescence of AIF (red) from non-ischemic CA1 (a) or 72 h after global ischemia (b–d). AAV–Cps (c, d) or the empty vector (b) was infused 14 d before ischemia, and brain sections were double-label immunostained for AIF (red) and HA (green, d). Note that majority of CA1 neurons lost normal localization of AIF after ischemia (b, arrows), but AIF translocation was rare in Cps-overexpressed CA1 (c, d, arrows). Scale bars, 50 μm. E, Western blots of CA1 whole-cell extracts or subcellular fractions after sham operation (S) or at 36 and 72 h after ischemia. AAV–Cps infection inhibited ischemia-induced truncation and nuclear translocation of AIF. F, Quantitative analysis of AIF nuclear translocation 72 h after ischemia, based on the immunostaining experiments described in D. **p < 0.01 versus empty vector group or non-infection group; n = 5 per group. G, Semiquantitative analysis of AIF nuclear translocation at 36 and 72 h after ischemia, based on Western blots. *p < 0.05, **p < 0.01 versus empty AAV-infected brains; n = 6 per group. H, I, Cps expression promoted CA1 cell survival after global ischemia. Viable cells (H) and DNA-damaged cells (I) in CA1 of the dorsal hippocampus were quantified stereologically at 7 d after ischemia or sham operation after AAV–Cps or empty vector infection. **p < 0.01 versus non-infected or empty vector-infected brains; n = 9 per group.
AIF knockdown is neuroprotective in CA1 against transient global ischemia
The final objective of this study was to determine whether AIF knockdown confers neuroprotection in CA1 against transient global ischemia. A mixture of AAV—enhanced GFP with AAV–AIFt or AAV–AIFs in the ratio 1:5 was infused into the CA1 sector using the convection technique, and the effect of gene infection on AIF expression was determined at 14 d after AAV infusion using both immunofluorescent staining (n = 5 per group) and Western blot analysis (n = 6 per group). AAV–AIFt, but not AAV–AIFs, significantly reduced the level of AIF expression in CA1 neurons (Fig. 10A,B). To determine the effect of AIF knockdown on CA1 cell death, transient global ischemia (n = 9 per group) or sham operation (n = 6 per group) was performed at 14 d after AAV infusion, and histology was assessed at 7 d after ischemia using stereological cell counting. Neither AAV–AIFt nor AAV–AIFs resulted in significant cell loss or DNA damage in CA1 at 21 d after vector infusion in sham control brains. In brains subjected to transient global ischemia, CA1 cell death (Fig. 10C) (supplemental data 5B, available at www.jneurosci.org as supplemental material) and DNA damage (Fig. 10D) were significantly attenuated by AAV–AIFt. In separate experiments (n = 4 per group), pulse-field gel electrophoresis was performed to detect large-scale DNA fragmentation, a hallmark of AIF-induced genomic DNA degradation, in the CA1 sector at 3 d after global ischemia (Fig. 10E). AAV–AIFt, but not AAV–AIFs, reduced ischemia-induced large-scale DNA fragmentation.
Figure 10.
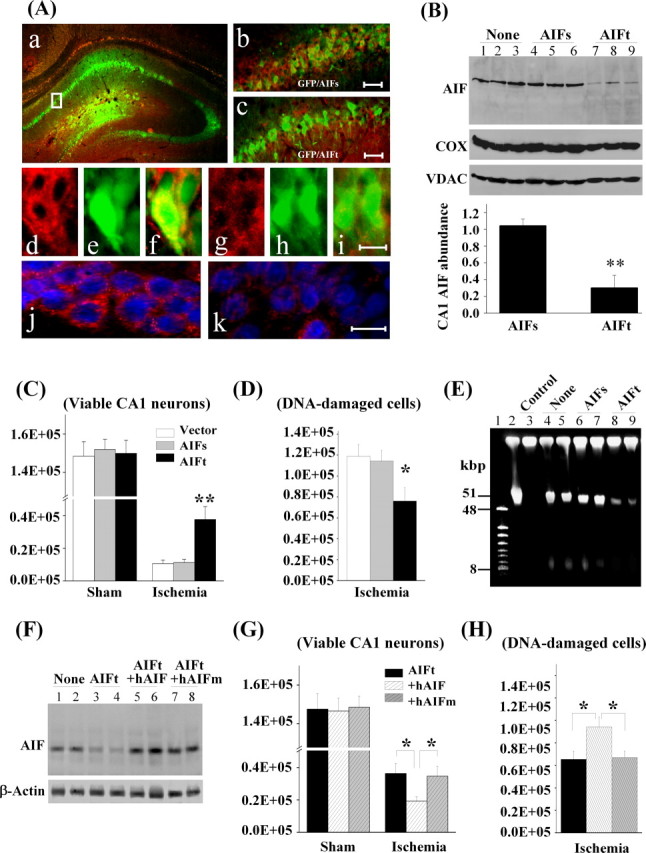
AIF knockdown is neuroprotective in CA1 against transient global ischemia. A, CA1 was coinfected for 14 d with AAV–GFP and the AIF-targeting sequence AAV–AIFt (a, c) or the AIF scramble sequence AAV–AIFs (b). d–f show double-label immunofluorescence of AIF (red) and GFP (green) in AAV–AIFs/AAV–GFP-infected CA1 neurons; g–i show reduced AIF expression (red) in AAV–AIFt/AAV–GFP-infected CA1 neurons. j and k show double-label immunofluorescence of AIF (red) and Hoechst 33258 (blue) in AAV–AIFs- and AAV–AIFt-infected CA1, respectively. B, Western blots show the expression levels of AIF in CA1, either non-infected (lanes 1–3) or at 14 d after infection with AAV–AIFs (lanes 4–6) or AAV–AIFt (lanes 7–9). The graph illustrates the relative changes of AIF expression in CA1 after AAV–AIFt infection. **p < 0.01 versus AAV–AIFs-infected brains; n = 6 per group. C, D, Knockdown of AIF expression promoted CA1 cell survival after transient global ischemia. Viable cells (C) and DNA-damaged cells (D) in CA1 of the dorsal hippocampus were quantified stereologically, respectively, at 7 d after ischemia or sham operation after AAV–AIFs or AAV–AIFt infection. *p < 0.05, **p < 0.01 versus non-infected or AAV–AIFs-infected brains; n = 9 per group. E, Knockdown of AIF expression attenuated ischemia-induced large-scale DNA fragmentation in the CA1 sector, determined using DNA pulse-field gel electrophoresis at 3 d after global ischemia. Lanes 1, 2, Size markers; lane 3, non-ischemic control; lanes 4, 5, non-infected ischemic brains; lanes 6, 7, AAV–AIFs-infected ischemic brains; lanes 8, 9, AAV–AIFt-infected ischemic brains. Note that ischemia-induced formation of both 50 and 10 kbp fragments was reduced in AAV–AIFt-infected brains. F, Western blots show the expression levels of AIF in CA1, either non-infected (lanes 1, 2) or at 14 d after infection with AAV–AIFt (lanes 3, 4), coinfection with AAV–AIFt/hAIF (lanes 5, 6), or coinfection with AIFt/hAIFm (lanes 7, 8). G, H, Overexpression of human AIF, but not the calpain-resistant hAIFm, ameliorated the prosurvival effect of AIF knockdown after transient global ischemia. Viable cells (G) and DNA-damaged cells (H) in CA1 of the dorsal hippocampus were quantified stereologically, respectively, at 7 d after ischemia (n = 10 per group) or sham operation (n = 6 per group). *p < 0.05 versus con-infection of AAV–AIFt/AAV–hAIF.
To determine whether overexpression of an shRNA-resistant AIF could restore the susceptibility of CA1 to transient global ischemia, a mixture of AAV–AIFt with vectors carrying either the wild-type human AIF (AAV–hAIF) or the mutant (L101/103G) human AIF (AAV–hAIFm) in the ratio 1:1 was infused into the CA1 sector. As determined 14 d after AAV infusion, either AAV–hAIF or AAV–hAIFm restored AIF expression levels in AAV–AIFt-infected CA1 (n = 6 per group) (Fig. 10F). Moreover, coinfection with AAV–hAIF significantly attenuated but did not completely eliminate the prosurvival effect of AAV–AIFt in CA1 at 7 d after transient global ischemia (Fig. 10G,H). In contrast, con-infection with AAV–hAIFm had no significant effect (Fig. 10G,H).
Discussion
The role of AIF nuclear translocation as a caspase-independent cell death pathway has been established recently in several neuronal models of ischemic-relevant injury (Cregan et al., 2002; Yu et al., 2002; Culmsee et al., 2005; Zhu et al., 2007). However, the signaling mechanism underlying AIF release in neurons after ischemia is not fully understood. We report here that calpain I is a direct activator for AIF release in ischemic neurons. The main findings of this study include the following: (1) a calpain-generated truncated form of AIF is released and translocated into the nucleus after ischemia; (2) calpain activation is required for AIF release after ischemia, because overexpression of the calpain inhibitor calpastatin or knockdown of the expression of calpain I in neurons inhibits AIF release; (3) calpain-dependent truncation of AIF near its N terminus is essential for AIF release from mitochondria, because the calpain-resistant mutant AIF is resistant to ischemia-induced release; and (4) inhibition of the pro-death effect of AIF by overexpressing calpastatin or knocking down AIF expression confers neuroprotection against ischemia-induced cell death. Together, these observations suggest that calpain I activation is a novel signaling mechanism that directly contributes to mitochondrial AIF release in ischemic neuronal injury.
Using the siRNA-mediated gene knockdown approach, we confirmed the obligatory role of AIF release in ischemia-induced neuronal cell death in both in vitro (OGD) and in vivo (transient global ischemia) models. The specificity of the AIF siRNA effect was supported in the current study, because transfection of scrambled siRNA sequences or AIF-unrelated siRNA sequences (GFP) failed to confer any neuroprotection against ischemia. Moreover, transfection of AIF cDNA in AIF-deficient neurons significantly restored OGD-induced cell death. These results are in agreement with recent studies demonstrating the resistance of AIF-deficient neurons from Harlequin mice to ischemic injury in both in vitro and in vivo models (Culmsee et al., 2005; Zhu et al., 2007).
Although the results based on Harlequin mice confirmed the pro-death role of AIF in ischemic injury, a caveat with using these mice is that the AIF deficiency is also associated with a neurodegenerative phenotype in brain and increased neuronal cell death in response to certain oxidative stressors (Klein et al., 2002). The adverse effect of AIF deficiency on the survival of certain neuronal populations in the brain has now been attributed to compromised respiratory chain complex I activity and impaired oxidative phosphorylation (Vahsen et al., 2004; Cheung et al., 2006). It has been shown in AIF-knock-out mouse embryonic stem cells, AIF-knockdown HeLa cells, and the Harlequin mice that the mitochondrial complex I activity is reduced to ∼50% of normal levels (Vahsen et al., 2004). In the present study, modest decreases in complex I (7.6%) and complex III (12.3%) were observed in AIF-knockdown neurons, and it appeared that reduced AIF expression had an insignificant neurodegenerative effect on cultured neurons or on hippocampal neurons in the brain. One possible explanation for the discrepancy between this study and others may be that different cell types may rely to different degrees on AIF for the maintenance of complex I in mitochondria. Thus, different cell types or organ tissues may have different thresholds for tolerating AIF loss. Supporting evidence for this assumption is that compromise of complex I activity was not found in the heart and liver of the Harlequin mice despite the same levels of AIF deficiency among different organs (Klein et al., 2002). However, muscle-specific complete loss of AIF leads to mitochondrial dysfunction, skeletal muscle atrophy, and dilated cardiomyopathy (Joza et al., 2005).
The main focus of the current study was to elucidate the signaling mechanism responsible for AIF translocation after neuronal ischemia. To this end, we found that a calpain-generated truncated AIF (57 kDa) was translocated from mitochondria to the nucleus in OGD-challenged neurons. In a search for the specific protease that produced the truncated AIF after ischemia, we found that calpain I, but not calpain II, caspases or lysosomal enzymes could generate the 57 kDa AIF from the recombinant AIF (62 kDa) in a cell-free system. It was further found in the in vitro assays that calpain I was fully capable of truncating and releasing AIF from mitochondria. These results are consistent with recently reported findings in isolated brain or liver mitochondria (Liou et al., 2005; Polster et al., 2005). We also found that the chaperone protein HSP10 is an endogenous binding partner for AIF in the mitochondria of normal neurons and that the interaction between AIF and HSP10 was decreased in the presence of activated calpain I. Thus, the interaction of AIF with HSP10 may be a mechanism by which AIF is retained within the mitochondria in normal cells.
Calpain, also known as calcium-activated neutral protease, is one of the most versatile molecules contributing to a variety of physiological processes in cells (Suzuki et al., 1992) and also is involved in various pathological conditions such as ischemic and traumatic brain injuries (Lee et al., 1991; Bartus et al., 1994; Saatman et al., 1996; Neumar et al., 2003). Of the best characterized calpain isoforms in neurons, calpain I (μ-calpain) and calpain II (m-calpain) require the presence of micromolar and millimolar Ca2+ ions, respectively, for their activation and subsequent translocation to their target substrates (Suzuki et al., 1992; Basse et al., 1993). To date, many substrates have been identified for calpain, and some are participants in cell death pathways (Gao and Dou, 2000; M. Chen et al., 2001). The current study, for the first time, investigates the role of calpain activation in AIF release in the context of ischemic neuronal injury, and the results suggest that AIF is a key pro-death substrate for calpain I in ischemic neurons. Supported by several lines of evidence described here, including the neuroprotective effect by knockdown of calpain I and the creation of a calpain-resistant AIF mutant, calpain I-induced truncation of AIF appears to be critical for its dissociation with HSP10 and subsequent release from the mitochondria in OGD neurons. In further support of this notion, overexpression of the wild-type AIF, but not the calpain-resistant mutant AIF, in AIF-deficient neurons was found to restore ischemic cell death in both in vitro and in vivo models.
It is indeed intriguing that calpain I, classically considered to be a cytosolic protein, gains access to the mitochondrial inner-membrane-bound AIF. In agreement with the recent report by Garcia et al. (2005), we detected calpain I in purified mitochondria from normal brain tissues and cultured neurons. We also found that calpain I is normally localized in the intermembrane space; however, during OGD, it moves to the inner membrane of mitochondria. This translocation of calpain I was found to coincide with increased calpain activity in the mitochondria, including their inner membrane. These results suggest that, in ischemic neurons, activated calpain I (from cytosolic and/or mitochondrial origin) may translocate to the mitochondrial inner membrane, leading to the truncation and release of AIF.
The other pro-death substrates identified for calpain thus far include Bid and Bax. Both are proapoptotic members of the Bcl-2 family and important regulators of the mitochondrial death pathway. Either Bid or Bax can be cleaved by calpain to an active fragment capable of compromising mitochondrial membrane integrity (Gao and Dou, 2000; Mandic et al., 2002). However, it has been controversial whether Bid or Bax is involved in AIF release during apoptosis (Arnoult et al., 2002; van Loo et al., 2002). In the present study, we found that activated calpain I could induce AIF release from isolated mitochondria in the absence of Bid and Bax (Bid/Bax knock-out). Moreover, despite the potency of both Bid and Bax in releasing cytochrome c, neither of them could induce AIF release from isolated mitochondria without the addition of calpain I. Nonetheless, the activation of Bid could enhance AIF release from mitochondria induced by calpain I. These results suggest that Bid or Bax is not essential for AIF release in ischemic neurons but may serve as a cofactor in calpain-induced AIF release.
Two recent studies identified PARP-1 activation and formation of poly(ADP-ribose) polymers as another mechanism leading to AIF release in models of NMDA neurotoxicity and DNA damage (Andrabi et al., 2006; Yu et al., 2006). This signaling pathway could also have important implications in ischemia because of the well established role of PARP-1 in mediating ischemic cell death. Whether the PARP-1 and calpain I pathways are interrelated in the context of cerebral ischemia-induced AIF release is unknown but will be worthwhile to explore in future studies. Indeed, PARP-1 activation and calcium signaling, including calpain activation, may engage in crosstalk at several different levels. For instance, calcium is an important cofactor in reactive oxygen species-induced PARP-1 hyperactivation (Karczewski et al., 1999; Bentle et al., 2006), whereas PARP-1 activation could further disrupt calcium homeostasis, leading to mitochondrial calcium overload (Yang et al., 2006). Interestingly, PARP-1 is a direct substrate for calpain I and undergoes calpain-dependent cleavage under certain conditions of neuronal apoptosis (Boland and Campbell, 2003), which could possibly form a feedback loop limiting the effect of PARP-1 activity.
In summary, the data presented in this report delineate calpain I activation as a direct mechanism triggering AIF release after ischemic neuronal injury. Because AIF release and its nuclear translocation constitute an important caspase-independent cell death pathway in cerebral ischemia, novel therapeutic interventions aiming at inhibiting calpain I activity and preventing AIF release may offer favorable neuroprotective effects without interfering with the physiological functions of AIF within mitochondria.
Footnotes
This work was supported by National Institutes of Health–National Institute of Neurological Disorders and Stroke Grants NS43802, NS44178, and NS36736 (J.C.), American Heart Association (AHA) Established Investigator Award 240135N (J.C.), and AHA Beginning Grant-in-Aid (G.C.). J.C. and S.H.G. were also supported in part by the Geriatric Research, Education, and Clinical Center, Veterans Affairs, Pittsburgh Health Care System (Pittsburgh, PA). We thank Dr. David Nicholls (The Buck Institute for Aging Research, Novato, CA) for sharing his expertise in assessing mitochondrial functions. We thank Carol Culver for editorial assistance and Pat Strickler for secretarial support.
References
- Alano CC, Ying W, Swanson RA. Poly(ADP-ribose) polymerase-1-mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition. J Biol Chem. 2004;279:18895–18902. doi: 10.1074/jbc.M313329200. [DOI] [PubMed] [Google Scholar]
- Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, Sasaki M, Klaus JA, Otsuka T, Zhang Z, Koehler RC, Hurn PD, Poirier GG, Dawson VL, Dawson TM. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci USA. 2006;103:18308–18313. doi: 10.1073/pnas.0606526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnoult D, Parone P, Martinou JC, Antonsson B, Estaquier J, Ameisen JC. Mitochondrial release of apoptosis-inducing factor occurs downstream of cytochrome c release in response to several proapoptotic stimuli. J Cell Biol. 2002;159:923–929. doi: 10.1083/jcb.200207071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankiewicz KS, Eberling JL, Kohutnicka M, Jagust W, Pivirotto P, Bringas J, Cunningham J, Budinger TF, Harvey-White J. Convection-enhanced delivery of AAV vector in parkinsonian monkeys; in vivo detection of gene expression and restoration of dopaminergic function using pro-drug approach. Exp Neurol. 2000;164:2–14. doi: 10.1006/exnr.2000.7408. [DOI] [PubMed] [Google Scholar]
- Bartus RT, Baker KL, Heiser AD, Sawyer SD, Dean RL, Elliott PJ, Straub JA. Postischemic administration of AK275, a calpain inhibitor, provides substantial protection against focal ischemic brain damage. J Cereb Blood Flow Metab. 1994;14:537–544. doi: 10.1038/jcbfm.1994.67. [DOI] [PubMed] [Google Scholar]
- Basse F, Gaffet P, Rendu F, Bienvenue A. Translocation of spin-labeled phospholipids through plasma membrane during thrombin- and ionophore A23187-induced platelet activation. Biochemistry. 1993;32:2337–2344. doi: 10.1021/bi00060a027. [DOI] [PubMed] [Google Scholar]
- Bentle MS, Reinicke KE, Bey EA, Spitz DR, Boothman DA. Calcium-dependent modulation of poly(ADP-ribose) polymerase-1 alters cellular metabolism and DNA repair. J Biol Chem. 2006;281:33684–33696. doi: 10.1074/jbc.M603678200. [DOI] [PubMed] [Google Scholar]
- Berman SB, Hastings TG. Dopamine oxidation alters mitochondrial respiration and induces permeability transition in brain mitochondria: implications for Parkinson's disease. J Neurochem. 1999;73:1127–1137. doi: 10.1046/j.1471-4159.1999.0731127.x. [DOI] [PubMed] [Google Scholar]
- Boland B, Campbell V. Beta-Amyloid (1–40)-induced apoptosis of cultured cortical neurons involves calpain-mediated cleavage of poly-ADP-ribose polymerase. Neurobiol Aging. 2003;24:179–186. doi: 10.1016/s0197-4580(02)00060-x. [DOI] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- Cande C, Cohen I, Daugas E, Ravagnan L, Larochette N, Zamzami N, Kroemer G. Apoptosis-inducing factor (AIF): a novel caspase-independent death effector released from mitochondria. Biochimie. 2002;84:215–222. doi: 10.1016/s0300-9084(02)01374-3. [DOI] [PubMed] [Google Scholar]
- Cao G, Pei W, Lan J, Stetler RA, Luo Y, Nagayama T, Graham SH, Yin XM, Simon RP, Chen J. Caspase-activated DNase/DNA fragmentation factor 40 mediates apoptotic DNA fragmentation in transient cerebral ischemia and in neuronal cultures. J Neurosci. 2001;21:4678–4690. doi: 10.1523/JNEUROSCI.21-13-04678.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao G, Luo Y, Nagayama T, Pei W, Stetler RA, Graham SH, Chen J. Cloning and characterization of rat caspase-9: implications for a role in mediating caspase-3 activation and hippocampal cell death after transient cerebral ischemia. J Cereb Blood Flow Metab. 2002;22:534–546. doi: 10.1097/00004647-200205000-00005. [DOI] [PubMed] [Google Scholar]
- Cao G, Clark RS, Pei W, Yin W, Zhang F, Sun FY, Graham SH, Chen J. Translocation of apoptosis-inducing factor in vulnerable neurons after transient cerebral ischemia and in neuronal cultures after oxygen-glucose deprivation. J Cereb Blood Flow Metab. 2003;23:1137–1150. doi: 10.1097/01.WCB.0000087090.01171.E7. [DOI] [PubMed] [Google Scholar]
- Cao G, Xiao M, Sun F, Xiao X, Pei W, Li J, Graham SH, Simon RP, Chen J. Cloning of a novel Apaf-1-interacting protein: a potent suppressor of apoptosis and ischemic neuronal cell death. J Neurosci. 2004;24:6189–6201. doi: 10.1523/JNEUROSCI.1426-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Stetler RA, Cao G, Pei W, O'Horo C, Yin XM, Chen J. Characterization of the rat DNA fragmentation factor 35/Inhibitor of caspase-activated DNase (Short form). The endogenous inhibitor of caspase-dependent DNA fragmentation in neuronal apoptosis. J Biol Chem. 2000;275:38508–38517. doi: 10.1074/jbc.M003906200. [DOI] [PubMed] [Google Scholar]
- Chen H, Hu CJ, He YY, Yang DI, Xu J, Hsu CY. Reduction and restoration of mitochondrial DNA content after focal cerebral ischemia/reperfusion. Stroke. 2001;32:2382–2387. doi: 10.1161/hs1001.097099. [DOI] [PubMed] [Google Scholar]
- Chen J, Nagayama T, Jin K, Stetler RA, Zhu RL, Graham SH, Simon RP. Induction of caspase-3-like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J Neurosci. 1998;18:4914–4928. doi: 10.1523/JNEUROSCI.18-13-04914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, He H, Zhan S, Krajewski S, Reed JC, Gottlieb RA. Bid is cleaved by calpain to an active fragment in vitro and during myocardial ischemia/reperfusion. J Biol Chem. 2001;276:30724–30728. doi: 10.1074/jbc.M103701200. [DOI] [PubMed] [Google Scholar]
- Cheung EC, Melanson-Drapeau L, Cregan SP, Vanderluit JL, Ferguson KL, McIntosh WC, Park DS, Bennett SA, Slack RS. Apoptosis-inducing factor is a key factor in neuronal cell death propagated by Bax-dependent and Bax-independent mechanisms. J Neurosci. 2005;25:1324–1334. doi: 10.1523/JNEUROSCI.4261-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung EC, Joza N, Steenaart NAE, McClellan KA, Neuspiel M, McNamara S, MacLaurin JG, Rippstein P, Park DS, Shore GC, McBride HM, Penninger JM, Slack RS. Dissociating the dual roles of apoptosis-inducing factor in maintaining mitochondrial structure and apoptosis. EMBO J. 2006;25:4061–4073. doi: 10.1038/sj.emboj.7601276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CT, Zhu JH, Cao G, Signore A, Wang S, Chen J. Apoptosis inducing factor mediates caspase-independent 1-methyl-4-phenylpyridinium toxicity in dopaminergic cells. J Neurochem. 2005;94:1685–1695. doi: 10.1111/j.1471-4159.2005.03329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregan SP, Fortin A, MacLaurin JG, Callaghan SM, Cecconi F, Yu SW, Dawson TM, Dawson VL, Park DS, Kroemer G, Slack RS. Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J Cell Biol. 2002;158:507–517. doi: 10.1083/jcb.200202130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuerrier D, Moldoveanu T, Davies PL. Determination of peptide substrate specificity for μ-calpain by a peptide library-based approach. J Biol Chem. 2005;280:40632–40641. doi: 10.1074/jbc.M506870200. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Zhu C, Landshamer S, Becattini B, Wagner E, Pellecchia M, Blomgren K, Plesnila N. Apoptosis-inducing factor triggered by poly(ADP-ribose) polymerase and Bid mediates neuronal cell death after oxygen-glucose deprivation and focal cerebral ischemia. J Neurosci. 2005;25:10262–10272. doi: 10.1523/JNEUROSCI.2818-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L, Zhang X, Han YY, Burke NA, Kochanek PM, Watkins SC, Graham SH, Carcillo JA, Szabo C, Clark RS. Intra-mitochondrial poly(ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J Biol Chem. 2003;278:18426–18433. doi: 10.1074/jbc.M301295200. [DOI] [PubMed] [Google Scholar]
- Fujimura M, Morita-Fujimura Y, Kawase M, Copin JC, Calagui B, Epstein CJ, Chan PH. Manganese superoxide dismutase mediates the early release of mitochondrial cytochrome C and subsequent DNA fragmentation after permanent focal cerebral ischemia in mice. J Neurosci. 1999;19:3414–3422. doi: 10.1523/JNEUROSCI.19-09-03414.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao G, Dou QP. N-terminal cleavage of bax by calpain generates a potent proapoptotic 18-kDa fragment that promotes bcl-2-independent cytochrome C release and apoptotic cell death. J Cell Biochem. 2000;80:53–72. doi: 10.1002/1097-4644(20010101)80:1<53::aid-jcb60>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Garcia M, Bondada V, Geddes JW. Mitochondrial localization of mu-calpain. Biochem Biophys Res Commun. 2005;338:1241–1247. doi: 10.1016/j.bbrc.2005.10.081. [DOI] [PubMed] [Google Scholar]
- Graham SH, Chen J. Programmed cell death in cerebral ischemia. J Cereb Blood Flow Metab. 2001;21:99–109. doi: 10.1097/00004647-200102000-00001. [DOI] [PubMed] [Google Scholar]
- Gundersen HJ. Stereology of arbitrary particles. A review of unbiased number and size estimators and the presentation of some new ones, in memory of William R. Thompson. J Microsc. 1986;143:3–45. [PubMed] [Google Scholar]
- Joza N, Oudit GY, Brown D, Benit P, Kassiri Z, Vahsen N, Benoit L, Patel MM, Nowikovsky K, Vassault A, Backx PH, Wada T, Kroemer G, Rustin P, Penninger JM. Muscle-specific loss of apoptosis-inducing factor leads to mitochondrial dysfunction, skeletal muscle atrophy, and dilated cardiomyopathy. Mol Cell Biol. 2005;25:10261–10272. doi: 10.1128/MCB.25.23.10261-10272.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski JM, Peters JG, Noordhoek J. Prevention of oxidant-induced cell death in Caco-2 colon carcinoma cells after inhibition of poly(ADP-ribose) polymerase and Ca2+ chelation: involvement of a common mechanism. Biochem Pharmacol. 1999;57:19–26. doi: 10.1016/s0006-2952(98)00286-x. [DOI] [PubMed] [Google Scholar]
- Kim TH, Zhao Y, Ding WX, Shin JN, He X, Seo YW, Chen J, Rabinowich H, Amoscato AA, Yin XM. Bid-cardiolipin interaction at mitochondrial contact site contributes to mitochondrial cristae reorganization and cytochrome c release. Mol Biol Cell. 2004;15:3061–3072. doi: 10.1091/mbc.E03-12-0864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Frankel WN, Bronson RT, Ackerman SL. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature. 2002;419:367–374. doi: 10.1038/nature01034. [DOI] [PubMed] [Google Scholar]
- Lee KS, Frank S, Vanderklish P, Arai A, Lynch G. Inhibition of proteolysis protects hippocampal neurons from ischemia. Proc Natl Acad Sci USA. 1991;88:7233–7237. doi: 10.1073/pnas.88.16.7233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou AK, Zhou Z, Pei W, Lim TM, Yin XM, Chen J. BimEL up-regulation potentiates AIF translocation and cell death in response to MPTP. FASEB J. 2005;19:1350–1352. doi: 10.1096/fj.04-3258fje. [DOI] [PubMed] [Google Scholar]
- Mandic A, Viktorsson K, Strandberg L, Heiden T, Hansson J, Linder S, Shoshan MC. Calpain-mediated Bid cleavage and calpain-independent Bak modulation: two separate pathways in cisplatin-induced apoptosis. Mol Cell Biol. 2002;22:3003–3013. doi: 10.1128/MCB.22.9.3003-3013.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagayama T, Sinor AD, Simon RP, Chen J, Graham SH, Jin K, Greenberg DA. Cannabinoids and neuroprotection in global and focal cerebral ischemia and in neuronal cultures. J Neurosci. 1999;19:2987–2995. doi: 10.1523/JNEUROSCI.19-08-02987.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumar RW, Meng FH, Mills AM, Xu YA, Zhang C, Welsh FA, Siman R. Calpain activity in the rat brain after transient forebrain ischemia. Exp Neurol. 2001;170:27–35. doi: 10.1006/exnr.2001.7708. [DOI] [PubMed] [Google Scholar]
- Neumar RW, Xu YA, Gada H, Guttmann RP, Siman R. Cross-talk between calpain and caspase proteolytic systems during neuronal apoptosis. J Biol Chem. 2003;278:14162–14167. doi: 10.1074/jbc.M212255200. [DOI] [PubMed] [Google Scholar]
- Polster BM, Basanez G, Etxebarria A, Hardwick JM, Nicholls DG. Calpain I induces cleavage and release of apoptosis-inducing factor from isolated mitochondria. J Biol Chem. 2005;280:6447–6454. doi: 10.1074/jbc.M413269200. [DOI] [PubMed] [Google Scholar]
- Roberts-Lewis JM, Savage MJ, Marcy VR, Pinsker LR, Siman R. Immunolocalization of calpain I-mediated spectrin degradation to vulnerable neurons in the ischemic gerbil brain. J Neurosci. 1994;14:3934–3944. doi: 10.1523/JNEUROSCI.14-06-03934.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saatman KE, Murai H, Bartus RT, Smith DH, Hayward NJ, Perri BR, McIntosh TK. Calpain inhibitor AK295 attenuates motor and cognitive deficits following experimental brain injury in the rat. Proc Natl Acad Sci USA. 1996;93:3428–3433. doi: 10.1073/pnas.93.8.3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanftner LM, Sommer JM, Suzuki BM, Smith PH, Vijay S, Vargas JA, Forsayeth JR, Cunningham J, Bankiewicz KS, Kao H, Bernal J, Pierce GF, Johnson KW. AAV2-mediated gene delivery to monkey putamen: evaluation of an infusion device and delivery parameters. Exp Neurol. 2005;194:476–483. doi: 10.1016/j.expneurol.2005.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz C. Towards more readily comprehensible procedures in disector stereology. J Neurocytol. 1997;26:707–710. doi: 10.1023/a:1018506113256. [DOI] [PubMed] [Google Scholar]
- Sugawara T, Fujimura M, Morita-Fujimura Y, Kawase M, Chan PH. Mitochondrial release of cytochrome c corresponds to the selective vulnerability of hippocampal CA1 neurons in rats after transient global cerebral ischemia. J Neurosci. 1999;19:RC39. doi: 10.1523/JNEUROSCI.19-22-j0002.1999. 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Saido TC, Hirai S. Modulation of cellular signals by calpain. Ann NY Acad Sci. 1992;674:218–227. doi: 10.1111/j.1749-6632.1992.tb27490.x. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Yokota H, Jover T, Cappuccio I, Calderone A, Simionescu M, Bennett MV, Zukin RS. Ischemic preconditioning: neuronal survival in the face of caspase-3 activation. J Neurosci. 2004;24:2750–2759. doi: 10.1523/JNEUROSCI.5475-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahsen N, Cande C, Briere JJ, Benit P, Joza N, Larochette N, Mastroberardino PG, Pequignot MO, Casares N, Lazar V, Feraud O, Debili N, Wissing S, Engelhardt S, Madeo F, Piacentini M, Penninger JM, Schagger H, Rustin P, Kroemer G. AIF deficiency compromises oxidative phosphorylation. EMBO J. 2004;23:4679–4689. doi: 10.1038/sj.emboj.7600461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Loo G, Saelens X, Matthijssens F, Schotte P, Beyaert R, Declercq W, Vandenabeele P. Caspases are not localized in mitochondria during life or death. Cell Death Differ. 2002;9:1207–1211. doi: 10.1038/sj.cdd.4401101. [DOI] [PubMed] [Google Scholar]
- White MJ, DiCaprio MJ, Greenberg DA. Assessment of neuronal viability with Alamar blue in cortical and granule cell cultures. J Neurosci Methods. 1996;70:195–200. doi: 10.1016/s0165-0270(96)00118-5. [DOI] [PubMed] [Google Scholar]
- Widlak P, Li LY, Wang X, Garrard WT. Action of recombinant human apoptotic endonuclease G on naked DNA and chromatin substrates: cooperation with exonuclease and DNase I. J Biol Chem. 2001;276:48404–48409. doi: 10.1074/jbc.M108461200. [DOI] [PubMed] [Google Scholar]
- Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakovlev AG, Ota K, Wang G, Movsesyan V, Bao WL, Yoshihara K, Faden AI. Differential expression of apoptotic protease-activating factor-1 and caspase-3 genes and susceptibility to apoptosis during brain development and after traumatic brain injury. J Neurosci. 2001;21:7439–7446. doi: 10.1523/JNEUROSCI.21-19-07439.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang KT, Chang WL, Yang PC, Chien CL, Lai MS, Su MJ, Wu ML. Activation of the transient receptor potential M2 channel and poly(ADP-ribose) polymerase is involved in oxidative stress-induced cardiomyocyte death. Cell Death Differ. 2006;13:1815–1826. doi: 10.1038/sj.cdd.4401813. [DOI] [PubMed] [Google Scholar]
- Ye H, Cande C, Stephanou NC, Jiang S, Gurbuxani S, Larochette N, Daugas E, Garrido C, Kroemer G, Wu H. DNA binding is required for the apoptogenic action of apoptosis inducing factor. Nat Struct Biol. 2002;9:680–684. doi: 10.1038/nsb836. [DOI] [PubMed] [Google Scholar]
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- Yu SW, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM, Dawson VL. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad Sci USA. 2006;103:18314–18319. doi: 10.1073/pnas.0606528103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Chen J, Graham SH, Du L, Kochanek PM, Draviam R, Guo F, Nathaniel PD, Szabo C, Watkins SC, Clark RS. Intranuclear localization of apoptosis-inducing factor (AIF) and large scale DNA fragmentation after traumatic brain injury in rats and in neuronal cultures exposed to peroxynitrite. J Neurochem. 2002;82:181–191. doi: 10.1046/j.1471-4159.2002.00975.x. [DOI] [PubMed] [Google Scholar]
- Zhu C, Qiu L, Wang X, Hallin U, Cande C, Kroemer G, Hagberg H, Blomgren K. Involvement of apoptosis-inducing factor in neuronal death after hypoxia-ischemia in the neonatal rat brain. J Neurochem. 2003;86:306–317. doi: 10.1046/j.1471-4159.2003.01832.x. [DOI] [PubMed] [Google Scholar]
- Zhu C, Wang X, Huang Z, Qiu L, Xu F, Vahsen N, Nilsson M, Eriksson PS, Hagberg H, Culmsee C, Plesnila N, Kroemer G, Blomgren K. Apoptosis-inducing factor is a major contributor to neuronal loss induced by neonatal cerebral hypoxia-ischemia. Cell Death Differ. 2007;14:775–784. doi: 10.1038/sj.cdd.4402053. [DOI] [PubMed] [Google Scholar]