

Abstract
Synapses need to encode a wide dynamic range of action potential frequencies. Essential vesicle priming proteins of the Munc13 (mammalian Unc13) family play an important role in adapting vesicle supply to variable demand and thus influence short-term plasticity characteristics and synaptic function. Structure–function analyses of Munc13s have identified a “catalytic” C-terminal domain and several N-terminal modulatory domains, including a diacylglycerol/phorbol ester [4β-phorbol-12, 13-dibutyrate (PDBu)] binding C1 domain. Although still allowing basal priming, a Munc13-1 C1 domain mutation (H567K) prevents PDBu induced potentiation of evoked transmitter release, leads to strong depression during trains of synaptic activity, and causes perinatal lethality in mice. To understand the mechanism of C1 domain-mediated modulation of Munc13 function, we examined how PDBu increases neurotransmitter release. Analyses of osmotically induced release as well as Ca2+ triggered and spontaneous release showed that PDBu increases the vesicular release rate without affecting the size of the readily releasable vesicle pool, linking C1 domain activation to a lowering of the energy barrier for vesicle fusion. PDBu binding-deficient mutant Munc13-1H567K synapses mirrored the vesicular release properties of PDBu-potentiated wild-type synapses, indicating that Munc13-1H567K is a gain-of-function mutant, which conformationally mimics the PDBu-activated state of Munc13-1. We propose a PKC analogous two-state model of regulation of Munc13s, in which the basal state of Munc13s is disinhibited by C1 domain activation into a state of facilitated vesicle release, regardless of whether the release is spontaneous or action potential triggered.
Keywords: exocytosis, presynaptic mechanisms, release probability, phorbol, patch clamp, neurotransmission
Introduction
Synaptic vesicle fusion with the presynaptic active zone membrane is the key step in neurotransmitter release. Before action potential-triggered fusion, vesicles need to dock to the active zone and become primed. Priming dramatically increases the propensity for vesicular release by lowering of the energy barrier for fusion. This enables the vesicles to efficiently use the action potential-induced Ca2+ transient for timed exocytosis. Only a fraction of synaptic vesicles is primed, and this fraction is defined as the readily releasable pool (RRP). Because of their unique functional state, primed vesicles can be readily quantified by application of hypertonic sucrose pulses. The hypertonic solution provides a Ca2+-independent mechanistic driving force that triggers within a few seconds the fusion of all RRP vesicles. The integral of the transient inward current produced by the concomitant release of glutamate is then a direct measure for the number of vesicles in the RRP in excitatory synapses (Rosenmund and Stevens, 1996).
Munc13s (mammalian Unc13) are presynaptic active zone proteins and essential priming factors. Munc13-deficient synapses fail to release vesicles during action potentials and sucrose stimuli, as well as spontaneously, whereas vesicle docking to the active zone is essentially normal (Aravamudan et al., 1999; Augustin et al., 1999; Richmond et al., 2001; Varoqueaux et al., 2002). Whereas the basic priming activity of Munc13-1 has been mapped to its C terminus (Basu et al., 2005; Madison et al., 2005; Stevens et al., 2005), the Munc13 N terminus has several modulatory domains, including a phorbol ester [4β-phorbol-12, 13-dibutyrate (PDBu)]/diacyglycerol (DAG) binding C1 domain. Although not essential for priming, proper C1 domain function is essential for survival in mice (Rhee et al., 2002). A single point mutation (H567K) in the Munc13-1 C1 domain abolishes phorbol ester binding, eliminates subsequent synaptic potentiation, and leads to dramatic changes in short-term plasticity characteristics. The RRP size is reduced in Munc13-1H567K mutant neurons, but evoked response amplitudes are unaltered, resulting in a relatively higher vesicular release probability (Pvr) compared with wild-type (WT) neurons (Rhee et al., 2002). Based on these results, we hypothesized that the RRP comprises high and low Pvr subpools, and PDBu preferentially targets the low Pvr vesicles to increase their release probability. Synapses with C1 domain mutant Munc13-1H567K would thus lack the low Pvr pool, be insensitive to PDBu, and simultaneously acquire a high Pvr phenotype. Apart from a mechanism that increases Pvr (Rhee et al., 2002; Rosenmund et al., 2002), it has been proposed that PDBu may also increase the RRP size in hippocampal cultured neurons (Stevens and Sullivan, 1998; Virmani et al., 2005).
To clarify the role of Munc13s in the PDBu-induced potentiation of release, we studied the effect of PDBu stimulation on Ca2+-dependent and -independent forms of release and used the Munc13-1H567K knock-in (KI) C1 domain mutant mice to link the observed behavior to Munc13-1 C1 domain function.
Materials and Methods
Cell culture.
Microdot neuronal cultures require previous preparation of astrocyte feeder wells. Clean 30 mm coverslips were placed in the culture plate wells and coated uniformly with 0.15% liquefied agarose gel (as an unfavorable cell-attachment substrate) using a sterile cotton swab. Once dry, a 1:4 mixture of rat-tail collagen and poly-d-lysine (PDL) was applied on top of the agarose using a custom-made dotted rubber stamp to form “microdots” on which cells readily attach. Four to 5 d before culturing the neurons, astrocytes were plated on the agarose–collagen–PDL-coated coverslips, so that they formed a confluent monolayer of “microislands” for the autapses to grow. Once confluent, the astrocytes were treated with antimitotic 8 μm 5-fluoro-2′-deoxyuridine and 20 μm uridine (FUDR) to halt the glial proliferation. The Munc13-1H567K KI mice die within 1–2 h after birth. Hence, newborn KI mice and their Munc13-1 WT and heterozygote (Het) littermates, both in the Munc13-2 knock-out background, were used for the primary neuronal cultures. Pups were decapitated according to the rules of the state animal welfare committee. Brains were removed and cleaned of meninges and vascular tissue, and hippocampi were dissected out in cold physiological HBSS. The hippocampal tissues of the desired genotypes were enzymatically dissociated with 2 U/ml papain in DMEM for 60 min at 37°C. After enzyme inactivation at 37°C for 5 min using a solution of albumin, trypsin inhibitor, and 5% FCS, the tissue was transferred to Neurobasal A medium containing B-27, 50 IU/ml penicillin, and 50 μg/ml streptomycin and gently triturated by passing through a narrow pipette tip. The cell suspension was plated onto FUDR-treated astrocyte feeder wells to a final cell density of 400–800 cells/cm2.
Solutions.
Patch-pipette solutions included the following: 135 mm KCl, 10 mm HEPES, 1 mm EGTA, 4.6 mm MgCl2, 4 mm Na-ATP, 15 mm creatine phosphate, and 50 U/ml phosphocreatine kinase, pH 7.3, 300 mOsm. The extracellular medium contained the following (in mm): 140 NaCl, 2.4 KCl, 10 HEPES, 10 glucose, 4 CaCl2, and 4 MgCl2, pH 7.3 (305 mOsm). Hypertonic solutions for determining the RRP were prepared by using 250, 500, 750, and 1000 mm sucrose in control external solution. Miniature EPSCs (mEPSCs) were measured in the standard external solution. Frequencies and amplitudes of false-positive events were regularly monitored by applying external solution containing 3 mm kynurenic acid for 2 s every 10 s.
Experimental setup.
Thick-walled borosilicate glass patch pipettes were pulled using a multistep puller (P-87; Sutter Instruments, Novato, CA), and pipette resistance ranged from 2 to 3 MΩ. All solutions were applied using a fast-flow system that provides reliable and precise solution exchanges with time constants of ∼20–30 ms (Pyott and Rosenmund, 2002). Currents were recorded using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA). Series resistance was 70–75% compensated. Data acquisition was controlled by Clampex 8 (Molecular Devices) on an IBM personal computer. Data were filtered using a low-pass eight-pole Bessel filter at a rate of 5 kHz.
Electrophysiological controls.
Electrophysiological recordings were performed at room temperature. The entire present dataset was acquired from three separate cultures, and the recorded values were pooled. Care was taken to record each day from a relatively equal number of cells per group for the three genotypes in question to prevent any ambiguities attributable to the developmental stage between the groups. The phorbol ester experiments were always paired, with the same protocols being recorded from each individual cell once in the absence of PDBu (1 μm) and once in its presence. This ensured lower variability in the sample set. For the sucrose gradient experiments, the order of the stimulation sequence (500, 250, and 1000 mm) and the application of PDBu were alternated, so that half of the cells were recorded in PDBu first, followed by washout and recording in control conditions and vice versa, to control for rundown. For control experiments to get a reliable estimate of nonspecific conductance during sucrose application, we used kynurenic acid (3 mm) or NBQX (3 μm) with the sucrose solution. To test how PDBu changes the speed of onset of responses to hypertonic solutions, we performed control experiments with added potassium chloride (30 mm). The resulting development of outward current was used to establish the time course of solution exchange for every flow pipe used and for all concentrations of hypertonic sucrose solutions.
Data analysis.
Data were analyzed using AXOGRAPH software [version 4.9 (Molecular Devices) or AxographX (courtesy of Dr. John Clements, Axograph Scientific, Sydney, Australia)]. The sucrose response was normalized by its own area after subtracting the steady-state component, and the integral after “baselining” at onset was used to calculate the maximum slope of the sucrose-induced release. mEPSCs were detected by a template based event detection algorithm package. The release rate of EPSCs was estimated using a deconvolution algorithm, AxographX (Axograph Scientific). Graphs were plotted using Kaleidagraph software (Synergy Software, Reading, PA). Paired Student's t test was applied to determine statistical significance of p < 0.05.
Results
To study the role of C1 domain activation in neurotransmitter release, we directly compared three mouse genotypes belonging to the same litter: Munc13-1 WT and mice expressing one (Het) or two (KI) alleles of the Munc13-1H567K mutation. All mice were Munc13-2-deficient to prevent any contribution of the Munc13-2 paralog. Most experiments were performed on glutamatergic autaptic hippocampal neurons 9–16 d in vitro. We also performed a separate set of experiments using wild-type neurons containing both Munc13-1 and Munc13-2 to compare excitatory and inhibitory synaptic transmission. To activate the C1 domain of Munc13-1, we used acute application of the agonist PDBu (1 μm) and compared synaptic parameters before and during PDBu stimulation.
Phorbol ester increases the rate of hypertonicity-induced vesicular release without increasing the RRP size
Munc13s play an essential role in synaptic vesicle priming, and the C1 domain of Munc13-1 is required for acute potentiation of EPSCs by phorbol esters (Rhee et al., 2002). These findings support the idea that phorbol esters potentiate release by increasing the RRP (Stevens and Sullivan, 1998). Application of hypertonic solutions has been used to detect the RRP (Rosenmund and Stevens, 1996). Acute application of sucrose using a fast-flow system leads to the activation of two kinetically distinct components: a transient inward current signifying the release of all fusion competent vesicles, followed by a steady-state inward current component at the stage when the rate of pool refilling and the rate of release are at equilibrium. The area of the inward transient after subtracting the steady-state component represents the total amount of charge of the RRP. Normalization of this charge by the mean charge of the mEPSC renders the total number of vesicles in the RRP.
Our study shows that the total number of fusion-competent vesicles, as defined by the 500 mm sucrose response, in the Munc13-1H567K knock-in mutant neurons is significantly reduced to 2299 ± 300 vesicles (n = 48), amounting to a 44% drop in RRP size compared with the WT average of 4109 ± 772 vesicles (n = 37) (p < 0.05, WT vs KI). This is in agreement with our previous study that used the same Munc13-1H567K mutation in the Munc13-2 WT background (Rhee et al., 2002). The heterozygotes show influences of both the native and mutant Munc13-1 proteins and can release on average 2763 ± 305 vesicles (n = 43) (p < 0.1, Het vs KI) (Fig. 1B,C). After a 60 s recovery from the sucrose-mediated pool depletion, we treated a subset of these neurons with PDBu (1 μm) for 2 min and then applied the same concentration of sucrose to study the effect of phorbol esters on the RRP. We found that PDBu treatment does not change the size of the RRP in any of the groups under consideration (4092 ± 873, n = 28, WT; 2814 ± 384, n = 29, Het; 2467 ± 399, n = 31, KI; all p values >0.5, WT vs WT + PDBu, Het vs Het + PDBu, KI vs KI + PDBu).
Figure 1.
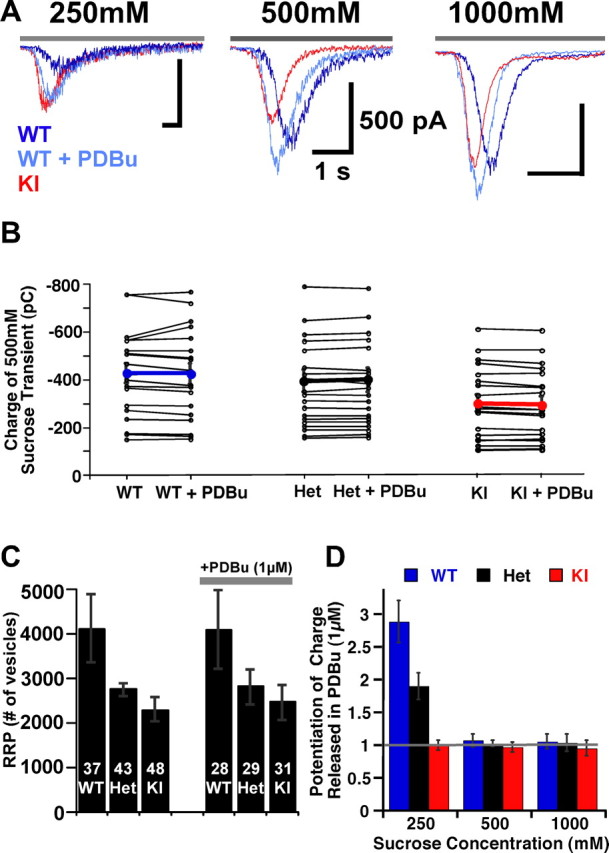
PDBu-mediated C1 domain activation does not affect the pool size. A, Exemplary raw traces of responses to 250 mm (left), 500 mm (middle), and 1000 mm (right) sucrose pulses in naive and PDBu (1 μm)-treated hippocampal autaptic neurons from Munc13-1 WT (dark blue), WT + PDBu (light blue), and Munc13-1H567K KI (red) mice. Gray bar above each trace indicates the duration of sucrose application. B, Distribution of charge of the inward transient response to a 5 s application of 500 mm sucrose, in the presence and absence of PDBu in 19 individual neurons from WT (left), Het (middle), and KI (right) groups. Thick lines indicate the mean values for WT in blue, Het in black, and KI in red. C, Bar plot showing the mean number of vesicles in the RRP calculated by dividing the charge of the sucrose (500 mm) inward transient by the corresponding mEPSC charge. The number of cells is indicated in white above each corresponding group. Error bars in this and the following figures indicate the SEM. D, Potentiation of response charge during PDBu application at 250, 500, and 1000 mm sucrose stimuli.
Application of doubled concentrations of sucrose (1000 mm, for 3 s) does not increase the charge released any further above the level achieved by 500 mm sucrose (4235 ± 669, WT; 4263 ± 725, WT + PDBu, n = 22; 3252 ± 398, Het; 3108 ± 484; Het + PDBu, n = 23; 3095 ± 490, KI; 2799 ± 551, KI + PDBu; n = 19), regardless of whether PDBu is present or not (Fig. 1D). This indicates that 500 mm sucrose is sufficiently strong a stimulus to cause the entire RRP to be released, and PDBu does not potentiate the pool size. Although stimuli of 500 mm or higher sucrose do not change the number of vesicles released, we can use hypertonic solutions of intermediate concentrations to test for changes in sensitivity of vesicles to the stimulus. We applied 250 mm sucrose (10 s) and quantified the transient component of the response, as a measure for the fraction of the pool released. PDBu increases the amount of release induced by 250 mm sucrose by almost threefold in the case of WT (2.88 ± 0.32 times naive WT; n = 22) and twofold in heterozygote (1.89 ± 0.20 times naive Het; n = 23) synapses (Fig. 1D). The same submaximal stimulus applied in the presence of PDBu to the KI mutant neurons caused no change in the charge of the 250 mm sucrose-induced inward transient (0.99 ± 0.08 times naive KI; n = 19). However, to our surprise, the fraction of the pool released by naive KI neurons is equivalent to that of PDBu-treated WT synapses (72–81% of their RRP) (Fig. 2D). We note that, although the RRP of KI mutants is reduced to 2299 vesicles (p < 0.05), the number of vesicles released by 250 mm in the KI group (2000 ± 460 vesicles) exceeded that of the naive WT synapses (1100 ± 300 vesicles).
Figure 2.
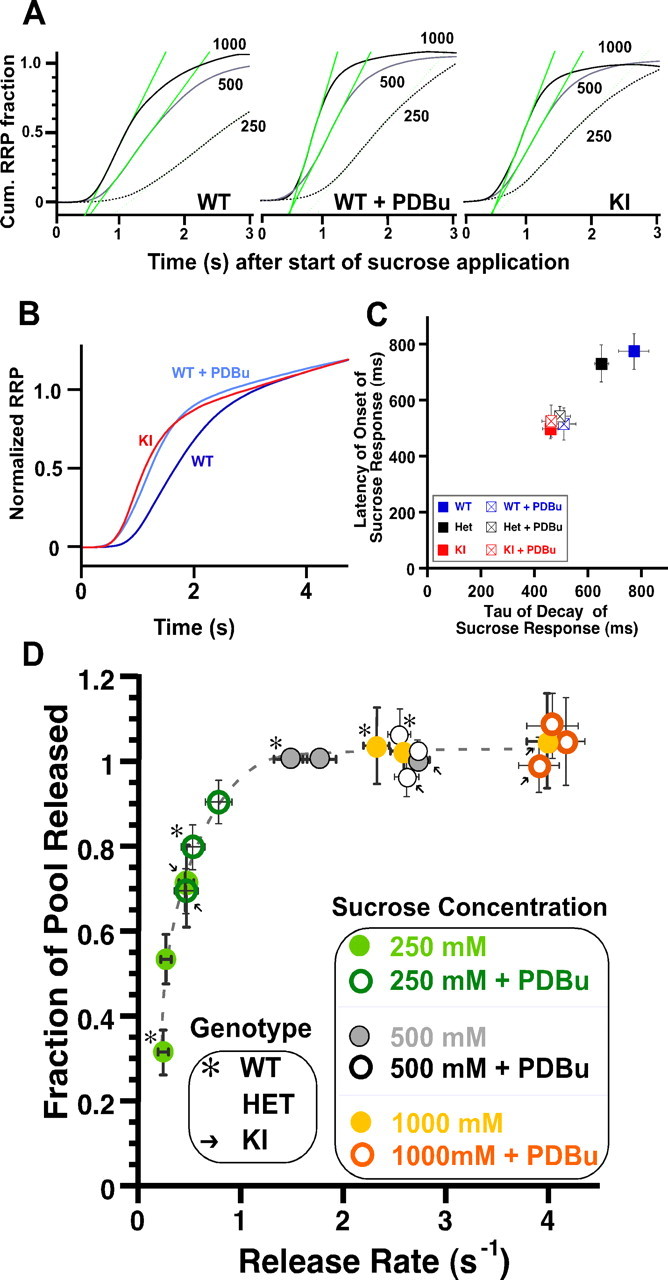
C1 domain activation and H567K mutation increase osmotically induced vesicular release rates. A, Inverted mean current integrals for the three gradients of sucrose (1000 mm in black solid, 500 mm in gray solid, and 250 mm in black dotted lines) in WT (left), WT + PDBu (middle), and KI (right) normalized by their respective RRPs. The green lines depict the maximum slopes signifying the rate of release (s−1) induced by that concentration of sucrose. B, Average normalized current integrals showing the release time course of the RRP during 500 mm sucrose applications in WT (dark blue), WT + PDBu (light blue), and KI (red). C, Correlation between time constant of decay of the sucrose (500 mm) response and latency of onset of the response from the start of sucrose application. D, Summary plot of the fraction of RRP released as a function of the maximal vesicular release rates. Data are from the three genotypes (WT marked with *, n = 22; Het unmarked, n = 23; and KI marked with →, n = 19) with (open symbols) or without (filled symbols) 1 μm PDBu present and at the three sucrose concentrations 250 mm (green), 500 mm (gray), and 1000 mm (orange). All values are normalized to the RRP size (as defined by the response to 500 mm sucrose in the absence of PDBu) to determine the fraction of the pool released. Note that the data for C1 domain activation appears to mimic a virtual increase in hypertonic pressure, indicating an increased responsiveness to the stimuli.
These results show that activation of the Munc13-1 C1 domain increases the number of vesicles released only when the hypertonic stimulus is submaximal and does not cause any additional potentiation of responses during saturating osmotic challenges. This indicates that C1 domain activation does not affect the RRP per se but lowers the energy threshold for fusion of vesicles within the pool. The standard operational definition of RRP is the number of vesicles that represent the transient release activity during short application of highly hypertonic solutions (e.g., 500 mm sucrose) (Rosenmund and Stevens, 1996). At such high concentrations, PDBu will not increase the charge released, but it will at submaximal concentrations. Therefore, neither increased responses to submaximal sucrose concentrations nor increases in the peak amplitude of sucrose responses are evidence for a PDBu-induced increase in RRP. Accurate detection of RRP with hypertonic solutions may be impeded when solution exchange methods are used that cause a gradient in sucrose concentration over space or time during application. This may interfere with distinguishing between changes in RRP size and fusion efficacy and may explain different results from different studies in the same experimental system (Stevens and Sullivan, 1998; Virmani et al., 2005).
These results also provide insights into the state of primed vesicles in the C1 domain mutant synapses: the H567K is a gain-of-function mutation that causes the mutant synapses to mirror the PDBu-stimulated state of WT synapses in their sensitivity to submaximal osmotic stimuli. Because PDBu does not affect RRP size, we can conclude that acute activation of the Munc13 C1 domain is independent of the priming function of Munc13. Hence, mutations that lead to disruption of C1 domain PDBu binding should not affect RRP size. However, the absolute RRP size in the KI neurons is reduced by 44%, which is puzzling. Initially, we explained this via a two-pool model, in which the mutant suffers a selective elimination of a low Pvr, PDBu-sensitive subpool of vesicles. Given our finding that the mutation is a gain-of-function mutation and that, at intermediate sucrose concentrations, it is able to release more vesicles than wild-type synapses challenges the validity of our initial two-pool model in the context of PDBu modulation of vesicle release. We therefore consider alternative possibilities to explain the reduced pool size in the mutant neurons. The loss of some primed vesicles may be related to interference with the neighboring Munc13 N-terminal RIM (Rab3-interacting molecule)-binding C2A domain (Brose et al., 1995; Lu et al., 2006), which is more directly involved in the early steps of priming. Munc13 N-terminus deletion (Betz et al., 2001) and mutations in Munc13-binding regions of RIM (Dulubova et al., 2005) and RIM-interacting regions in Munc13 C2A domain (Andrews-Zwilling et al., 2006) (J. Basu, C. Rosenmund, J. Lu, and J. Rizo, unpublished observations) causes up to 50% reduction in pool size. Alternatively, the constitutively increased release activity in the KI mutant may have lead to a homeostatic downregulation of RRP size.
C1 domain activation of Munc13-1 leads to accelerated vesicle release kinetics
If the Munc13-1 C1 domain activation increases release as a result of a lowered energy barrier for fusion, we would expect the kinetics of the sucrose response to be accelerated and the onset time shortened. In support of this, as is apparent for the raw traces in Figure 1A, the peak of the sucrose responses in PDBu are larger and shifted to the left compared with the control conditions.
To test the lowered energy barrier model, we set out to quantify the time course of release of responses to hypertonic stimuli. To quantify the release rate of individual vesicles, we normalized the individual responses in 250, 500, and 1000 mm sucrose of each cell to its corresponding RRP (the value of the response to 500 mm). Then we integrated the normalized response and quantified the maximal slope as a measure for peak release rate. We corrected this slope for the amount of vesicles that had already been depleted (Fig. 2A). In the WT naive neurons, 250 mm sucrose releases individual vesicles at a peak rate of 0.21 ± 0.02 s−1, whereas this rate increased approximately sevenfold in 500 mm sucrose (1.51 ± 0.06 s−1; n = 22) and 11-fold (2.34 ± 0.07 s−1) with 1000 mm sucrose (Fig. 2A,D). Thus, as expected, the release rate increases with the amount of osmotic energy supplied in a dose-dependent manner. Comparison with the subsequent sucrose response in the presence of PDBu showed that the release rate doubled in WT neurons when 250 mm sucrose was applied, whereas the KI group showed rates similar to the WT + PDBu group and did not change during application of PDBu (0.47 ± 0.03 s−1 in KI; 0.53 ± 0.06 s−1 in WT + PDBu; 0.47 ± 0.06 in KI + PDBu). Even at the two higher concentrations of sucrose, the release rate remained 30–40% higher in the C1 activated WT and in the Munc13-1H567K KI mutant group compared with the naive WT group (500 mm sucrose: 2.55 ± 0.10 s−1, WT + PDBu, n = 22; 2.76 ± 0.09 s−1, KI, n = 19; and 1000 mm: 4.04 ± 0.14 s−1, WT + PDBu; 3.99 ± 0.12 s−1, KI). This shows that activation of the Munc13-1 C1 domain accelerates vesicular release in general, consistent with the model of a lowered energy barrier for fusion. In addition, as seen previously, the Munc13-1H567K C1 mutant mimics PDBu-mediated C1 domain activation (Fig. 2B,D).
A lowered energy barrier should also decrease the onset time of the sucrose response. We quantified the delay between start of application of 500 mm sucrose and the onset of the response by extrapolating from the rising phase of the sucrose response the point of intercept with the zero baseline. We analyzed the acceleration of the response onset because of C1 domain activation by two means. First, we averaged the actual time taken by each response in the various groups and then calculated the difference between their mean values of onset. This provided an ∼250–300 ms acceleration of RRP release onset in the naive KI groups compared with naive WT (Fig. 2C). Second, we used the intrinsic relative shift in onset of the response seen in the WT group before and after PDBu was applied. This method does not allow for proper cross comparison between the three genotypes but provides superior signal-to-noise ratios within a group, because it is not sensitive to cell-to-cell variability. We found that activation of the C1 domain accelerated onset of release at 500 mm sucrose by 207 ± 37 ms, whereas as expected, no acceleration was observed in the KI group.
The third form of kinetic characterization focused on decay kinetics of the sucrose response. An acceleration of release in the C1 domain-activated state should result in accelerated decay kinetics attributable to faster pool depletion. We fitted a single exponential to the decay and found that the decay kinetics were accelerated in the WT + PDBu and in the KI group (WT, 772 ± 56 ms; WT + PDBu, 508 ± 47 ms; KI, 458 ± 29 ms; KI + PDBu, 462 ± 36 ms) (Fig. 2C). In addition, both onset of release and the time constant of decay correlated well among the different groups.
Together, the detailed analysis of the kinetics of the hypertonic sucrose-induced release after PDBu treatment or in KI neurons revealed a secretion behavior that reflects a lowered energy barrier for fusion. In Figure 2D, we summarize the data (from Figs. 1D, 2A) by correlating release rates with the fraction of pool released under conditions of variable sucrose concentrations, variable genotypes, and presence or absence of PDBu. The trend of the data indicates that, across the above conditions, increasing osmolarity and activating the C1 domain result in similar changes, an accelerated fusion rate in vesicles derived from the RRP. We generally measure the RRP as a charge integral of the transient inward current in response to sucrose from which the steady-state component is subtracted. At steady state, release and refilling are at equilibrium. The transient represents the RRP, as long as the release rate is much faster than the refilling rate, and this can be seen with the fraction of RRP reaching 1 at concentrations of 500 and 1000 mm sucrose. At concentrations of sucrose at which the release rate is close to the refilling rate (e.g., at 250 mm), the transient current component is a small fraction of RRP and displays high sensitivity to changes in release rate. At such a sensitive range, changes in energy barrier for fusion would be reflected in potentiation of vesicular fusion rates coupled with an increase in the fraction of the pool released. So, with both of these parameters, maximal release rate and fraction of pool released, we probe the same change in fusion willingness during C1 domain activation. As depicted in green for 250 mm sucrose in Figure 2D, activation of the C1 domain by PDBu or H567K mutation lowers the overall energy barrier for fusion of vesicles, so many more vesicles overcome the detection limiting refilling rate and hence with the same submaximal stimulus a far greater fraction of vesicles from the pool (70–90%) can be measured, releasing at an increased rate (∼0.5–1 s−1). Conversely, for saturating concentrations of sucrose (500 and 1000 mm), all vesicles release at a rate (∼1.5–2 s−1) that far exceeds the refilling rate, and the entire RRP is detected in the sucrose transient. So any shift in the energy barrier for fusion would be detected in terms of increases in the peak release rate of vesicles (∼2.5–4 s−1) and not the number of vesicles as represented by the plateau phase of the plot in Figure 2D.
Separate experiments that looked at paired-pulse sucrose response showed significantly faster recovery in PDBu-treated WT neurons compared with untreated neurons (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). This finding is in agreement with the finding of Stevens and Sullivan (1998) and could reflect a secondary function of C1 domain activation on the rate at which the RRP is replenished. Conversely, the faster recovery of the hypertonic response could also be the result of the increased sensitivity of the vesicle to be detected by the same stimulus. Although there is no formal evidence, a lowered energy barrier for fusion attributable to C1 domain activation would increase the chances of vesicles that are partially primed to fuse in response to the same stimulus. Furthermore, considering that vesicles from the KI mutant have a lower energy barrier for fusion, one would expect faster recovery compared with wild type. However, in our previous paper (Rhee et al., 2002), the H567K mutant pool recovered at the same rate as the wild-type RRP. In light of the observation that the KI mutant is defective in priming, there is a possibility that, although vesicular release rate is faster than the wild type, the recovery of the pool after depletion is offset to the wild-type level.
Finally, the fact that hypertonic solution releases vesicles in a Ca2+-independent manner implies that above modification in fusion efficacy is independent of Ca2+ triggering. To confirm this notion, we analyzed spontaneous release, another form of Ca2+ independent release.
C1 domain activation increases the frequency and rate of spontaneous release
We measured spontaneous mEPSCs in our three genotypes in the presence and absence of PDBu (1 μm). Data were acquired for a period of 60 s (six episodes, 10 s each) for each condition and genotype. Each 10 s period included a 2 s application of saturating concentrations (3 mm) of the AMPA receptor antagonist kynurenic acid to block all postsynaptic responses (Fig. 3A, top trace). This served as an internal control for detection of “false-positive” spontaneous events (Wojcik et al., 2004).
Figure 3.
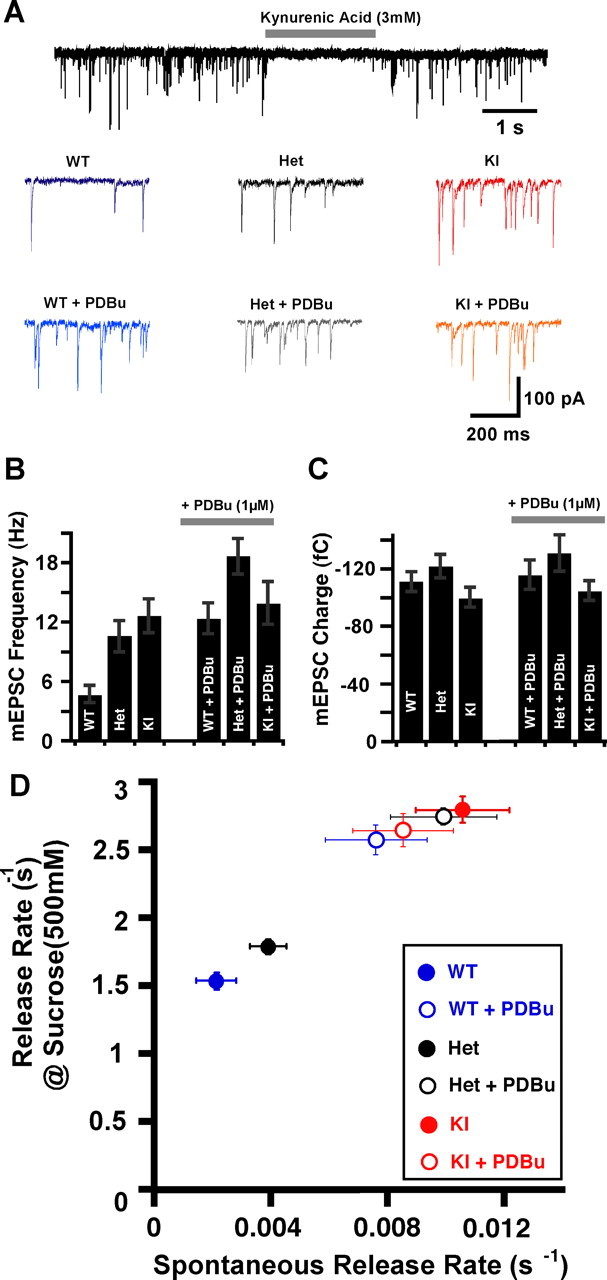
Increase in the frequency and rate of release of spontaneous events during C1 activation. A, Top, Trace showing the 10 s mEPSC acquisition protocol with a 2 s period of kynurenic acid (3 mm) for background noise detection. Bottom, A 500 ms span of raw traces showing the spontaneous activity in the three genotypes WT (blue), Het (black), and KI (red) and in the presence of PDBu (light blue, gray, and orange, respectively). B, Bar plot of absolute mEPSC frequencies in KI, Het, and KI without (left) and with (right) PDBu present. C, mEPSC charge in the same groups and conditions as in B. Charge released during a single spontaneous event (n = 40, WT; n = 55, Het; n = 49, KI; n = 28, WT + PDBu; n = 29, Het + PDBu; and n = 31, KI + PDBu). D, Strong correlation between spontaneous release rate and 500 mm sucrose maximal induced release rate in the three groups and two conditions.
We found that, compared with the naive WT group, the absolute frequency of mEPSCs is more than doubled in the phorbol-“resistant” mutant neurons from Munc13-1H567K KI mice and in the phorbol ester-treated WT neurons (Fig. 3B). We did not observe any difference in the charge, amplitude, and shape (half-width) of mEPSC events among the three different genotypes, regardless of whether they were treated with PDBu or not (Fig. 3A,C).
We then calculated the spontaneous release rate by dividing mEPSC frequency by the number of vesicles in the RRP. We found that vesicles from naive WT neurons have the lowest spontaneous release rate of 0.0022 ± 0.0007 s−1 (n = 37), corresponding to a mean dwell time of 454 s before individual vesicles fuse. The spontaneous release rate can be potentiated up to four times (0.0076 ± 0.0017 s−1; n = 28), with 1 μm PDBu. The KI mutant already exhibits similarly elevated release rates (0.0105 ± 0.0016 s−1; n = 48), and treating them with PDBu is ineffective (0.0085 ± 0.0017 s−1; n = 31, KI + PDBu). The heterozygotes have an intermediate spontaneous release rate of 0.0039 ± 0.0016 s−1 (n = 43), which is increased to 0.0099 ± 0.0018 s−1 (n = 29) during C1 domain activation by PDBu (Fig. 3D).
The increased basal release activity during C1 domain activation, just as in the case of responses to hypertonic stimuli, finds its simplest explanation in a lowered energy barrier for vesicle fusion. Consistent with this is the observation that both forms of release, although differing in their absolute rates by three orders of magnitude, showed very strong correlation in behavior when we considered the activation state of the Munc13-1 C1 domain (Fig. 3D). We next wanted to test whether the observed increases in Ca2+-triggered release during phorbol ester treatment (Fig. 4) may thus be the result of the same phenomenon underlying the shift in Ca2+-independent release.
Figure 4.
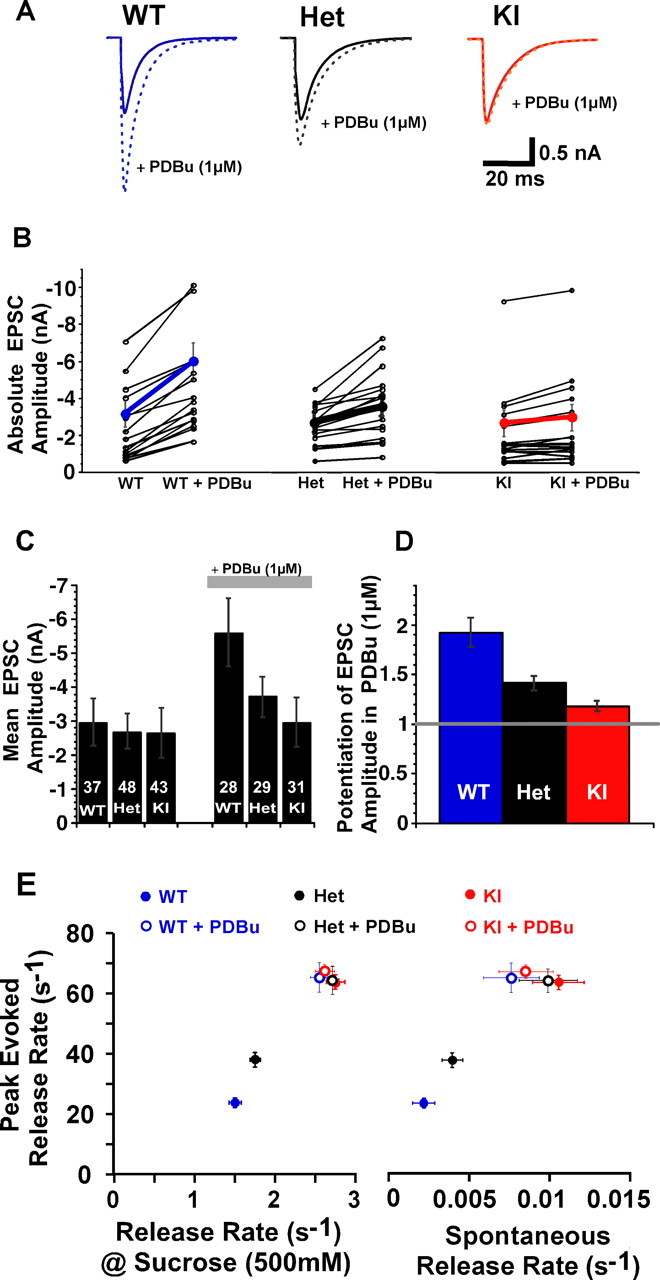
C1 domain activation increases the rates of all forms of release. A, Exemplary EPSC traces from WT (blue), Het (black), and KI (red) groups and the effect of PDBu (dotted lines). B, Scatter plot of 19 individual neurons. C, D, Bar plots of PDBu-induced potentiation of EPSCs. The absolute amplitudes in naive neurons from the three groups are identical (C, left). E, Correlation of vesicular release rates during all the three forms of release: Ca2+ evoked (y-axis), 500 mm sucrose induced (x-axis, left), and spontaneous (x-axis, right). The peak evoked release rate was calculated by deconvolution of individual EPSC responses by their corresponding mEPSCs after normalizing the EPSC by the RRP of that cell. The osmotically induced release rate was derived from the mean of the maximum slope of 500 mm sucrose response integrals normalized by the corresponding RRP and corrected for by taking into account the fraction of the pool already depleted at the peak of the response. Spontaneous release rate was obtained from the average ratio of the frequency of mEPSCs of each cell to its RRP.
C1 domain activation increases the rate of Ca2+-triggered release in correlation with Ca2+-independent release rates
We evoked EPSCs with 2-ms-long, 70 mV depolarizations at a frequency of 0.2 Hz. We found, in agreement with our previous findings in Munc13-1H567K mutant neurons on the Munc13-2 WT background (Rhee et al., 2002), that all three experimental groups have similar EPSC amplitudes (2.95 ± 0.69 nA, n = 37, WT; 2.68 ± 0.52 nA, n = 43, Het; and 2.64 ± 0.71 nA, n = 48, KI) (Fig. 1B). The H567K C1 domain mutation does not change the EPSC charge (24.7 ± 6.9 pC, n = 37, WT; 25.1 ± 5.0 pC, n = 43, Het; and 25.2 ± 6.8 pC, n = 48, KI), indicating that both synchronous and asynchronous releases are essentially unaltered in these mutant mice. We applied PDBu in control external solution for 1 min to the neurons and monitored the evoked responses over time using the same 0.2 Hz stimulus paradigm. The effect of phorbol esters was obvious in the first response after PDBu application, and the responses reached a maximum with the second or third response. We quantified the change of EPSC amplitude and charge attributable to phorbol esters and found that, in WT, responses increased almost twofold in the case of amplitude (1.92 ± 0.15; n = 28) (Fig. 1A,C) and more than twofold in the case of charge (2.48 ± 0.22; n = 28). In contrast, the Munc13-1H567K mutants seemed to be unaffected by phorbol esters (amplitude, 2.95 ± 0.71 nA; charge, 29.77 ± 6.54 pC; n = 31, KI + PDBu). The neurons derived from heterozygous mice, expressing both Munc13-1 WT and the H567K C1 domain mutant proteins, show intermediate responses to phorbol ester stimulation (1.42 ± 0.06, normalized amplitude; 1.56 ± 0.08, normalized charge; n = 29, Het + PDBu). The intermediate phenotype of the heterozygous group shows that both the WT and mutant proteins act independently and with overall similar efficacy.
To determine the rate at which vesicles are released at the peak of an evoked response, we normalized the peak amplitude of the EPSC by the corresponding charge of the RRP and deconvolved the response with the mEPSC shape (Rhee et al., 2005). Munc13-1 WT containing neurons that release individual vesicles occur at a peak rate of 23.5 ± 1.3 s−1 (n = 37), whereas heterozygous neurons have a peak release rate of 37.8 ± 2.1 s−1 (n = 43) and the KI mutants release at almost three times the WT rate, at 63.5 ± 2.1 s−1 (n = 48). Phorbol esters potentiate the WT and heterozygote EPSC release rates to a level similar to that seen in the Munc13-1H567K C1 domain mutant (65.4 ± 4.9 s−1, n = 28, WT + PDBu; 64.4 ± 3.9 s−1, n = 29, Het + PDBu). The vesicular release rate of the PDBu-treated KI group was unchanged compared with its non-PDBu control state (67.4 ± 2.2; n = 31, KI + PDBu; p > 0.1). Although all three forms of release (spontaneous, hypertonic, and Ca2+ triggered) vary more than four orders of magnitude in absolute release rates, changes in release as a function of genotype or as a function of activity of the Munc13-1 C1 domain were highly correlated (Fig. 4E). This strong correlation between spontaneous, osmotically induced, and evoked release processes coupled with the fact that Munc13 deletion results in loss of all forms of neurotransmitter release (Varaqueaux et al., 2002), suggest that they all undergo similar forms of modulation. This strongly speaks for a common constitutive mechanism in which vesicles for spontaneous and activity-dependant release are derived from essentially one pool and argues against the idea proposed by Virmani et al. (2005) that phorbol esters selectively regulate a so-called “activity-dependent recycling pool” and spare spontaneous vesicle recycling.
Enhancement of vesicular release probability with C1 domain activation
Above data taken together imply that Munc13-1 C1 domain activation induces an increase of EPSC amplitude without changing RRP size, hence increasing Pvr. The Pvr value can be computed by dividing the charge of the EPSC by the charge of the RRP, and, as expected, the overall Pvr is increased during Munc13-1 C1 activation (Fig. 5A). Whereas in WT Munc13-1 neurons, vesicles initially release with a probability of 11.4 ± 1.2%, phorbol ester-induced activation of the Munc13-1 C1 domain increases Pvr to 17.7 ± 2.5%. Again consistent with the previous measurements, the C1 domain mutant mimics the activated state by showing a high Pvr of 18.6 ± 1.6%. Pvr in these synapses was insensitive to any additional modification by PDBu (18.0 ± 1.5%), and furthermore the heterozygous group fell between the WT and KI groups (Het naive, 14.4 ± 1.2%; Het + PDBu, 18.2 ± 2.4%).
Figure 5.
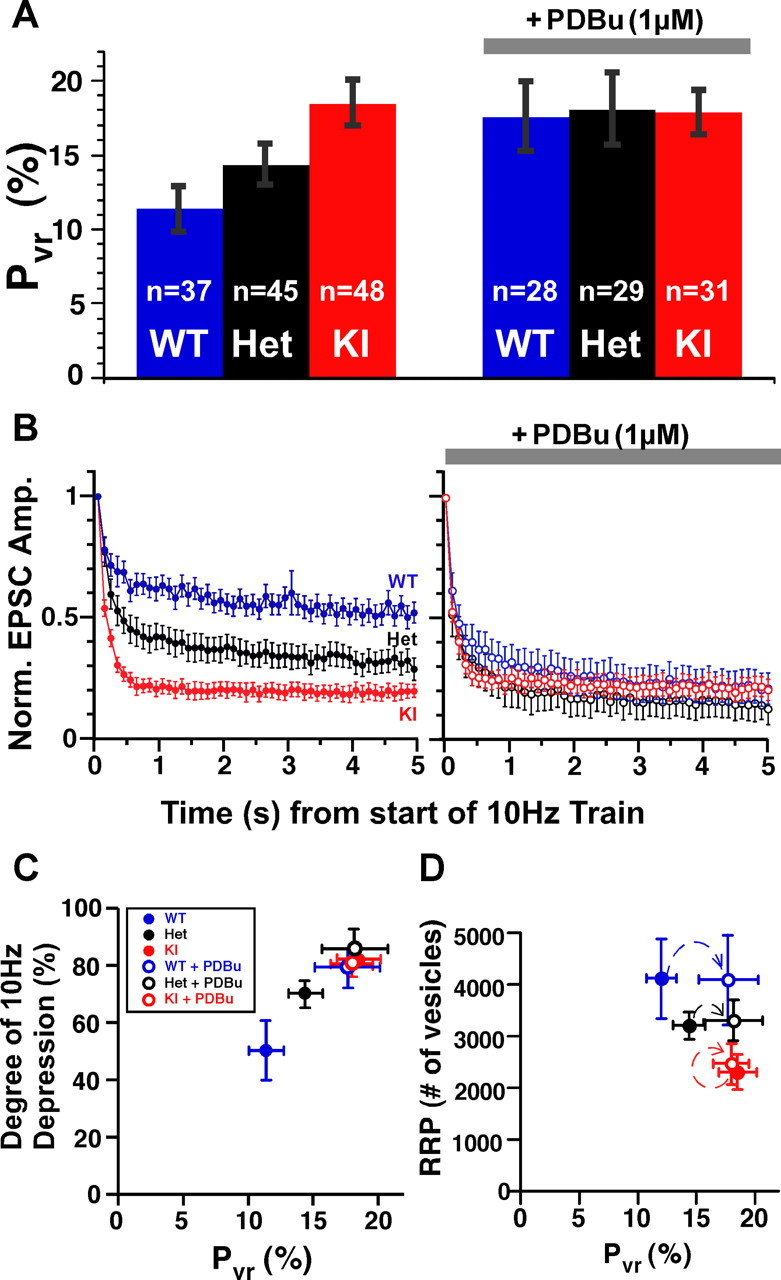
Munc13-1 C1 domain increases vesicular release probability. A, Bar plot quantifying the vesicular release probability (Pvr, percentage) showing that the activation of the C1 domain and H567K mutation lead to an increased Pvr. The KI mutant does not show any additional increase in Pvr during PDBu treatment. B, Normalized synaptic responses from wild-type (blue), heterozygote (black), and KI mutant (red) neurons during 10 Hz spike train (5 s) in the absence (left, filled circles) and presence (right, open circles) of PDBu (1 μm). C, Plot of the degree of depression during the 10 Hz spike train as a function of Pvr for each group examined in the presence and absence of PDBu. D, Plot of RRP size versus Pvr before (filled symbols) and after (open symbols) PDBu application. D, Same groups as in C.
Short-term plasticity characteristics are strongly affected by modulation of Pvr. For example, pure wild-type neurons that show 13–32% facilitation of EPSC amplitudes when stimulated with paired pulses at intervals of 20–40 ms depress in the presence of PDBu by ∼35–40% (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The Munc13-1H567K/Munc13-2 WT expressing neurons showed levels of paired-pulse depression similar to the Munc13-1/Munc13-2 WT neurons treated with PDBu (Rhee et al., 2002). Furthermore, the time course of the change in EPSC amplitude during 10 Hz action potential trains (50 stimuli) followed similar patterns. Moderate steady-state depression in the naive wild-type neurons was converted to strongly depressing time courses in the presence of PDBu or in the Munc13-1H567K KI mutant neurons. We quantified the degree of depression by comparing the average amplitudes of the last 10 responses at the end of the train with the average steady-state amplitude before the train. The EPSC amplitudes in WT neurons undergo 50.1 ± 9.7% (n = 22) depression, whereas Het animals show a 69.9 ± 4.3% (n = 29) decline and the KI mutants depress by 81.9 ± 2.7% (n = 39) during the 10 Hz spike train (Fig. 5B). PDBu increases the level of depression quite severely in Munc13-1 WT and heterozygote responses to 79.5 ± 6.4 and 85.9 ± 5.9% of their starting amplitudes, respectively (Fig. 5C). The already elevated level of spike train depression in the H567K mutant does not change in the presence of PDBu (80.6 ± 3.8%, KI + PDBu). The 10 Hz depression pattern of phorbol ester-treated WT and Het cells is almost duplicated in the KI mutant group, indicating that the activity-dependent short-term plasticity characteristics of the Munc13-1 C1 domain activated and mutated states are practically equivalent. We then correlated Pvr with the degree of spike train depression to illustrate how initial release probability determines synaptic depression (Fig. 5C). Increases in Pvr attributable to C1 domain activation have, however, no impact on the RRP (Fig. 5D).
Regulation of release rate of GABAergic vesicles
To study whether the potentiation of vesicular release via the activation of the C1 domain of Munc13-1 is a general phenomenon, we performed a comparative study of the effects of PDBu on pure wild-type GABAergic and glutamatergic hippocampal neurons. We first noted that potentiation of PSCs were very similar in the GABAergic inhibitory cells and glutamatergic excitatory neurons (1.86 ± 0.09, n = 129, EPSC, glutamatergic; 1.79 ± 0.13, n = 51, IPSC, GABAergic) (Fig. 6A,D). Similar to excitatory neurons, the RRP size of inhibitory neurons were also not potentiated by PDBu treatment (Fig. 6D), but their release kinetics were accelerated (Fig. 6B,C). However, the calculated Pvr for naive inhibitory neurons was twice as high compared with their glutamatergic counterparts (18.5 ± 1.2%, naive GABAergic; 7.8 ± 0.7%, naive glutamatergic), and the kinetics of sucrose-mediated release was 37% faster in the GABAergic cells (2.01 ± 0.12 s−1) compared with the glutamatergic neurons (1.46 ± 0.10 s−1) (Fig. 6E). PDBu causes Pvr and osmotically induced release rate in both types of neurons to potentiate in a similar manner by 1.7- to 2.1-fold. These results have important implications. First, hippocampal inhibitory neurons appear to undergo similar changes in release during stimulation of the Munc13 C1 domain as glutamatergic neurons. Second, the intrinsically increased basal fusion rate and release probability seen in inhibitory cells is independent of Ca2+ influx, because the effect on evoked responses is paralleled by an increase in release rate for the sucrose response. This in turn implies that inhibitory vesicles have an intrinsically lower threshold of vesicular fusion, independent of Munc13 and the Ca2+-triggering step, but their regulation via C1 domain activation of Munc13s is similar to that of excitatory neurons.
Figure 6.
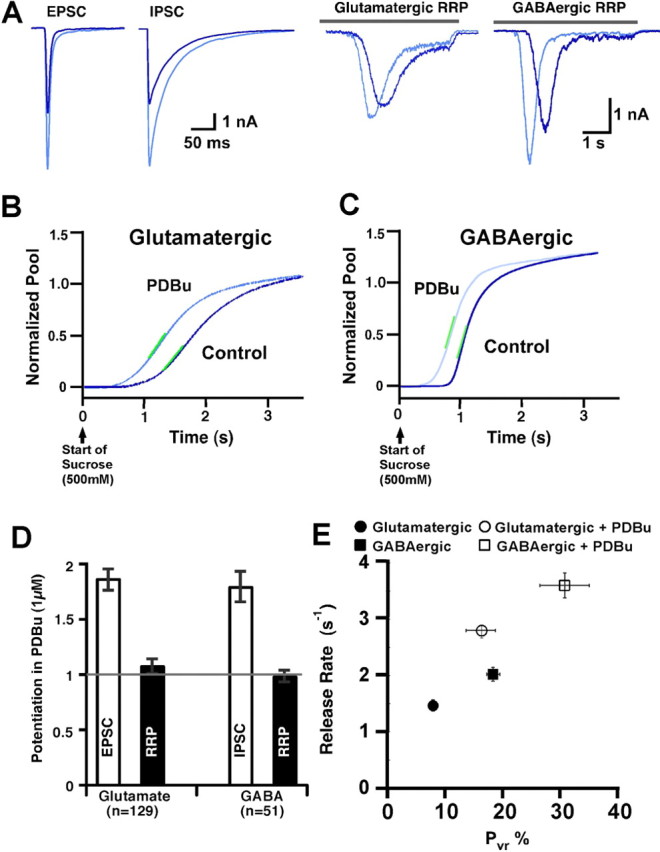
Comparison of release rates between glutamatergic and GABAergic hippocampal neurons. A, Typical raw traces of evoked (left) and 500 mm sucrose-induced (right) responses from wild-type glutamatergic and GABAergic neurons in the absence and presence of PDBu (1 μm). Average normalized sucrose response integrals from excitatory (B) and inhibitory (C) neurons from control (dark blue) to PDBu-treated (light blue) conditions. Note the faster basal release kinetics in the inhibitory neurons. E, Bar plot of PSC potentiation and the lack of potentiation of RRP in glutamatergic (n = 129) and GABAergic (n = 51) hippocampal neurons. E, Plot correlating the mean peak rate of vesicular release induced by 500 mm sucrose in hippocampal excitatory glutamatergic (circles) and inhibitory GABAergic (squares) autapses as a function of their average Pvr in their naive (filled symbols) and C1 domain-activated (open symbols) states.
Discussion
A major motivation for this study was to clarify the effect of C1 domain activation of Munc13s on vesicle priming and release probability. Although the mechanism of PDBu-induced presynaptic potentiation has been studied extensively, the strength of our approach is the ability to combine pharmacological (PDBu) and genetic (Munc13-1H567K) tools to interfere with C1 domain function of Munc13-1. Autaptic neurons uniquely allow simultaneous quantification of all forms of individual vesicular release. This study establishes that correlating different modes of release and analyzing their kinetics are useful methods to study putative alterations in vesicle fusion barrier.
Our main findings are that activation of the Munc13 C1 domain by PDBu potentiates the rate of spontaneously (increase of mEPSC frequency), hyperosmotically induced (faster response kinetics), or Ca2+-triggered (increased Pvr) release, without altering RRP size. In addition to their role in priming, our study suggests that Munc13s are post-priming regulators of synaptic vesicle exocytosis. We also find that the PDBu-binding-deficient Munc13-1H567K C1 domain mutant mimics the C1 domain-activated state, redefining it as a gain-of-function mutation.
PDBu as a regulator of the energy barrier for vesicle fusion
The effect of phorbol esters on synaptic transmission has been extensively studied and usually lead to an increase in presynaptic release. The fact that Munc13s are vesicle-priming factors and major targets for DAG/PDBu-mediated modulation of synaptic release (Augustin et al., 1999; Rhee et al., 2002; Rosenmund et al., 2002) favors at first glance a mechanism through regulation of RRP size. In support, overexpression of Munc13-1 enhances neurotransmitter release at Xenopus neuromuscular junctions (Betz et al., 1998) and increases the exocytotic burst in chromaffin cells (Smith, 1999; Ashery et al., 2000; Stevens et al., 2005). We found that PDBu affects via Munc13-1 vesicular release rates on a timescale of seconds, without changing RRP size. Although this is strongly supported by independent pool measurements using Ca2+ uncaging in the calyx of Held (Lou et al., 2005), there are two other studies using essentially the same system and hypertonic solution-based assay as our study, which suggest otherwise (Stevens and Sullivan, 1998; Virmani et al., 2005). As we discussed in Results, technical variability coupled with deviation from standard RRP definitions may account for these discrepancies. There may also be an effect of PDBu on presynaptic release and RRP size that depends on PKC. This component has, however, distinct kinetics, because it can be reliably revealed after 30 min preincubations with PKC inhibitors (Stevens and Sullivan, 1998; Capogna et al., 1999).
Our finding that activation of Munc13 C1 domain causes a left shift of the sensitivity to sucrose (Fig. 2B) indicates that activation of the C1 domain lowers the energy threshold for fusion of vesicles. More vesicles can be released with the same stimulus strength in case of low osmotic challenges, and all of the vesicles fuse more rapidly when faced with saturating concentrations of sucrose. This also implies that Munc13 C1 domain activation impacts vesicular release after vesicles have been primed to form the RRP, and hence Munc13s are not only priming proteins but also play a role as post-priming modulators of neurotransmitter release. In the calyx of Held preparation (Lou et al., 2005), PDBu potentiates evoked and spontaneous transmitter release by increasing the apparent Ca2+ sensitivity of vesicle fusion through allosteric regulation of fusion “willingness,” possibly via a lowering of the energy barrier. High-frequency stimulation in hippocampal cultures increases exocytotic rates by a possible lowering of the barrier for vesicle fusion during augmentation (Stevens and Wesseling, 1999). Both of these findings are complementary to our study, in that they describe a potentiation of the release apparatus without changing RRP. Furthermore, we believe that these and our observations are describing the same fundamental process, involving a conformational change in the N terminus of Munc13s. In the case of augmentation, it includes among other likely processes the Ca2+ and activity-dependent stimulation of N-terminal Munc13 domains by Ca2+/calmodulin (Junge et al., 2004), and by DAG (Rhee et al., 2002; Rosenmund et al., 2002).
Molecular mechanism of C1 domain activation and Munc13 disinhibition
Munc13s contain PKC-, Chimaerin-, and RasGRP-like “typical” DAG- and phorbol ester-binding C1 domains with two β-sheets, a short C-terminal α-helix, and two Zn2+ binding sites (Hurley et al., 1997; Shen et al., 2005; Colon-Gonzalez and Kazanietz, 2006). Based on PKC models (Zhang et al., 1995; Xu et al., 1997) and β2 Chimaerin structural data (Canagarajah et al., 2004), DAG receptors exist in a soluble inactive conformation in which the C1 domain hydrophobic ridge engages in intramolecular interactions. This sterically blocks the respective catalytic domain, directly or indirectly. Binding to phorbol esters or DAG break these inhibitory intramolecular interactions, thus activating the proteins. In their activated state, these proteins are deeply embedded into the membrane. Based on these allosteric activation models, we propose a mechanism of bimodal activity of Munc13s with two functional states (Fig. 7): a naive basal activity state in which the C1 domain binds to the Munc13 C terminus inhibiting the catalytic MUN domain, and an active potentiated state in which PDBu/DAG binding to C1 domain disrupt intramolecular interactions and disinhibit the catalytic domain (Fig. 7). The basal folded configuration of Munc13 possibly favors the binding of C2A to RIM and formation of the priming-relevant tripartite complex. PDBu/DAG have been known to increase membrane association of C1 domains. Under conditions of increased DAG levels, e.g., during high-frequency activity, or after external application of PDBu, C1 domain interactions with the membrane is favored, and this now competes with the intramolecular interactions of the protein, causing a conformational change that induces the activated state. In vivo, the other Ca2+-, calmodulin-, and phospholipid-targeted Munc13 N-terminal domains [C2B (J. S. Rhee, O. Shin, C. Rosenmund, N. Brose, T. Südhof, unpublished results); CaM (Junge et al., 2004)] may act in concert with the C1 domain to boost the catalytic activity of Munc13. Our model of disinhibition is strongly supported by the gain-of-function phenotype of the Munc13-1H567K C1 domain mutant. The histidine (H567) residue is a key residue in the C1 domain for chelating a central divalent Zn2+. We believe that the H567K mutation destroys the Zn2+-finger motif, not only preventing PDBu/DAG binding (Betz et al., 1998) but also interfering with the C1 domain-dependent intramolecular interactions. This leads to a constitutively disinhibited state of Munc13. Indeed, in the case of Chimaerins, the corresponding C1 domain mutation disrupts the Zn2+-finger motif and fully activates the Rac/Rho GTPase cascade (A. Betz and N. Brose, unpublished results). However, as a consequence of bypassing the basal conformation of Munc13, the KI mutant may have suffered interference with the N-terminal priming function.
Figure 7.

A two-state model for Munc13 activity. In the naive wild-type synapses, Munc13 exists in a folded configuration (top) in which intramolecular interactions between the N and C termini sterically interfere with the catalytic MUN domain. Such a basal “inhibited” state of Munc13 limits the vesicular release probability and rates by maintaining the energy barrier for fusion at a high level (shown in black). Interaction of Munc13 C2A with the RIM/Rab3 complex may have a stabilizing effect on the priming state. During binding of C1 domain with DAG/PDBu, the Munc13 molecule undergoes a conformational change that disrupts the intramolecular interactions and releases the catalytic domain from its inhibited condition to an activated state. The activation of Munc13 lowers the energy threshold for vesicular fusion, resulting in a potentiation of the kinetics of all forms of Ca2+ -dependent and -independent release. This process of enhancing the fusion willingness of vesicles comes at a stage after priming, possibly via downstream interactions of the MUN domain and the SNARE complex, and therefore impacts fusion probability without changing the RRP size. The Munc13-1H567K mutation leads to release probability and release rates in neurons that mimic the values observed in the C1 domain-activated WT, strongly suggesting that this gain-of-function mutation is a conformational mimic of the disinhibited “activated” state of Munc13. The constitutively open conformation may also have a negative impact on the upstream RIM/Rab3 interaction that normally helps in vesicle priming.
Functional implications of Munc13-1 as a post-priming regulator
The dual mode of Munc13 function equips synapses with the ability to effectively regulate vesicular release probability. The basal autoinhibitory state may not only be useful in providing “reserve activity,” but it also leads to lower spontaneous release activity, which in turn allows for a better discrimination of “noise” produced by spontaneous release as against evoked synaptic input to the postsynaptic neuron. The basal state of Munc13s may also help stabilize priming as such, as we discussed in our RRP data section, in that the gain-of-function Munc13-1H567K mutant might have interfered with N-terminal priming functions.
In their activated state, Munc13s lower the energy barrier for vesicle fusion such that vesicles can be released at twofold to fourfold higher rates. When action potentials arrive at high frequencies, the higher Pvr will help to meet the higher demand for neurotransmitter release. During a step after priming, the catalytic MUN domain, once relieved of the hindrance from the C1 domain, may alter the state of the SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) complex to regulate the rate and probability of vesicle fusion (Fig. 7, activated state). There is evidence that Munc13 interacts directly (Betz et al., 1997; Sassa et al., 1999) or indirectly (Basu et al., 2005) with the regulatory N-terminal domain of the SNARE syntaxin-1. Syntaxin-1 can exist in an open and closed conformation, which regulates the state of assembly of the SNARE complex (Dulubova et al., 1999; Margittai et al., 2003). The conformational switch in syntaxin-1 is thought to involve Munc13 (Brose et al., 2000; Richmond et al., 2001). However, recent findings indicating that the open syntaxin can restore priming but not stimulus-evoked secretion in Unc13 mutants (McEwen et al., 2006) support the idea of an additional post-priming role of Munc13s. Similar to the functional properties of activated Munc13 synapses, syntaxin-1 open-form mutant synapses also show an increased Pvr, accelerated release rates when stimulated with hypertonic solutions, and higher spontaneous release (S. Gerber, J. C. Rah, C. Rosenmund, and T. Südhof, unpublished data). This indicates that 'opening of Syntaxin 1′ and 'opening of Munc13′ have functionally equivalent impacts on the release machinery. Primed synaptic vesicles may possibly be equipped with SNARE complexes with variable fusogenic potentials depending on the associated Munc13 conformation. The energy barrier for fusion and ultimately the efficiency of spontaneous and Ca2+-triggered release are exponents of these different activity states taken together.
Footnotes
This work was supported by a National Institutes of Health/National Institute of Neurological Disorders and Stroke Research Grant NS051262 (C.R.). We thank Hui Deng, Dr. Ralf B. Nehring, and Hongmei Chen for technical support and Dr. Erwin Neher for discussion and comments on this manuscript.
References
- Andrews-Zwilling YS, Kawabe H, Reim K, Varoqueaux F, Brose N. Binding to Rab3A-interacting molecule RIM regulates the presynaptic recruitment of Munc13-1 and ubMunc13-2. J Biol Chem. 2006;281:19720–19731. doi: 10.1074/jbc.M601421200. [DOI] [PubMed] [Google Scholar]
- Aravamudan B, Fergestad T, Davis WS, Rodesch CK, Broadie K. Drosophila UNC-13 is essential for synaptic transmission. Nat Neurosci. 1999;2:965–971. doi: 10.1038/14764. [DOI] [PubMed] [Google Scholar]
- Ashery U, Varoqueaux F, Voets T, Betz A, Thakur P, Koch H, Neher E, Brose N, Rettig J. Munc13-1 acts as a priming factor for large dense-core vesicles in bovine chromaffin cells. EMBO J. 2000;19:3586–3596. doi: 10.1093/emboj/19.14.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustin I, Rosenmund C, Südhof TC, Brose N. Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature. 1999;400:457–461. doi: 10.1038/22768. [DOI] [PubMed] [Google Scholar]
- Basu J, Shen N, Dulubova I, Lu J, Guan R, Guryev O, Grishin NV, Rosenmund C, Rizo J. A minimal domain responsible for Munc13 activity. Nat Struct Mol Biol. 2005;12:1017–1018. doi: 10.1038/nsmb1001. [DOI] [PubMed] [Google Scholar]
- Betz A, Okamoto M, Benseler F, Brose N. Direct interaction of the rat unc-13 homologue Munc13-1 with the N terminus of syntaxin. J Biol Chem. 1997;272:2520–2526. doi: 10.1074/jbc.272.4.2520. [DOI] [PubMed] [Google Scholar]
- Betz A, Ashery U, Rickmann M, Augustin I, Neher E, Südhof TC, Rettig J, Brose N. Munc13-1 is a presynaptic phorbol ester receptor that enhances neurotransmitter release. Neuron. 1998;21:123–136. doi: 10.1016/s0896-6273(00)80520-6. [DOI] [PubMed] [Google Scholar]
- Betz A, Thakur P, Junge HJ, Ashery U, Rhee JS, Scheuss V, Rosenmund C, Rettig J, Brose N. Functional interaction of the active zone proteins Munc13-1 and RIM1 in synaptic vesicle priming. Neuron. 2001;30:183–196. doi: 10.1016/s0896-6273(01)00272-0. [DOI] [PubMed] [Google Scholar]
- Brose N, Hoffman K, Hata Y, Sudhof TC. Mammalian homologues of Caenorhabditis elegans unc-13 gene define novel family of C2-domain proteins. J Biol Chem. 1995;270:25273–25280. doi: 10.1074/jbc.270.42.25273. [DOI] [PubMed] [Google Scholar]
- Brose N, Rosenmund C, Rettig J. Regulation of transmitter release by Unc-13 and its homologues. Curr Opin Neurobiol. 2000;10:303–311. doi: 10.1016/s0959-4388(00)00105-7. [DOI] [PubMed] [Google Scholar]
- Canagarajah B, Leskow FC, Ho JY, Mischak H, Saidi LF, Kazanietz MG, Hurley JH. Structural mechanism for lipid activation of the Rac-specific GAP, beta2-chimaerin. Cell. 2004;119:407–418. doi: 10.1016/j.cell.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Capogna M, Fankhauser C, Gagliardini V, Gahwiler BH, Thompson SM. Excitatory synaptic transmission and its modulation by PKC is unchanged in the hippocampus of GAP-43-deficient mice. Eur J Neurosci. 1999;11:433–440. doi: 10.1046/j.1460-9568.1999.00450.x. [DOI] [PubMed] [Google Scholar]
- Colon-Gonzalez F, Kazanietz MG. C1 domains exposed: From diacylglycerol binding to protein-protein interactions. Biochim Biophys Acta. 2006;1761:827–837. doi: 10.1016/j.bbalip.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Dulubova I, Sugita S, Hill S, Hosaka M, Fernandez I, Sudhof T, Rizo J. A conformational switch in syntaxin during exocytosis: role of Munc18. EMBO J. 1999;18:4372–4382. doi: 10.1093/emboj/18.16.4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulubova I, Lou X, Lu J, Huryeva I, Alam A, Schneggenburger R, Sudhof TC, Rizo J. A Munc13/RIM/Rab3 tripartite complex: from priming to plasticity. EMBO J. 2005;24:2839–2850. doi: 10.1038/sj.emboj.7600753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JH, Newton AC, Parker PJ, Blumberg PM, Nishizuka Y. Taxonomy and function of C1 protein kinase C homology domains. Protein Sci. 1997;6:477–480. doi: 10.1002/pro.5560060228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junge HJ, Rhee JS, Jahn O, Varoqueaux F, Spiess J, Waxham MN, Rosenmund C, Brose N. Calmodulin and Munc13 form a Ca2+ sensor/effector complex that controls short-term synaptic plasticity. Cell. 2004;118:389–401. doi: 10.1016/j.cell.2004.06.029. [DOI] [PubMed] [Google Scholar]
- Lou X, Scheuss V, Schneggenburger R. Allosteric modulation of the presynaptic Ca2+ sensor for vesicle fusion. Nature. 2005;435:497–501. doi: 10.1038/nature03568. [DOI] [PubMed] [Google Scholar]
- Lu J, Machius M, Dulubova I, Dai H, Sudhof TC, Tomchick DR, Rizo J. Structural basis for a Munc13-1 homodimer to Munc13-1/RIM heterodimer switch. PLoS Biol. 2006;4:e192. doi: 10.1371/journal.pbio.0040192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison JM, Nurrish S, Kaplan JM. UNC-13 interaction with syntaxin is required for synaptic transmission. Curr Biol. 2005;15:2236–2242. doi: 10.1016/j.cub.2005.10.049. [DOI] [PubMed] [Google Scholar]
- Margittai M, Fasshauer D, Jahn R, Langen R. The Habc domain and the SNARE core complex are connected by a highly flexible linker. Biochemistry. 2003;42:4009–4014. doi: 10.1021/bi027437z. [DOI] [PubMed] [Google Scholar]
- McEwen JM, Madison JM, Dybbs M, Kaplan JM. Antagonistic regulation of synaptic vesicle priming by Tomosyn and UNC-13. Neuron. 2006;51:303–315. doi: 10.1016/j.neuron.2006.06.025. [DOI] [PubMed] [Google Scholar]
- Pyott SJ, Rosenmund C. The effects of temperature on vesicular supply and release in autaptic cultures of rat and mouse hippocampal neurons. J Physiol (Lond) 2002;539:523–535. doi: 10.1113/jphysiol.2001.013277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee J-S, Betz A, Pyott S, Reim K, Varoqueaux F, Augustin I, Hesse D, Südhof T, Takahashi M, Rosenmund C, Brose N. b-Phorbol ester and diacylglycerol-induced augmentation of transmitter release is mediated by Munc13s and not by PKC. Cell. 2002;108:121–133. doi: 10.1016/s0092-8674(01)00635-3. [DOI] [PubMed] [Google Scholar]
- Rhee JS, Li LY, Shin OH, Rah JC, Rizo J, Sudhof TC, Rosenmund C. Augmenting neurotransmitter release by enhancing the apparent Ca2+ affinity of synaptotagmin 1. Proc Natl Acad Sci USA. 2005;102:18664–18669. doi: 10.1073/pnas.0509153102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond JE, Weimer RM, Jorgensen EM. An open form of syntaxin bypasses the requirement for UNC-13 in vesicle priming. Nature. 2001;412:338–341. doi: 10.1038/35085583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Sigler A, Augustin I, Reim K, Brose B, Rhee J-S. Differential control of vesicle priming and short-term plasticity by Munc13 isoforms. Neuron. 2002;33:411–425. doi: 10.1016/s0896-6273(02)00568-8. [DOI] [PubMed] [Google Scholar]
- Sassa T, Harada S, Ogawa H, Rand JB, Maruyama IN, Hosono R. Regulation of the UNC-18–Caenorhabditis elegans syntaxin complex by UNC-13. J Neurosci. 1999;19:4772–4777. doi: 10.1523/JNEUROSCI.19-12-04772.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen N, Guryev O, Rizo J. Intramolecular occlusion of the diacylglycerol-binding site in the C1 domain of munc13-1. Biochemistry. 2005;44:1089–1096. doi: 10.1021/bi0476127. [DOI] [PubMed] [Google Scholar]
- Smith C. A persistent activity-dependent facilitation in chromaffin cells is caused by Ca2+ activation of protein kinase C. J Neurosci. 1999;19:589–598. doi: 10.1523/JNEUROSCI.19-02-00589.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Sullivan JM. Regulation of the readily releasable vesicle pool by protein kinase C. Neuron. 1998;21:885–893. doi: 10.1016/s0896-6273(00)80603-0. [DOI] [PubMed] [Google Scholar]
- Stevens CF, Wesseling JF. Augmentation is a potentiation of the exocytotic process. Neuron. 1999;22:139–146. doi: 10.1016/s0896-6273(00)80685-6. [DOI] [PubMed] [Google Scholar]
- Stevens DR, Wu ZX, Matti U, Junge HJ, Schirra C, Becherer U, Wojcik SM, Brose N, Rettig J. Identification of the minimal protein domain required for priming activity of Munc13-1. Curr Biol. 2005;15:2243–2248. doi: 10.1016/j.cub.2005.10.055. [DOI] [PubMed] [Google Scholar]
- Varoqueaux F, Sigler A, Rhee JS, Brose N, Enk C, Reim K, Rosenmund C. Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13-mediated vesicle priming. Proc Natl Acad Sci USA. 2002;99:9037–9042. doi: 10.1073/pnas.122623799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virmani T, Ertunc M, Sara Y, Mozhayeva M, Kavalali ET. Phorbol esters target the activity-dependent recycling pool and spare spontaneous vesicle recycling. J Neurosci. 2005;25:10922–10929. doi: 10.1523/JNEUROSCI.3766-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik SM, Rhee JS, Herzog E, Sigler A, Jahn R, Takamori S, Brose N, Rosenmund C. An essential role for vesicular glutamate transporter 1 (VGLUT1) in postnatal development and control of quantal size. Proc Natl Acad Sci USA. 2004;101:7158–7163. doi: 10.1073/pnas.0401764101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu RX, Pawelczyk T, Xia TH, Brown SC. NMR structure of a protein kinase C-gamma phorbol-binding domain and study of protein-lipid micelle interactions. Biochemistry. 1997;36:10709–10717. doi: 10.1021/bi970833a. [DOI] [PubMed] [Google Scholar]
- Yawo H. Protein kinase C potentiates transmitter release from the chick ciliary presynaptic terminal by increasing the exocytotic fusion probability. J Physiol (Lond) 1999;515:169–180. doi: 10.1111/j.1469-7793.1999.169ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Kazanietz MG, Blumberg PM, Hurley JH. Crystal structure of the cys2 activator-binding domain of protein kinase C delta in complex with phorbol ester. Cell. 1995;81:917–924. doi: 10.1016/0092-8674(95)90011-x. [DOI] [PubMed] [Google Scholar]