

Abstract
Amyotrophic lateral sclerosis (ALS) is a debilitating neurodegenerative disorder that results in the progressive loss of motoneurons (MNs) in the CNS. Several survival and death mechanisms of MNs have been characterized and it has been determined that MNs do not appear to mount a complete stress response, as determined by the lack of heat shock protein 70 (Hsp70) upregulation after several stress paradigms. Hsp70 has been shown to confer neuroprotection and the insufficient availability of Hsp70 may contribute to MNs' susceptibility to death in ALS mice. In this study, recombinant human Hsp70 (rhHsp70) was intraperitoneally injected three times weekly, beginning at postnatal day 50 until endstage, to G93A mutant SOD1 (G93A SOD1) mice. The administration of rhHsp70 was effective at increasing lifespan, delaying symptom onset, preserving motor function and prolonging MN survival. Interestingly, injected rhHsp70 localized to skeletal muscle and was not readily detected in the CNS. Treatment with rhHsp70 also resulted in an increased number of innervated neuromuscular junctions compared with control tissue. Together these results suggest rhHsp70 may delay disease progression in the G93A SOD1 mouse via a yet to be identified peripheral mechanism.
Keywords: motoneuron, denervation, NMJ, axonopathy, neurodegeneration, SOD1
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder affecting both upper and lower motoneurons (MNs), resulting in gradual muscle weakening and loss of MN function, ultimately leading to paralysis and death of afflicted individuals (Julien, 2001). Although 90% of cases are sporadic, mutations in the gene for superoxide dismutase-1 (SOD1) have been linked to ∼20% of familial cases (Rosen, 1993). The identification of these mutations has led to the development of experimental mouse models that recapitulate many features of the MN disease observed in humans (Pioro and Mitsumoto, 1995; Morrison et al., 1998).
Although the initial insult triggering sporadic ALS remains undetermined, several hallmark pathological features have been identified, including increased oxidative stress and free radical production, glutamate excitotoxicity, axonal transport deficits, protein aggregation and peripheral denervation (Cluskey and Ramsden, 2001; Fischer et al., 2004). Each of the proposed mechanisms thought to contribute to ALS onset and/or progression are events that compromise the cell's functionality and can be considered stressful. One of the most common cellular mechanisms to survive a stressful condition is the heat shock response, characterized by an increase in the transcription of a subset of genes resulting in the production of inducible heat shock proteins (Ritossa, 1996). Many environmental and physiological stressors can elicit a heat shock response, including those which occur in ALS.
Previous studies have shown that MNs are not capable of mounting a normal stress response, as characterized by the inability to readily upregulate heat shock protein 70 (Hsp70) (Batulan et al., 2003; Robinson et al., 2005). MNs from mice transfected to express mutant SOD1 (mSOD1) and overexpress Hsp70 have increased survival and reduced aggregate formation in culture (Bruening et al., 1999). Others have suggested that because of the widespread presence of mSOD1 in ALS, there is insufficient Hsp70 to protect the cells from stressful insults because it is bound to mSOD1 (Okado-Matsumoto and Fridovich, 2002). However, when G93A mutant SOD1 (G93A SOD1) mice were crossed with mice that overexpress Hsp70, there was not an increase in lifespan (Liu et al., 2005). These results suggest that an endogenous increase of Hsp70 in all cells is not sufficient to mitigate disease progression.
The addition of exogenous recombinant human Hsp70 (rhHsp70) confers increased embryonic chick MN survival in vitro after trophic factor deprivation and oxidative stress (Robinson et al., 2005, 2007). Data from our lab also produced comparable in vitro results when mouse embryonic MNs were subjected to trophic factor withdrawal and supplemented with rhHsp70 (our unpublished observation). Systemic administration of rhHsp70 to the developing chick's chorioallontoic membrane was capable of maintaining MN survival during the period of developmental programmed cell death (Robinson et al., 2005). These results provided evidence that rhHsp70 may be able to rescue MNs in vivo.
To determine whether similar effects on MN survival could be achieved in a mouse model of ALS, G93A SOD1 mice were injected intraperitoneally with rhHsp70. The results of our study suggest that rhHsp70 increases lifespan, delays symptom onset and preserves locomotor function in G93A SOD1 mice. Interestingly, the recombinant protein was not detected in the spinal cord or other CNS regions; however, it was detected in muscle. Lumbar MN survival was increased and more innervated NMJs were present in treated animals, suggesting a peripheral mode of action.
Materials and Methods
Animals.
All animal experiments were approved by the Wake Forest University Animal Care and Use Committee. Wild type females and G93A SOD1 males [B6SJL-TgN (SOD1-G93A) 1Gur], obtained from The Jackson Laboratory (Bar Harbor, ME), were bred to generate G93A SOD1 mice. Mice were weaned at postnatal day 21 (P21), separated by sex, and genotyped. DNA was obtained from tail clips and extracted using alkaline lysis (Truett et al., 2000). Throughout the study there were not any dramatic differences in lifespan between litters. This suggests that there were not any dramatic changes in transgene levels between litters that would result in abnormal variance in lifespan.
Treatments.
In the G93A SOD1 mouse, hindlimb tremor and weakness is observed at ∼P90 with death occurring at ∼P120 (Gurney et al., 1996). Other studies have begun experimental treatments in these animals at or approximately at P50 (Gurney et al., 1996; Li et al., 2000; Canton et al., 2001; Azari et al., 2003; Kaspar et al., 2003; Azari et al., 2005; Schutz et al., 2005; Chritin et al., 2006). For this study, treatment and behavioral analysis were started at P50. On P50, mice were given ad libitum access to Riluzole in their water at a concentration of 100 μg/ml with the solution changed three times weekly (Gurney et al., 1996). rhHsp70-treated mice were injected three times weekly to recapitulate the access to fresh Riluzole treated water three times weekly. Nontransgenic littermates [wild-type (WT)] and G93A SOD1 mice were intraperitoneally injected with rhHsp70 (Assay Designs, Ann Arbor, MI; catalog #ESP-555; 20 μg diluted in 100 μl sterile saline). In our previous study we found that 10 μg of rhHsp70 was effective for delaying naturally occurring MN death in chick embryos (Robinson et al., 2005). After accounting for weight and volume of the yolk and embryo, 20 μg was estimated to be a comparable dose for P50 mice. Intraperitoneal injections of bovine serum albumin (BSA; 20 μg in 100 μl sterile saline), and saline alone were used as negative controls. Because there were not any survival promoting effects of either BSA or saline alone, and there was no statistical difference between the two groups, both groups were combined into the single “control” group. A subset of animals was administered rhHsp70 beginning on day 30 or 90 to determine whether earlier or later initiation of treatment would influence lifespan. At P30, mSOD1 containing cytoplasmic aggregates have been detected in MNs of G93A SOD1 mice (Johnston et al., 2000) and as mentioned previously, P90 is their approximate age of symptom onset (Gurney et al., 1996). Because the mean survival varies between male and female G93A SOD1 mice (Heiman-Patterson et al., 2005), we included similar numbers of each gender in each treatment group.
Behavioral assays.
Disease progression was evaluated on each treatment day with weight determination, a leg extension test, and performance on the rotorod (San Diego Instruments, San Diego, CA) as described previously (Gurney et al., 1996; Wong et al., 1998). As animals progress toward disease end stage, muscle atrophy, resulting in weight loss occurs (Kieran et al., 2004). End stage disease is considered when the mouse cannot right itself after 30 s when placed on its side (Gurney et al., 1996). When mice are suspended by the tail, the hind limbs are normally extended, whereas failure of extension and/or shaking of the hind limbs are considered signs of symptom onset (Gurney et al., 1996). The rotorod test examines motor coordination and may be sensitive to MN dysfunction and degeneration (Crawley, 1999); the rod was rotated at a gradually accelerating speed up to 11 rpm over a 2 min interval, and the animals' latency to fall was recorded.
Histology.
Mice were killed at P90, P120, and at end stage, and the spinal cords were harvested and processed for MN counts. Briefly, mice were transcardially perfused with PBS (0.15 m NaCl, 2.98 mm Na2HPO4.7H2O, 1.03 mm KH2PO4) followed by Bouin's fixative. The lumbar region of the spinal cord was removed and embedded in paraffin. 12 μm sections were cut, and stained with a 5% thionin solution (Chu-Wang and Oppenheim, 1978). Only healthy MNs were counted in every tenth section of the lumbar spinal cord using a well established reliable method that has been validated against an optical fractionater unbiased stereological counting method. Healthy MNs are those that lie completely in the section with a nucleolus and normal MN morphology (Clarke and Oppenheim, 1995). At least five animals were included in each treatment group. Means and SEM were determined for each group.
Spinal cord glial activation.
Lumbar spinal cord tissue was dissected from mice at P90, fresh frozen in liquid nitrogen and later processed for Western blotting. Protein concentration was determined using a modified Lowry assay (Lowry et al., 1951). Once protein concentration was determined, 25 μg of spinal cord was resolved on a 12% polyacrylamide gel, and subjected to standard Western blotting procedures described below.
rhHsp 70 localization.
Within 30 min of their final injection of rhHsp70, mice were anesthetized with a ketamine/xylazine solution, decapitated, and their organs removed and fresh frozen in liquid nitrogen. Each organ was homogenized in nondenaturing lysis buffer containing 50 mm Tris-HCl pH7.5, 150 mm NaCl, 1% NP-40, and 1 mm EDTA, as well as 30 μl of a protease inhibitor mixture (Sigma, St. Louis, MO; P-2714). The samples were centrifuged at 3000 RPM, filtered through a 0.45 μm syringe filter and separated into aliquots. Protein concentration was determined as described previously. Equal amounts of protein were added to ATP-conjugated agarose beads (Sigma; A2767), and eluted with Tris Acetate buffer containing 3 mm ATP. Eluted proteins were TCA precipitated (Guzhova et al., 2001), resolved on a 7.5% polyacrylamide gel, and subjected to standard Western blotting procedures described below. We also tried to localize the rhHsp70 by immunohistochemistry; however, the human specific Hsp70 antibody (Hytest, Turku, Finland) was not specific for this application.
Hsp levels in lumbar spinal cord.
Lumbar spinal cord tissue was dissected from mice at P90 and processed similar to those described above. Once protein concentration was determined, 25 μg of spinal cord was resolved on a 10% polyacrylamide gel, and subjected to standard Western blotting procedures described below.
Detection rhHsp70 and BSA antibodies.
Animals were injected with rhHsp70 or BSA for 4 weeks, after which serum was collected and the protein concentration was determined as previously described. 50 μg per lane of rhHsp70 or BSA was resolved on a 12% polyacrylamide gel and standard Western blotting was performed. Serum from each injected mouse was used as the primary antibody.
Western blotting.
Once protein was resolved on a polyacrylamide gel and transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA), the membrane was blocked overnight at 4°C in 5% milk prepared in Tris-buffered saline with 0.1% Tween-20 (TBS-T). The following day, the membrane was incubated with primary antibody overnight, at 4°C. The primary antibodies used for glial cell activation were glial fibrillary acidic protein (GFAP; RA22101; Neuromics, Minneapolis, MN) and ionized calcium binding adaptor molecule 1 (Iba1; 016-20001; Wako, Neuss, Germany). The primary antibody used for the localization blots was an anti-human Hsp70 antibody (Hytest) at a dilution of 1:2000. The primary antibodies used for spinal cord Hsp levels were Hsp90 (SPA-830), Hsf-1 (SPA-901), Hsp70 (SPA-820), and Hsp40 (SPA-400), all from Assay Designs. The primary antibody used to probe for antibodies to rhHsp70 or BSA was mouse serum collected from each treatment group at a concentration of 15 μg/200 μl. Where applicable, mouse anti-actin (MAB1501; Calbiochem, La Jolla, CA) was used to verify equal loading.
The secondary antibodies, HRP-conjugated donkey anti-mouse IgG or HRP-conjugated donkey anti-rabbit IgG (Jackson ImmunoResearch, West Grove, PA), were applied for 1 h at room temperature. The membranes were washed in TBS-T and developed using the Super Signal West Pico ECL kit (Pierce, Rockford, IL).
Hindlimb NMJs.
For counting innervated hindlimb skeletal muscle neuromuscular junctions (NMJs), immunohistochemistry was performed on the medial gastrocnemious muscle of mice at P90. Animals were perfused with 2% paraformaldehyde in PBS. The muscles were dissected out and immersion fixed in the same fixative overnight at 4°C. Tissue was rinsed twice with PBS and placed in 30% sucrose for at least 72 h at 4°C. The muscles were sucrose embedded and cut at 40 μm. Antigen retrieval was achieved using a SDS pretreatment (Brown et al., 1996) and the sections were stained for the vesicular acetylcholine transporter (VAChT; Santa Cruz Biotechnology, Santa Cruz, CA) protein and α-bungarotoxin (α-BTX; Invitrogen, Eugene, OR) (Maeda et al., 2004). Some sections were also colabeled with NF-H and NF-L (Millipore). The percentage of innervated NMJs was determined by counting at least 115 NMJs in each treatment group using previously established counting criteria (Gould et al., 2006).
Statistical analysis.
Statistical analysis (Graph Pad Instat 3) in this study was performed by generating a mean value followed by formulating the SEM. Statistical significance was determined by running an ANOVA, followed by the Tukey-Kramer multiple comparisons post hoc test.
Results
rhHsp70 treatment at P120
To test the hypothesis that administration of rhHsp70 could delay MN death, prolong locomotor function and increase lifespan in the G93A SOD1 mouse model of ALS, mice were intraperitoneally injected with 20 μg of rhHsp70 three times per week beginning at P50. This dose had no adverse effects on WT mice after 9 months of treatment as these animals displayed normal hind-limb splay, locomotor ability, and weight gain/maintenance. The internal organs showed no gross abnormalities at autopsy (data not shown). rhHsp70 treated G93A SOD1 mice showed a significant delay in symptom onset when compared with the riluzole-treated or control groups (Fig. 1a). Riluzole, a glutamate antagonist and currently the only FDA approved treatment for ALS, was used in these experiments as a comparative measure to our treatment regime. The mice also demonstrated improved locomotor ability compared with the other groups (Fig. 1b). Finally, at P120, 100% of the rhHsp70-treated mice were alive whereas several animals in the riluzole and untreated groups had reached endstage (Fig. 1c).
Figure 1.

a, Average age of symptom onset is significantly delayed in rhHsp70 injected animals (n = 7) compared with Riluzole treated (n = 12) and untreated mice (n = 11; ***p ≤ 0.001). Age of symptom onset was determined when mice could not fully extend and/or shaking of the hind limbs. Statistical significance was determined using an ANOVA followed by the Tukey-Kramer multiple comparisons post hoc test. b, At the time of perfusion, P120, rhHsp70 treated mice (n = 7) had increased locomotor ability compared with both riluzole treated (n = 12) and untreated mice (n = 11; ***p ≤ 0.001). Statistical analysis was determined in the same manner as described previously. c, When mice were killed at P120, only 67% of the riluzole treated (n = 12) and 55% of the untreated (n = 11) animals had not reached endstage. One hundred percent of the rhHsp70 treated animals (n = 7) survived until this age.
rhHsp70 increases the number of surviving large MNs
As treatment with rhHsp70 increased lifespan and delayed symptom onset at P120, we wanted to determine whether this was related to an increase in MN survival. At P120, no significant difference in the total number of lumbar MNs was observed (Table 1); however, we did note that of the surviving MNs, those in the rhHsp70 group were significantly larger in size when compared with untreated animals (162.38 μm2 ± 7.77 vs 117.04 μm2 ± 15.08; *p ≤ 0.05). This result may represent a delay in the death of MNs and indicate that rhHsp70 treatment preserves MN size. When examined at an earlier time point (P90), MN survival was increased in the rhHsp70 treated G93A SOD1mice compared with control untreated mice (Table 1).
Table 1.
Motoneuron counts
| Hsp70 treated | Untreated | Riluzole treated | Wild type | |
|---|---|---|---|---|
| MN data at P90 | 2862 ± 160.36* | 2360 ± 105.02 | ||
| MN data at P120 | 1897 ± 122.49 | 1686 ± 210.63 | 1834 ± 83.94 | 3170 ± 100.40 |
Data are mean ± SEM. n = 5. *p ≤ 0.05.
rhHsp70 increases lifespan and delays symptom onset in mSOD1 mice
Although these initial results were encouraging, we repeated the study using a larger sample size. Injection of BSA or saline did not affect animal survival and was similar to untreated animals and therefore these groups were combined. Symptom onset was defined as a failure to fully extend and/or shaking of the hind limbs. Treatment with rhHsp70 resulted in a significant delay in symptom onset compared with Riluzole treated and untreated mice (Fig. 2a). Treatment with rhHsp70 also resulted in a significant increase in lifespan compared with untreated or Riluzole treated animals (Fig. 2b). The curves on the Kaplan Meier graph were similar suggesting that rhHsp70 treatment delayed symptom onset, but did not alter disease progression once symptoms are exhibited. We also monitored symptom progression on each treatment day in each treatment group using the leg extension test, rotorod performance and weight loss. rhHsp70 treatment delayed, but did not appear to alter these indicators of disease (data not shown).
Figure 2.

a, rhHsp70 treatment delays symptom onset in G93A mice. Mice treated with rhHsp70 (n = 20) resulted in delayed onset of leg tremors and hindlimb paralysis compared with the Riluzole treated (n = 15) and untreated mice (n = 17). Symptom onset occurred at P107.18 vs P97.22 vs P95.95, respectively (***p ≤ 0.001). Statistical significance was determined using an ANOVA followed by the Tukey–Kramer multiple comparisons post hoc test. b, Injection of rhHsp70 extends lifespan of G93A mice. Shown is a Kaplan–Meier Survival curve illustrating the increased probability of survival of rhHsp70 treated at P50 (n = 20) compared with Riluzole treated (n = 15) or untreated animals (n = 28). The increase in survival for rhHsp70 treated mice was >9 d (P136.75 vs P127.43; **p ≤ 0.01) compared with untreated mice and (P136.75 vs P128; *p ≤ 0.05) compared with riluzole treated animals. Statistical significance was determined as previously described. c, rhHsp70 delays symptom onset in G93A mice. Earlier injections, starting at P30 (n = 7), of rhHsp70 did not significantly enhance the delay of symptom onset compared with animals treated at P50 (n = 20). However, there was significant delay of symptom onset of P30 injected animals compared with those injected at P90 (n = 7; *p ≤ 0.05) and untreated animals (n = 17; *p ≤ 0.05). Statistical significance was determined as previously described. d, Earlier injections of rhHsp70 further increased lifespan of G93A mice. Mice were injected starting at P30, P50, and P90. Protein injections beginning at P30 (n = 7) significantly increased lifespan when compared with treatment beginning at P90 (*p ≤ 0.05; n = 7) and untreated mice (n = 28; **p ≤ 0.01). The average lifespan of animals with treatment beginning at P30 (n = 7) increased survival (P142.57 vs P136.75), not significantly, compared with treatment at P50 (n = 20). Statistical significance was determined as described previously.
Because other studies have begun various treatments of G93A SOD1 mice both earlier and later than P50 (Zhu et al., 2002; Kieran et al., 2004), we wanted to determine whether earlier or later treatment with rhHsp70 had a differential effect on symptom onset and lifespan. Earlier treatments delayed symptom onset, but there was no difference in the delay between animals that started rhHsp70 at P30 versus P50 (Fig. 2c). Likewise, there was an extension of survival, with animals treated at P30 living the longest (Fig. 2d). There was no significant effect on survival when treatment was started at P90 (Fig. 2d).
No increase in glial cell activation in the spinal cord
Glial cells have been suggested to contribute to disease progression in mSOD1 mouse models of ALS (Hall et al., 1998; Sargsyan et al., 2005; Boillee et al., 2006). To determine whether the increase in MN survival observed in rhHsp70 treated mice was caused by an inhibition of glial cell activation, lumbar spinal cord tissue was examined for potential changes in expression of two prominent proteins present in astrocytes (GFAP) and microglia (Iba1). The proteins have been shown to have increased expression when these cells become activated. There was no overt change in expression of GFAP between tissues collected from G93A SOD1 and WT mice and treatment with rhHsp70 did not alter this result. Whereas Iba1 expression was increased in tissue from G93A SOD1mice compared with WT mice, there was no marked change in expression between rhHsp70 treated and untreated animals (Fig. 3). Although not exhaustive, these results suggest that administration of rhHsp70 does not attenuate glial cell activation.
Figure 3.

Injection of rhHsp70 did not result in overt changes in glial cell activation. Lumbar spinal cord tissue was collected from treated and untreated G93A SOD1 or WT mice on P90. Twenty-five micrograms of protein per lane was loaded into each lane and Western blotting was performed to assess relative levels of GFAP and Iba1. When normalized to actin, quantification of the bands confirmed no change in expression (data not shown).
Injected protein localizes in the musculature
To determine whether the injected protein could localize to the CNS or musculature, we used an antibody that specifically recognizes human Hsp70 to distinguish between the endogenous mouse Hsp70 and rhHsp70. The injected protein was detected in the skeletal muscle but not in the brain or spinal cord (Fig. 4a). Using known amounts of recombinant protein, we determined that at least 200 ng of rhHsp70 is needed to be recognized by the extraction procedure and antibody (data not shown). These data suggest that either there were undetectable amounts of rhHsp70 in the CNS or that it may not cross the blood–brain barrier, and therefore may be acting through a peripheral mechanism.
Figure 4.

a, Injected rhHsp70 localizes in the hindlimb skeletal muscle. Results of Western blots revealed the presence of the injected protein in the hindlimb skeletal muscle and not in CNS tissue. rhHsp70 in skeletal muscle was eluted from ATP agarose beads, TCA precipitated, resolved on a 7.5% polyacrylamide gel and transferred to a PVDF membrane. Protein injection results in antibody production by the mice. b, c, Mice injected with either rhHsp70 (b) or BSA (c) produced antibodies to the injected protein. Serum collected from animals injected with either rhHsp70 or BSA was used as the primary antibody on Western blots generated from polyacrylamide gels loaded with either protein.
Because it has been reported that Hsp70 associates with mSOD1 in detergent-insoluble aggregates (Shinder et al., 2001; Okado-Matsumoto and Fridovich, 2002), and because overexpression of Hsp70 has been shown to reduce aggregate formation and MN death because of mSOD1 toxicity in vitro, we solubilized a detergent-insoluble protein fraction in 8M urea from the cortex, hindlimb skeletal muscle, lumbar spinal cord and liver and attempted to detect rhHsp70 (Shinder et al., 2001). Although both endogenous Hsp70 and mSOD1 were found in these samples, rhHsp70 was undetectable (data not shown).
Exogenous Hsp 70 injections results in antibody production
Injection of recombinant proteins often results in an antibody response. Furthermore, Hsp70 has been shown to reduce inflammation (McGeer and McGeer, 2002; van Eden et al., 2005), a process thought to contribute to ALS pathology. To determine whether injection of rhHsp70 resulted in antibody production in response to foreign protein, sera from mice was collected and used as antibodies for Western blot analysis. Mice treated with rhHsp70 expressed antibodies to the protein; however, mice treated with BSA also raised antibodies to the injected protein (Fig. 4b,c). Because treatment with BSA had no effect on symptom onset or survival, it is unlikely that antibody production itself accounts for the effects of rhHsp70.
Administration of rhHsp70 results in increased NMJ innervation
Neuromuscular junction denervation and dying back of axons versus MN loss is responsible for the onset of clinical symptoms in ALS (Fischer et al., 2004; Gould et al., 2006). Because rhHsp70 localizes in the muscle and rhHsp70 treated mice had improved motor performance, we examined NMJs in medial gastrocnemious muscles of treated and untreated animals. We found that treatment with rhHsp70 resulted in an increased number of innervated NMJs compared with control muscle (Fig. 5a,b). This result supports the theory that the injected rhHsp70 's survival promoting effect is through a peripheral mode of action.
Figure 5.
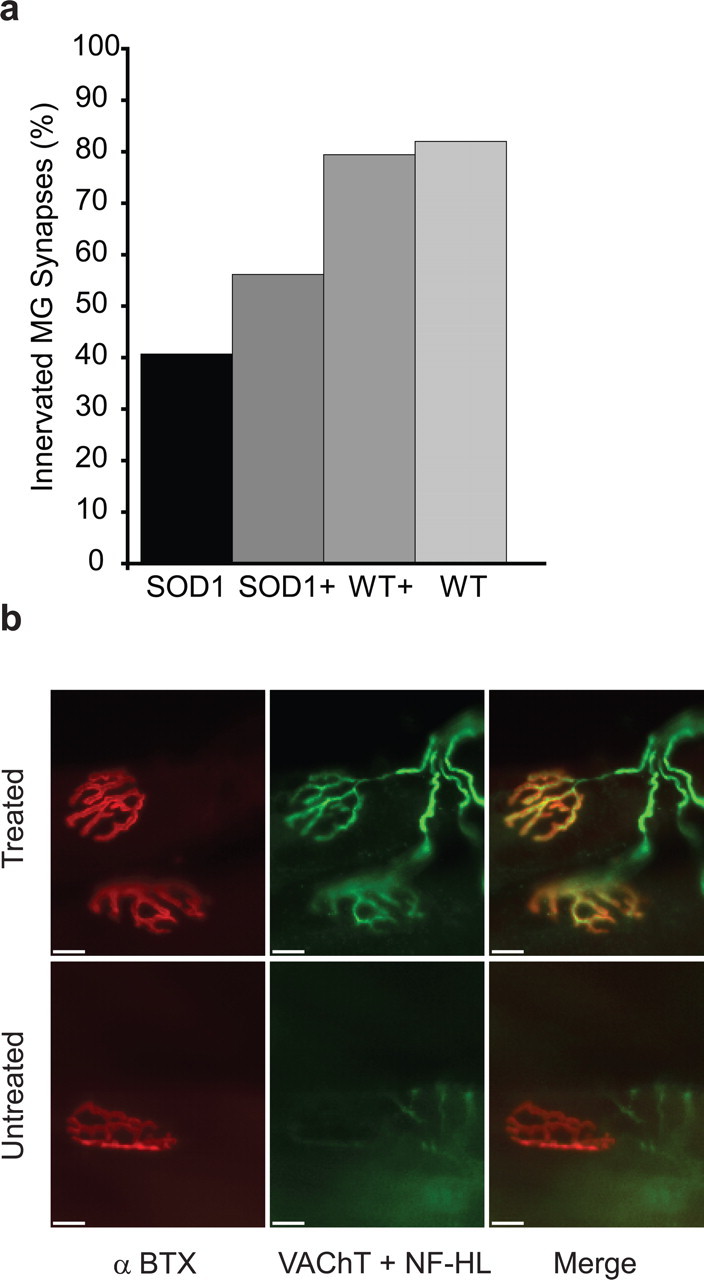
a, Injection of rhHsp70 resulted in an increased percentage of innervated NMJs in the MG at P90. Mice were injected with rhHsp70 beginning at P50 and killed at P90. Transgenic mSOD1 mice had a higher percentage of innervated NMJs at this age (+ denotes rhHsp70 treated animals). Both mSOD1 groups had a lower percent innervation compared with both wild type groups. There was no difference between the wild type groups. b, Photomicrographs of NMJs in treated and untreated animals. The two NMJs remain innervated in the treated animal, whereas in the untreated animal many NMJs showed absence of or partial innervation. nAChR clusters with corresponding VAChT and neurofilament H+L are labeled. Photographs of NMJs were taken at 20×. Scale bars: 20 μm.
No change in spinal cord Hsp levels
Endogenous Hsp levels have been reported to be reduced or unchanged in mSOD1 mice and produced mixed results pertaining to increased lifespan in G93A SOD1 mice (Kieran et al., 2004; Liu et al., 2005). Both in vitro and in vivo studies have provided evidence indicating that increased expression of multiple Hsps increases MN survival (Kieran et al., 2004; Batulan et al., 2006), whereas the overexpression of Hsp70 alone did not significantly increase lifespan of G93A SOD1 mice (Liu et al., 2005). Western blot analysis of homogenized spinal cords of P90 animals did not reveal marked changes in Hsp40, 70, 90 or Hsf-1 expression in either treated or untreated G93A SOD1 mice (Fig. 6). These data provide evidence that the mechanism of action of rhHsp70 does not induce an endogenous increase of Hsp levels in MNs.
Figure 6.

At P90, rhHsp70 treatment did not elevate endogenous Hsp or Hsf-1 levels in the lumbar spinal cord. Twenty-five micrograms of protein per lane was loaded into each lane and Western blotting was performed to assess relative levels of different Hsps and Hsf-1.
Discussion
The data presented here show that rhHsp70 treated mice have delayed symptom onset, the ability to run longer on the rotorod later in life, and increased lifespan. The rhHsp70 treated animals have increased number of lumbar MNs on P90, but by endstage (∼P120) this difference has disappeared; although soma size of MNs remained larger in surviving lumbar MNs of treated mice at P120. This suggests that treatment with rhHsp70 may maintain MNs in a functional state for a longer period before death, slowing their death and resulting in preserved motor performance and lifespan.
When treatment is started at P50, the increase in survival we observed is comparable with that seen in previous studies that used different treatments and delivery methods but also began treatment ∼P50 (Azari et al., 2003; Raoul et al., 2005; Schutz et al., 2005). Our results also suggest that an even earlier initiation of treatment (P30) increases survival by almost a week compared with P50. We also examined the effects of Riluzole in our mouse model of ALS, and in contrast to a previous report (Gurney et al., 1996) Riluzole failed to alter pathophysiology.
Intraperitoneal injection of arimoclomol, a coinducer of Hsps, beginning at P35 has been shown to significantly extend the survival of G93A SOD1 mice (Kieran et al., 2004). Upregulation of multiple Hsps through activation of Hsf1 in a primary cell culture model of ALS has been shown to be highly neuroprotective (Batulan et al., 2006). In contrast, G93A SOD1 mice that ubiquitously overexpress Hsp70 alone did not result in extended lifespan (Liu et al., 2005). These results, together with our findings, raise questions regarding the role of Hsps in promoting MN survival in response to expression of the mSOD1 protein. Hsp70 may have unique and distinct functions depending on its extracellular or intracellular location. These studies suggest that intracellularly, multiple Hsps must have increased expression to confer a survival promoting effect, whereas increased expression of Hsp70 alone is not sufficient. Another possibility is that constitutively increased Hsp70 expression negatively regulates the cell's ability to increase expression at time of stress (Manzerra et al., 1997). MNs have a high constitutive expression of Hsp/c70 and do not readily upregulate this particular Hsp in response to stress (Manzerra and Brown, 1992, 1996; Manzerra et al., 1997; Batulan et al., 2003; Robinson et al., 2005). In the in vivo studies treatment with arimoclomol or genetic manipulation resulted in an increase in endogenous Hsp levels in the spinal cord (Kieran et al., 2004; Liu et al., 2005). However, spinal cord Western blots from our animals did not show significant changes in Hsp levels, suggesting that our injected rhHsp70 protein is working through a different mechanism to prolong survival.
In a beginning attempt to determine the mechanism of action of rhHsp70, we collected soluble protein extracts from a variety of tissue sources. These data indicate that injected rhHsp70 localizes primarily to skeletal muscle. However, it is unclear whether rhHsp70 is present and active in other tissues (i.e., brain and spinal cord), but is below the limits of detection of our assay. The localization of rhHsp70 in skeletal muscle is consistent with a role for rhHsp70 in the maintenance of muscle or muscle innervation. Furthermore, when we determined whether glial cell activation was altered in rhHsp70 treated animals, we found no overt changes. Together with the results of localization of the protein in muscle and not CNS, these data suggest a potential peripheral mode of action for the survival promoting effect of the injected protein.
The denervation of hindlimb muscles has been previously reported to be an early event in the pathogenesis of ALS mice (Fischer et al., 2004; Gould et al., 2006). Hsp70 has been reported to be present at the NMJ in a trimeric complex with Cysteine string protein (Csp) and tetratricopeptide that promotes ATPase activity at the synapse (Tobaben et al., 2001). The function of this complex has also been postulated to aid in the maintenance of active synapses, to include the renaturing of synaptic proteins (Tobaben et al., 2001; Fernandez-Chacon et al., 2004). Csp is a 34 kDa secretory vesicle protein containing a J domain (Braun et al., 1996). In an attempt to further localize rhHsp70, we performed immunoprecipitation experiments to examine whether rhHsp70 interacts with Csp that proved to be inconclusive (data not shown). However, at P90 more NMJs were innervated in the medial of animals treated with rhHsp70 compared with controls.
Our examination of NMJs in untreated G93A SOD1 animals revealed denervation localized to the presynaptic portion of the synapse. These observations were in concert with previous reports (Frey et al., 2000; Fischer et al., 2004; Schaefer et al., 2005; Gould et al., 2006). NMJ innervation was preserved in rhHsp70 treated animals. Localization of the protein in muscle further suggests that the injected protein may confer its protective effect at the NMJ, although the precise mechanism is not known at this time. Axonal transport deficits in G93A SOD1 mice (Williamson and Cleveland, 1999) may then account for the inability of Hsp70 overexpression in the cell body to delay disease progression (Liu et al., 2005). Hsps are capable of being released by multiple cell types and taken up by others (Tytell et al., 1986; Hightower and Guidon, 1989; Sheller et al., 1998; Guzhova et al., 2001; Tytell, 2005). Furthermore, there are also reports of endogenous Hsp70 being upregulated in several cell types and released into the bloodstream after exercise induced stress; however, there is much debate whether Hsp70 is released from skeletal muscle in this model (Walsh et al., 2001; Febbraio et al., 2002a,b, 2004; Campisi et al., 2003; Lancaster et al., 2004). Hsp70 release does not appear to occur in healthy muscle, but stress that results in damage shows increased serum levels of Hsp70 (Febbraio et al., 2002a). It is possible that availability of the rhHsp70 protein at the NMJ can prolong function and innervation if muscle tissue does not readily release the protein and axonal transport deficits prevent even increased levels in the cell body from being transported to the NMJ. Exogenous delivery of the protein would circumvent these obstacles and be able to confer a protective effect.
ALS pathogenesis has been shown to be accompanied by an inflammatory response that has provided the basis for several studies analyzing the effects of anti-inflammatory agents on the survival of G93A SOD1 mice (McGeer and McGeer, 2002; Kiaei et al., 2005, 2006). Hsps have been shown to be upregulated and to help prevent tissue damage resulting from inflammatory disease conditions (van Eden et al., 2005). As expected, we found that rhHsp70 injection resulted in antibody production. This was expected because injected proteins (e.g., in solution) are reported to induce an antibody response to the protein in patients (Heise et al., 2005). To determine whether survival in our study was increased because of an immune response that potentially reduced CNS inflammation, control animals were injected with BSA. Similar to injection of rhHsp70, injection of BSA into G93A SOD1 mice also resulted in the production of antibodies, but did not increase animal survival or delay symptom onset. These data suggest that rhHsp70 may be prolonging survival and delaying symptom onset through a different noninflammatory mechanism.
A previous study suggested that combinations of therapeutic agents may be more effective than single agents in altering pathophysiology in the ALS mouse (Kriz et al., 2003). We tested a combination of rhHsp70 and recombinant mouse vascular endothelial growth factor (VEGF; R&D systems, Minneapolis, MN) beginning treatment at P50. Our work agrees with previous reports that VEGF treatment prolonged survival of G93A SOD1 mice (Zheng et al., 2004); however, the combination of VEGF and rhHsp70 did not delay symptom onset or prolong survival compared with VEGF alone (data not shown). Our results indicate that further studies are necessary to determine which combination of therapeutic agents may be effective in this mouse model.
In summary, the present study provides evidence that chronic administration of rhHsp70 delays symptom onset and extends the survival of G93A SOD1 mice; however, further study is needed to determine the dose of rhHSP70 that will produce maximal beneficial effects. The effect of rhHsp70 may be mediated by its effects on helping to maintain MN innervation skeletal muscle. Because NMJ integrity is comprised very early in G93A SOD1 mice, it is plausible that this may be an underlying cause of subsequent locomotor deficits and motor neuron death. Attenuating NMJ denervation may prove to be an effective therapeutic target in ALS.
Footnotes
This work was supported by a minority supplement to National Institutes of Health Grants NS36081 and NS46615, and by funds from the Wake Forest University School of Medicine Brian White ALS Foundation (C.E.M.). We thank Sharon Vinsant and Laura Delbono for technical assistance.
References
- Azari MF, Lopes EC, Stubna C, Turner BJ, Zang D, Nicola NA, Kurek JB, Cheema SS. Behavioural and anatomical effects of systemically administered leukemia inhibitory factor in the SOD1(G93A G1H) mouse model of familial amyotrophic lateral sclerosis. Brain Res. 2003;982:92–97. doi: 10.1016/s0006-8993(03)02989-5. [DOI] [PubMed] [Google Scholar]
- Azari MF, Profyris C, Le Grande MR, Lopes EC, Hirst J, Petratos S, Cheema SS. Effects of intraperitoneal injection of Rofecoxib in a mouse model of ALS. Eur J Neurol. 2005;12:357–364. doi: 10.1111/j.1468-1331.2004.00987.x. [DOI] [PubMed] [Google Scholar]
- Batulan Z, Shinder GA, Minotti S, He BP, Doroudchi MM, Nalbantoglu J, Strong MJ, Durham HD. High threshold for induction of the stress response in motor neurons is associated with failure to activate HSF1. J Neurosci. 2003;23:5789–5798. doi: 10.1523/JNEUROSCI.23-13-05789.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batulan Z, Taylor DM, Aarons RJ, Minotti S, Doroudchi MM, Nalbantoglu J, Durham HD. Induction of multiple heat shock proteins and neuroprotection in a primary culture model of familial amyotrophic lateral sclerosis. Neurobiol Dis. 2006;24:213–225. doi: 10.1016/j.nbd.2006.06.017. [DOI] [PubMed] [Google Scholar]
- Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Braun JE, Wilbanks SM, Scheller RH. The cysteine string secretory vesicle protein activates Hsc70 ATPase. J Biol Chem. 1996;271:25989–25993. doi: 10.1074/jbc.271.42.25989. [DOI] [PubMed] [Google Scholar]
- Brown D, Lydon J, McLaughlin M, Stuart-Tilley A, Tyszkowski R, Alper S. Antigen retrieval in cryostat tissue sections and cultured cells by treatment with sodium dodecyl sulfate (SDS) Histochem Cell Biol. 1996;105:261–267. doi: 10.1007/BF01463929. [DOI] [PubMed] [Google Scholar]
- Bruening W, Roy J, Giasson B, Figlewicz DA, Mushynski WE, Durham HD. Up-regulation of protein chaperones preserves viability of cells expressing toxic Cu/Zn-superoxide dismutase mutants associated with amyotrophic lateral sclerosis. J Neurochem. 1999;72:693–699. doi: 10.1046/j.1471-4159.1999.0720693.x. [DOI] [PubMed] [Google Scholar]
- Campisi J, Leem TH, Greenwood BN, Hansen MK, Moraska A, Higgins K, Smith TP, Fleshner M. Habitual physical activity facilitates stress-induced HSP72 induction in brain, peripheral, and immune tissues. Am J Physiol Regul Integr Comp Physiol. 2003;284:R520–R530. doi: 10.1152/ajpregu.00513.2002. [DOI] [PubMed] [Google Scholar]
- Canton T, Bohme GA, Boireau A, Bordier F, Mignani S, Jimonet P, Jahn G, Alavijeh M, Stygall J, Roberts S, Brealey C, Vuilhorgne M, Debono MW, Le Guern S, Laville M, Briet D, Roux M, Stutzmann JM, Pratt J. RPR 119990, a novel alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid antagonist: synthesis, pharmacological properties, and activity in an animal model of amyotrophic lateral sclerosis. J Pharmacol Exp Ther. 2001;299:314–322. [PubMed] [Google Scholar]
- Chritin M, Savasta M, Besson G. Benefit of tianeptine and morphine in a transgenic model of familial amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2006;7:32–37. doi: 10.1080/14660820510012022. [DOI] [PubMed] [Google Scholar]
- Chu-Wang IW, Oppenheim RW. Cell death of motoneurons in the chick embryo spinal cord. II. A quantitative and qualitative analysis of degeneration in the ventral root, including evidence for axon outgrowth and limb innervation prior to cell death. J Comp Neurol. 1978;177:59–85. doi: 10.1002/cne.901770106. [DOI] [PubMed] [Google Scholar]
- Clarke PG, Oppenheim RW. Neuron death in vertebrate development: in vitro methods. Methods Cell Biol. 1995;46:277–321. [PubMed] [Google Scholar]
- Cluskey S, Ramsden DB. Mechanisms of neurodegeneration in amyotrophic lateral sclerosis. Mol Pathol. 2001;54:386–392. [PMC free article] [PubMed] [Google Scholar]
- Crawley JN. Behavioral phenotyping of transgenic and knockout mice: experimental design and evaluation of general health, sensory functions, motor abilities, and specific behavioral tests. Brain Res. 1999;835:18–26. doi: 10.1016/s0006-8993(98)01258-x. [DOI] [PubMed] [Google Scholar]
- Febbraio MA, Steensberg A, Walsh R, Koukoulas I, van Hall G, Saltin B, Pedersen BK. Reduced glycogen availability is associated with an elevation in HSP72 in contracting human skeletal muscle. J Physiol (Lond) 2002a;538:911–917. doi: 10.1113/jphysiol.2001.013145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Febbraio MA, Ott P, Nielsen HB, Steensberg A, Keller C, Krustrup P, Secher NH, Pedersen BK. Exercise induces hepatosplanchnic release of heat shock protein 72 in humans. J Physiol (Lond) 2002b;544:957–962. doi: 10.1113/jphysiol.2002.025148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Febbraio MA, Mesa JL, Chung J, Steensberg A, Keller C, Nielsen HB, Krustrup P, Ott P, Secher NH, Pedersen BK. Glucose ingestion attenuates the exercise-induced increase in circulating heat shock protein 72 and heat shock protein 60 in humans. Cell Stress Chaperones. 2004;9:390–396. doi: 10.1379/CSC-24R1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Chacon R, Wolfel M, Nishimune H, Tabares L, Schmitz F, Castellano-Munoz M, Rosenmund C, Montesinos ML, Sanes JR, Schneggenburger R, Sudhof TC. The synaptic vesicle protein CSP alpha prevents presynaptic degeneration. Neuron. 2004;42:237–251. doi: 10.1016/s0896-6273(04)00190-4. [DOI] [PubMed] [Google Scholar]
- Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185:232–240. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Frey D, Schneider C, Xu L, Borg J, Spooren W, Caroni P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci. 2000;20:2534–2542. doi: 10.1523/JNEUROSCI.20-07-02534.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould TW, Buss RR, Vinsant S, Prevette D, Sun W, Knudson CM, Milligan CE, Oppenheim RW. Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J Neurosci. 2006;26:8774–8786. doi: 10.1523/JNEUROSCI.2315-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME, Cutting FB, Zhai P, Doble A, Taylor CP, Andrus PK, Hall ED. Benefit of vitamin E, riluzole, and gabapentin in a transgenic model of familial amyotrophic lateral sclerosis. Ann Neurol. 1996;39:147–157. doi: 10.1002/ana.410390203. [DOI] [PubMed] [Google Scholar]
- Guzhova I, Kislyakova K, Moskaliova O, Fridlanskaya I, Tytell M, Cheetham M, Margulis B. In vitro studies show that Hsp70 can be released by glia and that exogenous Hsp70 can enhance neuronal stress tolerance. Brain Res. 2001;914:66–73. doi: 10.1016/s0006-8993(01)02774-3. [DOI] [PubMed] [Google Scholar]
- Hall ED, Oostveen JA, Gurney ME. Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. Glia. 1998;23:249–256. doi: 10.1002/(sici)1098-1136(199807)23:3<249::aid-glia7>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Heiman-Patterson TD, Deitch JS, Blankenhorn EP, Erwin KL, Perreault MJ, Alexander BK, Byers N, Toman I, Alexander GM. Background and gender effects on survival in the TgN(SOD1-G93A)1Gur mouse model of ALS. J Neurol Sci. 2005;236:1–7. doi: 10.1016/j.jns.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Heise T, Bott S, Tusek C, Stephan JA, Kawabata T, Finco-Kent D, Liu C, Krasner A. The effect of insulin antibodies on the metabolic action of inhaled and subcutaneous insulin: a prospective randomized pharmacodynamic study. Diabetes Care. 2005;28:2161–2169. doi: 10.2337/diacare.28.9.2161. [DOI] [PubMed] [Google Scholar]
- Hightower LE, Guidon PT., Jr Selective release from cultured mammalian cells of heat-shock (stress) proteins that resemble glia-axon transfer proteins. J Cell Physiol. 1989;138:257–266. doi: 10.1002/jcp.1041380206. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Dalton MJ, Gurney ME, Kopito RR. Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2000;97:12571–12576. doi: 10.1073/pnas.220417997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julien JP. Amyotrophic lateral sclerosis. unfolding the toxicity of the misfolded. Cell. 2001;104:581–591. doi: 10.1016/s0092-8674(01)00244-6. [DOI] [PubMed] [Google Scholar]
- Kaspar BK, Llado J, Sherkat N, Rothstein JD, Gage FH. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science. 2003;301:839–842. doi: 10.1126/science.1086137. [DOI] [PubMed] [Google Scholar]
- Kiaei M, Kipiani K, Chen J, Calingasan NY, Beal MF. Peroxisome proliferator-activated receptor-gamma agonist extends survival in transgenic mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2005;191:331–336. doi: 10.1016/j.expneurol.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Kiaei M, Petri S, Kipiani K, Gardian G, Choi DK, Chen J, Calingasan NY, Schafer P, Muller GW, Stewart C, Hensley K, Beal MF. Thalidomide and lenalidomide extend survival in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci. 2006;26:2467–2473. doi: 10.1523/JNEUROSCI.5253-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieran D, Kalmar B, Dick JR, Riddoch-Contreras J, Burnstock G, Greensmith L. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat Med. 2004;10:402–405. doi: 10.1038/nm1021. [DOI] [PubMed] [Google Scholar]
- Kriz J, Gowing G, Julien JP. Efficient three-drug cocktail for disease induced by mutant superoxide dismutase. Ann Neurol. 2003;53:429–436. doi: 10.1002/ana.10500. [DOI] [PubMed] [Google Scholar]
- Lancaster GI, Moller K, Nielsen B, Secher NH, Febbraio MA, Nybo L. Exercise induces the release of heat shock protein 72 from the human brain in vivo. Cell Stress Chaperones. 2004;9:276–280. doi: 10.1379/CSC-18R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Ona VO, Guegan C, Chen M, Jackson-Lewis V, Andrews LJ, Olszewski AJ, Stieg PE, Lee JP, Przedborski S, Friedlander RM. Functional role of caspase-1 and caspase-3 in an ALS transgenic mouse model. Science. 2000;288:335–339. doi: 10.1126/science.288.5464.335. [DOI] [PubMed] [Google Scholar]
- Liu J, Shinobu LA, Ward CM, Young D, Cleveland DW. Elevation of the Hsp70 chaperone does not effect toxicity in mouse models of familial amyotrophic lateral sclerosis. J Neurochem. 2005;93:875–882. doi: 10.1111/j.1471-4159.2005.03054.x. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Maeda M, Ohba N, Nakagomi S, Suzuki Y, Kiryu-Seo S, Namikawa K, Kondoh W, Tanaka A, Kiyama H. Vesicular acetylcholine transporter can be a morphological marker for the reinnervation to muscle of regenerating motor axons. Neurosci Res. 2004;48:305–314. doi: 10.1016/j.neures.2003.11.008. [DOI] [PubMed] [Google Scholar]
- Manzerra P, Brown IR. Expression of heat shock genes (hsp70) in the rabbit spinal cord: localization of constitutive and hyperthermia-inducible mRNA species. J Neurosci Res. 1992;31:606–615. doi: 10.1002/jnr.490310404. [DOI] [PubMed] [Google Scholar]
- Manzerra P, Brown IR. The neuronal stress response: nuclear translocation of heat shock proteins as an indicator of hyperthermic stress. Exp Cell Res. 1996;229:35–47. doi: 10.1006/excr.1996.0341. [DOI] [PubMed] [Google Scholar]
- Manzerra P, Rush SJ, Brown IR. Tissue-specific differences in heat shock protein hsc70 and hsp70 in the control and hyperthermic rabbit. J Cell Physiol. 1997;170:130–137. doi: 10.1002/(SICI)1097-4652(199702)170:2<130::AID-JCP4>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. Inflammatory processes in amyotrophic lateral sclerosis. Muscle Nerve. 2002;26:459–470. doi: 10.1002/mus.10191. [DOI] [PubMed] [Google Scholar]
- Morrison BM, Morrison JH, Gordon JW. Superoxide dismutase and neurofilament transgenic models of amyotrophic lateral sclerosis. J Exp Zool. 1998;282:32–47. [PubMed] [Google Scholar]
- Okado-Matsumoto A, Fridovich I. Amyotrophic lateral sclerosis: a proposed mechanism. Proc Natl Acad Sci USA. 2002;99:9010–9014. doi: 10.1073/pnas.132260399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pioro EP, Mitsumoto H. Animal models of ALS. Clin Neurosci. 1995;3:375–385. [PubMed] [Google Scholar]
- Raoul C, Abbas-Terki T, Bensadoun JC, Guillot S, Haase G, Szulc J, Henderson CE, Aebischer P. Lentiviral-mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALS. Nat Med. 2005;11:423–428. doi: 10.1038/nm1207. [DOI] [PubMed] [Google Scholar]
- Ritossa F. Discovery of the heat shock response. Cell Stress Chaperones. 1996;1:97–98. doi: 10.1379/1466-1268(1996)001<0097:dothsr>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MB, Tidwell JL, Gould T, Taylor AR, Newbern JM, Graves J, Tytell M, Milligan CE. Extracellular heat shock protein 70: a critical component for motoneuron survival. J Neurosci. 2005;25:9735–9745. doi: 10.1523/JNEUROSCI.1912-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MB, Taylor AR, Gifondorwa DJ, Tytell M, Milligan CE. Exogenous Hsc70, but not thermal preconditioning, confers protection to motoneurons subjected to oxidative stress. Dev Neurobiol. 2007 doi: 10.1002/dneu.20550. in press. [DOI] [PubMed] [Google Scholar]
- Rosen DR. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;364:362. doi: 10.1038/364362c0. [DOI] [PubMed] [Google Scholar]
- Sargsyan SA, Monk PN, Shaw PJ. Microglia as potential contributors to motor neuron injury in amyotrophic lateral sclerosis. Glia. 2005;51:241–253. doi: 10.1002/glia.20210. [DOI] [PubMed] [Google Scholar]
- Schaefer AM, Sanes JR, Lichtman JW. A compensatory subpopulation of motor neurons in a mouse model of amyotrophic lateral sclerosis. J Comp Neurol. 2005;490:209–219. doi: 10.1002/cne.20620. [DOI] [PubMed] [Google Scholar]
- Schutz B, Reimann J, Dumitrescu-Ozimek L, Kappes-Horn K, Landreth GE, Schurmann B, Zimmer A, Heneka MT. The oral antidiabetic pioglitazone protects from neurodegeneration and amyotrophic lateral sclerosis-like symptoms in superoxide dismutase-G93A transgenic mice. J Neurosci. 2005;25:7805–7812. doi: 10.1523/JNEUROSCI.2038-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheller RA, Smyers ME, Grossfeld RM, Ballinger ML, Bittner GD. Heat-shock proteins in axoplasm: high constitutive levels and transfer of inducible isoforms from glia. J Comp Neurol. 1998;396:1–11. doi: 10.1002/(sici)1096-9861(19980622)396:1<1::aid-cne1>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Shinder GA, Lacourse MC, Minotti S, Durham HD. Mutant Cu/Zn-superoxide dismutase proteins have altered solubility and interact with heat shock/stress proteins in models of amyotrophic lateral sclerosis. J Biol Chem. 2001;276:12791–12796. doi: 10.1074/jbc.M010759200. [DOI] [PubMed] [Google Scholar]
- Tobaben S, Thakur P, Fernandez-Chacon R, Sudhof TC, Rettig J, Stahl B. A trimeric protein complex functions as a synaptic chaperone machine. Neuron. 2001;31:987–999. doi: 10.1016/s0896-6273(01)00427-5. [DOI] [PubMed] [Google Scholar]
- Truett GE, Heeger P, Mynatt RL, Truett AA, Walker JA, Warman ML. Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT) Biotechniques. 2000;29:52, 54. doi: 10.2144/00291bm09. [DOI] [PubMed] [Google Scholar]
- Tytell M. Release of heat shock proteins (Hsps) and the effects of extracellular Hsps on neural cells and tissues. Int J Hyperthermia. 2005;21:445–455. doi: 10.1080/02656730500041921. [DOI] [PubMed] [Google Scholar]
- Tytell M, Greenberg SG, Lasek RJ. Heat shock-like protein is transferred from glia to axon. Brain Res. 1986;363:161–164. doi: 10.1016/0006-8993(86)90671-2. [DOI] [PubMed] [Google Scholar]
- van Eden W, van der Zee R, Prakken B. Heat-shock proteins induce T-cell regulation of chronic inflammation. Nat Rev Immunol. 2005;5:318–330. doi: 10.1038/nri1593. [DOI] [PubMed] [Google Scholar]
- Walsh RC, Koukoulas I, Garnham A, Moseley PL, Hargreaves M, Febbraio MA. Exercise increases serum Hsp72 in humans. Cell Stress Chaperones. 2001;6:386–393. doi: 10.1379/1466-1268(2001)006<0386:eishih>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson TL, Cleveland DW. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci. 1999;2:50–56. doi: 10.1038/4553. [DOI] [PubMed] [Google Scholar]
- Wong PC, Rothstein JD, Price DL. The genetic and molecular mechanisms of motor neuron disease. Curr Opin Neurobiol. 1998;8:791–799. doi: 10.1016/s0959-4388(98)80123-2. [DOI] [PubMed] [Google Scholar]
- Zheng C, Nennesmo I, Fadeel B, Henter JI. Vascular endothelial growth factor prolongs survival in a transgenic mouse model of ALS. Ann Neurol. 2004;56:564–567. doi: 10.1002/ana.20223. [DOI] [PubMed] [Google Scholar]
- Zhu S, Stavrovskaya IG, Drozda M, Kim BY, Ona V, Li M, Sarang S, Liu AS, Hartley DM, Wu du C, Gullans S, Ferrante RJ, Przedborski S, Kristal BS, Friedlander RM. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]