

Abstract
Developing neurons have greater capacity in experience-dependent plasticity than adult neurons but the molecular mechanism is not well understood. Here we report a developmentally regulated long-term synaptic plasticity through actin-dependent activation of presynaptic silent synapses in cultured hippocampal neurons. Live FM 1-43 imaging and retrospective immunocytochemistry revealed that many presynaptic boutons in immature neurons are functionally silent at resting conditions, but can be converted into active ones after repetitive neuronal stimulation. The activation of presynaptic silent synapses is dependent on L-type calcium channels and protein kinase A (PKA)/PKC signaling pathways. Moreover, blocking actin polymerization with latrunculin A and cytochalasin B abolishes long-term increase of presynaptic functional boutons induced by repetitive stimulation, whereas actin polymerizer jasplakinolide increases the number of active boutons in immature neurons. In mature neurons, however, presynaptic boutons are mostly functional and repetitive stimulation did not induce additional enhancement. Quantitative immunostaining with phalloidin revealed a significant increase in axonal F-actin level after repetitive stimulation in immature but not mature neurons. These results suggest that actin-dependent activation of presynaptic silent synapses contributes significantly to the long-term synaptic plasticity during neuronal development.
Keywords: actin, activity, synaptic plasticity, presynaptic, silent synapse, hippocampus
Introduction
Long-term synaptic plasticity involves both functional and morphological changes of synapses (Bailey and Kandel, 1993; Collingridge and Bliss, 1995; Nicoll and Malenka, 1995; Hansel et al., 2001). In the CNS, postsynaptic silent synapses have been identified as showing only NMDA but not AMPA receptors, and activation of these postsynaptically silent synapses has been demonstrated to contribute significantly to long-term synaptic plasticity (Isaac et al., 1995; Liao et al., 1995; Durand et al., 1996; Wu et al., 1996; Choi et al., 2000). Presynaptic silent synapses have also been reported, which are possibly caused by a very low probability of synaptic vesicle release (Gasparini et al., 2000; Hanse and Gustafsson, 2001). The molecular mechanisms underlying activation of presynaptic silent synapses and their contribution to long-term synaptic plasticity are widely unknown.
Recent studies suggest an active role of actin in activity-dependent synaptic plasticity (Colicos et al., 2001; Okamoto et al., 2004). Actin is an essential cytoskeleton protein playing a vital role in axon guidance, synapse development, and synaptic plasticity (Matus, 2000; Dent and Gertler, 2003; Dillon and Goda, 2005). Actin can be found in two forms, globular (G-actin) and filamentous (F-actin), with F-actin being polymerized from many G-actin monomers (Dent and Gertler, 2003). Actin is enriched in both presynaptic nerve terminals and postsynaptic dendritic spines to regulate presynaptic and postsynaptic functions (Fischer et al., 2000; Chang and De Camilli, 2001; Dillon and Goda, 2005). In presynaptic terminals, F-actin surrounds synaptic vesicle clusters and regulates multiple steps during synaptic vesicle cycling and trafficking (Kuromi and Kidokoro, 1998; Cole et al., 2000; Morales et al., 2000; Shupliakov et al., 2002; Sakaba and Neher, 2003; Sankaranarayanan et al., 2003). In postsynaptic dendrites, actin may be directly linked to postsynaptic density and regulates AMPA receptor clustering and the maintenance of long-term potentiation (LTP) (Allison et al., 1998; Shen et al., 1998; Kim and Lisman, 1999; Krucker et al., 2000; Star et al., 2002; Ackermann and Matus, 2003; Fukazawa et al., 2003; Matsuzaki et al., 2004; Okamoto et al., 2004). Actin is also a critical requisite for new synapse formation and for the maintenance of immature synapses (Antonova et al., 2001; Colicos et al., 2001; Zhang and Benson, 2001; Zito et al., 2004; Wang et al., 2005).
We report here a novel function of actin in converting presynaptic silent synapses into functional ones during long-term synaptic plasticity induced by repetitive-spaced stimulation in hippocampal synapses. Live FM imaging and retrospective immunocytochemistry revealed many presynaptically silent synapses in immature neurons, which can be activated by repetitive stimulation. Actin polymerization appears to play a critical role in the activation of presynaptic silent synapses. Inhibition of actin polymerization with latrunculin A or cytochalasin B blocked, whereas promoting actin polymerization with jasplakinolide increased, the number of active boutons in immature neurons. The degree of actin polymerization after repetitive stimulation was significantly increased in immature but not mature neurons, suggesting that activity-dependent actin polymerization is crucial during developmental regulation of long-term synaptic plasticity.
Materials and Methods
Cell culture.
Microisland cultures of hippocampal neurons were prepared similarly to those in previous reports (Deng and Chen, 2003; Chen et al., 2004). Briefly, hippocampal CA1–CA3 regions were dissected from newborn rat pups [postnatal day 0 (P0)-P1], quickly subdivided into small cubes (<1 mm3), and incubated in a digestion solution consisting of 0.05% Trypsin-EDTA and supplemented with 20 mm glucose plus 25 U/ml DNAase. The small tissue blocks were then mechanically dissociated in a solution consisting of HBSS, 5 mm HEPES, 20 mm d-glucose, 20% horse serum, and 25 U/ml DNAase. The cells were resuspended after centrifugation and plated at a medium density (4000–6000 cells/cm2) onto microislands containing a monolayer of astrocytes. The culture medium contained 500 ml of MEM (Invitrogen, Eugene, OR), 5% FBS (HyClone, Logan, UT), 10 ml of B-27 supplement (Invitrogen), 100 mg of NaHCO3, 20 mm d-glucose, 0.5 mm l-glutamine, and 25 U/ml penicillin/streptomycin. Cultures were maintained at 37°C in a 5% CO2-humidified incubator, and 50% of the culture medium was replaced three times in the first week. Cultured neurons were used in ∼3 weeks. In this study, we define immature neurons as cultured for 7–11 d after plating, and mature neurons as 18–22 d after plating.
Electrophysiology.
Whole-cell recordings were performed in voltage-clamp mode using a MultiClamp 700A amplifier (Molecular Devices, Union City, CA) (Deng and Chen, 2003). The recording chamber was continuously perfused with a bath solution consisting of (in mm) 128 NaCl, 30 glucose, 25 HEPES, 5 KCl, 2 CaCl2, 1 MgCl2, pH 7.3, with NaOH, via a Warner (Hamden, CT) VC-6 drug delivery system. The 90 mm KCl solution with equal molar replacement of NaCl by KCl was used to serve as repetitive stimulation (2 min 90 K+ followed by 8 min bath solution, and repeated 6 times) (for protocol, see Fig. 1A) (Wu et al., 2001b). To record miniature EPSCs (mEPSCs), TTX (0.5 μm) and bicuculline (BIC, 20 μm) were added into the bath solution to block action potentials and GABAergic events. Patch pipettes were pulled from borosilicate glass and had resistances of 2–4 MΩ when filled with internal pipette solution, which consisted of the following (in mm): 135 KCl, 10 Tris-phosphocreatine, 2 EGTA, 10 HEPES, 4 MgATP, 0.5 Na2GTP, pH 7.3, with KOH. The series resistance was typically 10–20 MΩ and partially compensated by 30–50%. The membrane potential was held at −70 mV. Data were acquired using pClamp 9 software, sampled at 10 kHz, and filtered at 1 kHz. Off-line data analysis of mEPSCs was performed using MiniAnalysis software (Synaptosoft, Decator, GA). Experiments were performed at room temperature. Student’s t test was used for statistical analysis for mini events.
Figure 1.
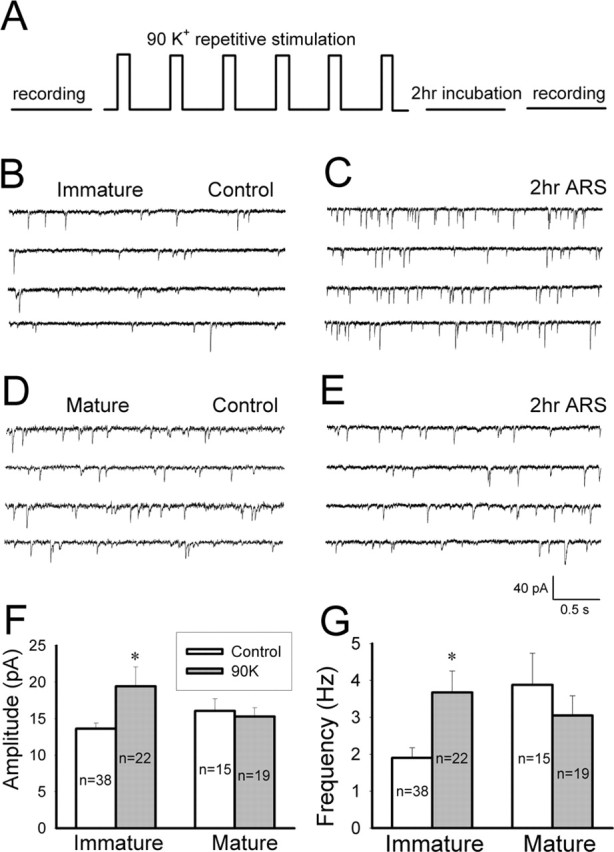
Repetitive stimulation induces long-term enhancement of synaptic transmission in immature but not mature hippocampal neurons. A, Diagram showing the experimental protocol. mEPSCs were recorded before and 2 h after six repetitive stimuli with 90 mm KCl solution. B, C, Representative traces illustrating mEPSCs recorded in immature neurons (7–11 d in culture) in the presence of TTX (0.5 μm) and BIC (20 μm) in control (B) and 2 h ARS (C). D, E, mEPSC traces in mature neurons (18–22 d in culture) in control (D) and 2 h after repetitive stimulation (E). F, Bar graphs showing that repetitive stimulation induced a significant increase in the average mEPSC amplitude in immature neurons (p < 0.003) but not mature neurons (p > 0.7). G, Bar graphs showing a significant increase in the mEPSC frequency in immature neurons (p < 0.03) but not mature neurons (p > 0.4) after repetitive stimulation. Error bars indicate SE. *p < 0.05.
FM 1-43 imaging and analysis.
Images were acquired using an inverted Nikon (Tokyo, Japan) TE 2000-S microscope equipped with a Hamamatsu (Hamamatsu City, Japan) ORCA 100 cooled CCD camera. FM 1-43 dye was loaded into nerve terminals by immersion in 90 mm KCl solution plus FM dye (10 μm) for 2 min and then washing in normal bath for 6 min before taking the first FM staining image (for protocol, see Fig. 2A). The FM signal was then destained by exposure to two sequential dye-free 90 mm KCl pulses and the second destained fluorescence image was taken. The subtracted image (FM1-stain − FM2-destain) represents the activity-dependent functional synaptic vesicle turnover in control nerve terminals. After a total of six repetitive 90 K+ stimuli, which also served to stain and destain FM 1-43 dye, neurons were returned to the incubator in their original culture medium at 37°C for 2 h. Then, the same neurons imaged before were relocated and the FM staining (FM3) and destaining (FM4) procedures were repeated as before. The subtracted fluorescence image (FM3-stain − FM4-destain) 2 h after repetitive stimulation measures any long-term change in presynaptic function. The FM signal was quantified using the algorithm of SimplePCI imaging software (Compix, Pittsburgh, PA) and the background noise in the nonsynaptic area was subtracted (Chen et al., 2003). The quantification of FM-labeled boutons included three steps. The first step was to apply enhancement to the images by activating Laplacian and smooth functions. The second step was to identify objects by setting a threshold so that all visually identifiable boutons are assigned as regions of interest, although the number of regions of interest in nonsynaptic area is minimal. The third step was to quantify objects by setting another threshold to further remove the tiny nonspecific dots in the nonsynaptic area (usually 3–5 pixels). We always used the same settings to quantify FM signal in the whole imaging field under the control condition and 2 h after repetitive stimulation, and the ratio of the two values represents long-term presynaptic changes. The number of FM-labeled boutons per imaging field ranged from hundreds, in immature neurons, to thousands, in mature neurons. Our analysis might underestimate the number of boutons in the densely innervated area where two boutons might appear to be merged as one bouton. Therefore, the ratio of increase in immature neurons could be an underestimation because repetitive stimulation increased the density of boutons. In addition to counting the FM-labeled active bouton number, the total integrated FM intensity in the bouton area was also quantified to assess the overall presynaptic functional changes. Because the same imaging field was compared before and after repetitive stimulation or drug treatment, the paired Student’s t test was used for all statistical analysis of FM signal.
Figure 2.

Repetitive stimulation induces long-term enhancement of presynaptic function in immature but not mature neurons. A, Experimental protocol for FM 1-43 imaging and repetitive 90 K+ stimulation. Four fluorescence images were acquired (arrows, 1–4) for subtraction and subsequent imaging analysis. FMcontrol = FM1-stain − FM2-destain; FM2 h ARS = FM3-stain − FM4-destain. B, C, Phase images of the same immature neurons before (B) and 2 h after repetitive stimulation (C). Scale bar: (in B) B–E, K–N, 25 μm. D, E, Subtracted FM 1-43 images of the same neurons (corresponding to B and C) before (D) and 2 h after (E) repeated stimulation. F–H, Enlarged FM images from control (F, enlarged from D) and after stimulation (G, enlarged from E), as well as their merged picture (H). Scale bar: (in F) F–H, O–Q, 2.5 μm. I–J, Quantitative analysis showing a significant increase in the integrated FM intensity (I; n = 11; p < 0.01, paired Student’s t test) and FM-labeled active bouton number (J; n = 11; ∗∗p < 0.01) after repetitive stimulation in immature neurons. The FM intensity and active bouton number after repetitive stimulation were normalized to the control value unless otherwise stated. K, L, Phase images of the same mature neuron before (K) and 2 h after (L) repeated stimulation. M, N, Subtracted FM 1-43 images of the mature neuron before (M) and 2 h after (N) repeated stimulation. O–Q, Enlarged FM images from control (O, enlarged from M), after repetitive stimulation (P, enlarged from N), and merged picture (Q). R, S, Quantification of changes in the integrated FM intensity (R; n = 11; p > 0.1, paired t test) and the active bouton number (S; n = 11; p > 0.1) after repetitive stimulation in mature neurons. Error bars indicate SE.
Immunocytochemistry.
Presynaptic nerve terminals were identified with mouse monoclonal antibodies specific for synaptophysin (1:200; Chemicon, Temecula, CA) and SV2 (1:2000; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA), and glutamatergic postsynaptic puncta were identified using rabbit polyclonal antibody specific for PSD-95 (1:100; Zymed, South San Francisco, CA). Immunostaining was performed after live FM 1-43 imaging, with or without repetitive stimulation. Neurons were rinsed in PBS for three times and fixed for 12 min in 4% paraformaldehyde, pH 7.4. Coverslips were then rinsed three times in PBS and primary antibodies were added together with 0.15% saponin blocking solution, incubating overnight at 4°C. Then, coverslips were rinsed three times in PBS with 0.15% saposin for 15 min. Subsequently, samples were incubated for 45 min in anti-mouse Cy3 conjugated or anti-rabbit Alexa 488-conjugated secondary antibodies. For phalloidin staining, we incubate neurons in Alexa 488-conjugated phalloidin (1:3000; Invitrogen) for 45 min. Coverslips were then rinsed six times in PBS with 0.15% saponin for 15 min, and then mounted with mounting solution (50% glycerol, 50% 0.1 m NaHCO3, pH 7.4). Fluorescence signal was visualized on a Zeiss (Oberkochen, Germany) Axioplan 2 microscope. Fluorescence images were acquired by OpenLab software and analyzed with SimplePCI software, similar to that described above for FM 1-43 imaging. Phalloidin intensity was quantified along tau1-labeled axons (∼100 μm per neuron, >1,000 μm in total length), and background noise in the neighboring area was subtracted (Chen et al., 2003).
Drugs.
CNQX, AP5, bicuculline, and nocodazole were purchased from Tocris (Ellisville, MO). Nimodipine, TTX, H89, and GF109203x, KT5720, and calphostin C were purchased from Sigma (St. Louis, MO). Latrunculin A, cytochalasin B, and jasplakinolide were purchased from Invitrogen. All of the drugs were freshly diluted in experimental solutions or culture medium to final concentrations before experiments.
Results
Repetitive-spaced stimulation induces long-term synaptic plasticity in immature but not mature hippocampal neurons
We applied repetitive-spaced stimulation to induce long-term synaptic plasticity in hippocampal cultures with 90 mm KCl solution (2 min, repeated six times), which depolarizes membrane potential to ∼0 mV and induces large calcium influx in nerve terminals and soma/dendrites (Wu et al., 2001a). After control recordings and repetitive stimulation, neurons on coverslips were returned to culture incubator for 2 h and then taken out for re-examination of long-term synaptic changes (Fig. 1A). We first analyzed the frequency and amplitude of mEPSCs before and 2 h after repetitive stimulation (ARS) to monitor long-term changes of glutamatergic neurotransmission (Fig. 1). Interestingly, the repetitive stimulation induced long-lasting enhancement of both the frequency and amplitude of mEPSCs in immature but not mature neurons, suggesting a developmental change of synaptic plasticity in hippocampal cultures (Fig. 1). Immature neurons are defined here as 7–11 d in culture, a time period when the rate of synaptogenesis is very fast (Hsia et al., 1998; Cottrell et al., 2000; Friedman et al., 2000; Renger et al., 2001), whereas mature neurons are cultured for 18–22 d. mEPSCs were recorded in the presence of TTX (0.5 μm) and the GABAA receptor blocker BIC (20 μm). In immature neurons, the average amplitude of mEPSCs increased from 13.6 ± 0.8 pA (n = 38) in control to 19.4 ± 2.6 pA (n = 22; p < 0.04, Student’s t test) after repetitive stimulation, whereas the frequency increased from 1.9 ± 0.3 Hz (n = 38) in control to 3.7 ± 0.6 Hz (p < 0.01; n = 22) after stimulation. Analysis of mEPSC kinetics revealed no significant difference in the decay time of mEPSCs before and after repetitive stimulation (control, 6.1 ± 0.3 ms, n = 9; after stimulation, 5.4 ± 0.5 ms, n = 9; p > 0.19), but the rising time decreased significantly after stimulation (control, 0.78 ± 0.08 ms, n = 9; after stimulation, 0.57 ± 0.06 ms, n = 9; p < 0.04). These results suggest that both presynaptic glutamate release and postsynaptic glutamate receptor responses in immature neurons are significantly enhanced after repetitive stimulation. In mature neurons, however, the same repetitive stimulation did not induce significant changes in either the mEPSC frequency (control, 3.9 ± 0.9 Hz, n = 15; after stimulation, 3.1 ± 0.5 Hz, n = 19; p > 0.4) or the amplitude (control, 16.1 ± 1.7 pA, n = 15; after stimulation, 15.3 ± 1.2 pA, n = 19; p > 0.7) (Fig. 1F,G). Thus, repetitive stimulation induces a long-term synaptic plasticity in hippocampal cultures that is significantly regulated during neuronal development.
Repetitive stimulation increases presynaptic functional boutons in immature neurons
We used live FM 1-43 imaging to investigate the underlying presynaptic mechanism of the long-term synaptic plasticity induced by repetitive stimulation (Betz and Bewick, 1992; Ryan et al., 1993; Ma et al., 1999). We used microisland cultures on dotted coverslips to examine activity-dependent FM puncta in the same imaging field before and 2 h after repetitive stimulation. Unlike electrophysiology recordings, which are performed on different neurons before and after stimulation, live FM imaging is noninvasive and allows imaging of the same neurons to accurately measure presynaptic changes after stimulation. The paired Student’s t test was used for imaging statistical analysis. The imaging protocol is illustrated in Figure 2A. The first 90 K+ stimulation solution contained fluorescent dye FM 1-43 (10 μm) to label active presynaptic boutons, which uptake FM dye through vesicle endocytosis. After 2 min staining and 6 min washing with bath solution, a fluorescent image was taken (image 1 for staining). Then, five repeated dye-free 90 K+ stimulations were used to destain the FM signal. A fluorescent image after two dye-free 90 K+ stimulations was taken as the destained image (image 2 for destaining). The subtracted FM image (FM1 − FM2) represents the efficiency of synaptic vesicle cycling in control condition. After a total of six repetitive 90 K+ stimuli, neurons were returned to culture incubator for 2 h and the previous imaged field was located back for the second round of staining-destaining procedure (Fig. 2A). Figure 2, B and C, illustrates an imaging field of immature neurons (phase images) before and 2 h after repetitive stimulation. No apparent change in the overall neuronal morphology was observed after stimulation as judged by eyes. However, the subtracted FM images showed significantly enhanced FM signal after repetitive stimulation (Fig. 2D–H). The FM-labeled puncta greatly increased from 42 per 100 μm dendrite in control (Fig. 2D) to 76 after repetitive stimulation (Fig. 2E). Group analysis revealed a 3.6-fold increase in the total integrated FM intensity (n = 11; p < 0.01) and a 2.6-fold increase in the active bouton number (p < 0.01) after repetitive stimulation (Fig. 2I,J). The quantification is based on the total FM-labeled bouton number in the whole imaging field, which increased from an average of 165 ± 27 (n = 11) active boutons per imaging field in control to 433 ± 87 boutons after repetitive stimulation. For clear illustration of the increase after stimulation, both integrated FM intensity and active bouton number were normalized to the control level before stimulation.
In contrast to immature neurons, the same repetitive stimulation protocol did not change the presynaptic function of mature neurons (Fig. 2K–Q). The morphology of mature neurons was also similar between control and 2 h after repetitive stimulation (Fig. 2K,L). The FM signal was not substantially changed after repetitive stimulation in mature neurons, with a bouton density at 149 per 100 μm dendrite in the control condition (Fig. 2M) and 168 after repetitive stimulation (Fig. 2N). Group analysis (paired t test) showed that neither the total FM intensity (n = 11; p > 0.1) nor the active bouton number (p > 0.1) was significantly altered after repetitive stimulation (Fig. 2R,S). The total number of active boutons per imaging field in mature neurons only changed slightly from 1649 ± 582 (n = 11) before repetitive stimulation to 2303 ± 536 after repetitive stimulation. Note that there is a 10-fold increase in the total number of functional boutons per imaging field in mature neurons compared with immature neurons in resting condition. Thus, in accordance with the electrophysiological analysis, the FM imaging experiments indicate that repetitive stimulation induces presynaptic long-term enhancement in immature but not mature neurons, supporting the notion that long-term synaptic plasticity is developmentally regulated.
To test whether multiple repetitive stimuli are necessary to induce long-term presynaptic enhancement in immature neurons, we examined the effect of single-spaced stimulation (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). No significant changes in the integrated FM intensity (p > 0.59; n = 6) or active bouton number (p > 0.25; n = 6) were found 2 h after single-spaced stimulation. Thus, multiple spaced stimuli are required for long-term synaptic plasticity.
Immature synapses are presynaptically silent but become active after repetitive stimulation
We next examined whether the increase of active boutons comes from activation of previously existing synapses or from the formation of new synapses. To distinguish these two possibilities, live FM imaging, which identifies activity-dependent functional boutons, was followed with retrospective immunostaining of synaptophysin, which identifies all presynaptic boutons including both active and silent ones (Fig. 3). For immature neurons without repetitive stimulation, many presynaptic boutons labeled with synaptophysin lacked FM 1-43 staining, suggesting that these synaptophysin-labeled boutons are physically existing but functionally silent (Fig. 3A–C). After repetitive stimulation, most of the synaptophysin-labeled boutons were stained with FM 1-43, suggesting that some previously silent boutons are now converted into active ones (Fig. 3D–G). Quantitative analysis revealed that in the control condition, the ratio of FM-labeled functional boutons over the total synaptophysin-positive boutons is 0.59 ± 0.04 (n = 4). After repetitive stimulation, the ratio increased to 0.92 ± 0.03 (n = 4; p < 0.001, Student’s t test), indicating a significant increase of functional boutons. In contrast, mature neurons showed highly correlated staining of synaptophysin and FM 1-43 puncta regardless of repetitive stimulation, with the ratio of FM-labeled boutons over total synaptophysin-labeled boutons being 0.85 ± 0.02 (n = 4) in control, and 0.90 ± 0.04 (n = 4; p > 0.3) after repetitive stimulation (Fig. 3H–N). These results suggest that the majority of presynaptic boutons of mature neurons are already active before stimulation.
Figure 3.

Retrospective immunocytochemistry reveals presynaptic silent boutons in immature but not mature neurons. A, Activity-labeled FM 1-43 images in control immature neurons. B, Retrospective immunostaining of synaptophysin at the same field shown in A. C, Overlaid image of A and B showing that many synaptophysin-labeled boutons are not stained with FM 1-43. D, E, FM 1-43 images before (D) and 2 h after repetitive stimulation (E) in immature neurons. F, Retrospective immunostaining of synaptophysin at the same field shown in D and E. G, Overlaid image of E and F showing most of the synaptophysin-labeled boutons are now stained with FM 1-43 after repetitive stimulation. H, FM 1-43-labeled presynaptic boutons in control mature neurons. I, Synaptophysin-labeled presynaptic boutons. J, Overlaid image of H and I showing well correlated synaptophysin and FM puncta in mature neurons under the control condition. K, L, Presynaptic boutons labeled by FM 1-43 before (K) and 2 h after repetitive stimulation (L) in mature neurons. M, Synaptophysin-stained presynaptic boutons. N, Overlaid image of L and M. Scale bars, 15 μm.
We next examined changes of the presynaptic and postsynaptic components simultaneously in immature neurons. Again, FM imaging was used to label functional presynaptic boutons, and retrospective double immunostaining of presynaptic marker SV2 and postsynaptic marker PSD-95 were used to label the total number of presynaptic and postsynaptic puncta (Fig. 4). Under the control condition, FM-labeled puncta were only a small fraction of SV2-labeled puncta, confirming that many SV2-labeled presynaptic boutons are functionally silent (Fig. 4A,C,E). Most of the FM-labeled puncta also overlaid with PSD-95 puncta, suggesting that these puncta represent functional synapses (Fig. 4A,B,D). After repetitive stimulation, the number of FM-labeled active boutons significantly increased and the majority of them correlate very well with PSD-95 and SV2 puncta (Fig. 4F–J). To quantify the relative changes of presynaptic versus postsynaptic puncta after repetitive stimulation, the puncta numbers of FM 1-43 and PSD-95 were normalized to that of SV2. The number of FM 1-43 puncta increased significantly from 40% in control (n = 12) to 90% after repetitive stimulation (n = 9) (p < 0.001), whereas the number of PSD-95 puncta only increased slightly from 80 to 90% without statistical significance (p > 0.2) (Fig. 4K). Thus, repetitive stimulation converts presynaptic silent synapses into functional ones in immature neurons, which may contribute significantly to long-term synaptic plasticity.
Figure 4.

Comparison of repetitive stimulation-induced changes of presynaptic versus postsynaptic puncta in immature neurons. A–C, Immature neurons under the control condition showing FM 1-43-labeled functional presynaptic boutons (A), PSD-95-labeled postsynaptic puncta (B), and SV2-labeled presynaptic puncta (C) in the same field. D, E, Overlaid images for FM 1-43 labeling together with PSD-95 staining (D), or with SV2 staining (E). F–H, Immature neurons after repetitive stimulation, illustrating FM 1-43-labeled functional presynaptic boutons (F), PSD-95-labeled postsynaptic puncta (G), and SV2-labeled presynaptic puncta (H). I, J, Overlaid images for FM 1-43 labeling with PSD-95 staining (I), or with SV2 staining (J). Scale bar, 10 μm. K, Quantification of changes of presynaptic versus postsynaptic puncta induced by repetitive stimulation. Data are normalized to the SV2-labeled puncta number. The FM 1-43-labeled active bouton number significantly increased from 40% in control (n = 12) to 90% after repetitive stimulation (n = 9; ***p < 0.001). Error bars indicate SE.
Activation of presynaptic silent synapses depends on L-type Ca2+ channels and PKA/PKC signaling pathways
The L-type voltage-sensitive Ca2+ channel is a major type of Ca2+ entry pathway implicated in long-term synaptic plasticity and stabilization of dendritic spines (Bading et al., 1993; Wu et al., 2001a). The strong membrane depolarization during 90 K+ stimulation may be sufficient to activate L-type Ca2+ channels and induce large Ca2+ influx to trigger long-term synaptic enhancement. Indeed, we found that blocking L-type Ca2+ channels essentially abolished the long-term synaptic plasticity induced by repetitive stimulation in immature neurons (Fig. 5). When L-type Ca2+ channel blocker nimodipine (10 μm) was present during repetitive 90 K+ stimulation, neither the mEPSC amplitude (18.4 ± 2.0 pA in control, n = 12; 19.3 ± 2.0 pA after stimulation, n = 14; p > 0.8) nor the frequency (1.95 ± 0.72 Hz in control, n = 12; 1.97 ± 0.38 Hz after stimulation, n = 14; p > 0.7) increased after repetitive stimulation (Fig. 5A,B), underscoring a critical role of L-type Ca2+ channels in long-term synaptic plasticity.
Figure 5.
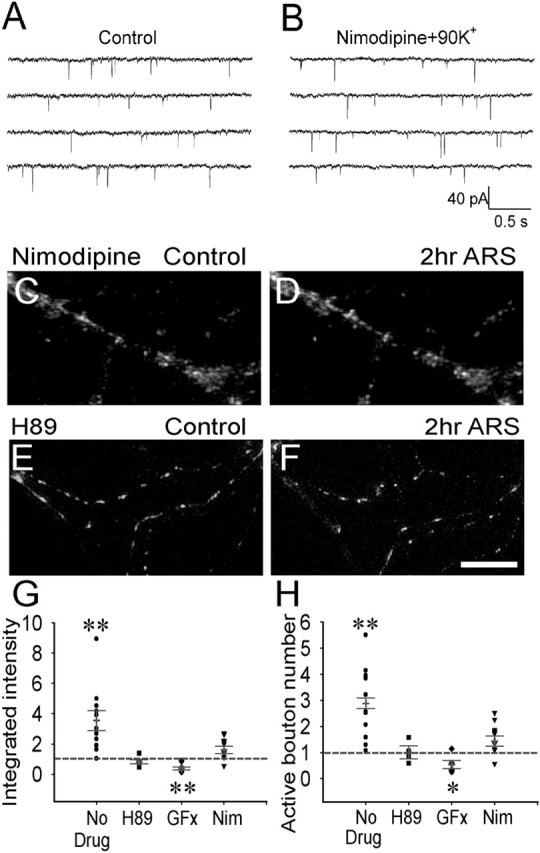
Dependence on L-type Ca2+ channels and PKA/PKC signaling pathways of the presynaptic enhancement. A, B, Typical mEPSC recordings in immature neurons under control and after repetitive stimulation in the presence of nimodipine (10 μm). Both the amplitude and frequency of mEPSCs did not show any significant change after nimodipine treatment (n = 12–14; p > 0.7). C, D, Subtracted FM 1-43 images of immature neurons before (C) and 2 h after repetitive stimulation in the presence of nimodipine (D). E, F, Subtracted FM 1-43 images of immature neurons before (E) and 2 h after repetitive stimulation (F) in the presence of PKA inhibitor H89 (1 μm). Scale bar: (in F) C–F, 15 μm. G, H, Quantitative analysis showing the effect of nimodipine, H89, and PKC inhibitor GF109203x (5 μm) on changes of presynaptic functional boutons after repetitive stimulation. Both nimodipine and H89 abolished the increase of the integrated FM intensity (G) and the active bouton number (H) after repetitive stimulation. GF109203x treatment decreased the fluorescence intensity (n = 7; p < 0.01) and the active bouton number (n = 7; p < 0.03) after repetitive stimulation, suggesting that PKC is important in maintaining normal synaptic functions. Error bars indicate SE. *p < 0.05; **p < 0.01.
To further investigate the function of L-type Ca2+ channels in activity-dependent presynaptic plasticity, we examined FM-labeled puncta changes before and after repetitive stimulation in the presence of nimodipine in immature neurons (Fig. 5C,D). With nimodipine treatment, repetitive stimulation no longer induces any significant increase of FM intensity (n = 11; p > 0.34) (Fig. 5G), nor the active bouton number (n = 11; p > 0.10) (Fig. 5H). Thus, L-type Ca2+ channels may play an important role in the activation of presynaptic silent synapses after repetitive stimulation.
Ca2+ influx activates downstream effectors such as protein kinases, which have been demonstrated to be important players in long-term synaptic plasticity (Malinow et al., 1988; Abel et al., 1997; Malenka and Nicoll, 1999; Xia and Storm, 2005). Here, we examined the effects of two important protein kinases, PKA and PKC, during activity-dependent presynaptic plasticity in immature neurons (Fig. 5E–H). In the presence of PKA inhibitor H89 (1 μm), the FM 1-43 staining pattern did not show a significant change after repetitive stimulation (Fig. 5E,F). Quantitative analysis revealed that after H89 treatment, the fluorescence intensity and active bouton number both had little change (n = 8; p > 0.14 for fluorescence intensity, and p > 0.48 for bouton number) after repetitive stimulation (Fig. 5G,H). In contrast, when neurons were treated with PKC inhibitor GF109203x (5 μm), both the fluorescence intensity (n = 7; p < 0.01) and active bouton number (n = 7; p < 0.03) of immature neurons decreased significantly after repetitive stimulation (Fig. 5G,H). This is confirmed with a second PKC inhibitor calphostin C (100 nm), which reduced the active bouton number to about half (the ratio after stimulation/control = 0.51 ± 0.03; n = 8; p < 0.001). We further examined the treatment of PKC inhibitors on functional boutons under normal conditions without repetitive stimulation. The number of functional boutons was significantly decreased by both GF109203x (0.48 ± 0.03; n = 15; p < 0.001) and calphostin C treatment (0.55 ± 0.07; n = 12; p < 0.001) under control conditions. Thus, both PKA and PKC are required for the activation of presynaptic silent synapses after repetitive stimulation in immature neurons. PKC may also be critical in maintaining normal presynaptic functions under resting conditions.
We next analyzed whether activation of glutamate receptors is required for activation of presynaptic silent synapses induced by repetitive 90 K+ stimulation in immature neurons (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Antagonists of AMPA (CNQX, 10 μm) and NMDA receptors (AP5, 50 μm) were applied during repetitive stimulation. We first analyzed mEPSCs before and after repetitive stimulation in the presence of AP5 and CNQX (supplemental Fig. 2A,B, available at www.jneurosci.org as supplemental material). Blocking glutamate receptors abolished the increase of the amplitude of mEPSCs induced by repetitive stimulation (control, 14.3 ± 1.3 pA, n = 18; stimulation with CNQX/AP5, 19.0 ± 4.1 pA, n = 24; p > 0.2), consistent with the important role of glutamate receptors in postsynaptic long-term plasticity. However, the frequency of mEPSCs was still significantly increased after repetitive stimulation in the presence of CNQX/AP5 (control, 1.8 ± 0.3 Hz, n = 18; stimulation with CNQX/AP5, 4.9 ± 0.9 Hz, n = 24; p < 0.004). To further examine the effect of glutamate receptors on presynaptic plasticity in immature neurons, FM imaging was conducted to analyze changes of presynaptic functional boutons before and after repetitive stimulation in the presence of CNQX/AP5. In agreement with electrophysiological experiments, repetitive stimulation induced a significant increase in both the integrated FM intensity (p < 0.01; n = 13) and active bouton number (p < 0.02; n = 13) (supplemental Fig. 2C–F, available at www.jneurosci.org as supplemental material). These studies suggest that activation of presynaptic silent synapses is dependent on L-type Ca2+ channels and PKA/PKC signaling pathways, whereas postsynaptic enhancement is dependent on glutamate receptor activation.
Actin plays a critical role in activating presynaptic silent synapses
Actin is thought to play an important role in synaptic remodeling (Matus, 2000; Colicos et al., 2001; Star et al., 2002; Sankaranarayanan et al., 2003; Wang et al., 2005). Here, we found that actin polymerization is critical in converting presynaptic silent synapses into functional ones induced by repetitive stimulation in immature neurons (Fig. 6). We first recorded mEPSCs in immature neurons after repetitive stimulation in the absence or presence of latrunculin A (5 μm), a strong actin depolymerizer widely used to study actin function in synaptic vesicle cycling (Morales et al., 2000; Sankaranarayanan et al., 2003; Richards et al., 2004). The frequency of mEPSCs was significantly reduced after latrunculin A treatment (90 K+ stimulation, 5.11 ± 1.01 Hz, n = 14; 90 K+ plus latrunculin A, 1.87 ± 0.37 Hz, n = 17; p < 0.01), but the amplitude was not greatly affected (21.8 ± 3.2 pA without latrunculin A, n = 14; 16.6 ± 2.1 pA with latrunculin A, n = 17; p > 0.1) (Fig. 6A,B). We next tested whether latrunculin A and another weaker actin depolymerizer cytochalasin B (4 μm) have any effect on mEPSCs of immature neurons under basal conditions without 90 K+ stimulation. Latrunculin A (5 μm) significantly affected basal mEPSCs, whereas cytochalasin B had no significant effect (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). The average amplitude of mEPSCs was 24.3 ± 1.9 pA (n = 15) in the control condition, dropped to 18.7 ± 3.4 pA (n = 15; p > 0.1) after cytochalasin B treatment, and significantly decreased to 15.5 ± 2.6 pA (n = 16; p < 0.02) after latrunculin A treatment. The mEPSC frequency was 0.62 ± 0.16 Hz (n = 15) in control, 0.35 ± 0.07 Hz (n = 15; p > 0.1) after cytochalasin B treatment, and 0.12 ± 0.02 Hz (n = 16; p < 0.001) after latrunculin A treatment. The strong effect of latrunculin A on basal mEPSCs of immature neurons suggests that actin polymerization is required for functional integrity of immature synapses (Zhang and Benson, 2001). However, the mild effect of cytochalasin B on basal release makes it better suited for studying actin function in synaptic plasticity.
Figure 6.
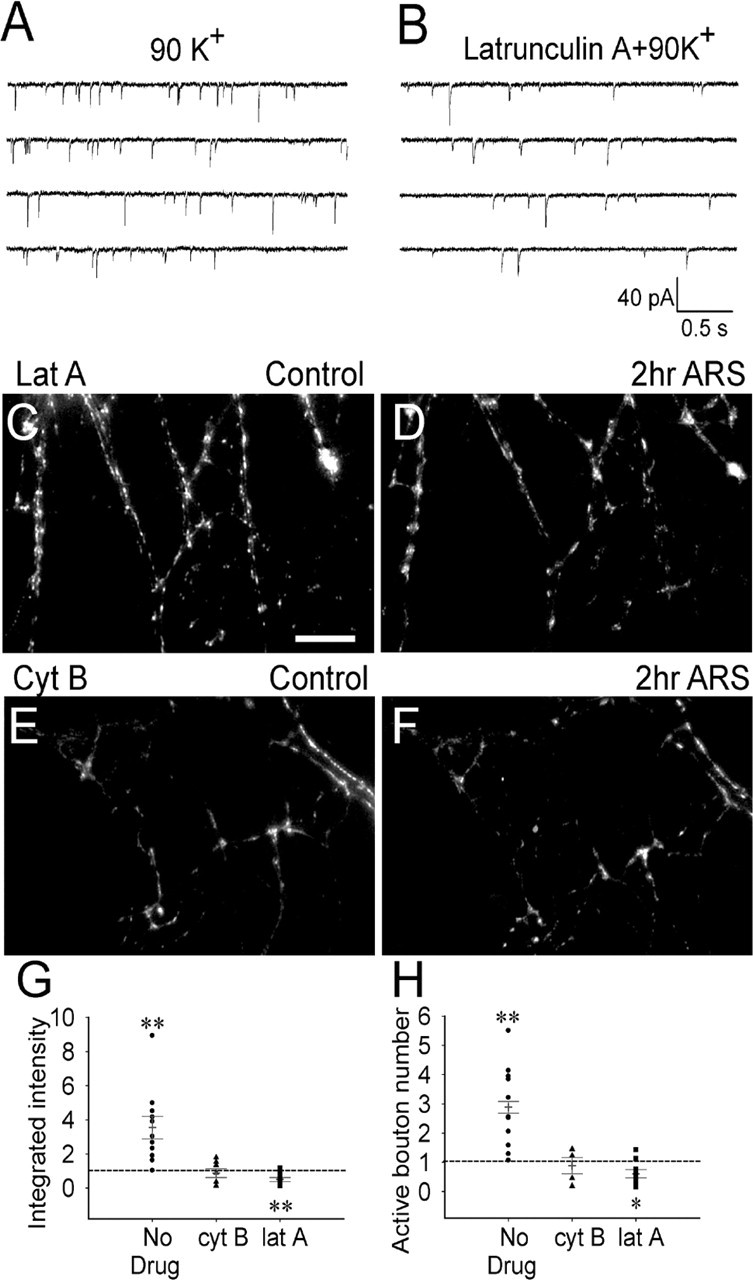
Inhibition of actin polymerization abolishes presynaptic long-term enhancement in immature neurons. A, B, mEPSCs of immature neurons recorded after repetitive 90 K+ stimulation in the absence (A) or presence of latrunculin A (5 μm) (B). Latrunculin A treatment did not affect the amplitude of mEPSCs (n = 14–17; p > 0.1) but reduced the frequency significantly (p < 0.01). C, D, Subtracted FM 1-43 images of immature neurons before (C) and 2 h after (D) repeated stimulation with latrunculin A treatment. E, F, Subtracted FM 1-43 images of immature neurons before (E) and 2 h after (F) repeated stimulation with cytochalasin B (4 μm) treatment. Scale bar: (in C) C–F, 20 μm. G, H, Quantification of changes in the integrated fluorescence intensity (G) and active bouton number (H) after blocking actin polymerization. Cytochalasin B treatment abolished repetitive stimulation-induced increase of the integrated FM intensity (p > 0.13; n = 8) and the active bouton number (p > 0.16; n = 8), whereas latrunculin A significantly decreased the integrated FM intensity (**p < 0.01; n = 10) and the active bouton number (p < 0.03; n = 10). Error bars indicate SE. *p < 0.05.
To investigate the function of actin in presynaptic plasticity, FM imaging was used to assay presynaptic changes after repetitive stimulation with or without inhibition of actin polymerization (Fig. 6C–H). Consistent with the electrophysiological data, latrunculin A (5 μm) not only abolished the increase of FM staining induced by repetitive stimulation, but also induced a significant reduction in both the integrated FM intensity (p < 0.01; n = 10) and active bouton number (p < 0.03; n = 10), further supporting the notion that F-actin is critical in the maintenance of young synapses (Zhang and Benson, 2001). In contrast, cytochalasin B (4 μm) only abolished the increase of integrated FM intensity (p > 0.13; n = 8) and active bouton number (p > 0.16; n = 8) induced by repetitive stimulation without causing any additional decrease compared with the control (Fig. 6E–H). Similar to the effect after repetitive stimulation, latrunculin A treatment also reduced the functional bouton number after single stimulation (the ratio after treatment/control = 0.44 ± 0.06; n = 10; p < 0.003, paired t test), but cytochalasin B did not show a significant effect (the ratio after treatment/control = 1.01 ± 0.13; n = 8; p > 0.9).
Actin but not microtubule is critical for presynaptic long-term plasticity
To test whether actin polymerization is sufficient to induce presynaptic long-term plasticity, we treated immature neurons with the actin polymerizer jasplakinolide (100 nm, 30 min) and examined presynaptic changes after single-spaced stimulation (Fig. 7A–D). As shown in supplementary Figure 1, single-spaced stimulation alone did not induce long-term changes of FM staining. However, the same single-spaced stimulation paradigm after pretreatment with jasplakinolide resulted in long-term increase of the integrated FM intensity (p < 0.01; n = 13) and active bouton numbers (p < 0.01; n = 13), suggesting that actin polymerization alone may be sufficient in enhancing presynaptic function (Fig. 7A–D). In support of this conclusion, we found that jasplakinolide treatment alone without any stimulation significantly increased the mEPSC frequency (control, 0.26 ± 0.02 Hz, n = 14; after jasplakinolide, 0.36 ± 0.04 Hz, n = 13; p < 0.05) but not the amplitude (control, 12.0 ± 0.7 pA, n = 14; after jasplakinolide, 11.3 ± 1.0 pA, n = 13; p > 0.5). Consistent with our finding, it has been demonstrated that jasplakinolide treatment alone can recruit actin to synaptic terminals and occlude further activity-dependent recruitment (Sankaranarayanan et al., 2003).
Figure 7.
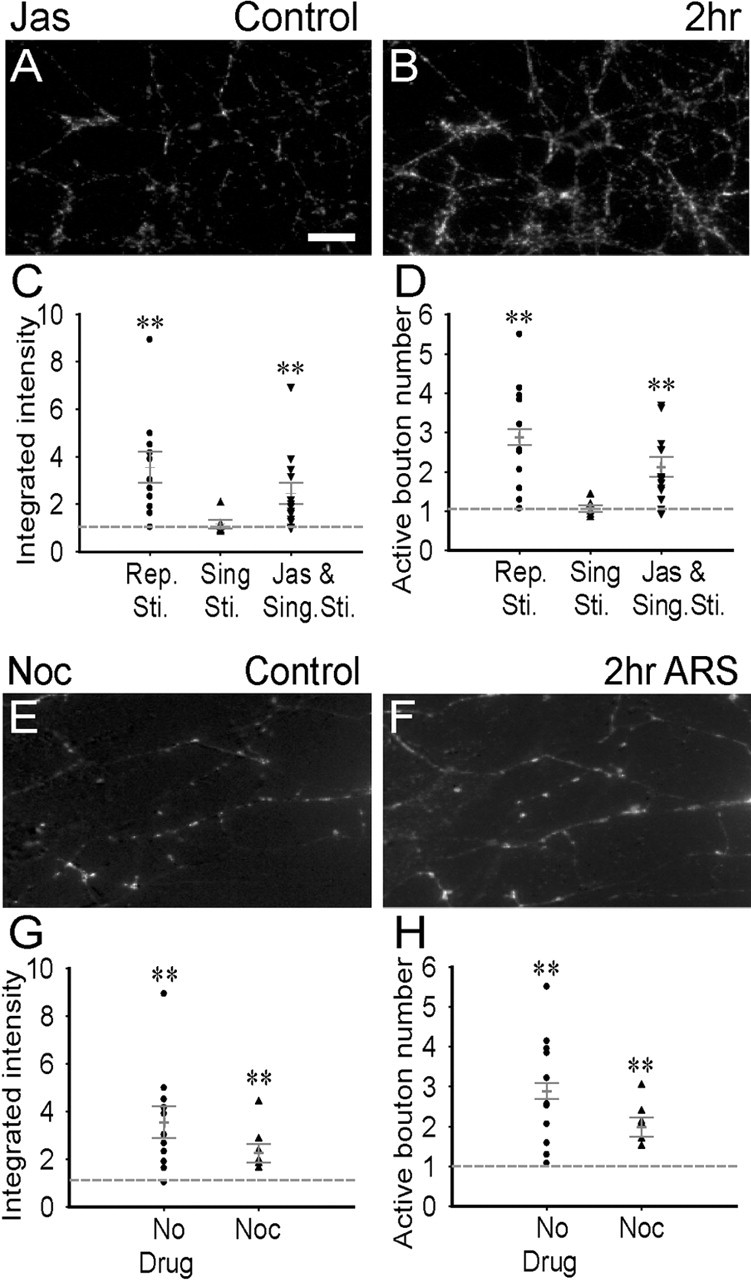
Actin but not microtubule polymerization is critical to presynaptic long-term enhancement in immature neurons. A, B, Subtracted FM 1-43 images of immature neurons pretreated with actin polymerizer jasplakinolide (100 nm, 30 min) before (A) and 2 h after (B) single-spaced stimulation. C, D, Quantitative analysis showing a significant increase in the integrated FM intensity (C; p < 0.01; n = 13) and active bouton number (D; p < 0.01; n = 13) after jasplakinolide treatment. E, F, Subtracted FM 1-43 images of immature neurons pretreated with microtubule depolymerizer nocodazole (10 μm, 30 min) before (E) and 2 h after (F) repetitive stimulation. G, H, Quantitative analysis showing that after nocodazole treatment, repetitive stimulation still increased the integrated FM intensity (G; **p < 0.01; n = 8) and the active bouton number (H; p < 0.02; n = 8) in immature neurons. Scale bar: (in A) A, B, E, F, 20 μm. Error bars indicate SE.
Microtubule is another important cytoskeleton element and also reported to play a role in synaptic remodeling in certain types of synapses (Langford, 1995; Ruiz-Canada et al., 2004). To test the function of microtubules in presynaptic plasticity, immature neurons were treated with nocodazole (10 μm) before and during repetitive stimulation to depolymerize microtubules, as used in previous studies (Yabe et al., 1999; Gibney and Zheng, 2003) (Fig. 7E–H). In contrast to actin depolymerizer cytochalasin B or latrunculin A, nocodazole did not block the increase of the integrated FM intensity (p < 0.01; n = 8) nor the active bouton number (p < 0.02; n = 8) induced by repetitive stimulation. Thus, it is the polymerization of actin but not microtubule that plays a critical role in long-term presynaptic enhancement.
Repetitive stimulation increases actin polymerization in immature but not mature axons
To understand why our repetitive stimulation paradigm induced long-term synaptic plasticity in immature but not mature neurons, we examined actin polymerization induced by single versus multiple 90 K+ stimulation in both immature and mature neurons (Fig. 8). The degree of actin polymerization was quantified by immunostaining of fluorescently labeled phalloidin, which binds selectively to F-actin and widely used as an index for actin polymerization. Coimmunostaining with Tau1 antibody was used to label axons. In immature neurons, phalloidin intensity was weak after single stimulation, but increased by twofold after repetitive stimulation (single stimulation, 817 ± 138 arbitrary unit; after repetitive stimulation,1648 ± 83; n = 5; p < 0.002) (Fig. 8A–D). In contrast, in mature neurons, phalloidin intensity was already strong with single stimulation, and not further increased after repetitive stimulation (single stimulation, 2016 ± 139; after repetitive stimulation, 2148 ± 83; n = 5; p > 0.44) (Fig. 8E–H). Together with the experiments using actin polymerizer and depolymerizer described above, our data suggest that actin dynamics undergo a significant change during neuronal maturation, which in turn regulates long-term synaptic plasticity during neuronal development.
Figure 8.

Repetitive stimulation increases actin polymerization in immature but not mature neurons. A, B, Immature neurons coimmunostained with axon marker tau1 (A) and F-actin marker phalloidin (B) after single 90 K+ stimulation. C, D, Coimmunostaining of tau1 (C) and phalloidin (D) in immature neurons after repetitive stimulation. Repetitive stimulation induced a significant increase of phalloidin intensity in tau1-labeled axons of immature neurons (p < 0.001; n = 10). E, F, Mature neurons stained with tau1 (E) and phalloidin (F) after single stimulation. G, H, Mature neurons stained with tau1 (G) and phalloidin (H) after repetitive stimulation. Scale bar: (in A) A–H, 5 μm. No significant change in phalloidin intensity was found in mature axons after repetitive stimulation (p > 0.44; n = 10). I, Simplified model illustrating an important role of actin polymerization in the activation of presynaptic silent boutons induced by repetitive stimulation.
Discussion
The major finding of this work is that actin-dependent activation of presynaptic silent synapses significantly contributes to long-term synaptic plasticity in developing hippocampal neurons. A combination of FM imaging and retrospective immunostaining experiments indicate that many physically existing presynaptic boutons in immature neurons are functionally silent at resting conditions. Repetitive neuronal stimulation substantially increases actin polymerization and simultaneously activates presynaptic silent boutons in immature neurons. In mature neurons, however, the F-actin level under the control condition is already high and the repetitive stimulation does not change the F-actin level nor enhance synaptic transmission. These data suggest a critical role of actin in regulating synaptic plasticity during neuronal development.
Actin-dependent activation of presynaptic silent boutons
It has been demonstrated that activation of postsynaptic silent synapses contributes to long-term synaptic plasticity (Isaac et al., 1995; Liao et al., 1995; Durand et al., 1996; Wu et al., 1996), but much less is known about the activation of presynaptic silent synapses. A previous study showed that bath application of cAMP in hippocampal cultures increased the number of presynaptic functional boutons, suggesting the importance of the PKA signaling pathway in the activation of presynaptic silent synapses (Ma et al., 1999). We investigated presynaptic silent synapses by combining live FM 1-43 imaging, which labels only functional presynaptic boutons, together with synaptophysin/SV2 immunostaining to label all pre-existing synaptic terminals. Taking advantage of microisland cultures on marked coverslips, we examined the same imaging field under three consecutive conditions, which are control, 2 h after repetitive stimulation, and followed by retrospective immunostaining (Figs. 3, 4). We found that in immature neurons, only a fraction of synaptophysin- or SV2-labeled boutons are capable of activity-dependent FM uptake in the control condition, suggesting that the majority of existing boutons are functionally silent. However, after repetitive stimulation, most of the synaptophysin- or SV2-labeled boutons are now capable of FM staining, indicating a functional conversion. We do not exclude the possibility that some functional boutons that appeared after repetitive stimulation are de novo formation of new presynaptic puncta. Some of the FM-labeled puncta may be mobile vesicle clusters, which will pause and form synapses later (Ahmari et al., 2000; Friedman et al., 2000; Krueger et al., 2003). It is possible that postsynaptic actin polymerization may play a role in activating presynaptic silent synapses as well through retrograde signaling (Wang et al., 2005). We should also point out that the absolute number of active boutons depends on the threshold setting of the imaging analysis. Therefore, some boutons with a very low rate of vesicle turnover may not be detected. We have optimized our automatic detection system so that the software-detected boutons are the best match with eye-detected boutons (see Materials and Methods). In addition, to offset for the threshold detection, we also quantified the total FM intensity in the bouton area. Similar to the bouton number change, the total FM intensity also increased substantially after repetitive stimulation in immature neurons.
Consistent with previous findings (Ma et al., 1999), we found that PKA and PKC signaling pathways, together with L-type Ca2+ channels, are required for the activation of presynaptic silent synapses. Furthermore, we have obtained several lines of evidence supporting the role of actin polymerization in the activation of presynaptic silent synapses: first, inhibition of actin polymerization with latrunculin A significantly decreased the mEPSC frequency after repetitive stimulation. Second, cytochalasin B and latrunculin A abolished the increase of active boutons after repetitive stimulation. Third, promoting actin polymerization by jasplakinolide increased the active bouton number. Fourth, repetitive stimulation induced a twofold increase of F-actin in immature axons. According to these data, we propose a simplified model to depict the actin-dependent activation of presynaptic silent boutons, as illustrated in Figure 8I. In immature neurons under the control condition, many presynaptic boutons are functionally silent because of a low level of polymerized F-actin. After repetitive stimulation, the level of F-actin is significantly increased (Fig. 8I, right arrow) to allow certain structural rearrangement so that synaptic vesicles are now accessible to the release zone and start exo-endocytosis. In contrast, latrunculin A may cause disassembly of F-actin into G-actin (Fig. 8I, left arrow) and convert active boutons into silent ones. The strong effects of latrunculin A and PKC inhibitors on basal mEPSCs suggest that both F-actin and PKC are required for maintaining normal synaptic functions in developing synapses, and their effects on synaptic plasticity should be cautiously interpreted.
Our finding that actin polymerization is critical in activating presynaptic silent synapses is consistent with recent studies suggesting an active role of actin in synaptic transmission and plasticity (Dillon and Goda, 2005). A prominent characteristic of actin is its activity-dependent dynamics. Electrical stimulation induces presynaptic actin condensation and postsynaptic actin enlargement in dendritic spines (Colicos et al., 2001; Sankaranarayanan et al., 2003; Okamoto et al., 2004). In accordance with these actin dynamic changes, we found that the total F-actin level in axons increased by twofold after repetitive stimulation in immature neurons, supporting the notion that activity shifts the F-actin/G-actin equilibrium toward F-actin (Okamoto et al., 2004). In association with an increase of F-actin level after repetitive stimulation, presynaptic silent boutons are also transformed into functional ones in immature neurons. The strong correlation between actin polymerization and the number of active boutons in immature neurons suggests that actin polymerization may act as a main gating mechanism toward presynaptic plasticity (Antonova et al., 2001; Colicos et al., 2001; Wang et al., 2005). Therefore, actin not only plays an important role in morphological plasticity, but also plays a critical role in functional plasticity in immature neurons.
Presynaptic versus postsynaptic contribution to long-term synaptic plasticity
Postsynaptic modification of glutamate receptors has long been recognized in the expression and maintenance of LTP (Collingridge and Bliss, 1995; Nicoll and Malenka, 1995). Our results suggest that both presynaptic and postsynaptic mechanisms are involved in long-term synaptic plasticity. The presynaptic enhancement is reflected by a long-term increase of mEPSC frequency and active bouton number after repetitive stimulation, in accordance with previous studies (Malgaroli et al., 1995; Ryan et al., 1996; Ma et al., 1999; Zakharenko et al., 2001). Activation of glutamate receptors and L-type Ca2+ channels has been implied in both presynaptic and postsynaptic long-term plasticity (Johnston et al., 1992; Malgaroli and Tsien, 1992; Nicoll and Malenka, 1995; Zakharenko et al., 2001; Niikura et al., 2004). We found that the postsynaptic potentiation of mEPSC amplitude after repetitive 90 K+ stimulation can be blocked by either L-type Ca2+ channel blocker or glutamate receptor antagonists (Fig. 5, supplemental Fig. 2, available at www.jneurosci.org as supplemental material), suggesting multiple signaling pathways involved. However, the presynaptic enhancement of mEPSC frequency and FM staining was only blocked by nimodipine but not AP5/CNQX, suggesting an unconventional presynaptic plasticity. We propose that the repetitive 90 K+ stimulation may induce a large Ca2+ influx into nerve terminals, which directly activates down-stream cell-signaling pathways such as protein phosphorylation and actin polymerization to trigger long-term plasticity (Bito et al., 1996; Wu et al., 2001b).
Actin and developmentally regulated long-term synaptic plasticity
One interesting result of this study is that repetitive 90 K+ stimulation induces long-term synaptic changes in immature but not mature hippocampal neurons. This difference between immature and mature neurons may be caused by activity-dependent presynaptic development. Actin is a major component of the cytoskeleton proteins, and plays a critical role in neurite growth and axon guidance (Dent and Gertler, 2003). Previous work found that actin exerts differential effects in maintaining immature versus mature synaptic structures (Zhang and Benson, 2001). Our studies further demonstrate that actin polymerization is functionally linked to synaptic plasticity during neuronal maturation. Therefore, in immature synapses, actin not only serves as a scaffolding cytoskeletal protein to maintain synaptic structures, but also actively regulates synaptic vesicle cycling. After neuronal maturation, actin may shift to a more scaffolding function to allow other proteins such as synapsin 1a or PSD-95 to regulate presynaptic and postsynaptic functions (Allison et al., 2000; Zhang and Benson, 2001; Schoch et al., 2002; Sankaranarayanan et al., 2003).
Our finding that actin-dependent activation of presynaptic silent synapses is prominent in immature but not mature neurons supports the notion that developing neurons are more plastic than mature neurons (Durand et al., 1996; Wu et al., 1996; Choi et al., 2000; Gasparini et al., 2000; Hanse and Gustafsson, 2001; Renger et al., 2001). Although it is possible that mature neurons in culture are less sensitive to certain type of neuronal stimulation, the lack of increase of active boutons in mature neurons after repetitive stimulation is consistent with previous studies showing no significant change in the total number of presynaptic puncta after LTP induction (Zakharenko et al., 2001; Fukazawa et al., 2003). Thus, long-term plasticity in mature synapses may be expressed mainly by enhancement of presynaptic release efficiency and postsynaptic receptor responses, whereas in immature synapses, activation of presynaptic and postsynaptic silent synapses contributes significantly to long-term synaptic plasticity.
Footnotes
This work was supported by National Science Foundation Grant 0236429 (G.C.). We thank Dr. Richard Ordway and Dr. Matthew Whim for careful reading of this manuscript and insightful comments. Min Jiang provided technical help for cell cultures.
References
- Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R (1997). Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell 88:615–626. [DOI] [PubMed] [Google Scholar]
- Ackermann M, Matus A (2003). Activity-induced targeting of profilin and stabilization of dendritic spine morphology. Nat Neurosci 6:1194–1200. [DOI] [PubMed] [Google Scholar]
- Ahmari SE, Buchanan J, Smith SJ (2000). Assembly of presynaptic active zones from cytoplasmic transport packets. Nat Neurosci 3:445–451. [DOI] [PubMed] [Google Scholar]
- Allison DW, Gelfand VI, Spector I, Craig AM (1998). Role of actin in anchoring postsynaptic receptors in cultured hippocampal neurons: differential attachment of NMDA versus AMPA receptors. J Neurosci 18:2423–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison DW, Chervin AS, Gelfand VI, Craig AM (2000). Postsynaptic scaffolds of excitatory and inhibitory synapses in hippocampal neurons: maintenance of core components independent of actin filaments and microtubules. J Neurosci 20:4545–4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonova I, Arancio O, Trillat AC, Wang HG, Zablow L, Udo H, Kandel ER, Hawkins RD (2001). Rapid increase in clusters of presynaptic proteins at onset of long-lasting potentiation. Science 294:1547–1550. [DOI] [PubMed] [Google Scholar]
- Bading H, Ginty DD, Greenberg ME (1993). Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science 260:181–186. [DOI] [PubMed] [Google Scholar]
- Bailey CH, Kandel ER (1993). Structural changes accompanying memory storage. Annu Rev Physiol 55:397–426. [DOI] [PubMed] [Google Scholar]
- Betz WJ, Bewick GS (1992). Optical analysis of synaptic vesicle recycling at the frog neuromuscular junction. Science 255:200–203. [DOI] [PubMed] [Google Scholar]
- Bito H, Deisseroth K, Tsien RW (1996). CREB phosphorylation and dephosphorylation: a Ca(2+)- and stimulus duration-dependent switch for hippocampal gene expression. Cell 87:1203–1214. [DOI] [PubMed] [Google Scholar]
- Chang S, De Camilli P (2001). Glutamate regulates actin-based motility in axonal filopodia. Nat Neurosci 4:787–793. [DOI] [PubMed] [Google Scholar]
- Chen G, Harata N, Tsien RW (2004). Paired-pulse depression of unitary quantal amplitude at single hippocampal synapses. Proc Natl Acad Sci USA 101:1063–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Deng LB, Maeno-Hikichi Y, Lai MZ, Chang SH, Chen G, Zhang JF (2003). Formation of an endophilin-Ca2+ channel complex is critical for clathrin-mediated synaptic vesicle endocytosis. Cell 115:37–48. [DOI] [PubMed] [Google Scholar]
- Choi S, Klingauf J, Tsien RW (2000). Postfusional regulation of cleft glutamate concentration during LTP at “silent synapses.”. Nat Neurosci 3:330–336. [DOI] [PubMed] [Google Scholar]
- Cole JC, Villa BR, Wilkinson RS (2000). Disruption of actin impedes transmitter release in snake motor terminals. J Physiol (Lond) 525:579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colicos MA, Collins BE, Sailor MJ, Goda Y (2001). Remodeling of synaptic actin induced by photoconductive stimulation. Cell 107:605–616. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Bliss TV (1995). Memories of NMDA receptors and LTP. Trends Neurosci 18:54–56. [PubMed] [Google Scholar]
- Cottrell JR, Dube GR, Egles C, Liu G (2000). Distribution, density, and clustering of functional glutamate receptors before and after synaptogenesis in hippocampal neurons. J Neurophysiol 84:1573–1587. [DOI] [PubMed] [Google Scholar]
- Deng LB, Chen G (2003). Cyclothiazide potently inhibits gamma-aminobutyric acid type A receptors in addition to enhancing glutamate responses. Proc Natl Acad Sci USA 100:13025–13029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent EW, Gertler FB (2003). Cytoskeletal dynamics and transport in growth cone motility and axon guidance. Neuron 40:209–227. [DOI] [PubMed] [Google Scholar]
- Dillon C, Goda Y (2005). The actin cytoskeleton: integrating form and function at the synapse. Annu Rev Neurosci 28:25–55. [DOI] [PubMed] [Google Scholar]
- Durand GM, Kovalchuk Y, Konnerth A (1996). Long-term potentiation and functional synapse induction in developing hippocampus. Nature 381:71–75. [DOI] [PubMed] [Google Scholar]
- Fischer M, Kaech S, Wagner U, Brinkhaus H, Matus A (2000). Glutamate receptors regulate actin-based plasticity in dendritic spines. Nat Neurosci 3:887–894. [DOI] [PubMed] [Google Scholar]
- Friedman HV, Bresler T, Garner CC, Ziv NE (2000). Assembly of new individual excitatory synapses: time course and temporal order of synaptic molecule recruitment. Neuron 27:57–69. [DOI] [PubMed] [Google Scholar]
- Fukazawa Y, Saitoh Y, Ozawa F, Ohta Y, Mizuno K, Inokuchi K (2003). Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron 38:447–460. [DOI] [PubMed] [Google Scholar]
- Gasparini S, Saviane C, Voronin LL, Cherubini E (2000). Silent synapses in the developing hippocampus: lack of functional AMPA receptors or low probability of glutamate release? Proc Natl Acad Sci USA 97:9741–9746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibney J, Zheng JQ (2003). Cytoskeletal dynamics underlying collateral membrane protrusions induced by neurotrophins in cultured Xenopus embryonic neurons. J Neurobiol 54:393–405. [DOI] [PubMed] [Google Scholar]
- Hanse E, Gustafsson B (2001). Quantal variability at glutamatergic synapses in area CA1 of the rat neonatal hippocampus. J Physiol (Lond) 531:467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansel C, Linden DJ, D’Angelo E (2001). Beyond parallel fiber LTD: the diversity of synaptic and non-synaptic plasticity in the cerebellum. Nat Neurosci 4:467–475. [DOI] [PubMed] [Google Scholar]
- Hsia AY, Malenka RC, Nicoll RA (1998). Development of excitatory circuitry in the hippocampus. J Neurophysiol 79:2013–2024. [DOI] [PubMed] [Google Scholar]
- Isaac JT, Nicoll RA, Malenka RC (1995). Evidence for silent synapses: implications for the expression of LTP. Neuron 15:427–434. [DOI] [PubMed] [Google Scholar]
- Johnston D, Fisher R, Gray R (1992). Voltage-gated calcium channels in adult hippocampal neurons. Ion Channels 3:39–62. [DOI] [PubMed] [Google Scholar]
- Kim CH, Lisman JE (1999). A role of actin filament in synaptic transmission and long-term potentiation. J Neurosci 19:4314–4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krucker T, Siggins GR, Halpain S (2000). Dynamic actin filaments are required for stable long-term potentiation (LTP) in area CA1 of the hippocampus. Proc Natl Acad Sci USA 97:6856–6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger SR, Kolar A, Fitzsimonds RM (2003). The presynaptic release apparatus is functional in the absence of dendritic contact and highly mobile within isolated axons. Neuron 40:945–957. [DOI] [PubMed] [Google Scholar]
- Kuromi H, Kidokoro Y (1998). Two distinct pools of synaptic vesicles in single presynaptic boutons in a temperature-sensitive Drosophila mutant, shibire. Neuron 20:917–925. [DOI] [PubMed] [Google Scholar]
- Langford GM (1995). Actin- and microtubule-dependent organelle motors: interrelationships between the two motility systems. Curr Opin Cell Biol 7:82–88. [DOI] [PubMed] [Google Scholar]
- Liao D, Hessler NA, Malinow R (1995). Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature 375:400–404. [DOI] [PubMed] [Google Scholar]
- Ma L, Zablow L, Kandel ER, Siegelbaum SA (1999). Cyclic AMP induces functional presynaptic boutons in hippocampal CA3-CA1 neuronal cultures. Nat Neurosci 2:24–30. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA (1999). Long-term potentiation–a decade of progress? Science 285:1870–1874. [DOI] [PubMed] [Google Scholar]
- Malgaroli A, Tsien RW (1992). Glutamate-induced long-term potentiation of the frequency of miniature synaptic currents in cultured hippocampal neurons. Nature 357:134–139. [DOI] [PubMed] [Google Scholar]
- Malgaroli A, Ting AE, Wendland B, Bergamaschi A, Villa A, Tsien RW, Scheller RH (1995). Presynaptic component of long-term potentiation visualized at individual hippocampal synapses. Science 268:1624–1628. [DOI] [PubMed] [Google Scholar]
- Malinow R, Madison DV, Tsien RW (1988). Persistent protein kinase activity underlying long-term potentiation. Nature 335:820–824. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H (2004). Structural basis of long-term potentiation in single dendritic spines. Nature 429:761–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matus A (2000). Actin-based plasticity in dendritic spines. Science 290:754–758. [DOI] [PubMed] [Google Scholar]
- Morales M, Colicos MA, Goda Y (2000). Actin-dependent regulation of neurotransmitter release at central synapses. Neuron 27:539–550. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Malenka RC (1995). Contrasting properties of two forms of long-term potentiation in the hippocampus. Nature 377:115–118. [DOI] [PubMed] [Google Scholar]
- Niikura Y, Abe K, Misawa M (2004). Involvement of L-type Ca2+ channels in the induction of long-term potentiation in the basolateral amygdala-dentate gyrus pathway of anesthetized rats. Brain Res 1017:218–221. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y (2004). Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci 7:1104–1112. [DOI] [PubMed] [Google Scholar]
- Renger JJ, Egles C, Liu G (2001). A developmental switch in neurotransmitter flux enhances synaptic efficacy by affecting AMPA receptor activation. Neuron 29:469–484. [DOI] [PubMed] [Google Scholar]
- Richards DA, Rizzoli SO, Betz WJ (2004). Effects of wortmannin and latrunculin A on slow endocytosis at the frog neuromuscular junction. J Physiol (Lond) 557:77–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Canada C, Ashley J, Moeckel-Cole S, Drier E, Yin J, Budnik V (2004). New synaptic bouton formation is disrupted by misregulation of microtubule stability in aPKC mutants. Neuron 42:567–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan TA, Reuter H, Wendland B, Schweizer FE, Tsien RW, Smith SJ (1993). The kinetics of synaptic vesicle recycling measured at single presynaptic boutons. Neuron 11:713–724. [DOI] [PubMed] [Google Scholar]
- Ryan TA, Ziv NE, Smith SJ (1996). Potentiation of evoked vesicle turnover at individually resolved synaptic boutons. Neuron 17:125–134. [DOI] [PubMed] [Google Scholar]
- Sakaba T, Neher E (2003). Involvement of actin polymerization in vesicle recruitment at the calyx of Held synapse. J Neurosci 23:837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan S, Atluri PP, Ryan TA (2003). Actin has a molecular scaffolding, not propulsive, role in presynaptic function. Nat Neurosci 6:127–135. [DOI] [PubMed] [Google Scholar]
- Schoch S, Castillo PE, Jo T, Mukherjee K, Geppert M, Wang Y, Schmitz F, Malenka RC, Sudhof TC (2002). RIM1alpha forms a protein scaffold for regulating neurotransmitter release at the active zone. Nature 415:321–326. [DOI] [PubMed] [Google Scholar]
- Shen K, Teruel MN, Subramanian K, Meyer T (1998). CaMKIIbeta functions as an F-actin targeting module that localizes CaMKIIalpha/beta heterooligomers to dendritic spines. Neuron 21:593–606. [DOI] [PubMed] [Google Scholar]
- Shupliakov O, Bloom O, Gustafsson JS, Kjaerulff O, Low P, Tomilin N, Pieribone VA, Greengard P, Brodin L (2002). Impaired recycling of synaptic vesicles after acute perturbation of the presynaptic actin cytoskeleton. Proc Natl Acad Sci USA 99:14476–14481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Star EN, Kwiatkowski DJ, Murthy VN (2002). Rapid turnover of actin in dendritic spines and its regulation by activity. Nat Neurosci 5:239–246. [DOI] [PubMed] [Google Scholar]
- Wang HG, Lu FM, Jin I, Udo H, Kandel ER, de Vente J, Walter U, Lohmann SM, Hawkins RD, Antonova I (2005). Presynaptic and postsynaptic roles of NO, cGK, and RhoA in long-lasting potentiation and aggregation of synaptic proteins. Neuron 45:389–403. [DOI] [PubMed] [Google Scholar]
- Wu G, Malinow R, Cline HT (1996). Maturation of a central glutamatergic synapse. Science 274:972–976. [DOI] [PubMed] [Google Scholar]
- Wu GY, Deisseroth K, Tsien RW (2001a). Spaced stimuli stabilize MAPK pathway activation and its effects on dendritic morphology. Nat Neurosci 4:151–158. [DOI] [PubMed] [Google Scholar]
- Wu GY, Deisseroth K, Tsien RW (2001b). Activity-dependent CREB phosphorylation: convergence of a fast, sensitive calmodulin kinase pathway and a slow, less sensitive mitogen-activated protein kinase pathway. Proc Natl Acad Sci USA 98:2808–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Storm DR (2005). The role of calmodulin as a signal integrator for synaptic plasticity. Nat Rev Neurosci 6:267–276. [DOI] [PubMed] [Google Scholar]
- Yabe JT, Pimenta A, Shea TB (1999). Kinesin-mediated transport of neurofilament protein oligomers in growing axons. J Cell Sci 112:3799–3814. [DOI] [PubMed] [Google Scholar]
- Zakharenko SS, Zablow L, Siegelbaum SA (2001). Visualization of changes in presynaptic function during long-term synaptic plasticity. Nat Neurosci 4:711–717. [DOI] [PubMed] [Google Scholar]
- Zhang W, Benson DL (2001). Stages of synapse development defined by dependence on F-actin. J Neurosci 21:5169–5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zito K, Knott G, Shepherd GM, Shenolikar S, Svoboda K (2004). Induction of spine growth and synapse formation by regulation of the spine actin cytoskeleton. Neuron 44:321–334. [DOI] [PubMed] [Google Scholar]