

Abstract
Dysfunction of the 140 aa protein α-synuclein plays a central role in Lewy body disorders, including Parkinson’s disease, as well as in multiple system atrophy. Here, we show that the expression of truncated human α-synuclein(1–120), driven by the rat tyrosine hydroxylase promoter on a mouse α-synuclein null background, leads to the formation of pathological inclusions in the substantia nigra and olfactory bulb and to a reduction in striatal dopamine levels. At the behavioral level, the transgenic mice showed a progressive reduction in spontaneous locomotion and an increased response to amphetamine. These findings suggest that the C-terminal of α-synuclein is an important regulator of aggregation in vivo and will help to understand the mechanisms underlying the pathogenesis of Lewy body disorders and multiple system atrophy.
Keywords: aggregation, behavior, tyrosine hydroxylase, fibril, dopamine, nigrostriatal, Parkinson, α-synuclein
Introduction
Parkinson’s disease (PD) is the most common movement disorder. Neuropathologically, it is defined by nerve cell loss in several brain regions, including the substantia nigra, and by the presence of Lewy bodies and Lewy neurites (Goedert, 2001; Braak et al., 2003). Abundant Lewy bodies and Lewy neurites in cerebral cortex are also the defining neuropathological characteristics of dementia with Lewy bodies (DLB), a common late-life dementia. Ultrastructurally, Lewy bodies and Lewy neurites are composed of filamentous and granular material (Forno, 1996). Missense mutations (A30P, E46K, and A53T) and multiplications of the α-synuclein gene cause dominantly inherited forms of PD and DLB (Polymeropoulos et al., 1997; Krüger et al., 1998; Singleton et al., 2003; Chartier-Harlin et al., 2004; Ibanez et al., 2004; Zarranz et al., 2004), and α-synuclein is the major component of the filamentous inclusions of Lewy bodies and Lewy neurites (Spillantini et al., 1997, 1998a). The correlation between Lewy body formation and neurodegeneration suggests that the aggregation of α-synuclein is an important event during disease pathogenesis. Therefore, dissection of the mechanisms underlying inclusion formation in vivo is essential for understanding disease pathogenesis. In this respect, several mouse lines have been generated that express wild-type or mutant human α-synuclein from several different promoters (Kahle et al., 2000; Masliah et al., 2000; van der Putten et al., 2000; Matsuoka et al., 2001; Rathke-Hartlieb et al., 2001; Giasson et al., 2002; Lee et al., 2002; Neumann et al., 2002; Richfield et al., 2002).
Overexpression of wild-type human α-synuclein was found to lead to the formation of granular deposits and subtle biochemical abnormalities (Masliah et al., 2000). Subsequent work showed that mice overexpressing A30P and A53T human α-synuclein developed accumulation of α-synuclein in cell bodies and dystrophic neurites in several brain regions (van der Putten et al., 2000; Giasson et al., 2002; Lee et al., 2002; Neumann et al., 2002), with one study (Giasson et al., 2002) documenting the presence of filamentous α-synuclein deposits in cerebellum and spinal cord. However, none of these studies, which used either the Thy1 or the prion protein promoter, resulted in the accumulation of α-synuclein in dopaminergic nerve cells of the substantia nigra. Furthermore, expression of wild-type or mutant human α-synuclein from the tyrosine hydroxylase (TH) promoter did not result in the formation of pathological inclusions (Matsuoka et al., 2001; Rathke-Hartlieb et al., 2001; Richfield et al., 2002).
Previous work has shown that Lewy body extracts are enriched in C-terminally truncated α-synuclein (Baba et al., 1998; Tofaris et al., 2003) and that human α-synuclein lacking the C-terminal 20 amino acids [α-synuclein(1–120) (α-Syn120)] assembles into filaments in vitro faster than either wild-type or mutant protein (Crowther et al., 1998; Serpell et al., 2000; Murray et al., 2003). It is therefore possible that truncation of α-synuclein could accelerate aggregation in a similar manner in vivo, resulting in neuropathological changes at an earlier age. Here, we investigated the effects of this modification by generating transgenic mice expressing human α-Syn120 from the rat TH promoter.
Materials and Methods
Generation of transgenic mouse lines.
Transgene constructs were generated using the rat TH promoter (see Fig. 1A). The TH cassette (a kind gift from Dr. H. van der Putten, Novartis, Basel, Switzerland) (Min et al., 1994) directs high expression in dopaminergic and other TH-positive neurons. Human α-synuclein(1–120) DNA was generated from the wild-type cDNA (Jakes et al., 1994) by PCR amplification, and a Kozak consensus sequence was introduced upstream of the initiation codon. The expression construct was generated by subcloning human α-synuclein(1–120) DNA into the unique EcoRI site of the expression vector. Before microinjection, vector sequences were removed by digestion with NotI. Founders and transgenic progeny were identified by PCR analysis of lysates from tail biopsies using two sets of primers. Transgenic lines were established as C57BL/6J × CBA/ca hybrids and subsequently backcrossed to C57BL/6J, which lacks α-synuclein (Specht and Schoepfer, 2001).
Fig. 1.

Expression of α-Syn120 in transgenic mouse brain. A, Schematic diagram of the construct: human α-synuclein(1–120) DNA was cloned downstream of the rat TH promoter. B, Immunoblot probed for α-synuclein (antibody Syn-1) and TH. The tissues were obtained from 6-week-old transgenic mice and littermate controls. Cx, Cerebral cortex; Cb, cerebellum; OB, olfactory bulb; SN, substantia nigra. No endogenous α-synuclein was detected at 19 kDa. C, Expression levels of α-Syn120 in olfactory bulb of transgenic mice compared with full-length α-synuclein in olfactory bulb from C57BL/6 mice. D, E, Immunoblotting (antibody Syn-1) of extracts from olfactory bulb (D) and substantia nigra (E) at 6, 12, and 24 months showed stable expression of the transgene. rS, Recombinant full-length human α-synuclein.
Immunoblotting.
Total protein concentrations in tissue lysates were measured using the biocinchoninic acid assay (BCA; Pierce, Rockford, IL), and equal amounts were run on 12 or 15% SDS-PAGE. Proteins were transferred electrophoretically onto nitrocellulose membranes (Millipore, Bedford, MA). The membranes were blocked with 4% milk and incubated with the appropriate primary antibodies. Bound antibodies were then visualized using peroxidase-conjugated secondary antisera (1:2000) and enhanced chemiluminescence (PerkinElmer Life Sciences, Boston, MA).
Immunohistochemistry.
Mice were anesthetized with an intraperitoneal injection of xylazine and ketamine or pentabarbitone, followed by intracardial perfusion with TBS and 4% ice-cold (w/v) paraformaldehyde. The brain was dissected and postfixed in 4% paraformaldehyde before being transferred to a 30% sucrose solution. Sections (15 or 30 μm) were cut on a freezing microtome, and immunohistochemistry was performed on free-floating sections. Incubations were done in TBS containing 0.25% Tween 20, and the sections were quenched for 20 min in 20% methanol/1.5% H2O2. When rabbit antisera were used, sections were preincubated with 3% goat serum for 1 h and incubated with primary antibody overnight at room temperature in 1% goat serum, followed by incubation with biotinylated goat anti-rabbit antibody (1:200; Vector Laboratories, Burlingame, CA) in 1% goat serum for 1 h and avidin–biotin peroxidase complex for 30 min (ABC Elite; Vector Laboratories). Staining was visualized using 3,3-diaminobenzidine (DAB; Sigma, St. Louis, MO) as the chromogen. When monoclonal antibodies were used, sections were processed as above, except that biotinylated horse anti-mouse secondary antibody and the mouse-on-mouse kit (Vector Laboratories) were used to minimize background staining. For double-immunofluorescence analysis of α-synuclein, TH, and CD11b, Alexa Fluor 488-conjugated (green) and Alexa Fluor 594-conjugated (red) secondary antibodies (Invitrogen, Eugene, OR) were used, and the sections were mounted using a glycerol-based anti-fading kit (Invitrogen). A panel of anti-α-synuclein antibodies was used. Antiserum PER4 recognizes the C-terminal region of α-synuclein (Spillantini et al., 1998a), whereas antiserum PER7 was raised against recombinant human α-synuclein(1–120) (Jakes et al., 1999). They recognize mouse and human α-synucleins. Syn-1 (Transduction Laboratories, Lexington, KY) and LB509 (Zymed, San Francisco, CA) are monoclonal antibodies, which recognize residues 91–99 and 115–122 of human α-synuclein, respectively (Jakes et al., 1999; Perrin et al., 2003). Syn-1 recognizes both mouse and human α-synucleins, whereas LB509 is specific for the human protein. Antibody Syn h119 (a kind gift from Dr. V.M.-Y. Lee, University of Pennsylvania, Philadelphia, PA) is a monoclonal antibody specific for residues 71–82 of α-synuclein, which recognizes the mouse and human proteins (Mishizen-Eberz et al., 2003). Syn204 (Biomeda, Foster City, CA) is a human-specific monoclonal antibody against the amino terminus of α-synuclein (Giasson et al., 2000). Monoclonal anti-TH antibody was purchased from Chemicon (Temecula, CA). Monoclonal anti-CD11b was purchased from Serotec (Oxford, UK). SMI-31 (Sternberger Monoclonals, Lutherville, MD) is a monoclonal antibody that recognizes phosphorylated neurofilaments.
Thioflavin S staining.
Fixed mouse brain sections (15 μm) were incubated with 1% thioflavin S (Sigma) for 10 min and washed sequentially in 70% and 50% ethanol (4 min each), followed by a 3 min wash in water. Sections were left to dry for 20 min in the dark, before being mounted in glycerol-based anti-fading kit (Invitrogen) containing 1 ng/μl 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI).
Electron microscopy.
Mice were anesthetized with an intraperitoneal injection of xylazine and ketamine and perfused intracardially with PBS, followed by 4% cold paraformaldehyde and 0.25% glutaraldehyde. The brain was dissected and postfixed in the same solution for another 2 h. For immunoelectron microscopy, fixed specimens (1 mm3) were washed in distilled water and placed in TBS, pH 7.6. Sections (50 μm thick) were cut using a vibratome. Immunogold labeling with antibody Syn-1 (1:100) was performed using 10 nm gold particles (Goldmark Biologicals, Phillipsburg, NJ) conjugated to goat anti-mouse IgG (diluted 1:10). Sections were postfixed in 2.5% glutaraldehyde, followed by 1% osmium tetroxide, dehydrated through a graded alcohol series, and embedded in epoxy resin. Semithin sections were stained with toluidine blue. Ultrathin sections were contrasted with lead citrate and uranyl acetate and scanned with electron microscopy.
Sequential extraction of α-synuclein.
Extraction of α-synuclein was performed as described previously (Tofaris et al., 2003). Tissues were homogenized on ice in 5 vol of TBS+ [50 mm Tris-HCl, pH 7.4, 175 mm NaCl, 5 mm EDTA, 0.1 mm PMSF, 1 mmN-ethyl-maleimide, plus complete proteasome inhibitor cocktail (Roche Diagnostics, Mannheim, Germany)] and spun for 30 min at 120,000 × g at 4°C. The resulting supernatants corresponded to the TBS+ soluble fraction. The pellets were homogenized in TBS+ containing 1% Triton X-100, followed by centrifugation. The pellets were in turn homogenized in TBS+ containing 1 m sucrose, and, after centrifugation, floating myelin was discarded. The resulting pellets were extracted with radioimmunoprecipitation assay (RIPA) buffer (50 mm Tris-HCl, pH 7.4, 175 mm NaCl, 5 mm EDTA, 1% NP-40, and 0.5% sodium deoxycholate) and 0.1% SDS, followed by centrifugation. The detergent-insoluble pellets were solubilized in 8 m urea/5% SDS. In some experiments, the 1% Triton step was omitted, and TBS extraction was directly followed by RIPA buffer treatment.
HPLC.
Striatal monoamine levels were compared between transgenic and littermate control mice at 1, 3, 6, 9, and 12 months of age. The anterior striatum was dissected on ice, weighed, and homogenized in 0.1 m cold perchloric acid. Homogenates were centrifuged twice at 15,000 rpm for 5 min at 4°C, and the supernatants were diluted 1:10 in 0.1 m perchloric acid. They were stored at −80°C for 2 d. On the day of analysis, the supernatants were thawed and mixed with 3,4-dihydroxybenzylamine as an internal standard. Levels of dopamine, homovanillic acid, and 5-hydroxytryptamine (5-HT) were determined by HPLC combined with electrochemical detection, as described previously (O’Connell et al., 1991).
Behavioral testing.
Twenty-three α-Syn120 transgenic and 25 wild-type control mice were assessed for spontaneous locomotor activity (LMA) and the effectiveness of d-amphetamine sulfate to modulate LMA at 6 and 18 months of age (n = 12 and 13 for wild-type mice at 6 and 18 months of age and n = 12 and 11 for transgenic mice at 6 and 18 months of age, respectively). Mice were individually placed into test chambers [for a description of the apparatus, see Isles et al. (2004)] for 30 min before administration of 1 mg/kg (i.p.) d-amphetamine sulfate or 0.7% saline vehicle and LMA, and then recorded for an additional 120 min. The mice were given two sessions, 7 d apart, and drug treatments were counterbalanced across groups and ages. All procedures were conducted in accordance with the requirements of the United Kingdom Animals (Scientific Procedures) Act 1986 (behavioral testing). Spontaneous LMA, designated as the total number of beam breaks made during the 30 min preinjection phase from the first LMA session was analyzed by two-way ANOVA with the factors GENOTYPE (control, transgenic) and AGE (6 and 18 months) and habituation to the chambers, examined by dividing the data into 5 min time bins, by three-way ANOVA with factors GENOTYPE, AGE, and repeated measure of TIME (six 5 min time bins). Total activity after drug treatment was subjected to a three-way ANOVA with factors GENOTYPE, AGE, and TREATMENT (vehicle, amphetamine). The percentage increase in LMA for each 30 min block after drug treatment was calculated relative to the LMA in the final 10 min of the preceding 30 min habituation session. These data were analyzed by four-way ANOVA with factors GENOTYPE, AGE, TREATMENT, and repeated measure of TIME (four 30 min time bins).
Results
Expression of human α-Syn120
Five founders were obtained, three of which transmitted the transgene in a Mendelian manner. By immunoblotting using antibody Syn-1, two lines expressed high levels of α-Syn120 in the nigrostriatal pathway and the olfactory bulb, brain regions expressing high levels of TH. One of these lines was used for additional characterization (Fig. 1B). Relative to immunoreactivity for TH, the expression of α-Syn120 was higher in the olfactory bulb than in the substantia nigra, and high levels of expression were detected in both regions at all ages examined (Fig. 1D,E). When compared with the substantia nigra and the olfactory bulb of wild-type C57BL/6 mice with endogenous α-synuclein, transgenic α-Syn120 was present at lower levels than the endogenous protein (Fig. 1C). However, it has to be considered that although most neurons in the wild-type mouse express α-synuclein, transgenic α-Syn120 is only expressed in TH neurons. Antibodies PER4 and LB509, which are specific for the last 20 amino acids of α-synuclein (Jakes et al., 1999), failed to recognize the transgenic protein. As expected (Specht and Schoepfer, 2001), no mouse α-synuclein was detected in either transgenic lines or littermate controls. Heterozygous transgenic mice were used for this study.
Immunohistochemical distribution of human α -Syn120
The cellular localization of α-Syn120 was studied by single- and double-labeling immunohistochemistry. Strong immunoreactivity was present in the somatodendritic compartment of the vast majority of TH-positive neurons from 6 weeks of age onwards. Within the nigrostriatal pathway of young mice, staining for α-Syn120 closely followed TH staining throughout the cell soma and around the nucleus, extending into dendritic processes (Fig. 2). Colocalization of the two proteins was observed in 90% of cells. α-Syn120 was also present in the corresponding terminal fields, as indicated by punctate staining in the striatum (Fig. 3a). However, when stained with an antibody against TH, axons of transgenic mice appeared with focal varicose swellings in contrast to littermate controls. Using antiphosphorylated neurofilament antibodies, axons appeared to have a larger diameter in transgenic mice (Fig. 3b,c,h–j). In the olfactory bulb, in contrast, α-Syn120 accumulated in inclusion-like structures at all ages examined (Fig. 3d,f). As a result, its staining pattern did not completely overlap with the more diffuse distribution of TH (Fig. 3e) and was different from the staining observed with the same anti-α-synuclein antibody in C57BL/6 control mice expressing the endogenous protein (Fig. 3g). TH-positive neurons in the locus ceruleus were also immunoreactive for α-Syn120 (data not shown).
Fig. 2.

Cellular localization of α-Syn120 in the substantia nigra of transgenic mice. a–d, Adjacent coronal sections of the substantia nigra from 6-month-old transgenic (a, b) and littermate control (c, d) mice were stained for TH and α-synuclein (Syn-1). TH-positive neurons from transgenic mice (a) showed somal expression of α-Syn120 (b). A similar region from littermate control mice was positive for TH (c) but negative for α-synuclein (d), as would be expected in mice with a null α-synuclein background. e, f, Immunofluorescence microscopy of the substantia nigra using anti-TH (e, red) and anti-α-synuclein (f, green) antibodies showed colocalization (g, yellow). Scale bar: (in a) a–d, 200 μm; e–g, 250 μm.
Fig. 3.
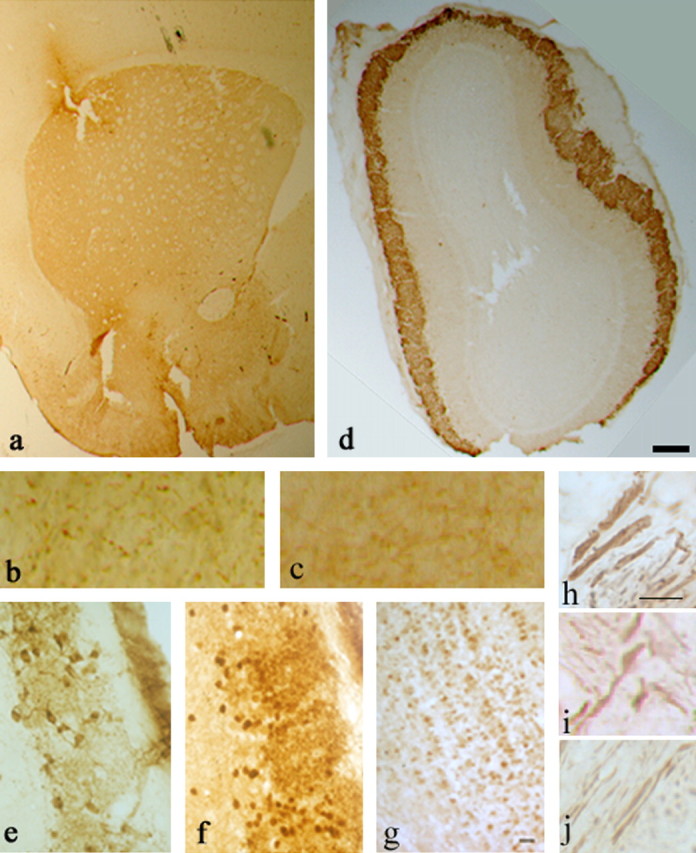
Histological characterization of α-Syn120 in the striatum and the olfactory bulb. a, Punctate neuropil localization of α-Syn120 in the striatum was consistent with enrichment at synaptic nerve terminals. b, c, TH staining showed enlarged nerve cell processes in striatum of transgenic (b) but not in littermate control (c) mice. h–j, Abnormal axonal morphology was confirmed by phosphorylated neurofilament staining with SMI 31 antibody in transgenic (h, i) compared with littermate control (j) mice. α-Syn120 immunoreactivity (syn-1 antibody) in the olfactory bulb (d) from a transgenic mouse. e, At a higher magnification, TH staining appeared diffusely distributed along the somatodendritic compartment. Abundant α-Syn120-syn-1-positive inclusions (f) were present in the olfactory bulb at all ages examined, showing a different distribution of the protein compared with endogenous full-length α-synuclein in C57BL/6 mice stained with the same antibody (g). Scale bars: a, d, 600 μm; (in h) h–j, 5 μm; (in g) b, c, e, f, 150 μm.
Staining for human α-Syn120 in substantia nigra as a function of age
Transgenic mice were studied at 3, 6, and 12–14 months of age (n = 3 per time point). At 3 months (Fig. 4a), α-Syn120 accumulated in the somatodendritic compartment, where its staining pattern resembled that of TH. By 12–14 months of age, pathological changes were detected (Fig. 4b–j). They included dense, shrunken perikarya (Fig. 4b), beaded or dystrophic processes (Fig. 4c,e), as well as inclusions that were either perinuclear (Fig. 4d,j, arrows) or very localized (Fig. 4f,g, arrowheads). Double immunostaining for both TH and α-synuclein confirmed the localization of dense inclusions within the cytoplasm of dopaminergic neurons (Fig. 4g). Granular deposits and vacuolation of the cytoplasm were also detected in older mice (Fig. 4h–j). Furthermore, there was an increase in microglial cells associated with affected brain regions, indicative of an inflammatory response (Fig. 5>A,B). There was no significant cell loss by cell counting of TH-positive neurons in the substantia nigra using stereology (data not shown). Staining was observed with α-synuclein antibodies Syn-1, Syn h119, and PER7, but not with PER4 or LB509. Inclusions were not stained with anti-neurofilament or anti-ubiquitin antibodies (data not shown).
Fig. 4.

Histological characterization of α-Syn120 in the substantia nigra. a, At 3 months of age, α-Syn120 was diffusely distributed through the somatodendritic compartment. b, At 14 months, α-Syn120 immunoreactivity revealed the presence of pyknotic perikarya. c, e, Between 11 and 14 months, a number of pathological profiles was observed, which included beaded (c) or swollen (e) processes. Perinuclear aggregates (d and j, arrows) or dense inclusions (f, arrowheads) within TH neurons (g, arrowhead, double-stained neurons with anti-TH blue and Syn1 brown) are shown. Vacuolation of the cytoplasm (arrows h, i) was evident at this stage (h–j). The following α-synuclein antibodies were used: Syn-1, a, b, e–i; PER7, c, d; SYN h119, j. Scale bar, 60 μm.
Fig. 5.

Microglial activation. Double-staining immunofluorescence with TH (red) and CD11b (green) shows increased microglial cell numbers in the SN of transgenic mice (A) compared with littermate controls (B).
Electron microscopy
Pathological profiles were investigated by electron microscopy and immunogold labeling of olfactory bulb using antibody Syn-1. The α-Syn120 aggregates had a mixed granular and fibrillar morphology (Fig. 6a). A subset of inclusions in olfactory bulb and substantia nigra was thioflavin S-positive, consistent with the electron microscopic findings (Fig. 6b,c). Occasional inclusions were present in 3-month-old mice, and their numbers increased with age.
Fig. 6.

Ultrastructure of α-Syn120 aggregates. a, Immunoelectron microscopy (antibody Syn-1) of the olfactory bulb from 7-month-old transgenic mice revealed granular and filamentous deposits. Arrows point to filamentous structures. b, c, Thioflavin S fluorescence in olfactory bulb (b) and substantia nigra (c). Nuclei were visualized with DAPI (blue fluorescence). Scale bar: a, 250 nm.
Sequential extraction of human α-Syn120
α-Synuclein was sequentially extracted from olfactory bulb and substantia nigra of 6-month-old transgenic mice using TBS, 1% Triton X-100, RIPA buffer, and 8 m urea/5% SDS (Fig. 7). A protein band with an apparent molecular mass of 12 kDa was detected with antibody Syn-1 in the TBS and Triton X-100 fractions. It corresponds to monomeric α-Syn120. When the RIPA-insoluble material was treated with urea, in addition to a high-molecular weight smear, a band corresponding to monomeric truncated α-Syn120 was found in substantia nigra and olfactory bulb. The smear and the monomeric α-Syn120 band present in the urea fraction were stained by antibodies Syn-1, Syn204, and PER 7 (Fig. 7). The substantia nigra from a case of PD was extracted in parallel and the urea-soluble material is shown (Fig. 7). In control experiments, full-length α-synuclein extracted from age-matched C57BL/6 mice with endogenous protein was detected in the soluble, but not the urea fraction (Fig. 7). Furthermore, no staining was detected in the soluble or urea-treated extracts from littermate controls (data not shown).
Fig. 7.

Sequential extraction of α-Syn120 from substantia nigra and olfactory bulb. Substantia nigra and olfactory bulb tissues from 6-month-old transgenic mice were sequentially extracted with Tris-HCl, Triton X-100, RIPA buffer, and urea, followed by immunoblotting with antibodies Syn-1 (a–c), Syn204 (d), and PER7 (e). a, The urea-soluble material from the substantia nigra of a PD patient was used as a positive control. In age-matched C57BL/6 wild-type mice with endogenous α-synuclein, the protein was detected only in soluble fractions (c) but not in urea extracts (U in c, CU in d, e). CU, Urea extract from control wild-type mice with endogenous α-synuclein. The asterisk indicates the position of the monomeric α-Syn120 band in the urea-soluble fractions. SN, Substantia nigra; U, urea; T, Tris-HCl; R, RIPA buffer; T/T, Triton X-100; OB, olfactory bulb.
Neurochemical changes
The impact of human α-Syn120 expression on dopamine synthesis and turnover was assessed in the anterior striatum of transgenic mice at 1, 3, 6, 9, and 12 months of age (n = 4 per group). When compared with age-matched littermate controls, although striatal dopamine levels were the same at 1 month, a statistically significant 30% reduction (p < 0.0001, F = 32.8, R2 = 0.653) was detected at later time points in the transgenic mice (Fig. 8). This was paralleled by a 30% reduction in the dopamine metabolite homovanillic acid (Fig. 8). Measurement of 5-HT levels showed no significant difference between transgenic and littermate control mice (Fig. 8).
Fig. 8.
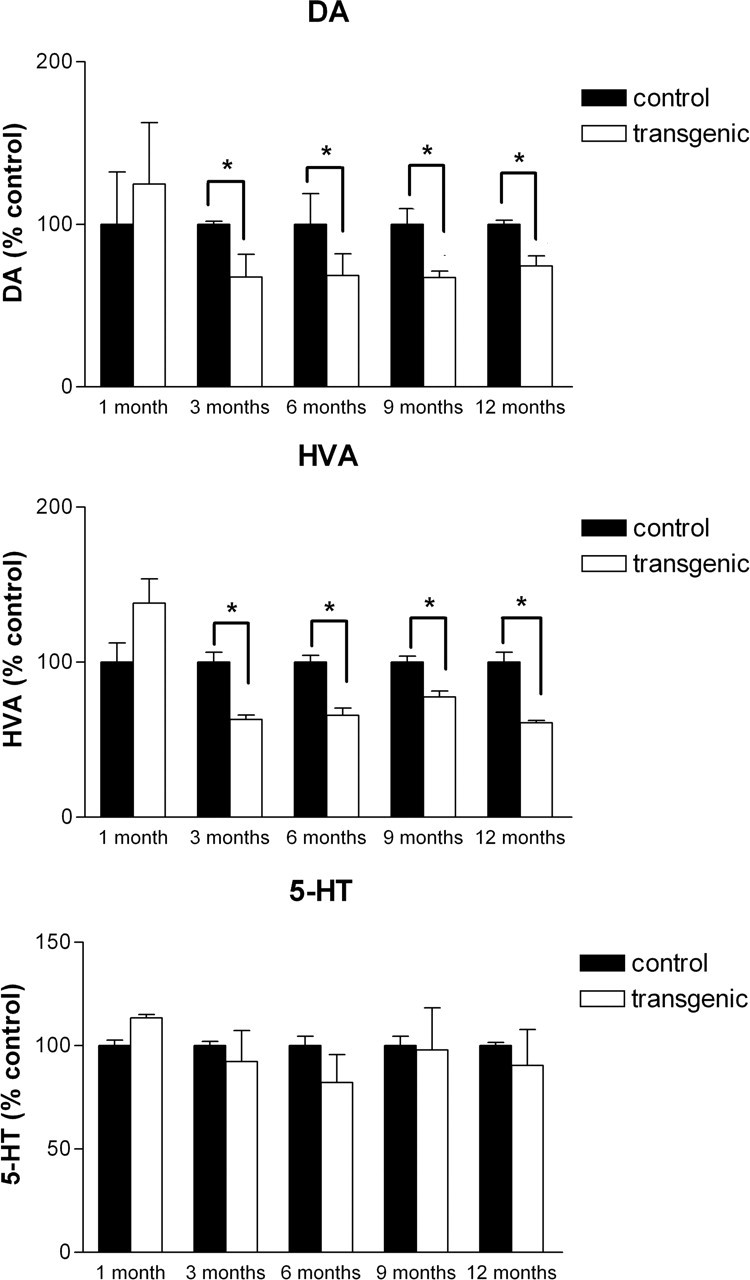
Striatal dopamine (DA), homovanillic acid (HVA), and 5-HT levels in transgenic and littermate control mice at different ages. Top panel, A significant reduction (p < 0.0001) in striatal dopamine levels was present in transgenic mice at 3, 6, 9, and 12 months of age but not in 1-month-old animals. Middle panel, This was paralleled by a similar reduction (p < 0.003) in the levels of homovanillic acid. Bottom panel, Striatal 5-HT levels did not differ significantly between control and transgenic mice. Error bars represent SEM.
Spontaneous locomotor activity
The effect of α-Syn120 expression on spontaneous activity of 6- and 18-month-old transgenic mice is shown in Figure 9, A and B. At 6 months, there was no significant difference in total activity between wild-type and transgenic mice, whereas at 18 months, transgenic mice made significantly fewer beam breaks during the 30 min test than age-matched controls (AGE × GENOTYPE interaction, F(1,44) = 6.6, p < 0.02); this was observed against a general reduction in locomotor activity by the older group of mice (effect of AGE, F(1,44) = 15.2, p < 0.001). The pattern of reduced activity in 18-month-old transgenic mice was consistent throughout the session [Fig. 9B, no AGE × TIME × GENOTYPE interaction, F(5,220) = 0.3, not significant (n.s.)]. However, all mice showed equivalent levels of habituation to the environment (effect of TIME, F(5,220) = 28.8, p < 0.001), and the older mice made consistently fewer beam breaks in each 5 min time bin (AGE × TIME interaction, F(5,220) = 5.9, p < 0.001).
Fig. 9.

Spontaneous and amphetamine-induced activity in 6- and 18-month-old α-Syn120 transgenic and control mice. A, Eighteen-month-old transgenic mice showed reduced levels of spontaneous activity, measured as infrared beam breaks, compared with equivalent aged wild-type mice, and, overall, the older mice showed a reduction in activity relative to 6 month-old subjects. B, When examined in more detail, all of the groups of mice habituated to the environment equally during the 30 min test. After 30 min habituation, mice received injections of either saline vehicle or 1 mg/kg d-amphetamine intraperitoneally, counterbalanced over two sessions 7 d apart, and activity was monitored for an additional 120 min. Amphetamine induced increased activity in all mice, with a peak response 60 min after injection (n = 11 and 13 for 6-month-old, and n = 12 and 12 for 18-month-old transgenic and control mice, respectively). C, Compared with wild-type mice at both ages and 6-month-old α-Syn120 transgenic mice, 18-month-old α-Syn120 mice exhibited a significantly enhanced response to amphetamine relative to the preinjection activity performance.
Effects of amphetamine
After the 30 min habituation sessions, control and α-Syn120 transgenic mice were given an injection of either saline vehicle or amphetamine (Fig. 9C). When the drug treatment effects were calculated as a percentage of the final 10 min of the preinjection habituation session, 18-month-old transgenic mice exhibited an enhanced response to amphetamine relative to age-matched controls and 6-month-old transgenic mice and controls (effect of AGE × TREATMENT × GENOTYPE, F(1,88) = 6.8, p < 0.02). Overall, amphetamine yielded a significant increase in locomotor activity (effect of TREATMENT, F(1,88) = 58.3, p < 0.001) that peaked 60 min after administration (TIME × DRUG interaction, F(3,264) = 15.3, p < 0.001). There were no genotype-related differences in the total number of beam breaks made in the 2 h after drug or vehicle injection (data not shown, effect of GENOTYPE, F(1,88) = 1.8, n.s.), although 18-month-old mice made significantly fewer beam breaks than 6-month-old mice (effect of AGE, F(1,88) = 12.5, p < 0.001). There was no interaction between genotype, age, and drug treatment (F(1,88) = 0.1, n.s.).
Discussion
α-Synuclein is a presynaptic protein, which gives a characteristic punctate staining in rodent and human brain (Jakes et al., 1994). Similar findings were obtained for α-Syn120 in the striatum and olfactory bulb, indicating that it was transported like the full-length protein. However, the progressive accumulation of α-Syn120 in the somatodendritic compartment was pathological and associated with the accumulation of microglia and inflammation. In 3-month-old transgenic mice, α-synuclein showed a diffuse somatodendritic distribution; by 12–14 months, perikarya in the substantia nigra were shrunken or contained cytoplasmic inclusions, and axons in the striatum contained varicose swellings and had an enlarged diameter, resembling the pathological profiles in PD (Duda et al., 2002; Neumann et al., 2004).
In the olfactory bulb, α-Syn120-positive inclusions were already present by 3 months of age. This is reminiscent of human brain, where Lewy bodies and Lewy neurites appear in the olfactory bulb before the substantia nigra (Braak et al., 2003). Olfactory dysfunction precedes clinical motor signs in PD (Katzenschlager and Lees, 2004). Electron microscopy and immunogold labeling of α-Syn120 deposits revealed the presence of granular and fibrillar structures in the olfactory bulb. Furthermore, some inclusions in substantia nigra and olfactory bulb were thioflavin S-positive, consistent with the presence of aggregated α-Syn120. By sequential extraction, we detected insoluble α-Syn120 aggregates in the olfactory bulb and the substantia nigra of transgenic mice. Their limited solubility was similar to that of α-synuclein extracted from PD and DLB brains (Campbell et al., 2000; Tofaris et al., 2003), although there was less insoluble material than in the human conditions. Furthermore, although in other reports the insoluble material appeared as a smear containing distinct bands as well as monomeric α-synuclein (Kahle et al., 2001), this was not the case here, where only one band corresponding to monomeric α-Syn 120 was present in the insoluble fraction. This difference could be attributable to the different methods of extraction, the nature of the transgenic protein (truncated versus full-length) or the amount of insoluble aggregates and their distribution, which was limited to substantia nigra and olfactory bulb in the α-Syn120 transgenic mice (Kahle et al., 2001; Giasson et al., 2002). The α-Syn120 inclusions were usually ubiquitin negative, consistent with the extensive evidence indicating that the ubiquitination of α-synuclein in Lewy bodies and Lewy neurites is a late event (Spillantini et al., 1998a; Tofaris et al., 2001, Sampathu et al., 2003; Tofaris et al., 2003). To our knowledge, this is the first transgenic mouse model with mixed filamentous and granular α-synuclein aggregates in dopaminergic neurons. It shows that truncated human protein expressed on a α-synuclein null background can induce inclusion formation in vivo.
The neurochemical effects of human α-Syn120 expression in the striatum were assessed in mice up to 12 months of age. Although at 1 month there was no difference in dopamine levels between transgenic mice and littermate controls, a significant reduction in dopamine and homovanillic acid, but not 5-HT, was detected from 3 months onwards. These findings indicate a specific dysfunction of the dopaminergic system, in agreement with the differential expression of α-Syn120, and are consistent with neurochemical changes in PD, which is characterized by a deficiency in striatal dopamine and homovanillic acid (Hornykiewicz, 1998). Unlike in PD, there was no evidence of significant nerve cell death in our model. It is conceivable that the reduction in dopamine levels is best explained by a metabolic dysfunction resulting from the early accumulation of α-Syn120 in presynaptic nerve terminals in the striatum rather than by changes at the cell body level in the substantia nigra. In support of the latter is the temporal correlation between the presence of dilated striatal terminals and dopamine deficiency and the evidence from studies in human brain, which showed that, in PD, although striatal dopamine is decreased by 80%, nigral neurons are only reduced by 50% (Hornykiewicz, 1998).
The behavioral analysis, which revealed a progressive reduction in spontaneous locomotor activity and an increased response to amphetamine, was consistent with transgene-induced effects on dopamine-mediated functions. Similar findings have previously been reported in animal models of PD after chemical and genetic lesions, which reduce presynaptic dopamine (Cai et al., 2002; Marazziti et al., 2004). Furthermore, expression of full-length mutant, but not wild-type, α-synuclein under the control of the TH promoter led to a significant decrease in locomotor activity with aging (Thiruchelvam et al., 2004), suggesting that C-terminal truncation of the protein can have a pathological phenotype similar to that of a disease-causing missense mutation. The enhanced response to amphetamine is reminiscent of dopamine “supersensitivity,” a postsynaptic adaptation related to the chronic lowering of basal dopamine levels in terminal regions, especially the striatum. The synaptic mechanisms underlying this phenomenon can involve increased dopamine receptor numbers or be related to changes in binding efficiencies and/or downstream signal transduction pathways (Cai et al., 2002; Gerfen et al., 2002). Although several other transgenic mouse models of α-synuclein expression have been produced, they showed accumulation of α-synuclein in brain regions other than the substantia nigra (Giasson al., 2002), or the inclusions were granular and nuclear (Masliah et al., 2000), or there were no inclusions (Thiruchelvam et al., 2004). Our model is the first to demonstrate a direct link between granular and filamentous α-synuclein cytoplasmic pathology confined to the dopaminergic system and a progressive behavioral deficit without involvement of areas of motor function (i.e., corticospinal tract and anterior horn cells), which were affected in other models (Masliah et al., 2000; van der Putten et al., 2000; Giasson et al., 2002; Neumann et al., 2002). These data, together with our immunohistochemical and neurochemical findings, raise the possibility that behavioral deficits may predate overt neurodegeneration and point to nerve terminals as possible anatomical substrates for the primary insult in α-synucleinopathies.
The above findings show that mice expressing human α-Syn120 in dopaminergic neurons recapitulate many changes characteristic of human Lewy body diseases. The major limitations of our model are the small number of fibrils, the absence of significant cell death, and the lack of endogenous full-length α-synuclein. One of our aims was to produce a model whereby the effect of aggregated human α-synuclein could be investigated without interference from endogenous mouse protein. A previous in vitro study has reported that the fibrillization of human α-synuclein is inhibited in the presence of the mouse protein (Rochet et al., 2000). However, truncated human α-synuclein can cross-seed the fibrillation of full-length protein in vitro (Murray et al., 2003). It will therefore be important to determine whether the presence of full-length protein influences the pathological and behavioral phenotype detected in α-Syn120 mice.
C-terminal truncation of α-synuclein is considered by some to play a role in the pathogenesis of Lewy body diseases and the degeneration of dopaminergic nerve cells (Tofaris et al., 2003; Li et al., 2005; Liu et al., 2005). C-terminally truncated α-synuclein forms filaments at a faster rate than the full-length protein (Crowther et al., 1998; Serpell et al., 2000; Murray et al., 2003). Furthermore, C-terminally truncated α-synuclein has been detected in Lewy bodies in human diseases and in the brains of transgenic mice expressing mutant human α-synuclein (Giasson et al., 2002; Lee et al., 2002; Tofaris et al., 2003). How truncated α-synuclein can be generated in cells, under pathological conditions, is currently unclear; the proteasome could be involved (Tofaris et al., 2001; Liu et al., 2003). Isolated α-synuclein filaments from human brain are made of the full-length protein, suggesting that truncation may occur after assembly (Spillantini et al., 1998a,b; Crowther et al., 2000; Takao et al., 2004). This notwithstanding, it is clear that the C-terminal region of α-synuclein, which binds dopamine derivatives (Norris et al., 2005), is a negative regulator of self-assembly. Therefore, modifications in this region, such as oxidation, nitration, and phosphorylation (Hashimoto et al., 1999; Giasson et al., 2000; Fujiwara et al., 2002), may well influence the propensity of α-synuclein to aggregate in vivo in a way similar to truncation. The same is true of molecules that bind to the monomeric protein. Thus, polyamines have been shown to promote the aggregation of α-synuclein through binding to its C-terminal region (Antony et al., 2003; Fernandez et al., 2004). Other positively charged molecules may act in a similar way (Goers et al., 2003).
In conclusion, the present work describes the production and characterization of a novel transgenic mouse model for α-synuclein aggregation. Expression of human α-Syn120 under the control of the TH promoter led to the formation of pathological inclusions in substantia nigra and olfactory bulb, as revealed by morphological and biochemical studies. This was accompanied by a reduction in dopamine levels in the striatum, a progressive reduction in spontaneous locomotor activity, and an increased response to amphetamine. Our findings suggest that the C-terminal region of α-synuclein is an important regulator of aggregation.
Footnotes
This work was supported by the United Kingdom Alzheimer’s Research Trust, the Parkinson’s Disease Society, the Biotechnology and Biological Sciences Research Council, and the Medical Research Council. G.K.T. was supported by an Overseas Research Scholarship award, a Trinity College Scholarship, and the MB/PhD program of Cambridge University. P.G.R. has a studentship from European Union Marie Curie Actions RTN-NSR number 504636. B.G. was supported by United States Public Health Service Grant P30 AG10133. S.L.L. is the recipient of an Alzheimer’s Research Trust fellowship. We thank B. Allen and R. Raha-Chowdhury for help, H. van der Putten for the TH promoter construct, and V.M.-Y. Lee for antibody Syn h119.
References
- Antony T, Hoyer W, Cherny D, Heim G, Jovin TM, Subramaniam V (2003). Cellular polyamines promote the aggregation of α-synuclein. J Biol Chem 278:3235–3240. [DOI] [PubMed] [Google Scholar]
- Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, Trojanowski JQ, Iwatsubo T (1998). Aggregation of α-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am J Pathol 152:879–884. [PMC free article] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24:197–211. [DOI] [PubMed] [Google Scholar]
- Cai G, Wang HY, Friedman E (2002). Increased dopamine receptor signaling and dopamine receptor-G protein coupling in denervated striatum. J Pharmacol Exp Ther 302:1105–1112. [DOI] [PubMed] [Google Scholar]
- Campbell BCV, Li QL, Culvenor JG, Jäkälä P, Cappai R, Beyreuther K, Masters CL, McLean CA (2000). Accumulation of insoluble α-synuclein in dementia with Lewy bodies. Neurobiol Dis 7:192–200. [DOI] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destee A (2004). α-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364:1167–1169. [DOI] [PubMed] [Google Scholar]
- Crowther RA, Jakes R, Spillantini MG, Goedert M (1998). Synthetic filaments assembled from C-terminally truncated α-synuclein. FEBS Lett 436:309–312. [DOI] [PubMed] [Google Scholar]
- Crowther RA, Daniel SE, Goedert M (2000). Characterisation of isolated α-synuclein filaments from substantia nigra of Parkinson’s disease brain. Neurosci Lett 292:128–130. [DOI] [PubMed] [Google Scholar]
- Duda JE, Giasson BI, Mabon ME, Lee VMY, Trojanowski JQ (2002). Novel antibodies to α-synuclein show abundant striatal pathology in Lewy body diseases. Ann Neurol 52:205–210. [DOI] [PubMed] [Google Scholar]
- Fernandez CO, Hoyer W, Zweckstetter M, Jares-Erijman EA, Subramaniam V, Griesinger C, Jovin TM (2004). NMR of α-synuclein-polyamine complexes elucidates the mechanism and kinetics of induced aggregation. EMBO J 23:2039–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T (2002). α-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol 4:160–164. [DOI] [PubMed] [Google Scholar]
- Forno LS (1996). Neuropathology of Parkinson’s disease. J Neuropathol Exp Neurol 55:259–272. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Miyachi S, Paletzki R, Brown P (2002). D1 dopamine receptor supersensitivity in the dopamine-depleted striatum results from a switch in the regulation of ERK1/2/MAP kinase. J Neurosci 22:5042–5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Murray IVJ, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VMY (2000). Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions. Science 290:985–989. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VMY (2002). Neuronal α-synucleinopathy with severe movement disorder in mice expressing A53T human α-synuclein. Neuron 34:521–533. [DOI] [PubMed] [Google Scholar]
- Goedert M (2001). Alpha-synuclein and neurodegenerative diseases. Nature Rev Neurosci 2:492–501. [DOI] [PubMed] [Google Scholar]
- Goers J, Uversky VN, Fink AL (2003). Polycation-induced oligomerization and accelerated fibrillation of human α-synuclein in vitro. Protein Sci 12:702–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto M, Takeda A, Hsu LJ, Takenouchi T, Masliah E (1999). Role of cytochrome c as a stimulator of α-synuclein aggregation in Lewy body disease. J Biol Chem 274:28849–28852. [DOI] [PubMed] [Google Scholar]
- Hornykiewicz O (1998). Biochemical aspects of Parkinson’s disease. Neurology 51:S2–S9. [DOI] [PubMed] [Google Scholar]
- Ibanez P, Bonnet AM, Débarges B, Lohmann E, Tison F, Pollak P, Agid Y, Dürr A, Brice A (2004). Causal relation between α-synuclein gene duplication and familial Parkinson’s disease. Lancet 364:1169–1171. [DOI] [PubMed] [Google Scholar]
- Isles AR, Humby T, Walters E, Wilkinson LS (2004). Common genetic effects on variation in impulsivity and activity in mice. J Neurosci 24:6733–6740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakes R, Spillantini MG, Goedert M (1994). Identification of two distinct synucleins from human brain. FEBS Lett 345:27–32. [DOI] [PubMed] [Google Scholar]
- Jakes R, Crowther RA, Lee VMY, Trojanowski JQ, Iwatsubo T, Goedert M (1999). Epitope mapping of LB509, a monoclonal antibody directed against human α-synuclein. Neurosci Lett 269:13–16. [DOI] [PubMed] [Google Scholar]
- Kahle PJ, Neumann M, Ozmen L, Muller V, Jacobsen H, Schindzielorz A, Okochi M, Leimer U, van Der Putten H, Probst A, Kremmer E, Kretzschmar HA, Haass C (2000). Subcellular localization of wild-type and Parkinson’s disease-associated mutant α-synuclein in human and transgenic mouse brain. J Neurosci 20:6365–6373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle PJ, Neumann M, Ozmen L, Muller V, Odoy S, Okamoto N, Jacobsen H, Iwatsubo T, Trojanowski JQ, Takahashi H, Wakabayashi K, Bogdanovic N, Riederer P, Kretzschmar HA, Haass C (2001). Selective insolubility of α-synuclein in human Lewy body diseases is recapitulated in a transgenic mouse model. Am J Pathol 159:2215–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzenschlager R, Lees AJ (2004). Olfaction and Parkinson’s syndromes: its role in differential diagnosis. Curr Opin Neurol 17:417–423. [DOI] [PubMed] [Google Scholar]
- Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schöls L, Riess O (1998). Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat Genet 18:106–108. [DOI] [PubMed] [Google Scholar]
- Lee MK, Stirling W, Xu Y, Xu X, Qui D, Mandir AS, Dawson TM, Copeland NG, Jenkins NA, Price DL (2002). Human α-synuclein-harboring familial Parkinson’s disease-linked Ala-53-Thr mutation causes neurodegenerative disease with α-synuclein aggregation in transgenic mice. Proc Natl Acad Sci USA 99:8968–8973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, West N, Colla E, Pletnikova O, Troncoso JC, Marsh L, Dawson TM, Jäkälä P, Hartmann T, Price DL, Lee MK (2005). Aggregation promoting C-terminal truncation of α-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations. Proc Natl Acad Sci USA 102:2162–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C-W, Corboy MJ, DeMartino GN, Thomas PJ (2003). Endoproteolytic activity of the proteasome. Science 299:408–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CW, Giasson BI, Lewis KA, Lee VM, Demartino GN, Thomas PJ (2005). A precipitating role for truncated α-synuclein and the proteasome in alpha-synuclein aggregation: implications for pathogenesis of Parkinson disease. J Biol Chem 280:22670–22678. [DOI] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L (2000). Dopaminergic loss and inclusion body formation in α-synuclein mice: implications for neurodegenerative disorders. Science 287:1265–1269. [DOI] [PubMed] [Google Scholar]
- Marazziti D, Golini E, Mandillo S, Magrelli A, Witke W, Matteoni R, Tocchini-Valentini GP (2004). Altered dopamine signaling and MPTP resistance in mice lacking the Parkinson’s disease-associated GPR37/parkin-associated endothelin-like receptor. Proc Natl Acad Sci USA 101:10189–10194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka Y, Vila M, Lincoln S, McCormack A, Picciano M, LaFrancois J, Yu X, Dickson D, Langston WJ, McGowan E, Farrer M, Hardy J, Duff K, Przedborski S, Di Monte DA (2001). Lack of nigral pathology in transgenic mice expressing human α-synuclein driven by the tyrosine hydroxylase promoter. Neurobiol Dis 8:535–539. [DOI] [PubMed] [Google Scholar]
- Min N, Joh TH, Kim KS, Peng C, Son JH (1994). 5′ upstream DNA sequence of the rat tyrosine hydroxylase gene directs high-level and tissue-specific expression to catecholaminergic neurons in the central nervous system of transgenic mice. Mol Brain Res 27:281–289. [DOI] [PubMed] [Google Scholar]
- Mishizen-Eberz AJ, Guttmann RP, Giasson BI, Day GA, Hodara R, Ischiropoulos H, Lee VMY, Trojanowski JQ, Lynch DR (2003). Distinct cleavage patterns of normal and pathologic forms of α-synuclein by calpain I in vitro. J Neurochem 86:836–847. [DOI] [PubMed] [Google Scholar]
- Murray IV, Giasson BI, Quinn SM, Koppaka V, Axelsen PH, Ischiropoulos H, Trojanowski JQ, Lee VMY (2003). Role of α-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry 42:8530–8540. [DOI] [PubMed] [Google Scholar]
- Neumann M, Kahle PJ, Giasson BI, Ozmen L, Borroni E, Spooren W, Müller V, Odoy S, Fujiwara H, Hasegawa M, Iwatsubo T, Trojanowski JQ, Kretzschmar HA, Haass C (2002). Misfolded proteinase K-resistant hyperphosphorylated α-synuclein in aged transgenic mice with locomotor deterioration and in human α-synucleinopathies. J Clin Invest 110:1429–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Müller V, Kretzschmar HA, Haass C, Kahle PJ (2004). Regional distribution of proteinase K-resistant α-synuclein correlates with Lewy body disease stage. J Neuropathol Exp Neurol 63:1225–1235. [DOI] [PubMed] [Google Scholar]
- Norris EH, Giasson BI, Hodara R, Xu S, Trojanowski JQ, Ischiropoulos H, Lee VMY (2005). Reversible inhibition of α-synuclein fibrillization by dopaminochrome-mediated conformational alterations. J Biol Chem 280:21212–21219. [DOI] [PubMed] [Google Scholar]
- O’Connell MT, Portas CM, Sarna GS, Curzon G (1991). Effect of p-chlorophenylalanine on release of 5-hydroxytryptamine from the rat frontal cortex in vivo. Br J Pharmacol 102:831–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin RJ, Payton JE, Barnett DH, Wraight CL, Woods WS, George JM (2003). Epitope mapping and specificity of the anti-α-synuclein monoclonal antibody Syn-1 in mouse brain and cultured cell lines. Neurosci Lett 349:133–135. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanasiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, DiIorio G, Golbe LI, Nussbaum RL (1997). Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 276:2045–2047. [DOI] [PubMed] [Google Scholar]
- Rathke-Hartlieb S, Kahle PJ, Neumann M, Ozmen L, Haid S, Okochi M, Haass C, Schulz JB (2001). Sensitivity to MPTP is not increased in Parkinson’s disease-associated mutant α-synuclein transgenic mice. J Neurochem 77:1181–1184. [DOI] [PubMed] [Google Scholar]
- Richfield EK, Thiruchelvan MJ, Cory-Slechta DA, Wuertzer C, Gainetdinov RR, Caron MG, Di Monte DA, Federoff HJ (2002). Behavioral and neurochemical effects of wild-type and mutated human α-synuclein in transgenic mice. Exp Neurol 175:35–48. [DOI] [PubMed] [Google Scholar]
- Rochet JC, Conway KA, Lansbury PT (2000). Inhibition of fibrillization and accumulation of prefibrillar oligomers in mixtures of human and mouse α-synuclein. Biochemistry 39:10619–10626. [DOI] [PubMed] [Google Scholar]
- Sampathu DM, Giasson BI, Pawlyk AC, Trojanowski JQ, Lee VMY (2003). Ubiquitination of α-synuclein is not required for formation of pathological inclusions in α-synucleinopathies. Am J Pathol 163:91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA (2000). Fiber diffraction of synthetic α-synuclein filaments shows amyloid-like cross-β conformation. Proc Natl Acad Sci USA 97:4897–4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K (2003). α-Synuclein locus triplication causes Parkinson’s disease. Science 302:841. [DOI] [PubMed] [Google Scholar]
- Specht CG, Schoepfer R (2001). Deletion of the α-synuclein locus in a subpopulation of C57BL/6J inbred mice. BMC Neurosci 2:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997). α-Synuclein in Lewy bodies. Nature 388:839–840. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998a). α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci USA 95:6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M (1998b). Filamentous α-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett 251:205–208. [DOI] [PubMed] [Google Scholar]
- Takao M, Ghetti B, Yoshida H, Piccardo P, Narain Y, Murrell JR, Vidal R, Glazier BS, Jakes R, Tsutsui M, Spillantini MG, Crowther RA, Goedert M, Koto A (2004). Early-onset dementia with Lewy bodies. Brain Pathol 14:137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiruchelvam MJ, Powers JM, Cory-Slechta DA, Richfield EK (2004). Risk factors for dopaminergic neuron loss in human α-synuclein transgenic mice. Eur J Neurosci 19:845–854. [DOI] [PubMed] [Google Scholar]
- Tofaris GK, Layfield R, Spillantini MG (2001). α-Synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett 509:22–26. [DOI] [PubMed] [Google Scholar]
- Tofaris GK, Razzaq A, Ghetti B, Lilley KS, Spillantini MG (2003). Ubiquitination of α-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function. J Biol Chem 278:44405–44411. [DOI] [PubMed] [Google Scholar]
- van der Putten H, Wiederhold KH, Probst A, Barbieri S, Mistl C, Danner S, Kauffmann S, Hofele K, Spooren WP, Ruegg MA, Lin S, Caroni P, Sommer B, Tolnay M, Bilbe G (2000). Neuropathology in mice expressing human α-synuclein. J Neurosci 20:6021–6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG (2004). The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 55:164–173. [DOI] [PubMed] [Google Scholar]