

Abstract
The amyloid precursor protein (APP) is critical in the pathogenesis of Alzheimer's disease. The question of its normal biological function in neurons, in which it is predominantly located at synapses, is still unclear. Using autaptic cultures of hippocampal neurons, we demonstrate that hippocampal neurons lacking APP show significantly enhanced amplitudes of evoked AMPA- and NMDA-receptor-mediated EPSCs. The size of the readily releasable synaptic vesicle pool was also increased in neurons lacking APP, whereas the release probability was not affected. In addition, the analysis of spontaneous miniature synaptic currents revealed an augmented frequency in neurons lacking APP, whereas the amplitude of miniature synaptic currents was not found to be altered. Together, these findings strongly indicate that lack of APP increases the number of functional synapses. This hypothesis is further supported by morphometric immunohistochemical analysis revealing an increase of synaptophysin-positive puncta per cultured APP knock-out neuron. In conclusion, lack of APP affects synapse formation and transmission in cultured hippocampal neurons.
Keywords: amyloid precursor protein, Alzheimer's disease, synaptic transmission, amyloid, synaptic vesicle, patch clamp
Introduction
Alzheimer's disease (AD) is the most common form of dementia in the elderly. The pathology of AD is characterized by a progressive neuronal dysfunction, reactive gliosis, and the deposition of amyloid-β (Aβ) plaques in the brain. These mainly consist of Aβ peptides 1–40 and 1–42, which are derived from proteolysis of the amyloid precursor protein (APP), a type I transmembrane protein. The duration and severity of cognitive impairment in AD patients is closely associated with synaptic loss (Terry et al., 1991), leading to the notion that synaptic dysfunction is a critical element in the pathogenesis of AD (Selkoe, 2002). Given the pivotal role of APP in AD pathogenesis, it is therefore essential to understand its physiological function, particularly its possible involvement in synaptic transmission.
The proteolytic processing pathways leading to the formation of Aβ from APP have been well characterized (Selkoe, 2000). APP is delivered to the surface membrane in which it is subjected to proteolytic processing by α-secretase. APP molecules that fail to be cleaved by α-secretase can be internalized into endocytic compartments and subsequently cleaved by β-secretase and γ-secretase to generate Aβ. A fraction of Aβ peptides is also generated in the Golgi apparatus and, to a lesser extent, the endoplasmic reticulum. Aβ peptides generated in the Golgi and in recycling compartments are secreted into the extracellular space (Greenfield et al., 1999). Aβ peptides are generated only by the cleavage of APP and not by the cleavage of other APP protein family members such as Apl-1 in Caenorhabditis elegans (Daigle and Li, 1993), Appl in Drosophila (Rosen et al., 1989), APP-like protein 1 (APLP1) (Wasco et al., 1992) and APLP2 (Wasco et al., 1993; Slunt et al., 1994) in mammals, which all lack the Aβ domain. It follows that the function of the Aβ domain has to be different from the function of other domains of the APP family member proteins (Herms et al., 2004). The Aβ sequence is the least conserved part between the human and mouse APP sequences. Mouse APP (when compared with human) is processed poorly by β-secretase, resulting in approximately threefold lower amounts of Aβ peptide. This suggests that Aβ peptides may be less critical in mouse than in human brain (De Strooper et al., 1995). This is one possible explanation why mice lacking APP only have subtle phenotypes, including impairment in behavior and long-term potentiation in old age (Zheng et al., 1995; Dawson et al., 1999; Phinney et al., 1999; Seabrook et al., 1999). Recently, however, convincing evidence was presented showing that APP processing has a physiological role in synaptic transmission in mice (Kamenetz et al., 2003). Neuronal activity was found to positively modulate the secretion of Aβ peptides. In turn, Aβ was found to suppress excitatory synaptic transmission. It was suggested that Aβ generation may be part of a negative feedback mechanism that controls neuronal excitability. If this is true, then lack of APP and consequently a total lack in the production of Aβ peptides should have a physiological effect on synaptic transmission as well. To validate this hypothesis, we studied synaptic transmission in autaptic hippocampal cultures from APP knock-out (KO) mice. This preparation allows a detailed analysis of presynaptic and postsynaptic parameters of synaptic transmission.
Materials and Methods
APP knock-out mice.
The APP knock-out mice used in this study were described originally by Li et al. (1996). The wild-type (WT) mice used in this study are descendants of an F1 generation mouse produced by interbreeding the original parental strain (129Ola and C57BL/6 mice) used to generate the APP knock-out mice initially. Littermates were generated by crossing heterozygous C57BL/6J × 129/Ola APP+/− mice. For genotyping from tail biopsies, a three-primer PCR was used amplifying for the WT allele a 650 bp fragment with primers UM44 (5′-GAGACGAGGACGCTCAGTCCTAGGG-3′) and UM42 (5′-ATCACCTGGTTCTAATCAGAGGCCC-3′) flanking exon 17 and, for the mutant allele, a 430 bp fragment with primers UM42 located in intron 17 and P3-hygro (5′-CGAGATCAGCAGCCTCTGTTCCACA-3′) derived from the phosphoglycerate kinase–hygro cassette, respectively.
Cell culture.
Microisland cultures of mouse hippocampal neurons were prepared and maintained as described previously (Bekkers and Stevens, 1991; Rosenmund et al., 1993, 1995). In brief, astrocyte feeder wells enriched in type 1 astrocytes (>95%) were first prepared from postnatal day 1 mouse hippocampal tissue. After dissection and removal of the chorioid plexus and meninges, the material was dissociated and trypsinized. Cells were grown in culture medium consisting of DMEM with 4.5 g/liter glucose (Pan, Aidenbach, Germany), 5% fetal bovine serum (Pan), 100 IU/ml penicillin/streptomycin (Invitrogen, Karlsruhe, Germany), N2 supplement (Invitrogen), Glutamax (Invitrogen), and Mito+ serum extender (BD Biosciences, Bedford, MA) at their indicated concentrations. Cells were kept at 37°C in a 10% CO2-enriched atmosphere. Once a monolayer was achieved, cells were shaken for 12 h so that only astrocytes remained attached to the flask bottom. Astrocytes were trypsinized and replated onto microdot-coated glass coverslips. Hippocampi were dissected from newborn mice pups and digested in DMEM solution containing 0.2 mg/ml cysteine, 100 mm CaCl2, 50 mm EDTA, and 10 U/ml papain (Roche, Mannheim, Germany) at 37°C. Digestion was stopped after 60 min, and tissue was gently triturated using fire-polished Pasteur pipettes. Cell suspensions, diluted to 5–8 × 103 cells/ml, were then added to the prepared astrocyte feeder wells (see above). To prevent excessive proliferation of astrocytes, the anti-mitotic agent 5-fluoro-2′-deoxyuridine (8.1 mm) was applied to the cultures on the day after neuronal plating. APP knock-out and wild-type hippocampal neurons were cultured on astrocytic feeder layers prepared from wild-type mice. We did not use APP knock-out astrocytes (which would have had the advantage of an exclusion of astrocytic-derived Aβ) because we did not know whether the trophic function of astrocytes in hippocampal neuronal cultures would suffer from lack of APP expression in astrocytes. For the γ-secretase inhibitor experiment, 1 μm N-[N.(3,5-difluorophenacetyl-l-alanyl)]-S-phenylglycine t-butyl ester (DAPT) (Calbiochem Merck, Darmstadt, Germany) was added to the neuronal cultures on the day of neuronal plating. Medium was partly exchanged every week and new DAPT was added.
For the Aβ incubation studies, freshly diluted monomeric Aβ42 (10 nm; Bachem, Bubendorf, Switzerland) was added to the autaptic cultures, and electrophysiological measurements were performed after 48 h.
For the medium exchange experiment, APP knock-out medium was replaced by conditioned wild-type medium 1 week after neuronal plating and the following week.
Electrophysiology.
For each experiment, approximately equal numbers of cells from the respective genotypes were measured in parallel and blindly on the same day in vitro [18–20 d in vitro (DIV)]. Whole-cell recordings were performed using an EPC 9 amplifier (HEKA Elektronik, Lambrecht/Pfalz, Germany). The standard extracellular medium contained the following: 140 mm NaCl, 2.4 mm KCl, 10 mm HEPES, 10 mm glucose, 4 mm CaCl2, 4 mm MgCl2, and 15 μm bicuculline, 300 mOsm, pH 7.3. NMDA EPSCs were measured at 4 mm external Ca2+ (except for 2.7 mm in the experiment with MK-801 (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine hydrogen maleate) in the presence of 5 μm 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) and 10 μm glycine and in the absence of external Mg2+. Internal pipette recording solution consisted of the following: 10 mm NaCl, 125 mm K-gluconate, 1 mm EGTA, 4.6 mm MgCl2, 4 mm Na2ATP, 15 mm creatine phosphate, and 20 U/ml phosphocreatine kinase, 300 mOsm, pH 7.3. Data were filtered at 2 kHz and acquired at 10 kHz using Pulse 8.5 software (HEKA Elektronik). Patch electrodes had resistances of 3–4 MΩ. Series resistance and capacity of the electrodes were compensated; only recordings with access resistance below 15 MΩ and with an initial membrane potential less than −50 mV (measured in the current-clamp mode shortly after cell access was obtained) were included in the analysis. Access resistance and cell capacities (5–25 pF) were compensated. Cells were voltage clamped at −70 mV, except for 1 ms depolarizations to 0 mV to elicit an action potential-like stimulus. The rise time constant (10–90%) of the synaptic currents was determined by fitting a linear function to the activating current, and the decay time constant was determined by fitting a single exponential function to the current decay. Detection of miniature EPSCs (mEPSCs) was usually performed for at least 60 s in presence of 200 nm tetrodotoxin (TTX) (Biotrend, Köln, Germany). Data were analyzed with a template detection program (Axograph 4.1; Molecular Devices, Palo Alto, CA). Threshold for detection was set to 3.5 times the baseline SD. Captured mEPSCs of individual cells were averaged to determine mean amplitude and charge per cell. Application of hypertonic solution for pool size determination was made by pulsed application of 500 mm sucrose to the extracellular solution for 4 s using a fast perfusion system (Warner Instruments, Hamden, CT). Evoked responses and responses to hypertonic sucrose solutions were always recorded successively from the same cell. The readily releasable vesicle pool (RRP) per cell was calculated by dividing the charge of the transient sucrose response with the mean mEPSC charge of the same cell, if available. To further determine the mean vesicular release probability (Pvr), the number of vesicles released by a single action potential-like stimulus was divided by the number of vesicles in the RRP. Recordings and data analysis were performed blindly on each sample.
Immunocytochemistry.
Hippocampal cultures of 18–20 DIV were used for immunocytochemistry. For fluorescent microscopic determinations of synaptic density in neurons, cultures were fixed for 10 min with 4% paraformaldehyde, blocked, and permeabilized with PBS containing 2% BSA, 10% goat serum (DakoCytomation, Hamburg, Germany), and 0.3% Triton X-100 (Calbiochem, La Jolla, CA) for 30 min. Cells were incubated with rabbit polyclonal anti-microtubule-associated protein 2 (MAP-2) (1:100; Chemicon, Hampshire, UK) for 60 min, washed with PBS, and incubated with FITC-conjugated goat anti-rabbit IgG (1:100) for another 60 min. After another washing step, fluorescent-labeled mouse monoclonal anti-synaptophysin (1:1000; Synaptic Systems, Göttingen, Germany) was added, incubated for 60 min, and finally fixed in fluorescent mounting medium (DakoCytomation). Images were acquired on an Axiovert 200M microscope (Zeiss, Oberkochen, Germany) with a 63× LD Achroplan objective and Axiovision 4.5 software (Zeiss). For quantification of the total dendritic length and total amount of synapses, analySIS software (Soft Imaging System, Münster, Germany) was used. The summary graph represents four independent experiments using different culture preparations from APP knock-out animals and littermate controls.
All chemicals were obtained from Sigma-Aldrich (Deisenhofen, Germany), unless otherwise noted.
Immunohistochemistry.
Six hemispheres (three of each genotype) of 20-d-old and 11-month-old APP knock-out animals and littermate controls were removed and fixed in 4% paraformaldehyde overnight. Coronal 50 μm free-floating sections were prepared with a VT 1000S vibratome (Leica, Wetzlar, Germany) and immunostained with anti-synaptophysin and indocarbo-cyanine 3 (Cy3)-conjugated streptavidin. Briefly, sections were placed in PBS, blocked with antibody diluent (Ventana Medical Systems, Illkirch, France), and then incubated overnight at 4°C with the polyclonal antibody against synaptophysin (1:300; Chemicon). This step was followed by incubation in biotinylated swine anti-rabbit IgG (1:150; DakoCytomation), and finally the sections were fluorescence labeled with Cy3-conjugated streptavidin (1:25; Dianova, Hamburg, Germany). The sections were mounted on precoated glass slides (Super-frost-plus; Menzel, Braunschweig, Germany) and covered (Coverquick; Labonord, Templemars, France). All sections were processed simultaneously under the same conditions. The immunofluorescent labeling protocol was repeated three times to assess the reproducibility of results. Immunolabeled brain sections were viewed with a Zeiss 40×/0.8 IR Achroplan objective on a Zeiss Axioskop 2FS MOT LSM510 confocal microscope.
Cy3 was excited with a helium/neon laser at 543 nm, and the emitted fluorescence was detected with a long-pass, 560 nm emission filter. Frames of 230 × 230 μm size were scanned with 0.22 μm/pixel, and the pinhole diameter was set to an area unit of 1. The z distance of each z stack ranged from 10 to 15 μm and consisted of 20–30, 0.5 μm spaced images.
Synaptophysin levels were assessed in the stratum radiatum and stratum moleculare of the hippocampus, in three coronal sections per animal. Two serial optical z stacks (one for each marker) of the synaptophysin-labeled structures were collected from each section. All series of z stacks from each section were maximum intensity projected and acquired under standard conditions maintaining the same gain, aperture, and black level settings. This method of analysis was evaluated previously by Masliah et al. (1990a,b, 1991). The digitized images were then analyzed regarding the signal intensity and density (covered area of immunohistochemical-positive objects) of synaptophysin-labeled structures (PictureAnalyzer Software; Bjarne Krebs, Zentrum für Neuropathologie und Prion Forschung, Munich, Germany). A threshold was set so that all objects with an intensity value below the threshold were set equal to the background value. For each mouse, the values obtained for the stratum radiatum and stratum moleculare of the hippocampus were averaged.
Western blot.
For measurement of the expression of synaptophysin in hippocampus, coronal brain sections were collected on glass slides and Nissl stained. The hippocampal region was then separately lysed and subjected to SDS-PAGE as reported previously (Krebs et al., 2006). After electrotransfer of proteins to the polyvinylidene fluoride membrane, immunoblotting was followed by the incubation with primary antibody against synaptophysin (1:5000; Chemicon) and β-actin (1:5000, actin I-19; Santa Cruz Biotechnology, Heidelberg, Germany) overnight. The visualization was performed with a secondary antibody coupled to alkaline phosphatase (polyclonal goat anti-mouse and anti-rabbit Ig/alkaline phosphatase; DakoCytomation) and chromogene nitroblue tetrazolium salt with 5-bromo-4-chloro-3-indolylphosphate. Western blots were scanned with a high-resolution flatbed scanner, and the density of bands was determined using Total Lab version 2.01 software (Nonlinear Dynamics, Newcastle-upon-Tyne, UK).
Statistical analysis.
All error bars indicate the SEM. All results are reported as ±SEM. A value <0.05 was considered as statistically significant (Student's t test).
Results
Evoked neurotransmitter release in wild-type and APP knock-out neurons
To determine whether loss of APP leads to alterations in neurotransmitter release, patch-clamp recordings were made from cultured primary hippocampal neurons. Microisland cultures were used to induce the formation of autaptic synaptic connections on individual neurons (Bekkers and Stevens, 1991). Synaptic transmission could then be evoked by briefly depolarizing a voltage-clamped neuron, while using the same patch pipette to measure the resulting EPSCs. The advantage of this method over analysis of synaptic transmission in conventional hippocampal neuronal cultures or brain slice preparations is that this approach results in the stimulation of all synapses of a single neuron. Thus, the evocation of the postsynaptic current is not dependent on the stimulus intensity at all. Therefore, the synaptic response does not need to be normalized to stimulus intensity, giving a more accurate measurement of the synaptic strength of single neurons. Using this approach, we concentrated our analysis on glutamatergic neurons because they are much more abundant than inhibitory GABAergic cells in autaptic cultures. We thus recorded AMPA- and NMDA-receptor-mediated EPSCs from 24–36 WT and 23–33 APP KO neurons from three to five cultures. All analyses were performed simultaneously on knock-out and control cultures derived from littermates to avoid culture-specific artifacts. No differences were observed in the resting membrane potential between wild-type (59 ± 3 mV; n = 11) and APP knock-out (60 ± 4 mV; n = 11) autaptic hippocampal neurons at 18–20 d in culture.
We then tested the action potential-evoked neurotransmitter release. Synaptic responses were evoked by a brief somatic depolarization (1 ms depolarization from −70 mV holding potential to 0 mV) and measured as peak inward currents a few ms after action potential induction (Fig. 1A,B). The amplitude of the AMPA-mediated EPSCs was enhanced ∼1.7-fold in APP knock-out cells compared with control littermate neurons (2.33 ± 0.34 nA for WT and 4.05 ± 0.26 nA for KO; p < 0.05) (Fig. 1C). Similarly, NMDA-receptor-mediated EPSCs, analyzed by blocking AMPA receptors using CNQX (5 μm), revealed a 47% increased amplitude in APP knock-out neurons (1.46 ± 0.19 nA for WT and 2.14 ± 0.24 nA for KO; p < 0.05) (Fig. 1C). No significant differences were observed in the kinetics (rise time and decay time constant τ) of both AMPA- and NMDA-receptor-mediated EPSCs between APP knock-out neurons and control cells (Table 1).
Figure 1.
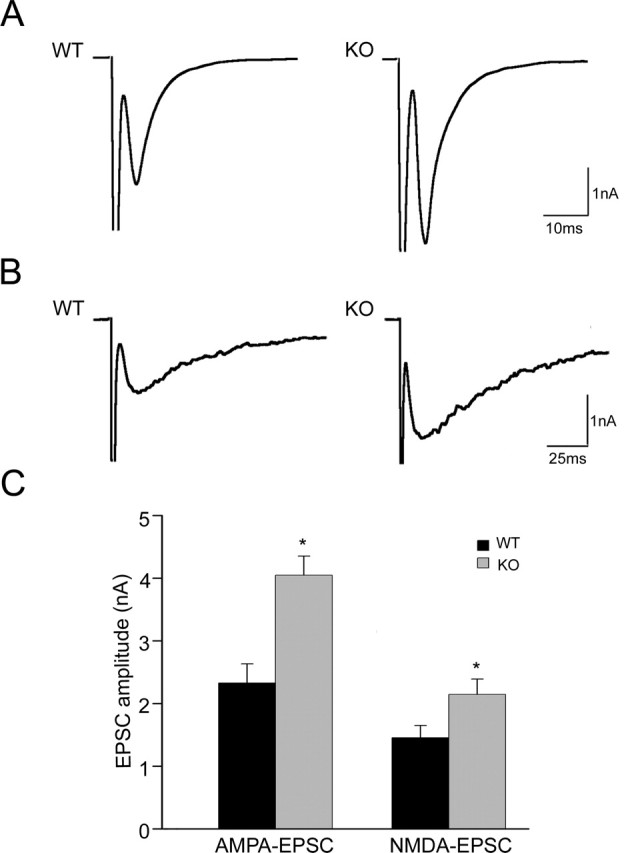
Evoked neurotransmitter release in WT and APP KO neurons. A, Representative AMPA EPSC traces from wild-type and APP knock-out neurons. B, Representative NMDA EPSC traces from wild-type and APP knock-out neurons. C, Mean AMPA and NMDA EPSC amplitudes in neurons from wild-type (n = 24 and n = 35, respectively) and APP knock-out (n = 23 and n = 33, respectively) mice (∗p < 0.05).
Table 1.
Summary of evoked EPSC kinetics
| Rise time (ms) | τ (ms) | |
|---|---|---|
| AMPA EPSC | ||
| WT | 1.88 ± 0.14 | 5.87 ± 0.38 |
| KO | 2.30 ± 0.18 | 6.63 ± 0.32 |
| NMDA EPSC | ||
| WT | 7.94 ± 0.71 | 69.76 ± 5.11 |
| KO | 7.33 ± 0.63 | 58.03 ± 5.40 |
Data are expressed as mean ± SEM; n = 20–31 cells per genotype.
Thus, the strength of excitatory synaptic transmission was found to be enhanced in autaptic hippocampal neurons lacking APP. Factors possibly affecting this change include an increase in postsynaptic responsiveness, an alteration in presynaptic release parameters, and a higher number of synapses.
Spontaneous neurotransmitter release in wild-type and APP knock-out neurons
As an indicator of postsynaptic responsiveness, we next examined spontaneous mEPSCs (Fig. 2A,B). We found that the frequency of mEPSCs was ∼2.4-fold higher in APP knock-out neurons than in controls (3.2 ± 0.6 s−1 for WT and 7.6 ± 1.1 s−1 for KO; p < 0.05) (Fig. 2C). The mean mEPSC amplitude, however, was not affected (23.7 ± 1.2 pA for WT and 22.9 ± 1.0 pA for KO; p > 0.05) (Fig. 2C), indicating that the responsiveness and number of postsynaptic receptors is not altered by a lack of APP. In addition, we approximated the number of vesicles released by an action potential-like stimulus by dividing the charge of evoked responses by that of a single mEPSC. For those neurons in which both EPSCs and mEPSCs were measured, the quantity of vesicles released during an EPSC was determined and found to be almost twice as high as in neurons lacking APP (306 ± 67 for WT and 593 ± 78 for KO; p < 0.05) (Fig. 2C).
Figure 2.

Spontaneous neurotransmitter release in WT and APP KO neurons. A, B, Representative mEPSC traces from wild-type and APP knock-out neurons in the presence of 200 nm TTX. C, Left, mEPSC frequency in neurons from wild-type (n = 25) and APP knock-out (n = 23) mice. Middle, mEPSC amplitude in neurons from wild-type (n = 25) and APP knock-out (n = 23) mice. Right, Estimated EPSC vesicle number per evoked EPSC in wild-type (n = 10) and APP knock-out (n = 11) neurons (∗p < 0.05).
Readily releasable vesicle pool and release probability in wild-type and APP knock-out neurons
Presynaptic release is thought to depend on the size of the RRP and release probability (Pr). We therefore performed a detailed analysis of the sizes of RRPs in neurons of APP knock-out mice and their littermate controls. The fact that the mEPSC rates were significantly different between the two genotypes already suggested that the size of the RRP pool is changed in the absence of APP. The sizes of RRPs were quantified by measuring the response of cells lacking APP and their wild-type controls to pulsed application of 500 mm hypertonic sucrose solution for 3–4 s. This treatment induces the release of the entire pool of readily releasable vesicles, which in turn leads to a transient inward current followed by a steady current component (Bekkers and Stevens, 1991; Rosenmund and Stevens, 1996). We calculated the amount of RRP vesicles by dividing the total charge of the transient current component after application of hypertonic sucrose by the charge of a single mEPSC measured from the same cell. Neurons lacking APP showed significantly more releasable vesicles in their RRP compared with control cells (5.36 ± 0.87 × 103 for WT and 16.35 ± 2.94 × 103 for KO; p < 0.005) (Fig. 3A). No differences were observed in the basal rate of RRP refilling, which was estimated by averaging the ratio of the charge of a second hypertonic sucrose response over the initial sucrose response measured 3.5 s before (0.52 ± 0.02 for WT and 0.57 ± 0.03 for KO) (Fig. 3B).
Figure 3.
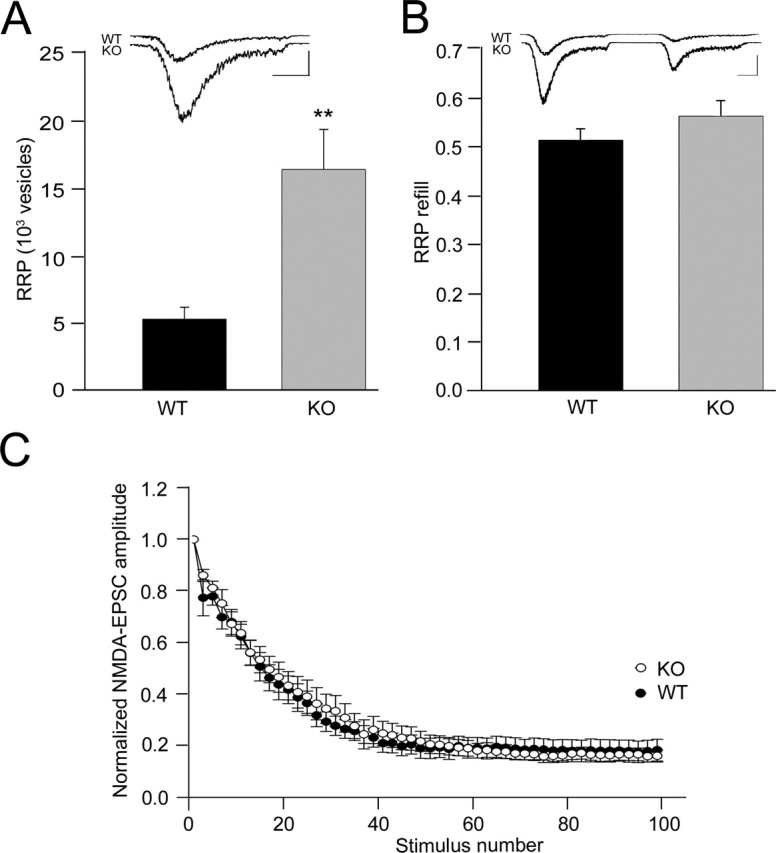
Estimated RRP, RRP refilling, and release probability in WT and APP KO neurons. A, Estimated RRP vesicles for wild-type (n = 10) and APP knock-out (n = 11) mice. Inset shows representative traces. Calibration: 1 s, 0.5 nA. ∗∗ p < 0.005. B, Average ratio of the second hypertonic sucrose response to the initial sucrose response measured 4 s before wild-type (n = 14) and APP knock-out (n = 18) mice. Inset shows sample traces. Calibration: 1 s, 0.5 nA). C, Analysis of MK-801 blocking rate in wild-type (n = 13) and APP knock-out (n = 10) mice. The average NMDA EPSC amplitude normalized to the first response in the presence of MK-801 (5 μm) is plotted against evoked EPSC stimulus number.
To assess whether the efficiency of the action potential-induced vesicle release is influenced by lack of APP, we analyzed Pvr. We determined Pvr by dividing the number of vesicles released during an action potential by the number of vesicles in the RRP. We found that Pvr did not vary significantly between neurons from control animals (5.4 ± 0.5%; n = 10) and from animals lacking APP (4.2 ± 0.5%; n = 11; p > 0.05). The Pvr values obtained are similar to the values obtained in other studies on autaptic hippocampal neurons (Reim et al., 2001). The synaptic release probability Pr depends on the number of fusion-competent vesicles per synapse and Pvr. Observing a significant difference only in the RRP size but not in Pvr between the two genotypes, we wanted to rule out a change in synaptic release probability Pr. We estimated Pr by analyzing the progressive block of NMDA-receptor-mediated synaptic currents in the presence of MK-801, which reflects the release probability across all synapses of a given neuron (Rosenmund et al., 1993). APP knock-out and control cells showed the typical amplitude decay in the presence of MK-801 (Fig. 3C). In summary, these results indicate that presynaptic vesicles seem to release efficiently and that the increase in synaptic responses in APP knock-out autapses can be attributed to the increase in RRP.
Prolonged repetitive and high-frequency stimulation in wild-type and APP knock-out neurons
In additional electrophysiological experiments, we analyzed the response of wild-type and APP knock-out neurons to prolonged repetitive and high-frequency stimulation (Fig. 4A,C).
Figure 4.
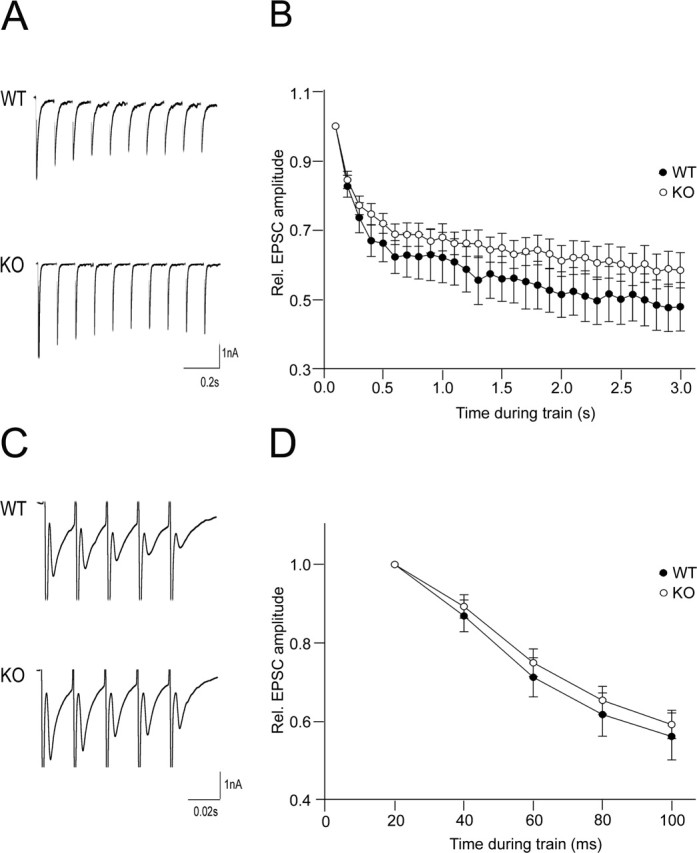
Prolonged repetitive and high-frequency stimulation of WT and APP KO neurons. A, Sample recordings of the first 10 responses in a 10 Hz train. B, Average EPSC amplitudes of wild-type (n = 19) or APP knock-out (n = 16) neurons during 10 Hz train normalized to the size of first response. C, Sample recordings of five responses in a 50 Hz train. D, Average EPSC amplitudes of wild-type (n = 22) and APP knock-out (n = 23) cells during 50 Hz train normalized to the size of first response.
Repetitive stimulation gives insights into the nature of the release probability, Pr, and the size of the RRP in the central synapse (Schneggenburger et al., 1999). For prolonged repetitive stimulation, we used a 10 Hz protocol and, for high-frequency stimulation, a 50 Hz protocol at 4 mm [Ca2+]out. Both in control and in APP knock-out neurons, the relative EPSC amplitude decreases gradually during repetitive stimulation at 10 Hz (Fig. 4B). In cells lacking APP, the decay of the relative EPSC amplitude is slightly reduced compared with control cells, without however reaching a significant level. Similar properties were observed in evoked responses elicited by high-frequency stimulation at 50 Hz (Fig. 4D).
γ-Secretase inhibitor enhances evoked and spontaneous neurotransmitter release in WT neurons
To determine whether the increase in EPSC amplitude of APP knock-out neurons is caused by loss of γ-secretase cleavage products, we blocked γ-secretase activity in wild-type neurons by addition of 1 μm DAPT. DAPT-treated wild-type neurons showed significantly higher AMPA EPSC responses compared with untreated neurons (2.76 ± 0.30 nA for control WT and 3.66 ± 0.30 nA for DAPT-treated WT neurons; p < 0.05) (Fig. 5A). The frequency of spontaneous neurotransmitter release was also found to be increased in DAPT-treated wild-type neurons (4.0 ± 1.1 s−1 for control WT and 10.2 ± 1.8 s−1 for DAPT-treated WT neurons; p < 0.05) (Fig. 5B). Both results indicate that the APP knock-out phenotype can be mimicked by adding DAPT to wild-type neurons.
Figure 5.
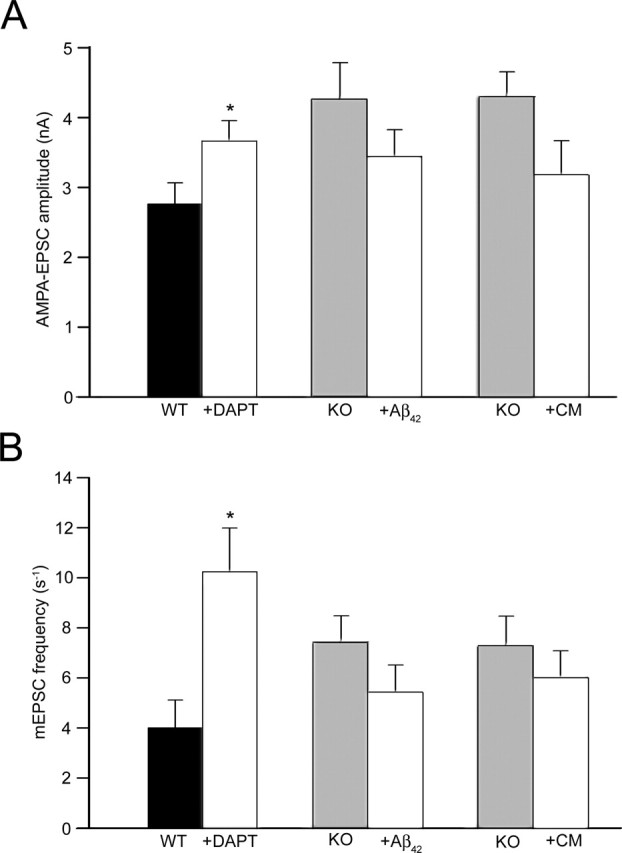
Evoked and spontaneous neurotransmitter release in neurons after incubation with DAPT, Aβ42, or conditioned medium (CM). A, AMPA EPSC amplitude in neurons from control wild-type (n = 15) compared with wild-type neurons treated with DAPT (n = 15) or control APP knock-out neurons (n = 15) compared with APP knock-out neurons treated with either Aβ42 (n = 15) or CM from wild-type cultures (n = 15). B, mEPSC frequency in control wild-type neurons (n = 15) compared with wild-type neurons treated with DAPT (n = 15) or control APP knock-out neurons (n = 15) compared with APP knock-out neurons treated with either Aβ42 (n = 15) or CM from wild-type cultures (n = 15) (∗p < 0.05).
Addition of Aβ42 or conditioned medium slightly decreases evoked and spontaneous neurotransmitter release in APP knock-out neurons
If the DAPT effect of mimicking the APP knock-out phenotype is mediated by a decreased production and extracellular release of Aβ peptides, then monomeric Aβ42 or conditioned medium of wild-type cells should negatively affect synaptic function in APP knock-out cells. Indeed, APP knock-out neurons treated with Aβ42 (10 nm) showed a slight but not significant reduction in AMPA EPSC responses compared with untreated APP knock-out neurons (4.25 ± 0.53 nA for control KO and 3.43 ± 0.40 nA for Aβ-treated KO neurons; p > 0.05) (Fig. 5A). The frequency of spontaneous neurotransmitter release was also slightly lower in APP knock-out neurons treated with Aβ42 (7.5 ± 1.0 s−1 for control KO and 5.4 ± 1.1 s−1 for Aβ42-treated KO cells; p > 0.05) (Fig. 5B). The incubation of APP knock-out neurons with conditioned wild-type medium revealed similar results. Both the amplitude of evoked EPSCs (4.29 ± 0.36 nA for control KO and 3.18 ± 0.49 nA for KO neurons treated with conditioned medium; p > 0.05) (Fig. 5A) and the frequency of the spontaneous neurotransmitter release were reduced, but not to statistically significant levels, in APP knock-out cells treated with conditioned medium (7.3 ± 1.2 s−1 for control KO and 6.0 ± 1.1 s−1 for KO cells treated with conditioned medium; p > 0.05) (Fig. 5B). In summary, extracellularly applied Aβ or conditioned medium from wild-type neurons have the tendency to reduce both the amplitude and the frequency of excitatory synaptic currents in autaptic hippocampal cultures of APP knock-out mice, without reaching significant values other than the effect of DAPT on wild-type neurons.
Increase in synapse formation in APP knock-out neurons
Based on the increase in evoked EPSCs and larger RRP in APP knock-out autapses, we next turned to assessing whether the absence of APP alters the actual number of synapses. Therefore, we performed immunofluorescence labeling using antibodies against MAP-2 and synaptophysin, a well characterized presynaptic marker (Fig. 6A) (Augustin et al., 1999). We first evaluated total dendritic length and found a 29% increase in dendritic length in APP knock-out neurons (1929 ± 113 μm for WT and 2486 ± 101 μm for KO; p < 0.0005) (Fig. 5B). We subsequently determined the total number of synapses per cell and observed significantly more synapses in cells lacking APP (209 ± 13 for WT and 312 ± 20 for KO; p < 0.0005) (Fig. 6B). However, the number of synaptophysin-positive puncta per 10 μm dendrite was not affected by loss of APP (1.19 ± 0.07 for WT, n = 55; 1.25 ± 0.06 for KO, n = 62; p > 0.05). Together, these results suggest that, in the absence of APP, neurons form more dendrites and consequently exhibit an enhanced formation of synapses. This can explain the previous results showing an increase in evoked EPSC amplitude and the size of the RRP in APP knock-out neurons as well as the enhanced mEPSC frequency.
Figure 6.
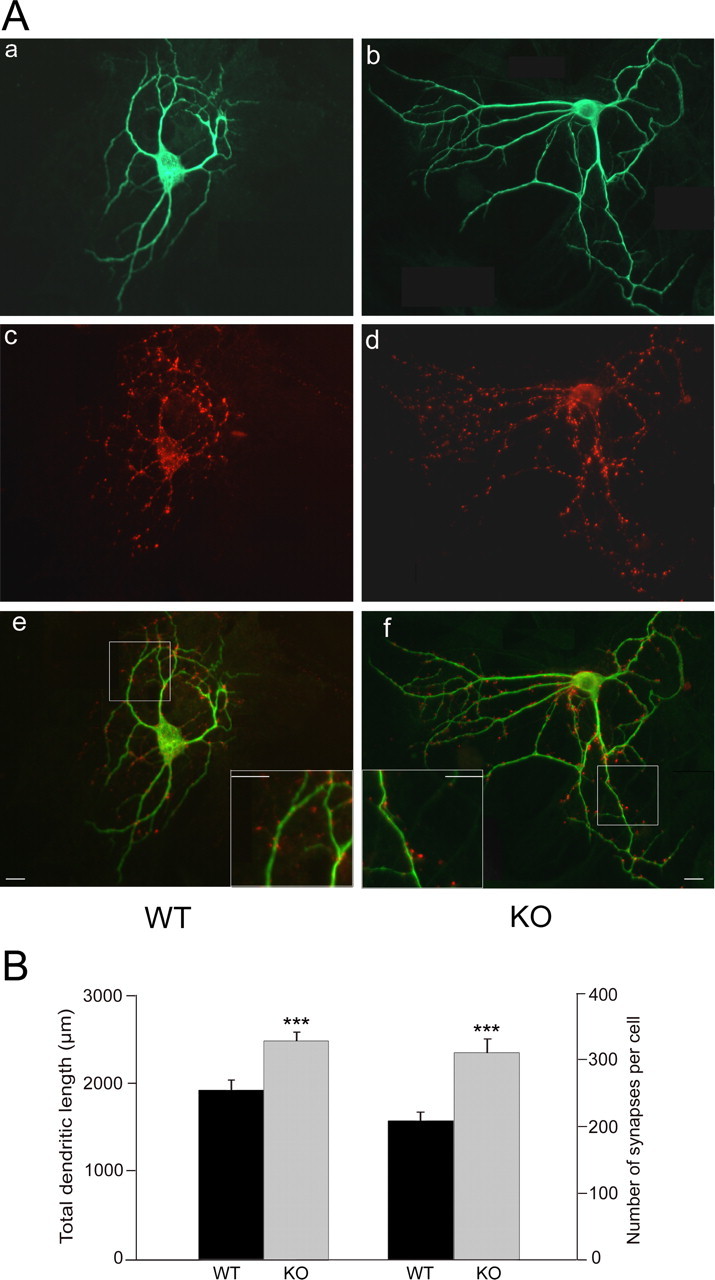
Structural analysis of synapses in microisland cultures from WT and APP KO neurons. A, Immunostains of autaptic neurons. Aa, Ab, Staining for the dendritic marker MAP-2 to assess the size of wild-type and APP knock-out neurons. Ac, Ad, Staining of the same culture for synaptophysin to assess the number of synapses. Ae, Af, Merged picture of MAP-2 and synaptophysin staining. Scale bar, 25 μm. B, Left, Total dendritic length of wild-type (n = 55) and APP knock-out (n = 62) neurons (left). Right, Average number of synapses of wild-type (n = 55) and APP knock-out (n = 62) neurons (∗∗∗p < 0.0005).
Staining intensity and expression of synaptophysin in hippocampus of wild-type and APP knock-out mice
To demonstrate that the increase in synapse formation in cultured APP knock-out neurons is also present in vivo, we performed immunohistochemistry on floating sections of young (20 d) and old (11 months) wild-type and APP knock-out animals. We then analyzed synaptophysin levels in the stratum radiatum and the stratum moleculare of the hippocampus by confocal microscopy (Fig. 7A). The average staining intensity of synaptophysin-positive presynaptic terminals in the stratum radiatum was 17% higher in 20-d-old mice lacking APP compared with wild-type mice (77.04 ± 3.05% for WT and 90.13 ± 1.95% for KO; p < 0.005) (Fig. 7B), whereas no significant difference could be observed in 11-month-old mice (85.77 ± 2.19% for WT and 88.50 ± 3.44% for KO; p > 0.05) (Fig. 7B). Evaluation of the average synaptophysin-staining intensity in the stratum moleculare showed an enhanced staining intensity in 20-d-old APP knock-out mice (75.87 ± 3.66% for WT and 92.81 ± 1.00% for KO; p < 0.005) (Fig. 7C). No difference in staining intensity in the stratum moleculare could be found in 11-month-old animals (69.73 ± 4.58% for WT and 72.90 ± 4.97% for KO; p > 0.05) (Fig. 7C). Altogether, these results are consistent with an enhanced synapse formation in young APP knock-out hippocampal neurons.
Figure 7.
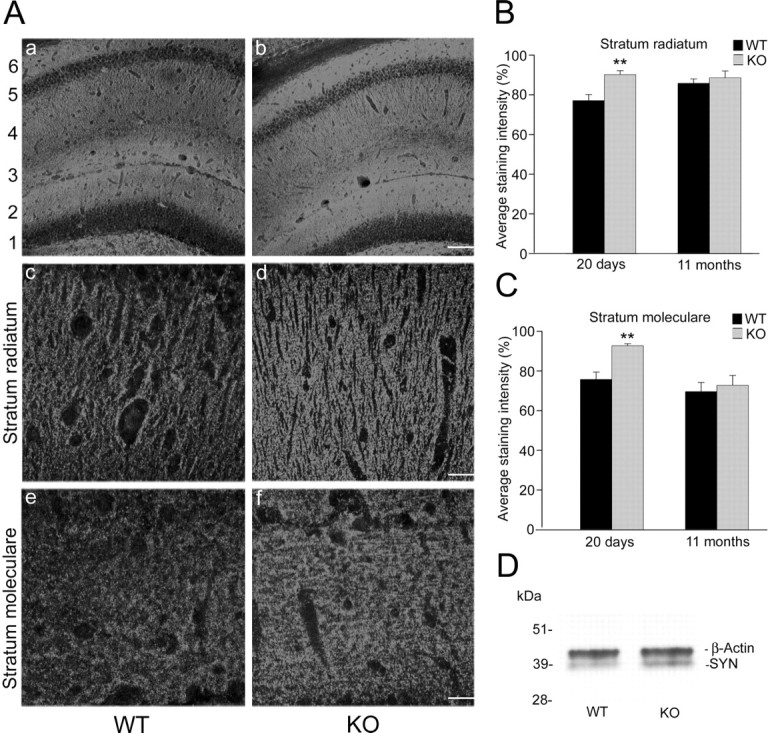
Synaptophysin staining of sections derived from wild-type and APP knock-out mice. A, Synaptophysin-positive presynaptic terminals in mouse hippocampus. Aa, Ab, Low-magnification view of wild-type and APP knock-out hippocampus from 20-d-old mice: 1, stratum granulosum; 2, stratum moleculare; 3, stratum lacunosum; 4, stratum radiatum; 5, stratum pyramidale; 6, stratum oriens. Scale bar, 100 μm. Ac, Ad, Synaptophysin staining of the stratum radiatum from 20-d-old wild-type and APP knock-out mice. Ae, Af, Synaptophysin-positive presynaptic terminals in the stratum moleculare of 20-d-old wild-type and APP knock-out mice. Scale bar, 25 μm. B, Average staining intensity in the stratum radiatum of wild-type (n = 18 areas, 9 sections) and APP knock-out (n = 18 areas, 9 sections) mice at the age of 20 d and 11 months, respectively. C, Average staining intensity in the stratum moleculare of wild-type (n = 18 areas, 9 sections) and APP knock-out (n = 18 areas, 9 sections) mice at the age of 20 d and 11 months, respectively (∗∗p < 0.005). D, Representative Western blot analysis of synaptophysin (SYN; bottom band) and β-actin (top band) expression in hippocampal brain sections of wild-type (left lane) and APP knock-out (right lane) mice.
To corroborate the histological difference of synaptophysin expression in wild-type and APP knock-out mice, we performed Western blots from the hippocampal region. Figure 7D shows a typical blot of the hippocampus of a wild-type (left lane) and an APP knock-out (right lane) mouse. Synaptophysin (SYN), detected at 38 kDa (bottom band), gave a stronger signal in the sample of the APP knock-out mouse. The equal loading of tissue was controlled by β-actin (42 kDa; top band) staining. The relative expression of synaptophysin compared with β-actin was determined by densitometric measurements and showed significantly higher levels in APP knock-out samples (0.38 ± 0.04 for WT, n = 3; 0.44 ± 0.04 for KO, n = 3; p < 0.05).
Discussion
Our study has primarily shown that excitatory synaptic transmission in cultured hippocampal neurons lacking APP is strongly enhanced. Both the amplitude of evoked excitatory synaptic currents as well as the frequency of spontaneous synaptic currents were found to be increased. In line with these findings, we observed a significantly enhanced pool size of synaptic vesicles per neuron, whereas the synaptic and vesicular release probability remained unchanged. Quantitative immunohistochemical analysis of cultured autaptic neurons and brain slices from 3-week-old mice further supported the notion that the above electrophysiological observations are most likely attributable to a physical increase in the number of functional synapses per neuron. This conclusion is very much in line with morphometric studies on cultured neurons in APP knock-out mice that noted an enhanced neuritic outgrowth (Young and Selkoe, 2005). An enhanced neuritic growth and consequently an increased number of synapses could also explain the enhanced glutamate sensitivity and toxicity in mice lacking APP (Steinbach et al., 1998).
The finding of enhanced synapse formation did not extend to hippocampal slice preparations derived from 11-month-old animals. In line with previous studies on adult APP knock-out brains (Seabrook et al., 1999; Heber et al., 2000), we observed no significant enhancement of the synaptophysin staining in aged mice. Indeed, previous electrophysiological studies performed on slice preparations of adult APP knock-out mice using extracellular stimulation electrodes to induce synaptic vesicle release revealed no significant alterations in the synaptic strength in adult APP knock-out mice (Dawson et al., 1999; Phinney et al., 1999; Seabrook et al., 1999).
What could be the reason why loss of APP significantly affects only the synapse number in young age but not in adult animals? Basically, two mechanisms have to be considered: either loss of APP accelerates synapse formation during development, or an enhanced synapse formation in young APP knock-out mice is compensated by accelerated synapse elimination during later development. We cannot exclude the first alternative, but it does not seem to be very likely. Synapse formation in autaptic cultures of wild-type neurons reaches its plateau during the third week in culture (Zhang and Benson, 2001), and we studied the neurons at the end of the third week. Rather, we suggest that the physiological mechanisms that modulate synaptic density during late brain development, namely activity-dependent synaptic elimination, convert the APP knock-out phenotype to the wild-type phenotype via those adaptive mechanisms (Cohen-Cory, 2002).
How can a lack of APP enhance the number of synapses per cultured neuron? Interestingly, a recent electrophysiological study on cultured neurons of presenilin-1 (PS1) knock-out mice revealed findings very similar to ours (Parent et al., 2005). Here an enhanced mEPSC frequency as well as enhanced synaptic formation were observed. Because PS1 is part of the γ-secretase complex, critically involved in APP cleavage and Aβ generation, it is tempting to speculate that the electrophysiological observations we obtained in APP knock-out hippocampal neurons may actually be caused by a lack of γ-secretase-derived APP cleavage products, namely Aβ or the APP intracellular domain (AICD). Here we show that wild-type neurons cultured in the presence of the γ-secretase blocker DAPT indeed reveal significant increases in both the amplitude of evoked EPSCs and the frequency of spontaneous neurotransmitter release. Thus, the APP knock-out phenotype can be mimicked by adding DAPT to cultured wild-type neurons. Whether this is mediated by AICD or intracellular or extracellular Aβ cannot be answered from our results with certainty. The involvement of AICD, however, does not very likely account for the phenotype we observed in hippocampal neurons lacking APP because APP knock-out mice do not lack AICD completely. There are two proteins expressed in APP knock-out mice that are very similar to APP, APLP1 and APLP2 (Zheng et al., 1995). These proteins are cleaved by γ-secretase as well and give rise to AICD; however, these proteins lack the Aβ domain. Therefore, we suppose that it is unlikely that a reduced amount of AICD is responsible for the alterations we observed in neurons lacking APP.
It is more likely that the effect is mediated by Aβ peptides. To study this, we applied synthetic monomeric Aβ42 at a low concentration (10 nm) as well as conditioned medium derived from wild-type autaptic cultures onto APP knock-out autaptic cultures. Indeed, we observed a depression of the evoked EPSC amplitude and the mEPSC frequency by extracellular Aβ but not reaching significant levels as obtained by applying DAPT to wild-type cultures. This may have several reasons. First, DAPT may block the production of Aβ close to the cell membrane, the location in which Aβ may bind to certain receptors at the cell membrane involved in synapse formation. When Aβ is applied to the cell culture medium, it gets highly diluted and its concentration at the synaptic plasma membrane becomes much lower. Second, we applied monomeric Aβ only for 2 d and in low, physiological concentrations [10 nm (Haass et al., 1992; Seubert et al., 1992; Shoji et al., 1992)] to avoid being confronted with effects attributable to Aβ oligomerization or aggregation. Those explanations may also hold true for the conditioned medium. Aβ released from wild-type cells becomes highly diluted within the medium and may also oligomerize or aggregate to some extent, which further reduces the concentration of Aβ monomers.
Together, therefore, our results strongly indicate that Aβ has an important physiological role in modulating synaptic formation and function. Support for this novel idea is increasing. For instance, it was shown recently that APP overexpression in hippocampal slice preparations depressed both AMPA- and NMDA-receptor-mediated synaptic currents and reduced the miniature EPSC frequency (Kamenetz et al., 2003). Moreover, the expression of APP mutant constructs that do not give rise to Aβ was not found to induce these phenomena, which again implies that Aβ generation is indeed critical. There was no indication that AICD is involved because the expression of APP/APLP2 fusion constructs that lack the Aβ domain was not found to affect synaptic transmission. It may be the function of Aβ to reduce excitatory synaptic transmission via a modulation of the number of functional synapses, thus limiting neuronal excitability. This would be beneficial because it reduces both glutamate excitotoxicity and cellular energy demands. Whether it is primarily the Aβ monomers, Aβ oligomers, or Aβ fibrils that are involved physiologically is very difficult to assess. Judging from our results based on the comparison of APP knock-out mouse and wild-type mouse cultured neurons of very low density (<10,000 cells per well), conditions in which appreciable oligomer formation is highly unlikely, the effect of Aβ on synaptic formation is most likely mediated by Aβ monomers.
The mechanisms by which Aβ modulates neurite outgrowth and the number of functional synapses is still unknown. One possible mechanism may be related to the observation that the unaggregated, nonpathological form of Aβ modulates the function of calcium and potassium channels within the cell membrane (Price et al., 1998; Ramsden et al., 2001, 2002). This could in turn influence neurite outgrowth and synapse formation. The motility of dendritic filopodia, for example, is modulated by depolarization-induced calcium influx (Jontes et al., 2000).
How can the proposed physiological function of monomeric Aβ in regulating synaptic formation be related to Alzheimer's disease? It is well known that APP is highly expressed under all kinds of neuronal injury. Under those conditions, Aβ may reduce excitatory synaptic transmission via a modulation of the number of functional excitatory synapses, thus limiting excitotoxicity. Chronically enhanced levels of Aβ, however, attributable to repetitive mild neuronal injuries of any kind including hypoxia or trauma, may either directly alter synaptic transmission via a gain-of-function mechanism or lead to the formation of toxic Aβ aggregates or oligomers. These Aβ aggregates or soluble oligomers may further alter synaptic transmission and cellular excitability by altering the function of voltage-gated calcium channels (Price et al., 1998; Ramsden et al., 2002), certain types of potassium channels (Colom et al., 1998; Yu et al., 1998), AMPA receptors (Chang et al., 2006), NMDA receptors (Snyder et al., 2005), and the α7 nicotinic receptor (Wang et al., 2000; Dineley et al., 2001) or by the formation of calcium-permeable membrane pores (Arispe et al., 1993; Hirakura et al., 2000; Kawahara et al., 2000; Kourie et al., 2001; Kayed et al., 2004; Demuro et al., 2005). Alternatively, as suggested by Kamenetz et al. (2003), neurons may lose their sensitivity to Aβ-induced synaptic depression during aging, for example, via the modulation of AMPA-receptor-mediated synaptic currents (Kamenetz et al., 2003; Almeida et al., 2005), which may cause a persistently elevated neuronal activity known to be toxic.
The concept of a physiological function of Aβ, previously proposed also by Plant et al. (2003) and Ramsden et al. (2002), may hence have an important impact on therapeutic approaches that aim at reducing Aβ production in AD patients.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Sonderforschungsbereich 596 (J.H.). We are very thankful to Michael Mansour for helping to establish the autaptic culture, his expertise in the analysis of synaptic currents, and helpful discussions throughout the project. We thank Ulrike Müller for providing the APP knock-out mice, Doris Schechinger for excellent technical assistance, and Neville Vassallo for critical reading of this manuscript.
References
- Almeida CG, Tampellini D, Takahashi RH, Greengard P, Lin MT, Snyder EM, Gouras GK (2005). Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol Dis 20:187–198. [DOI] [PubMed] [Google Scholar]
- Arispe N, Pollard HB, Rojas E (1993). Giant multilevel cation channels formed by Alzheimer disease amyloid-β protein [AβP-(1–40)] in bilayer membranes. Proc Natl Acad Sci USA 90:10573–10577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustin I, Rosenmund C, Sudhof TC, Brose N (1999). Munc13–1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature 400:457–461. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF (1991). Excitatory and inhibitory autaptic currents in isolated hippocampal neurons maintained in cell culture. Proc Natl Acad Sci USA 88:7834–7838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang EH, Savage MJ, Flood DG, Thomas JM, Levy RB, Mahadomrongkul V, Shirao T, Aoki C, Huerta PT (2006). AMPA receptor downscaling at the onset of Alzheimer's disease pathology in double knockin mice. Proc Natl Acad Sci USA 103:3410–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Cory S (2002). The developing synapse:construction and modulation of synaptic structures and circuits. Science 298:770–776. [DOI] [PubMed] [Google Scholar]
- Colom LV, Diaz ME, Beers DR, Neely A, Xie WJ, Appel SH (1998). Role of potassium channels in amyloid-induced cell death. J Neurochem 70:1925–1934. [DOI] [PubMed] [Google Scholar]
- Daigle I, Li C (1993). apl-1, a Caenorhabditis elegans gene encoding a protein related to the human beta-amyloid protein precursor. Proc Natl Acad Sci USA 90:12045–12049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson GR, Seabrook GR, Zheng H, Smith DW, Graham S, O'Dowd G, Bowery BJ, Boyce S, Trumbauer ME, Chen HY, Van der Ploeg LH, Sirinathsinghji DJ (1999). Age-related cognitive deficits, impaired long-term potentiation and reduction in synaptic marker density in mice lacking the beta-amyloid precursor protein. Neuroscience 90:1–13. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Simons M, Multhaup G, Van Leuven F, Beyreuther K, Dotti CG (1995). Production of intracellular amyloid-containing fragments in hippocampal neurons expressing human amyloid precursor protein and protection against amyloidogenesis by subtle amino acid substitutions in the rodent sequence. EMBO J 14:4932–4938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG (2005). Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem 280:17294–17300. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD (2001). β-Amyloid activates the mitogen-activated protein kinase cascade via hippocampal α7 nicotinic acetylcholine receptors: in vitro and in vivo mechanisms related to Alzheimer's disease. J Neurosci 21:4125–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield JP, Tsai J, Gouras GK, Hai B, Thinakaran G, Checler F, Sisodia SS, Greengard P, Xu H (1999). Endoplasmic reticulum and trans-Golgi network generate distinct populations of Alzheimer beta-amyloid peptides. Proc Natl Acad Sci USA 96:742–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, Lieberburg I, Koo EH, Schenk D, Teplow DB (1992). Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature 359:322–325. [DOI] [PubMed] [Google Scholar]
- Heber S, Herms J, Gajic V, Hainfellner J, Aguzzi A, Rulicke T, von Kretzschmar H, von Koch C, Sisodia S, Tremml P, Lipp HP, Wolfer DP, Muller U (2000). Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J Neurosci 20:7951–7963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herms J, Anliker B, Heber S, Fuhrmann M, Kretzschmar H, Sisodia S, Muller U (2004). Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP-family members. EMBO J 23:4106–4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakura Y, Yiu WW, Yamamoto A, Kagan BL (2000). Amyloid peptide channels: blockade by zinc and inhibition by Congo red (amyloid channel block). Amyloid 7:194–199. [DOI] [PubMed] [Google Scholar]
- Jontes JD, Buchanan J, Smith SJ (2000). Growth cone and dendrite dynamics in zebrafish embryos: early events in synaptogenesis imaged in vivo. Nat Neurosci 3:231–237. [DOI] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R (2003). APP processing and synaptic function. Neuron 37:925–937. [DOI] [PubMed] [Google Scholar]
- Kawahara M, Kuroda Y, Arispe N, Rojas E (2000). Alzheimer's beta-amyloid, human islet amylin, and prion protein fragment evoke intracellular free calcium elevations by a common mechanism in a hypothalamic GnRH neuronal cell line. J Biol Chem 275:14077–14083. [DOI] [PubMed] [Google Scholar]
- Kayed R, Sokolov Y, Edmonds B, McIntire TM, Milton SC, Hall JE, Glabe CG (2004). Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J Biol Chem 279:46363–46366. [DOI] [PubMed] [Google Scholar]
- Kourie JI, Henry CL, Farrelly P (2001). Diversity of amyloid beta protein fragment [1–40]-formed channels. Cell Mol Neurobiol 21:255–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs B, Kohlmannsperger V, Nolting S, Schmalzbauer R, Kretzschmar HA (2006). A method to perform Western blots of microscopic areas of histological sections. J Histochem Cytochem 54:559–565. [DOI] [PubMed] [Google Scholar]
- Li Z-W, Stark G, Götz J, Rülicke T, Müller U, Weissmann C (1996). Generation of mice with a 200-kb amyloid precursor protein gene deletion by Cre recombinase-mediated site-specific recombination in embryonic stem cells. Proc Natl Acad Sci USA 93:6158–6162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Terry RD, Alford M, DeTeresa R (1990a). Quantitative immunohistochemistry of synaptophysin in human neocortex: an alternative method to estimate density of presynaptic terminals in paraffin sections. J Histochem Cytochem 38:837–844. [DOI] [PubMed] [Google Scholar]
- Masliah E, Terry RD, Mallory M, Alford M, Hansen LA (1990b). Diffuse plaques do not accentuate synapse loss in Alzheimer's disease. Am J Pathol 137:1293–1297. [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Terry RD, Alford M, DeTeresa R, Hansen LA (1991). Cortical and subcortical patterns of synaptophysinlike immunoreactivity in Alzheimer's disease. Am J Pathol 138:235–246. [PMC free article] [PubMed] [Google Scholar]
- Parent AT, Barnes NY, Taniguchi Y, Thinakaran G, Sisodia SS (2005). Presenilin attenuates receptor-mediated signaling and synaptic function. J Neurosci 25:1540–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phinney AL, Calhoun ME, Wolfer DP, Lipp HP, Zheng H, Jucker M (1999). No hippocampal neuron or synaptic bouton loss in learning-impaired aged beta-amyloid precursor protein-null mice. Neuroscience 90:1207–1216. [DOI] [PubMed] [Google Scholar]
- Plant LD, Boyle JP, Smith IF, Peers C, Pearson HA (2003). The production of amyloid β peptide is a critical requirement for the viability of central neurons. J Neurosci 23:5531–5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price SA, Held B, Pearson HA (1998). Amyloid beta protein increases Ca2+ currents in rat cerebellar granule neurones. NeuroReport 9:539–545. [PubMed] [Google Scholar]
- Ramsden M, Plant LD, Webster NJ, Vaughan PF, Henderson Z, Pearson HA (2001). Differential effects of unaggregated and aggregated amyloid beta protein (1–40) on K+ channel currents in primary cultures of rat cerebellar granule and cortical neurones. J Neurochem 79:699–712. [DOI] [PubMed] [Google Scholar]
- Ramsden M, Henderson Z, Pearson HA (2002). Modulation of Ca2+ channel currents in primary cultures of rat cortical neurones by amyloid beta protein (1–40) is dependent on solubility status. Brain Res 956:254–261. [DOI] [PubMed] [Google Scholar]
- Reim K, Mansour M, Varoqueaux F, McMahon HT, Sudhof TC, Brose N, Rosenmund C (2001). Complexins regulate a late step in Ca2+-dependent neurotransmitter release. Cell 104:71–81. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Martin-Morris L, Luo LQ, White K (1989). A Drosophila gene encoding a protein resembling the human beta-amyloid protein precursor. Proc Natl Acad Sci USA 86:2478–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF (1996). Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron 16:1197–1207. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Clements JD, Westbrook GL (1993). Nonuniform probability of glutamate release at a hippocampal synapse. Science 262:754–757. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Feltz A, Westbrook GL (1995). Calcium-dependent inactivation of synaptic NMDA receptors in hippocampal neurons. J Neurophysiol 73:427–430. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, Neher E (1999). Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron 23:399–409. [DOI] [PubMed] [Google Scholar]
- Seabrook GR, Smith DW, Bowery BJ, Easter A, Reynolds T, Fitzjohn SM, Morton RA, Zheng H, Dawson GR, Sirinathsinghji DJ, Davies CH, Collingridge GL, Hill RG (1999). Mechanisms contributing to the deficits in hippocampal synaptic plasticity in mice lacking amyloid precursor protein. Neuropharmacology 38:349–359. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ (2000). Toward a comprehensive theory for Alzheimer's disease. Hypothesis: Alzheimer's disease is caused by the cerebral accumulation and cytotoxicity of amyloid beta-protein. Ann NY Acad Sci 924:17–25. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ (2002). Alzheimer's disease is a synaptic failure. Science 298:789–791. [DOI] [PubMed] [Google Scholar]
- Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schlossmacher M, Whaley J, Swindlehurst C, McCormack R, Wolfert R, Selkoe D, Lieberburg I, Schenk D (1992). Isolation and quantification of soluble Alzheimer's beta-peptide from biological fluids. Nature 359:325–327. [DOI] [PubMed] [Google Scholar]
- Shoji M, Golde TE, Ghiso J, Cheung TT, Estus S, Shaffer LM, Cai XD, McKay DM, Tintner R, Frangione B (1992). Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science 258:126–129. [DOI] [PubMed] [Google Scholar]
- Slunt HH, Thinakaran G, von Koch C, Lo AC, Tanzi RE, Sisodia SS (1994). Expression of a ubiquitous, cross-reactive homologue of the mouse beta-amyloid precursor protein (APP). J Biol Chem 269:2637–2644. [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P (2005). Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci 8:1051–1058. [DOI] [PubMed] [Google Scholar]
- Steinbach JP, Muller U, Leist M, Li ZW, Nicotera P, Aguzzi A (1998). Hypersensitivity to seizures in beta-amyloid precursor protein deficient mice. Cell Death Differ 5:858–866. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R (1991). Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30:572–580. [DOI] [PubMed] [Google Scholar]
- Wang HY, Lee DH, D'Andrea MR, Peterson PA, Shank RP, Reitz AB (2000). Beta-amyloid(1–42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer's disease pathology. J Biol Chem 275:5626–5632. [DOI] [PubMed] [Google Scholar]
- Wasco W, Bupp K, Magendantz M, Gusella JF, Tanzi RE, Solomon F (1992). Identification of a mouse brain cDNA that encodes a protein related to the Alzheimer disease-associated amyloid beta protein precursor. Proc Natl Acad Sci USA 89:10758–10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasco W, Gurubhagavatula S, Paradis MD, Romano DM, Sisodia SS, Hyman BT, Neve RL, Tanzi RE (1993). Isolation and characterization of APLP2 encoding a homologue of the Alzheimer's associated amyloid beta protein precursor. Nat Genet 5:95–100. [DOI] [PubMed] [Google Scholar]
- Young TL, Selkoe DJ (2005). Molecular rescue studies on role of APP family members in neuronal development. Soc Neurosci Abstr 31:712.2. [Google Scholar]
- Yu SP, Farhangrazi ZS, Ying HS, Yeh CH, Choi DW (1998). Enhancement of outward potassium current may participate in beta-amyloid peptide-induced cortical neuronal death. Neurobiol Dis 5:81–88. [DOI] [PubMed] [Google Scholar]
- Zhang W, Benson DL (2001). Stages of synapse development defined by dependence on F-actin. J Neurosci 21:5169–5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Jiang M, Trumbauer ME, Sirinathsinghji DJ, Hopkins R, Smith DW, Heavens RP, Dawson GR, Boyce S, Conner MW, Stevens KA, Slunt HH, Sisoda SS, Chen HY, Van der Ploeg LH (1995). Beta-amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell 81:525–531. [DOI] [PubMed] [Google Scholar]