

Abstract
Mutants of human superoxide dismutase-1 (hSOD1) cause amyotrophic lateral sclerosis (ALS), and mitochondria are thought to be primary targets of the cytotoxic action. The high expression rates of hSOD1s in transgenic ALS models give high levels of the stable mutants G93A and D90A as well as the wild-type human enzyme, significant proportions of which lack Cu and the intrasubunit disulfide bond. The endogenous murine SOD1 (mSOD1) also lacks Cu and is disulfide reduced but is active and oxidized in mice expressing the low-level unstable mutants G85R and G127insTGGG. The possibility that the molecular alterations may cause artificial loading of the stable hSOD1s into mitochondria was explored. Approximately 10% of these hSOD1s were localized to mitochondria, reaching levels 100-fold higher than those of mSOD1 in control mice. There was no difference between brain and spinal cord and between stable mutants and the wild-type hSOD1. mSOD1 was increased fourfold in mitochondria from high-level hSOD1 mice but was normal in those with low levels, suggesting that the Cu deficiency and disulfide reduction cause mitochondrial overloading. The levels of G85R and G127insTGGG mutant hSOD1s in mitochondria were 100- and 1000-fold lower than those of stable mutants. Spinal cords from symptomatic mice contained hSOD1 aggregates covering the entire density gradient, which could contaminate isolated organelle fractions. Thus, high hSOD1 expression rates can cause artificial loading of mitochondria. Unstable low-level hSOD1s are excluded from mitochondria, indicating other primary locations of injury. Such models may be preferable for studies of ALS pathogenesis.
Keywords: amyotrophic lateral sclerosis, mitochondria, superoxide dismutase, transgenic mice, Cu, disulfide bond
Introduction
Amyotrophic lateral sclerosis (ALS) is a disease characterized by motor neuron degeneration, resulting in progressive paralysis and death. Approximately 10% of ALS cases are familial (Haverkamp et al., 1995), and, in some of these, the disease is linked to mutations in the CuZn-superoxide dismutase (SOD1) gene (Rosen et al., 1993). Overall, ∼5% of all cases with ALS show SOD1 mutations, and >100 such mutations have been identified (Andersen et al., 2003). SOD1 is a ubiquitously expressed antioxidant enzyme located in the cytosol, in the mitochondrial intermembrane space (IMS) (Weisiger and Fridovich, 1973), and possibly also in the nucleus and peroxisomes (Crapo et al., 1992). The mutations confer a cytotoxic gain of function to the enzyme (Gurney et al., 1994; Andersen et al., 1995). The nature of the cytotoxicity and also the primary subcellular location of targets remain to be established.
Based on studies in both transgenic mice (Bowling et al., 1993; Higgins et al., 2002, 2003; Mattiazzi et al., 2002, 2004; Liu et al., 2004; Kirkinezos et al., 2005; Vijayvergiya et al., 2005) and cultured cells (Rizzardini et al., 2005; Cozzolino et al., 2006), mitochondria have been suggested to be the primary targets. There are, however, complicating circumstances in the murine ALS models. To cause disease within the short lifespan of the mouse, ALS-linked human SOD1s (hSOD1s) have to be expressed at rates that are at least 20-fold higher than that of the endogenous murine SOD1 (mSOD1) (Jonsson et al., 2006). This results in steady-state levels of wild-type hSOD1 and stable mutants such as G93A and D90A that are 17- to 24-fold higher than those of the mSOD1. Most of this hSOD1 lacks SOD activity because of insufficient Cu charging. Large proportions also carry a reduced C57–C146 intrasubunit disulfide bond. The endogenous mSOD1 in these mice is disulfide reduced to a similar extent and probably also incompletely Cu charged. In contrast, the levels of the unstable mutants G85R and G127insTGGG (G127X) in transgenic models are equal to or lower than that of the endogenous mSOD1 (Jonsson et al., 2004, 2006). They also lack both SOD activity and disulfide bond. Conversely, the mSOD1 is fully active and disulfide oxidized (Jonsson et al., 2006).
Loss of the prosthetic metals and reduction of the disulfide bond promotes the entry of SOD1 into the mitochondrial IMS (Okado-Matsumoto and Fridovich, 2002; Field et al., 2003). The high expression rates of hSOD1 in the transgenic models thus have effects on the molecular forms of SOD1s that should increase the proportion of species amenable to mitochondrial entry. Thus, SOD1 might be loaded into mitochondria in a nonphysiological manner, causing effects that are potentially misleading for our understanding of human ALS pathogenesis. To explore this, we used somewhat divergent procedures to study the presence of human and murine SOD1 in brain and spinal cord mitochondria from transgenic mice expressing stable and unstable forms of hSOD1. The occurrence and localization of hSOD1 aggregates in symptomatic mice was also examined.
Materials and Methods
Transgenic mice.
Transgenic mice expressing the G93A mutant (G93AGur) and wild-type hSOD1 (Gurney et al., 1994) were obtained from The Jackson Laboratory (Bar Harbor, ME). Mice expressing the G85R mutant of hSOD1 were obtained from Dr. Don Cleveland (Ludwig Institute for Cancer Research and Department of Neurosciences, University of California at San Diego, La Jolla, CA) (Bruijn et al., 1997), and mice expressing the G127X (Jonsson et al., 2004) and D90A mutants (P. A. Jonsson, K. S. Graffmo, T. Brännström, P. Nilsson, P. M. Andersen, and S. L. Marklund, unpublished observations) of hSOD1 were generated in our laboratory. The mice were deemed terminally ill when, because of limb weakness, they could no longer reach the food in the cages. This occurred for the D90A, G85R, G127X, and G93AGur mice at an average age of 407, 345, 250, and 124 d, respectively. The presymptomatic D90A, G85R, and G127X mice used in the study were ∼100 d old, whereas the G93AGur mice were ∼50 d old.
Subcellular fractionation by density gradient ultracentrifugation.
Mice were killed by intraperitoneal injections of pentobarbital. The brains and spinal cords were immediately cut out, weighed, and gently homogenized in a Dounce glass-pestle homogenizer at 4°C in 25 vol of mitochondrial buffer (0.21 m mannitol, 0.11 m sucrose, 10 mm K HEPES, pH 7.2, 1 mm EGTA, and the Complete antiproteolytic cocktail without EDTA (Roche Diagnostics, Basel, Switzerland). Brains were homogenized with 4 + 8 strokes and spinal cords with 12 + 2 strokes using loose-fit and tight-fit pestles, respectively. This procedure was optimized for maximal recovery of intact mitochondria. Liver and kidney were analyzed for comparison and handled as for brain. The homogenates were then passed through a 5 μm nylon mesh by centrifugation at 20 × g at 4°C for 10 min. The filtrates were supplied with iodixanol (OptiPrep; Axis-Shield PoC, Oslo, Norway) to a final concentration of 8%. The filtrates (0.5–1 ml) were loaded on top of continuous 9 ml 12–38% iodixanol gradients in mitochondrial buffer and centrifuged at 100,000 × g at 4°C for 90 min in a swinging-bucket rotor (type 45 Ti; Beckman Coulter, Fullerton, CA). The tubes were punctured with a cannula, tapped from the bottom in 500 μl portions into tubes containing the Complete antiproteolytic cocktail, and, if not analyzed immediately, stored at −80°C.
Separation of mitochondria and peroxisomes.
A filtrate of a brain homogenate from a 100-d-old D90A mouse was divided into two aliquots. One aliquot was supplied with 6 mm CaCl2, 5 mm succinate, and 5 mm K2HPO4 and incubated at room temperature for 5 min to cause permeability transition pore opening and swelling of the mitochondria. The aliquots were then subjected to density gradient ultracentrifugation.
Dot-blot assay for aggregates.
Fractions from the separations were filtered through 0.15 μm cellulose acetate membranes (Schleicher and Schuell, Dassel, Germany) in a dot-blot apparatus by a method modified from Wang et al. (2002). The major difference from the published procedure was that the homogenates were supplied with 0.1% SDS and gently sonicated before the initial 800 × g centrifugation instead of after. This increased the sensitivity of the assay 25-fold.
Detection of detergent-resistant aggregates.
Brain and spinal cord filtrates were split into two aliquots. To one aliquot, 0.5% Nonidet P-40 (NP-40) was added, followed by sonication for 30 s. The other aliquot served as control, and both were subjected to density gradient centrifugation as described above.
Gel chromatography.
Brains and spinal cords were homogenized with an Ultraturrax followed by sonication in 25 vol of PBS, pH 7.0, containing Complete without EDTA and 20 mm iodoacetamide, followed by centrifugation at 20,000 × g for 30 min. The iodoacetamide was added to quench free thiol groups in proteins and low-molecular-weight compounds. The supernatants were subjected to chromatography in 1 × 30 cm Superdex 75 columns (GE Healthcare, Uppsala, Sweden) in PBS with 5 mm iodoacetamide. The elution rate was 45 ml/h, and the eluate was collected in 0.3 ml fractions. The SOD activities (Marklund, 1976) and hSOD1 protein contents of the fractions were determined.
Marker enzyme activity assays.
For analysis of succinate dehydrogenase (SDH), samples were incubated with 100 mm Na HEPES, pH 7.4, 1 mm EDTA, 10 mm succinic acid, and 0.08% MTT for 90 min. Three volumes of 2-propanol were then added, followed by centrifugation at 14,000 × g for 10 min. The absorbance of the supernatants at 550 nm was then measured. Acid phosphatase was determined with p-nitrophenyl phosphate as substrate. For lactate dehydrogenase (LD) activity analysis, the CytoTox96 kit from Promega (Madison, WI) was used.
Immunoblotting and quantification.
Samples were separated by regular SDS-PAGE and then electroblotted onto polyvinylidene difluoride membranes (GE Healthcare, Uppsala, Sweden). Blots were probed with antibodies and chemiluminescence was generated using ECL-advance (GE Healthcare, Buckinghamshire, UK). Bands were visualized by using a Chemidoc XRS imager and Quantity One software (Bio-Rad, Hercules, CA). For quantification of hSOD1 by immunoblotting, wild-type hSOD1 with concentration determined by quantitative amino acid analysis was used as original standard (Marklund et al., 1997). Murine SOD1 was quantified against mSOD1 in a brain homogenate from a control mouse; this was calibrated against the human standard with an antibody directed against a sequence (amino acids 131–153) that is equal in human and murine SOD1. The primary antibodies used for detection of human and murine SOD1 were polyclonal rabbit antibodies raised against keyhole limpet hemocyanin-coupled peptides corresponding to amino acids 24–39 (hSOD1-specific) or 24–36 (mSOD1-specific) (Jonsson et al., 2004). The amounts of G85R and G127X hSOD1s in the gradients were very low, and these fractions were therefore concentrated 10-fold. This was accomplished by precipitation with 10% trichloroacetic acid at 4°C overnight, washing of the precipitated protein with acetone, and dissolving in a smaller volume of SDS-PAGE sample buffer. The lamin A/C, Tim23, and β-COP (Golgi β-coatomer protein) antibodies were obtained from BD Biosciences (Franklin Lakes, NJ), PMP70 (70 kDa peroxisomal membrane protein) was from Affinity BioReagents (Golden, CO) or Zymed (San Francisco, CA), and the synaptotagmin antibody was from Stressgen (Victoria, British Columbia, Canada).
Results
Design of the density gradient separations
The separation of organelles from the CNS is challenging. Neurons in particular, but also glia, possess abundant trees of cellular processes. During tissue disruption, these may close to vesicles, in which organelles and other cellular components residing in the processes may be trapped. The tissue is also rich in myelin and other lipid materials, which tend to form sticky interfaces in density gradients. The addition of 8% iodixanol caused the lipid materials to remain dispersed in the loaded filtrate during the ultracentrifugation. To reduce the risk of artificial association between components, the tissues were homogenized in a relatively large buffer volume, and pelletings by centrifugation was avoided. We also avoided discontinuous gradients, in which there might be a risk that components with partially overlapping densities could be artificially cofractionated.
The use of a standard protocol to remove nondisrupted tissue and cells, debris, and nuclei (two 10-min 1000 × g centrifugations) resulted in a recovery in the combined supernatants of only ∼70% of the hSOD1 of presymptomatic G127X spinal cord homogenates. Because we aimed at a global view of the distribution of SOD1, alternative methods were tested. The filtration of the whole homogenates through 5 μm mesh nylon filters resulted in average recoveries of >90% of the hSOD1, 85% of the mitochondrial marker SDH, and ∼35% of the DNA. Of the mitochondria in the filtrates, generally only 40–60% entered the density gradient during ultracentrifugation (Fig. 1A). The mitochondria in the top fractions of the tubes are probably trapped in the vesicles and lipid materials. Supporting this notion, mitochondria in a filtrate of murine liver (a tissue without abundant cellular processes) entered the gradient to >90% (Fig. 1A). The locations of markers for organelles and other components are shown in Figure 1, B and C. Only peroxisomes show a major overlap with mitochondria. Of the DNA that passes through the 5 μm nylon mesh, small peaks with high density (tubes 5 and 6) can be found after ultracentrifugation. Generally, >90% is present in the top three tubes, probably mainly representing disrupted nuclei.
Figure 1.
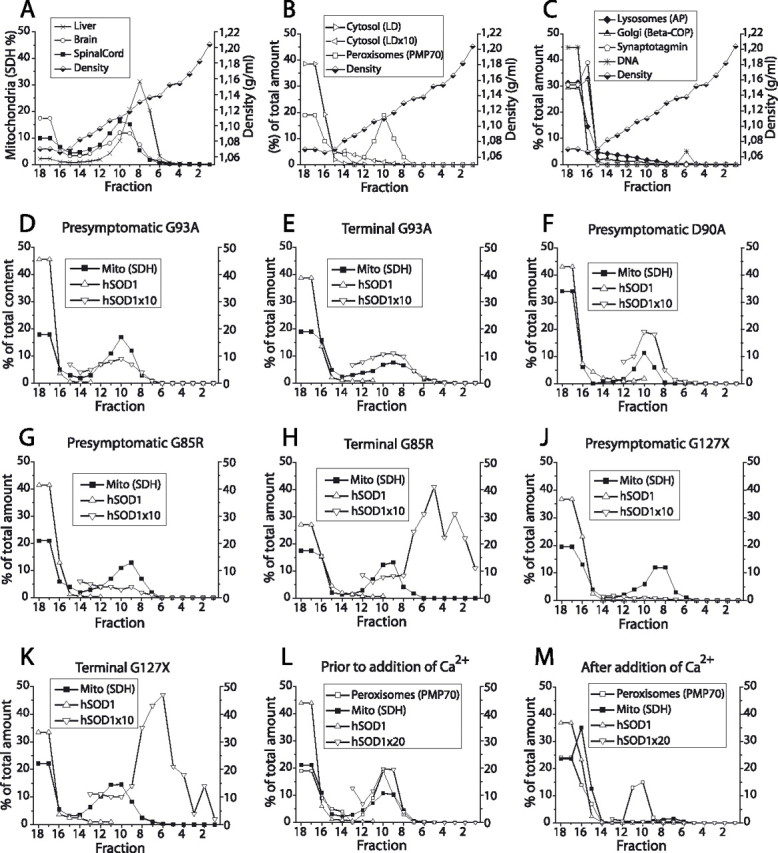
Distribution of hSOD1 and organelles in density gradient ultracentrifugations. Tissue homogenization, preparation of gradients, ultracentrifugation, and analysis of fractions were performed as described in Materials and Methods. A, Comparison of fractionation of mitochondria from brain, spinal cord, and liver. B, C, Location of markers for organelles in separations of spinal cord homogenates. Synaptotagmin is a marker for synaptic vesicles and plasma membrane. D–K, Relative distributions of hSODs and the mitochondrial marker SDH in spinal cord extracts from different transgenic models in presymptomatic and terminal stages of disease, as indicated in the framed insets. Note that the relative amounts of hSODs in the gradients have been multiplied by 10 to make the data easier to visualize. L, M, Ca2+ and succinate were added to a filtrate of a brain homogenate from a D90A mouse to cause permeability transition pore opening and mitochondrial swelling. L and M show density gradient ultracentrifugations of aliquots of the filtrate either untreated or treated with Ca2+ plus succinate. AP, Acid phosphatase; β-COP, Golgi β-coatomer protein; Mito (SDH), the mitochondrial marker SDH; PMP70, 70 kDa peroxisomal membrane protein.
Large proportions of the stable high-level hSOD1s cofractionate with mitochondria in transgenic mice
Filtrates of homogenates from spinal cords and brain were subjected to density gradient centrifugation. In spinal cords from presymptomatic G93AGur (Figs. 1D, 2A), D90A (Fig. 1F), and wild-type hSOD (data not shown) transgenic mice, significant proportions of the hSOD1 entered the density gradient and showed distribution patterns similar to that of the mitochondrial marker SDH. Filtrates of brains from these mice showed virtually identical patterns (Fig. 1L). In terminally ill mice, no major differences compared with the presymptomatic samples could be discerned in the G93A mice (Fig. 1E) and the other high-level hSOD1 transgenic mice (data not shown).
Figure 2.
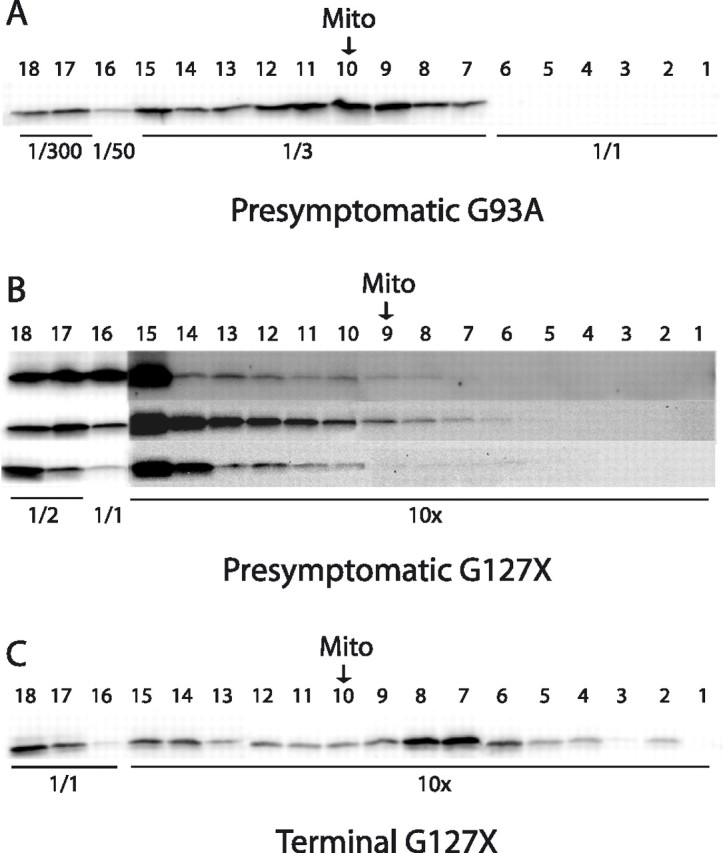
Examples of Western immunoblots for localization of hSOD1 in tapped gradient fractions. To allow visualization in the same blot, the fractions were diluted or concentrated as indicated. The arrows indicate the location of the peak of the mitochondrial marker SDH (Mito). A, Density gradient centrifugation of a spinal cord extract from a presymptomatic G93A mouse; see also Figure 1D. Density gradient centrifugations of spinal cords from three different presymptomatic (B) and one terminally ill (C) G127X mice; see also Figure 1, J and K. The blots of the 10-fold concentrated fractions from the presymptomatic mice were overexposed to visualize the very faint bands.
In spinal cords (Fig. 1G) and brains (data not shown) from presymptomatic G85R mice, much smaller proportions of the hSOD1 entered the density gradients, but there were always peaks coinciding with the mitochondrial SDH peaks. In spinal cord (Figs. 1J, 2B) and brain samples (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material) from G127X mice, even less hSOD1 entered the density gradient, and there were no peaks coinciding with the peaks of the mitochondrial marker SDH.
In spinal cords from terminally ill G85R (Fig. 1H) and G127X (Figs. 1K, 2C) mice, large proportions of the hSOD1 entered the density gradient. These dense materials were heterogeneous, covering the whole gradient, but with the major parts located at higher densities than any of the organelle markers. The patterns do not suggest any particular mitochondrial location. These dense materials probably represent hSOD1-containing aggregates (see also below). In brain homogenates from the terminally ill G85R (data not shown) and G127X mice, more hSOD1 entered the gradients than was the case for presymptomatic mice, but the amounts were 10-fold lower than those seen in the spinal cords (supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material).
Because of the retention of half of the mitochondrial marker in the applied filtrate (together with all SOD1 in solution), it was not possible to directly calculate the amounts of SOD1 that were associated with the mitochondria. To obtain an estimate of the maximal amount of SOD1 that could reside in mitochondria, the amount of hSOD1 in the three fractions of the density gradient with the highest level of mitochondrial marker (SDH) was estimated (mostly fractions 9–11). The ratio of hSOD1/SDH in these fractions was then multiplied by the SDH activities found in all 18 tubes (Fig. 3A). These calculations indicated that very large proportions of the hSOD1s were associated with mitochondria: ∼10% in spinal cords and brains from G93AGur and D90A mice and a few percent less in wild-type hSOD1 transgenic mice. The proportions were much lower in the G85R mice and, in particular, in G127X mice (Fig. 3A). The differences in absolute levels were even more remarkable: mitochondria in spinal cords and brains from wild-type hSOD1, G93AGur, and D90A mice appear to contain ∼100-fold as much hSOD1 as mitochondria from G85R mice and 1000-fold as much as those in G127X mice. The levels are also 100-fold higher than those of mSOD1 in mitochondria of control mice (Fig. 3B).
Figure 3.

Quantification of amounts and proportions of SOD1 associated with mitochondria in spinal cord (black columns) and brain (gray columns). The amount of hSOD1 in the three fractions in the gradient with the highest levels of the mitochondrial marker SDH (mostly fractions 9–11) was determined. The ratio of hSOD1/SDH in these was then multiplied by the SDH activities in all 18 fractions to estimate the total amount of hSOD1 associated with mitochondria. Both absolute and relative (percentage) amounts of hSOD1 associated with mitochondria per gram wet weight of tissue are presented. A, Amounts of hSOD1 associated with mitochondria in the different transgenic mouse strains. B, Corresponding results for mSOD1 in the transgenic strains and in nontransgenic control C57BL/6 mice. The error bars represent SDs. wt, Wild type; Sp.c., spinal cord.
Increased loading of mSOD1 into CNS mitochondria in the high-level hSOD1 transgenic mice
The mSOD1 is disulfide reduced to a similar extent as the hSOD1s in the G93AGur, D90A, and wild-type hSOD1 mice and likely also incompletely Cu charged (Jonsson et al., 2006). No disulfide-reduced mSOD1 was seen, and the enzyme was fully active in the G85R and G127X transgenic mice (Jonsson et al., 2006). Thus, there should also be differences in the charging of CNS mitochondria with mSOD1 between the transgenic models expressing high and low levels of hSOD1. Murine SOD1 was specifically determined in the separations and generally appeared to distribute similarly to the hSOD1s. We made similar calculations for mSOD1 as were done for hSOD1 in Figure 3A. In accordance with the predictions, the proportions apparently associated with mitochondria (8–10%) were similar to those for hSOD1 in the high-level mice, whereas the proportions in G85R and G127X mice were similar to those seen in nontransgenic control mice (2–3%) (Fig. 3B).
In liver and kidney of the high-level hSOD1 transgenic mice, the enzymes are essentially fully active, and the proportions of disulfide-reduced enzyme are much lower (Jonsson et al., 2006). For comparison, we therefore performed some density gradient studies of these organs (n = 2 or 3 of each). Calculated as in Figure 3, the proportions of mSOD1 maximally associated with mitochondria were 1.2, 1.4, and 0.7% in nontransgenic control, G93A, and G85R hSOD1 transgenic mice, respectively. The corresponding proportions of hSOD1 were 0.8 and 0.4% in the G93A and G85R mice, respectively, thus in all cases lower than in the CNS. In the kidney, we found slightly higher figures, 1.8% mSOD1 and 2.4% hSOD1 in G93A mice and 3.5% mSOD1 and 1.2% hSOD1 in G85R mice (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). These somewhat lower levels in peripheral organs are in accord with findings in previous studies (Mattiazzi et al., 2002; Vijayvergiya et al., 2005).
Separation of mitochondria and peroxisomes
There was a relatively large overlap between the peaks of the markers for mitochondria, peroxisomes, and SOD1 in the separations (Fig. 1A,B). Although mitochondria account for much larger relative volumes of CNS tissue than peroxisomes, we cannot formally assign the hSOD to the former in the present study. We therefore attempted to separate the organelles and follow the distribution of hSOD1. A filtrate of a brain homogenate from a 100-d-old D90A mouse was divided into two aliquots. To one aliquot, Ca2+ and succinate were added to cause permeability transition pore opening and swelling of the mitochondria, which should lead to a reduced density. Both extracts were then subjected to density gradient ultracentrifugation. Compared with the control filtrate, the treatment led to an 87% reduction in mitochondrial marker and a 90% reduction in hSOD1 entering the gradient (Fig. 1L,M). The amount of the peroxisomal marker in the gradient was essentially unaffected. Thus, the hSOD1 that cofractionates with mitochondria in the gradient is also primarily located in that organelle.
Heterogeneous hSOD1 aggregates in terminally ill mice
Inclusions and aggregates of hSOD1 are found in cells in motor areas of terminally ill transgenic mice (Bruijn et al., 1998; Johnston et al., 2000; Jonsson et al., 2004, 2006) and in human ALS cases carrying hSOD1 mutations (Jonsson et al., 2004). The dense hSOD1 fractions from spinal cords of terminally ill G85R and G127X transgenic mice that enter the density gradients thus probably represent aggregates (Fig. 1H,K). Such materials are difficult to observe against the copious background of soluble hSOD1 in mitochondria in the G93A, D90A, and wild-type hSOD1 transgenic mice. We therefore used a filter trap assay to demonstrate hSOD1 aggregates. Whereas only small amounts of aggregates were found in spinal cords from presymptomatic G93A, D90A, and G85R mice, much more could be demonstrated in the terminally ill mice (Figs. 4A–C). In G127X mice, aggregates were also seen in presymptomatic mice (Fig. 4D). These localized to the top fractions (15–18) and probably represent the protoaggregates shown previously to be present throughout life in this model (Jonsson et al., 2004). The aggregates in symptomatic mice were widely dispersed over the density gradients. Their densities overlapped with the density of the mitochondrial marker, but the patterns did not in any case suggest a particular localization to that or to other organelles. The aggregates from the low-level hSOD1 transgenic mice showed higher densities than those from the high-level mice (Fig. 4A–D).
Figure 4.

Detection of hSOD1 aggregates in density gradient separations of spinal cord homogenates from presymptomatic and terminally ill mice, as indicated by the framed insets and in the figures. In each figure, the largest amount of aggregates found in a fraction was set to 100, and the results for all other fractions have been presented relative to that. A–D, Filter trap assay for hSOD1 aggregates. E, F, Filtrates of spinal cord extracts were treated with 0.5% of the detergent NP-40 and sonicated before density gradient ultracentrifugation. SDH and hSOD1 were analyzed in the tapped fractions. Mito (SDH), The mitochondrial marker SDH.
As another means of demonstrating hSOD1 aggregates, spinal cord filtrates were divided into two aliquots, and one was supplied with 0.5% NP-40 followed by sonication. Both were then subjected to density gradient ultracentrifugation. SOD1 aggregates are relatively resistant to solubilization by NP-40 (Jonsson et al., 2004). This treatment caused all markers for organelles to disappear from the gradient and to be localized in the top two to three tubes (shown for mitochondria in Fig. 4E,F). In spinal cord from a presymptomatic D90A mouse (Fig. 4E), ∼0.5% of the hSOD1 entered the gradient (tubes 14 to 1), whereas in a terminal D90A mouse, 7.5% did so (Fig. 4F). Similar results were found for G93A mice (data not shown). In the spinal cord from a terminally ill G127X mouse, the figures for hSOD1 entering the gradient were almost equal in untreated filtrates and filtrates treated with NP-40: 26 versus 23%, respectively (data not shown). As for the aggregates demonstrated by the filter assay, those sonicated with NP-40 showed a wide density distribution.
Gel chromatography of tissue extracts
The structure of the SOD1 protein influences both the entry into and retention by the mitochondria (Okado-Matsumoto and Fridovich, 2002; Field et al., 2003). Extracts of brains and spinal cords from the various mouse strains were therefore examined by gel chromatography, and the hSOD1s in the two organs showed analogous patterns in all cases. In the D90A, G93A, and wild-type hSOD1 mice, >95% of the hSOD1 protein peaked at the same elution volume as the hSOD1 enzymatic activity of the extracts and of the mSOD1 activity of extracts from nontransgenic control mice (data not shown). These hSOD1s are thus primarily dimeric. In contrast, >95% of the hSOD1 in spinal cord from a 100-d-old G85R mouse formed a peak at a higher elution volume than the activity of the background mSOD1 (Fig. 5A). There was also a small peak that eluted just before the dimeric mSOD and an even smaller peak at the void volume of the column (not shown in the figure). Thus, G85R hSOD1 is mainly monomeric in the CNS. Extracts from G127X transgenic mice show two major bands when examined by Western immunoblotting (Jonsson et al., 2004): one at 17 kDa, apparently the monomer, and one at 33 kDa, which carries a posttranslational modification of unknown nature. The 33 kDa band elutes before the activity of the dimeric mSOD1 (Fig. 5B). The elution volume of the 17 kDa band, however, was between those of the SOD activity of the dimeric mSOD1 and the G85R “monomer.” The G127X hSOD1 molecule, in which some of the areas that are involved in the dimer contacts are truncated, is however unlikely to be dimeric. Possibly the G127X subunit has a different, more linearized conformation and is therefore more excluded from the gel particles than a more globular G85R subunit.
Figure 5.

Gel chromatography of spinal cord homogenates from G85R and G127X mice. The hSOD1 in the eluted fractions was analyzed by Western immunoblot, and the resulting relative distributions (percentage) are shown in the figures. The actual immunoblots are shown under the figures. For comparison, the SOD activity of the dimeric mSOD1 is also shown. A, Results for a G85R spinal cord. B, G127X spinal cord. In the immunoblot, both the 17 kDa monomer and a 33 kDa hSOD1 complex of unknown composition are shown.
Discussion
The presence of hSOD1 in mitochondria does not correlate with injury
The amounts of hSOD1 in mitochondria were very high in mice expressing the stable ALS-linked mutants G93A and D90A and the wild-type hSOD1 (Fig. 3A). The levels were equally high in spinal cords, which are afflicted by injury in the murine models, as in brains that show much less alterations. There was no significant increase in hSOD1 in spinal cord mitochondria as the mice became symptomatic (Fig. 1E). Mitochondria from mice expressing G85R and G127X hSOD1s contained 100- and 1000-fold less hSOD1, respectively, again without any significant differences between spinal cords and brains. In these mice, the amounts of hSOD1 cofractionating with mitochondria increased in the terminal phase, but this increase seems to be explained by the appearance of the heterogeneous aggregates (Figs. 1H,K, 2C, 4C,D). From these results, we conclude that the injury to the motor neurons is not explained by the presence of hSOD1 per se in spinal cord mitochondria.
The results are at variance with some previous studies, in which specific accumulation of mutant but not wild-type hSOD1 in spinal cord mitochondria was found, as well as marked increases with time (Liu et al., 2004). The reasons for the different outcomes may reside in the techniques used for isolation of mitochondria and the appearance of aggregates in symptomatic mice.
Excessive hSOD1 loading of mitochondria is an artifact caused by extreme overexpression of hSOD1
The high expression rates in the murine transgenic ALS models lead to very high protein levels of stable mutants such as G93A, D90A, and the wild-type hSOD1, which in turn become Cu charged to only minor extent and partially disulfide reduced. The mSOD1 in these mice appears to be altered similarly, whereas it is fully active and disulfide oxidized in mice expressing the unstable low-level mutants G85R and G127X (Jonsson et al., 2006). These structural alterations of SOD1 in the high-level transgenic mice might artificially favor mitochondrial entry (Okado-Matsumoto and Fridovich, 2002; Field et al., 2003). The copious mitochondrial loading of hSOD1 in G93A, D90A, and wild-type hSOD1 transgenic mice support this conjecture (Fig. 3A). This loading of mitochondria is not a property that is specific to hSOD1. The proportions of mSOD1 and hSOD1 in mitochondria (∼10%) were similar in G93A, D90A, and wild-type hSOD1 transgenic mice (Fig. 3A,B). The amounts of mSOD1 in mitochondria of G85R and G127X transgenic mice were fourfold lower and similar to that found in nontransgenic control mice (Fig. 3B). We conclude that the mitochondrial overloading is caused by both the high levels and the molecular alterations of SOD1 in transgenic models in which stable hSOD1 variants are expressed.
In the CNS, the G85R mutant hSOD1 lacks Cu, the intrasubunit disulfide bond (Jonsson et al., 2006), and is monomeric (Fig. 5A). G85R can bind Cu, and, when Cu charged, the enzyme shows full SOD activity. G85R has a low affinity for Cu, however, and cannot retain the metal when competing with other ligands in the CNS (Jonsson et al., 2006). Although the degree of Zn charging is unknown, larger proportions of G85R than of the D90A and G93A and wild-type hSOD1s would be expected to be in a conformation that allows passage through the mitochondrial outer membrane. Still, a fourfold lower proportion appeared to be associated with mitochondria. It is possible that any G85R hSOD1 entering the mitochondrion is not efficiently retained in the IMS. Disulfide oxidation, charging with the prosthetic metals and dimerization in the IMS, would conceivably trap the hSOD1. The fact that such forms of G85R hSOD1 are not detected in the tissue could thus be an explanation for the relatively low levels. Copper chaperone for SOD1 (CCS) promotes the retention of SOD1 in yeast mitochondria. Both C57 and C146 seem to be important for the effect, whereas Cu binding is not. SOD1 mutated at positions 50 and 51 to prevent dimerization does not form heterodimers with CCS, and the retention of such SOD1 proteins in the IMS of yeast mitochondria is not promoted by CCS (Field et al., 2003). The structure of the G85R hSOD1 may be so distorted that both the homodimerization and the heterodimerization with CCS is precluded.
Is G127X mutant hSOD1 excluded from mitochondria?
In presymptomatic mice, very little G127X hSOD1 entered the density gradient and there was no patterning at the peak of the mitochondrial marker SDH that would suggest a colocalization (Figs. 1J, 2B). The cytosol marker LD showed a similar downhill slope into the gradient (Fig. 1B). The proportion of LD present in the three tubes with the highest SDH contents was approximately as large as the proportion of G127X hSOD1, both ∼0.15% of the total amounts in the density gradient separations. G127X hSOD1 that is present in the cytosol in the CNS should at least partially distribute similarly to that of LD in the density gradients. Also of note is the presence of G127X protoaggregates in presymptomatic mice (Fig. 4D) and the abundance of denser aggregates as the mice become symptomatic (Figs. 1K, 4D) (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). Parts of the G127X hSOD1 from presymptomatic mice that enter the gradient could represent such materials. We conclude that the estimates of G127X hSOD1 presence in mitochondria of Figure 3A are too high and that the mutant protein may well lack any association with mitochondria.
The densities of terminal hSOD1 aggregates overlap with those of all organelles
In terminally ill mice, large amounts of aggregates could be demonstrated by the filter trap and NP-40 assays (Fig. 4). In presymptomatic mice, the levels were very low, except in the G127X mice. The densities of the terminal hSOD1 aggregates covered the entire density range of the present study and showed no peaks that would suggest preferential presence in any specific type of organelle, including the mitochondrion.
Any method for isolation of organelles based on centrifugation and density differences carries the risk of cofractionation of the heterogeneous inclusions/aggregates. This could lead to false conclusions regarding the overall localization of hSOD1 and also regarding the physical properties of hSOD1 in organelles.
Could the loading of mitochondria cause artifacts?
Mitochondria from transgenic mice expressing G93A, D90A, and wild-type hSOD1 contain 100-fold more hSOD1 than the content of mSOD1 in control mice. The mitochondrial loading should differ between cell types in the CNS; why, the actual levels in some should be even higher. Such extreme levels might influence the structure and function of affected mitochondria. Mitochondrial swelling and widespread vacuolar pathology have been found in spinal cords of mice expressing the D90A and G93A mutant as well as wild-type hSOD1 transgenic mice (Jaarsma et al., 2000, 2001; Jonsson et al., 2004, 2006) and the likewise high-level G37A mice (Wong et al., 1995). In contrast, such alterations are absent in mice expressing SOD1 mutations G85R and G127X (Bruijn et al., 1997; Jonsson et al., 2004) and also in ALS patients with and without SOD1 mutations. The vacuolar pathology is thus probably an artifact caused by the extreme hSOD1 loading of mitochondria. The motor neuron injury in murine models in which the stable mutants are expressed could be a combination of effects of the mitochondrial hSOD1 overloading and other mechanisms more relevant to human ALS. Functional defects in mitochondria derived from transgenic mice have been reported in many studies (Higgins et al., 2002, 2003; Mattiazzi et al., 2002; Kirkinezos et al., 2005). Most of these studies have been performed in G93A mice, and the mitochondrial overloading may have contributed to the pathology. Alterations have, however, also been noted in symptomatic G85R mice (Damiano et al., 2006). In this regard, a recent study has suggested that injury caused by mutant hSOD1s could be initiated elsewhere in the cells and cause mitochondrial dysfunction and oxidative stress via amplification loops originating in the cytosol (Cozzolino et al., 2006).
The >100 different hSOD1 mutations so far detected are likely to cause ALS by essentially the same mechanism. The levels of different mutant hSODs in spinal cords vary >200-fold in ALS patients and 50-fold in the transgenic models carrying different mutations (Jonsson et al., 2004, 2006). A common cytotoxic ALS-causing conformational species of SOD1 is thus likely to exist in very low concentrations in the tissue. This as yet unidentified species would constitute larger proportions of the low-level unstable mutants G85R and G127X than of mutants that form high steady-state levels. The cellular distributions of the former might thus better inform on the primary location(s) of injury. In this respect, the present findings of low levels of G85R, and in particular G127X hSOD1s, associated with mitochondria would strongly argue against this organelle being the primary target.
In intervention studies, the G93A ALS model is convenient and is used almost exclusively. Some of the therapeutic effects detected in these mice may, however, be the result of protection against effects caused by the mitochondrial overloading. The ALS models with low levels of SOD1 may be preferable for intervention studies and studies of disease mechanisms.
Footnotes
This work was supported by the Swedish Science Council, the Swedish Brain Fund/Hållsten Fund, the Swedish Medical Society, including the Björklund Fund for ALS Research, the Swedish Association of Persons with Neurological Disabilities, Västerbotten County Council, The Kempe Foundations, and Konung Gustaf V's and Drottning Victorias Foundation. We thank Eva Bern, Ingalis Fransson, Karin Hjertkvist, Ann-Charlott Nilsson, Karin Wallgren, and Agneta Öberg for technical assistance and Dr D. W. Cleveland for supplying the G85R SOD1 transgenic mice.
References
- Andersen PM, Nilsson P, Ala-Hurula V, Keranen ML, Tarvainen I, Haltia T, Nilsson L, Binzer M, Forsgren L, Marklund SL (1995). Amyotrophic lateral sclerosis associated with homozygosity for an Asp90Ala mutation in CuZn-superoxide dismutase. Nat Genet 10:61–66. [DOI] [PubMed] [Google Scholar]
- Andersen PM, Sims KB, Xin WW, Kiely R, O'Neill G, Ravits J, Pioro E, Harati Y, Brower RD, Levine JS, Heinicke HU, Seltzer W, Boss M, Brown RH Jr (2003). Sixteen novel mutations in the Cu/Zn superoxide dismutase gene in amyotrophic lateral sclerosis: a decade of discoveries, defects and disputes. Amyotroph Lateral Scler Other Motor Neuron Disord 2:62–73. [DOI] [PubMed] [Google Scholar]
- Bowling AC, Schulz JB, Brown RH Jr, Beal MF (1993). Superoxide dismutase activity, oxidative damage, and mitochondrial energy metabolism in familial and sporadic amyotrophic lateral sclerosis. J Neurochem 61:2322–2325. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW (1997). ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 18:327–338. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW (1998). Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281:1851–1854. [DOI] [PubMed] [Google Scholar]
- Cozzolino M, Ferri A, Ferraro E, Rotilio G, Cecconi F, Carri MT (2006). Apaf1 mediates apoptosis and mitochondrial damage induced by mutant human SOD1s typical of familial amyotrophic lateral sclerosis. Neurobiol Dis 21:69–79. [DOI] [PubMed] [Google Scholar]
- Crapo JD, Oury T, Rabouille C, Slot JW, Chang LY (1992). Copper,zinc superoxide dismutase is primarily a cytosolic protein in human cells. Proc Natl Acad Sci USA 89:10405–10409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damiano M, Starkov AA, Petri S, Kipiani K, Kiaei M, Mattiazzi M, Beal MF, Manfredi G (2006). Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J Neurochem 96:1349–1361. [DOI] [PubMed] [Google Scholar]
- Field LS, Furukawa Y, O'Halloran TV, Culotta VC (2003). Factors controlling the uptake of yeast copper/zinc superoxide dismutase into mitochondria. J Biol Chem 278:28052–28059. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX (1994). Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264:1772–1775. [DOI] [PubMed] [Google Scholar]
- Haverkamp LJ, Appel V, Appel SH (1995). Natural history of amyotrophic lateral sclerosis in a database population. Validation of a scoring system and a model for survival prediction. Brain 118:707–719. [DOI] [PubMed] [Google Scholar]
- Higgins CM, Jung C, Ding H, Xu Z (2002). Mutant Cu, Zn superoxide dismutase that causes motoneuron degeneration is present in mitochondria in the CNS. In: J Neurosci 22: pp. RC215 (1–6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins CM, Jung C, Xu Z (2003). ALS-associated mutant SOD1G93A causes mitochondrial vacuolation by expansion of the intermembrane space and by involvement of SOD1 aggregation and peroxisomes. BMC Neurosci 4:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaarsma D, Haasdijk ED, Grashorn JA, Hawkins R, van Duijn W, Verspaget HW, London J, Holstege JC (2000). Human Cu/Zn superoxide dismutase (SOD1) overexpression in mice causes mitochondrial vacuolization, axonal degeneration, and premature motoneuron death and accelerates motoneuron disease in mice expressing a familial amyotrophic lateral sclerosis mutant SOD1. Neurobiol Dis 7:623–643. [DOI] [PubMed] [Google Scholar]
- Jaarsma D, Rognoni F, van Duijn W, Verspaget HW, Haasdijk ED, Holstege JC (2001). CuZn superoxide dismutase (SOD1) accumulates in vacuolated mitochondria in transgenic mice expressing amyotrophic lateral sclerosis-linked SOD1 mutations. Acta Neuropathol 102:293–305. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Dalton MJ, Gurney ME, Kopito RR (2000). Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA 97:12571–12576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson PA, Ernhill K, Andersen PM, Bergemalm D, Brannstrom T, Gredal O, Nilsson P, Marklund SL (2004). Minute quantities of misfolded mutant superoxide dismutase-1 cause amyotrophic lateral sclerosis. Brain 127:73–88. [DOI] [PubMed] [Google Scholar]
- Jonsson PA, Graffmo KS, Andersen PM, Brannstrom T, Lindberg M, Oliveberg M, Marklund SL (2006). Disulphide-reduced superoxide dismutase-1 in CNS of transgenic amyotrophic lateral sclerosis models. Brain 129:451–464. [DOI] [PubMed] [Google Scholar]
- Kirkinezos IG, Bacman SR, Hernandez D, Oca-Cossio J, Arias LJ, Perez-Pinzon MA, Bradley WG, Moraes CT (2005). Cytochrome c association with the inner mitochondrial membrane is impaired in the CNS of G93A-SOD1 mice. J Neurosci 25:164–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lillo C, Jonsson PA, Vande VC, Ward CM, Miller TM, Subramaniam JR, Rothstein JD, Marklund S, Andersen PM, Brannstrom T, Gredal O, Wong PC, Williams DS, Cleveland DW (2004). Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 43:5–17. [DOI] [PubMed] [Google Scholar]
- Marklund S (1976). Spectrophotometric study of spontaneous disproportionation of superoxide anion radical and sensitive direct assay for superoxide dismutase. J Biol Chem 251:7504–7507. [PubMed] [Google Scholar]
- Marklund SL, Andersen PM, Forsgren L, Nilsson P, Ohlsson PI, Wikander G, Oberg A (1997). Normal binding and reactivity of copper in mutant superoxide dismutase isolated from amyotrophic lateral sclerosis patients. J Neurochem 69:675–681. [DOI] [PubMed] [Google Scholar]
- Mattiazzi M, D'Aurelio M, Gajewski CD, Martushova K, Kiaei M, Beal MF, Manfredi G (2002). Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem 277:29626–29633. [DOI] [PubMed] [Google Scholar]
- Mattiazzi M, Vijayvergiya C, Gajewski CD, DeVivo DC, Lenaz G, Wiedmann M, Manfredi G (2004). The mtDNA T8993G (NARP) mutation results in an impairment of oxidative phosphorylation that can be improved by antioxidants. Hum Mol Genet 13:869–879. [DOI] [PubMed] [Google Scholar]
- Okado-Matsumoto A, Fridovich I (2002). Amyotrophic lateral sclerosis: a proposed mechanism. Proc Natl Acad Sci USA 99:9010–9014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzardini M, Mangolini A, Lupi M, Ubezio P, Bendotti C, Cantoni L (2005). Low levels of ALS-linked Cu/Zn superoxide dismutase increase the production of reactive oxygen species and cause mitochondrial damage and death in motor neuron-like cells. J Neurol Sci 232:95–103. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, van den Bergh R (1993). Mutations in Cu/Zn superoxide-dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362:59–62. [DOI] [PubMed] [Google Scholar]
- Vijayvergiya C, Beal MF, Buck J, Manfredi G (2005). Mutant superoxide dismutase 1 forms aggregates in the brain mitochondrial matrix of amyotrophic lateral sclerosis mice. J Neurosci 25:2463–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Xu G, Borchelt DR (2002). High molecular weight complexes of mutant superoxide dismutase 1: age-dependent and tissue-specific accumulation. Neurobiol Dis 9:139–148. [DOI] [PubMed] [Google Scholar]
- Weisiger RA, Fridovich I (1973). Mitochondrial superoxide dismutase. Site of synthesis and intramitochondrial localization. J Biol Chem 248:4793–4796. [PubMed] [Google Scholar]
- Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL (1995). An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 14:1105–1116. [DOI] [PubMed] [Google Scholar]