

Abstract
A glial glutamate transporter, GLAST, is expressed abundantly in Bergmann glia and plays a major role in glutamate uptake at the excitatory synapses in cerebellar Purkinje cells (PCs). It has been reported that a higher percentage of PCs in GLAST-deficient mice are multiply innervated by climbing fibers (CFs) than in the wild-type (WT) mice, and that CF-mediated EPSCs with small amplitude and slow rise time, designated as atypical slow CF-EPSCs, are observed in these mice. To clarify the mechanism(s) underlying the generation of these atypical CF-EPSCs, we used (2S,3S)-3-[3-(4-methoxybenzoylamino)benzyloxy]aspartate (PMB-TBOA), an inhibitor of glial glutamate transporters. After the application of PMB-TBOA, slow-rising CF-EPSCs were newly detected in WT mice, and their rise and decay kinetics were different from those of conventional fast-rising CF-EPSCs but similar to those of atypical CF-EPSCs in GLAST-deficient mice. Furthermore, both slow-rising CF-EPSCs in the presence of PMB-TBOA in WT mice and atypical CF-EPSCs in GLAST-deficient mice showed much greater paired-pulse depression compared with fast-rising CF-EPSCs. In addition, both of them were more markedly inhibited by γ-d-glutamyl-glycine, a low-affinity competitive antagonist of AMPA receptors. These results indicated that both of these types of EPSCs were mediated by a low concentration of glutamate released from neighboring CFs. Based on all of these findings, we suggest that glial transporters prevent glutamate released from a single CF from spilling over to neighboring PCs other than the synaptically connected PC, and play an essential role in the maintenance of the functional one-to-one relationship between CFs and PCs.
Keywords: glutamate transporter, GLAST, multiple innervation, climbing fiber, Purkinje cell, EPSC
Introduction
Glutamate transporters regulate excitatory synaptic transmission by rapid removal of released glutamate. To date, five subtypes of glutamate transporters have been cloned: GLAST (Storck et al., 1992; Tanaka, 1994), GLT1 (Pines et al., 1992), excitatory amino acid carrier 1 (EAAC1) (Kanai and Hediger, 1992), excitatory amino acid transporter 4 (EAAT4) (Fairman et al., 1995), and EAAT5 (Arriza et al., 1997). GLAST and GLT1 are expressed in glial cells (Rothstein et al., 1994; Lehre et al., 1995), whereas EAAC1 and EAAT4 are expressed in neurons (Furuta et al., 1997; Dehnes et al., 1998). EAAT5 is present in glia and neurons of the retina (Eliasof et al., 1998). In the cerebellar cortex, both GLAST and EAAT4 are abundantly expressed as predominant glial and neuronal transporters, respectively (Danbolt, 2001). GLAST is located on the plasma membranes of Bergmann glial processes surrounding the excitatory synapses in Purkinje cells (PCs) (Rothstein et al., 1994; Chaudhry et al., 1995), whereas EAAT4 is concentrated in extrasynaptic regions at excitatory synapses in PCs (Tanaka et al., 1997; Dehnes et al., 1998). This different distribution of GLAST and EAAT4 at the electron microscopic level suggests that these transporters serve distinct functions in excitatory synapses on PCs.
Glial glutamate transporters, especially GLAST, have been shown to remove most of the released glutamate at climbing fiber (CF) synapses in PCs (Otis et al., 1997; Wadiche and Jahr, 2001). In fact, removal of released glutamate is retarded in GLAST-deficient mice (Takayasu et al., 2005) or as a result of the inefficient glial uptake induced by the retraction of astrocytic processes (Iino et al., 2001). Furthermore, such selective inhibitions of glial glutamate uptake appear to cause persistent multiple CF innervation in PCs (Watase et al., 1998; Iino et al., 2001).
Watase et al. (1998) reported that a higher percentage of PCs in GLAST-deficient mice were multiply innervated compared with that in the wild-type (WT) mice. They also found the presence of two distinct types of CF-mediated EPSCs (CF-EPSCs) with small amplitudes in the mutant mice. One had a fast rise time similar to multiple CF-EPSCs in young WT mice. The other, designated as atypical CF-EPSCs, had an extremely slow rise time. In this study, we aimed to clarify the mechanism(s) underlying the generation of atypical CF-EPSCs by using a newly synthesized, nontransportable inhibitor of glutamate uptake, (2S,3S)-3-[3-(4-methoxybenzoylamino)benzyloxy]aspartate (PMB-TBOA) obtained by introducing amido substitutions on the benzyl ring of l-threo-β-benzyloxyaspartate (Shimamoto et al., 2004). The application of 200 nm PMB-TBOA, which inhibited glial glutamate transporters almost completely with a minor effect on neuronal ones (Takatsuru et al., 2006), caused slow-rising CF-EPSCs in WT mice, whose properties were similar to those of atypical CF-EPSCs in GLAST-deficient mice. Our results suggest that the atypical CF-EPSCs in GLAST-deficient mice are generated by a low concentration of glutamate released from neighboring CFs. Glial glutamate transporters are likely to play an essential role in the maintenance of the functional one-to-one relationship between CFs and PCs.
Materials and Methods
Generation of GLAST-deficient mice.
GLAST-deficient mice were produced as described previously (Watase et al., 1998). Both the WT and GLAST-deficient mice used were of C57BL/6 genetic background and were maintained under standard conditions of feeding and lighting (12 h light/dark cycle, 22°C). For each experiment, all littermates were processed, and their individual genotype was determined a posteriori by PCR as described previously (Watase et al., 1998). All experiments were performed according to the guidelines approved by the Animal Care and Experimentation Committee of Gunma University.
Brain slice preparation.
Parasagittal cerebellar slices of 200–250 μm thickness were prepared from Wistar rats [ages, 13–17 d; postnatal day 13 (P13)–P17] or WT or GLAST-deficient mice (P28–P76) using a microslicer (DTK-1000; Dosaka EM, Kyoto, Japan) as described previously (Iino et al., 2001). The slices were maintained at room temperature after an initial 1 h of incubation until needed for recording. The solution used for slice storage and the recording solution consisted of the following (in mm): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 d-glucose, saturated with 95% O2 and 5% CO2. During recordings, the external solution was maintained at 32°C with a solution in-line heater (SH-27A; Warner Instruments, Hamden, CT).
Electrophysiology.
PCs and Bergmann glia were visualized by using an upright microscope (Axioskop; Zeiss, Oberkochen, Germany) with a 60× water-immersion objective and equipped with Nomarski optics. Whole-cell patch-clamp recordings were performed in the presence of 100 μm picrotoxin to block GABAA receptors. The internal solution for recording of AMPA receptor (AMPAR)-mediated EPSCs in PCs contained the following (in mm): 150 Cs-gluconate, 8 NaCl, 2 Mg-ATP, 10 HEPES, 0.1 spermine, and 5 lidocaine N-ethyl bromide (QX-314), pH 7.2. CF-evoked responses in Bergmann glia, that is, AMPAR-mediated currents and synaptically activated transporter currents (STCs) were recorded at −80 mV in the absence and presence of 20 μm NBQX, respectively, with a similar internal solution, except that K-gluconate was substituted for Cs-gluconate. The resistances of patch pipettes filled with these internal solutions were 2–4 MΩ and 4–5 MΩ for recording of PCs and Bergmann glia, respectively. CFs and parallel fibers (PFs) were activated by applying square pulses (10–100 μA, 100 μs) with a concentric bipolar tungsten electrode placed in the granule cell layer and the molecular layer, respectively. CF-EPSCs were considered as slow-rising or atypical if their 10–90% rise time was longer than 1 ms. To search for multiple innervation by CFs in a tested PC, the stimulating electrode was moved systematically in the granule cell layer, and the stimulus intensity was gradually increased at each stimulation site. The current responses were recorded with an EPC-8 amplifier (HEKA Elektronik, Lambrecht, Germany), and the pCLAMP system (version 7; Molecular Devices, Foster City, CA) was used for data acquisition and analysis. EPC-9 amplifier (HEKA Elektronik) was used for dual recordings from a pair of PCs. Origin (version 5.5; Microcal Software, Northampton, NC), Pulse/Pulsefit (HEKA Elektronik), and Igor Pro (WaveMetrics, Lake Oswego, OR) programs were also used for data analysis. AMPAR-mediated EPSCs and STCs were filtered at 3 kHz and digitized at 10 kHz; metabotropic glutamate receptor (mGluR)-mediated synaptic currents were filtered at 1 kHz and digitized at 2 kHz. The amplitude-weighted decay time constant of EPSCs was defined as the integrated EPSC after the peak divided by the peak amplitude and was used as a single parameter measure to represent the kinetics of decay of the EPSC. Unless otherwise noted, values are given as mean ± SEM. Student's t test was performed for statistical analysis, and differences were considered significant at p < 0.05.
Drugs and chemicals.
PMB-TBOA was prepared as described previously (Shimamoto et al., 2004). 2,3-Dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX), dl-threo-β-benzyloxyaspartate (dl-TBOA), (2S)-3-{[(15)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl}(phenylmethyl)phosphinic acid (CGP55845), d-(−)-2-amino-5-phosphonovalerate (APV), (rs)-α-methyl-3-carboxymethylphenylglycine (MCPG), (±)-4-(4-aminophenyl)-1,2-dihydro-1-methyl-2-propylcarbamoyl-6,7-methylenedioxyphthalazine (SYM 2206), γ-d-glutamyl-glycine (γ-DGG), and cyclothiazide (CTZ) were purchased from Tocris Bioscience (Bristol, UK). Picrotoxin was from Wako Pure Chemical Industries (Osaka, Japan), and QX-314 was from Sigma (St. Louis, MO). All drugs were dissolved in distilled water or DMSO at a concentration of 1–10 mm. The final concentrations of the drugs indicated in the Results section were obtained by diluting the stock solutions in the recording external solution. The final DMSO concentration was always <0.1%. All solutions containing drugs were bath applied at a rate of 2–3 ml/min that resulted in solution exchange within 2–3 min.
Results
Inhibition of glial glutamate transporters by PMB-TBOA
The novel antagonist of glutamate transporters, PMB-TBOA, specifically blocks GLAST and GLT1 at extremely low concentrations (Shimamoto et al., 2004). PMB-TBOA was shown to inhibit STCs evoked by CF stimulation in Bergmann glia in mice in a dose-dependent manner with the IC50 value of 46.9 nm. The bath application of 200 nm PMB-TBOA suppressed STCs in Bergmann glia in mice almost completely, whereas it reduced STCs evoked by CF stimulation in PCs only by 9.6% (Takatsuru et al., 2006). In this study, we obtained similar results in Bergmann glia and PCs in cerebellar slices of young rats (P13–P17) (see supplemental Fig. S1, available at www.jneurosci.org as supplemental material). The IC50 values of PMB-TBOA for STCs in Bergmann glia and PCs were 52.0 and 651 nm, respectively. PMB-TBOA at 200 nm blocked STCs in Bergmann glia almost completely (by 97.8 ± 1.4% inhibition of control amplitude; n = 7), whereas it inhibited STCs in PCs only weakly (by 7.8 ± 1.8% inhibition of control amplitude; n = 11). This indicates that PMB-TBOA at 200 nm is useful to block glial glutamate transporters with only a small effect on neuronal transporters.
In the presence of 200 nm PMB-TBOA, CF stimulation elicited small currents (13.2 ± 5.7 pA; n = 4) in an all-or-none manner in the late phase in 4 of 10 PCs tested (supplemental Fig. S1E,F, available at www.jneurosci.org as supplemental material). These late currents had their peaks at several hundred milliseconds after the stimulus and were obviously different from the STCs with their peaks within 20 ms after the stimulus. These currents were increased by 200 μm dl-TBOA and completely blocked by 1 mm MCPG, an mGluR antagonist (supplemental Fig. S1E,F, available at www.jneurosci.org as supplemental material). They are most likely to be mGluR-mediated EPSCs (mGluR-EPSCs) evoked by CF stimulation (Dzubay and Otis, 2002). In contrast, the peak amplitude of CF-evoked STCs in PCs was not affected by MCPG. Furthermore, 50 μm APV, an inhibitor of NMDA receptors, and 5 μm CGP55845, an inhibitor of GABAB receptors, exerted no influence on the effects of PMB-TBOA on STCs in PCs.
Slow-rising CF-EPSCs under conditions in which glial glutamate uptake is inhibited
We first examined the properties of CF-EPSCs in cerebellar slices from WT and GLAST-deficient mice at similar ages (P28–P76). CFs were stimulated in the granule cell layer 60–100 μm away from the recorded PCs, and the stimulus intensity was gradually increased. In all PCs tested in WT mice, CF-EPSCs were elicited in an all-or-none manner with one discrete step, and they had fast-rising kinetics and large amplitude in the normal solution (Fig. 1A, top traces). The application of 200 nm PMB-TBOA prolonged the weighted decay time constant of these conventional CF-EPSCs significantly in WT mice because of the retarded removal of synaptically released glutamate attributable to the lack of function of glial glutamate transporters (Takatsuru et al., 2006). However, this treatment exerted no influence on either rise time or peak amplitude of these CF-EPSCs. Interestingly, small, slow-rising CF-EPSCs often occurred in PCs tested in the presence of 200 nm PMB-TBOA. These slow-rising CF-EPSCs were clearly distinguished from fast-rising ones by their 10–90% rise times of >1 ms. These results implied that CF-EPSCs had multiple discrete steps that occurred in an all-or-none manner in the presence of 200 nm PMB-TBOA (Fig. 1A, middle traces). It has been reported that significantly a higher percentage of PCs are multiply innervated by CFs in GLAST-deficient mice than in WT mice and that two distinct types of small-amplitude CF-EPSCs occur in GLAST-deficient mice (Watase et al., 1998). One has a fast rise time and is similar to WT multiple CF-EPSCs, whereas the other, designated as atypical CF-EPSCs, has a slow rise time, which is not seen in WT mice. We confirmed the occurrence of such slow-rising CF-EPSCs in GLAST-deficient mice (Fig. 1A, bottom traces) and noted that their shapes were similar to those of slow-rising CF-EPSCs in the presence of 200 nm PMB-TBOA in WT mice. In WT mice, the mean amplitude and 10–90% rise time of fast-rising and large CF-EPSCs were 1126 ± 139 pA and 0.43 ± 0.03 ms in CF-EPSCs in the control condition (n = 5), whereas the corresponding values were 152.8 ± 38.9 pA and 5.4 ± 1.0 ms for slow-rising CF-EPSCs (n = 9) and 1208 ± 119 pA and 0.44 ± 0.02 ms for fast-rising CF-EPSCs in the presence of 200 nm PMB-TBOA (n = 7). In contrast, they were 125.8 ± 19.8 pA and 4.3 ± 0.2 ms for atypical CF-EPSCs (n = 10), and 1150 ± 113 pA and 0.46 ± 0.03 ms for fast-rising CF-EPSCs in GLAST-deficient mice (n = 7). Thus, the atypical CF-EPSCs in GLAST-deficient mice were similar in both amplitude and 10–90% rise time to slow-rising CF-EPSCs in the presence of PMB-TBOA in WT mice (Fig. 1B).
Figure 1.

Slow-rising CF-EPSCs under conditions in which glial glutamate uptake is inhibited. A, CF-EPSCs in a WT PC innervated by a single CF under the control condition (top traces) and those in the presence of 200 nm PMB-TBOA in the same PC (middle traces), in which small, slow-rising CF-EPSCs were additionally detected. Similar atypical CF-EPSCs were detected in GLAST-deficient mice (bottom traces). One to three traces are superimposed at each threshold of the stimulus intensity. The right graphs show EPSC amplitude (Amp.) against the stimulation (Stim.) intensity for the left traces, respectively. Each point indicates the peak amplitude of EPSC. With gradually increasing stimulus intensity, all EPSCs were elicited discretely. Stimuli were applied at 0.1 Hz. Holding potential was −10 mV. B, Summary of measurements of peak amplitude (left graph) and 10–90% rise time (right graph) of WT CF-EPSCs in the control condition (n = 5; Control), slow-rising (n = 9; Slow) and fast-rising CF-EPSCs (n = 7; Fast) in the presence of 200 nm PMB-TBOA, and atypical (n = 10; Atypical) and fast-rising (n = 7; Mutant fast) CF-EPSCs in GLAST-deficient mice. All CF-EPSCs whose 10–90% rise times were slower than 1 ms were grouped as slow-rising CF-EPSCs in WT mice and atypical CF-EPSCs in GLAST-deficient mice, respectively.
To detect the occurrence of slow-rising CF-EPSCs more systematically, the electrode for the stimulation of CFs was moved by ∼20 μm steps, and the stimulus intensity was gradually increased at each stimulation site at 0.1 Hz (Hashimoto et al., 2001). At some stimulus sites, slow-rising CF-EPSCs were evoked at lower stimulus intensity than fast-rising CF-EPSCs, whereas vice versa at other stimulus sites in the same PCs. By means of moving the stimulating electrode, the chance to elicit slow-rising CF-EPSCs by a lower stimulus intensity than that necessary to elicit fast-rising CF-EPSCs increased. Interestingly, once the fast-rising CF-EPSC was evoked, no slow-rising current was detected as an additional component at higher stimulus intensities. In 40 (80.0%) of 50 PCs tested in WT mice, slow-rising CF-EPSC(s) in addition to a fast-rising, large CF-EPSC were observed in the presence of 200 nm PMB-TBOA. Of these 40 cells, 20, 14, and 6 PCs had one, two, and three discrete steps of slow-rising CF-EPSCs, respectively (Fig. 2A, top traces; B). In contrast, all PCs tested had only a single fast-rising CF-EPSC in the normal solution.
Figure 2.
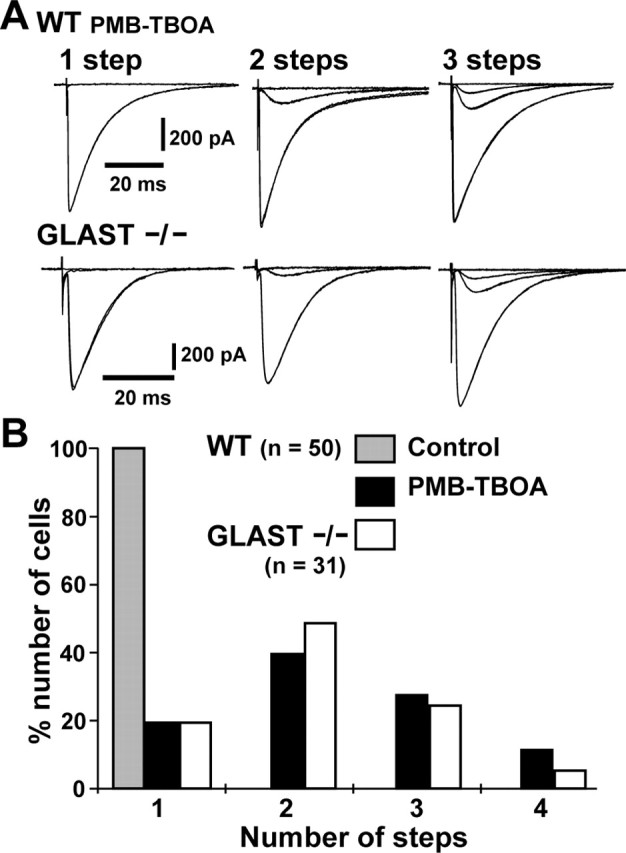
Multiple discrete steps of CF-EPSCs induced by inhibition of glial glutamate uptake. A, Examples of a single fast-rising plus multiple slow-rising CF-EPSCs in the presence of 200 nm PMB-TBOA in WT mice (top traces) and a single fast-rising plus multiple atypical CF-EPSCs in GLAST-deficient mice (bottom traces). Two or three traces are superimposed at each threshold of the stimulus intensity. Holding potential was −10 mV. B, Summary histogram showing the distribution of the number of discrete steps of CF-EPSCs. In WT mice, 1–3 steps of slow-rising CF-EPSCs were detected in the presence of PMB-TBOA in 80.0% (40 of 50) of PCs tested (black columns) in addition to a fast-rising EPSC. In the normal solution, all PCs had only a single-step fast-rising CF-EPSC (gray column). In GLAST-deficient mice, one to three discrete steps of atypical CF-EPSCs were detected in 80.1% (25 of 31) of PCs. The distribution of discrete steps of GLAST-deficient CF-EPSCs (white columns) was similar to that of WT CF-EPSCs in the presence of PMB-TBOA.
Using the same method, we examined the occurrence of atypical CF-EPSCs in GLAST-deficient mice. The atypical CF-EPSCs were detected in 25 (80.1%) of 31 PCs of GLAST-deficient mice. Of these 25 cells, 15, 8, and 2 PCs had one, two, and three discrete steps of atypical CF-EPSCs, respectively (Fig. 2A, bottom traces; B). Thus, the incidence of atypical CF-EPSCs in GLAST-deficient mice was similar to that of slow-rising EPSCs in the presence of PMB-TBOA in WT mice.
In this study, all PCs tested had only one fast-rising CF-EPSC in GLAST-deficient mice, whereas multiple fast-rising CF-EPSCs have been reported to occur in ∼15% of PCs in GLAST-deficient mice (Watase et al., 1998). One possible reason for this discrepancy might be that we used mice older than those used by previous authors.
Paired-pulse depression of slow-rising CF-EPSCs
Paired-pulse depression (PPD) is a characteristic feature of CF-PC synapses, which is attributed to a decreased transmitter release from presynaptic terminals in response to the second pulse of a paired stimulus (Konnerth et al., 1990). The magnitude of PPD of atypical CF-EPSCs in GLAST-deficient mice has been shown to be significantly greater than that of the WT and mutant fast-rising CF-EPSCs (Watase et al., 1998). Slow-rising CF-EPSCs in the presence of PMB-TBOA also showed greater PPD (Fig. 3A). When the depression ratio was plotted as a function of interpulse interval between 10 and 5000 ms, both the magnitude and time course of PPD of slow-rising CF-EPSCs were clearly different from those of fast-rising CF-EPSCs but were similar to those of atypical CF-EPSCs in GLAST-deficient mice (Fig. 3B). This result again supports the notion that slow-rising CF-EPSCs in the presence of PMB-TBOA in WT mice and atypical CF-EPSCs in GLAST-deficient mice are attributable to common mechanism(s). Furthermore, this result confirms that slow-rising CF-EPSCs in the presence of PMB-TBOA are elicited by stimulating CFs and not by activating mossy fiber–granule cell–PF circuits, because PF-evoked EPSCs display clear paired-pulse facilitation regardless of the presence or absence of PMB-TBOA (Fig. 4C).
Figure 3.

Paired-pulse depression of CF-EPSCs. A, Single-sweep examples of paired-pulse depression (interpulse interval of 50 ms) of CF-EPSC in control condition (Control), fast-rising (Fast) and slow-rising (Slow) CF-EPSCs in the presence of 200 nm PMB-TBOA in WT mice, and fast-rising (Mutant fast) and atypical (Atypical) CF-EPSCs in GLAST-deficient mice. B, Summary graph showing the magnitude and time course of paired-pulse depression of CF-EPSCs, of which example traces are shown in (A) (Control, n = 5, gray triangles; Fast, n = 5, black circles; Slow, n = 6, black squares; Mutant fast, n = 7, white circles; Atypical, n = 6, white squares). The second response was expressed as a percentage of the response to the first pulse and was plotted as a function of interpulse interval.
Figure 4.

Pharmacology and voltage dependency of slow-rising EPSCs and comparison of rise times between slow-rising CF-EPSC and PF-EPSCs. A, Slow-rising CF-EPSCs in the presence of 200 nm PMB-TBOA in WT mice (top traces) and atypical CF-EPSCs in GLAST-deficient mice (bottom traces) before and during the application of 100 μm SYM 2206, an AMPAR-specific antagonist. B, Example traces (top) of slow-rising CF-EPSCs in PMB-TBOA in WT mice recorded at various membrane potentials between −80 and + 60 mV in 20 mV steps, and their current–voltage relationship (bottom). C, Example traces of slow-rising CF-EPSCs and PF-EPSCs in the presence of 200 nm PMB-TBOA in a WT PC. A paired-pulse stimulus was given, of which the interpulse interval was 50 ms. The inset shows scaled traces of the first EPSCs. D, Summary plot of the 10–90% rise time of slow-rising CF-EPSCs and PF-EPSCs as shown in C (5.2 ± 0.4 vs 1.4 ± 0.1 ms; n = 12; p < 0.001). Each pair of slow-rising CF-EPSC and PF-EPSC in C and D was recorded in the same PC clamped at −70 mV. E, Example traces of atypical CF-EPSC and PF-EPSC in a GLAST-deficient mouse. The interpulse interval was 50 ms, and the inset shows scaled traces of the first EPSCs. F, Summary plot of the 10–90% rise time of atypical CF-EPSCs and PF-EPSCs as shown in E (4.0 ± 0.2 vs 1.4 ± 0.1 ms; n = 10; p < 0.001). Each pair of atypical CF-EPSC and PF-EPSC in E and F was recorded in the same PC clamped at −70 mV.
Synaptic AMPARs mediate slow-rising currents
Although CF-EPSCs are mediated primarily by AMPARs, PCs also possess a variety of other glutamate receptors, including mGluRs (Masu et al., 1991), NMDA receptors (Thompson et al., 2000), and kainate receptors (KARs) (Wisden and Seeburg, 1993; Huang et al., 2004). In particular, the activation of mGluRs elicits extremely slow EPSCs in PCs (Batchelor and Garthwaite, 1997; Dzubay and Otis, 2002). Therefore, we first used the mGluR antagonist MCPG and the non-NMDA receptor antagonist, NBQX to determine the contributions of the respective receptor to the slow-rising CF-EPSC in the presence of PMB-TBOA in WT mice and atypical CF-EPSCs in GLAST-deficient mice. The application of 1 mm MCPG had no effect on these slow CF-EPSCs (n = 4, slow-rising CF-EPSC; n = 4, mutant atypical CF-EPSC), whereas all slow-rising CF-EPSCs were blocked completely by 10 μm NBQX (n = 6, slow-rising CF-EPSC; n = 6, mutant atypical CF-EPSC). We next tested whether KARs underlie these slow CF-EPSCs by using a selective AMPAR antagonist, SYM 2206. The application of 100 μm SYM 2206, a concentration that blocks AMPARs but attenuates KAR-mediated currents by <20% (Pelletier et al., 1996; Li et al., 1999), completely abolished slow-rising CF-EPSCs in 200 nm PMB-TBOA in WT mice (n = 4) (Fig. 4A, WT) as well as atypical CF-EPSCs in GLAST-deficient mice (n = 4; Fig. 4A; GLAST −/−). Furthermore, slow-rising CF-EPSCs in PMB-TBOA had a reversal potential close to 0 mV and a linear current–voltage relationship at membrane potentials from −80 to +60 mV (n = 3) (Fig. 4B). Similar results were obtained in atypical CF-EPSCs for GLAST-deficient mice (n = 3; data not shown). These results indicate that almost all components of both PMB-TBOA-induced slow-rising CF-EPSCs and mutant atypical CF-EPSCs are mediated by AMPARs.
Slow-rising currents are not attributable to dendritic filtering
Synaptic currents generated in distal synapses and measured at the soma are substantially filtered and distorted (Johnston and Brown, 1983; Spruston et al., 1993). PF synapses are located on the distal dendrites of PCs, whereas CF synapses are located at more proximal dendrites (Eccles et al., 1967). Such a distinct electrotonic length from soma to the site of the synapse is one of the reasons for the slower 10–90% rise time of PF-mediated EPSCs (PF-EPSCs) compared with those of CF-EPSCs (Llano et al., 1991; Aiba et al., 1994; Kano et al., 1995, 1997). In fact, in PCs lacking glutamate receptor δ2 (GluRδ2), additional CF synapses at distal dendrites cause atypical CF-EPSCs with slow rise time (Hashimoto et al., 2001; Ichikawa et al., 2002). In addition, the passive membrane properties would affect the rise time of EPSCs (Llano et al., 1991). Therefore, we compared the rise time of the slow-rising CF-EPSCs and PF-EPSCs in the same PC. The slow-rising CF-EPSCs in the presence of PMB-TBOA and mutant atypical CF-EPSCs were elicited by the stimulation of CFs in the granule cell layer, whereas PF-EPSCs were elicited in the same PCs by the stimulation of PFs in the molecular layer 60–100 μm away from the recoded PCs. The 10–90% rise times of slow-rising and mutant atypical CF-EPSCs were considerably slower than those of PF-EPSCs, respectively [5.2 ± 0.4 ms for slow-rising CF-EPSCs, 1.4 ± 0.1 ms for PF-EPSCs, n = 12, p < 0.001 (Fig. 4C,D); 4.0 ± 0.2 ms for mutant atypical CF-EPSCs, 1.4 ± 0.1 ms for mutant PF-EPSCs, n = 10, p < 0.001 (Fig. 4E,F)]. Considering the normal anatomy of the GLAST-deficient cerebellum (Watase et al., 1998), we can safely conclude that the slow rise times of both PMB-TBOA-induced and mutant atypical CF-EPSCs are not attributable simply to dendritic filtering.
Difference in glutamate concentration between slow- and fast-rising CF-EPSCs
The results described above suggest that both pharmacologically induced slow-rising EPSCs in WT mice and atypical CF-EPSCs in GLAST-deficient mice are attributed to a common mechanism, that is, an inadequate uptake of released glutamate by glial transporters. It is likely that low concentrations of glutamate that escaped from the uptake by the glial transporters spilled over to neighboring PCs and activated their AMPARs. If so, these receptors would encounter lower concentrations of glutamate during a synaptic event than those located in an active synapse. We investigated the differences in the concentration of glutamate transient between slow- and fast-rising CF-EPSCs using pharmacological tools (Fig. 5).
Figure 5.

Difference in glutamate concentration between slow- and fast-rising CF-EPSCs. A, C, Example traces showing the effect of 100 μm CTZ (A) or 1 mm γ-DGG (C) on the WT CF-EPSC in control condition (Control), slow-rising (Slow) and fast-rising (Fast) CF-EPSCs in the presence of 200 nm PMB-TBOA, and atypical (Atypical) and fast-rising (Mutant fast) CF-EPSCs in GLAST-deficient mice. Holding potential was −70 mV in the experiments of slow-rising EPSCs and atypical CF-EPSCs, and −10 mV in the other experiments in A and C. B, D, Summary of relative peak amplitudes (Amp.) in the presence of 100 μm CTZ (B) or 1 mm γ-DGG (D) of WT CF-EPSCs in the control condition (n = 7; Control), slow-rising (n = 5; Slow) and fast-rising CF-EPSCs (n = 5; Fast) in the presence of 200 nm PMB-TBOA, and atypical (n = 7; Atypical) and fast-rising (n = 6; Mutant fast) CF-EPSCs in GLAST-deficient mice. The peak amplitude of each EPSC before the application of CTZ or γ-DGG was taken as a reference in estimating the relative amplitude.
We first tested the effects of CTZ (100 μm), which greatly reduces desensitization and increases the affinity of glutamate for AMPARs (Patneau et al., 1993; Yamada and Tang, 1993; Partin et al., 1994). Based on the latter property, CTZ markedly increases AMPAR currents mediated by lower concentrations of glutamate (Dzubay and Jahr, 1999). As was expected, the addition of 100 μm CTZ markedly increased the amplitude of slow-rising CF-EPSCs in PMB-TBOA to 374 ± 53% (n = 6) and that of atypical CF-EPSCs in GLAST-deficient mice to 560 ± 46% (n = 7). In contrast, 100 μm CTZ increased the amplitude of fast-rising CF-EPSCs to only 128 ± 4.8% in the presence of PMB-TBOA in WT mice (n = 5) and that of fast-rising EPSCs to 125 ± 4.0% in GLAST-deficient mice (n = 6) (Fig. 5A,B).
We next used a low-affinity competitive antagonist of non-NMDA receptors, γ-DGG (1 mm), which competes more effectively against lower concentrations of glutamate than higher ones (Watkins, 1991; Liu et al., 1999; Wadiche and Jahr, 2001). The efficacy of γ-DGG is determined by the nonequilibrium interaction between the transient presence of glutamate and the continuous presence of γ-DGG (Clements et al., 1992). The application of 1 mm γ-DGG markedly decreased the amplitude of slow-rising CF-EPSCs to 30.0 ± 1.9% in PMB-TBOA in WT mice (n = 5) and that of atypical EPSCs to 32.0 ± 2.6% in GLAST-deficient mice (n = 6). In contrast, 1 mm γ-DGG decreased the amplitude of fast-rising CF-EPSCs to only 85.0 ± 4.8% in the presence of PMB-TBOA in WT mice (n = 5) and that of fast-rising EPSCs to 83.0 ± 5.2% in GLAST-deficient mice (n = 5) (Fig. 5C,D).
CTZ and γ-DGG had no effect on the amplitude and kinetics of STCs evoked by CF stimulation in PCs in matured WT mice (P35–P42) (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). The same result has also been shown in young rats (Dzubay and Jahr, 1999; Wadiche and Jahr, 2001). This indicates that these drugs exert no influence on glutamate release at CF-PC synapses and that their effects on the amplitude of CF-EPSCs are attributed exclusively to their actions on postsynaptic AMPARs. Thus, we conclude that AMPARs underlying the slow-rising CF-EPSCs are exposed to lower concentrations of glutamate than those underlying the fast-rising CF-EPSCs.
Lowering glutamate release probability preferentially reduces slow-rising CF-EPSCs
The activation of AMPARs at the peak of conventional fast-rising CF-EPSCs is saturated by an excessive amount of glutamate from CF terminals with the high release probability (Wadiche and Jahr, 2001; Harrison and Jahr, 2003). If slow-rising CF-EPSCs are mediated by glutamate that is released from CFs innervating neighboring PCs and spills over to AMPARs of the PC tested, the activation of AMPARs would not reach the saturation level at the peak, and therefore the amplitude of slow-rising CF-EPSCs would be more sensitive to release probability than that of conventional fast-rising CF-EPSCs. We next examined this possibility. According to the Ca2+ dependency of release probability at CF-PC synapses, lowering extracellular Ca2+ from 2.0 to 1.0 or 0.5 mm (and simultaneously increasing Mg2+ from 1.0 to 2.0 or 2.5 mm, respectively) reduced the peak amplitude of CF-EPSCs (Fig. 6A). The peak amplitude of CF-EPSCs in 0.5 mm Ca2+ relative to that in 2 mm Ca2+ was 45.0 ± 4.0% (n = 5) in conventional CF-EPSCs and 7.0 ± 0.7% (n = 7) and 39.0 ± 3.0% (n = 7) in slow- and fast-rising CF-EPSCs in the presence of 200 nm PMB-TBOA in WT mice, respectively. Figure 6B summarizes the preferential reduction of the relative peak amplitude of slow-rising CF-EPSCs by lowering the Ca2+ concentration. Such a preferential Ca2+ sensitivity of slow-rising CF-EPSCs supports the notion that they arise from the activation of AMPARs by glutamate released from the neighboring active sites.
Figure 6.
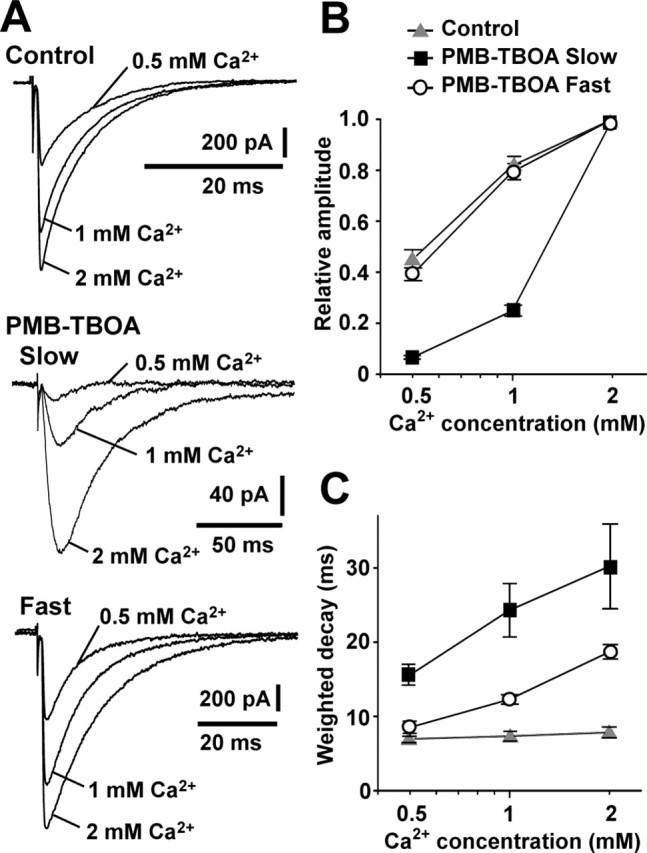
Differential effects of lowering glutamate release probability on slow-rising and fast-rising CF-EPSCs. A, Example traces showing the effect of lowering extracellular Ca2+ from 2.0 to 1.0 or 0.5 mm on WT CF-EPSCs (Control) and slow-rising (Slow) and fast-rising (Fast) CF-EPSCs in the presence of 200 nm PMB-TBOA. B, C, Summary of peak amplitudes (B) and weighted decay time constants (C) of WT CF-EPSCs (n = 5; gray triangles) and slow-rising (n = 7; black squares) and fast-rising (n = 7; open circles) CF-EPSCs in the presence of 200 nm PMB-TBOA under the different extracellular Ca2+ conditions (0.5, 1, and 2 mm). Peak amplitude in 0.5 and 1 mm Ca2+ is represented as a value relative to that in 2 mm Ca2+. Holding potential was −70 mV in the experiments of slow-rising CF-EPSCs and −10 mV in the other experiments in A–C.
It has been shown that the suppression of glial glutamate transporters by 200 nm PMB-TBOA prolonged the decay of fast-rising CF-EPSCs (Takatsuru et al., 2006). This decay prolonged by PMB-TBOA was markedly speeded by lowering the extracellular Ca2+ concentration. This was also the case for slow-rising CF-EPSCs (Fig. 6C). Thus, under the conditions in which there was a lack of the functions of glial glutamate transporters, the decay kinetics of both fast- and slow-rising CF-EPSCs depended on the release probability of glutamate.
dl-TBOA, a nonselective inhibitor of glutamate uptake, also induces slow-rising CF-EPSCs
If PMB-TBOA induces slow-rising CF-EPSCs by inhibiting glial glutamate uptake, they would also be visible or even more prominent when all glutamate transporters are inhibited nonselectively by dl-TBOA (Shimamoto et al., 1998). We next tested this possibility. In the presence of 200 μm dl-TBOA, slow-rising CF-EPSCs also occurred in an all-or-none manner in WT mice in 9 of 11 PCs tested (Fig. 7A). The magnitude of PPD and the time course of recovery from PPD for these CF-EPSCs were similar to those in the presence of 200 nm PMB-TBOA (Fig. 7B,C). These CF-EPSCs were completely inhibited by SYM 2206 (Fig. 7D), indicating that they were also mediated by AMPARs (Fig. 7D). The mean amplitude of slow-rising CF-EPSCs in the presence of dl-TBOA was 185.2 ± 17.1 pA (n = 9). This value was slightly larger than that in the presence of 200 nm PMB-TBOA (152.8 ± 38.9 pA, n = 9), but there was no significant difference between them (Fig. 7E, left). The 10–90% rise time of slow-rising CF-EPSCs in the presence of dl-TBOA was 11.3 ± 0.9 ms (n = 9), being significantly longer than that in the presence of 200 nm PMB-TBOA (5.4 ± 1.0 ms; n = 9; p < 0.001) (Fig. 7E, middle). Furthermore, the weighted decay time constant was also longer in the presence of dl-TBOA (66.0 ± 10.3 ms; n = 9) than that in the presence of 200 nm PMB-TBOA (29.5 ± 2.9 ms; n = 9; p < 0.001) (Fig. 7E, right). Thus, a high concentration of dl-TBOA induced the slow-rising CF-EPSCs more prominently than 200 nm PMB-TBOA.
Figure 7.

Slow-rising CF-EPSCs in the presence of dl-TBOA. A, Slow-rising and fast-rising CF-EPSCs in a WT PC in the presence of 200 μm dl-TBOA. All EPSCs were elicited discretely with gradually increasing stimulus (Stim.) intensity. Two or three traces are superimposed at each threshold of the stimulus intensity. The right graph shows EPSC amplitude (Amp.) against the stimulus intensity for the left traces. B, Paired-pulse depression of slow-rising CF-EPSCs in the presence of dl-TBOA evoked at interpulse intervals indicated at the top of the traces. C, The mean ratio of PPD of slow-rising CF-EPSCs in the presence of dl-TBOA (n = 5). The second response was expressed as a percentage of the response to the first pulse and was plotted as a function of interpulse interval. D, Complete inhibition of slow-rising CF-EPSCs in the presence of dl-TBOA by 100 μm SYM 2206. E, Comparison of the mean values of the amplitude, 10–90% rise time, and weighted decay time constant of slow-rising CF-EPSCs in the presence of dl-TBOA (n = 9) with the corresponding values of slow-rising CF-EPSCs in the presence of PMB-TBOA (n = 9). The 10–90% rise time and the weighted decay time constant of slow-rising CF-EPSCs in the presence of dl-TBOA were significantly longer than those in the presence of PMB-TBOA (*p < 0.001). There was no significant difference in the amplitude between these two drugs (p = 0.286)
Simultaneous recordings of CF-EPSCs from two adjacent PCs in the presence of PMB-TBOA
To confirm the notion that CF stimulation would induce a fast-rising EPSC in the directly innervated PC and simultaneously slow-rising EPSCs in neighboring PCs when the function of glial glutamate transporters is inhibited, we finally performed simultaneous recordings of CF-EPSCs from 13 pairs of two adjacent PCs. When the intensity of CF stimulation was gradually increased in the presence of 200 nm PMB-TBOA in slices of WT mice, conventional fast-rising EPSCs in one cell and slow-rising EPSCs in the other cell were elicited simultaneously in all-or-none manner in five pairs of PCs (Fig. 8A). The simultaneous occurrence of slow-rising CF-EPSCs with different amplitudes was also seen in three pairs of PCs (Fig. 8B). Thus, we observed the coincidental EPSCs with the identical stimulus thresholds in 8 of 13 pairs of adjacent PCs (61.5%).
Figure 8.

Simultaneous recordings of CF-EPSCs from a pair of adjacent PCs in the presence of PMB-TBOA. A, Example traces of CF-EPSCs from two adjacent PCs (Cell 1 and Cell 2) in the presence of 200 nm PMB-TBOA. CF stimulation with the same intensity elicited simultaneously fast-rising EPSCs in one PC (Cell 1) and slow-rising EPSCs in the other PC (Cell 2). Both EPSCs occurred in an all-or-none manner with one discrete step. B, Example traces from another pair of PCs in the presence of PMB-TBOA. CF stimulation elicited simultaneously small slow-rising EPSCs in one PC (Cell 1) and larger slow-rising EPSCs in the other PC (Cell 2). Two or three traces are superimposed at each stimulus intensity. The right graphs show the EPSC amplitude (Amp.) against the stimulus (Stim.) intensity for the left traces of paired recordings (Cell 1, black circles; Cell 2, open circles), respectively.
Discussion
In this study, we showed that slow-rising CF-EPSCs appeared in WT mice in the presence of PMB-TBOA, a potent antagonist of glial glutamate transporters. These slow-rising CF-EPSCs were similar to atypical CF-EPSCs in GLAST-deficient mice (Watase et al., 1998) in their kinetics, the magnitude of PPD, and pharmacological properties. These results strongly suggest that both of these types of slow CF-EPSCs are caused by the indirect activation of AMPARs by low concentrations of glutamate released from neighboring CFs, as schematically summarized in supplemental Fig. S3 (available at www.jneurosci.org as supplemental material).
Glutamate spillover
It has been demonstrated in many synapses that the inhibition of uptake of synaptically released transmitter causes spillover of transmitter to neighboring synapses (Rossi and Hamann, 1998; Carter and Regehr, 2000; Diamond, 2001; Clark and Cull-Candy, 2002; DiGregorio et al., 2002; Takayasu et al., 2004). The results shown in this study can also be reasonably explained by the hypothesis that glutamate spillover causes both pharmacologically induced slow-rising and mutant atypical CF-EPSCs. First of all, both EPSCs exhibited a greater magnitude of PPD compared with the conventional fast-rising CF-EPSCs. This greater magnitude of PPD was reminiscent of that of CF-evoked currents in Bergmann glia (Dzubay and Jahr, 1999; Matsui and Jahr, 2004). In CF-EPSCs elicited by the first CF stimulation, the activation of postsynaptic receptors is saturated by an excessive amount of glutamate. Therefore, the amplitude of the CF-EPSCs does not accurately represent the amount of synaptically released glutamate, and the magnitude of PPD is underestimated (Harrison and Jahr, 2003). In contrast, unsaturated postsynaptic responses, such as CF-evoked currents in Bergmann glia or CF-EPSCs in PCs with a reduced synaptic glutamate transient, would reflect the amount of released glutamate more faithfully, and consequently the greater magnitude of PPD would be observed (Dzubay and Jahr, 1999; Harrison and Jahr, 2003). The slow-rising CF-EPSCs detected in this study would be unsaturated postsynaptic responses elicited by lower concentrations of glutamate that diffused out of the releasing sites and therefore would exhibit a greater magnitude of PPD. The lower concentration of glutamate transient of slow-rising CF-EPSCs is also supported by the more marked effects of CTZ and γ-DGG and reducing presynaptic glutamate release probability on slow-rising CF-EPSCs compared with conventional fast-rising CF-EPSCs.
The cerebellar architecture is normal in GLAST-deficient mice (Watase et al., 1998). In this study, slow-rising CF-EPSCs were induced pharmacologically even in WT mice. Therefore, glutamate yielding slow-rising CF-EPSCs would not come from atypical CF terminals distinctive to GLAST-deficient mice but rather from ordinary CF terminals. In the adult cerebellum, each PC is innervated by a single CF. This one-to-one relationship is preceded by a developmental stage with redundant innervation of PCs by multiple CFs (Crépel, 1982; Hashimoto and Kano, 2005). One possibility is that glutamate released from free CF terminals degenerating during a period of CF pruning would become detectable because of the inhibition of glial glutamate uptake. This CF pruning in mice occurs during the second and third postnatal weeks and is completed by approximately P21. However, slow-rising currents in this study were detectable in more mature stages of both WT and GLAST-deficient mice (P28–P76). Therefore, this possibility seems unlikely. Another possibility is that glutamate spilled over from normal terminals or ectopic release sites of CFs (Matsui and Jahr, 2003, 2004) innervating neighboring PCs could activate AMPARs on recorded PCs (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). A remarkable feature of the dendritic tree of PCs is that the extremely profuse branches are distributed widely (to ∼250 μm) in the transverse plane of the folium but are very restricted (to ∼10 μm) in the longitudinal plane (Eccles et al., 1967). In the molecular layer, such flat dendritic sheets are compactly arranged in parallel with the transverse plane, providing an opportunity for more intimate interdigitation of dendritic sheets of neighboring PCs (Eccles et al., 1967). In addition, the glutamate transient at CF-PC synapses reaches a very high concentration (Wadiche and Jahr, 2001; Harrison and Jahr, 2003). Accordingly, trans-cellular spillover of glutamate onto adjacent PCs in the longitudinal plane may occur easily when glutamate uptake is hindered by inhibiting the functions of glial glutamate transporters. This notion is strongly supported by the result obtained by simultaneous recordings of CF-EPSCs from a pair of adjacent PCs in which CF stimulation with the same threshold elicited conventional fast-rising CF-EPSCs in one cell and simultaneously slow-rising EPSCs in the other cell (Fig. 8A).
In this experiment, once the fast-rising CF-EPSC was evoked, no slow-rising current was detected as an additional component at higher stimulus intensities. This observation suggests that slow- and fast-rising CF-EPSCs are mediated by common receptors, and thus glutamate that spilled over to generate slow-rising currents may predominantly activate AMPARs on CF synapses of the recorded PC, the activation of which would be nearly saturated during the occurrence of fast-rising CF-EPSCs (Wadiche and Jahr, 2001; Harrison and Jahr, 2003).
Differences between pharmacological inhibition and GLAST mutation
GLT1, another type of glial glutamate transporter, is also expressed in Bergmann glia, although the number of molecules of GLT1 is <15% of that of GLAST molecules (Lehre and Danbolt, 1998). PMB-TBOA at 200 nm inhibited both GLAST and GLT1 almost completely (Shimamoto et al., 2004), whereas GLT1 on Bergmann glia remains functional in GLAST-deficient mice (Takatsuru et al., 2006). Therefore, the removal of released glutamate would be retarded more severely by the application of 200 nm PMB-TBOA than in the mutant mice. In fact, slow-rising CF-EPSCs in the presence of PMB-TBOA had a slightly larger amplitude and slower rise time than mutant atypical ones, suggesting that the released glutamate in the presence of PMB-TBOA could spill over more abundantly and activate more numerous AMPARs, including those located at more distant sites (Fig. 1B). This also accounts for the more marked effect of CTZ on the amplitude of atypical CF-EPSCs in GLAST-deficient mice than that on slow-rising CF-EPSCs in the presence of PMB-TBOA in WT mice (Fig. 5A,B).
Other possible mechanisms that could produce the slow-rising EPSCs
mGluR activation generates slow synaptic currents not only at PF synapses but also at CF synapses (Dzubay and Otis, 2002), which could affect slow-rising CF-EPSCs. Indeed, we found that mGluR-EPSCs were evoked by CF stimulation in some cells in the presence of 200 nm PMB-TBOA. However, their amplitude and rise time was extremely small (∼10 pA at −80 mV) and did not affect the estimation of slow-rising EPSCs (supplemental Fig. S1E,F, available at www.jneurosci.org as supplemental material). It is also possible that PMB-TBOA affects the activating and/or desensitization kinetics of AMPARs and slows down the rise time of CF-EPSCs. Alternatively, slow-rising currents could arise from asynchronous release of glutamate caused by PMB-TBOA. However, these possibilities are unlikely because PMB-TBOA had no effect on the rise time of fast CF-EPSCs.
Functional role of glial glutamate transporters
The occurrence of multiple discrete CF-EPSCs during the application of PMB-TBOA in WT mice (2.7 steps on the average) and in GLAST-deficient mice (2.3 steps on the average) indicates that each PC is activated by one or two neighboring CFs other than the CF innervating the PC concerned under the conditions in which the uptake of glutamate by glial transporters is hindered. The one-to-one relationship at CF-PC synapses has been considered to be important for the cerebellar control of motor function, because motor coordination is impaired in many kinds of mutated mice in which multiple CF innervation persists abnormally in adulthood (Aiba et al., 1994; Chen et al., 1995; Kashiwabuchi et al., 1995; Offermanns et al., 1997). All of the targeted molecules examined in these mutants except for GLAST were critical for the elimination of CF synapse during cerebellar development, although we cannot exclude the possibility that the developmental elimination of CF innervation is delayed by the deletion of GLAST gene during the fourth postnatal week because multiple fast-rising CF-EPSCs were observed in 15% of the GLAST-deficient mice at P24–P50 in a previous report (Watase et al., 1998). However, the apparent multiple CF innervation in GLAST-mutant mice can be reproduced in the wild-type mature cerebellum solely by inhibiting glial glutamate uptake. This suggests that GLAST plays a more important role in the maintenance of the functional one-to-one relationship than in the elimination of CF synapses at CF-PC synapses. GLAST-deficient mice have been shown to be able to manage simple coordinated motor tasks but fail to perform more challenging tasks such as staying on a quickly rotating rod (Watase et al., 1998). This impairment of motor coordination would be attributed, at least partly, to the trans-cellular spillover of glutamate among neighboring PCs during CF activation.
Footnotes
This work was supported by Grants-in-Aid 14208096 for Scientific Research (A) and 17700364 for Young Scientist (B) from the Japan Society for the Promotion of Sciences and Grant-in-Aid 15082202 for Scientific Research on Priority Areas on “Elucidation of glia-neuron network mediated information processing systems” from the Ministry of Education, Culture, Sports, Science, and Technology, Japan. We thank Drs. Yasuhiko Saito and Wataru Kakegawa for helpful suggestions and discussions of this manuscript.
References
- Aiba A, Kano M, Chen C, Stanton ME, Fox GD, Herrup K, Zwingman TA, Tonegawa S (1994). Deficient cerebellar long-term depression and impaired motor learning in mGluR1 mutant mice. Cell 79:377–388. [PubMed] [Google Scholar]
- Arriza JL, Eliasof S, Kavanaugh MP, Amara SG (1997). Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proc Natl Acad Sci USA 94:4155–4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchelor AM, Garthwaite J (1997). Frequency detection and temporally dispersed synaptic signal association through a metabotropic glutamate receptor pathway. Nature 385:74–77. [DOI] [PubMed] [Google Scholar]
- Carter AG, Regehr WG (2000). Prolonged synaptic currents and glutamate spillover at the parallel fiber to stellate cell synapse. J Neurosci 20:4423–4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry FA, Lehre KP, van Lookeren Campagne M, Ottersen OP, Danbolt NC, Storm-Mathisen J (1995). Glutamate transporters in glial plasma membranes: highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron 15:711–720. [DOI] [PubMed] [Google Scholar]
- Chen C, Kano M, Abeliovich A, Chen L, Bao S, Kim JJ, Hashimoto K, Thompson RF, Tonegawa S (1995). Impaired motor coordination correlates with persistent multiple climbing fiber innervation in PKC gamma mutant mice. Cell 83:1233–1242. [DOI] [PubMed] [Google Scholar]
- Clark BA, Cull-Candy SG (2002). Activity-dependent recruitment of extrasynaptic NMDA receptor activation at an AMPA receptor-only synapse. J Neurosci 22:4428–4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JD, Lester RA, Tong G, Jahr CE, Westbrook GL (1992). The time course of glutamate in the synaptic cleft. Science 258:1498–1501. [DOI] [PubMed] [Google Scholar]
- Crépel F (1982). Regression of functional synapses in the immature mammalian cerebellum. Trends Neurosci 5:266–269. [Google Scholar]
- Danbolt NC (2001). Glutamate uptake. Prog Neurobiol 65:1–105. [DOI] [PubMed] [Google Scholar]
- Dehnes Y, Chaudhry FA, Ullensvang K, Lehre KP, Storm-Mathisen J, Danbolt NC (1998). The glutamate transporter EAAT4 in rat cerebellar Purkinje cells: a glutamate-gated chloride channel concentrated near the synapse in parts of the dendritic membrane facing astroglia. J Neurosci 18:3606–3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS (2001). Neuronal glutamate transporters limit activation of NMDA receptors by neurotransmitter spillover on CA1 pyramidal cells. J Neurosci 21:8328–8338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGregorio DA, Nusser Z, Silver RA (2002). Spillover of glutamate onto synaptic AMPA receptors enhances fast transmission at a cerebellar synapse. Neuron 35:521–533. [DOI] [PubMed] [Google Scholar]
- Dzubay JA, Jahr CE (1999). The concentration of synaptically released glutamate outside of the climbing fiber-Purkinje cell synaptic cleft. J Neurosci 19:5265–5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzubay JA, Otis TS (2002). Climbing fiber activation of metabotropic glutamate receptors on cerebellar Purkinje neurons. Neuron 36:1159–1167. [DOI] [PubMed] [Google Scholar]
- Eccles JC, Ito M, Szentagothai J (1967). In: The cerebellum as a neuronal machine New York: Springer.
- Eliasof S, Arriza JL, Leighton BH, Kavanaugh MP, Amara SG (1998). Excitatory amino acid transporters of the salamander retina: identification, localization, and function. J Neurosci 18:698–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairman WA, Vandenberg RJ, Arriza JL, Kavanaugh MP, Amara SG (1995). An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature 375:599–603. [DOI] [PubMed] [Google Scholar]
- Furuta A, Martin LJ, Lin CL, Dykes-Hoberg M, Rothstein JD (1997). Cellular and synaptic localization of the neuronal glutamate transporters excitatory amino acid transporter 3 and 4. Neuroscience 81:1031–1042. [DOI] [PubMed] [Google Scholar]
- Harrison J, Jahr CE (2003). Receptor occupancy limits synaptic depression at climbing fiber synapses. J Neurosci 23:377–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, Kano M (2005). Functional differentiation of multiple climbing fiber inputs during synapse elimination in the developing cerebellum. Neuron 38:785–796. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Ichikawa R, Takechi H, Inoue Y, Aiba A, Sakimura K, Mishina M, Hashikawa T, Konnerth A, Watanabe M, Kano M (2001). Roles of glutamate receptor δ2 subunit (GluRδ2) and metabotropic glutamate receptor subtype 1 (mGluR1) in climbing fiber synapse elimination during postnatal cerebellar development. J Neurosci 21:9701–9712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YH, Dykes-Hoberg M, Tanaka K, Rothstein JD, Bergles DE (2004). Climbing fiber activation of EAAT4 transporters and kainate receptors in cerebellar Purkinje cells. J Neurosci 24:103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa R, Miyazaki T, Kano M, Hashikawa T, Tatsumi H, Sakimura K, Mishina M, Inoue Y, Watanabe M (2002). Distal extension of climbing fiber territory and multiple innervation caused by aberrant wiring to adjacent spiny branchlets in cerebellar Purkinje cells lacking glutamate receptor δ2. J Neurosci 22:8487–8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iino M, Goto K, Kakegawa W, Okado H, Sudo M, Ishiuchi S, Miwa A, Takayasu Y, Saito I, Tsuzuki K, Ozawa S (2001). Glia-synapse interaction through Ca2+-permeable AMPA receptors in Bergmann glia. Science 292:926–929. [DOI] [PubMed] [Google Scholar]
- Johnston D, Brown TH (1983). Interpretation of voltage-clamp measurements in hippocampal neurons. J Neurophysiol 50:464–486. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Hediger MA (1992). Primary structure and functional characterization of a high-affinity glutamate transporter. Nature 360:467–471. [DOI] [PubMed] [Google Scholar]
- Kano M, Hashimoto K, Chen C, Abeliovich A, Aiba A, Kurihara H, Watanabe M, Inoue Y, Tonegawa S (1995). Impaired synapse elimination during cerebellar development in PKCγ mutant mice. Cell 83:1223–1231. [DOI] [PubMed] [Google Scholar]
- Kano M, Hashimoto K, Kurihara H, Watanabe M, Inoue Y, Aiba A, Tonegawa S (1997). Persistent multiple climbing fiber innervation of cerebellar Purkinje cells in mice lacking mGluR1. Neuron 18:71–79. [DOI] [PubMed] [Google Scholar]
- Kashiwabuchi N, Ikeda K, Araki K, Hirano T, Shibuki K, Takayama C, Inoue Y, Kutsuwada T, Yagi T, Kang Y, Aizawa S, Mishina M (1995). Impairment of motor coordination, Purkinje cell synapse formation, and cerebellar long-term depression in GluRδ2 mutant mice. Cell 81:245–252. [DOI] [PubMed] [Google Scholar]
- Konnerth A, Llano I, Armstrong CM (1990). Synaptic currents in cerebellar Purkinje cells. Proc Natl Acad Sci USA 87:2662–2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehre KP, Danbolt NC (1998). The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain. J Neurosci 18:8751–8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehre KP, Levy LM, Ottersen OP, Storm-Mathisen J, Danbolt NC (1995). Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J Neurosci 15:1835–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Wilding TJ, Kim SJ, Calejesan AA, Huettner JE, Zhuo M (1999). Kainate-receptor-mediated sensory synaptic transmission in mammalian spinal cord. Nature 397:161–164. [DOI] [PubMed] [Google Scholar]
- Liu G, Choi S, Tsien RW (1999). Variability of neurotransmitter concentration and nonsaturation of postsynaptic AMPA receptors at synapses in hippocampal cultures and slices. Neuron 22:395–409. [DOI] [PubMed] [Google Scholar]
- Llano I, Marty A, Armstrong CM, Konnerth A (1991). Synaptic- and agonist-induced excitatory currents of Purkinje cells in rat cerebellar slices. J Physiol (Lond) 434:183–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masu M, Tanabe Y, Tsuchida K, Shigemoto R, Nakanishi S (1991). Sequence and expression of a metabotropic glutamate receptor. Nature 349:760–765. [DOI] [PubMed] [Google Scholar]
- Matsui K, Jahr CE (2003). Ectopic release of synaptic vesicles. Neuron 40:1173–1183. [DOI] [PubMed] [Google Scholar]
- Matsui K, Jahr CE (2004). Differential control of synaptic and ectopic vesicular release of glutamate. J Neurosci 24:8932–8939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offermanns S, Hashimoto K, Watanabe M, Sun W, Kurihara H, Thompson RF, Inoue Y, Kano M, Simon MI (1997). Impaired motor coordination and persistent multiple climbing fiber innervation of cerebellar Purkinje cells in mice lacking Gαq. Proc Natl Acad Sci USA 94:14089–14094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis TS, Kavanaugh MP, Jahr CE (1997). Postsynaptic glutamate transport at the climbing fiber-Purkinje cell synapse. Science 277:1515–1518. [DOI] [PubMed] [Google Scholar]
- Partin KM, Patneau DK, Mayer ML (1994). Cyclothiazide differentially modulates desensitization of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor splice variants. Mol Pharmacol 46:129–138. [PubMed] [Google Scholar]
- Patneau DK, Vyklicky L Jr, Mayer ML (1993). Hippocampal neurons exhibit cyclothiazide-sensitive rapidly desensitizing responses to kainate. J Neurosci 13:3496–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier JC, Hesson DP, Jones KA, Costa AM (1996). Substituted 1,2-dihydrophthalazines: potent, selective, and noncompetitive inhibitors of the AMPA receptor. J Med Chem 39:343–346. [DOI] [PubMed] [Google Scholar]
- Pines G, Danbolt NC, Bjoras M, Zhang Y, Bendahan A, Eide L, Koepsell H, Storm-Mathisen J, Seeberg E, Kanner BI (1992). Cloning and expression of a rat brain l-glutamate transporter. Nature 360:464–467. [DOI] [PubMed] [Google Scholar]
- Rossi DJ, Hamann M (1998). Spillover-mediated transmission at inhibitory synapses promoted by high affinity α6 subunit GABAA receptors and glomerular geometry. Neuron 20:783–795. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW (1994). Localization of neuronal and glial glutamate transporters. Neuron 13:713–725. [DOI] [PubMed] [Google Scholar]
- Shimamoto K, Lebrun B, Yasuda-Kamatani Y, Sakaitani M, Shigeri Y, Yumoto N, Nakajima T (1998). dl-threo-β-benzyloxyaspartate, a potent blocker of excitatory amino acid transporters. Mol Pharmacol 53:195–201. [DOI] [PubMed] [Google Scholar]
- Shimamoto K, Sakai R, Takaoka K, Yumoto N, Nakajima T, Amara SG, Shigeri Y (2004). Charactarization of novel l-threo-β-benzyloxyaspartate derivatives, potent blockers of the glutamate transporters. Mol Pharmacol 65:1008–1015. [DOI] [PubMed] [Google Scholar]
- Spruston N, Jaffe DB, Williams SH, Johnston D (1993). Voltage- and space-clamp errors associated with the measurement of electrotonically remote synaptic events. J Neurophysiol 70:781–802. [DOI] [PubMed] [Google Scholar]
- Storck T, Schulte S, Hofmann K, Stoffel W (1992). Structure, expression, and functional analysis of a Na+-dependent glutamate/aspartate transporter from rat brain. Proc Natl Acad Sci USA 89:10955–10959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takatsuru Y, Takayasu Y, Iino M, Nikkuni O, Ueda Y, Tanaka K, Ozawa S (2006). Roles of glial glutamate transporters in shaping EPSCs at the climbing fiber-Purkinje cell synapses. Neurosci Res 54:140–148. [DOI] [PubMed] [Google Scholar]
- Takayasu Y, Iino M, Ozawa S (2004). Roles of glutamate transporters in shaping excitatory synaptic currents in cerebellar Purkinje cells. Eur J Neurosci 19:1285–1295. [DOI] [PubMed] [Google Scholar]
- Takayasu Y, Iino M, Kakegawa W, Maeno H, Watase K, Wada K, Yanagihara D, Miyazaki T, Komine O, Watanabe M, Tanaka K, Ozawa S (2005). Differential roles of glial and neuronal glutamate transporters in Purkinje cell synapses. J Neurosci 25:8788–8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka J, Ichikawa R, Watanabe M, Tanaka K, Inoue Y (1997). Extra-junctional localization of glutamate transporter EAAT4 at excitatory Purkinje cell synapses. NeuroReport 8:2461–2464. [DOI] [PubMed] [Google Scholar]
- Tanaka K (1994). Pharmacological characterization of a cloned rat glutamate transporter (GluT-1). Brain Res Mol Brain Res 21:167–170. [DOI] [PubMed] [Google Scholar]
- Thompson CL, Drewery DL, Atkins HD, Stephenson FA, aChazot PL (2000). Immunohistochemical localization of N-methyl-d-aspartate receptor NR1, NR2A, NR2B and NR2C/D subunits in the adult mammalian cerebellum. Neurosci Lett 283:85–88. [DOI] [PubMed] [Google Scholar]
- Yamada KA, Tang CM (1993). Benzothiadiazides inhibit rapid glutamate receptor desensitization and enhance glutamatergic synaptic currents. J Neurosci 13:3904–3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadiche JI, Jahr CE (2001). Multivesicular release at climbing fiber-Purkinje cell synapses. Neuron 32:301–313. [DOI] [PubMed] [Google Scholar]
- Watase K, Hashimoto K, Kano M, Yamada K, Watanabe M, Inoue Y, Okuyama S, Sakagawa T, Ogawa S, Kawashima N, Hori S, Takimoto M, Wada K, Tanaka K (1998). Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur J Neurosci 10:976–988. [DOI] [PubMed] [Google Scholar]
- Watkins JC (1991). Some chemical highlights in the development of excitatory amino acid pharmacology. Can J Physiol Pharmacol 69:1064–1075. [DOI] [PubMed] [Google Scholar]
- Wisden W, Seeburg PH (1993). A complex mosaic of high-affinity kainate receptors in rat brain. J Neurosci 13:3582–3598. [DOI] [PMC free article] [PubMed] [Google Scholar]