

Abstract
Expression of synaptic plasticity involves the translation of mRNA into protein and, probably, active protein degradation via the proteasome pathway. Here, we report on the rapid activation of synthesis and degradation of a probe protein with the induction of long-term potentiation (LTP) in the hippocampal Schaffer collateral CA1 pathway. The proteasome inhibitor MG132 significantly reduced the field EPSP slope potentiation and LTP maintenance without acutely affecting basal synaptic transmission. To visualize protein dynamics, CA1 pyramidal cells of hippocampal slices were transfected with Semliki Forest virus particles expressing a recombinant RNA. This RNA contained the coding sequence for a degradable green fluorescence protein with a nuclear localization signal (NLS-d1EGFP) followed by a 3′- untranslated region dendritic targeting sequence. NLS-d1EGFP fluorescence remained stable in the low-frequency test stimulation but increased with LTP induction in the cell body and in most dendritic compartments of CA1 neurons. Applying anisomycin, a protein synthesis inhibitor, caused NLS-d1EGFP levels to decline; a proteasome inhibitor MG132 reversed this effect. In the presence of anisomycin, LTP induction accelerated the degradation of NLS-d1EGFP. When both inhibitors were present, NLS-d1EGFP levels remained unaffected by LTP induction. Moreover, LTP-induced acceleration of NLS-d1EGFP synthesis was blocked by rapamycin, which is consistent with the involvement of dendritic mammalian target of rapamycin in LTP-triggered translational activity.
Our results clearly demonstrate that LTP induction not only leads to a rapid increase in the rate of protein synthesis but also accelerates protein degradation via the proteasome system.
Keywords: LTP, hippocampus, protein degradation, protein synthesis, proteasome, imaging
Introduction
Synaptic plasticity represents one of the cellular mechanisms suitable for lasting adaptation of network activities during memory formation (Bliss and Collingridge, 1993). Remodeling of synaptic compartments involves a variety of processes, including protein phosphorylation (Malinow et al., 1989; Roberson et al., 1999), protein translocation (Krucker et al., 2000), and enhanced translational activity (Stanton and Sarvey, 1984; Deadwyler et al., 1987; Frey et al., 1988, 1996; Mochida et al., 2001; Behnisch et al., 2004). Translational activity occurs mainly in the soma, but there is compelling evidence that activity-induced protein synthesis also takes place in the dendritic processes of hippocampal neurons (Tiedge and Brosius, 1996; Aakalu et al., 2001; Job and Eberwine, 2001; Steward and Schuman, 2001; Jiang and Schuman, 2002; Bradshaw et al., 2003). Sequences mediating the targeting of mRNAs to dendrites are often found in the 3′-untranslated region (UTR) (Tiedge et al., 1999) as, for instance, in transcripts encoding microtubule-associated protein 2 (MAP2) (Garner et al., 1988; Kindler et al., 1996; Huang et al., 2003; Cristofanilli et al., 2004). Dendritic translation of a subset of mRNAs that play an important role in the induction of protein synthesis-dependent forms of long-term potentiation (LTP) is regulated by the mammalian target of rapamycin (mTOR), a serine-threonine kinase that participates in the activation of ribosomal p70S6K (Raught et al., 2001; Tang et al., 2002; Cammalleri et al., 2003).
Complementing enhanced protein synthesis, activity-activated protein degradation has emerged as a mechanism to control synaptic protein composition (Colledge et al., 2003; Bingol and Schuman, 2004; Hegde, 2004). Strong evidence for an active involvement of the proteasome system in synaptic plasticity has been provided by studies on sensory neuron synapses of Aplysia (Hegde et al., 1997; Chain et al., 1999) and on neuromuscular junctions of Drosophila (DiAntonio et al., 2001; Speese et al., 2003). In these systems, the proteasome system was found to impinge on protein kinase A- and mitogen-activated protein kinase (MAPK)-dependent signaling pathways (Hegde et al., 1993; Chain et al., 1999). In hippocampal neurons, proteins of the proteasome complex are localized in the soma and dendrites near synapses (Ehlers, 2003; Patrick et al., 2003). Recent evidence in cultured hippocampal cells indicated that AMPA-induced internalization of glutamate receptor 1 (GluR1) and GluR2 requires protein degradation via the proteasome system (Colledge et al., 2003; Patrick et al., 2003). Inhibition of active protein degradation leads to deficits in behavioral and LTP performance (Jiang et al., 1998; Foley et al., 2000; Lopez-Salon et al., 2001).
The goal of this study was to investigate whether, and how, induction of LTP in the hippocampal CA1 region affects the balance between protein synthesis and degradation. We found that protein degradation is required for expression of late-phase LTP. Using a viral reporter construct to directly monitor the spatiotemporal dynamics of proteolytic degradation and protein synthesis, we demonstrated that LTP induction could trigger alterations in the concentration of a reporter protein by increasing translational and proteasome-mediated degradation.
Materials and Methods
Generation of the reporter construct.
The pd1EGFP vector (Clontech, Mountain View, CA) carries the coding sequence for destabilized enhanced green fluorescence protein (EGFP), d1EGFP, which is short-lived because of a PEST sequence derived from ornithine decarboxylase and directs the protein to the proteolytic proteasome pathway (Li et al., 1998; Olmo et al., 1999). To reduce the diffusion of somatically synthesized d1EGFP into dendrites, a basic nuclear localization signal (MGPKKKRKV) (Krebber and Silver, 2000) was fused to the N-terminal end of d1EGFP to yield a degradable EGFP with a nuclear localization signal (pNLS-d1EGFP). For this, the following oligonucleotides were annealed and inserted between the EcoRI and BamHI sites of pd1EGFP.
Sense: 5′-AATTCATGGGGCCCAAGAAGAAACGCAAAGTG-3′. Antisense: 5′-GATCCACTTTGCGTTTCTTCTTGGGCCCCATG-3′. A dendritic targeting element derived from the 3′-UTR of a MAP2 cDNA (Kindler et al., 1996; Blichenberg et al., 1999) (kind gift from Dr. Stefan Kindler, Institut für Humangenetik, Hamburg, Germany) was inserted as a NotI-HpaI fragment into the corresponding sites downstream of the NLS-d1EGFP coding sequence. The resulting cDNA, nls-d1EGFP-MAP2 3′-UTR, was subcloned as a BglII-HpaI fragment between the compatible BamHI and SmaI sites of the Semliki Forest vector pSFV1 (Invitrogen, Carlsbad, CA).
Preparation of Semliki Forest particles and transfection of CA1 neurons.
Semliki Forest particles were prepared as described by Ehrengruber (2002) and Ehrengruber et al. (1999, 2001) using the pSFV-Helper2 for structural proteins. After in vitro transcription, both the RNAs of pSFV-nls-d1EGFP-MAP2 3′-UTR and pSFV-Helper 2 were cotransfected into Chinese hamster ovary-K1 (CHO-K1) cells with 1,2-dimyristyloxypropyl-3-dimethyl-hydroxy ethyl ammonium bromide and cholesterol (DMRIE-C; Invitrogen) according to the supplier manual. After 48 h, the culture medium containing the budded particles was harvested. After ultracentrifugation through 10% sucrose, the pellet was resolved in tris-buffered solution overnight at 4°C. The titer [T = (P × D)/V] of the initial viral suspension was calculated by counting the number of fluorescent CHO cells (P), the volume of the platted viral suspension (V), and the reciprocal of virus dilution (D). The obtained titer ranged between 107 and 108 PFU/ml. Aliquots of the particles were stored at −80°C after shock freezing. Three hours before injection into the hippocampal slices, the particles were activated by chymotrypsin and further diluted with TTX containing modified Gey's balanced salt solution (mGBSS) (see below) to get a titer of ∼105 PFU/ml.
Hippocampal slice preparations.
Hippocampal slices were prepared from 20- to 25-d-old male Wistar rats (SHOE; Institute Breeding Stock, Magdeburg, Germany) as described previously (Leutgeb et al., 2003; Behnisch et al., 2004). The animals were maintained in accordance with institutional, state, and federal government regulations (Land Sachsen-Anhalt, Germany). Efforts were made to minimize the number of animals used as well as any suffering. Acutely prepared hippocampal slices were placed on an anopore membrane (NUNC, Wiesbaden, Germany) in carbogenated artificial CSF (ACSF) for 2 h. After the incubation, the hippocampal slices were transferred directly to the recording chamber. For viral transfections, the slices were placed on anopore membranes, supplied with 1 ml slice culture medium solution (Leutgeb et al., 2003), and incubated in humidified carbogen atmosphere (95% O2/5% CO2) at 34°C for 2 h. After recovery, a hippocampal slice on the anapore membrane was transferred to a chamber filled with 1 ml of 4°C cold mGBSS (pH 7.3 at ambient air) containing 600 nm TTX, 130 mm NaCl, 4.9 mm KCl, 1.5 mm CaCl2, 0.3 mm MgSO4, 11 mm MgCl2x6H2O, 0.2 mm KH2PO4, 0.8 mm Na2HPO4, 5 mm glucose, 22 mm HEPES, and 2.2 mm NaHCO3. The infectious particles were diluted in TTX containing mGBSS and two to three times of 0.5–1 μl were injected in the stratum pyramidale of CA1 region using glass pipettes with a tip diameter of ∼10 μm connected through a paraffin oil-filled plastic tube with a 50 μl Hamilton syringe. After a local replicon solution application, membranes with slices were placed into 35 mm dishes containing 1 ml of culture medium and left in humidified carbogen atmosphere at 34°C for 12–18 h. Usually, several CA1 neurons per slice were transfected and expressed d1EGFP (see Fig. 1A). Transfected slices exhibited normal synaptic transmission, stable baseline responses, and expressed LTP (see Fig. 1D).
Figure 1.

Inhibition of proteasome activity decreases the magnitude of short-term and late-phase LTP in hippocampal slices. A, Shown are ensemble averages for experiments with HFS in control ACSF (filled circles; n = 11) and in the presence of 10 μm MG132 (n = 6). HFS consisted of three 100 Hz trains delivered at 10 min intervals after time point zero. The inset shows representative fEPSPs for the time points indicated. B, Baseline recordings in control ACSF (filled circles; n = 10) and in the presence of 10 μm MG132 (n = 5). The arrows indicate the times of HFS, and the horizontal bar represents the time of drug application.
Electrophysiology.
After 12–18 h of incubation, we transferred slices to a submerged type recording chamber mounted on an upright microscope equipped with a Leica laser-scanning microscope (DM LFSA SP1; Leica, Bensheim, Germany). A piece of the membrane with slice was punched out using a small taper pin punch and a rubber cushion. During this procedure, the slices were covered with mGBSS containing TTX (1 μm) to reduce mechanically induced spiking activity. The submerged hippocampal slices were perfused continuously with carbogenated ACSF (in mm: 110 NaCl, 5 KCl, 2.50 CaCl2, 1.50 MgSO4, 1.24 KH2PO4, 20 glucose, 27.4 NaHCO3, pH 7.3; 32°C) with a flow rate of 5 ml/min.
The field EPSPs (fEPSPs) were recorded from the stratum radiatum of the CA1 area by use of glass micropipettes filled with ACSF. Schaffer collateral fibers were stimulated by biphasic rectangular current pulses (300 μs/phase; A385; WPI, Berlin, Germany) in a range of 15–30 μA using a glass pipette localized in the stratum radiatum. The electrodes were placed in close proximity to a fluorescent neuron at a distance of ∼100 μm from the stratum pyramidale (see Fig. 1A). The LTP induction paradigm consisted of 3 × 100 Hz bursts for 1 s with a 10 min interburst interval. We evoked and recorded fEPSPs every minute throughout the experiments. Compared with field potentials recorded from acute slices, the amplitude of the signals from 12–18 h incubated slices were approximately three to four times smaller (see Fig. 1D). For the recording of fEPSPs, we used the amplifier AxoPatch 200B and analog/digital converter Axon Digidata 1322A and the acquisition software Clampex 8.0 (Axon, Chicago, IL). Data analyses were performed using Clampfit 8.0 and Statistica 5.5 to allow the measurement of initial fEPSP slope and the calculation of descriptive (average; SEM) and nonparametric statistics (Mann–Whitney U test). Initial fEPSP slopes were calculated from the average of five subsequent responses.
The protein synthesis inhibitors anisomycin and rapamycin and the proteasome inhibitor MG132 (product number M-1157; A.G. Scientific, San Diego, CA) were bath applied. Anisomycin (Sigma, Munich, Germany) was dissolved freshly before use in ACSF. Rapamycin (product number R-5000; LC Laboratories, Woburn, MA) and MG132 were prepared as a stock solution in DMSO and then diluted in ACSF to the desired concentration. The final concentration of DMSO did not exceed 0.01%. Drug experiments were interleaved with drug-free controls.
Imaging.
The fluorescence of d1EGFP expressing CA1 neurons was measured from summated (averaged) confocal image stacks (eight to nine planes) acquired at 10 min intervals. The distance between the first and the last plane was between 50 and 70 μm. Fluorescence intensity values [averages over regions of interest (ROIs) as indicated] were calculated for every time point, expressed as percentage change from baseline, and normalized against background intensity values.
Results
Late-phase LTP is blocked by proteasome inhibitor MG132
It was shown recently that AMPA-induced internalization of glutamate receptor 1 (GluR1) and GluR2 in hippocampal cell cultures is inhibited by the cell-permeable peptide aldehyde Cbz-leu-leu-leucinal (MG132) (Colledge et al., 2003; Bingol and Schuman, 2004). MG132 acts as a proteasome substrate and transition-state inhibitor and thereby inhibits the chymotrypsin-like activity of proteasome in a micromolar concentration range (Lee and Goldberg, 1998). To investigate the role of proteasome activity for LTP in hippocampal slices, we bath applied MG132 (10 μm) during baseline fEPSP recordings 30 min before induction of LTP and washed out the drug 20 min after the last tetanization train (Fig. 1). Compared with control recordings, the proteasome inhibitor reduced the magnitude of fEPSP slope potentiation within the first minutes after tetanization (fifth minute, 170 ± 5.83 and 145.4 ± 4.14%, p < 0.05, control and drug, respectively). Moreover, in the presence of MG132, the potentiated field potentials were not maintained and reached baseline fEPSP slope values after 200 min (n = 6). The normalized mean fEPSP slopes measured 120 and 240 min after LTP induction were 125.2 ± 3.61 and 103.2 ± 5.12%, respectively (Fig. 1A, open circles). Corresponding slope values of control experiments (n = 11) were 152.8 ± 5.26 and 140.5 ± 5.57% (Fig. 1A, filled circles) and were significantly different at all time points after the fifth minute (p < 0.05). Importantly, application of MG132 without tetanization did not affect baseline fEPSPs within 240 min (i.e., virtually identical mean normalized fEPSP values were obtained at time points −30, 0, 120, and 240 min, respectively, 100.7 ± 1.12, 98.2 ± 1.07, 97.7 ± 1.96, and 95.0 ± 1.35%) (Fig. 1B). These findings suggest that both early and late-phase LTP specifically depend on proteasome activity.
LTP and imaging of protein dynamics in virus transfected hippocampal slices
The concentration of a protein alters over time if either the rate of de novo synthesis or the rate of degradation changes. To monitor the spatiotemporal dynamics of activity-induced protein synthesis and protein degradation at the level of single neurons, we expressed a fluorescent protein reporter system in hippocampal slices. Twelve to 18 h after transfection, CA1 pyramidal cells expressed the fluorescent protein in soma and dendrites (Fig. 2A,B). Stable baseline recordings and the expression of long-lasting LTP in nontransfected (Fig. 2C) and transfected (Fig. 2D) slices demonstrated the viability of this preparation. Furthermore, maintenance of LTP was compromised by the translational blocker anisomycin similar as described previously (Tsokas et al., 2005) (Fig. 2C).
Figure 2.
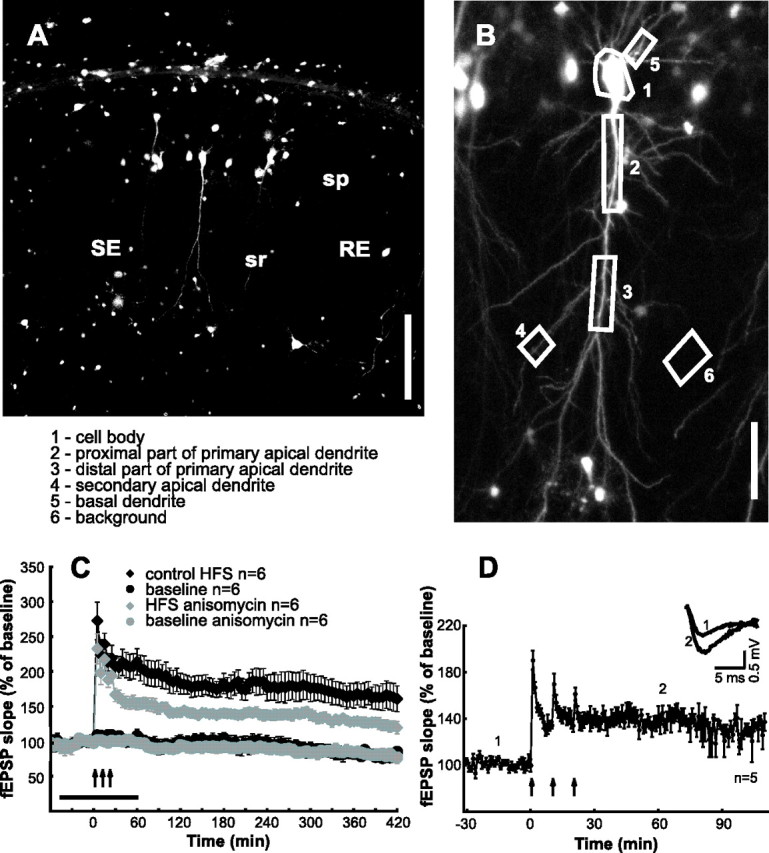
LTP in hippocampal slices transfected with reporter construct. A, Fluorescence image of CA1 region of hippocampal slice 18 h after transfection. The CA1 pyramidal cells in stratum pyramidale (sp) can be identified by basal and apical dendrites. Stimulation (SE) and recording (RE) pipettes are localized in stratum radiatum (sr). Scale bar, 300 μm. B, Z-projection image of a single CA1 neuron with description of ROIs (see A) used for analysis. Scale bar, 85 μm. C, Stable baseline recordings, expression of long-lasting LTP, and the effect of anisomycin on LTP maintenance in slices maintained for >24 h. Ensemble averages for experiments with HFS (n = 6), baseline recordings (n = 6), HFS in the presence of anisomycin (n = 6; horizontal bar), and baseline in the presence of anisomycin (n = 6) are shown. D, Electrophysiological recordings in transfected slices. Ensemble averages for experiments with HFS (n = 5) are shown. The inset shows representative fEPSPs for the time points indicated.
Induction of LTP induces protein synthesis
We monitored reporter fluorescence at 10 min intervals during low-frequency test stimulation (0.017 Hz) in ROIs outlining the cell body, portion of the proximal apical and basal dendrites, the distal portion of the proximal dendrite, and secondary apical dendrites (Fig. 2B). Induction of LTP (3 × 100 Hz stimulation for 1 s every 10 min) caused significant increases in the level of fluorescence intensity at all cellular compartments except for some distal secondary dendrites (Fig. 3A1–A3). Mean fluorescence intensities of proximal dendritic compartments remained stable in control experiments without tetanization (Fig. 3B1–B3) (n = 7; 0.2 ± 0.79, 1.7 ± 0.82, and −1.8 ± 1.43% at −40, 0, and 80 min, respectively), thus reflecting balanced rates of d1EGFP synthesis and degradation.
Figure 3.

HFS activates synthesis of reporter protein. A1, B1, Fluorescence images of pyramidal cells transfected with reporter construct. Left baseline fluorescence, middle, and right color-coded changes from baseline fluorescence before (middle) and 60 min after (right) HFS (A1) or with continued baseline recordings (B1). Colors indicate the following: decrease, blue; no change, green; increase, red. Scale from −50 to +50%. A2, B2, Time course of fluorescence levels in ROIs from a cell subjected to HFS (A2) and a control cell without HFS (B2). ROIs are indicated in A1 and B1. A3, B3, Ensemble average of reporter protein levels from ROIs as defined in Figure 2B. Signals from secondary apical dendrites (distant from the stimulation electrode) that exhibited decreasing fluorescence levels after HFS were pooled together and represented by gray symbols in A3. Scale bars, 70 μm.
For each cell (n = 8), fluorescence signals from several ROIs (as defined in Fig. 2B) were measured and averaged over all cells. Decreasing fluorescence signals that were systematically observed in some distal secondary dendrites were pooled separately. The pooled data (Fig. 3A3) showed that fluorescence levels began to increase immediately after the first tetanization train and continued to increase up to the end of the recording period. A different portion of the dendrites of the neurons exhibited different rates of fluorescence increase. The largest fluorescence increase (∼20%; n = 8) was observed in the distal parts of primary dendrites and distal parts of secondary dendrites. In the soma, increases of fluorescence intensity were somehow less steep and slightly delayed when compared with the fluorescence intensity dynamics found in primary dendrites. The decrease in fluorescence intensity that was seen in some dendritic compartments after tetanization trains (Fig. 2A3, gray symbols) may be caused by increased protein degradation. The next set of experiments was designed to test this possibility.
Induction of LTP induces proteasome-dependent protein degradation
To investigate protein degradation in the absence of de novo synthesis, we used anisomycin, a blocker for translational activity. Within a few minutes after application of anisomycin (25 μm), d1EGFP fluorescence levels started to decline in each ROI (Fig. 4A). The rate of decrease in intensity of fluorescence was constant. Maximum intensities occurred at proximal dendrites and in the cell body. Normalized fluorescence values for time points −40, 0, and 80 min were 0.1 ± 0.78, −2.7 ± 1.19, and −10.5 ± 1.12%. Induction of LTP [high-frequency stimulation (HFS); 3 × 100 Hz stimulation for 1 s every 10 min] dramatically accelerated the rate of loss in d1EGFP fluorescence mean normalized fluorescence values determined at −40 min, immediately before HFS, and 80 min after LTP induction were 0.7 ± 0.78, −2.0 ± 1.50, and −19.6 ± 4.13% (Fig. 4B). Application of MG132 (10 μm) 15 min before tetanization reduced, but not abolished, the HFS-induced increase in reporter fluorescence (Fig. 4C, compare with Fig. 3A). The reduced increases in reporter fluorescence in the presence of MG132 are consistent with the reduction of short-term and long-term fEPSP potentiation under the same conditions (Fig. 1). Application of MG132 along with anisomycin had no effect on reporter fluorescence, and probe protein levels remained constant after HFS (Fig. 4D). This indicates that the HFS-induced decrease in probe protein levels (demonstrated in the presence of anisomycin) is caused by an activation of the proteasome pathway. Importantly, time-lapse imaging of protein dynamics directly shows that activation of proteasome-mediated degradation occurs very rapidly after tetanization of the Schaffer collateral CA1 synapses (Fig. 4B).
Figure 4.

HFS activates protein degradation. A–F, The diagrams show ensemble averages of the time course of reporter protein levels under different experimental conditions. Horizontal bars indicate drug application times. A, Anisomycin (25 μm; n = 4). B, HFS in the presence of anisomycin. C, HFS in the presence of MG132 (10 μm). D, HFS in the presence of MG132 and anisomycin.
HFS-induced synthesis and proteolytic degradation of proteins in dendrites
The HFS-induced rise in reporter fluorescence occurred at dendritic sites at least as rapidly as in the cell body (Fig. 3A3). This along with the nuclear localization signal of the reporter protein (that limits diffusion of the protein from the cell body to the dendrite) indicates the induction of rapid dendritic protein synthesis (Tsokas et al., 2005). We therefore tested the protein synthesis inhibitor rapamycin, which interferes with the mTOR pathway involved in dendritic LTP-associated protein synthesis (Raught et al., 2001; Tang et al., 2002; Bradshaw et al., 2003; Cammalleri et al., 2003; Tsokas et al., 2005). In the presence of 200 nm rapamycin and without tetanization, no change of d1EGFP fluorescence intensities was detectable (n = 6) (Fig. 5A). The values were 0.5 ± 0.59, 1.1 ± 0.57, and −0.1 ± 2.44% for the time points −40, 0, and 80 min, respectively. Stable baseline fluorescence levels likely resulted from protein synthesis that was not acutely dependent on rapamycin-sensitive machineries and that occurred either in the dendrites or in the cell soma. Regardless of the origin of these baseline levels, HFS in the presence of rapamycin induced a decrease of fluorescence intensity immediately after the first train (n = 13) (Fig. 5B). The whole dendritic tree responded in a similar manner to LTP induction trains, whereas the signal from the cell body was somewhat delayed. Fluorescence values for the proximal dendrites were 1.1 ± 0.31, −3.4 ± 1.04, and −15.3 ± 2.25% taken −40, 0, and 80 min, respectively.
Figure 5.

Inhibition of translation by rapamycin and prevention of LTP expression by AP-5 prevent HFS-induced protein synthesis but not protein degradation. A, Rapamycin (200 nm; n = 6) applied during baseline recordings. B, HFS in the presence of rapamycin (n = 13). C, HFS in the presence of AP-5 (n = 9).
Induction of LTP at Schaffer collateral CA1 synapses requires the activation of NMDA receptors, and this activation is an important trigger for HFS-induced protein synthesis (Bliss and Collingridge, 1993; Malenka and Nicoll, 1993; Nguyen and Kandel, 1997; Behnisch et al., 2004). We confirmed that this is the case also under our present experimental conditions. As illustrated in Figure 5C, HFS applied in the presence of AP-5 did not trigger protein synthesis (compare with Fig. 2A). In contrast, HFS-triggered protein degradation was still present when NMDA receptors were blocked (Fig. 5C).
Discussion
Summary
We monitored synthesis and degradation of a fluorescent reporter protein after induction of LTP. Live imaging of protein levels along with pharmacological manipulations demonstrated that LTP induction not only leads to a rapid increase in the rate of protein synthesis but also requires accelerated degradation of proteins via the proteasome system.
LTP-associated protein synthesis
It is well established that activity-induced translation is required for long-lasting forms of LTP. Early evidence that protein synthesis was required for long-lasting forms of LTP was based on pharmacological inhibition of translation (Stanton and Sarvey, 1984; Deadwyler et al., 1987; Frey et al., 1988, 1996; Mochida et al., 2001; Behnisch et al., 2004). More recently, very rapid LTP-associated de novo synthesis of marker and reporter proteins was shown directly (Behnisch et al., 2004; Tsokas et al., 2005). It should be noted that the blockers of translational activity used in these studies (anisomycin, emetine, and rapamycin) all exert activities in addition to inhibition of translation. Anisomycin activates stress-activated protein kinase and MAPK. Anisomycin can also induce (at nanomolar concentrations) transcription of different genes, including immediate-early genes (Cano et al., 1994; Zinck et al., 1995; Hazzalin et al., 1998). The anti-translational activity of anisomycin, at the concentration used in this study, most likely disables the translation of these gene transcripts. Emetine and rapamycin do not share these additional actions of anisomycin. Unlike anisomycin, Rapamycin does not inhibit translation by binding to ribosomal subunits but interacts with the mTOR that in turn involves in regulation of translational activity. Therefore, rapamycin limits the activity-dependent increase in translational activity without acutely or completely inhibiting basal translational activity. Longer-lasting application of rapamycin may, however, compromise the translational machinery and thereby basal protein synthesis.
All of the drugs that were used in the previous studies and the two used in this study (anisomycin and rapamycin) inhibited HFS-triggered protein synthesis. HFS-induced protein synthesis required the activation of NMDA receptors (Fig. 5C) (Behnisch et al., 2004). This result indicates a specific association between protein synthesis stimulation and LTP expression, because increased synaptic or postsynaptic activity alone (induced by HFS in the presence of AP5) did not trigger LTP nor did it trigger synthesis of the reporter protein.
The stimulated protein synthesis was likely mediated through the rapamycin-sensitive mTOR pathway (Bradshaw et al., 2003; Kelleher et al., 2004a,b; Tsokas et al., 2005; Karpova et al., 2006). It is not clear whether all protein species that are synthesized after induction of LTP are directly involved in processes relevant for establishment and maintenance of LTP, are involved in components of synaptic remodeling that are not directly reflected in the fEPSP measure, or whether protein synthesis is upregulated nonspecifically (Kelleher et al., 2004a,b). Proteins that may be synthesized rapidly after induction of LTP may include those that are “captured” by “tagged” synapses (Frey and Morris, 1997). That translation of a reporter message is upregulated indicates that proteins not directly related to synaptic plasticity are also upregulated after induction of LTP.
LTP-associated protein degradation
We found that the maintenance of fEPSP potentiation was blocked by proteasome inhibitor MG132. A corresponding effect of MG132 was described for hippocampal long-term depression (Colledge et al., 2003). Our finding that MG132 also reduces early fEPSP potentiation is consistent with the idea of a fast proteasome-mediated degradation of scaffolding proteins soon after tetanization. With translational activity blocked, induction of long-lasting LTP increased MG132-sensitive degradation of the NLS-d1EGFP reporter protein, a result that directly demonstrated tetanization-triggered enhancement of protein degradation via the proteasome system.
Without inhibition of translation, reporter protein fluorescence increased at most, but not at all, subcellular compartments. This indicates that de novo protein synthesis prevails over degradation after LTP induction in most subcellular compartments. Remarkably, however, there were distinct regions (distal parts of lateral dendrites away from sites of synaptic stimulation) where protein degradation was detectable in LTP control experiments. The most likely explanation is that HFS did not induce protein synthesis in these dendritic regions and that induced degradation is more widespread. In addition, a higher density of proteasomes in the cell bodies than in dendrites (Patrick et al., 2003) might explain the observed strong HFS-induced decrease of somatic fluorescence.
Because AP-5 did not prevent HFS-triggered protein degradation (Fig. 5C), signal transduction cascades for activity-dependent protein synthesis and degradation may diverge at an early stage.
The fast activation of proteolytic activities is consistent with the involvement of proteasomes in gene expression (Lee and Goldberg, 1998) or in the modification of existing protein assemblies (Patrick et al., 2003). Indeed, HFS-induced phosphorylation of the transcription factor cAMP response element-binding protein (Leutgeb et al., 2005) depends on a functional proteasome system (Ehlers, 2003). In addition, several lines of evidence suggest that plasticity of spine morphology can appear tens of minutes after induction of LTP (Luscher et al., 2000; Yuste and Bonhoeffer, 2001; Bonhoeffer and Yuste, 2002) and that this process also involves the proteasome system (Ehlers, 2003).
In conclusion, we demonstrated that LTP induction induces protein synthesis and degradation of proteins via the proteasome and that the activation of the latter is necessary for LTP induction and maintenance.
Footnotes
This work was supported by Volkswagen Foundation Grant I/78 555. We thank Dr. Annette Gaertner (University College London), Dr. Thomas Dresbach (University of Heidelberg), and Dr. Michaela Kraus (Medical Research Council London) for initial methodical help with hippocampal transfection and Bonnie Lee La Madeleine (RIKEN Brain Science Institute, Japan) for manuscript editing.
References
- Aakalu G, Smith WB, Nguyen N, Jiang C, Schuman EM (2001). Dynamic visualization of local protein synthesis in hippocampal neurons. Neuron 30:489–502. [DOI] [PubMed] [Google Scholar]
- Behnisch T, Matsushita S, Knöpfel T (2004). Imaging of gene expression during long-term potentiation. NeuroReport 15:2039–2043. [DOI] [PubMed] [Google Scholar]
- Bingol B, Schuman EM (2004). A proteasome-sensitive connection between PSD-95 and GluR1 endocytosis. Neuropharmacology 47:755–763. [DOI] [PubMed] [Google Scholar]
- Blichenberg A, Schwanke B, Rehbein M, Garner CC, Richter D, Kindler S (1999). Identification of a cis-acting dendritic targeting element in MAP2 mRNAs. J Neurosci 19:8818–8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361:31–39. [DOI] [PubMed] [Google Scholar]
- Bonhoeffer T, Yuste R (2002). Spine motility. Phenomenology, mechanisms, and function. Neuron 35:1019–1027. [DOI] [PubMed] [Google Scholar]
- Bradshaw KD, Emptage NJ, Bliss TV (2003). A role for dendritic protein synthesis in hippocampal late LTP. Eur J Neurosci 18:3150–3152. [DOI] [PubMed] [Google Scholar]
- Cammalleri M, Lutjens R, Berton F, King AR, Simpson C, Francesconi W, Sanna PP (2003). Time-restricted role for dendritic activation of the mTOR-p70S6K pathway in the induction of late-phase long-term potentiation in the CA1. Proc Natl Acad Sci USA 100:14368–14373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano E, Hazzalin CA, Mahadevan LC (1994). Anisomycin-activated protein kinases p45 and p55 but not mitogen-activated protein kinases ERK-1 and -2 are implicated in the induction of c-fos and c-jun. Mol Cell Biol 14:7352–7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chain DG, Schwartz JH, Hegde AN (1999). Ubiquitin-mediated proteolysis in learning and memory. Mol Neurobiol 20:125–142. [DOI] [PubMed] [Google Scholar]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF, Scott JD (2003). Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron 40:595–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristofanilli M, Thanas S, Brosius J, Kindler S, Tiedge H (2004). Neuronal MAP2 mRNA: species-dependent differential dendritic targeting competence. J Mol Biol 341:927–934. [DOI] [PubMed] [Google Scholar]
- Deadwyler SA, Dunwiddie T, Lynch G (1987). A critical level of protein synthesis is required for long-term potentiation. Synapse 1:90–95. [DOI] [PubMed] [Google Scholar]
- DiAntonio A, Haghighi AP, Portman SL, Lee JD, Amaranto AM, Goodman CS (2001). Ubiquitination-dependent mechanisms regulate synaptic growth and function. Nature 412:449–452. [DOI] [PubMed] [Google Scholar]
- Ehlers MD (2003). Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci 6:231–242. [DOI] [PubMed] [Google Scholar]
- Ehrengruber MU (2002). Alphaviral vectors for gene transfer into neurons. Mol Neurobiol 26:183–201. [DOI] [PubMed] [Google Scholar]
- Ehrengruber MU, Lundstrom K, Schweitzer C, Heuss C, Schlesinger S, Gahwiler BH (1999). Recombinant Semliki Forest virus and Sindbis virus efficiently infect neurons in hippocampal slice cultures. Proc Natl Acad Sci USA 96:7041–7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrengruber MU, Hennou S, Bueler H, Naim HY, Deglon N, Lundstrom K (2001). Gene transfer into neurons from hippocampal slices: comparison of recombinant Semliki Forest virus, adenovirus, adeno-associated virus, lentivirus, and measles virus. Mol Cell Neurosci 17:855–871. [DOI] [PubMed] [Google Scholar]
- Foley AG, Hartz BP, Gallagher HC, Ronn LC, Berezin V, Bock E, Regan CM (2000). A synthetic peptide ligand of neural cell adhesion molecule (NCAM) IgI domain prevents NCAM internalization and disrupts passive avoidance learning. J Neurochem 74:2607–2613. [DOI] [PubMed] [Google Scholar]
- Frey U, Morris RG (1997). Synaptic tagging and long-term potentiation. Nature 385:533–536. [DOI] [PubMed] [Google Scholar]
- Frey U, Krug M, Reymann KG, Matthies H (1988). Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res 452:57–65. [DOI] [PubMed] [Google Scholar]
- Frey U, Frey S, Schollmeier F, Krug M (1996). Influence of actinomycin D, a RNA synthesis inhibitor, on long-term potentiation in rat hippocampal neurons in vivo and in vitro. J Physiol (Lond) 490:703–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner CC, Tucker RP, Matus A (1988). Selective localization of messenger RNA for cytoskeletal protein MAP2 in dendrites. Nature 336:674–677. [DOI] [PubMed] [Google Scholar]
- Hazzalin CA, Le Panse R, Cano E, Mahadevan LC (1998). Anisomycin selectively desensitizes signalling components involved in stress kinase activation and fos and jun induction. Mol Cell Biol 18:1844–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde AN (2004). Ubiquitin-proteasome-mediated local protein degradation and synaptic plasticity. Prog Neurobiol 73:311–357. [DOI] [PubMed] [Google Scholar]
- Hegde AN, Goldberg AL, Schwartz JH (1993). Regulatory subunits of cAMP-dependent protein kinases are degraded after conjugation to ubiquitin: a molecular mechanism underlying long-term synaptic plasticity. Proc Natl Acad Sci USA 90:7436–7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde AN, Inokuchi K, Pei W, Casadio A, Ghirardi M, Chain DG, Martin KC, Kandel ER, Schwartz JH (1997). Ubiquitin C-terminal hydrolase is an immediate-early gene essential for long-term facilitation in Aplysia. Cell 89:115–126. [DOI] [PubMed] [Google Scholar]
- Huang YS, Carson JH, Barbarese E, Richter JD (2003). Facilitation of dendritic mRNA transport by CPEB. Genes Dev 17:638–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Schuman EM (2002). Regulation and function of local protein synthesis in neuronal dendrites. Trends Biochem Sci 27:506–513. [DOI] [PubMed] [Google Scholar]
- Jiang YH, Armstrong D, Albrecht U, Atkins CM, Noebels JL, Eichele G, Sweatt JD, Beaudet AL (1998). Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron 21:799–811. [DOI] [PubMed] [Google Scholar]
- Job C, Eberwine J (2001). From the cover: identification of sites for exponential translation in living dendrites. Proc Natl Acad Sci USA 98:13037–13042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpova A, Sanna PP, Behnisch T (2006). Involvement of multiple phosphatidylinositol 3-kinase-dependent pathways in the persistence of late-phase long term potentiation expression. Neuroscience 137:833–841. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ III, Govindarajan A, Jung HY, Kang H, Tonegawa S (2004a). Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell 116:467–479. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ III, Govindarajan A, Tonegawa S (2004b). Translational regulatory mechanisms in persistent forms of synaptic plasticity. Neuron 44:59–73. [DOI] [PubMed] [Google Scholar]
- Kindler S, Muller R, Chung WJ, Garner CC (1996). Molecular characterization of dendritically localized transcripts encoding MAP2. Brain Res Mol Brain Res 36:63–69. [DOI] [PubMed] [Google Scholar]
- Krebber H, Silver PA (2000). Directing proteins to nucleus by fusion to nuclear localization signal tags. Methods Enzymol 327:283–296. [DOI] [PubMed] [Google Scholar]
- Krucker T, Siggins GR, Halpain S (2000). Dynamic actin filaments are required for stable long-term potentiation (LTP) in area CA1 of the hippocampus. Proc Natl Acad Sci USA 97:6856–6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, Goldberg AL (1998). Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol 8:397–403. [DOI] [PubMed] [Google Scholar]
- Leutgeb JK, Frey JU, Behnisch T (2003). LTP in cultured hippocampal-entorhinal cortex slices from young adult (P25–30) rats. J Neurosci Methods 130:19–32. [DOI] [PubMed] [Google Scholar]
- Leutgeb JK, Frey JU, Behnisch T (2005). Single cell analysis of activity-dependent cyclic AMP-responsive element-binding protein phosphorylation during long-lasting long-term potentiation in area CA1 of mature rat hippocampal-organotypic cultures. Neuroscience 131:601–610. [DOI] [PubMed] [Google Scholar]
- Li X, Zhao X, Fang Y, Jiang X, Duong T, Fan C, Huang CC, Kain SR (1998). Generation of destabilized green fluorescent protein as a transcription reporter. J Biol Chem 273:34970–34975. [DOI] [PubMed] [Google Scholar]
- Lopez-Salon M, Alonso M, Vianna MR, Viola H, Mello e Souza H, Izquierdo I, Pasquini JM, Medina JH (2001). The ubiquitin-proteasome cascade is required for mammalian long-term memory formation. Eur J Neurosci 14:1820–1826. [DOI] [PubMed] [Google Scholar]
- Luscher C, Nicoll RA, Malenka RC, Muller D (2000). Synaptic plasticity and dynamic modulation of the postsynaptic membrane. Nat Neurosci 3:545–550. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA (1993). NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci 16:521–527. [DOI] [PubMed] [Google Scholar]
- Malinow R, Schulman H, Tsien RW (1989). Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science 245:862–866. [DOI] [PubMed] [Google Scholar]
- Mochida H, Sato K, Sasaki S, Yazawa I, Kamino K, Momose-Sato Y (2001). Effects of anisomycin on LTP in the hippocampal CA1: long-term analysis using optical recording. NeuroReport 12:987–991. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Kandel ER (1997). Brief theta-burst stimulation induces a transcription-dependent late phase of LTP requiring cAMP in area CA1 of the mouse hippocampus. Learn Mem 4:230–243. [DOI] [PubMed] [Google Scholar]
- Olmo MT, Rodriguez-Agudo D, Medina MA, Sanchez-Jimenez F (1999). The pest regions containing C-termini of mammalian ornithine decarboxylase and histidine decarboxylase play different roles in protein degradation. Biochem Biophys Res Commun 257:269–272. [DOI] [PubMed] [Google Scholar]
- Patrick GN, Bingol B, Weld HA, Schuman EM (2003). Ubiquitin-mediated proteasome activity is required for agonist-induced endocytosis of GluRs. Curr Biol 13:2073–2081. [DOI] [PubMed] [Google Scholar]
- Raught B, Gingras AC, Sonenberg N (2001). The target of rapamycin (TOR) proteins. Proc Natl Acad Sci USA 98:7037–7044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD (1999). The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci 19:4337–4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speese SD, Trotta N, Rodesch CK, Aravamudan B, Broadie K (2003). The ubiquitin proteasome system acutely regulates presynaptic protein turnover and synaptic efficacy. Curr Biol 13:899–910. [DOI] [PubMed] [Google Scholar]
- Stanton PK, Sarvey JM (1984). Blockade of long-term potentiation in rat hippocampal CA1 region by inhibitors of protein synthesis. J Neurosci 4:3080–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O, Schuman EM (2001). Protein synthesis at synaptic sites on dendrites. Annu Rev Neurosci 24:299–325. [DOI] [PubMed] [Google Scholar]
- Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM (2002). A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci USA 99:467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiedge H, Brosius J (1996). Translational machinery in dendrites of hippocampal neurons in culture. J Neurosci 16:7171–7181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiedge H, Bloom FE, Richter D (1999). RNA, whither goest thou? Science 283:186–187. [DOI] [PubMed] [Google Scholar]
- Tsokas P, Grace EA, Chan P, Ma T, Sealfon SC, Iyengar R, Landau EM, Blitzer RD (2005). Local protein synthesis mediates a rapid increase in dendritic elongation factor 1A after induction of late long-term potentiation. J Neurosci 25:5833–5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T (2001). Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci 24:1071–1089. [DOI] [PubMed] [Google Scholar]
- Zinck R, Cahill MA, Kracht M, Sachsenmaier C, Hipskind RA, Nordheim A (1995). Protein synthesis inhibitors reveal differential regulation of mitogen-activated protein kinase and stress-activated protein kinase pathways that converge on Elk-1. Mol Cell Biol 15:4930–4938. [DOI] [PMC free article] [PubMed] [Google Scholar]