

Abstract
Muscle-specific tyrosine kinase receptor (MuSK) has been believed to be mainly expressed and functional in muscle, in which it mediates the formation of neuromuscular junctions. Here we show that MuSK is expressed in the brain, particularly in neurons, as well as in non-neuronal tissues. We also provide evidence that MuSK expression in the hippocampus is required for memory consolidation, because temporally restricted knockdown after training impairs memory retention. Hippocampal disruption of MuSK also prevents the learning-dependent induction of both cAMP response element binding protein (CREB) phosphorylation and CCAAT enhancer binding protein β (C/EBPβ) expression, suggesting that the role of MuSK during memory consolidation critically involves the CREB–C/EBP pathway. Furthermore, we found that MuSK also plays an important role in mediating hippocampal oscillatory activity in the theta frequency as well as in the induction and maintenance of long-term potentiation, two synaptic responses that correlate with memory formation. We conclude that MuSK plays an important role in brain functions, including memory formation. Therefore, its expression and role are broader than what was believed previously.
Keywords: MuSK, brain, long-term plasticity, memory, oscillatory response, C/EBP
Introduction
Muscle-specific tyrosine kinase receptor (MuSK) is a receptor tyrosine kinase first discovered in the Torpedo electric organ (Jennings et al., 1993) and subsequently cloned in rat, human (Valenzuela et al., 1995), mouse (Ganju et al., 1995), Xenopus (Fu et al., 1999), and chicken (Ip et al., 2000). Although some studies based on Northern blot analyses and in situ hybridizations have reported that MuSK mRNA is present in both developing and adult tissues of mouse, chicken, and Xenopus (Ganju et al., 1995; Fu et al., 1999; Ip et al., 2000), it has been generally accepted that this receptor is mainly expressed and functional only in muscle, in which it plays an essential role in neuromuscular junction (NMJ) formation (DeChiara et al., 1996; Sanes and Lichtman, 1999). Recently, however, MuSK has been reported to also be expressed in sperm (Kumar et al., 2006).
At the NMJ, MuSK is critically involved in redistributing the acetylcholine receptors (AChRs) in the postsynaptic apparatus. This function requires the interaction of MuSK with the heparan-sulfate proteoglycan agrin and additional proteins, which include rapsyn and other unknown components. Together these proteins form the agrin receptor complex (DeChiara et al., 1996; Sanes and Lichtman, 1999; Hoch, 2003). In muscle, the activation of MuSK via the ErbB–neuregulin pathway results in an increase of AChR expression and triggers the activation of signaling pathways that lead to postsynaptic differentiation (Sandrock et al., 1997; Burden, 2002). Both agrin and MuSK are essential for NMJ formation and specifically for AChR organization at the NMJs, because knock-out mice of agrin or MuSK do not form neuromuscular synapses, lack AChRs clustering, and die soon after birth (DeChiara et al., 1996; Gautam et al., 1996). Agrin is expressed in several tissues, including the CNS, in which it participates in synapse formation (Hoch et al., 1993; Stone and Nikolics, 1995; Ferreira, 1999; Smith and Hilgenberg, 2002). Despite agrin isoforms with high AChR aggregating activity being widespread throughout the brain, their receptor complex has not yet been identified (Smith and Hilgenberg, 2002).
In the brain, synaptogenesis and remodeling of existing synapses are abundant throughout development but also occur in adulthood after injury and during long-term synaptic plasticity-associated functions, including memory formation (Scheff et al., 2005; Waites et al., 2005).
New memories become stable through a process known as consolidation, which requires the activation of a cascade of gene expression that critically involves the cAMP response element binding protein (CREB) and CCAAT enhancer binding protein (C/EBP) transcription factors (Alberini, 1999) and is accompanied by modifications of synaptic structure and/or number (Bailey et al., 1996; O’Connell et al., 2000; Geinisman et al., 2001). These synaptic changes are regulated by modulatory neurotransmitter, hormonal, or neurohumoral systems (Power et al., 2000; Izquierdo and McGaugh, 2000), which include, among others, the nicotinic cholinergic system (Gold, 2003; Hogg et al., 2003).
Following up on the findings that MuSK had been detected in developing and adult tissues other than muscle (Ganju et al., 1995; Fu et al., 1999; Ip et al., 2000) and that agrin is present in the CNS (Smith and Hilgenberg, 2002), we investigated the expression and the functional role of MuSK in the brain.
Materials and Methods
Animals.
Adult male Long–Evans rats weighing between 200 and 250 g or 21- to 28-d-old male Long–Evans rats were used, as specified in Results. Animals were individually housed and maintained on a 12 h light/dark cycle. The behavioral experiments were performed during the light cycle. All rats were allowed ad libitum access to food and water. All protocols complied with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Mt. Sinai School of Medicine Animal Care Committees.
DNA amplifications.
Total RNA was isolated with TRIzol (Invitrogen, Carlsbad, CA), and reverse transcription was performed using oligo-dT. Five sets of primers were designed based on the sequence of rat muscle MuSK (GenBank accession number U34985) and used for PCR amplifications. Primers were as follows: starting from the 5′ end of MuSK sequence, forward (F) 1, 5′-TTACAGATGCTCACCCTGGT-3′; reverse (R) 1, 5′-CTTAATCCAGGACACGGATGG-3′; F2, 5′-CAAGCCATCCGTGTCCTGGAT-3′; R2, 5′-ACAGTAGCCTTTGCTTTCTT-3′; F3, 5′-AGTATAGCAGAATGGAGCAA-3′; R3, 5′-GGAAGGCAATGTGGTGAGGGT-3′; F4, 5′-CTGCCGAAGGAGGAGAGAGTG-3′; R4, 5′-GTTTCCATCAGCTTTGTAGTA-3′; F5, 5′-AGGAACATCTACTCCGCAGAC-3′; R5, 5′-TGAAAAGATCCTCCTGGGTG-3′. All reactions were run on an iCycler (Bio-Rad, Hercules, CA) with the following cycle parameters: 1 cycle of 95°C for 2 min, 40 cycles of 95°C for 30 s followed by 57°C for 30 s and 72°C for 1 min, and 1 final extension cycle of 72°C for 10 min. For the MuSK full-length open reading frame amplifications, we used the following: F, 5′-ATGAGAGAGCTCGTCAACAT-3′; R, 5′-TGAAAAGATCCTCCTGGGTG-3′. Amplification reactions were performed in the iCycler using the following parameters: 1 cycle of 95°C for 2 min, 40 cycles of 95°C for 40 s followed by 57°C for 1 min and 70°C for 3.5 min, and 1 final extension cycle of 70°C for 10 min. DNA sequencing was performed at W. M. Keck Biotechnology Laboratory at Yale University (New Haven, CT).
Hippocampal neuronal cell culture.
Hippocampal neuronal cell cultures (HNCs) were prepared from hippocampi of embryonic day 18 (E18) Long–Evans rats as described previously by Goslin and Banker (1991) with some modifications. Cells were dissociated by treatment with 0.25% trypsin for 15 min at 37°C followed by trituration through a Pasteur pipette. Cells were plated at a density of 1.8 × 104 cells/cm2 on poly-l-lysine-coated coverslips in minimum essential medium (Invitrogen; Carlsbad, CA) containing 10% horse serum. After 3 h, when cells had attached, coverslips were transferred to dishes containing Neurobasal medium supplemented with B-27 (Invitrogen), where they were maintained for the entire time of culture. Cells were used at 12–14 d in vitro (DIV).
Riboprobe synthesis and in situ hybridization.
Three cDNA fragments included between base pairs 148–588, 1664–1900, and 2349–2767 of the rat MuSK (GenBank accession number U34985), subcloned into the pPCR-Script Amp SK (+) plasmid (Stratagene, La Jolla, CA) were used. The linearized plasmids were purified and used as templates for antisense and sense digoxigenin (DIG)-labeled riboprobe synthesis (Roche Diagnostics, Indianapolis, IN). Transcription was performed using T3 or T7 polymerases (Roche Diagnostics), and the synthesized riboprobes were purified with ProbeQuant G-50 Micro Columns (GE Healthcare Bio-Sciences, Piscataway, NJ). DIG was visualized with anti-DIG antibody coupled to alkaline phosphatase (AP) and detected with AP Conjugate Substrate kit (Bio-Rad). Primary HNCs at 12–14 DIV were fixed with 4% paraformaldehyde (PFA) for 30 min at room temperature (RT). Cells were washed with PBS-diethyl pyrocarbonate (DEPC), permeabilized with 0.5% Triton X-100, and incubated twice for 10 min in PBS containing 0.1% active DEPC. The cells were then equilibrated twice for 10 min in 2× SSC. Prehybridization was performed at 56°C for 1 h with the hybridization buffer containing 50% formamide, 5× SSC, 2% blocking reagent (Roche Diagnostics), and 40 μg/ml salmon sperm DNA. Twenty-micrometer rat brain tissue sections were obtained with a cryostat after perfusion with 4% PFA in PBS. Sections were mounted on slides and postfixed with 4% PFA-DEPC for 15 min at RT. After rinsing with PBS-DEPC, sections were treated with 0.2 m HCl for 10 min and permeabilized with 0.5% Triton X-100 for 10 min at RT. Sections were finally equilibrated in 0.1 m triethanolamine (TEA) for 5 min and acetylated in freshly prepared 0.25% acetic anhydrite in 0.1 m TEA for 10 min. Sense and antisense riboprobes (200 ng/ml for cell cultures and 400 ng/ml for tissue sections) were diluted in hybridization buffer, denatured at 75°C for 10 min, and incubated with either HNCs or brain sections overnight at 56°C. The next day, the samples were rinsed with 2× SSC twice for 10 min at RT, treated with RNase A (20 μg/ml) for 30 min at 37°C, and washed sequentially with 2× SSC for 30 min at RT, 2× SSC for 1 h at 65°C, and 0.1× SSC for 1 h at 65°C. After rinsing with Tris-NaCl buffer (100 mm Tris, 150 mm NaCl), the samples were incubated with blocking buffer [1% blocking reagent (Roche Diagnostics) and 0.05% Tween 20 in Tris-NaCl buffer] for 30 min at 37°C. The hybridization was detected using AP-conjugated anti-DIG antibody (Roche Diagnostics) diluted in blocking buffer at a final concentration of 1:200 and added to cells for 3 h at RT. The reaction was revealed by nitroblue tetrazolium/5-bromo-4-chloro-indolyl-phosphate solution (Roche Diagnostics) diluted in 0.1 m Tris pH 9.5/0.1 m NaCl containing 1 mm levamisole incubated at RT in the dark for 3–8 d. Images were acquired with Zeiss (Oberkochen, Germany) Axioscope at the Mount Sinai School of Medicine Microscopy Shared Resource Facility.
Transfection of small interfering RNAs.
The day before transfection, rat MuSK-Myc-transfected human embryonic kidney 293 cells (HEK293M) (generously provided by Steven Burden, Skirball Institute of Biomolecular Medicine, New York University, New York, NY) were plated in six-well plates at 3 × 105 cells per well. Synthetic double-stranded small interfering RNA (siRNA) targeting MuSK (siRNA-MuSK: sense, 5′ CCAUCAGCUGGAUUGAAAAtt 3′; antisense, 5′ CCGUAUCGUAAGCAGUACUtt) and the nonsilencing siRNA used as negative control (sense, 5′ AGUACUGCUUACGAUACGGtt; antisense, 5′ CCGUAUCGUAAGCAGUACUtt) were generously provided by Ambion (Austin, TX). siRNAs were resuspended in RNase-free water at 100 μm. HEK293M cells were transfected with Lipofectamine 2000 (Invitrogen) and siRNAs (200 nm) according to the protocol provided by Invitrogen. Forty-eight hours after transfection, cells were harvested, lysed, and analyzed for their MuSK expression levels using quantitative Western blot analysis.
Western blot analysis.
Fifty micrograms of total protein extract/lane were resolved using 7.5% SDS-PAGE and analyzed by Western blot as described previously (Taubenfeld et al., 2001b). Primary antibodies were incubated in TBS overnight at 4°C. Primary antibodies: goat anti-MuSK N-terminal (1:300; R&D Systems, Minneapolis, MN), mouse anti-agrin AGR-520 (1:500; Stressgen, Victoria, CA), rabbit anti-GluR1 (1:2000; Chemicon Temecula, CA), rabbit anti-C/EBPβ (1:10,000; Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-pCREB (1:1000; Upstate Biotechnology, Lake Placid, NY), rabbit anti-CREB (1:1000; Cell Signaling Technology, Beverly, MA), mouse anti-nuclear pore protein 62 (NP62) (1:5000; Covance Research Products, Berkeley, CA), mouse anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:4000; Chemicon), and mouse anti-actin (1:15 × 104; Chemicon). Horseradish peroxidase-coupled specific secondary antibodies (1:4000) were incubated in TBS for 1 h at room temperature. The detection system used was ECL detection reagents (GE Healthcare Bio-Sciences). Recombinant extracellular domain of MuSK (R&D Systems) and recombinant insulin-like growth factor II (IGF-II) (Chemicon) were incubated at 250 ng/ml. Quantitative densitometric analysis was done using NIH Image software. Actin or GAPDH were used for sample normalization and data are expressed as mean percentage ± SEM of the 0 h− (100%) control mean values. Statistical analyses were performed as specified in Results. Data were analyzed with one-way ANOVA followed by either Newman–Keuls post hoc or Dunnett’s multiple-comparison tests. When two groups were compared, a t test was used.
Two-dimensional gel electrophoresis.
To compare MuSK proteins present in embryonic hippocampal and muscle protein extracts, we used two-dimensional (2D) electrophoresis followed by Western blotting with anti-MuSK antiserum (R&D Systems). Isoelectric focusing (IEF) was performed on an Ettan IPGphor II (GE Healthcare, Buckinghamshire, UK) using 7 cm broad-range nonlinear immobilized pH gradient (IPG) strips (pH 3–10; GE Healthcare). Briefly, protein extract from E18 brain or muscle were diluted in 7 m urea, 2 m thiourea, 4% CHAPS [3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfate], and 1% DTT (1,4-dithiothreitol). IPG buffer (pH 3–10; GE Healthcare) and DeStreak reagent (GE Healthcare) were added to the protein mixtures and loaded onto the IPG strips. Twelve hours later, IEF was performed at 20°C according to the following voltage-time program: 500 V for 30 min, 1000 V for 30 min, and 5000 V for 1 h. Before separation of proteins in the second dimension by SDS-PAGE, IPG strips were equilibrated in 1% DTT, followed by 4% iodoacetamide. Second dimension was performed in 7.5% SDS-PAGE gels. Immunoblotting was performed using the anti-MuSK antiserum as described above in Western blot analysis (R&D Systems).
Inhibitory avoidance training.
Inhibitory avoidance (IA) training was performed as described previously (Taubenfeld et al., 2001a). During training, each rat received a foot shock (0.6 mA for 2 s) in the dark compartment and was then returned to the home cage. Memory was tested by measuring the latency to enter the shock chamber. Foot shock was not administered on the retention test, and testing was terminated at 540 s. Training and testing procedures were performed blind to treatments. Different control groups were used in different experiments, as detailed in the Results section. Statistical analysis was performed using ANOVA followed by either Dunnett’s or Newman–Keuls post hoc tests as specified in Results. After testing, rats were anesthetized, and their hippocampi were rapidly dissected and frozen for Western blot or Northern blot analyses. Both hippocampi from each animal were pooled and homogenized; therefore, each hippocampal sample, unless otherwise specified, consisted of the lysate obtained from both hippocampi. The control conditions used for the IA training are specified in Results and consisted of temporally matched unpaired or nonshocked conditions. Unpaired controls received the exposure to the training apparatus in the same way as the trained rats but were not shocked in the dark chamber. They returned to their home cage and, 1 h later, were placed directly onto the grid floor of the dark side and shocked. Immediately afterward, they were returned to their home cage. These unpaired experiences of context and shock do not create an association between the two stimuli and, in fact, the animals that undergo this protocol never show IA memory when tested. The nonshocked controls consisted of rats that were exposed to the training apparatus in the same way as the trained rats but did not receive the shock in the dark chamber and were then returned to their home cages. Both control groups were killed at time points that matched those of trained rats.
Surgeries and oligodeoxynucleotide injections.
Bilateral hippocampal surgeries (4.0 mm posterior to bregma; 2.6 mm lateral from midline; 2.0 mm ventral) were performed as described by Taubenfeld et al. (2001a), MuSK antisense oligodeoxynucleotide (M-ODN) (5′-GAATGTTGACGAGCTCTCTCATG-3′), scrambled ODN (S-ODN) (5′-TACTATGGATCGTCTGCGCATAG-3′) or vehicle solution (PBS) were injected bilaterally into the hippocampi. M-ODN was specific for the sequence that includes MuSK translational starting site. S-ODN, which served as a control, contained the M-ODN base composition but in a randomized order and showed no homology to any sequence in the GenBank database, as confirmed by a BLAST (basic local alignment search tool) search. Both ODNs were phosphorothioated on the three terminal bases at each end to protect against nuclease degradation. ODNs were reverse phase cartridge-purified and purchased from Gene Link (Hawthorne, NY). ODN injections were performed as described previously (Taubenfeld et al., 2001a). Briefly, at the indicated time points before or after IA training, animals received bilateral injections (2 nmol/μl) of M-ODN, S-ODN, or vehicle (PBS, pH 7.4). As detailed in Results, either single or double injections were performed. Biotinylated M-ODN was purchased from Gene Link. To detect biotinylated M-ODN, animals were perfused with 4% PFA in PBS. Brains were postfixed overnight in the same fixative with 30% sucrose. Forty-micrometer coronal sections were stained using an Immunopure ABC kit according to the manufacturer’s protocol (Pierce, Rockville, IL).
TaqMan real-time quantitative reverse transcription-PCR.
Hippocampal total RNA was extracted with TRIzol (Invitrogen) and reverse transcribed into cDNAs using Omniscript reverse transcriptase (Qiagen, Hilden, Germany). TaqMan 5′ nuclease fluorogenic quantitative PCR assays were performed using the TaqMan Universal PCR Master Mix kit (PE Applied Biosystems, Foster City, CA) according to manufacturer’s instructions. Probes and primers were designed using PrimerExpress3 software (PE Applied Biosystems). GAPDH (GenBank accession number M17701) was used as a reference gene for comparative analysis. The following dye combinations for probe generation were used for detection and data normalization, respectively: 6-carboxy-fluorescein (FAM) (reporter for MuSK), hexachlorofluoresceine (HEX) (reporter for GAPDH), and BHQ1 (black hole quencher 1, nonfluorescent quencher). Amplifications were generated by using the following primers and hybridization probes: MuSK-F, 5′-TGCGCCTATGTTGGAGCAA-3′; MuSK-R, 5′-CCTCTGCTCTCTCGCACATG-3′; probe, 5′/FAM/TGCCTGCAGACAGACCCAGC. GAPDH-F, 5′-GAACATCATCCCTGCATCCA-3′; GAPDH-R, 5′-CCAGTGAGCTTCCCGTTCA-3′; probe, 5′-/HEX/CTTGCCCACAGCCTTGGCAGCA). The cycle threshold method (CT) was chosen to determine the relative quantification of gene expression in trained and control rats, as described in PE Applied Biosystems User Bulletin Number 2 (P/N 4303859). Before comparative analysis, a validation experiment using GAPDH was performed to determine whether the amplification efficiencies of the target and reference gene are approximately equal. The amplification reactions were performed using 1 μl of cDNA, TaqMan universal PCR Master Mix (PE Applied Biosystems), and 100 nm of fluorogenic internal probe. Three TaqMan assays with triplicates were performed for each cDNA sample. All reactions were run on a GenAmp ABI 7900 sequence detection system with the following cycle parameters: 1 cycle of 50°C for 2 min followed by 95°C for 10 min and 40 cycles of 95°C for 15 s followed by 60°C for 1 min. Relative quantification was expressed as fold change values in the 20 h+ group or 20 h− group compared with the 0 h− group. Data represent the mean fold change ± SEM of three experiments run for each experimental group. Statistical analysis was performed using Student’s t test.
Northern blot analysis.
Northern blot analyses were performed as described previously (Taubenfeld et al., 2001b). The rat agrin probe consisted of a 366 bp fragment that represents a region common to the agrin isoforms (nucleotides 5492–5878; GenBank accession number M64780). The same membrane was first hybridized with the agrin probe and then stripped and rehybridized with a full-length rat cyclophilin cDNA that was used as a control for normalization. Probes were labeled with random oligonucleotides primers (Prime-It II kit; Stratagene) and [α-32P]dCTP (GE Healthcare). Quantitative densitometry analysis was performed using NIH Image software. Data were expressed as mean percentage ± SEM of the 0 h− (100%) control mean values. Statistical analyses were performed using one-way ANOVA followed by Newman–Keuls post hoc test.
Electrophysiology.
In all electrophysiology experiments, rats were injected bilaterally with ODNs, so that one hippocampus received M-ODN and the other S-ODN (randomly assigned). Like in the behavioral experiments, we performed double injections in each hippocampus, with the second injection occurring 8 h after the first. The animals were killed 24 h after the first set of ODN injections. The experimenter was blind to the assignment of injection sites. Acute hippocampal slices were prepared as described previously (Tsokas et al., 2005). Briefly, the rats were deeply anesthetized with halothane and decapitated. The brain was rapidly removed and placed in ice-cold artificial CSF (ACSF) containing the following (in mm; all from Sigma-Aldrich, St. Louis, MO): 118 NaCl, 3.5 KCl, 1.3 MgSO4, 3.5 CaCl2, 1.25 NaH2PO4, 24 NaHCO3, and 15 glucose, bubbled with 95% O2/5% CO2. The hippocampus was then rapidly dissected out and transverse 500-μm-thick slices were cut from its middle third using a tissue chopper at an ambient temperature of 4°C. The slices were maintained in an interface chamber (ACSF and humidified 95% O2/5% CO2 atmosphere) at room temperature (22–25°C) for at least 90 min before use. For recording, the slices were transferred to a submersion recording chamber, in which they were perfused on a nylon mesh with preheated, oxygenated ACSF. Slice health was determined by delivering monophasic, constant-current stimuli (100 μs) with a bipolar stainless steel electrode (FHC, Bowdoinham, ME) placed in stratum radiatum of the CA3 region and measuring the evoked field EPSP (fEPSP) in the stratum radiatum of the CA1 region with electrodes filled with ACSF (Re 2 MΩ). fEPSPs were acquired by delivering stimuli at 0.033 Hz, and the signals were low-pass filtered at 3 kHz and digitized at 20 kHz. fEPSPs acquisition, as well as amplitude and maximum initial slope measurement were performed using either an Axobasic routine or pClamp 9 (Molecular Devices, Foster City, CA). After recording was completed, the slice was removed from the recording chamber and immediately frozen on microscope slides on dry ice, for use in Western blotting experiments.
For the carbachol experiments, slices were prepared from male Long–Evans rats aged 21–28 d and recorded (33–34°C) using a modification of the method of Williams and Kauer (1997). After the maximal fEPSP was determined, the delivery of stimuli in area CA3 was terminated, and slices were superfused continuously with ACSF containing 2.5 mm KCl and 50 μm carbachol (Sigma-Aldrich). Extracellular recordings were obtained from the CA1 region of such slices during a period of 30 min from the onset of carbachol treatment and stored using pClamp 9 (Molecular Devices) for subsequent off-line analysis. In our quantification of carbachol-induced oscillations, we considered only slices that were competent of producing field events in response to carbachol treatment. In our statistical analysis, we scored as “successes” only those slices that displayed regular periods of oscillatory activity, defined as at least five consecutive regularly spaced bursts, whereas all other slices were scored as “failures” (Williams and Kauer, 1997).
For the LTP experiments, slices were prepared from adult male Long–Evans rats, and the fEPSP was recorded at 30–31°C. LTP was induced by two 1-s-long trains of 100 Hz high-frequency stimulation (HFS), separated by 20 s, using a stimulus intensity that evoked an fEPSP corresponding to 75% of the maximal fEPSP. In all experiments, HFS was delivered when the basal EPSP had been stable for at least 30 min, and recordings were concluded 120 min after HFS. Data points were normalized to the final 10 min of the baseline period and are presented as group means ± SEs.
Results
MuSK is expressed in the brain, particularly in neurons, as well as in non-neuronal tissues
To determine whether MuSK is expressed in rat brain, we used reverse transcription-PCR amplifications of cDNAs obtained from adult hippocampus, cortex, cerebellum or HNCs with sets of primers that spanned the entire muscle MuSK sequence (GenBank accession number U34985) (Fig. 1A). Gel electrophoresis analyses revealed that each set of primers generated fragments of similar size across all cDNA preparations. For each cDNA, five overlapping PCR products of 439 bp, 507/222 bp, 717 bp, 723 bp, and 409 bp that covered the entire coding sequence of MuSK were obtained and sequenced. The analysis of these sequences revealed that the hippocampus, cortex, cerebellum and HNCs express two MuSK isoforms (Fig. 1A). One isoform was 2644 bp long, and its sequence was identical to an alternatively spliced MuSK transcript (A454), described previously in muscle of human (Valenzuela et al., 1995), mouse (Ganju et al., 1995), and rat (Hesser et al., 1999). Compared with the longest isoform cloned in muscle (U34985), A454 displays an alanine residue at position 454 that replaces an 8 aa deletion in the ectodomain. To our knowledge, no functional significance has yet been associated with this isoform.
Figure 1.
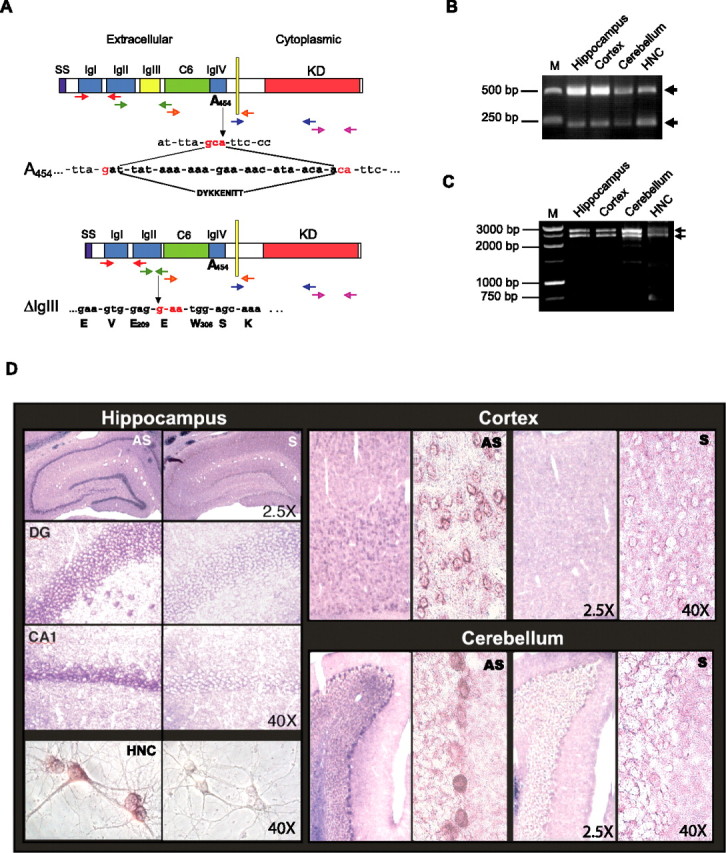
MuSK is expressed in adult rat brain. A–C, Cloning of MuSK in the brain. A, Schematic representation of MuSK isoforms expressed in the brain. SS, Signal sequence; IgI–IgIV, Ig-like domains; C6, cysteine rich domain; KD, kinase domain; E, glutamic acid; W, tryptophan; S, serine; V, valine; K, lysine. Arrows in different colors indicate the five sets of primers used for PCRs. B, Electrophoretic analysis of PCR amplifications generated with the second set of primers (shown in green in Fig. 1A). M, Molecular weight marker. C, Electrophoretic analysis of PCR amplifications with a set of primers that flanked the entire MuSK ORF. D, Musk mRNA distribution in adult rat brain and HNCs revealed by in situ hybridization. Representative examples of rat adult brain coronal sections and HNCs hybridized with antisense (AS) or sense (S) probes. DG, Dentate gyrus. CA1 regions of the hippocampus, somatosensory cortex, cerebellum, and HNCs are shown. Magnifications: 2.5 and 40×. Data are representative of results obtained from eight experiments.
The second MuSK transcript, also found in all brain regions and HNCs, was shorter (2359 bp) and, compared with the U34985 MuSK sequence, carried two distinctive features: the A454 substitution and a deletion of the third Ig-like domain. As shown in Figure 1 B, all PCR amplifications of cDNAs from the hippocampus, cortex, cerebellum, and HNCs, obtained using primers designed between positions 564 and 1071 (U34985), generated two bands of 507 and 222 bp, respectively, whose sequences corresponded to the two alternatively spliced MuSK transcripts. An isoform lacking the same IgIII-like domain (MuSK-ΔIgIII) had been found previously in denervated rat muscle by Hesser et al. (1999) and reported to mediate muscle AChRs clustering. However, to our knowledge this A454-ΔIgIII isoform has not been described previously.
These results were further confirmed by gel electrophoretic analysis and sequencing of full-length MuSK fragments generated by PCR with 5′ and 3′ primers that flanked the entire MuSK open reading frame (ORF) (Fig. 1C). Indeed, all cDNAs from the hippocampus, cortex, cerebellum and HNCs generated the amplification of two bands of 2644 and 2359 bp, respectively. Sequencing of the two hippocampal bands confirmed both alternatively spliced transcripts described above.
Next, the distribution of MuSK mRNA transcripts in the brain, and particularly in neurons, was investigated using in situ hybridizations. Adult brain sections and sister HNCs were hybridized with either MuSK antisense or sense (control) probes. Most brain regions, including the cortex, hippocampus, and cerebellum, as well as most neurons of HNCs, exhibited labeling with the antisense but not with the sense probe. As shown in Figure 1D, neuronal populations that selectively hybridized with the antisense probe included the following: in the hippocampus, most dentate gyrus granule cells, CA1, CA2 and CA3 pyramidal neurons and hilar cells; in the cortex, the majority of neuronal populations of most layers; and in the cerebellum, the Purkinje cells and some elements of the granule cell layer. These results were confirmed in several experiments by using three independent sequences as probes, which hybridized to different regions of the MuSK transcript (the N terminus, a region juxtaposed with the transmembrane domain, or the C terminus as detailed in Material and Methods).
Finally, the expression of MuSK protein levels in rat adult brain, HNCs, developing brain and muscle, as well as several non-neuronal tissues were investigated using Western blot analyses. For this study, we used a commercially available goat anti-MuSK antiserum (R&D Systems) raised against the purified recombinant rat MuSK extracellular domain (aa 22–453) and previously shown to recognize rat and mouse MuSK in Western blot after immunoprecipitation with other anti-MuSK antibodies (Finn et al., 2003). We further examined the reactivity of this antiserum against MuSK in Western blot to confirm its specificity. Thus, we performed several tests using three independent technical approaches. First, the antiserum was used in Western blot against the recombinant N-terminal portion of MuSK, which had been used as immunogen. Results depicted in Figure 2A showed that the antiserum reacted with the recombinant truncated MuSK. Second, the antiserum was used in Western blot to immunostain extracts obtained from either the HEK293 cell line stably transfected with the rat MuSK fused to a 10 aa Myc tag in position 249 (293M) or its relative nontransfected 293 control. As shown in Figure 2B, both anti-MuSK or anti-Myc antisera revealed a similar pattern in the 293M, which consisted of 2 bands of 100 and 75 kDa, respectively, that were absent in the nontransfected 293. This indicated that both bands correspond to the MuSK-Myc fusion protein and that the 75 kDa band likely represents a proteolytic fragment of the 100 kDa protein.
Figure 2.
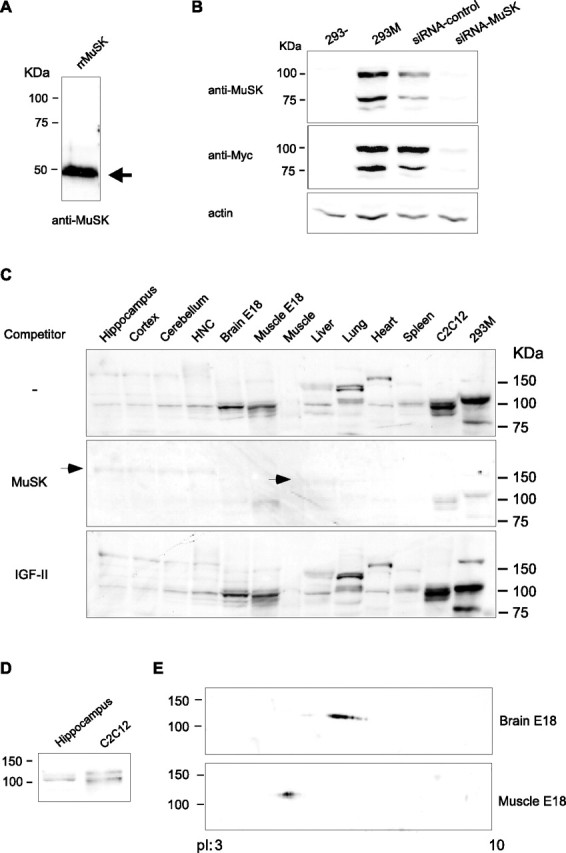
MuSK expression is detected in several tissues by Western blot analysis. A, B, Validation of anti-MuSK antiserum specificity. A, Western blot analysis of recombinant N-terminal portion of rat MuSK (rrMuSK). B, Western blot analysis of protein extracts obtained from nontransfected HEK293 cell line (293-), HEK293 transfected with rat MuSK tagged with the Myc sequence (293M), 293M treated with siRNA specific for MuSK (siRNA-MuSK) or with control, sense sequence (siRNA-control). The membrane was stained with anti-MuSK antiserum, stripped, and restained with anti-Myc antiserum. C, MuSK is expressed in several tissues. Western blot analysis was performed on extracts obtained from the indicated tissues. C2C12 and 293M were used as positive controls. Arrows indicate nonspecific bands that are not competed by the recombinant MuSK. D, At least two specific bands are detected with anti-MuSK antiserum in different tissues tested. Hippocampus and C2C12 are shown as representative examples. E, 2D gel analysis of extracts obtained from E18 brain and E18 muscle stained with anti-MuSK antibody. Muscle and brain MuSK isoforms show different isoelectric point (pI) migration.
To further confirm that both bands specifically recognized by the two antisera in the 293M were indeed MuSK polypeptides, we transfected these cells with constructs that either carried a MuSK-specific siRNA or a relative control sequence. As depicted in Figure 2B, quantitative Western blot analyses of extracts obtained from these transfections showed that the anti-MuSK but not the control sequence profoundly knocked down the expression of the 100 and 75 kDa polypeptides recognized by both anti-MuSK and anti-Myc antisera, confirming that these bands are indeed MuSK-Myc proteins. Thus, together, all of the results described above led us to conclude that the R&D Systems anti-MuSK antiserum specifically reacts to rat MuSK in Western blot.
This antiserum was then used in Western blot analyses to test for the presence of MuSK in protein extracts obtained from several tissues including hippocampus, cortex, cerebellum, 14 DIV HNCs, E18 brain (brain E18), E18 muscle (muscle E18), adult muscle, liver, lung, heart, and spleen. The C2C12 myotube (Yaffe and Saxel, 1977) and 293M cell lines were included as positive controls. Results showed that MuSK is ubiquitously expressed, although to different degrees. As shown in Figure 2C, MuSK was detected in several of these tissues. Its relatively strongest expression was found in E18 muscle and E18 brain extracts. A weaker, but still detectable, expression was evident in the various adult brain regions, in HNCs and the other tissues. The faintest relative expression appeared to be that of adult muscle. To further confirm the specificity of the anti-MuSK labeling, Western blot membranes containing the same extracts were immunostained with the anti-MuSK antiserum preincubated with an excess of either extracellular domain of recombinant MuSK or, as a control, an excess of the unrelated recombinant polypeptide IGF-II. As shown in Figure 2C, the 100 kDa band/s recognized by the anti-MuSK antiserum were competed by recombinant MuSK but not by recombinant IGF-II. Notably, some nonspecific bands at ∼130–150 kDa (indicated by arrows in Fig. 2C) were not competed. In contrast, in heart, lung, and spleen, additional bands >100 kDa were also competed by recombinant MuSK but not by recombinant IGF-II, suggesting that they may represent either cross-reacting proteins, MuSK isoforms complexed with other proteins, or posttranslationally modified MuSK isoforms that display a slower migration. RT-PCR amplifications of cDNAs obtained from all of the non-neuronal tissues used in the Western blot study confirmed the ubiquitous expression of MuSK (data not shown).
Interestingly, in some extracts, including hippocampus, cortex, cerebellum, and HNCs, like in previous Western blot studies performed in muscle and myotube extracts (Herbst and Burden, 2000), at least two closely migrating immunoreactive bands with a molecular weight of ∼100 kDa were detected (see representative examples in Fig. 2D). It is possible that these bands correspond to differentially expressed splicing forms and/or differentially phosphorylated isoforms of MuSK.
In agreement with the hypothesis that MuSK exists in multiple posttranslationally modified (likely to be phosphorylation) forms, a 2D PAGE analysis of E18 rat brain and E18 rat muscle extracts revealed that MuSK resolves in multiple spots separated along the isoelectric point gradient and that E18 brain MuSK is differentially modified compared with E18 muscle MuSK (Fig. 2E).
Together, all of the results described above demonstrate that MuSK is expressed not only in muscle but also in developing and adult brain, particularly in neurons, and in a variety of non-neuronal tissues. These data confirm and extend previous results (Fu et al., 1999; Ip et al., 2000; Kumar et al., 2006), showing that MuSK is expressed in tissues other than skeletal muscle, including liver, lung, heart, spleen, and sperm.
MuSK is required for memory formation
We next investigated whether MuSK in the brain plays a role during functions that involve synapse formation/remodeling; thus, we tested whether MuSK is required for memory formation. MuSK expression in the hippocampi of adult rats was knocked down by bilateral injections of MuSK antisense (M-ODN) through stereotactically implanted cannulas; S-ODN was used as a control. The effect of these treatments was determined on memory retention.
Similar to previous studies showing the distribution and stability of other hippocampally injected ODNs (Guzowski and McGaugh, 1997; Taubenfeld et al., 2001a), we found that M-ODN diffused throughout the entire dorsal hippocampus and its temporal stability was quite brief. In fact, M-ODN was detected 2 h, but not 8 h after injection (Fig. 3A). Therefore, we investigated the effect of either single or double injections of M-ODN, S-ODN, or vehicle (PBS) on memory retention tested 24 h after IA training.
Figure 3.

Knockdown of hippocampal MuSK blocks IA memory consolidation. A, Biotinylated M-ODN diffusion and relative concentrations after hippocampal injection. Representative sections of a rat brain at different bregma (left to right) and representative sections injected with biotinylated M-ODN and killed either 2 or 8 h after injection (−2.8 μm from bregma) are shown. Representative examples of three independent experiments. B, Injection of M-ODN or S-ODN, training and testing time points. C, Quantitative Western blot analyses of hippocampal extracts from trained rats that underwent the protocols shown in B. Blots were stained with anti-MuSK antibody stripped and restained with anti-GluR1 antibody. Staining for actin was used for normalization of both MuSK and GluR1 values. A representative blot per condition is shown. Graphs represent the densitometric analysis of all data. Data are expressed as a mean percentage ± SEM of the S-ODN/PBS (S/P) control group (100%). D, Memory retention expressed as mean latency ± SEM (in seconds) of trained rats that received the treatments shown in B. M, M-ODN; S, S-ODN.
As depicted in Figure 3B, single injections were administered either 8 h after training or 2 h before testing (22 h after training). Double injections, which were performed to increase the effect of the M-ODN treatment, involved delivery immediately after and 8 h after training (0 and 8 h). All rats were killed immediately after testing (24 h after training), and their hippocampal MuSK concentration was evaluated by quantitative Western blot analysis. As depicted in Figure 3C, at 24 h after training, hippocampal MuSK expression levels were reduced to 70.8% by the 8 h single M-ODN injection (n = 8), and even more profoundly, to 58.8% by the double M-ODN injection (n = 8). In contrast, the 22 h single M-ODN injection had no effect (92.7%; n = 8), compared with S-ODN (100%; n = 4) and PBS (100%; n = 4) controls performed at the same time points. Both S-ODN- and PBS-injected control groups showed very similar levels of MuSK expression. Indeed, if the PBS-injected groups were assigned the value of 100%, the single S-ODN injections at 8 or 22 h resulted in values of 96.3 ± 8.1 and 105.7 ± 3.0%, respectively. Hence, the PBS and S-ODN-injected groups were combined for statistical analysis.
To verify that M-ODN treatment specifically affected MuSK expression, the concentration of an unrelated receptor protein, the glutamate R1 AMPA subunit (GluR1), was determined on the same Western blot membranes. No change in GluR1 was found across samples. Both MuSK and GluR1 were normalized against the relative concentration of actin, which was also immunostained on the same membrane, and remained stable across samples relative to the equal concentrations loaded. An ANOVA among all treatment and time point groups showed a significant group effect (F(5,42) = 4.5; p < 0.01), and a Newman–Keuls post hoc comparison test revealed that MuSK levels were significantly lower in the hippocampi that received the 0 and 8 h double injection (p < 0.01) compared with all other groups. A single injection resulted in a consistent, although not significant, decrease. No significant changes were found in the expression of GluR1 (Fig. 3C). Thus, M-ODN selectively downregulates the expression of MuSK in a dose-dependent manner.
The effect of M-ODN treatment on memory retention is depicted in Figure 3D. A one-way ANOVA comparing latencies across all groups indicated a significant group effect (F(6, 82) = 30.1; p < 0.0001). Newman–Keuls post hoc test revealed that the 8 h single M-ODN injection significantly impaired memory retention (87.3 ± 19.5 s; n = 8) compared with S-ODN (n = 4) and PBS (n = 4) control groups (p < 0.01). As in the previous experiment, because no significant difference was observed between the S-ODN- (221.4 ± 33.3 s) and PBS-injected groups (210.5 ± 14.3 s), the two were combined (215.9 ± 16.9 s) for statistical analysis. The double-injection treatment of M-ODN more profoundly and significantly impaired memory retention (M-ODN: 61.6 ± 15.4 s, n = 8; S-ODN: 200.7 ± 32.3 s, n = 8, p < 0.001). In contrast, a single injection performed at 22 h after training had no effect on memory retention tested 2 h later (M-ODN: 182.7 ± 38.1 s, n = 8 compared with S-ODN/PBS: 181.9 ± 31.5 s, n = 8). Because no significant difference was observed between the S-ODN- (171.4 ± 62.1 s; n = 4) and PBS-injected groups (192.5 ± 26.3 s; n = 4), the two were combined for statistical analysis.
Together, these data indicate that blocking MuSK expression during the consolidation phase of memory impairs memory retention. Moreover, because the 22 h single injection did not affect memory tested 2 h later, our results also showed that M-ODN does not have toxic or nonspecific effects on the expression of the memory. This is also supported by the fact that the effect of the M-ODN on both MuSK expression and memory retention is dose dependent.
Thus, we concluded that MuSK plays an essential role during IA memory consolidation.
MuSK expression is critical for the activation of the CREB–C/EBP-dependent gene cascade required for IA memory consolidation
Which molecular mechanisms are affected by MuSK disruption during memory consolidation? Recent studies have shown that, in hippocampal neurons, treatment with the neuron-specific isoform of agrin, but not with its ubiquitously expressed counterpart, causes the phosphorylation of the transcription factor CREB (Ji et al., 1998). We have confirmed these results and found that, in addition to CREB activation, agrin treatment of HNCs also results in increased expression of C/EBPβ (K. Gagnidze and C. M. Alberini, unpublished data). Notably, the CREB–C/EBP-dependent pathway is known to be required for the consolidation of different kinds of memories, including IA (Yin and Tully, 1996; Silva et al., 1998; Alberini, 1999; Kandel, 2001; Taubenfeld et al., 2001a,b; Athos et al., 2002). In addition, previous work from our laboratory has shown that the activation of the CREB–C/EBP pathway in the hippocampus after IA training requires an intact fornix (Taubenfeld et al., 1999, 2001b), which is known to carry modulatory inputs to the hippocampus, among which the most prominent component is cholinergic. Hence, here we tested whether knocking down MuSK expression in the hippocampus influences the activation of CREB and the induction of C/EBPβ.
In the first set of experiments, we determined whether the knockdown of MuSK affects the learning-induced phosphorylation of CREB in Ser133 (pCREB), a posttranslational modification critical for the activation of this transcription factor. Thus, we tested the levels of pCREB in the hippocampi of trained and unpaired control rats that underwent either M-ODN or S-ODN injections in their hippocampi. As described in Materials and Methods, unpaired controls consisted of rats exposed to the training apparatus but shocked 1 h after exposure, so that an association between the context and the shock was never formed, and killed at the same time points as trained rats. Because hippocampal pCREB significantly increases immediately after IA training (Bernabeu et al., 1997; Taubenfeld et al., 1999, 2001b), to ensure that MuSK disruption was achieved at this time, we injected M-ODN or S-ODN bilaterally into the rat hippocampi 4 h before training and killed the animals 1 h after training (Fig. 4A). Hippocampal protein extracts were assessed for the concentration of pCREB using quantitative Western blot analyses. As depicted in Figure 4A, a one-way ANOVA showed a significant group effect (F(3,12) = 6.1; p < 0.01). Dunnett’s multiple-comparison test revealed that rats that received S-ODN injection and underwent IA training (S-ODN-train) had a significant increase in hippocampal pCREB (222.6 ± 47.5%; n = 4; p < 0.05) compared with unpaired S-ODN-injected rats (S-ODN-unp) (100.0 ± 17.6%; n = 4), confirming our previous findings (Taubenfeld et al., 1999, 2001b). This increase was completely blocked in trained rats that received M-ODN treatment (M-ODN-train) (128.5 ± 34.9; n = 4), and, in fact, in these rats hippocampal pCREB levels remained comparable with those of unpaired control rats injected with either S-ODN (100.0 ± 17.6%; n = 4) or M-ODN (M-ODN-unp) (85.0 ± 14.6%). To determine whether the pCREB blockade was attributable to decreased phosphorylation of preexisting CREB or a change in CREB expression, the same membranes were stripped and restained with anti-CREB antibody. As depicted in Figure 4A, CREB concentrations were similar across all samples, and no significant differences were found with a one-way ANOVA (S-ODN-unp, 100 ± 6.8%; S-ODN-train, 118.8 ± 13.1%; M-ODN-unp, 115.5 ± 7.5%; M-ODN-train, 122.2 ± 5.7%). Moreover, to confirm the selectivity of the M-ODN effect, the same membranes were stripped and stained for an unrelated nuclear protein, NP62. As shown in Figure 4A, NP62 also remained unchanged (S-ODN-unp, 100 ± 10.3%; S-ODN-train, 127.2 ± 12.8%; M-ODN-unp, 128.1 ± 9.9%; M-ODN-train, 118.7 ± 9.9%, no significant differences with a one-way ANOVA). In each experiment, pCREB, CREB, and NP62 levels were normalized against the relative concentration of actin, which was used as a loading control.
Figure 4.
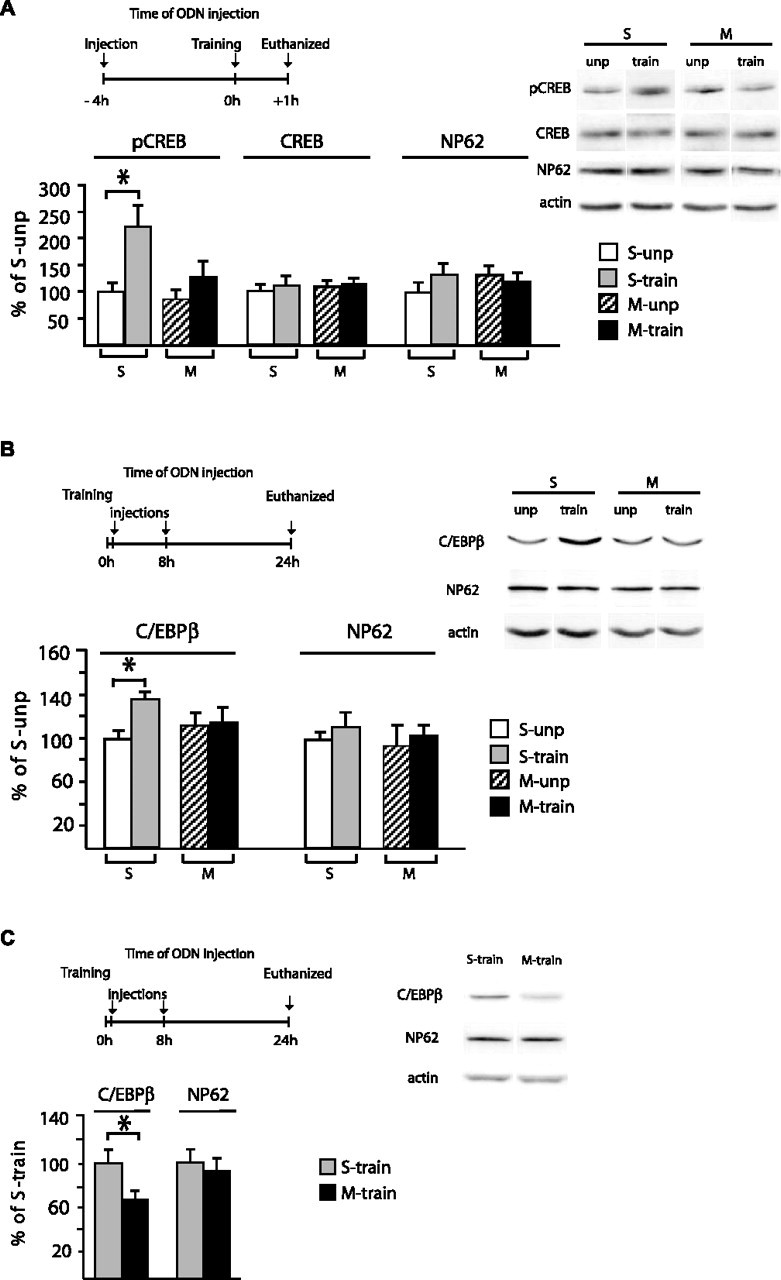
Hippocampal disruption of MuSK prevents the activation of the CREB–C/EBPβ pathway after training. A, Schematic representation of injection of M-ODN or S-ODN, training, and killing time points. Quantitative Western blot analysis of hippocampal extracts from untrained and trained rats that received the above treatments. Blots were stained with anti-pCREB (Ser133) antibody, stripped, and restained with anti-CREB, anti-NP62 antibodies and finally with anti-actin, which was used for normalization. A representative blot per condition is shown. Graphs represent the statistical densitometric analysis of all data. Data are expressed as a mean percentage ± SEM of the S-ODN (S) unpaired (S-unp) control group (100%). M, M-ODN; M-unp, M-ODN unpaired. B, Injection of M-ODN or S-ODN, training, and killing time points. Quantitative Western blot analysis of hippocampal extracts from unpaired and trained rats that received the above treatments. Blots were stained with anti-C/EBPβ antibody, stripped, and restained with anti-NP62 antibody and finally with anti-actin, which was used for normalization. A representative blot per condition is shown. Graphs represent the statistical densitometric analysis of all data. Data are expressed as a mean percentage ± SEM of the S-ODN unpaired control group (100%). C, Injection of M-ODN or S-ODN training and testing time points. Quantitative Western blot analysis of hippocampal extracts obtained from trained rats that received the above treatments (experiment described in Fig. 3). Blots were stained with anti-C/EBPβ antibody, stripped, and restained with anti-NP62 antibody and finally with anti-actin, which was used for normalization. A representative blot per condition is shown. Graphs represent the statistical densitometric analysis of all data. Data are expressed as a mean percentage ± SEM of the S-ODN trained group (100%).
We next tested whether hippocampal MuSK knockdown also affects C/EBPβ expression and/or its learning-dependent induction (Fig. 4B). Groups of rats underwent either IA training or unpaired behavioral protocol followed by a double injection at 0 and 8 h of either M-ODN or S-ODN. Twenty-four hours after training or context exposure, the rats were killed and their hippocampi processed for quantitative Western blot analyses. A one-way ANOVA showed a significant group effect (F(3,12) = 3.6; p < 0.05). Similar to the effect found on pCREB, rats that received S-ODN injection and underwent IA training had a significant increase in hippocampal C/EBPβ (138.5 ± 5.1%; n = 4; p < 0.05) compared with unpaired S-ODN-injected rats (100.0 ± 6.9%; n = 4) (p < 0.05, Dunnett’s multiple-comparison test), confirming our previous findings that training results in a significant induction of this transcription factor (Taubenfeld et al., 2001b). This increase was completely blocked in trained rats that received M-ODN treatment (115.4 ± 10.1; n = 4). In the hippocampi of M-ODN-treated rats, C/EBPβ levels were similar to those of unpaired control rats injected with either S-ODN (100.0 ± 6.9%; n = 4) or M-ODN (113.9 ± 10.2%; n = 4). To confirm the selectivity of the M-ODN effect on C/EBPβ, the same membranes were stripped and stained for NP62. As shown in Figure 4B, the concentration of this protein remained unchanged across samples (S-ODN-unp, 100 ± 4.1%; S-ODN-train, 114.9 ± 13.0%; M-ODN-unp, 89.9 ± 22.3%; M-ODN-train, 107.8 ± 8.7%). In each experiment, C/EBPβ and NP62 levels were normalized against the relative concentration of actin, which was used as a loading control.
Moreover, in agreement with these results, we found that the C/EBPβ hippocampal levels of the rats that were behaviorally analyzed in the previous experiment (Fig. 3D) were also significantly and selectively reduced by the double M-ODN injection (69.6 ± 9.1%; n = 8) compared with the double S-ODN injection (100% ± 10.4%; n = 8; t test, p < 0.05) (Fig. 4C). In contrast, the levels of NP62, used as unrelated control, remained unaffected (M-ODN, 93.8 ± 7.6%; S-ODN, 100 ± 12.3%). Both C/EBPβ and NP62 levels were normalized against the relative concentration of actin, which was used as loading control (Fig. 4C).
Together the results described above indicate that MuSK expressed in the hippocampus plays a key role in the learning-dependent activation of the CREB/C/EBPβ pathway that mediates memory consolidation.
MuSK is critically involved in cholinergic oscillatory responses in the CA3–CA1 synapse
Given the role of hippocampal MuSK on IA, we next addressed a possible role of MuSK in the cholinergic modulation of hippocampal synaptic activity. Studies from several groups have shown that the cholinergic input to the hippocampus underlies physiological theta rhythm in vivo (for review, see Bland and Colom, 1993), which has been implicated in diverse behavioral and cognitive functions, including learning and memory (Winson, 1978; Buzsaki, 2002). Other studies have demonstrated that the cholinergic agonist carbachol produces a patterned oscillatory behavior in acute hippocampal slices that resembles theta rhythm (Bland et al., 1988; MacVicar and Tse, 1989; Williams and Kauer, 1997) and that such theta-like activity can enhance synaptic plasticity in a bidirectional manner (Huerta and Lisman, 1995, 1996). We therefore tested whether MuSK is involved in the modulation of patterned cholinergic activity in the hippocampus.
We used the knockdown approach used in the experiments outlined above and targeted the hippocampus of 3- to 4-week-old rats (n = 4). We confirmed, as described below, that MuSK was selectively downregulated by injection of M-ODN through cannulas that had been stereotaxically implanted 1 week earlier. We examined the effects of this treatment on the extracellularly recorded oscillatory activity induced by carbachol in area CA1 of acute hippocampal slices obtained from such animals. To control for possible effects of the injection itself, in each animal we injected one hippocampus with M-ODN and the other hippocampus with control S-ODN. We adopt here the terminology of Williams and Kauer (1997) and refer to single events in the extracellular record as “field events” and to groups of these events as “bursts.” As shown in Figure 5, A (left) and B, in the majority of slices (6 of 9) obtained from hippocampi injected with S-ODN, bath application of 50 μm carbachol generated periodic bursts that occurred at approximately theta frequency. These were similar, in terms of intraburst frequency (5–11 Hz), mean interburst interval (17.1 ± 2.7 s; n = 6), and mean burst duration (5.2 ± 0.7 s; n = 6), to those observed by Williams and Kauer (1997) in naive hippocampal slices. In contrast, in the majority of slices (11 of 12) obtained from hippocampi injected with M-ODN, such field events were not organized as bursts but rather as unitary events. In most of these slices (6 of 11), the activity was periodic (pacemaking) and occurred at frequencies generally ranging from 0.5 to 1 Hz (Fig. 5A, right). The remainder showed little or no activity. The difference between S-ODN- and M-ODN-treated slices in terms of their ability to produce periodic bursts in response to carbachol treatment (summarized in Fig. 5B) was statistically significant, as determined by Fisher’s exact test (p < 0.05). Western immunoblots performed on pooled homogenates of the slices used in these experiments showed that the levels of MuSK protein were decreased to 68.9% in slices that had received M-ODN, relative to matched S-ODN-injected controls.
Figure 5.

MuSK plays a critical role in cholinergic oscillatory activity and long-term potentiation in the CA3–CA1 synapse. A, Left, Carbachol perfusion (50 μm) induced periodic bursts (top) recorded extracellularly in area CA1 of slices from S-ODN-injected hippocampi. Representative experiment of six. Expanded traces (bottom) show that each burst consisted of a series of field events, occurring at ∼10 Hz. Right, Hippocampal M-ODN injection disrupted cholinergic field oscillatory responses in the CA3–CA1 synapse, resulting in regularly spaced unitary events (top). Representative experiment of six; expanded trace shown below. Calibration: top, 0.2 mV, 60 s; bottom, 0.2 mV, 1 s. B, Summary data from all carbachol experiments. The majority (66.7%) of S-ODN slices displayed periodic bursting activity (left), whereas only 8.3% of the M-ODN slices exhibited such activity. The difference is statistically significant (*p < 0.01). Of those M-ODN slices showing nonbursting periodic responses (right), 54.5% displayed pacemaking (PACE) activity, with the remainder showing little or no such activity. Carbachol elicited pacemaking activity in only 11.1% of the S-ODN slices (right). INACT, Inactivity. C, D, Hippocampal M-ODN injection did not affect basal synaptic function. C, Input/output curve of M-ODN-treated slices was indistinguishable from that obtained from S-ODN-treated slices. D, There was no statistical difference between the degree of paired-pulse facilitation (PPF) produced in S-ODN and M-ODN slices. Data are expressed as mean percentage ± SEM. E, Hippocampal M-ODN injection impaired long-term potentiation induced by two trains of HFS (delivered at t = 0) in area CA1. In slices from hippocampi injected with S-ODN, HFS induced stable LTP that persisted for at least 2 h (filled circles). However, injection with M-ODN impaired LTP as early as 5–15 min post-HFS (for fEPSP slope measured over the 5–15 post-HFS period and normalized to baseline, S-ODN vs M-ODN, p < 0.01). After this period, LTP continued to decay in the M-ODN-treated slices relative to controls. The dashed line represents the normalized baseline value of 100%. Inset traces show superimposed sample fEPSPs recorded during the baseline period and 2 h after HFS in S-ODN-treated slices (left traces) or M-ODN-treated slices (right traces). Error bars represent SE. Calibration: 0.5 mV, 10 ms. F, Treatment with M-ODN did not affect the field potential evoked by HFS. Left, Slices were stimulated with two trains of HFS in the presence of 50 μm d-APV, and a second pair of trains was delivered 30 min after APV washout. The field potentials recorded during the second train from each set are shown here. Note the additional field negativity that was observed in the absence of APV, representing the NMDAR-mediated component of the field potential. Right, The AUCs for the potentials obtained in the presence and absence of APV were determined, and the NMDAR-mediated component was calculated by subtracting the +APV AUC from the −APV AUC (total AUC). Treatment with M-ODN affected neither the total AUC nor the NMDAR-mediated component. Calibration: 1 mV, 200 ms.
MuSK plays a critical role in hippocampal LTP
To determine the possible involvement of MuSK in hippocampal synaptic plasticity, we examined the LTP induced in the CA3→CA1 synapses of acute hippocampal slices obtained from adult rats (n = 3) that had received the same M-ODN or S-ODN treatments as those involved in the carbachol experiments outlined above. Quantitative Western immunoblotting experiments performed on the pooled slices used in these experiments verified the downregulation of MuSK specifically in those slices that had received M-ODN (68.9% of matched S-ODN-injected controls). Basal synaptic function was not compromised by M-ODN, because the input/output relationship (Fig. 5C) (p > 0.5, n = 6 per group, two-way ANOVA) and degree of paired-pulse facilitation (Fig. 5D) (p > 0.5, n = 5 per group, two-tailed t test) were indistinguishable from slices that had been treated with S-ODN (Fig. 5C, input/output curves). In addition, the spike threshold was unaffected by M-ODN (2.2 ± 0.3 mV, n = 6 for S-ODN vs 2.2 ± 0.3 mV, n = 6 for M-ODN; p > 0.5).
We used an HFS protocol that is sufficient to produce a stable LTP that can be maintained for at least 2 h (Osten et al., 1996; Tsokas et al., 2005). As shown in Figure 5E, M-ODN-injected slices showed an impairment of LTP relative to slices injected with S-ODN, which displayed robust LTP that lasted for at least 2 h (two-way ANOVA of EPSP slope at 110–120 min post-HFS, p < 0.0001; n = 6 for both S-ODN and M-ODN). In fact, the M-ODN effect on LTP was statistically significant as early as 5–15 min post-HFS (two-way ANOVA of EPSP slope, p < 0.0001; n = 6 for both S-ODN and M-ODN), indicating that MuSK contributes to the induction of LTP. However, MuSK also seems to be required for the late phase of LTP, because the potentiation continued to decay in the M-ODN-treated slices relative to S-ODN controls for the duration of the recording period. Thus, between 15 and 120 min after HFS, the size of LTP in M-ODN-treated slices declined by 50% (from 62 to 31%) compared with a decline of only 18% in the S-ODN controls (from 89 to 73%).
It is known that, subsequent to induction, two phases of LTP can be resolved: an early phase that involves only posttranslational mechanisms and that decays within 1 h under our conditions (Tsokas et al., 2005) and a protein synthesis-dependent phase (late LTP) that begins as early as 15 min after HFS and that can persist for hours (Frey et al., 1988; Osten et al., 1996; Tsokas et al., 2005). Our experiments indicate that normal expression of MuSK is required for the induction of LTP, because the antisense effect was apparent during the first 5–15 min after HFS. In addition, MuSK seems to contribute to the late phase of LTP, because the potentiation continued to decay in the antisense-treated slices relative to scrambled ODN controls for the duration of the recording period.
To determine whether the effect of M-ODN on LTP could reflect a change in the activation of NMDA receptors (NMDARs), which are required for LTP at the CA3–CA1 synapse (Harris et al., 1984), we analyzed the field potentials evoked by HFS. Slices obtained from three independent rats that received either M-ODN (n = 7) or S-ODN (n = 6) injection into their hippocampi, as described above, underwent two-train stimulations in the presence of 50 μm d-amino-phosphonovaleric acid (APV) to block NMDARs. As expected, no LTP was observed under these conditions. We then washed out APV for 30 min and delivered a second set of HFS trains. The field potential evoked after washout exhibited a slowly developing negativity beyond that seen in the presence of APV, and this APV-sensitive potential was more apparent in the second train, as reported previously (Blitzer et al., 1990) (Fig. 5F, left). The APV-sensitive component of the HFS-evoked potential was measured by subtracting the area under the curve (AUC) in the presence of APV (second train) from the AUC after washout (second train). As shown in Figure 5F (right), M-ODN treatment had no effect on the total field potential evoked in the absence of APV nor on the APV-sensitive component of the potential. Thus, the ability of MuSK antisense to inhibit LTP cannot be explained by an effect on NMDAR-dependent transmission.
MuSK expression is regulated after IA training
To determine whether MuSK expression is regulated after IA training, we performed real-time PCR analyses to quantify the levels of MuSK mRNAs in the hippocampi of trained versus control rats using TaqMan nuclease fluorigenic quantitative PCR assay. cDNAs were transcribed from pooled hippocampal extracts obtained from groups of rats that either underwent IA training and were killed 20 h later (20 h+; n = 10) or from control groups that were exposed to the context without shock administration and killed either immediately thereafter (0 h−; n = 10) or 20 h later (20 h−; n = 10). The 20 h time point was chosen because previous work showed that the activation of CREB (CREB phosphorylation in Ser133) as well as the induction and requirement for C/EBPβ during IA consolidation lasts for >20 h after training (Taubenfeld et al., 1999, 2001a,b).
As shown in Figure 6A, statistical analysis of these amplifications based on Student’s t test that compared 20 h+ versus 0 h− and 20 h− versus 0 h− showed that MuSK mRNA significantly increased in the hippocampi of trained rats compared with controls by a factor of 1.55 ± 0.02-fold.
Figure 6.

MuSK and agrin expression is increased in rat hippocampus after IA training. A, TaqMan-PCR analysis of MuSK on hippocampal extracts taken from rats trained in IA at 0 h−, 20 h−, and 20 h+. The concentration of mRNA was normalized against that of GAPDH. Data are expressed as mean fold change ± SEM of the ratio 20 h+:0 h− versus 20 h−:0 h− (∗p < 0.05). B, Quantitative Western blot analysis of hippocampal extracts taken from 0 h−, unpaired (unp), and IA trained (20 h+) individual rats. A representative blot per condition is shown. Graphs represent the statistical densitometric analysis of all data. Data were normalized against actin and are expressed as mean percent ± SEM of the 0 h− control mean values (100%). C, Quantitative Northern blot analysis of agrin expressed in hippocampal RNA extracts taken from 0 h−, unpaired, and 20 h+ rats. A representative blot per condition is shown. Graphs represent the statistical densitometric analysis of all data. The same membranes were also hybridized with a cyclophilin probe, which was used for normalization. Data are expressed as mean percentage ± SEM of the 0 h− control mean values (100%). ∗∗p < 0.01; ∗p < 0.05. D, Quantitative Western blot analysis of hippocampal extracts taken from 0 h−, unpaired, and 20 h+ rats. A representative blot per condition is shown. Graphs represent the statistical densitometric analysis of all data. Data were normalized against GAPDH and are expressed as mean percentage ± SEM of the 0 h− control mean values (100%), ∗p < 0.05.
We next determined whether the expression of MuSK protein underwent parallel increase after training and, for this investigation, we tested MuSK concentrations in the hippocampi of individual rats. Controls included two groups, 0 h− and unpaired. Retention tests before the rats were killed confirmed that they did not form any IA memory (19.9 ± 2.9 s). This latency, in fact, was significantly lower compared with that of trained rats (453.7 ± 44.6 s, p < 0.001, ANOVA followed by Neuman–Keuls multiple-comparison test) but similar to that of the 0 h− group (4.9 ± 1.2 s). As depicted in Figure 6B, quantitative Western blot analyses showed that, in trained rats, there was a trend toward an increase in MuSK protein level (n = 12; 130.9 ± 16.1%), which, however, was not significant compared with both control groups (0 h−, n = 12, 100.0 ± 8.4%; 20 h unpaired, n = 8, 105.2 ± 12.3%). Notably, the trend was observed in all experiments.
Agrin is upregulated after IA training
In the next sets of experiments, we tested whether agrin expression changes in the hippocampus after IA training. First, we determined agrin mRNA concentration in the hippocampi of trained or control groups of rats. Controls consisted of 0 h− and unpaired groups (Fig. 6C). Quantitative Northern blot and statistical analyses, performed with a one-way ANOVA (F(2,12) = 11.14; p < 0.01) followed by Newman–Keuls post hoc test, revealed that trained rats had a significant increase in agrin mRNA (175.3 ± 13.2%; n = 7) compared with 0 h− (100 ± 3.8%; n = 4; p < 0.01) and unpaired controls (108.6 ± 15.0%; n = 4; p < 0.05).
In the second set of experiments, we used quantitative Western blot analyses to determine whether agrin protein levels also increased. As shown in Figure 6D, the hippocampi of trained animals exhibited a marked induction of agrin protein (258.5 ± 38.4%, n = 8) compared with 0 h− and unpaired control groups (100 ± 7.2% and 100.5 ± 17.5%, n = 4 per group, respectively). One-way ANOVA showed a significant group effect (F(2,13) = 6.26; p < 0.05) and Newman–Keuls post hoc comparison test revealed that agrin levels were significantly higher in the hippocampi of trained rats compared with both control groups (p < 0.05 for both).
Discussion
This study provides evidence that MuSK is expressed in the brain, particularly in neurons as well as in several non-neuronal tissues. Hippocampal MuSK in the adult rat plays an essential role during the consolidation of IA memory, because its disruption after training impairs both memory retention and the activation of the underlying CREB–C/EBP-dependent pathway. Hippocampal MuSK disruption also significantly blocks the carbachol-induced oscillatory activity as well as both induction and maintenance of LTP. We propose that MuSK expressed in the hippocampus mediates memory consolidation via the synchronization of theta frequencies and recruitment of the CREB–C/EBP-dependent pathway.
Interestingly, in agreement with the role of MuSK in memory, patients suffering from myasthenia gravis (MG) have been reported to have memory deficits (Bohbot et al., 1997). Because antibody responses against both nicotinic receptors and MuSK have been found in MG patients (Hoch et al., 2001), it is possible that MuSK alterations in their brain may contribute to their CNS functional impairments.
MuSK is expressed in the brain as well as in non-neuronal tissues
Reverse transcription-PCR amplifications, in situ hybridization, Western blot analyses, and antisense treatments, all provide evidence that MuSK is expressed in the brain and particularly in neurons. In agreement with previous reports (Fu et al., 1999; Ip et al., 2000), MuSK also appears to be expressed in other tissues including liver, lung, heart, and spleen.
As shown by in situ hybridizations, the widespread expression of MuSK is evident in numerous neuronal populations across the hippocampus, cortex, and cerebellum. This suggests a potential function of MuSK in the context of the cholinergic network, which is known to play an important role in neuropsychic activities, such as learning, memory, arousal, sleep, and movement. In depth characterization of the expression of MuSK in relationship to that of cholinergic or other types of receptors in the brain will help to understand similarities and differences of MuSK roles in muscle and CNS.
Why did previous work indicate that MuSK was expressed selectively in muscle? The term muscle-specific tyrosine kinase receptor was initially proposed by Valenzuela et al. (1995), who, using Northern blot analysis, detected this transcript only in early muscle development, in muscle fibers after denervation and in NMJ but not in a variety of other adult rat tissues, including brain. Similar results using Northern blot analysis were obtained in mouse by Ganju et al. (1995), who, in the same study, also showed that in situ hybridization detected the expression of MuSK (Nsk2) transcripts in a number of developing tissues, including the nervous system. Like Valenzuela et al. (1995) and Ganju et al. (1995), we also found that Northern blot analysis is not sufficiently sensitive to detect physiologic concentrations of MuSK in a variety of tissues (data not shown).
In agreement, a relatively low level of expression is also found by Western blot analysis in adult brain and muscle, compared with higher levels in both tissues during development. Notably, low levels of expression do not preclude that MuSK is functionally relevant. In fact, even in adult muscle, in which MuSK plays a critical role at the NMJ (Kong et al., 2004; Hesser et al., 2006), neither the mRNA nor the protein has yet been detected by Northern or Western blot analyses. Therefore, it is likewise possible that, despite low levels of expression, MuSK plays a significant function in several adult tissues and structures, including the brain.
When compared with the longest isoform cloned from muscle, both MuSK isoforms expressed in the brain exhibit a 9 aa deletion with alanine substitution in the ectodomain (A454); moreover, the shortest isoform also presents a deletion of the IgIII domain (ΔIgIII). The A454 isoform is identical to the isoforms described in mouse myotubes by Ganju et al. (1995), in rat by Hesser et al. (1999), and in human by Valenzuela et al. (1995). The A454 protein lacks a potential N-linked glycosylation site; however, no functional significance has yet been associated with this deletion. Both A454 and ΔIgIII deletions seem to originate from alternative splicing of the longer sequence and, in fact, the ΔIgIII has also been found previously in muscle and shown to mediate muscle AChRs clustering similarly to the long isoform (Hesser et al., 1999). A transcript bearing both deletions, to our knowledge, has not been described previously.
Our Western blot analysis of brain extracts shows the presence of two distinct bands. As these bands migrate very closely, it is unclear whether they represent both the A454 and A454ΔIgIII isoforms. The molecular weight difference between these two isoforms, calculated on the basis of the deleted sequence (95 aa), is 10.3 kDa. However, posttranslational modifications may change the migration of the corresponding band; thus, the two proteins may ultimately migrate in more proximity to one another. It is also possible that one of the two isoforms is more abundantly expressed compared with the other and that the two closely migrating bands are generated by the same transcript that, however, underwent different posttranslational modification. In agreement, our 2D gel studies suggest that MuSK expressed in the brain undergoes posttranslational modification different from those of MuSK expressed in muscle. The nature of these modifications and the potentially differential roles of MuSK in brain and muscle remain to be determined.
Interestingly, a recent finding indicates that α3Na+/K+-ATPase (α3NKA) is a neuronal receptor for agrin in the brain (Hilgenberg et al., 2006). Whether MuSK expressed in the brain, like MuSK expressed in muscle, is part of the agrin receptor complex remains to be determined. Moreover, even in muscle, despite many efforts, the direct physical interaction between MuSK and agrin has not yet been established. In fact, ectopic expression of muscle MuSK had previously suggested that an additional component called muscle-specific accessory component (MASC), which is necessary to mediate agrin activation of MuSK, must exist as coreceptor for agrin at the NMJ (Glass et al., 1996). In light of these results, we are tempted to speculate that α3NKA, which directly binds to agrin, may represent MASC or its brain equivalent.
MuSK and memory consolidation
The antisense-mediated hippocampal knockdown of MuSK disrupts memory consolidation. The effect of M-ODN on memory retention at 24 h was evident after one injection administered 8 h after training and was even more profound with two injections, one administered immediately after training and the second 8 h later. In contrast, one M-ODN injection administered 2 h before the retention test had no effect, indicating that M-ODN is neither toxic nor caused nonspecific effects on memory expression. The biochemical analyses, performed in parallel on the hippocampi of the same animals, demonstrated a selective and dose-dependent decrease in MuSK expression. Moreover, MuSK knockdown also prevented the learning-induced activation of the CREB–C/EBP pathway, indicating that the role of MuSK in the hippocampus during memory consolidation involves this molecular cascade.
Interestingly, previous work from our laboratory has shown that the hippocampal CREB–C/EBPβ activation necessary for the consolidation of IA memory requires an intact fornix (Taubenfeld et al., 1999, 2001b). Fornix inputs to the hippocampus include cholinergic projections. Hence, a working model that we propose is that, after learning, the cholinergic input to the hippocampus leads to the activation of hippocampal MuSK, perhaps through agrin. MuSK activation would then produce the induction of the CREB–C/EBPβ-dependent gene cascade and hence, consequently, the consolidation of memory. The activation of the CREB–C/EBPβ-dependent cascade, via MuSK, would in turn regulate the expression of MuSK and agrin, which, together with additional late gene expression changes, would be critical for maintaining MuSK function and/or restoring the homeostasis of the system. In line with this model, MuSK expression has been shown to be autoregulated (Moore et al., 2001), and putative C/EBPβ and CREB binding sites are present in the promoter region of the MuSK gene (A. Garcia-Osta, unpublished observation) (Kim et al., 2003).
Despite its essential role during IA memory consolidation, we observed a small level of MuSK increase at 20 h after training. This small change in expression does not preclude the fundamental role of MuSK in long-term synaptic plasticity and memory formation and could be attributable to several reasons: (1) the change in MuSK protein expression may occur only in a small number of cells and therefore be too diluted in the whole hippocampal extracts; (2) the 20 h posttraining time point might not be the time of highest induction of MuSK protein. Finally, (3) the increase of MuSK protein may indeed be very small because, as suggested above in our working model, it simply reflects a learning-induced turnover regulation process that maintains the homeostatic pool of MuSK protein.
The role of MuSK in oscillatory behavior and synaptic plasticity
In slices treated with MuSK antisense, basal synaptic function appeared normal: the input/output relationships, spike thresholds, and paired-pulse facilitation of these slices were all indistinguishable from controls. However, MuSK downregulation produced a clear deficit on two electrophysiological measures that are relevant to learning.
First, normal MuSK expression is required for carbachol-induced oscillations in area CA1 of the hippocampus slice. In the hippocampus of behaving rats, ongoing motor activity and certain types of sensory stimulation are associated with theta activity, a large-amplitude synchronized oscillation at a frequency of ∼8–12 Hz (Buzsaki, 2002). Theta activity has been linked to synaptic plasticity and learning; for example, LTP in area CA1 is facilitated when the conditioning stimulation is timed to coincide with a particular phase of the theta oscillation (Hyman et al., 2003). Furthermore, the rate of acquisition for some behaviors is accelerated when training is given during periods of relatively intense oscillations (Berry and Thompson, 1978), and interventions that eliminate theta activity, such as lesions of the medial septum and fornix, can impair performance on hippocampus-based tasks (Givens and Olton, 1990; Taubenfeld et al., 1999). The precise role of theta activity in memory processing remains speculative, but it has been proposed that the oscillations serve a timing function, which governs the associativity of stimuli by regulating neuronal excitability at a characteristic frequency (Hasselmo et al., 2002). In agreement, rhythmically synchronized theta activity increases between the lateral amygdala and the CA1 after fear conditioning (Pape and Stork, 2003; Seidenbecher et al., 2003). Moreover, theta rhythms coordinate hippocampal–prefrontal interactions in a spatial memory task (Jones and Wilson, 2005), suggesting that theta rhythm may participate to both encoding and representing information integrated at the network level.
Both MuSK antisense and nicotinic antagonists tend to disorganize carbachol-induced oscillations, converting the periodic bursts to single field events that occur at 0.5–1 Hz or, in the case of MuSK antisense, sometimes producing an almost complete suppression of field events. Although this similarity suggests that MuSK might modulate hippocampal nicotinic transmission through effects on receptor clustering, it is notable that MuSK plays a pleiotropic role in signaling. For example, activated MuSK recruits phosphotyrosine-binding proteins to regulate transcription via the Ras and Rac/CDC42 pathways (Strochlic et al., 2005). Moreover, the cytoplasmic PDZ [postsynaptic density-95 (PSD-95)/Discs large/zona occludens-1] domain of MuSK suggests the possibility of a direct interaction with PSD-95, a scaffolding protein that is critical for the organization and function of the glutamatergic synapses whose activation generates theta activity (El-Husseini et al., 2000; Schnell et al., 2002).
In addition, in vivo MuSK knockdown impaired both induction and maintenance of hippocampal LTP. These results are in line with the behavioral experiments and strengthen the conclusion that MuSK plays a role in long-term synaptic plasticity underlying memory formation. The critical role of MuSK during LTP induction is in agreement with our working model described above, which proposes that MuSK plays an important role that begins during learning, and is also supported by the observation that MuSK is required for the activation of the CREB–C/EBP-dependent cascade. Furthermore, in the context of our behavioral experiments in which MuSK disruption was targeted after acquisition, the effect of antisense treatment on the maintenance of LTP is also of particular interest. Indeed, these results confirm the behavioral data indicating that the integrity of the memory depends on the role of MuSK during consolidation. Thus, MuSK expression seems to be important during both the initial phase of induction as well as the consolidation process of memory.
In conclusion, based on the results shown in this study, we propose that hippocampal MuSK mediates learning-induced synchronization activity, which requires an intact fornix, and is likely mediated by cholinergic input. This rhythmic activity would stabilize synaptic plasticity through the activation of the CREB–C/EBPβ pathway. This pathway, which includes MuSK expression homeostatic regulation, will then lead to memory consolidation.
Footnotes
*P.T. and G.P. contributed equally to this work.
This work was supported by National Institute of Mental Health Grant R01 MH65635 (C.M.A.) and by the Hirschl Trust Foundation. A.G.-O. received a Fulbright postdoctoral fellowship. We thank Steven Burden (Skirball Institute of Biomolecular Medicine, New York University, New York, NY) for generously providing the rat MuSK-myc transfected HEK293 cell line and helpful discussions; Ambion Inc. (Austin, Texas), Nitin Puri, and Chris Echeverri for generously providing the siRNAs; Deanna Benson for teaching us the HNC technique; Khatuna Gagnidze, Maria Milekic, Sophie Tronel, Bridget Vicinski, Anne Rocher, and Jose Moron-Concepcion for technical help; Kelsey Martin (University of California, Los Angeles, CA) and Stephen Taubenfeld for their comments on this manuscript; and Reginald Miller and the Center for Comparative Medicine and Surgery facility of Mount Sinai for technical support.
References
- Alberini CM (1999). Genes to remember. Exp Biol 202:2887–2891. [DOI] [PubMed] [Google Scholar]
- Athos J, Impey S, Pineda VV, Chen X, Storm DR (2002). Hippocampal CRE-mediated gene expression is required for contextual memory formation. Nat Neurosci 5:1119–1120. [DOI] [PubMed] [Google Scholar]
- Bailey CH, Bartsch D, Kandel ER (1996). Toward a molecular definition of long-term memory storage. Proc Natl Acad Sci USA 93:13445–13452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernabeu R, Cammarota M, Izquierdo I, Medina JH (1997). Involvement of hippocampal AMPA glutamate receptor changes and the cAMP/protein kinase A/CREB-P signalling pathway in memory consolidation of an avoidance task in rats. Braz J Med Biol Res 30:961–965. [DOI] [PubMed] [Google Scholar]
- Berry SD, Thompson RF (1978). Prediction of learning rate from the hippocampal electroencephalogram. Science 200:1298–1300. [DOI] [PubMed] [Google Scholar]
- Bland BH, Colom LV (1993). Extrinsic and intrinsic properties underlying oscillation and synchrony in limbic cortex. Prog Neurobiol 41:157–208. [DOI] [PubMed] [Google Scholar]
- Bland BH, Colom LV, Konopacki J, Roth SH (1988). Intracellular records of carbachol-induced theta rhythm in hippocampal slices. Brain Res 447:364–368. [DOI] [PubMed] [Google Scholar]
- Blitzer RD, Gil O, Landau EM (1990). Long-term potentiation in rat hippocampus is inhibited by low concentrations of ethanol. Brain Res 537:203–208. [DOI] [PubMed] [Google Scholar]
- Bohbot VD, Jech R, Bures J, Nadel L, Ruzicka E (1997). Spatial and nonspatial memory involvement in myasthenia gravis. J Neurol 244:529–532. [DOI] [PubMed] [Google Scholar]
- Burden SJ (2002). Building the vertebrate neuromuscular synapse. J Neurobiol 53:501–511. [DOI] [PubMed] [Google Scholar]
- Buzsaki G (2002). Theta oscillations in the hippocampus. Neuron 33:325–340. [DOI] [PubMed] [Google Scholar]
- DeChiara TM, Bowen DC, Valenzuela DM, Simmons MV, Poueymirou WT, Thomas S, Kinetz E, Compton DL, Rojas E, Park JS, Smith C, DiStefano PS, Glass DJ, Burden SJ, Yancopoulos GD (1996). The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell 85:501–512. [DOI] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS (2000). PSD-95 involvement in maturation of excitatory synapses. Science 290:1364–1368. [PubMed] [Google Scholar]
- Ferreira A (1999). Abnormal synapse formation in agrin-depleted hippocampal neurons. J Cell Sci 112:4729–4738. [DOI] [PubMed] [Google Scholar]
- Finn AJ, Feng G, Pendergast AM (2003). Postsynaptic requirement for Abl kinases in assembly of the neuromuscular junction. Nat Neurosci 6:717–723. [DOI] [PubMed] [Google Scholar]
- Frey U, Krug M, Reymann KG, Matthies H (1988). Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res 452:57–65. [DOI] [PubMed] [Google Scholar]
- Fu AK, Smith FD, Zhou H, Chu AH, Tsim KW, Peng BH, Ip NY (1999). Xenopus muscle-specific kinase: molecular cloning and prominent expression in neural tissues during early embryonic development. Eur J Neurosci 11:373–382. [DOI] [PubMed] [Google Scholar]
- Ganju P, Walls EJ, Brennan J, Reith AD (1995). Cloning and developmental expression of Nsk2, a novel receptor tyrosine kinase implicated in skeletal myogenesis. Oncogene 11:281–290. [PubMed] [Google Scholar]
- Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, Merlie JP, Sanes JR (1996). Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell 85:525–535. [DOI] [PubMed] [Google Scholar]
- Geinisman Y, Berry RW, Disterhoft JF, Power JM, Van der Zee EA (2001). Associative learning elicits the formation of multiple-synapse boutons. J Neurosci 21:5568–5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Givens BS, Olton DS (1990). Cholinergic and GABAergic modulation of medial septal area: effect on working memory. Behav Neurosci 104:849–855. [DOI] [PubMed] [Google Scholar]
- Glass DJ, Bowen DC, Stitt TN, Radziejewski C, Bruno J, Ryan TE, Gies DR, Shah S, Mattsson K, Burden SJ, DiStefano PS, Valenzuela DM, DeChiara TM, Yancopoulos GD (1996). Agrin acts via a MuSK receptor complex. Cell 85:513–523. [DOI] [PubMed] [Google Scholar]
- Gold PE (2003). Acetylcholine modulation of neural systems involved in learning and memory. Neurobiol Learn Mem 80:194–210. [DOI] [PubMed] [Google Scholar]
- Goslin K, Banker G (1991). Rat hippocampal neurons in low density cultures. In: Culturing nerve cells (Banker G, Goslin K, eds) pp. 251–278. London: MIT.
- Guzowski JF, McGaugh JL (1997). Anti-sense oligodeoxynucleotide-mediated disruption of hippocampal CREB protein levels impairs memory of a spatial task. Proc Natl Acad Sci USA 94:2693–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris EW, Ganong AH, Cotman CW (1984). Long-term potentiation in the hippocampus involves activation of N-methyl-D-aspartate receptors. Brain Res 323:132–137. [DOI] [PubMed] [Google Scholar]
- Hasselmo ME, Bodelon C, Wyble BP (2002). A proposed function for hippocampal theta rhythm: separate phases of encoding and retrieval enhance reversal of prior learning. Neural Comput 14:793–817. [DOI] [PubMed] [Google Scholar]
- Herbst R, Burden SJ (2000). The juxtamembrane region of MuSK has a critical role in agrin-mediated signaling. EMBO J [Erratum (2000) 19:1167] 19:67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesser BA, Sander A, Witzemann V (1999). Identification and characterization of a novel splice variant of MuSK. FEBS Lett 442:133–137. [DOI] [PubMed] [Google Scholar]
- Hesser BA, Henschel O, Witzemann V (2006). Synapse disassembly and formation of new synapses in postnatal muscle upon conditional inactivation of MuSK. Mol Cell Neurosci 31:470–480. [DOI] [PubMed] [Google Scholar]
- Hilgenberg LG, Su H, Gu H, O’Dowd DK, Smith MA (2006). Alpha3Na+/K+-ATPase is a neuronal receptor for agrin. Cell 125:359–369. [DOI] [PubMed] [Google Scholar]
- Hoch W (2003). Molecular dissection of neuromuscular junction formation. Trends Neurosci 26:335–337. [DOI] [PubMed] [Google Scholar]
- Hoch W, Ferns M, Campanelli JT, Hall ZW, Scheller RH (1993). Developmental regulation of highly active alternatively spliced forms of agrin. Neuron 11:479–490. [DOI] [PubMed] [Google Scholar]
- Hoch W, McConville J, Helms S, Newsom-Davis J, Melms A, Vincent A (2001). Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med 7:365–368. [DOI] [PubMed] [Google Scholar]
- Hogg RC, Raggenbass M, Bertrand D (2003). Nicotinic acetylcholine receptors: from structure to brain function. Rev Physiol Biochem Pharmacol 147:1–4. [DOI] [PubMed] [Google Scholar]
- Huerta PT, Lisman JE (1995). Bidirectional synaptic plasticity induced by a single burst during cholinergic theta oscillation in CA1 in vitro. Neuron 15:1053–1063. [DOI] [PubMed] [Google Scholar]
- Huerta PT, Lisman JE (1996). Low-frequency stimulation at the troughs of theta-oscillation induces long-term depression of previously potentiated CA1 synapses. J Neurophysiol 75:877–884. [DOI] [PubMed] [Google Scholar]
- Hyman JM, Wyble BP, Goyal V, Rossi CA, Hasselmo ME (2003). Stimulation in hippocampal region CA1 in behaving rats yields long-term potentiation when delivered to the peak of theta and long-term depression when delivered to the trough. J Neurosci 23:11725–11731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip FC, Glass DG, Gies DR, Cheung J, Lai KO, Fu AK, Yancopoulos GD, Ip NY (2000). Cloning and characterization of muscle-specific kinase in chicken. Mol Cell Neurosci 16:661–673. [DOI] [PubMed] [Google Scholar]
- Izquierdo I, McGaugh JL (2000). Behavioural pharmacology and its contribution to the molecular basis of memory consolidation. Behav Pharmacol 11:517–534. [DOI] [PubMed] [Google Scholar]
- Jennings CG, Dyer SM, Burden SJ (1993). Muscle-specific trk-related receptor with a kringle domain defines a distinct class of receptor tyrosine kinase. Proc Natl Acad Sci USA 90:2895–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Bose CM, Lesuisse C, Qiu D, Huang JC, Zhang Q, Rupp F (1998). Specific agrin isoforms induce cAMP response element binding protein phosphorylation in hippocampal neurons. J Neurosci 18:9695–9702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MW, Wilson MA (2005). Theta rhythms coordinate hippocampal-prefrontal interactions in a spatial memory task. PLoS Biol 3:2187–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER (2001). The molecular biology of memory storage: a dialogue between genes and synapses. Science 294:1030–1038. [DOI] [PubMed] [Google Scholar]
- Kim CH, Xiong WC, Mei L (2003). Regulation of MuSK expression by a novel signaling pathway. J Biol Chem 278:38522–38527. [DOI] [PubMed] [Google Scholar]
- Kong XC, Barzaghi P, Ruegg MA (2004). Inhibition of synapse assembly in mammalian muscle in vivo by RNA interference. EMBO Rep 5:183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Ferns MJ, Meizel S (2006). Identification of agrinSN isoform and muscle-specific receptor tyrosine kinase (MuSK) [corrected] in sperm. Biochem Biophys Res Commun [Erratum (2006) 344:453] 342:522–528. [DOI] [PubMed] [Google Scholar]
- MacVicar BA, Tse FW (1989). Local neuronal circuitry underlying cholinergic rhythmical slow activity in CA3 area of rat hippocampal slices. J Physiol (Lond) 417:197–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore C, Leu M, Muller U, Brenner HR (2001). Induction of multiple signaling loops by MuSK during neuromuscular synapse formation. Proc Natl Acad Sci USA 98:14655–14660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell C, Gallagher HC, O’Malley A, Bourke M, Regan CM (2000). CREB phosphorylation coincides with transient synapse formation in the rat hippocampal dentate gyrus following avoidance learning. Neural Plast 7:279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osten P, Valsamis L, Harris A, Sacktor TC (1996). Protein synthesis-dependent formation of protein kinase Mζ in long-term potentiation. J Neurosci 16:2444–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape HC, Stork O (2003). Genes and mechanisms in the amygdala involved in the formation of fear memory. Ann NY Acad Sci 985:92–105. [DOI] [PubMed] [Google Scholar]
- Power AE, Roozendaal B, McGaugh JL (2000). Glucocorticoids enhancement of memory consolidation in the rat is blocked by muscarinic receptor antagonism in the basolateral amygdala. Eur J Neurosci 12:3481–3487. [DOI] [PubMed] [Google Scholar]
- Sandrock AW Jr, Dryer SE, Rosen KM, Gozani SN, Kramer R, Theill LE, Fischbach GD (1997). Maintenance of acetylcholine receptor number by neuregulins at the neuromuscular junction in vivo. Science 276:599–603. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW (1999). Development of the vertebrate neuromuscular junction. Annu Rev Neurosci 22:389–442. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Hicks RR, Baldwin SA, Robinson S, Brackney C (2005). Synaptogenesis in the hippocampal CA1 field following traumatic brain injury. J Neurotrauma 22:719–732. [DOI] [PubMed] [Google Scholar]
- Schnell E, Sizemore M, Karimzadegan S, Chen L, Bredt DS, Nicoll RA (2002). Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc Natl Acad Sci USA 99:13902–13907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidenbecher T, Laxmi TR, Stork O, Pape HC (2003). Amygdalar and hippocampal theta rhythm synchronization during fear memory retrieval. Science 301:846–850. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Kogan JH, Frankland PW, Kida S (1998). CREB and memory. Rev Neurosci 21:127–148. [DOI] [PubMed] [Google Scholar]
- Smith MA, Hilgenberg LG (2002). Agrin in the CNS: a protein in search of a function? Neuroreport 13:1485–1495. [DOI] [PubMed] [Google Scholar]
- Stone DM, Nikolics K (1995). Tissue- and age-specific expression patterns of alternatively spliced agrin mRNA transcripts in embryonic rat suggest novel developmental roles. J Neurosci 15:6767–6778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strochlic L, Cartaud A, Cartaud J (2005). The synaptic muscle-specific kinase (MuSK) complex: new partners, new functions. Bioessays 27:1129–1135. [DOI] [PubMed] [Google Scholar]
- Taubenfeld SM, Wiig KA, Bear MF, Alberini CM (1999). A molecular correlate of memory and amnesia in the hippocampus. Nat Neurosci 2:309–310. [DOI] [PubMed] [Google Scholar]
- Taubenfeld SM, Milekic M, Monti B, Alberini CM (2001a). The consolidation of new but not reactivated memory requires hippocampal C/EBPβ. Nat Neurosci 4:813–818. [DOI] [PubMed] [Google Scholar]
- Taubenfeld SM, Wiig KA, Monti B, Dolan B, Pollonini G, Alberini CM (2001b). Fornix-dependent induction of hippocampal CCAAT enhancer-binding protein β and δ co-localizes with phosphorylated cAMP response element binding protein and accompanies long-term memory consolidation. J Neurosci 21:84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsokas P, Grace EA, Chan P, Ma T, Sealfon SC, Iyengar R, Landau EM, Blitzer RD (2005). Local protein synthesis mediates a rapid increase in dendritic elongation factor 1A after induction of late long-term potentiation. J Neurosci 25:5833–5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela DM, Stitt TN, DiStefano PS, Rojas E, Mattsson K, Compton DL, Nunez L, Park JS, Stark JL, Gies DR (1995). Receptor tyrosine kinase specific for the skeletal muscle lineage: expression in embryonic muscle, at the neuromuscular junction, and after injury. Neuron 15:573–584. [DOI] [PubMed] [Google Scholar]
- Waites CL, Craig AM, Garner CC (2005). Mechanisms of vertebrate synaptogenesis. Annu Rev Neurosci 28:251–274. [DOI] [PubMed] [Google Scholar]
- Williams JH, Kauer JA (1997). Properties of carbachol-induced oscillatory activity in rat hippocampus. J Neurophysiol 78:2631–2640. [DOI] [PubMed] [Google Scholar]
- Winson J (1978). Loss of hippocampal theta rhythm results in spatial memory deficit in the rat. Science 201:160–163. [DOI] [PubMed] [Google Scholar]
- Yaffe D, Saxel O (1977). Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature 270:725–727. [DOI] [PubMed] [Google Scholar]
- Yin YC, Tully T (1996). CREB and the formation of long-term memory. Curr Opin Neurobiol 6:264–268. [DOI] [PubMed] [Google Scholar]