

Abstract
Brain function is usually perceived as being performed by neurons with the support of glial cells, the network of blood vessels situated nearby serving simply to provide nutrient and to dispose of metabolic waste. Revising this view, we find from experiments on a rodent central white matter tract (the optic nerve) in vitro that microvascular endothelial cells signal persistently to axons using nitric oxide (NO) derived from the endothelial NO synthase (eNOS). The endogenous NO acts to stimulate guanylyl cyclase-coupled NO receptors in the axons, leading to a raised cGMP level which then causes membrane depolarization, apparently by directly engaging hyperpolarization-activated cyclic nucleotide-gated ion channels. The tonic depolarization and associated endogenous NO-dependent cGMP generation was absent in optic nerves from mice lacking eNOS, although such nerves responded to exogenous NO, with raised cGMP generation in the axons and associated depolarization. In addition to the tonic activity, exposure of optic nerves to bradykinin, a classical stimulator of eNOS in endothelial cells, elicited reversible NO- and cGMP-dependent depolarization through activation of bradykinin B2 receptors, to which eNOS is physically complexed. No contribution of other NO synthase isoforms to either the action of bradykinin or the continuous ambient NO level could be detected. The results suggest that microvascular endothelial cells participate in signal processing in the brain and can do so by generating both tonic and phasic NO signals.
Keywords: endothelial cell, nitric oxide synthase, cGMP, HCN channels, optic nerve, bradykinin
Introduction
Thoughts about computation in the brain usually center on the synapse, in which discrete chemical signals transmit information across the 20 nm gap between presynaptic and postsynaptic neuronal elements. Astrocytes, situated close by, are increasingly regarded as intimate players in neuronal processing (Volterra and Meldolesi, 2005). Conceptually and anatomically remote from this picture are the cells making up blood vessels, the most abundant of which are endothelial cells forming the capillaries. Endothelial cells produce bioactive molecules, such as nitric oxide (NO) and arachidonic acid metabolites, but these are usually considered to function only in the context of blood vessels themselves, for example by promoting vasodilatation (Faraci, 1993). However, any point in the brain is, at most, only approximately a cell diameter (∼25 μm) away from a capillary (Pawlik et al., 1981), raising the possibility that such molecules could also access neurons or glial cells and affect their function. To do so, they would need to diffuse the distance in active concentrations, and the recipient cells would need to possess the appropriate receptors.
In principle, NO could perform this function. NO is synthesized from l-arginine by NO synthase enzymes, two isoforms of which are constitutively expressed: neuronal NO synthase (nNOS) and endothelial NO synthase (eNOS). nNOS is present in almost all brain areas, mainly in subsets of neurons, and is typically activated by NMDA receptor stimulation (Garthwaite and Boulton, 1995; Christopherson and Bredt, 1997). eNOS was reported also to reside in some brain neurons (Dinerman et al., 1994), but the evidence for this conclusion has been repudiated (Demas et al., 1999); instead, various approaches indicate that eNOS is just expressed in endothelial cells (Stanarius et al., 1997; Blackshaw et al., 2003; Chan et al., 2004). Theoretically, the physical properties of NO, in particular its capacity for rapid anisotropic diffusion, mean that it could signal between endothelial cells and cellular elements located within the brain parenchyma (Tsoukias and Popel, 2003). Furthermore, NO receptors, which possess intrinsic guanylyl cyclase (GC) activity leading to raised cellular cGMP, are widely expressed in central neurons and in some glial cells (Ding et al., 2004).
The multiple sources and targets of NO present in most brain tissue, coupled with a shortage of methods for distinguishing between them, make the idea of endothelial cells communicating with neurons or glial cells difficult to test. As shown here, an exception is the optic nerve, which is an anatomically simple stretch of central white matter containing the axons of retinal ganglion cells, glial cells, and blood vessels. Previously, we observed that optic nerve axons are enriched in GC-coupled NO receptors (Garthwaite et al., 1999) and that application of high NO concentrations caused a biphasic axonal depolarization (Garthwaite et al., 2002). The large delayed phase of the depolarization was caused by metabolic stress, but an earlier depolarization persisted when this pathological effect was inhibited. On pursuing the mechanism of this rapid depolarizing response, the present study progressed to test the hypothesis that NO generated by microvascular endothelial cells influences axonal function.
Materials and Methods
Special chemicals.
1-Benzyl-3-(hydroxymethyl-2-furyl)indazole (YC-1) was supplied by the Chemistry Department at Wolfson Institute for Biomedical Research (London, UK). (Z)-1-[N-(3-Ammoniopropyl)-N-(n-propyl)-amino]/NO (PAPA/NO), 5-cyclopropyl-2-{1-(2-fluoro-benzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl}pyrimidin-4-ylamine (BAY 41-2272), N-[(3-aminomethyl)benzyl]acetamidine (1400W), vinyl-l-N-5-(1-imino-3-butenyl)-l-ornithine (l-VNIO), and N-propyl-l-arginine (NPA) were from Alexis (Nottingham, UK). Cilostamide, milrinone, 8-bromo-cAMP (8-Br-cAMP), 8-bromo-cGMP (8-Br-cGMP), forskolin, l-arginine, and isobutylmethylxanthine (IBMX) were from Sigma (Poole, UK). 1H-[1,2,4]Oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), l-nitroarginine, l-methylarginine, and 4-ethylphenylamino-1,2-dimethyl-6-methylaminopyrimidinium chloride (ZD7288) were from Tocris Bioscience (Avonmouth, UK). 9-(Tetrahydro-2-furanyl)-9H-purin-6-amine (SQ 22536) was from Calbiochem/Merck Biosciences (Nottingham, UK).
Optic nerves.
These were dissected from 10- to 11-d-old rats or 14- to 15-d-old mice, using methods described previously (Garthwaite et al., 2002). The smaller diameters of the developing nerves compared with those of the adults used previously were advantageous in facilitating electrophysiological recording and wash-in and wash-out of exogenously applied compounds. The eNOS−/− mice (SV129 and C57BL/6 background, homozygotes bred in-house) were given to us by Dr. Adrian Hobbs (Wolfson Institute for Biomedical Research). The wild-type (WT) mice were SV129/C57BL/6 F1 hybrids (Harlan, Bicester, UK), matching the background of the knock-outs.
Electrophysiology.
Potential changes were recorded at 37°C using the grease-gap technique as described previously (Garthwaite et al., 2002). The nerves were perfused continuously with a solution composed of the following (mm): 120 NaCl, 2 KCl, 2 CaCl2, 26 NaHCO3, 1.18 KH2PO4, 1.19 MgSO4, and 11 glucose (equilibrated with 95% O2, 5% CO2). Response amplitudes, when reversible, were quantified relative to the interpolated baseline, obtained by fitting a linear regression to 5 min periods on either side. Otherwise, measurements were made relative to the mean voltage during a 5 min period of preceding baseline. For statistical analysis, measurements were made in the same way on random stretches of baseline of the same duration to account for possible drift: positive peaks averaged 0.045 ± 0.009 mV (n = 5) and negative ones averaged −0.060 ± 0.006 mV (n = 7), with the average of both in the same records being 0.002 ± 0.019 (n = 8). Statistical comparisons with test data were performed against the appropriate one of these baseline values. All measurements were performed using Clampfit 8.0 (Molecular Devices, Palo Alto, CA). To check the stability of the recordings, responses to PAPA/NO (usually 1 μm) and/or K+ (1–3 mm, added as KCl) were obtained at the beginning and end of each experiment. In the figures, applications of compounds were for the duration shown by the horizontal bars.
The theory and practice of the method have been analyzed in detail previously (Leppanen and Stys, 1997). The resting membrane potential of optic nerve axons in vitro is close to −80 mV (Stys et al., 1997). To estimate the true magnitude of the membrane potential changes occurring in our experiments, we conducted calibration experiments based on the technique used beforehand (Leppanen and Stys, 1997). In this, voltage responses to 1 μm PAPA/NO were recorded (mean value, 0.44 ± 0.13 mV; n = 4) and then 130 mm K+ was perfused to drive the membrane potential to near 0 (mean recorded depolarization at steady state, 18.9 ± 2.4 mV; n = 4). The ratio of the two responses (K+/PAPA/NO) was very similar in the different nerves, ranging from 39 to 46 (mean, 43 ± 1.7), implying that the actual depolarization produced by this concentration of PAPA/NO was close to 2 mV. The sizes of other voltage changes can be estimated from this value.
Measurement of cGMP and cAMP.
The methods were broadly as described previously (Garthwaite et al., 2002). Nerves were preincubated for at least 1 h (at 37°C) in the above solution. Test nerves were incubated with IBMX (1 mm) for 10 min before addition of PAPA/NO, BAY 41-2272, YC-1, or forskolin. When l-arginine or inhibitors were used, they were added 5 min (l-arginine), 10 min (ODQ and l-nitroarginine), or 20 min (SQ 22536) before IBMX. After inactivating the tissues, cGMP was measured by radioimmunoassay and cAMP was measured using the Biotrack assay kit (Amersham Biosciences, Little Chalfont, UK).
cGMP immunohistochemistry.
For rat optic nerves, cGMP immunohistochemistry was performed on tissue from the same experiments in which cGMP levels were measured by radioimmunoassay and which were treated identically except that, at the end of the experiment, they were fixed in ice-cold, freshly depolymerized paraformaldehyde (4%) in 0.1 m phosphate buffer, pH 7.4, for 2 h, and then processed for frozen (10 μm) or resin (1 μm) sectioning and cGMP immunostaining as described previously (Garthwaite et al., 1999). Counterstaining was performed using Mayer’s hemalum for 15 s (frozen sections) or 5 min (resin sections). The experiments on mouse optic nerves followed the same procedures for the frozen rat sections but were processed for fluorescent microscopy as described previously (Garthwaite et al., 2005) and additionally stained for neurofilament-200 (1:500; monoclonal from Chemicon, Temecula, CA).
Other immunohistochemistry.
For whole-mount preparations of rat and mouse optic nerves, the tissues were fixed with 1% paraformaldehyde for 1 h at room temperature, washed with 0.1 m phosphate buffer for at least 1 h with three changes, incubated for 10 min with Tris-buffered saline containing 0.1% Triton X-100 (TBST), pH 7.6, at room temperature, and then blocked for 1 h with 10% normal donkey serum in TBST at 4°C. The nerves were then incubated with a mixture of sheep anti-nNOS (1:20,000; a gift from Dr. P. C. Emson, The Babraham Institute, Babraham, UK) and mouse anti-eNOS (1:200; BD Transduction Laboratories, Lexington, KY) in TBST, for 30–48 h. To eliminate the possibility of cross-reactivity, the nNOS antibody was preabsorbed with a lysate of Sf21 cells expressing recombinant human eNOS (Charles et al., 1993) at 20× the protein concentration (provided by N. Foxwell and Prof. I. G. Charles, Wolfson Institute for Biomedical Research). The results on rat optic nerve were the same with two other polyclonal nNOS antibodies (rabbit anti-nNOS from Zymed, San Francisco, CA; rabbit anti-nNOS from Santa Cruz Biotechnology, Santa Cruz, CA), but, in the mouse, the Zymed antibody gave high nonspecific staining. After incubation with the primary antibodies, the nerves were washed extensively for 12 h with several changes of TBST and then incubated overnight at 4°C with secondary antibodies (fluorescein-labeled donkey anti-sheep and rhodamine-labeled donkey anti-mouse, 1:200 dilution; both from Chemicon, Harrow, UK). After washing (TBST for 6–7 h with three changes), the nerves were placed on slides and mounted in Vectashield mounting medium containing the nuclear stain 4′,6′-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA), taking care not to impose downward pressure on the coverslip. Wide Z images through entire the nerve thickness were captured using a confocal microscope (TCS-DMRE-7; Leica, Cambridge, UK). Controls (primary antibody omitted) showed no visible staining in the rat optic nerve, but there was some nonspecific staining in blood vessels in mouse tissue.
Confocal image processing and analysis.
The confocal images illustrated were assembled in Adobe Photoshop CS and Adobe Illustrator CS (Adobe Systems, San Jose, CA). When comparisons between different tissues are presented in the same figure, all images were adjusted for color, brightness, and contrast using identical settings. For quantitation of cGMP immunostaining in the mouse tissue, three longitudinal sections (10 μm thick, ∼100 μm apart) from each nerve were viewed using 20× objective, and the field (1.3–2.4 × 105 μm2 depending on the section width) beginning 100 μm away from the cut end was captured; identical photomultiplier tube sensitivity values were used for all sections from all nerves. Relative grayscale pixel intensities per area of unmanipulated red–green–blue color images were measured using a computer-assisted image analyzer (AxioVision, version 4.4; Zeiss, Welwyn Garden City, UK). The mean fluorescence values of the sections from each nerve were pooled after subtraction of the mean background value obtained from control sections to give a single n value.
Western blotting.
For NO synthase detection, published methods were followed (Charles et al., 1993). Tissues (optic nerve or cerebellum) were prepared by freezing on dry ice/isopentane and were stored at −80°C until use; they were then ground to powder at −70°C, suspended in homogenization buffer containing 20 mm HEPES, 1 mm EDTA, 0.2 m sucrose, 5 mm DTT, and protease inhibitor cocktail. The eNOS standard was a lysate of Sf21 cells expressing the protein (see above). For hyperpolarization-activated, cyclic nucleotide-gated (HCN) channel detection, membrane fractions from optic nerve and cerebellum were prepared as described previously (Muller et al., 2003). The NOS antibodies were as above (at the same dilutions), and, for HCN channels, rabbit anti-rat antibodies were used (HCN-1 at 1:500, HCN-2 at 1:600, HCN-3 at 1:600, and HCN-4 at 1:400; all from Alomone Labs, Jerusalem, Israel).
Statistics.
Data are presented as means ± SEM; significance was analyzed using Student’s t test or Dunnett’s test for multiple comparisons, as appropriate.
Results
Depolarizing action of exogenous and endogenous NO
Because direct recording from the thin, myelinated optic nerve axons is not feasible, we used a standard extracellular gap technique for monitoring membrane potential changes in isolated optic nerves (Stys et al., 1993; Garthwaite et al., 2002). The technique has the advantages of providing sensitive, stable, noninvasive recording over many hours, all of which were essential for the experiments. Exposure of rat isolated optic nerves to the NONOate PAPA/NO gave rise to concentration-dependent depolarizations, peaking after 1–3 min and reversing on washing out the donor (Fig. 1a). The maximally active PAPA/NO concentration was 3 μm, which is much lower than that evoking pathological depolarization of optic nerve axons (1 mm) (Garthwaite et al., 2002). Accordingly, the NO-evoked depolarization (1 μm PAPA/NO) was unaffected by the sodium channel blocker tetrodotoxin (0.40 ± 0.02 and 0.35 ± 0.03 mV in the absence and presence of 200 nm tetrodotoxin, respectively; n = 3), which inhibits the pathological NO response (Garthwaite et al., 2002). Calibration of the response to 1 μm PAPA/NO suggests a true depolarization of ∼2 mV (see Materials and Methods).
Figure 1.

Depolarizing action of exogenous and endogenous NO on rat optic nerve. a, Concentration–response curve for PAPA/NO-induced depolarization (filled circles; n = 3–20). Inhibition of the responses to 1 and 10 μm PAPA/NO by 3 μm ODQ are also illustrated (filled squares; n = 3–4). Insets are typical responses to 1 and 3 μm PAPA/NO applied for the duration shown by the bars. b, Hyperpolarizing effect of l-nitroarginine (L-NNA). c, Depolarizing effect of l-arginine (L-Arg). d, Inhibition of l-arginine-induced depolarization by l-nitroarginine. e, Repeatable hyperpolarizations on perfusion of l-methylarginine (L-MeArg). f, l-Arginine overcomes hyperpolarization by l-methylarginine. g, d-Arginine fails to affect l-methylarginine-induced hyperpolarization. h, Summary data for experiments illustrated in b–g and, for ODQ, in Figure 3b (n = 3–9; ∗p < 0.03 and ∗∗p < 0.0002 vs the baseline; +p < 0.02 vs l-methylarginine control). Calibration bars are all as in g.
NO synthase inhibitors were used to determine whether there was any endogenous NO acting similarly. l-Nitroarginine (100 μm) caused a hyperpolarization (Fig. 1b,d,h), as did another inhibitor, l-methylarginine (100 μm) (Fig. 1e–h), results that are consistent with loss of a tonic NO-mediated depolarization. Conversely, the NO synthase substrate l-arginine (100 μm) produced a sustained depolarization (Fig. 1c,h) that could be eliminated by l-nitroarginine (Fig. 1d,h), indicating that l-arginine produces its effect by enhancing NO synthase activity. l-Methylarginine, being a reversible NO synthase inhibitor, could evoke hyperpolarizations repeatedly (Fig. 1e), and these responses were reversed by l-arginine (against which the inhibitor competes) but not by its inactive enantiomer d-arginine (Fig. 1f–h). The amplitudes of the hyperpolarizations seen after inhibiting NO synthase relative to the maximum response to exogenous NO (Fig. 1a) suggest that NO influences the axonal membrane potential over a range of ∼6 mV (see Materials and Methods).
Sources of endogenous NO
The foregoing results indicate that there is an endogenous source of NO that tonically depolarizes optic nerve axons. Immunoblotting showed that both nNOS and eNOS were present in the optic nerve (Fig. 2c,d). By immunocytochemistry, the only detectable staining for nNOS was in larger blood vessels on the surface of the nerve (Fig. 2a,b), whereas eNOS was found in all the vascular elements, most notably in the network of capillaries permeating the tissue (Fig. 2a). No eNOS staining was present in glial cells or axons. In the larger superficial vessels, nNOS and eNOS were frequently colocalized in endothelial cells (Fig. 2b), conforming to observations made on pial blood vessels (Seidel et al., 1997). As before with normal rat optic nerves incubated in vitro (Garthwaite et al., 2005), no inducible NO synthase (iNOS) could be detected (data not illustrated).
Figure 2.
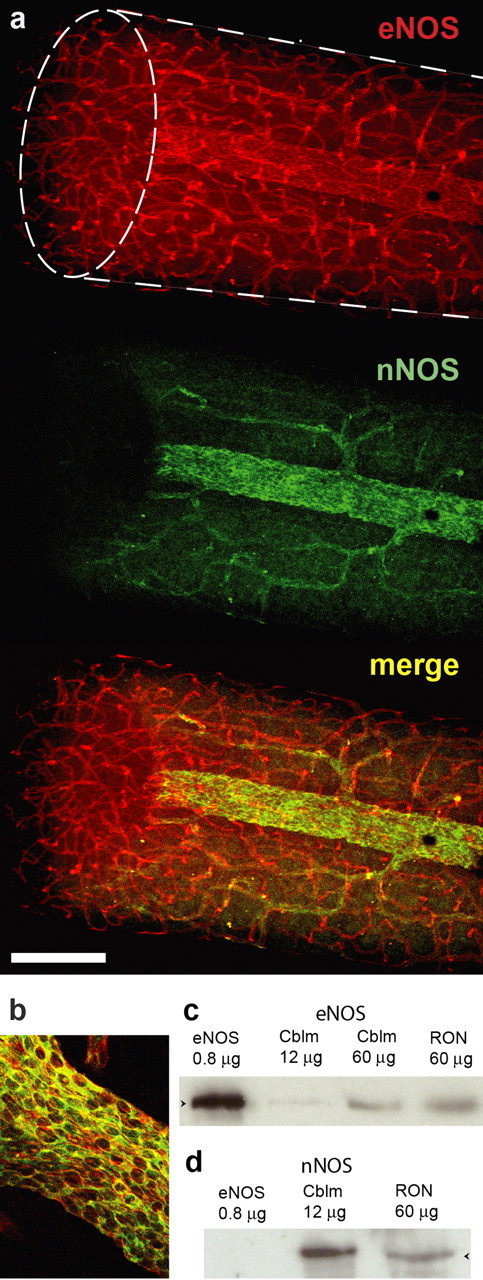
NO synthase isoform expression in rat optic nerve. a, Confocal microscopic images of eNOS (red) and nNOS (green) staining in whole-mount preparation, with the merged image shown underneath. The shape of the specimen is indicated with broken lines in the top image, the ellipse being the cut end; this shows that eNOS, but not nNOS, is located within the nerve. b, Merged image of eNOS (red) and nNOS (green) showing colocation in endothelial cells. The images (a, b) are representative of 12 nerves examined. c, d, Immunoblots for eNOS (c) and nNOS (d) in extracts of rat cerebellum (Cblm) and optic nerve (RON); the eNOS control (left lanes) was from lysates of Sf21 cells expressing recombinant human eNOS. Arrowheads in c and d correspond to molecular weights of 133 and 161 kDa, respectively. The blots are representative of five performed. Scale bar (in a): a, 200 μm; b, 70 μm.
Role of cGMP
ODQ (3–10 μm), the standard blocker of GC-coupled NO receptors (Garthwaite et al., 1995), caused a hyperpolarization (Figs. 1h, 3a,b), implying tonic NO receptor activity. In the presence of ODQ, the depolarization produced by PAPA/NO was inhibited (Figs. 1a, 3a), as was the hyperpolarizing effect of l-nitroarginine (Figs. 1h, 3b), suggesting that NO acts via GC-coupled receptors and subsequent cGMP formation. In support of this conclusion, the cGMP analog 8-Br-cGMP (0.1–1 mm) elicited concentration-dependent depolarizations (Fig. 3c) of a similar amplitude and time course to those produced by PAPA/NO (see below). As an additional test, we used the compounds BAY 41-2272 (1 μm) and YC-1 (60 μm), which, by reducing the rate at which NO dissociates from its receptors, potentiate NO-evoked GC activity (Friebe and Koesling, 1998; Koglin et al., 2002). Both compounds caused depolarizing responses that were slow to recover (Fig. 3d,e), particularly with YC-1. A slow recovery is expected from the mode of action of these compounds accentuated, in the case of YC-1, by concomitant phosphodiesterase inhibition (Galle et al., 1999). Depolarizations by the two agents were inhibited by 100 μm l-nitroarginine (Fig. 3d–f), indicating dependence on endogenous NO. ODQ was also inhibitory when tested against the response to YC-1 (Fig. 3f), signifying mediation by GC-coupled NO receptors.
Figure 3.

Role of cGMP in depolarizing effect of NO in rat optic nerve. a, Block of PAPA/NO-induced depolarization by ODQ. Summary data for this experiment are given in Figure 1a. b, ODQ produces a hyperpolarization and inhibits the hyperpolarizing action of l-nitroarginine (L-NNA; summary data for this experiment are given in Fig. 1h). c, Concentration-dependent depolarization by 8-Br-cAMP (squares; n = 3–4) and 8-Br-cGMP (circles; n = 3–5). d, e, Depolarizations by BAY 41-2272 (BAY) and YC-1 and their inhibition by l-nitroarginine (L-NNA). Calibration in d applies to all traces. f, Summary data for experiments shown in d and e (n = 3–6; ∗p < 0.02, ∗∗p < 0.01, †p < 0.03 by two-tailed t test compared with controls). g, Concentration–cGMP response curve for PAPA/NO in the absence (filled circles; n = 5–10) and presence (open circles; n = 3–5) of ODQ. h, l-Arginine, BAY 41-2272, and YC-1 enhance cGMP levels in the presence of the phosphodiesterase inhibitor IBMX and the inhibition by l-nitroarginine and ODQ (n = 3–6; ∗p < 0.05, ∗∗p < 0.01 vs IBMX control, Dunnett’s test; †p < 0.001 vs BAY 41-2272 or YC-1 controls, t test).
In parallel direct measurements performed in the presence of the nonspecific phosphodiesterase inhibitor IBMX, cGMP was elevated by l-arginine and reduced by l-nitroarginine or ODQ (Fig. 3h). In addition, PAPA/NO, at concentrations that depolarized the optic nerve, elevated cGMP levels and did so in an ODQ-sensitive manner (Fig. 3g). The EC50 of PAPA/NO for generating cGMP was shifted to the right (8 μm) compared with its EC50 for depolarization (0.6 μm). The sensitizers of GC-coupled NO receptors BAY 41-2272 and YC-1, at concentrations giving similar depolarization amplitudes to that induced by PAPA/NO (1 μm), gave similar cGMP responses (Fig. 3g,h), and these responses were inhibited by l-nitroarginine or ODQ (Fig. 3h). All of these results show that there is ongoing NO formation within the nerve and that endogenous and exogenous NO both act through GC-coupled receptors and subsequent cGMP accumulation to exert their depolarizing action.
cGMP immunocytochemistry provides a powerful method for locating the sites in which GC is active (de Vente et al., 1998). Basal cGMP in the optic nerve is too low to be detected with this technique, but agents such as BAY 41-2272 can be used to amplify the ongoing GC activity (attributable to endogenous NO) into the range at which the resulting cGMP can be visualized. In agreement with previous findings (Garthwaite et al., 1999), a maximally effective concentration of PAPA/NO (30 μm) produced intense cGMP immunostaining in axons but no staining of glial cells (Fig. 4a). The failure to observe glial cGMP staining in response to NO is not attributable to some technical problem because the method readily detects cGMP in optic nerve oligodendrocytes in response to agonists for membrane-bound GC-coupled atriopeptin receptors (G. Garthwaite, unpublished observation). In the presence of BAY 41-2272 (1 μm), robust cGMP staining was observed throughout the nerve, and, as with exogenous NO, the staining was localized to axons (Fig. 4b). The cGMP immunostaining caused by exposure to BAY 41-2272 was abolished by l-nitroarginine (Fig. 4c), confirming that it was attributable to endogenous NO. Quantitative analysis of cGMP in these experiments is given in Figure 3h.
Figure 4.
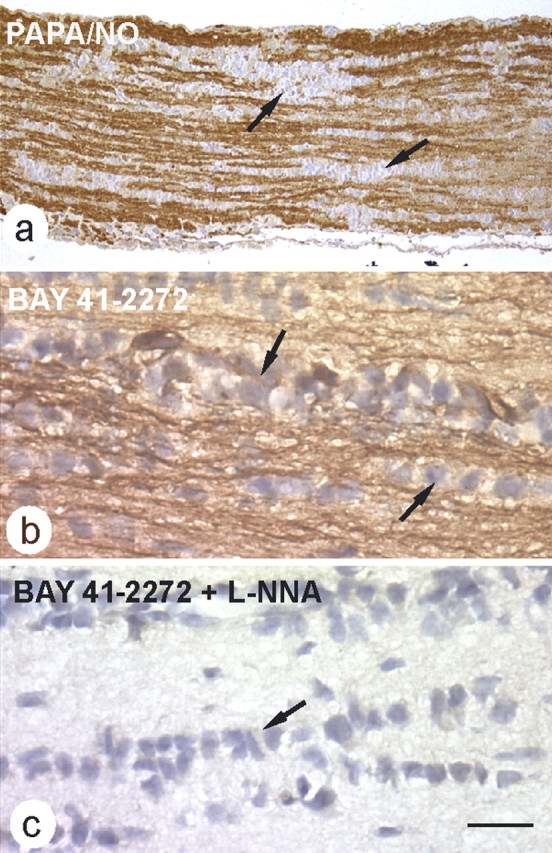
Immunostaining for cGMP in rat optic nerve. a, The nerve was exposed to 30 μm PAPA/NO (5 min) before being fixed and processed for cGMP immunocytochemistry, resulting in selective staining of axons (brown); glial cells were unstained. The image is representative of four nerves examined in this way (resin sections). b, c, cGMP immunocytochemistry on frozen sections of nerves exposed to BAY 41-2272 (1 μm) in the absence (b) and presence (c) of 100 μm l-nitroarginine (L-NNA) (n = 3 for each condition). Examples of rows of glial cells are indicated in a–c by arrows. Scale bar (in c): a, 81 μm; b, c, 13 μm.
Role of HCN channels
Among a number of possible cGMP transduction pathways engaged in axons, we chose to examine the role of HCN channels because they are present on optic nerve axons (Eng et al., 1990) and can be modulated by direct binding of cGMP or cAMP (Ludwig et al., 1998; Zagotta et al., 2003). HCN channels are activated at hyperpolarized membrane potentials and produce an inward (depolarizing) current; cyclic nucleotides shift the activation curve to the right by 5–25 mV depending on the channel composition, thereby generating depolarizing current at more positive voltages.
Immunoblotting showed that optic nerves express all four known HCN channel subunits (Fig. 5a), two of which (HCN-2 and HCN-4) confer high cyclic nucleotide-modulation of channel assemblies (Robinson and Siegelbaum, 2003). HCN channels can be blocked by Cs+ and by the organic inhibitor ZD7288. Cs+ (2–5 mm) was problematic in that it initially caused a hyperpolarization (0.60 ± 0.06 mV; n = 4), as expected for a block of HCN channels, but this rapidly reverted to a stable depolarization (0.8 ± 0.1 mV relative to baseline; n = 4), presumably attributable to block of potassium channels (Ghamari-Langroudi and Bourque, 2001). In concentrations selectively blocking HCN channels (Chevaleyre and Castillo, 2002), ZD7288 (1–10 μm) caused a sustained hyperpolarization and inhibited the depolarizing response to PAPA/NO or 8-Br-cGMP (Fig. 5b,e) to an extent similar to that seen in other preparations (Gasparini and DiFrancesco, 1997). The inhibition was unlikely to be attributable to the hyperpolarization itself because, as reported previously (Stys et al., 1993; Garthwaite et al., 2002), tetrodotoxin (1 μm) caused a hyperpolarization that was of similar amplitude (1.2 ± 0.05 vs 1.4 ± 0.08 mV with ZD7288; n = 4) but did not affect the response to PAPA/NO (see above). The cAMP analog 8-Br-cAMP (100–300 μm) depolarized the nerve similarly to the cGMP derivative (Fig. 5c) and was of similar potency (Fig. 3c). The adenylyl cyclase activator forskolin (100 nm) also generated a depolarization that was inhibited by ZD7288, as was the depolarization elicited by 8-Br-cAMP (Fig. 5c–e). These results all indicate that HCN channels sensitive to cyclic nucleotides are responsible for the NO-induced depolarization.
Figure 5.
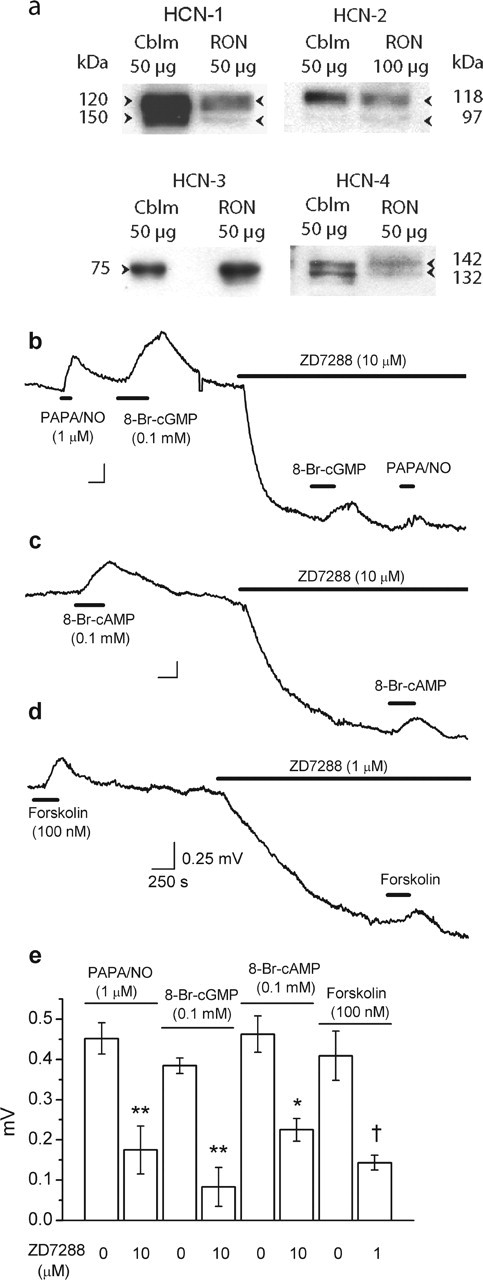
Role of HCN channels in NO-induced depolarization of rat optic nerve. a, Immunoblot for HCN channels (isoforms 1–4) in rat optic nerve (RON) compared with rat cerebellum (Cblm). Except for HCN-3, the blots show doublets of the indicated molecular weights, signifying glycosylated and nonglycosylated channel protein. Results are representative of two to three separate tissue preparations. b–d, ZD7288 causes a hyperpolarization and inhibits the depolarizing effect of PAPA/NO and 8-Br-cGMP (b), 8-Br-cAMP (c), and forskolin (d). Calibration in d applies to all traces. e, Summary data for the above experiments (n = 3–7; ∗p = 0.004, ∗∗p = 0.002, †p < 0.01 vs respective controls).
cAMP is often considered the preferred agonist for HCN channels (Ludwig et al., 1998; Zagotta et al., 2003). Consequently, measurements were performed to investigate possible crosstalk between cGMP and cAMP (Table 1). Forskolin (100 nm) raised optic nerve cAMP levels without affecting cGMP, and, conversely, PAPA/NO (1 or 10 μm) elevated cGMP without affecting cAMP. In addition, l-nitroarginine and ODQ reduced cGMP levels without changing the levels of cAMP. These results, which were obtained in the presence of the general phosphodiesterase inhibitor IBMX (1 mm), suggest that NO does not indirectly cause activation of adenylyl cyclase (or forskolin GC). To further investigate a possible role of adenylyl cyclase, we attempted to reduce its activity using the alleged inhibitor SQ 22536. At concentrations of 100 or 300 μm, however, SQ 22536 failed to reduce forskolin-evoked cAMP accumulation significantly, suggesting that it is of little use as an adenylyl cyclase inhibitor in the optic nerve. One way that cGMP could raise cAMP is by acting on phosphodiesterase-3, a cGMP-inhibited, cAMP phosphodiesterase. However, in contrast to the marked effect of IBMX, two different inhibitors of this enzyme (cilostamide and milrinone) did not affect forskolin-stimulated cAMP levels, indicating that inhibition of phosphodiesterase 3 (if present) is not capable of elevating cAMP levels in the optic nerve (Table 1). The results indicate that the levels of cGMP and cAMP vary independently.
Table 1.
cGMP and cAMP in rat optic nerve
| Treatment | cGMP (pmol/mg protein) | cAMP (pmol/mg protein) |
|---|---|---|
| Basal | 4.0 ± 0.5 (n = 6) | 6 ± 1 (n = 6) |
| Control; IBMX (1 mm) | 10 ± 0.5 (n = 13)* | 39 ± 2 (n = 17)* |
| l-NNA (100 μm) + IBMX | 5 ± 1 (n = 5)** | 45 ± 3 (n = 5) |
| ODQ (10 μm) + IBMX | 6 ± 1 (n = 7)** | 45 ± 2 (n = 5) |
| PAPA/NO (10 μm) + IBMX | 246 ± 37 (n = 9)*** | 44 ± 3 (n = 9) |
| PAPA/NO (1 μm) + IBMX | 91 ± 10 (n = 20)*** | 45 ± 3 (n = 20) |
| Forskolin (100 nm) + IBMX | 8 ± 2 (n = 4) | 100 ± 7 (n = 6)** |
| + SQ 22536 (100 μm) | 129 ± 4 (n = 3) | |
| + SQ 22536 (300 μm) | 74 ± 7 (n = 3) | |
| Forskolin (100 nm) | 10 ± 1 (n = 9) | |
| + IBMX (1 mm) | 108 ± 9 (n = 11)**** | |
| + cilostamide (100 μm) | 12 ± 4 (n = 3) | |
| + milrinone (10 μm) | 15 ± 6 (n = 3) |
*p < 0.001 versus basal;
**p < 0.003 and
***p < 0.001 versus IBMX controls;
****p < 0.001 versus forskolin control; all others are not significantly different from their respective controls.
Studies in eNOS knock-out mice
The results obtained using rat optic nerves suggest that eNOS in the microvasculature generates the tonic levels of NO that act on the axons, because this was the only source of NO found in the vicinity. However, a participation of nNOS, either in the surface blood vessel endothelium or below detection limits elsewhere, could not be excluded. To identify the active NO synthase isoform more rigorously, experiments were performed using mice with or without the eNOS gene. In optic nerves from WT mice, as in the rat, eNOS immunostaining was found only in endothelial cells, but, in contrast to the rat, no specific nNOS staining was apparent (Fig. 6a,b). This was not attributable to a species-related problem with the antibody because normal nNOS immunostaining was observed in mouse brain in the same experiments (result not illustrated). In the eNOS knock-out (eNOS−/−) mouse optic nerves, specific eNOS immunostaining was lost and nNOS staining remained undetectable (Fig. 6c,d), despite nNOS staining in eNOS−/− mouse brain being easily observed (result not illustrated). Just as in the rat, exposure to PAPA/NO (30 μm) caused cGMP to rise in the axons, and this response was similar in nerves from WT and eNOS−/− mice (Fig. 6e,g,n), showing preservation of the downstream NO signaling pathway regardless of the presence of eNOS. Importantly, however, cGMP accumulation in axons in response to BAY 41-2272 (which depends on endogenous NO) was present in WT mice but absent in eNOS−/− mice (Fig. 6f,h,n).
Figure 6.

Loss of tonic NO and of the associated depolarization in optic nerves from eNOS knock-out (eNOS−/−) mice. a–d, Confocal images of whole-mount preparations from WT (a, b) and eNOS knock-out (c, d) mice immunostained for eNOS (a, b) and nNOS (b, d). e–h, Optic nerves from wild-type (e, f) and eNOS knock-out (g, h) mice incubated for 5 min with 30 μm PAPA/NO (e, g) or BAY 41–2272 (f, h; 1 and 3 μm, respectively). Frozen sections were costained for neurofilament-200 (red), cGMP (green), and the nuclear stain DAPI (blue), and the images were merged. a–h are representative of three to five nerves examined in each condition. Scale bar (in d): a–d, 100 μm; e–h, 25 μm. i, k, Hyperpolarization by l-nitroarginine (L-NNA) in wild-type mouse optic nerves (i) and lack thereof in eNOS knock-outs (k). j, l, Depolarization by PAPA/NO and its inhibition by ZD7288 in wild-type (j) and eNOS knock-out (l) mouse optic nerve. Calibration in l applies to all traces. m, Summary data for the above experiments (i–l; n = 3–7; ∗p = 0.03 vs controls; +p = 0.001 vs baseline). n, Quantitation of cGMP immunostaining in wild-type and eNOS knock-out nerves in response to PAPA/NO (30 μm) and BAY 41-2272 (BAY; 1 μm in WT, 3 μm in eNOS knock-outs). ∗p = 0.0003 versus wild-type response (n = 3).
Corroborative findings were obtained in electrophysiological experiments. PAPA/NO (1 μm) elicited a depolarizing response from WT mouse optic nerves, and the HCN channel blocker ZD7288 caused a hyperpolarization and block of the response to PAPA/NO, as found in the rat (Fig. 6j,m). Optic nerves from eNOS−/− mice exhibited similar properties (Fig. 6l,m). The amplitude of the PAPA/NO response in the eNOS−/− tissue did not differ significantly from that in WT nerves (Fig. 6m), nor did the amplitude of the ZD7288-induced hyperpolarization (0.89 ± 0.14 and 0.84 ± 0.21 mV, respectively; n = 3), suggesting that the resting membrane potential in the mutants is not grossly different. Inhibition of endogenous NO synthesis with l-nitroarginine gave the usual hyperpolarization in WT optic nerves, although it had no effect in eNOS−/− optic nerves (Fig. 6k–m), signifying a lack of tonic NO.
Effect of bradykinin, an eNOS stimulator, in rat optic nerve
If NO derived from eNOS in endothelial cells signals tonically to axons, agonists that increase eNOS activity should elicit a depolarizing response through HCN channels and a corresponding increase in cGMP. The classical stimulator of endothelial cell NO production is bradykinin acting on B2 receptors, which exist in a complex with eNOS (Ju et al., 1998). As predicted, perfusion of bradykinin (300 nm) induced reproducible depolarizations peaking after 2–3 min and reversing on washing (Fig. 7). l-Nitroarginine (100 μm), ODQ (10 μm), the B2 antagonist HOE-140 (1 μm), and ZD7288 (10 μm) all inhibited the bradykinin-evoked depolarization (Fig. 7a–e), showing that bradykinin is acting through B2 receptors, NO synthase, cGMP formation, and HCN channels. In harmony with these findings, biochemical measurements showed that bradykinin increased the level of cGMP and that this response was inhibited by ODQ, l-nitroarginine, and HOE-140 (Fig. 7f). There was no effect of HOE-140 on the basal level of cGMP, although, as usual (Fig. 3h), it was reduced by l-nitroarginine (Fig. 7f). This, together with a lack of effect of HOE-140 on the membrane potential (Fig. 7c) indicates that bradykinin does not contribute to the tonic NO release in rat optic nerves, despite it being a good stimulator of NO generation therein.
Figure 7.

Effect of bradykinin and the roles of nNOS and iNOS in rat optic nerve. a–d, Inhibition of bradykinin (BK)-induced depolarization by l-nitroarginine (L-NNA), ODQ, HOE-140, and ZD7288. e, Summary data for experiments shown in a–d (n = 3–4; ∗p < 0.009, ∗∗p < 0.002, +p < 0.001, ++p < 0.00001 by two-tailed t test vs bradykinin controls). f, Bradykinin elevated cGMP, and this response was inhibited by l-nitroarginine, ODQ, and HOE-140 at the concentrations used in a–e (n = 4–5; ∗p < 0.01 compared with bradykinin control, +p < 0.01 compared with basal level by Dunnett’s test). All data were obtained in the presence of IBMX (1 mm). g, Representative trace showing that the nNOS inhibitor NPA did not cause a hyperpolarization (unlike l-nitroarginine) and did not affect the bradykinin-induced depolarization. h, Summary data for the effects of nNOS/iNOS inhibitors. None induced a significant hyperpolarization or affected the bradykinin-induced depolarization significantly, whereas l-nitroarginine retained its hyperpolarizing effect in their presence (n = 3–5; ∗p < 0.001 vs baseline).
Possible involvement of other NO synthase isoforms in rat optic nerve
The clear-cut results from WT and eNOS−/− mouse optic nerves, together with the more circumstantial evidence from the rat tissue, strongly point to eNOS being responsible for the tonic NO formation. For testing an involvement of nNOS specifically, nNOS−/− mice are problematic because of compensation by surviving enzymatically active splice variants (Eliasson et al., 1997). Instead, a pharmacological approach was adopted using rat optic nerves. In enzymatic studies, three NO synthase inhibitors, namely 1400W, l-VNIO, and NPA display good selectivity for nNOS over eNOS (Erdal et al., 2005). One of the compounds (1400W) also more potently blocks iNOS. In tissue studies performed in this laboratory, 1 μm 1400W, 0.1 μm l-VNIO, and 1 μm NPA all inhibited NMDA-stimulated cGMP accumulation in rat hippocampal slices by 80–90%, a response that is mediated by nNOS, but did not significantly affect acetylcholine-induced cGMP accumulation in strips of rat aorta, which is mediated by eNOS (Hopper, 2005). Being demonstrably selective in our hands, these concentrations were used in the present experiments. None of the inhibitors caused a significant hyperpolarization of the rat optic nerve, whereas the nonselective NO synthase inhibitor l-nitroarginine acted as normal in the same experiments (Fig. 7g,h). In addition, unlike l-nitroarginine (Fig. 7a,e), the selective nNOS/iNOS inhibitors failed to affect the depolarizing response to bradykinin (Fig. 7g,h). These results indicate that nNOS and iNOS do not contribute measurably to the tonic level of NO in the optic nerve nor to the elevation in NO brought about by bradykinin.
Discussion
We have provided evidence that microvascular endothelial cells can provide both tonic and phasic physiological signals to axons using NO derived from eNOS and that, in optic nerve, the message is transduced in the axons by cGMP and HCN channels, resulting in depolarization.
The majority of our studies used both electrophysiological recording to chart the NO-evoked changes in nerve function and direct measurement and localization of NO-evoked cGMP accumulation. The cGMP measurements constitute important tests of the role of this second messenger in NO signaling and also provide the most sensitive method available for monitoring changes in endogenous NO levels. All approaches gave coherent results. To summarize, the main evidence that eNOS generates the operative NO includes the presence and localization of the enzyme, the pharmacological properties of tonic NO release, the stimulation of NO release by bradykinin with the correct characteristics, and the absence of a detectable NO signal in eNOS knock-out mouse optic nerve. The latter result also argues strongly against a participation of other NO synthase isoforms, with additional evidence being their apparent absence (iNOS in rat optic nerve; nNOS in mouse optic nerve), unsuitable location (nNOS in rat optic nerve), and the lack of effect of inhibitors acting selectively on these isoforms. The only place in which eNOS was found was in blood vessels, which accords with the current view that eNOS mRNA and protein in the brain are confined to endothelial cells (Stanarius et al., 1997; Blackshaw et al., 2003; Chan et al., 2004).
It is well known from work on large blood vessels that eNOS in the vascular endothelium generates NO tonically and in concentrations sufficient to raise the level of cGMP in adjacent smooth muscle, causing relaxation (Martin et al., 1985; Gold et al., 1990). Although a Ca2+-dependent enzyme, eNOS is subject to additional regulation through multiple phosphorylation sites, the tonic activity being mainly attributable to phosphorylation by the protein kinase Akt, downstream of the phosphatidylinositol 3-kinase signaling pathway. Akt-mediated phosphorylation has the effect of stimulating eNOS activity 15- to 20-fold at resting levels of cytosolic Ca2+ (Fulton et al., 2001). By far, the majority of endothelial cells reside in capillaries, which also release active concentrations of NO tonically (Mitchell and Tyml, 1996). In preliminary experiments, we found that the tonic level of NO in rat optic nerve (using cGMP as a readout) is substantially reduced by inhibitors of phosphatidylinositol 3-kinase (wortmannin and LY294002 [2-(4-morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one)]), consistent with the established mechanism for tonic eNOS activity in endothelial cells also being the one functioning in optic nerve.
NO release from the geometrical arrangement of capillaries in a tissue is well suited to the generation of a global level of NO in the parenchyma (Tsoukias and Popel, 2003), in much the same way that capillaries provide the parenchyma with a relatively uniform concentration of oxygen. Of course, erythrocytes in the blood will inactivate endothelium-derived NO, but they do not do so sufficiently rapidly in vivo to prevent the NO acting on circulating leukocytes (Kubes et al., 1991) or on underlying vascular smooth muscle (Vallance et al., 1989) and so should not prevent NO accessing brain parenchyma in biologically active concentrations either.
Throughout the brain, nerve fibers are prominent sites of NO-evoked cGMP accumulation (de Vente et al., 1998). Various effects of NO on neurotransmitter release (Prast and Philippu, 2001) and other aspects of axon function (Ernst et al., 2000) have been described, but the underlying mechanisms remain essentially obscure. Our results provide evidence that endogenous NO can (via cGMP) influence the axonal membrane potential. It is already established that NO contributes to long-term changes in synaptic function in many different brain areas, but it is rare to observe acute electrical responses (Garthwaite and Boulton, 1995). However, no investigations of responses of axons appear to have been performed previously, except in relation to the pathological effects of high exogenous NO concentrations (Garthwaite et al., 2002).
Controlled biophysical investigation of the mechanism of action of the NO–cGMP pathway on ion channels in optic nerve axons cannot presently be performed because they are too small and are ensheathed with myelin. Although advantageous for the long-term, noninvasive recordings reported here, the electrophysiological technique we used, being extracellular, has clear limitations for investigating ionic mechanisms in depth. Although not central to the main conclusion (concerning endothelial cell–axon signaling), the results indicate that the NO-induced cGMP elevation in axons stimulates HCN channels to elicit membrane potential changes.
HCN channels are not generally regarded as targets for NO–cGMP signaling, mainly because cAMP appears to be ∼10-fold more potent an agonist for the channels than cGMP (∼1 vs ∼10 μm) (Ludwig et al., 1998; Zagotta et al., 2003). In several cell types, however, cGMP can rise to very high levels on exposure to NO, in some cases approaching 1 mm (Garthwaite, 2005), which is more than adequate to saturate the cyclic nucleotide binding site on HCN channels. This situation appears to apply here because the depolarizing effect of PAPA/NO was maximal at concentrations that produced less than half the maximal rise in cGMP (compare Figs. 1a, 3g). There are also precedents for NO and/or cGMP activating HCN channels in neurons (Pape and Mager, 1992; Ingram and Williams, 1996; Abudara et al., 2002; Pose et al., 2003; Kim et al., 2005), and the scope of NO for altering the membrane potential in optic nerve axons (estimated to be 6 mV) is very similar to the values reported in these previous studies. The similar depolarizing potencies of 8-Br-cAMP and 8-Br-cGMP in optic nerve, as also reported for sensory neurons (Ingram and Williams, 1996), indicates that the native channels may be relatively nondiscriminatory. Their molecular composition in optic nerve axons (as elsewhere) is not known, but HCN2 channels are present on the axons at the level of the retina (Muller et al., 2003).
Because HCN channels showing prominent cyclic nucleotide modulation (HCN-2 and HCN-4) are frequently expressed in axons in the brain (Notomi and Shigemoto, 2004), they may represent general targets for NO–cGMP signal transduction, leading to increased excitability and ability to conduct action potentials at high frequency (Robinson and Siegelbaum, 2003). The mechanism could help explain why block of NO synthase activity inhibits light-evoked compound action potentials in the optic nerve (Maynard et al., 1995), and it may have implications for the functioning of NO in synapses in which a small degree of presynaptic axon depolarization may have disproportionate effects on neurotransmitter release (Awatramani et al., 2005). In other locations, the NO–cGMP pathway could act on neuronal cell bodies or dendrites to affect their firing threshold or pattern (Pape and Mager, 1992; Abudara et al., 2002).
HCN channel activation is only one potential pathway for NO–cGMP signal transduction (Garthwaite and Boulton, 1995), and the more general conclusion from our results is that neuronal function may be acutely influenced by endothelial cells independently of their role in the vasculature. This is a relatively new concept in neurobiology, extending the intimate macromolecular interplay between axons and blood vessels taking place during development (Carmeliet and Tessier-Lavigne, 2005). It may also help explain some anomalous observations made in the past. For example, several studies concluded that eNOS is required for long-term potentiation of synaptic transmission in the hippocampus and elsewhere (Kantor et al., 1996; Son et al., 1996; Haul et al., 1999; Wilson et al., 1999). Although originally explained by eNOS being expressed in the pyramidal neurons (Dinerman et al., 1994), this has not been upheld by subsequent investigations by the same laboratory (Demas et al., 1999; Blackshaw et al., 2003) and others (Seidel et al., 1997; Stanarius et al., 1997; Topel et al., 1998). Access of biologically active concentrations of NO from eNOS in endothelial cells to neuronal structures allows the observations to be viewed in a new light. Specifically, endothelium-derived NO becomes a good candidate for maintaining the tonic level of NO needed for hippocampal long-term potentiation (Bon and Garthwaite, 2003) and, probably related to this, for sustaining the NMDA receptor-independent, but NO-dependent, level of cGMP found in this tissue under basal conditions (Chetkovich et al., 1993). Mice lacking eNOS exhibit various behavioral abnormalities (Demas et al., 1999; Frisch et al., 2000), altered neurotransmitter release or turnover (Kano et al., 1998; Frisch et al., 2000), and decreased neurogenesis (Reif et al., 2004; Chen et al., 2005). These observations may reflect alterations in vascular physiology or secondary genetic changes, but a deficiency in endothelial cell-to-neuron signaling mediated by NO now becomes plausible.
Finally, the results on the optic nerve show that the level of activity of the NO–cGMP pathway is such that the level of downstream signal transduction is poised approximately at the midpoint, so that increases and decreases in eNOS activity change cGMP levels and the membrane potential in opposite directions. The dynamic coupling of endothelial eNOS activity to alterations in neuronal function may help explain how various factors that change eNOS activity in vivo, such as physical exercise (Green et al., 2004) or estrogens (McNeill et al., 2002), alter brain activity.
Footnotes
The work was supported by The Wellcome Trust. We are grateful to Adrian Hobbs for providing the eNOS−/− mice, to Jan de Vente (Maastricht Univerity, Maastricht, The Netherlands) and Piers Emson (The Babraham Institute, Babraham, UK) for antibodies against cGMP and nNOS, respectively, to Neale Foxwell and Ian Charles for supplying lysates of eNOS-expressing Sf21 cells, and to Andrew Batchelor for help with the electrophysiology.
References
- Abudara V, Alvarez AF, Chase MH, Morales FR (2002). Nitric oxide as an anterograde neurotransmitter in the trigeminal motor pool. J Neurophysiol 88:497–506. [DOI] [PubMed] [Google Scholar]
- Awatramani GB, Price GD, Trussell LO (2005). Modulation of transmitter release by presynaptic resting potential and background calcium levels. Neuron 48:109–121. [DOI] [PubMed] [Google Scholar]
- Blackshaw S, Eliasson MJ, Sawa A, Watkins CC, Krug D, Gupta A, Arai T, Ferrante RJ, Snyder SH (2003). Species, strain and developmental variations in hippocampal neuronal and endothelial nitric oxide synthase clarify discrepancies in nitric oxide-dependent synaptic plasticity. Neuroscience 119:979–990. [DOI] [PubMed] [Google Scholar]
- Bon CL, Garthwaite J (2003). On the role of nitric oxide in hippocampal long-term potentiation. J Neurosci 23:1941–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, Tessier-Lavigne M (2005). Common mechanisms of nerve and blood vessel wiring. Nature 436:193–200. [DOI] [PubMed] [Google Scholar]
- Chan Y, Fish JE, D’Abreo C, Lin S, Robb GB, Teichert AM, Karantzoulis-Fegaras F, Keightley A, Steer BM, Marsden PA (2004). The cell-specific expression of endothelial nitric-oxide synthase: a role for DNA methylation. J Biol Chem 279:35087–35100. [DOI] [PubMed] [Google Scholar]
- Charles IG, Chubb A, Gill R, Clare J, Lowe PN, Holmes LS, Page M, Keeling JG, Moncada S, Riveros Moreno V (1993). Cloning and expression of a rat neuronal nitric oxide synthase coding sequence in a baculovirus/insect cell system. Biochem Biophys Res Commun 196:1481–1489. [DOI] [PubMed] [Google Scholar]
- Chen J, Zacharek A, Zhang C, Jiang H, Li Y, Roberts C, Lu M, Kapke A, Chopp M (2005). Endothelial nitric oxide synthase regulates brain-derived neurotrophic factor expression and neurogenesis after stroke in mice. J Neurosci 25:2366–2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetkovich DM, Klann E, Sweatt JD (1993). Nitric oxide synthase-independent long-term potentiation in area CA1 of hippocampus. NeuroReport 4:919–922. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE (2002). Assessing the role of Ih channels in synaptic transmission and mossy fiber LTP. Proc Natl Acad Sci USA 99:9538–9543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopherson KS, Bredt DS (1997). Nitric oxide in excitable tissues: physiological roles and disease. J Clin Invest 100:2424–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vente J, Hopkins DA, Markerink-Van Ittersum M, Emson PC, Schmidt HH, Steinbusch HW (1998). Distribution of nitric oxide synthase and nitric oxide-receptive, cyclic GMP-producing structures in the rat brain. Neuroscience 87:207–241. [DOI] [PubMed] [Google Scholar]
- Demas GE, Kriegsfeld LJ, Blackshaw S, Huang P, Gammie SC, Nelson RJ, Snyder SH (1999). Elimination of aggressive behavior in male mice lacking endothelial nitric oxide synthase. J Neurosci 19:RC30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinerman JL, Dawson TM, Schell MJ, Snowman A, Snyder SH (1994). Endothelial nitric oxide synthase localized to hippocampal pyramidal cells: implications for synaptic plasticity. Proc Natl Acad Sci USA 91:4214–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding JD, Burette A, Nedvetsky PI, Schmidt HH, Weinberg RJ (2004). Distribution of soluble guanylyl cyclase in the rat brain. J Comp Neurol 472:437–448. [DOI] [PubMed] [Google Scholar]
- Eliasson MJ, Blackshaw S, Schell MJ, Snyder SH (1997). Neuronal nitric oxide synthase alternatively spliced forms: prominent functional localizations in the brain. Proc Natl Acad Sci USA 94:3396–3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng DL, Gordon TR, Kocsis JD, Waxman SG (1990). Current-clamp analysis of a time-dependent rectification in rat optic nerve. J Physiol (Lond) 421:185–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdal EP, Litzinger EA, Seo J, Zhu Y, Ji H, Silverman RB (2005). Selective neuronal nitric oxide synthase inhibitors. Curr Top Med Chem 5:603–624. [DOI] [PubMed] [Google Scholar]
- Ernst AF, Gallo G, Letourneau PC, McLoon SC (2000). Stabilization of growing retinal axons by the combined signaling of nitric oxide and brain-derived neurotrophic factor. J Neurosci 20:1458–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraci FM (1993). Endothelium-derived vasoactive factors and regulation of the cerebral circulation. Neurosurgery 33:648–658. [DOI] [PubMed] [Google Scholar]
- Friebe A, Koesling D (1998). Mechanism of YC-1-induced activation of soluble guanylyl cyclase. Mol Pharmacol 53:123–127. [DOI] [PubMed] [Google Scholar]
- Frisch C, Dere E, Silva MA, Godecke A, Schrader J, Huston JP (2000). Superior water maze performance and increase in fear-related behavior in the endothelial nitric oxide synthase-deficient mouse together with monoamine changes in cerebellum and ventral striatum. J Neurosci 20:6694–6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulton D, Gratton JP, Sessa WC (2001). Post-translational control of endothelial nitric oxide synthase: why isn’t calcium/calmodulin enough? J Pharmacol Exp Ther 299:818–824. [PubMed] [Google Scholar]
- Galle J, Zabel U, Hubner U, Hatzelmann A, Wagner B, Wanner C, Schmidt HH (1999). Effects of the soluble guanylyl cyclase activator, YC-1, on vascular tone, cyclic GMP levels and phosphodiesterase activity. Br J Pharmacol 127:195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garthwaite G, Goodwin DA, Garthwaite J (1999). Nitric oxide stimulates cGMP formation in rat optic nerve axons, providing a specific marker of axon viability. Eur J Neurosci 11:4367–4372. [DOI] [PubMed] [Google Scholar]
- Garthwaite G, Goodwin DA, Batchelor AM, Leeming K, Garthwaite J (2002). Nitric oxide toxicity in CNS white matter: an in vitro study using rat optic nerve. Neuroscience 109:145–155. [DOI] [PubMed] [Google Scholar]
- Garthwaite G, Batchelor AM, Goodwin DA, Hewson AK, Leeming K, Ahmed Z, Cuzner ML, Garthwaite J (2005). Pathological implications of iNOS expression in central white matter: an ex vivo study of optic nerves from rats with experimental allergic encephalomyelitis. Eur J Neurosci 21:2127–2135. [DOI] [PubMed] [Google Scholar]
- Garthwaite J (2005). Dynamics of cellular NO-cGMP signaling. Front Biosci 10:1868–1880. [DOI] [PubMed] [Google Scholar]
- Garthwaite J, Boulton CL (1995). Nitric oxide signaling in the central nervous system. Annu Rev Physiol 57:683–706. [DOI] [PubMed] [Google Scholar]
- Garthwaite J, Southam E, Boulton CL, Nielsen EB, Schmidt K, Mayer B (1995). Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Mol Pharmacol 48:184–188. [PubMed] [Google Scholar]
- Gasparini S, DiFrancesco D (1997). Action of the hyperpolarization-activated current (Ih) blocker ZD 7288 in hippocampal CA1 neurons. Pflügers Arch 435:99–106. [DOI] [PubMed] [Google Scholar]
- Ghamari-Langroudi M, Bourque CW (2001). Ionic basis of the caesium-induced depolarisation in rat supraoptic nucleus neurones. J Physiol (Lond) 536:797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold ME, Wood KS, Byrns RE, Fukuto J, Ignarro LJ (1990). NG-methyl-l-arginine causes endothelium-dependent contraction and inhibition of cyclic GMP formation in artery and vein. Proc Natl Acad Sci USA 87:4430–4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DJ, Maiorana A, O’Driscoll G, Taylor R (2004). Effect of exercise training on endothelium-derived nitric oxide function in humans. J Physiol (Lond) 561:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haul S, Godecke A, Schrader J, Haas HL, Luhmann HJ (1999). Impairment of neocortical long-term potentiation in mice deficient of endothelial nitric oxide synthase. J Neurophysiol 81:494–497. [DOI] [PubMed] [Google Scholar]
- Hopper RA (2005). Nitric oxide and hippocampal synaptic plasticity. In: PhD thesis University of London.
- Ingram SL, Williams JT (1996). Modulation of the hyperpolarization-activated current (Ih) by cyclic nucleotides in guinea-pig primary afferent neurons. J Physiol (Lond) 492:97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju H, Venema VJ, Marrero MB, Venema RC (1998). Inhibitory interactions of the bradykinin B2 receptor with endothelial nitric-oxide synthase. J Biol Chem 273:24025–24029. [DOI] [PubMed] [Google Scholar]
- Kano T, Shimizu-Sasamata M, Huang PL, Moskowitz MA, Lo EH (1998). Effects of nitric oxide synthase gene knockout on neurotransmitter release in vivo. Neuroscience 86:695–699. [DOI] [PubMed] [Google Scholar]
- Kantor DB, Lanzrein M, Stary SJ, Sandoval GM, Smith WB, Sullivan BM, Davidson N, Schuman EM (1996). A role for endothelial NO synthase in LTP revealed by adenovirus-mediated inhibition and rescue. Science 274:1744–1748. [DOI] [PubMed] [Google Scholar]
- Kim HY, Kim SJ, Kim J, Oh SB, Cho H, Jung SJ (2005). Effect of nitric oxide on hyperpolarization-activated current in substantia gelatinosa neurons of rats. Biochem Biophys Res Commun 338:1648–1653. [DOI] [PubMed] [Google Scholar]
- Koglin M, Stasch JP, Behrends S (2002). BAY 41-2272 activates two isoforms of nitric oxide-sensitive guanylyl cyclase. Biochem Biophys Res Commun 292:1057–1062. [DOI] [PubMed] [Google Scholar]
- Kubes P, Suzuki M, Granger DN (1991). Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci USA 88:4651–4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppanen L, Stys PK (1997). Ion transport and membrane potential in CNS myelinated axons I. Normoxic conditions. J Neurophysiol 78:2086–2094. [DOI] [PubMed] [Google Scholar]
- Ludwig A, Zong X, Jeglitsch M, Hofmann F, Biel M (1998). A family of hyperpolarization-activated mammalian cation channels. Nature 393:587–591. [DOI] [PubMed] [Google Scholar]
- Martin W, Villani GM, Jothianandan D, Furchgott RF (1985). Selective blockade of endothelium-dependent and glyceryl trinitrate-induced relaxation by hemoglobin and by methylene blue in the rabbit aorta. J Pharmacol Exp Ther 232:708–716. [PubMed] [Google Scholar]
- Maynard KI, Yanez P, Ogilvy CS (1995). Nitric oxide modulates light-evoked compound action potentials in the intact rabbit retina. NeuroReport 6:850–852. [DOI] [PubMed] [Google Scholar]
- McNeill AM, Zhang C, Stanczyk FZ, Duckles SP, Krause DN (2002). Estrogen increases endothelial nitric oxide synthase via estrogen receptors in rat cerebral blood vessels: effect preserved after concurrent treatment with medroxyprogesterone acetate or progesterone. Stroke 33:1685–1691. [DOI] [PubMed] [Google Scholar]
- Mitchell D, Tyml K (1996). Nitric oxide release in rat skeletal muscle capillary. Am J Physiol 270:H1696–H1703. [DOI] [PubMed] [Google Scholar]
- Muller F, Scholten A, Ivanova E, Haverkamp S, Kremmer E, Kaupp UB (2003). HCN channels are expressed differentially in retinal bipolar cells and concentrated at synaptic terminals. Eur J Neurosci 17:2084–2096. [DOI] [PubMed] [Google Scholar]
- Notomi T, Shigemoto R (2004). Immunohistochemical localization of Ih channel subunits, HCN1–4, in the rat brain. J Comp Neurol 471:241–276. [DOI] [PubMed] [Google Scholar]
- Pape HC, Mager R (1992). Nitric oxide controls oscillatory activity in thalamocortical neurons. Neuron 9:441–448. [DOI] [PubMed] [Google Scholar]
- Pawlik G, Rackl A, Bing RJ (1981). Quantitative capillary topography and blood flow in the cerebral cortex of cats: an in vivo microscopic study. Brain Res 208:35–58. [DOI] [PubMed] [Google Scholar]
- Pose I, Sampogna S, Chase MH, Morales FR (2003). Mesencephalic trigeminal neurons are innervated by nitric oxide synthase-containing fibers and respond to nitric oxide. Brain Res 960:81–89. [DOI] [PubMed] [Google Scholar]
- Prast H, Philippu A (2001). Nitric oxide as modulator of neuronal function. Prog Neurobiol 64:51–68. [DOI] [PubMed] [Google Scholar]
- Reif A, Schmitt A, Fritzen S, Chourbaji S, Bartsch C, Urani A, Wycislo M, Mossner R, Sommer C, Gass P, Lesch KP (2004). Differential effect of endothelial nitric oxide synthase (NOS-III) on the regulation of adult neurogenesis and behaviour. Eur J Neurosci 20:885–895. [DOI] [PubMed] [Google Scholar]
- Robinson RB, Siegelbaum SA (2003). Hyperpolarization-activated cation currents: from molecules to physiological function. Annu Rev Physiol 65:453–480. [DOI] [PubMed] [Google Scholar]
- Seidel B, Stanarius A, Wolf G (1997). Differential expression of neuronal and endothelial nitric oxide synthase in blood vessels of the rat brain. Neurosci Lett 239:109–112. [DOI] [PubMed] [Google Scholar]
- Son H, Hawkins RD, Martin K, Kiebler M, Huang PL, Fishman MC, Kandel ER (1996). Long-term potentiation is reduced in mice that are doubly mutant in endothelial and neuronal nitric oxide synthase. Cell 87:1015–1023. [DOI] [PubMed] [Google Scholar]
- Stanarius A, Topel I, Schulz S, Noack H, Wolf G (1997). Immunocytochemistry of endothelial nitric oxide synthase in the rat brain: a light and electron microscopical study using the tyramide signal amplification technique. Acta Histochem 99:411–429. [DOI] [PubMed] [Google Scholar]
- Stys PK, Sontheimer H, Ransom BR, Waxman SG (1993). Noninactivating, tetrodotoxin-sensitive Na+ conductance in rat optic nerve axons. Proc Natl Acad Sci USA 90:6976–6980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stys PK, Lehning E, Saubermann AJ, LoPachin RM Jr (1997). Intracellular concentrations of major ions in rat myelinated axons and glia: calculations based on electron probe X-ray microanalyses. J Neurochem 68:1920–1928. [DOI] [PubMed] [Google Scholar]
- Topel I, Stanarius A, Wolf G (1998). Distribution of the endothelial constitutive nitric oxide synthase in the developing rat brain: an immunohistochemical study. Brain Res 788:43–48. [DOI] [PubMed] [Google Scholar]
- Tsoukias NM, Popel AS (2003). A model of nitric oxide capillary exchange. Microcirculation 10:479–495. [DOI] [PubMed] [Google Scholar]
- Vallance P, Collier J, Moncada S (1989). Nitric oxide synthesised from L-arginine mediates endothelium dependent dilatation in human veins in vivo. Cardiovasc Res 23:1053–1057. [DOI] [PubMed] [Google Scholar]
- Volterra A, Meldolesi J (2005). Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci 6:626–640. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Godecke A, Brown RE, Schrader J, Haas HL (1999). Mice deficient in endothelial nitric oxide synthase exhibit a selective deficit in hippocampal long-term potentiation. Neuroscience 90:1157–1165. [DOI] [PubMed] [Google Scholar]
- Zagotta WN, Olivier NB, Black KD, Young EC, Olson R, Gouaux E (2003). Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature 425:200–205. [DOI] [PubMed] [Google Scholar]