

Abstract
Organization and dynamic remodeling of postsynaptic density (PSD) are thought to be critical in postsynaptic signal transduction, but the underlying molecular mechanisms are not well understood. We show here that four major scaffolding molecules, PSD-95, GKAP, Shank, and PSD-Zip45, show distinct instability in total molecular content per synapse. Fluorescence recovery after photobleaching also confirmed their distinct turnover rates. Among the PSD molecules examined, PSD-95 was most stable, but its elimination did not influence the dynamics of its direct binding partner GKAP. Multiple interactions of scaffolding molecules with the actin cytoskeleton have suggested their importance in both maintenance and remodeling of the PSD. Indeed, acute pharmacological disruption of F-actin rapidly eliminated the dynamic fraction of GKAP, Shank, and PSD-Zip45, without changing synaptic localization of PSD-95. GKAP content in synapses increased after pharmacological enhancement of neuronal activity, whereas Shank and PSD-Zip45 content showed reduction. Inhibition of F-actin dynamics prevented activity-dependent redistribution of all three scaffolds. We also assessed involvement of glutamate receptors in the regulation of PSD dynamics. Genetic manipulations eliminating either NMDA receptors or metabotropic glutamate receptors did not primarily influence mobility of their binding scaffolds. These results collectively indicate a critical role of filamentous actin in determining the extent of dynamic reorganization in PSD molecular composition.
Keywords: postsynaptic density, actin cytoskeleton, synaptic plasticity, green fluorescent protein (GFP), hippocampus, time-lapse imaging
Introduction
Electron microscopic analysis of glutamatergic synapses revealed electron-dense thickening of postsynaptic membranes, which was termed postsynaptic density (PSD) (Palay, 1958). Biochemical analysis of PSD revealed presence of multiple membrane-anchored and cytoplasmic proteins containing a variety of domains for intermolecular association (Garner et al., 2000; Kennedy, 2000; Sheng and Sala, 2001). A majority of these PSD scaffolding proteins have an ability to interact either directly or indirectly with glutamate receptors. Changes in PSD scaffolding protein content may influence excitatory synaptic transmission through glutamate receptor redistribution (Stein et al., 2003; Ehrlich and Malinow, 2004). PSD scaffolding proteins also interact with multiple signaling molecules including protein kinases and phosphatases (Dalva et al., 2000; Gomez et al., 2002). Changes in the activity of these signaling molecules are directly coupled to modulation of signal transduction at synapses. Thus, it is likely that PSD contributes to local information processing at the glutamatergic synapses (Sheng and Kim, 2002).
Laminar organization of scaffolding proteins within the PSD (Valtschanoff and Weinberg, 2001; Petersen et al., 2003) and their distinct binding domains suggest the presence of multiple rules governing assembly/disassembly of different scaffolding proteins. To unravel this complexity, systematic analysis of individual scaffolding proteins with respect to their interaction with key organizers of the PSD, such as synaptic membranes and the actin cytoskeleton, will be informative. Among the PSD scaffolding proteins, PSD-95-family proteins [also known as membrane-associated guanylate kinases (MAGUKs)] stand out by their tight association with plasma membrane by palmitoylation (Craven et al., 1999; El-Husseini et al., 2002) and are thought to provide a framework beneath the plasma membrane with their direct binding with the NMDA receptors (NMDARs) (Kornau et al., 1995; Kim et al., 1997; Naisbitt et al., 1999; Sugiyama et al., 2005). In turn, maintenance of the PSD structure may also involve the actin cytoskeleton (Allison et al., 1998). Recent experiments indicate a variety of interactions between PSD scaffolding proteins and the actin cytoskeleton (Wyszynski et al., 1997; Bockers et al., 2001; Hering and Sheng, 2003). This indicates the possibility that intact actin meshwork is important in the maintenance and remodeling of the PSD composition. To test these possibilities, dynamics of multiple scaffolding proteins should be analyzed quantitatively, in combination with determination of their differential dependency on both the submembranous protein network and the actin cytoskeleton.
In this study, we analyzed dynamic properties of four scaffolding proteins with distinct domain organizations in living hippocampal neurons. Distinct kinetics of their turnover indicated the presence of multiple regulatory mechanisms. Although we found that glutamate receptors and PSD-95 play minor roles in the PSD maintenance per se, the actin cytoskeleton was essential in both maintenance and reorganization of the PSD. An important role of actin in structural plasticity of the dendritic spine has been proposed (Matus, 2000; Matsuzaki et al., 2004; Okamoto et al., 2004). Our study here indicates that the actin cytoskeleton also dynamically regulates the molecular organization of the PSD, and thereby integrates and modulates postsynaptic signal transduction.
Materials and Methods
Generation of recombinant adenoviruses and hippocampal cultures.
The generation and characterization of recombinant adenoviruses expressing enhanced green fluorescent protein (EGFP), PSD-95-EGFP, EGFP-PSD-Zip45, and EGFP-actin were described previously (Okabe et al., 1999, 2001b; Furuyashiki et al., 2002). GKAP N-terminally labeled with enhanced yellow fluorescent protein (EYFP), GKAP L694A mutant N-terminally labeled with EYFP, Shank2 N-terminally labeled with EGFP, and PSD-Zip45 C-terminally labeled with ECFP were constructed by inserting full-length cDNA (Kawashima et al., 1997; Usui et al., 2003) into pEYFP-C2, pEGFP-C2, and pECFP-N1 (Clontech, Mountain View, CA). Replication-deficient adenoviruses and hippocampal cultures from 17-d-old embryonic mice were prepared as described previously (Okabe et al., 1999).
Microinjection and chemical transfection.
Microinjection of expression plasmids for EGFP-tagged Shank and Shank-C (a deletion construct lacking the N-terminal half of the full-length Shank cDNA) was performed as described previously (Usui et al., 2003). Injected neurons were allowed to express proteins for 2 d before imaging experiments. Transfection of expression plasmids for EGFP-tagged PSD-Zip45 and PSD-Zip45 G89A [a point mutation in the EVH1 domain that abolishes interaction with the proline-rich domain of binding partners including Shank and type 1 metabotropic glutamate receptors (mGluRs)] was performed by using Effectene transfection reagent (Qiagen, Hilden, Germany) at 4 d after plating.
Immunocytochemistry and immunoblotting.
Cells were fixed in 2% paraformaldehyde in PBS for 25 min at room temperature or with methanol for 10 min at −20°C, blocked with 5% NGS, and incubated with mouse monoclonal anti-PSD-95 antibody (Affinity BioReagents, Golden, CO), mouse monoclonal anti-pan MAGUK proteins (Affinity BioReagents) (Sugiyama et al., 2005), mouse monoclonal anti-PSD-Zip45 antibody (Tadokoro et al., 1999), rabbit polyclonal anti-GKAP antibody (Usui et al., 2003), rabbit polyclonal anti-Shank antibody (Usui et al., 2003), rabbit polyclonal anti-NR1 antibody (Chemicon, Temecula, CA), rabbit polyclonal anti-NR2A antibody (Watanabe et al., 1998), rabbit polyclonal anti-NR2B antibody (Watanabe et al., 1998), rabbit polyclonal anti-synapsin I antibody (Chemicon), guinea pig polyclonal anti-vesicular glutamate transporter 1 antibody (Chemicon), and mouse monoclonal anti-SAP97 (synapse-associated protein with a molecular weight of 97 kDa) antibody (Nventa Biopharmaceuticals, Victoria, British Columbia, Canada). Primary antibodies were visualized with goat anti-mouse, anti-rabbit, or anti-guinea pig IgG conjugated to Cy3 (Jackson ImmunoResearch, West Grove, PA) or Alexa (Invitrogen, Carlsbad, CA). Rhodamine–phalloidin (Invitrogen) was applied to the cells fixed in 2% paraformaldehyde in PBS for 25 min and permeabilized in 0.1% Triton X-100 in PBS for 5 min. After 15 min of incubation, cells were rinsed with PBS and subsequent immunoreactions were performed. Immunoblotting was performed using rabbit polyclonal anti-PSD-95, rabbit polyclonal anti-GKAP, rabbit polyclonal anti-Shank, rat polyclonal anti-Homer (Chemicon), mouse monoclonal anti-α-tubulin (Seikagaku Corporation, Tokyo, Japan), and rabbit polyclonal anti-GFP (Invitrogen) antibodies. Primary hippocampal neurons were extracted with either NP-40 buffer (1% NP-40, 150 mm NaCl, 20 mm Tris-HCl, pH 8.0, and protease inhibitors) or radioimmunoprecipitation assay buffer (1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 150 mm NaCl, 50 mm Tris-HCl, pH 8.0, and protease inhibitors), and immunoprecipitation was performed by using protein-G-conjugated agarose (Santa Cruz Biotechnology, Santa Cruz, CA).
Pharmacological manipulation and electrophysiology.
Live cells were placed in a chamber containing Tyrode’s solution (in mm: 119 NaCl, 2.5 KCl, 2 Ca2+, 2 Mg2+, 25 HEPES, pH 7.4, and 30 glucose) for 30 min before the experiment. The chamber was maintained at 37°C by setting the temperature of the microscope stage at 37°C, the microscope objective lens at 39°C, and the chamber lid at 40°C. We applied a concentrated stock of latrunculin A (Invitrogen) or jasplakinolide (Invitrogen) in prewarmed Tyrode’s solution to the chamber. The final concentration of latrunculin A and jasplakinolide was 5 μm. Application of latrunculin A at a lower temperature (<35°C) was less effective in rapid disassembly of F-actin. The effect of latrunculin A was also reduced in the medium containing FBS. The concentrated stock of bicuculline (Sigma-RBI, St. Louis, MO) and 4-aminopyridine (4-AP; Sigma, St. Louis, MO) in prewarmed Tyrode’s solution was applied to the microscope chamber at the final concentrations of 50 and 500 μm, respectively. Whole-cell patch-clamp recordings from cultured hippocampal neurons were performed as described previously (Okabe et al., 1999). The concentrated stock of 2-bromopalmitate (50 mm in DMSO; Aldrich, Milwaukee, WI) was applied to culture dishes at the final concentration of 50 μm. The cells were assayed 12 h after application of 2-bromopalmitate. AMPA receptor blocker 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; Tocris, Ellisville, MO) and NMDAR blocker 2-amino-5-phosphonovaleric acid (APV; Tocris) were added at the concentrations of 10 and 100 μm, respectively.
RNA interference.
SAP97 and PSD-95 small interfering RNA (siRNA) sequences were as described previously (Nakagawa et al., 2004). We introduced a single nucleotide substitution in PSD-95 that reflects sequence divergence between mouse and rat. The oligonucleotides were subcloned into the pSilencer siRNA expression vector (Ambion, Austin, TX) and transfected by Effectene transfection reagent at 4 d after plating.
Microscopy.
Live cells were mounted in a chamber at 37°C with a continuous flow of humidified 5% CO2 to maintain the pH of the medium for long time-lapse experiments. Alternatively, culture medium was replaced with Tyrode’s solution, and live cells were mounted in a chamber at 37°C without a continuous gas flow. Images were obtained on a Fluoview confocal laser-scanning microscope (Olympus, Tokyo, Japan). A 60× oil-immersion lens (numerical aperture, 1.1) was used, and images were collected at an additional electric zoom factor from 3× to 6×. For long time-lapse imaging, multiple optical sections (z-spacing of 0.3–0.4 μm) were collected, and these images were recombined using a maximum-brightness operation. To improve efficiency of time-lapse observation, multiple neurons (three to five cells per experiments) on a coverslip 100–500 μm separated with each other were imaged with intervals of 10–20 min automatically by using a macro program running on Fluoview software that regulates movements of both the z-axis step controller and x/y-axis stage scanner. Fluorescence recovery after photobleaching (FRAP) experiments were performed using a macro program to control sequential image acquisition and delivery of a photobleaching laser pulse to the region of interest defined by acousto-optic tunable filters.
Image analysis.
Image analysis was performed using MetaMorph software (Universal Imaging Corporation, Downingtown, PA) as described previously (Ebihara et al., 2003). Twenty to fifty clusters were measured for each time-lapse sequence, and four or five independent time-lapse sequences were analyzed at specific developmental stages. The number of fluorescent clusters and the number of time-lapse sequences are as follows: 405 clusters from 16 time-lapse sequences for PSD-95; 390 clusters from 17 time-lapse sequences for GKAP; 365 clusters from 16 time-lapse sequences for Shank; 425 clusters from 16 time-lapse sequences for PSD-Zip45. We used a two-compartment model of protein diffusion for fast FRAP analysis. The difference of EGFP concentration between the dendritic shaft (C0) and the spine [C(t)] is proportional to the movement of EGFP molecules through the spine neck. This relationship can be written as follows:
where Q is the amount of EGFP in the spine head, k is the diffusion coefficient of EGFP, l is the spine neck length, and A is the cross-sectional area of the spine neck. From this equation we obtain the following:
where V is the volume of the spine head. This equation indicates that the recovery of fluorescence follows a single exponential curve and k is inversely proportional to the time constant.
Results
PSD proteins with distinct domain organizations reveal different cluster dynamics at synapses
To obtain a broad view on dynamic properties of PSD scaffolding proteins, we generated four fluorescent probes: EGFP-tagged PSD-95, EGFP-tagged PSD-Zip45 (Homer1c), EYFP-tagged GKAP (Kim et al., 1997), and EGFP-tagged Shank2 (Shank2 is also known as ProSAP1 and cortactin-binding protein 1) (Du et al., 1998; Boeckers et al., 1999; Naisbitt et al., 1999). PSD-95 is a major scaffolding protein interacting with NMDAR subunit NR2. PSD-Zip45 is a direct binding partner of type 1 mGluRs. GKAP can bind to both PSD-95- and Shank-family proteins. Identification of the interaction between Shank-family proteins and Homer-family proteins suggested that the PSD-95/GKAP/Shank/Homer assembly functions in linking NMDAR signal complex with mGluRs (Naisbitt et al., 1999; Tu et al., 1999; Sugiyama et al., 2005). Four EGFP/EYFP fusion proteins expressed in primary hippocampal neurons by recombinant adenoviruses formed clusters in dendritic spines, and these clusters were juxtaposed with presynaptic components (Fig. 1A).
Figure 1.
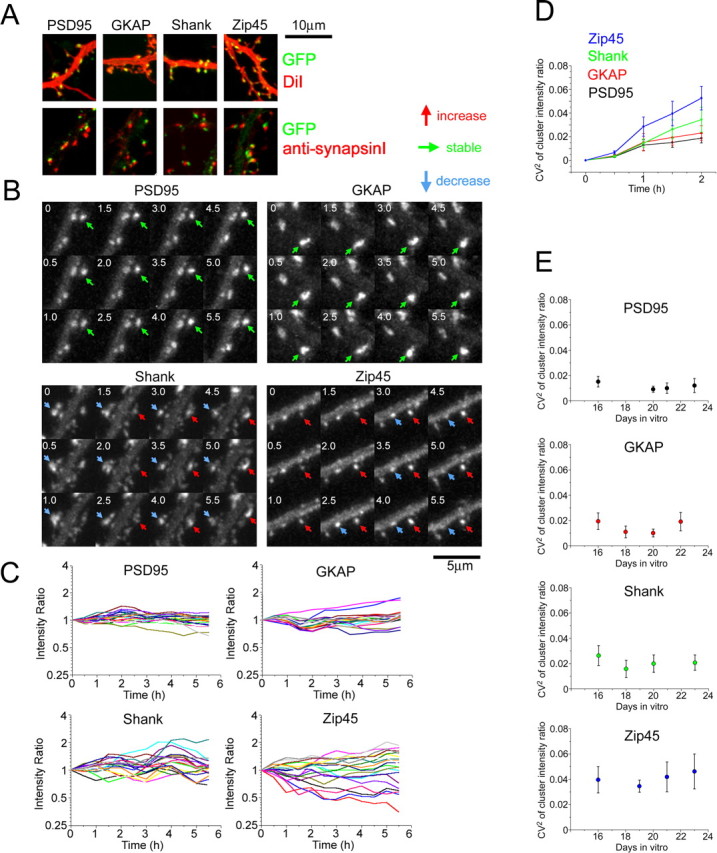
Differential behavior of four PSD scaffolding proteins. A, Correct targeting of four EGFP/EYFP-tagged scaffolding proteins to the postsynaptic sites. Hippocampal neurons were infected with recombinant adenoviruses. Visualization of dendritic morphology by application of DiI revealed localization of fluorescent clusters within dendritic filopodia and spines (top row). Synapsin I immunocytochemistry revealed an association of fluorescent clusters with presynaptic varicosities. Scale bar, 10 μm. B, Time-lapse imaging of EGFP/EYFP-tagged scaffolding proteins. Both the increase and decrease in fluorescence intensity of scaffolding protein clusters were observed (red and blue arrows). Stable clusters were abundant in PSD-95- and GKAP-expressing neurons (green arrows). Scale bar, 5 μm. C, Plots of relative fluorescence intensity of individual clusters against time. Data from 20 fluorescent clusters in neurons derived from the same batch of culture preparation at 16 d in vitro were presented. Fluorescence intensities at t = 0 h were normalized to 1, and a relative change of cluster intensities was plotted. D, A plot of the extent of cluster intensity dispersion (CV2 of cluster populations) against time. From the plots in C, we calculated CV2 in the intensity ratio of cluster population. Data points are the average of four time-lapse sequences at 16 d in vitro. E, Developmental profiles of cluster dynamics. CV2 increase within initial a 1.5 h observation period was calculated from plots similar to D and averaged for multiple time-lapse sequences at specific developmental stages (n = 4 or 5 for each developmental stage). Error bars indicate SEM.
A previous study showed that overexpression of Shank1 promoted the maturation of dendritic spines (Sala et al., 2001). Similar effects of EGFP-Shank overexpression on spine size were observed with a high titer of recombinant viruses. However, this effect was negligible in neurons infected with low-titer recombinant adenoviruses, which increased the amount of total Shank to 150 ± 6.3% (mean ± SEM for all data) of the control. We also showed previously that moderate overexpression of PSD-95-EGFP, EYFP-GKAP, EGFP-Shank, and EGFP-PSD-Zip45 does not induce structural enlargement of spines and accumulation of other PSD molecules (Okabe et al., 1999, 2001a; Sugiyama et al., 2005). To further characterize the effects of four overexpressing EGFP/EYFP fusion proteins, we measured AMPA receptor GluR1 and GluR2 immunoreactivities in the postsynaptic sites of neurons infected with high-titer recombinant adenoviruses (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). We noticed a positive correlation between the PSD-95 overexpression level and synaptic GluR1/GluR2 content. This observation is consistent with a previous analysis of PSD-95-overexpressing neurons in culture (El-Husseini et al., 2000). The effect should be negligible in our experimental condition, because the extent of PSD-95 overexpression for imaging experiments is in the lower range (120–160% of the control), in which increase in synaptic GluR1/GluR2 is estimated to be <10%. In contrast to PSD-95, little correlation was found between the amount of exogenous GKAP, Shank, and PSD-Zip45 scaffolding proteins and that of synaptic GluR1/GluR2.
Time-lapse confocal microscopy was performed on culture preparations 14–24 d after plating. At this stage, initial synaptogenesis was already completed and a continual remodeling of synaptic connections took place. Approximately 20% of PSD-95-EGFP clusters turned over within a 24 h period at this developmental stage (Okabe et al., 1999). In agreement with this, most of the PSD-95-EGFP clusters were stable throughout the observation period of 5 h (Fig. 1B). In contrast, a large number of EGFP-PSD-Zip45 clusters showed a dramatic increase and decrease in fluorescence intensity. Fluorescent clusters of GKAP and Shank showed an intermediate phenotype. A quantitative analysis of fluorescence intensity change in individual clusters was performed by plotting normalized cluster intensity against time (Fig. 1C). Although integrated fluorescence intensities of most PSD-95 clusters remained at constant levels, a rapid deviation of integrated fluorescence from the original value was found in PSD-Zip45 clusters. Instability of GKAP and Shank cluster fluorescence was intermediate between PSD-95 and PSD-Zip45. The square coefficient of variation (CV2) of normalized cluster intensity was calculated and was plotted against time (Fig. 1D). Again, this plot confirmed the intermediate dynamics of GKAP and Shank clusters compared with stable PSD-95 clusters and very dynamic PSD-Zip45 clusters. The value of CV2 at 1.5 h was taken as a representative index of cluster dynamics and was plotted against days after plating (Fig. 1E). The plots indicate that cluster dynamics is independent of neuronal maturation. Retrospective immunocytochemistry with anti-synapsin I antibody confirmed apposition of >95% of GFP clusters we analyzed with synapsin I-positive puncta in experiments with all four scaffolding proteins. The extent of fluctuation of cluster intensity showed little correlation with the maximal cluster intensity (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), indicating that large PSD clusters have similar dynamics as small clusters. Together, these results indicate that distinct kinetics is determined mainly by the intrinsic properties of each PSD molecule and is unaffected by either synaptic maturation or the extent of PSD clustering.
Differential turnover rates of PSD proteins quantified by FRAP analysis
Our quantitative time-lapse imaging experiments revealed distinct kinetics of individual PSD scaffolding proteins. The simplest explanation of the distinct kinetics is that underlying steady-state turnover rates of PSD molecules are different, possibly because of distinct binding affinities of each molecule to other scaffolds. This can be tested by measuring steady-state turnover of proteins in local cytoplasmic environment using FRAP. Interpretation of FRAP data in the dendritic spine, however, needs considerations for confinement of freely diffusible proteins within the spine head (Majewska et al., 2000). To estimate the extent of confinement, we performed FRAP analysis of soluble EGFP molecules (Fig. 2A,B). Fluorescence recovery of EGFP can be fitted by a single-exponential curve with a time constant of 0.771 ± 0.102 s. In a two-compartment model of diffusion, the diffusion coefficient is inversely proportional to a time constant of FRAP recovery (see Materials and Methods) and a diffusion coefficient of protein molecules with molecular weight of 23,000–340,000 has been reported to be in the range of 2.0–10.7 × 107 cm2/s (Truskey et al., 2004). From these considerations, we calculated that the upper limit for time constants of freely diffusible EGFP/EYFP fusion molecules must be 4.1 s, indicating that FRAP recovery attributable to diffusion is almost saturated after an initial period of 12 s. Indeed, our FRAP analysis of fusion molecules revealed a fast component of fluorescence recovery that could be fitted with a single exponential. However, these fractions were relatively minor in all four scaffolding proteins (13.3 ± 2.6% for PSD-95, 10.8 ± 1.2% for GKAP, 6.5 ± 2.0% for Shank, and 13.6 ± 2.7% for PSD-Zip45) (Fig. 2C), suggesting scarcity of diffusible PSD scaffolding proteins.
Figure 2.

FRAP analysis of EGFP and EGFP/EYFP-tagged scaffolding molecules. A, Fluorescence recovery curve of EGFP molecules after photobleaching of a single dendritic spine. B, Fluorescence images of an EGFP-filled dendritic spine during FRAP analysis. Scale bar, 2 μm. C, Fluorescence recovery curve of EGFP/EYFP-tagged scaffolding proteins within 12 s after photobleaching. The insets show the same plots with an enlarged vertical scale. D, Fluorescence recovery curve of EGFP/EYFP-tagged scaffolding proteins within 5 min after photobleaching. The baseline of fluorescence recovery was set to t = 20 s to eliminate the diffusible fraction identified in fast FRAP experiments shown in C. Scale bar, 1 μm. E, Fluorescence recovery curve of EYFP-tagged PSD-95 with or without ECFP-tagged PSD-Zip45. The presence of ECFP-tagged PSD-Zip45 did not alter dynamics of EYFP-tagged PSD-95. Error bars indicate SEM.
We next estimated the steady-state exchange rate between cytoplasm and the PSD structure by measuring the slow component of FRAP recovery (Fig. 2D). To eliminate the contribution of the diffusible fraction, we set an initial time point of data acquisition at 20 s after bleach-pulse application. This slow FRAP analysis revealed considerably distinct kinetics among four PSD molecules. The most dynamic molecule was PSD-Zip45, and 63.4 ± 6.9% of PSD-Zip45 molecules were already replaced by newly arrived molecules after 300 s. In contrast, exchange of PSD-95 molecules was only 13.9 ± 2.8% after 300 s. Here again, GKAP and Shank showed intermediate kinetics. Coexpression of YFP-tagged PSD-95 with CFP-tagged PSD-Zip45 did not alter recovery kinetics of PSD-95, suggesting higher dynamics of PSD-Zip45 is not a secondary effect of synapse modification by its overexpression (Fig. 2E). The observed differential FRAP kinetics is consistent with our time-lapse imaging experiments showing differential fluctuations in total cluster fluorescence. Thus, distinct PSD scaffolding proteins have different intrinsic mobility, which may determine the speed and extent of changes in scaffolding protein content per single synapses.
Synaptic localization of PSD-95 is not an absolute requirement for synaptic targeting and for determination of the turnover rate of other PSD scaffolds
The stability of PSD-95 molecules at synapses is consistent with their direct association with the lipid bilayer and suggests their potential role as a core organizer of the PSD. To test this possibility, we disrupted the assembly of PSD-95 and other MAGUK proteins using 2-bromopalmitate, an inhibitor of protein palmitoylation. Previous studies have shown that palmitoylation of PSD-95 is critical to its membrane association and clustering (El-Husseini et al., 2002). Treatment of hippocampal neurons with 2-bromopalmitate significantly reduced the amount of PSD-95 and its family proteins in synapses (Fig. 3A,B). Immunoblotting of primary neurons extracted under the mild detergent condition confirmed dissociation of PSD-95 from the postsynaptic sites after 2-bromopalmitate treatment (Fig. 4A). Extraction of primary neurons with an SDS-containing buffer and subsequent immunoprecipitation with an anti-GKAP antibody revealed reduced interaction of PSD-95 with GKAP after 2-bromopalmitate treatment (Fig. 4B). These results indicate that release from synaptic membranes after blockade of palmitoylation results in dissociation of PSD-95 from the postsynaptic protein complex.
Figure 3.

Unaltered dynamic properties of GKAP after pharmacological elimination of PSD-95 from the PSD. A, Immunocytochemistry of 2-bromopalmitate-treated neurons using anti-MAGUK (recognizing PSD-95, SAP97, SAP102, and Chapsyn-110 with similar sensitivity) and anti-PSD-95 antibodies. Scale bar, 5 μm. B, Quantitation of immunocytochemistry shown in A. Treatment with 2-bromopalmitate significantly reduced the amount of PSD-95 and related MAGUK proteins at synapses (∗∗∗p < 0.001). C, Presence of GKAP- and Shank-immunoreactive clusters after 2-bromopalmitate treatment and their association with a marker of excitatory presynaptic structure, vesicular glutamate transporter 1 (VGLUT1). Scale bar, 5 μm. D, FRAP analysis of EYFP-GKAP in 2-bromopalmitate-treated neurons showing unaltered dynamic properties of EYFP-GKAP. Error bars indicate SEM.
Figure 4.

Reduced interaction of PSD-95 and GKAP by 2-bromopalmitate treatment. A, Immunoblotting of proteins extracted from primary hippocampal neurons treated with 2-bromopalmitate (BrPal). After mild extraction of cells with NP-40 buffer and protein (pt) fractionation, PSD-95 was primarily in the pellet. Incubation with 2-bromopalmitate increased the amount of PSD-95 in the supernatant (sp). B, Immunoprecipitation (i.p.) of PSD-95 and GKAP from neurons treated with 2-bromopalmitate. Extraction of primary neurons with buffer containing SDS (radioimmunoprecipitation assay buffer) increased the solubility of PSD-95 in control condition. After immunoprecipitation with anti-GKAP antibody, the reduced amount of PSD-95 was observed in samples treated with 2-bromopalmitate.
To determine the effects of PSD-95 dissociation from the PSD on the localization of other scaffolding molecules, we next characterized localization of GKAP and Shank in 2-bromopalmitate-treated neurons. We observed little alterations in the distribution of GKAP- and Shank-immunopositive clusters, and quantitation of fluorescence intensity of both types of clusters showed no statistical differences (GKAP cluster: 105 ± 5.3%, p > 0.05; Shank cluster: 103 ± 4.8%, p > 0.05) (Fig. 3C). Furthermore, FRAP analysis of EYFP-GKAP, a direct binding partner of PSD-95, revealed unaltered recovery kinetics (Fig. 3D). Treatment with 2-bromopalmitate induces selective loss of synaptic AMPA receptors (El-Husseini et al., 2002) and can potentially affect GKAP dynamics. However, FRAP kinetics of neurons treated with 2-bromopalmitate in the presence of CNQX was not significantly different (35.5 ± 3.6% in normal culture medium, 33.9 ± 4.3% in 2-bromopalmitate, 34.6 ± 5.1% in CNQX, and 33.1 ± 4.1% in 2-bromopalmitate plus CNQX; percentage of recovery at t = 300 s).
Among MAGUK proteins, SAP97 does not have the N-terminal palmitoylation motif and can be resistant to 2-bromopalmitate treatment (Topinka and Bredt, 1998). We next attempted to reduce the expression level of SAP97 by RNA interference (RNAi) (Nakagawa et al., 2004). SAP97 RNAi specifically reduced SAP97-EGFP expression in fibroblast (Fig. 5A). Expression of SAP97 RNAi also caused profound knockdown of endogenous SAP97 proteins in neurons (Fig. 5B). When expression of SAP97 RNAi was combined with 2-bromopalmitate treatment, we observed enhancement of MAGUK protein elimination from the postsynaptic sites (Fig. 5C,E). However, the same treatment did not affect the amount of postsynaptic GKAP (Fig. 5D,E). These results collectively indicate the possibility that a major portion of PSD-95 and related MAGUK proteins at postsynaptic sites may not directly contribute to the ultimate synaptic accumulation of GKAP.
Figure 5.

Unaltered GKAP localization after combined treatment with SAP97 RNAi and 2-bromopalmitate. A, Cotransfection of SAP97 or PSD-95 RNAi vector with SAP97-EGFP in COS7 cells. Selective suppression of SAP97-EGFP was observed in cells expressing SAP97 siRNA. DIC, Differential interference contrast. B, Knockdown of endogenous SAP97 expression by SAP97 RNAi. The SAP97 RNAi vector was cotransfected with GFP. C, Enhanced reduction in synaptic MAGUK proteins by combined treatment of primary neurons with SAP97 RNAi and 2-bromopalmitate. D, Preserved synaptic clustering of GKAP after combined treatment with SAP97 RNAi and 2-bromopalmitate. E, Fluorescence quantitation of synaptic anti-MAGUK and anti-GKAP staining in neurons treated with SAP97 RNAi and/or 2-bromopalmitate. The amount of GKAP in synapses did not change after combined treatment with SAP97 RNAi and 2-bromopalmitate. Error bars indicate SEM. Scale bars: A, 150 μm; B–D, 10 μm. 2-Br-Pal, 2-Bromopalmitate.
Actin-dependent rapid disassembly of multiple PSD scaffolding proteins
Our pharmacological and genetic manipulation to eliminate major membrane-associated scaffolding proteins from PSD did not profoundly change dynamic properties of other PSD components. Therefore, we next assessed the role of actin cytoskeleton that presumably associates with PSD at its cytoplasmic face. Previous imaging experiments revealed rapid turnover of actin in dendritic spines (Star et al., 2002). To confirm this observation, we performed FRAP analysis of EGFP-actin (Fig. 6A,B). Fluorescence recovery of EGFP-actin was completed within 3 min (92.1 ± 6.4% at 300 s) and was inhibited by treatment with 5 μm jasplakinolide, an actin filament-stabilizing compound (−0.01 ± 1.1% at 300 s). Latrunculin A binds to G-actin and inhibits further polymerization of actin filaments. Latrunculin A treatment should be highly effective in eliminating dynamic F-actin from dendritic spines. However, previous studies indicated relatively slow kinetics of actin disassembly after application of latrunculin A (Allison et al., 1998; Okabe et al., 2001b; Usui et al., 2003). The rate of latrunculin A-induced actin disassembly increased significantly by modifying application protocol (see Materials and Methods). Under the modified condition, most of the EGFP-actin signal was eliminated from the spines within 5 min after latrunculin A treatment (Fig. 6C). The effect of latrunculin A on reduction in endogenous F-actin content was assessed by rhodamine–phalloidin staining (Fig. 6D). A significant reduction in phalloidin staining 3 min after latrunculin A treatment (13.4% of the control; p < 0.001, t test) indicates rapid turnover of filamentous actin in the spine, consistent with the FRAP results.
Figure 6.

Rapid turnover of actin in the postsynaptic cytoplasm. A, Time-lapse sequences of an EGFP-actin-expressing neuron during FRAP analysis. Rapid recovery of fluorescence was observed (arrow). B, Fluorescence recovery curve of EGFP-actin in single spines. Application of an actin stabilizer, jasplakinolide, completely suppressed rapid actin turnover. Error bars indicate SEM. C, Rapid elimination of EGFP-actin from dendritic spines by latrunculin A. Within 5 min after application of latrunculin A, most of the fluorescence derived from EGFP-actin was eliminated from the spines. D, Rhodamine–phalloidin staining of neurons with or without latrunculin A (lat) treatment for 3 min. Double staining with either a presynaptic marker (synapsin I) or a postsynaptic marker (MAGUK proteins) revealed preferential accumulation of F-actin at the postsynaptic sites (arrows). Scale bars, 5 μm.
We next analyzed the degree of PSD scaffolding protein disassembly after acute disruption of F-actin by latrunculin A. PSD-95 clusters were resistant to F-actin disruption, and fluorescence intensity showed little change (97.6 ± 4.0% at t = 15 min) (Fig. 7A,B). In contrast, PSD-Zip45 clusters reduced their fluorescence significantly (43.9 ± 7.0% at t = 15 min). A similar amount of fluorescence loss induced by actin depolymerization was observed in the case of GKAP and Shank (54.8 ± 6.6% at t = 15 min for GKAP and 66.5 ± 4.8% for Shank) (Fig. 7A,B). The loss of fluorescence in PSD-Zip45, GKAP, and Shank clusters was saturated within 3 min after latrunculin A application, and this rapid time course was comparable to the time course of F-actin disruption. Endogenous scaffolding proteins also showed rapid decreases in immunoreactivity after latrunculin A treatment (Fig. 7C) (76.0 ± 3.3, 72.4 ± 3.9, and 58.9 ± 5.1% of control preparations for GKAP, Shank, and PSD-Zip45, respectively). We asked whether elimination of PSD-95 by 2-bromopalmitate may influence the size of actin-dependent fraction of other scaffolds. However, application of 2-bromopalmitate before latrunculin A treatment did not increase the relative fraction of the actin-dependent pool of both GKAP and Shank (Fig. 7D), indicating that acute removal of MAGUK proteins from synaptic membranes is not sufficient to disrupt the protein organization of PSD scaffolds supported by F-actin.
Figure 7.

Rapid dissociation of multiple scaffolding proteins by disruption of F-actin. A, Time-lapse image sequences of neurons expressing EGFP/EYFP-tagged scaffolding proteins before and after application of latrunculin A. Latrunculin A induced the rapid decrease in fluorescence in GKAP, Shank, and PSD-Zip45 clusters. PSD-95 clusters did not change their fluorescence after latrunculin A treatment. Scale bar, 5 μm. B, Time course of relative fluorescence change in scaffolding protein clusters after latrunculin A application. C, Response of endogenous scaffolding proteins to latrunculin A administration. Neurons were fixed 10 min after application of latrunculin A, and total fluorescence intensity of clusters was measured after immunostaining of scaffolding proteins. A significant decrease in immunoreactivity for GKAP, Shank, and PSD-Zip45 was observed (∗∗∗p < 0.001). D, Loss of fluorescence intensity after latrunculin A administration in neurons pretreated with 2-bromopalmitate (2-Br-Pal). Previous application of 2-bromopalmitate did not enhance dissociation of EYFP-GKAP and EGFP-Shank molecules. E, Interaction of PSD-95, GKAP, and Shank after acute disruption of F-actin by latrunculin A (Lat). Cells were extracted with radioimmunoprecipitation assay buffer and immunoprecipitated (i.p.) with either anti-GKAP or anti-Shank antibody. Reduced interactions of Shank with both PSD-95 and GKAP were observed. In contrast, immunoprecipitation with anti-GKAP antibody revealed unaltered interaction of GKAP and PSD-95. F, Immunoprecipitation of EGFP-Shank by using anti-GFP antibody. After treatment with latrunculin A, cells were extracted with NP-40 buffer and immunoprecipitated. Latrunculin A treatment reduced the interaction between EGFP-Shank and Homer. Error bars indicate SEM.
To biochemically confirm that F-actin is essential to maintain the assembly of scaffolding proteins, primary neurons were exposed to latrunculin A for 5 min and the proteins were subsequently extracted for immunoprecipitation with anti-GKAP and anti-Shank antibodies. Actin disassembly reduced the amount of PSD-95 and GKAP incorporated into the complex precipitated by anti-Shank antibody (Fig. 7E). In contrast, the amount of PSD-95 in the complex precipitated by anti-GKAP was not altered. We next expressed EGFP-Shank by using recombinant adenoviruses and analyzed the amount of Homer in the EGFP–Shank-containing complex. Actin disassembly induced dissociation of Homer from the Shank-containing complex (Fig. 7F). Collectively, our immunoprecipitation experiments indicate that actin disassembly impairs multiple interactions between four PSD scaffolding proteins.
FRAP experiments revealed that ∼60% of PSD-Zip45, 40% of Shank, and 30% of GKAP are exchangeable within 300 s. If latrunculin A treatment induced disassembly of a dynamic subpopulation of scaffolding molecules, then the FRAP recovery curve after latrunculin A treatment should be altered. To test this, we performed FRAP analysis in the same neurons before and after latrunculin A treatment (Fig. 8). Time constants of fluorescence recovery were not altered, but the exchangeable fraction was significantly reduced (from 35.7 ± 4.4% to 19.1 ± 3.6% for GKAP, from 34.9 ± 6.9% to 18.4 ± 4.7% for Shank, and from 62.3 ± 1.2% to 15.9 ± 2.1% for PSD-Zip45 at t = 300 s). The result indicates that the dynamic fraction of multiple PSD proteins contains a significant pool that is sensitive to F-actin disruption.
Figure 8.
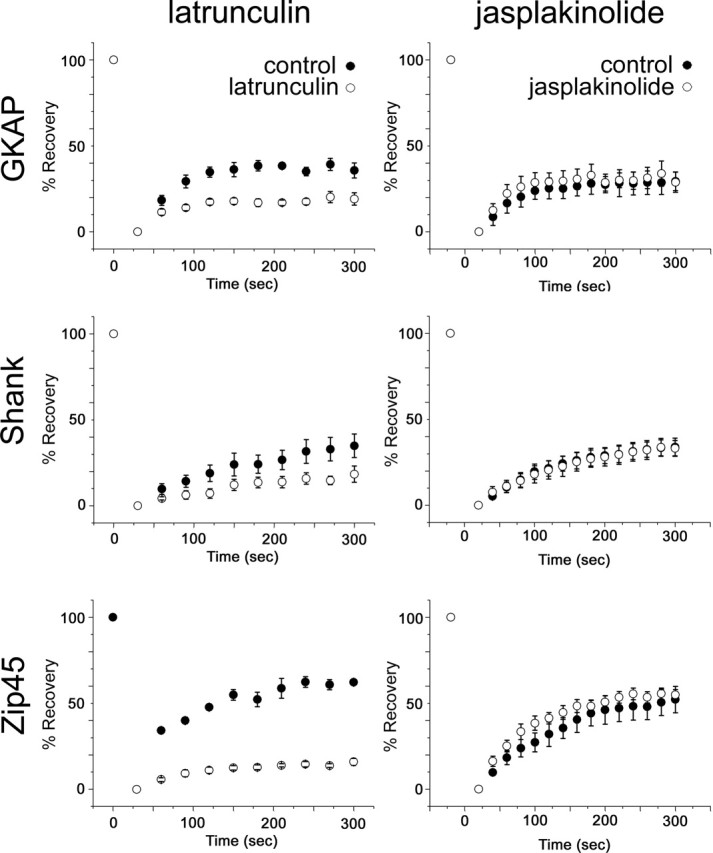
Effects of actin-depolymerizing and actin-stabilizing reagents on the dynamics of GKAP, Shank, and PSD-Zip45. Fluorescence recovery in PSDs after photobleaching was monitored for 5 min in the presence or absence of chemicals that depolymerize (latrunculin A) or stabilize (jasplakinolide) actin filaments. Although latrunculin A suppressed dynamics of all three scaffolding proteins, jasplakinolide did not induce suppression or enhancement of protein turnover at the synapses. Error bars indicate SEM.
The direct interaction of scaffolding proteins with F-actin was reported previously (Shiraishi et al., 1999), and it is possible that rapid turnover of scaffolding proteins is primarily dominated by the kinetics of actin turnover. To test whether a reduction in actin turnover indeed influences the kinetics of scaffolding proteins, we performed FRAP analysis in the presence of jasplakinolide. This treatment did not alter the size of the rapidly exchangeable pool of GKAP, Shank, and PSD-Zip45 (Fig. 8), indicating that mobile pools of PSD proteins, which are disrupted and eliminated from synapses by latrunculin A-induced actin disassembly, are retained after stabilization of F-actin without changing their dynamic properties.
Rapid redistribution of multiple scaffolding proteins by neuronal activity is dependent on F-actin
We next analyzed roles of actin turnover in the process of activity-dependent redistribution of scaffolding proteins. Application of 50 μm bicuculline with 500 μm 4-AP induces enhanced synaptic transmission and increases the spontaneous firing rate that sustained >10 min (Fig. 9A) (Hardingham et al., 2002). By using this synaptically evoked stimulation, we monitored distributional changes of actin, PSD-95, GKAP, Shank, and PSD-Zip45 (Fig. 9B,C). The response of EGFP-actin was complex, with rapid clustering at the postsynaptic sites within 5 min and their subsequent dissociation. Consistent with our previous observation (Okabe et al., 2001b), bicuculline/4-AP treatment did not induce redistribution of PSD-95 (94.76 ± 5.20%). In contrast, the identical stimulation protocol induced rapid dissociation of PSD-Zip45 clusters (85.32 ± 5.5% at t = 15 min). Bicuculline/4-AP treatment has been shown previously to induce synchronous bursts of action potentials resulting in plateau-type calcium elevation (Hardingham et al., 2002), which favors dispersion of PSD-Zip45 (Okabe et al., 2001b). The response of Shank to bicuculline/4-AP treatment was similar to PSD-Zip45 (85.20 ± 5.1% at t = 15 min). The behavior of GKAP was distinct from PSD-Zip45 and Shank. Accumulation of GKAP to the postsynaptic sites took place within 6 min after the application of bicuculline/4-AP, and this redistribution was not transient (118.0 ± 6.8% at t = 15 min). These results indicate that independent rules apply to activity-dependent redistribution of scaffolding proteins. Importantly, application of jasplakinolide prevented both accumulation of GKAP (94.2 ± 1.6% at t = 15 min) and dissociation of Shank and PSD-Zip45 (Shank, 97.9 ± 3.0%; PSD-Zip45, 98.6 ± 1.8%; at t = 15 min), indicating an essential role of actin turnover in the activity-dependent reorganization of scaffolding proteins (Fig. 9C).
Figure 9.
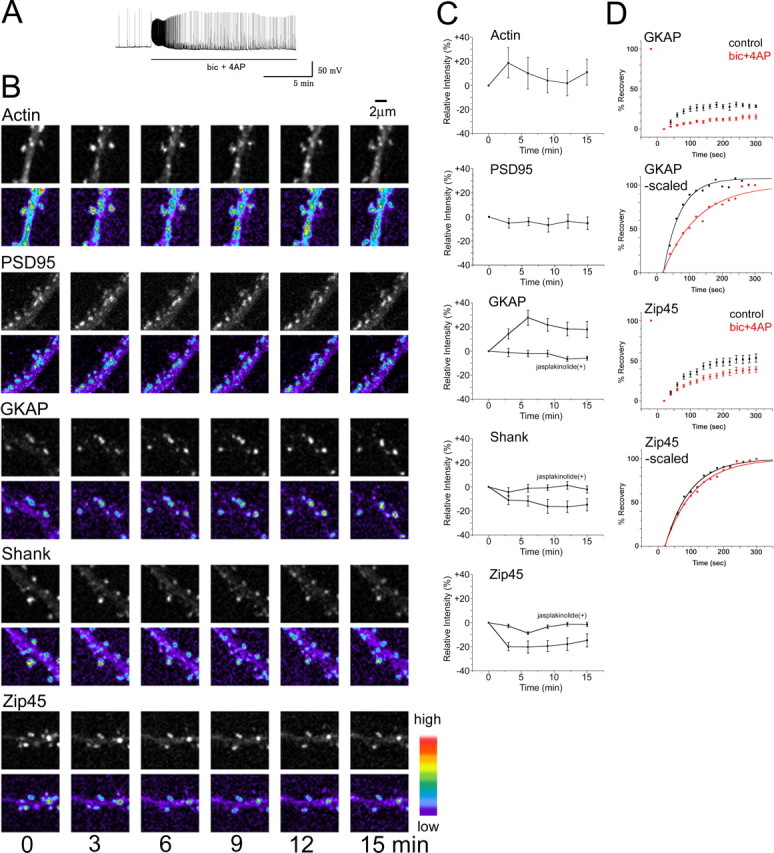
Activity-dependent redistribution of multiple PSD proteins and their altered dynamics. A, Enhancement of spike firing by treatment of neurons with bicuculline/4-AP (from 5.17/min to 27.53/min; average of 15 min period). B, Rapid remodeling of PSD proteins after application of bicuculline/4-AP. Enhancement of synaptic activity by bicucullin/4-AP treatment induced the increase in GKAP, decrease in Shank and PSD-Zip45, and transient increase in actin at the postsynaptic sites. Scale bar, 2 μm. C, Quantitation of fluorescence change after bicuculline/4-AP treatment. Previous incubation of cells with the actin-stabilizing reagent jasplakinolide completely suppressed redistribution of GKAP, Shank, and PSD-Zip45. D, Suppression of scaffolding protein dynamics by bicuculline/4-AP treatment. FRAP analysis of neurons exposed to bicuculline/4-AP revealed significant suppression of fluorescence recovery of EYFP-GKAP and EGFP-PSD-Zip45. When recovery curves were normalized for the amount of exchangeable fraction within 300 s, an altered time constant of GKAP turnover after bicuculline/4-AP treatment was evident. Error bars indicate SEM. bic, bicuculline.
To what extent does basal synaptic activity affect the turnover of scaffolding proteins? When intrinsic NMDAR-dependent activity was acutely suppressed by APV, there was no change in the kinetics of scaffolding protein turnover revealed by FRAP analysis (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Relatively low spiking rates of excitatory neurons in our culture system (0.28 Hz) (Okabe et al., 1999) may explain the small effect of suppressing endogenous NMDAR activity on scaffolding protein dynamics.
Does enhanced neuronal activity suppress dynamics of scaffolding proteins? To answer this question, we performed FRAP analysis of GKAP and PSD-Zip45 before and 10–20 min after bicuculline/4AP treatment. At this time point, the GKAP and PSD-Zip45 amount in the PSD reached a steady state, and this condition allowed us to monitor scaffolding protein turnover by FRAP. Enhanced neuronal activity significantly suppressed dynamics of both GKAP and PSD-Zip45 (from 28.6 ± 1.7% recovery at t = 300 s to 12.1 ± 1.9% for GKAP; from 49.9 ± 6.5% recovery at t = 300 s to 31.4 ± 3.2% for PSD-Zip45) (Fig. 9D). However, analysis of recovery kinetics suggested that underlying mechanisms of suppression are not uniform. The time constant of PSD-Zip45 fluorescence recovery after bicuculline/4AP treatment was not profoundly altered (76.5 ± 2.4 s before treatment, 90.5 ± 3.4 s after treatment), suggesting that activity does not modulate affinity of PSD-Zip45 to its binding partner. In contrast, the time constant of GKAP fluorescence recovery increased significantly after bicuculline/4AP treatment (50.4 ± 3.3 s before treatment, 105.2 ± 6.6 s after treatment). The increase in the time constant suggests a lower on/off-rate of GKAP. Thus, neuronal activity may lead to increased affinity of GKAP to its binding partner.
Dynamics of scaffolding protein mutants defective in their interaction with other PSD proteins
PSD scaffolding proteins contain multiple binding motifs, and their interactions with other scaffolds and cytoskeletal proteins are thought to be important in the postsynaptic accumulation. Previous studies have shown precise mapping of synapse targeting domains of these molecules, but the role of individual binding domains for the stabilization of protein dynamics has not been characterized. Therefore, we expressed the GKAP L694A mutant, Shank-C mutant (deletion of the N-terminal domain), and PSD-Zip45 G89A mutant (Fig. 10A) and measured their FRAP recovery kinetics. We have shown previously postsynaptic accumulation of the GKAP L694A mutant (Usui et al., 2003), which does not bind to the PDZ [PSD-95/Discs large/zona occludens-1] domain of Shank. FRAP experiments revealed pronounced acceleration of GKAP L694A turnover (Fig. 10B), suggesting the importance of interaction with other PDZ-containing scaffolding proteins, possibly Shank, in GKAP stabilization. Shank-C mutant lacking the N-terminal PDZ domain showed moderate accumulation to the postsynaptic sites compared with the wild-type Shank molecules. FRAP analysis of postsynaptic Shank-C molecules revealed less prominent acceleration of turnover (Fig. 10C). This result suggests that stabilization of Shank is less dependent on its PDZ domain. We have shown previously the importance of the EVH1 domain of PSD-Zip45 in synaptic targeting (Usui et al., 2003). The point mutation of G89A suppresses the ability of the EHV1 domain to interact with the proline-rich domain of synaptic proteins including Shank and mGluRs. PSD-Zip45 G89A still accumulated, albeit less efficiently, in the postsynapse and showed accelerated FRAP kinetics (Fig. 10D). Without interaction via the EVH1 domain, most of the PSD-Zip45 molecules were replaced by newly arrived molecules 5 min after photobleaching. Does enhanced FRAP turnover of mutated GKAP and PSD-Zip45 molecules correlate with their dependency on F-actin? We measured scaffolding protein disassembly after acute disruption of F-actin by latrunculin A. The extent of fluorescence reduction was larger in GKAP L694A and PSD-Zip45 G89A mutants compared with the wild-type proteins (Fig. 10E). The results indicate that scaffolding proteins with mutations in their binding motifs can be targeted to the postsynapse but are less stable and more dependent on F-actin.
Figure 10.
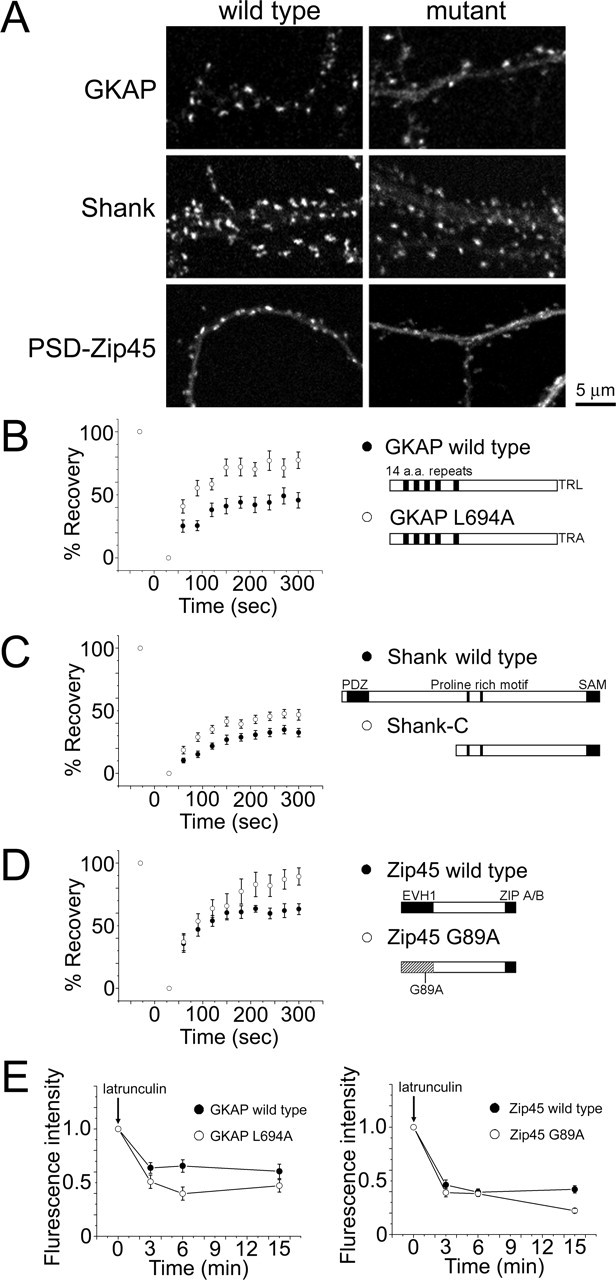
Dynamic properties of GKAP, Shank, and PSD-Zip45 molecules containing mutations that block interaction with other scaffolding proteins. A, Distribution of mutated forms of GKAP, Shank, and PSD-Zip45 tagged with EGFP within dendrites. The extent of synaptic clustering is reduced in all mutants compared with the wild-type proteins. Scale bar, 5 μm. B–D, FRAP kinetics of GKAP L694A (B), Shank-C (C), and PSD-Zip45 G89A (D). Marked enhancement of FRAP kinetics was observed in GKAP L694A and PSD-Zip45 G89A. E, Enhancement of latrunculin A-dependent loss of GKAP and PSD-Zip45 by introduction of mutations disrupting their interactions with other scaffolding proteins. Error bars indicate SEM.
Treatment of neurons with bicuculline/4AP induced accumulation of GKAP and suppression of its dynamics in synapses. To see whether the GKAP L694A mutant shows a behavior similar to wild-type GKAP, we quantitated the synaptic content of GKAP L694A before and after bicuculline/4AP treatment. The GKAP L694A mutant showed similar kinetics and extent of synaptic accumulation after stimulation (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). Furthermore, the extent of mobility suppression of GKAP L694A after bicuculline/4AP treatment was similar to that of wild-type GKAP. These results indicate that enhanced activity influences GKAP localization and mobility through a pathway independent of GKAP interaction via its C-terminal PDZ-binding motif.
Lack of destabilization of PSD scaffolds in either NR1- or mGluR5-deficient neurons
Is the actin-independent fraction of scaffolding molecules stabilized by their interaction with membrane receptors localized at synapses? To test this possibility, we examined possible roles of the interactions of PSD-95 and PSD-Zip45 with either NMDARs or mGluRs in regulating scaffolding protein dynamics at synapses. To eliminate the NMDAR complex from synaptic sites, neurons taken from NR1 mutant mice were cultured (Forrest et al., 1994). Immunocytochemistry of NR1−/− neurons revealed a large reduction in NR2A and NR2B to virtually background levels at the synaptic sites (3.2 ± 0.23% for NR2A, 4.5 ± 0.33% for NR2B; n = 4), in addition to complete elimination of NR1 (supplemental Fig. 5A, available at www.jneurosci.org as supplemental material). Despite a significant reduction in NMDAR subunits, immunoreactivity of PSD-95 was indistinguishable from wild-type culture (95 ± 4.4%; n = 4). FRAP analysis of PSD-95-EGFP also revealed unaltered kinetics of PSD-95 turnover (Fig. 11).
Figure 11.

Unaltered dynamic properties of PSD-95 and PSD-Zip45 in neurons lacking NMDARs or mGluR5. FRAP analysis of PSD-95 in NR1−/− neurons and that of PSD-Zip45 in mGluR5−/− neurons. The fluorescence recovery curve of PSD-95 and PSD-Zip45 did not change by genetically removing either NMDARs or mGluR5. Scale bar, 2 μm. Error bars indicate SEM.
A major postsynaptic group 1 mGluR in CA1 pyramidal neurons is mGluR5. We therefore prepared hippocampal neuronal culture from mGluR5 mutant mice (Lu et al., 1997). Distribution of PSD-Zip45, Shank, and PSD-95 clusters in mGluR5−/− neurons was indistinguishable from wild-type neurons (supplemental Fig. 5B, available at www.jneurosci.org as supplemental material). Fluorescence intensity of PSD-Zip45 clusters in mGluR5−/− neurons was comparable to that in wild-type neurons (92 ± 6.7%; n = 4). Furthermore, FRAP analysis of EGFP-PSD-Zip45 revealed unaltered recovery kinetics (Fig. 11). Thus, absence of two types of glutamate receptors failed to influence localization and dynamics of PSD scaffolding proteins, despite the presence of direct binding motifs to the receptors in these PSD proteins.
Discussion
In this study, we presented several lines of evidence indicating distinct dynamics for different PSD proteins. Time-lapse analysis of individual fluorescent clusters revealed the rate of redistribution in the following order: PSD-Zip45 > Shank > GKAP > PSD-95. FRAP analysis of the steady-state exchange rate of scaffolding proteins confirmed the differential dynamics. The fact that two independent experimental approaches revealed the same order in kinetic properties strongly supports the existence of regulatory mechanisms specific to individual molecules. Our subsequent analyses indeed revealed several mechanisms, which were based on either differential dependency on the actin cytoskeleton or intermolecular interactions of scaffolding proteins via specific binding domains.
The affinity of PSD molecules to their binding partners, together with the stability of binding partners themselves, is an important parameter regulating the turnover rate within PSD. FRAP analysis of four scaffolding proteins revealed the presence of a dynamic fraction with the time constants of several minutes. This fraction represents molecules weakly attached to the PSD and/or associated with dynamic cytoskeletal components. Our pharmacological analysis identified critical involvement of dynamic actin cytoskeleton in the maintenance of this exchangeable fraction. Previous studies indicated a possible involvement of actin cytoskeleton in proper localization of both glutamate receptors and a subset of scaffolding molecules (Allison et al., 1998; Usui et al., 2003; Nonaka et al., 2006). In this study, we extended these observations by capturing and quantifying the rapid redistribution of PSD proteins after acute disassembly of F-actin. The observed disassembly of scaffolding proteins was significant and occurred to a variable degree in three distinct protein species. The same disassembly protocol of F-actin also dissociated PSD-95, GKAP, and Homer from the Shank-containing protein complex isolated by immunoprecipitation. Thus, we conclude that the presence of F-actin is critical in the integration and maintenance of multiple scaffolding proteins in the PSD.
Biochemical analysis revealed the interaction of Shank and Homer proteins with a variety of molecules involved in the actin cytoskeletal network. Shank interacts with α-fodrin (Bockers et al., 2001), cortactin (Hering and Sheng, 2003), and Abp1 (Qualmann et al., 2004). Both fodrin and Abp1 interact with F-actin. Cortactin can bind to both F-actin and Arp2/3 complex (Weed et al., 2000). Homer-family proteins also reported to bind with F-actin (Shiraishi et al., 1999) and an actin-binding protein, drebrin (Mizutani et al., 1999). Although these biochemical data are consistent with the idea that exchangeable fractions of Shank and Homer proteins interact with dynamic actin cytoskeleton, the basic scaffolding structure involved in the linkage between the Shank–Homer complex and actin cytoskeleton is not yet clear. Both cortactin and drebrin have been shown to be involved in the morphogenesis of spines (Hayashi and Shirao, 1999; Hering and Sheng, 2003), suggesting their ability to interact with and reorganize spine F-actin. Specific inhibition of cortactin and drebrin activity should be performed to evaluate their roles in linking PSD scaffolds with F-actin.
FRAP experiments also revealed immobile fractions of PSD-95, GKAP, Shank, and Homer. These fractions represent molecules tightly associated to a stable structure, which is resistant to actin depolymerization. Elimination of NMDARs, mGluR5 receptors, and MAGUK proteins did not significantly alter clustering and FRAP kinetics of PSD-95, Homer, and GKAP, respectively. What are the anchoring mechanisms of PSD-95, Homer, and GKAP if their interactions with glutamate receptors and MAGUKs are not essential? In the absence of NMDARs, PSD-95 can interact with other synaptic molecules including transmembrane AMPA receptor regulatory proteins and a cell adhesion molecule, neuroligin. In the case of GKAP, interaction with Shank is another major candidate for its synaptic localization and stabilization. However, L694A mutation of GKAP, which disrupts interaction with Shank, did not block synaptic targeting. This result is consistent with a previous analysis of synapse targeting domains of SAPAP, a splice isoform of GKAP (Yao et al., 2003). Although GKAP L694A showed synaptic localization, we found that this point mutation significantly increased the turnover rate of GKAP. These observations suggest that although GKAP interaction with Shank is not essential for its synaptic localization, it enhances GKAP stability within the PSD. The EVH1 domain of PSD-Zip45/Homer1 potentially interacts with a variety of molecules including dynamin 3 and drebrin, which are shown to be localized in the postsynapse (Mizutani et al., 1999; Gray et al., 2003). We observed a marked reduction in PSD-Zip45 accumulation by its mutation in the EVH1 domain, indicating importance of EVH1 domain-dependent interaction for its proper synaptic localization. Enhanced FRAP kinetics suggests additional role of EVH1 domain in PSD-Zip45 stabilization. It is likely that both synaptic localization and stabilization of Homer is mediated by EVH1-dependent binding to postsynaptic molecules other than mGluR5.
Fluorescence imaging and FRAP analysis of EGFP-tagged actin revealed short-term remodeling of the actin network in dendritic spines (Fischer et al., 1998; Star et al., 2002). Multiple forms of actin reorganization can be induced by different stimulation protocols (Halpain et al., 1998; Colicos et al., 2001; Furuyashiki et al., 2002). Although basic turnover mechanisms of actin molecules and its regulation by synaptic activity have been clarified, how actin reorganization is linked to subsequent alterations of synaptic structure and function is not well understood. Here, we provided strong evidence of F-actin involvement in the remodeling of the PSD molecular composition after synaptic activation. We hypothesize that the cytoplasmic side of the PSD is in contact with F-actin, which undergoes rapid remodeling and assembly/disassembly. Our results suggest that the presence of this dynamic interaction between F-actin and a fraction of PSD scaffolds is essential in both maintenance and remodeling of the PSD. It is less likely that disassembly of F-actin is a prerequisite for dissociation of scaffolding proteins, because scaffolding protein turnover measured by FRAP was normal when F-actin was pharmacologically stabilized. Instead, a network of F-actin present in spines, either under rapid assembly/disassembly or stabilized, may function as a loosely tethered platform with which dynamic scaffolding proteins are repetitively associated/dissociated. An interesting point is that this platform itself is highly dynamic and rapidly changes its size by activity (Okamoto et al., 2004). Multiple mechanisms should be present for the activity-dependent PSD reorganization, because PSD-Zip45 and GKAP redistribute in opposite directions. In the FRAP experiment of PSD-Zip45, we observed a reduction in a dynamic fraction with a small change in recovery kinetics. Reduction in the binding capacity of scaffolds may explain this observation. On the other hand, the FRAP experiment of GKAP revealed a marked increase in the time constant. This observation is consistent with the increased affinity of GKAP to its binding partners in the PSD. After application of bicuculline/4-AP, freely diffusible GKAP may be modified, possibly through posttranslational modifications, and recruited to its binding partners. Although Shank is a candidate molecule for regulated GKAP binding, the GKAP L694A mutant, which is defective in its interaction with Shank via the C-terminal PDZ-binding domain, can still change its distribution after bicuculline/4-AP treatment and increase its stability. GKAP has been shown to interact with S-SCAM (Hirao et al., 1998), nArgBP2 (Kawabe et al., 1999), and a dynein light chain (Naisbitt et al., 2000) with domains distinct from the C-terminal PDZ-binding motif. It is likely that posttranslational modifications of some of these interaction domains may be involved in activity-dependent stabilization of GKAP.
In summary, we presented evidence of several regulatory mechanisms of scaffolding protein assembly that act on their steady-state exchange with local diffusible pools. This finding is compatible with a recent model of gradual recruitment of PSD molecules from local diffusible pools during synaptogenesis (Bresler et al., 2004). The experimental evidence presented here also indicated an essential role of F-actin in both maintenance and remodeling of the PSD. This interaction between F-actin and the PSD may be important in transforming transient remodeling of the cytoskeletal system in response to synaptic activity into long-lasting alterations of the submembranous protein complex that regulates glutamate receptor function and distribution.
Footnotes
This work was supported by grants from the Ministry of Education, Science, Sports, Culture, and Technology of Japan, the Solution Oriented Research for Science and Technology of Japan Science and Technology Agency, and the Human Frontier Science Program. We thank Drs. M. Watanabe and M. Inui for antibodies, Dr. T. Curran for NR1-null mice, Dr. J. Roder for mGluR5-null mice, Dr. H. Okado and T. Urushido for preparation of recombinant adenoviruses, I. Kawabata for preparation of primary hippocampal neurons, and Drs. T. Ebihara and K. Yamamoto for establishment of mice colonies.
References
- Allison DW, Gelfand VI, Spector I, Craig AM (1998). Role of actin in anchoring postsynaptic receptors in cultured hippocampal neurons: differential attachment of NMDA versus AMPA receptors. J Neurosci 18:2423–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockers TM, Mameza MG, Kreutz MR, Bockmann J, Weise C, Buck F, Richter D, Gundelfinger ED, Kreienkamp HJ (2001). Synaptic scaffolding proteins in rat brain. Ankyrin repeats of the multidomain Shank protein family interact with the cytoskeletal protein alpha-fodrin. J Biol Chem 276:40104–40112. [DOI] [PubMed] [Google Scholar]
- Boeckers TM, Kreutz MR, Winter C, Zuschratter W, Smalla KH, Sanmarti-Vila L, Wex H, Langnaese K, Bockmann J, Garner CC, Gundelfinger ED (1999). Proline-rich synapse-associated protein-1/cortactin binding protein 1 (ProSAP1/CortBP1) is a PDZ-domain protein highly enriched in the postsynaptic density. J Neurosci 19:6506–6518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresler T, Shapira M, Boeckers T, Dresbach T, Futter M, Garner CC, Rosenblum K, Gundelfinger ED, Ziv NE (2004). Postsynaptic density assembly is fundamentally different from presynaptic active zone assembly. J Neurosci 24:1507–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colicos MA, Collins BE, Sailor MJ, Goda Y (2001). Remodeling of synaptic actin induced by photoconductive stimulation. Cell 107:605–616. [DOI] [PubMed] [Google Scholar]
- Craven SE, El-Husseini AE, Bredt DS (1999). Synaptic targeting of the postsynaptic density protein PSD-95 mediated by lipid and protein motifs. Neuron 22:497–509. [DOI] [PubMed] [Google Scholar]
- Dalva MB, Takasu MA, Lin MZ, Shamah SM, Hu L, Gale NW, Greenberg ME (2000). EphB receptors interact with NMDA receptors and regulate excitatory synapse formation. Cell 103:945–956. [DOI] [PubMed] [Google Scholar]
- Du Y, Weed SA, Xiong WC, Marshall TD, Parsons JT (1998). Identification of a novel cortactin SH3 domain-binding protein and its localization to growth cones of cultured neurons. Mol Cell Biol 18:5838–5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebihara T, Kawabata I, Usui S, Sobue K, Okabe S (2003). Synchronized formation and remodeling of postsynaptic densities: long-term visualization of hippocampal neurons expressing postsynaptic density proteins tagged with green fluorescent protein. J Neurosci 23:2170–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Malinow R (2004). Postsynaptic density 95 controls AMPA receptor incorporation during long-term potentiation and experience-driven synaptic plasticity. J Neurosci 24:916–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS (2000). PSD-95 involvement in maturation of excitatory synapses. Science 290:1364–1368. [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Dakoji S, Sweeney N, Zhou Q, Prange O, Gauthier-Campbell C, Aguilera-Moreno A, Nicoll RA, Bredt DS (2002). Synaptic strength regulated by palmitate cycling on PSD-95. Cell 108:849–863. [DOI] [PubMed] [Google Scholar]
- Fischer M, Kaech S, Knutti D, Matus A (1998). Rapid actin-based plasticity in dendritic spines. Neuron 20:847–854. [DOI] [PubMed] [Google Scholar]
- Forrest D, Yuzaki M, Soares HD, Ng L, Luk DC, Sheng M, Stewart CL, Morgan JI, Connor JA, Curran T (1994). Targeted disruption of NMDA receptor 1 gene abolishes NMDA response and results in neonatal death. Neuron 13:325–338. [DOI] [PubMed] [Google Scholar]
- Furuyashiki T, Arakawa Y, Takemoto-Kimura S, Bito H, Narumiya S (2002). Multiple spatiotemporal modes of actin reorganization by NMDA receptors and voltage-gated Ca2+ channels. Proc Natl Acad Sci USA 99:14458–14463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner CC, Nash J, Huganir RL (2000). PDZ domains in synapse assembly and signalling. Trends Cell Biol 10:274–280. [DOI] [PubMed] [Google Scholar]
- Gomez LL, Alam S, Smith KE, Horne E, Dell’Acqua ML (2002). Regulation of A-kinase anchoring protein 79/150-cAMP-dependent protein kinase postsynaptic targeting by NMDA receptor activation of calcineurin and remodeling of dendritic actin. J Neurosci 22:7027–7044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray NW, Fourgeaud L, Huang B, Chen J, Cao H, Oswald BJ, Hemar A, McNiven MA (2003). Dynamin 3 is a component of the postsynapse, where it interacts with mGluR5 and Homer. Curr Biol 13:510–515. [DOI] [PubMed] [Google Scholar]
- Halpain S, Hipolito A, Saffer L (1998). Regulation of F-actin stability in dendritic spines by glutamate receptors and calcineurin. J Neurosci 18:9835–9844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H (2002). Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci 5:405–414. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Shirao T (1999). Change in the shape of dendritic spines caused by overexpression of drebrin in cultured cortical neurons. J Neurosci 19:3918–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering H, Sheng M (2003). Activity-dependent redistribution and essential role of cortactin in dendritic spine morphogenesis. J Neurosci 23:11759–11769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirao K, Hata Y, Ide N, Takeuchi M, Irie M, Yao I, Deguchi M, Toyoda A, Sudhof TC, Takai Y (1998). A novel multiple PDZ domain-containing molecule interacting with N-methyl-d-aspartate receptors and neuronal cell adhesion proteins. J Biol Chem 273:21105–21110. [DOI] [PubMed] [Google Scholar]
- Kawabe H, Hata Y, Takeuchi M, Ide N, Mizoguchi A, Takai Y (1999). nArgBP2, a novel neural member of ponsin/ArgBP2/vinexin family that interacts with synapse-associated protein 90/postsynaptic density-95-associated protein (SAPAP). J Biol Chem 274:30914–30918. [DOI] [PubMed] [Google Scholar]
- Kawashima N, Takamiya K, Sun J, Kitabatake A, Sobue K (1997). Differential expression of isoforms of PSD-95 binding protein (GKAP/SAPAP1) during rat brain development. FEBS Lett 418:301–304. [DOI] [PubMed] [Google Scholar]
- Kennedy MB (2000). Signal-processing machines at the postsynaptic density. Science 290:750–754. [DOI] [PubMed] [Google Scholar]
- Kim E, Naisbitt S, Hsueh YP, Rao A, Rothschild A, Craig AM, Sheng M (1997). GKAP, a novel synaptic protein that interacts with the guanylate kinase- like domain of the PSD-95/SAP90 family of channel clustering molecules. J Cell Biol 136:669–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornau HC, Schenker LT, Kennedy MB, Seeburg PH (1995). Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science 269:1737–1740. [DOI] [PubMed] [Google Scholar]
- Lu YM, Jia Z, Janus C, Henderson JT, Gerlai R, Wojtowicz JM, Roder JC (1997). Mice lacking metabotropic glutamate receptor 5 show impaired learning and reduced CA1 long-term potentiation (LTP) but normal CA3 LTP. J Neurosci 17:5196–5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewska A, Tashiro A, Yuste R (2000). Regulation of spine calcium dynamics by rapid spine motility. J Neurosci 20:8262–8268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H (2004). Structural basis of long-term potentiation in single dendritic spines. Nature 429:761–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matus A (2000). Actin-based plasticity in dendritic spines. Science 290:754–758. [DOI] [PubMed] [Google Scholar]
- Mizutani A, Shiraishi Y, Mikoshiba K, Furuichi T (1999). Characterization of cupidin-binding proteins. Soc Neurosci Abstr 25:805–807. [Google Scholar]
- Naisbitt S, Kim E, Tu JC, Xiao B, Sala C, Valtschanoff J, Weinberg RJ, Worley PF, Sheng M (1999). Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron 23:569–582. [DOI] [PubMed] [Google Scholar]
- Naisbitt S, Valtschanoff J, Allison DW, Sala C, Kim E, Craig AM, Weinberg RJ, Sheng M (2000). Interaction of the postsynaptic density-95/guanylate kinase domain-associated protein complex with a light chain of myosin-V and dynein. J Neurosci 20:4524–4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Futai K, Lashuel HA, Lo I, Okamoto K, Walz T, Hayashi Y, Sheng M (2004). Quaternary structure, protein dynamics, and synaptic function of SAP97 controlled by L27 domain interactions. Neuron 44:453–467. [DOI] [PubMed] [Google Scholar]
- Nonaka M, Doi T, Fujiyoshi Y, Takemoto-Kimura S, Bito H (2006). Essential contribution of the ligand-binding βB/βC loop of PDZ1 and PDZ2 in the regulation of postsynaptic clustering, scaffolding, and localization of postsynaptic density-95. J Neurosci 26:763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe S, Kim HD, Miwa A, Kuriu T, Okado H (1999). Continual remodeling of postsynaptic density and its regulation by synaptic activity. Nat Neurosci 2:804–811. [DOI] [PubMed] [Google Scholar]
- Okabe S, Miwa A, Okado H (2001a). Spine formation and correlated assembly of presynaptic and postsynaptic molecules. J Neurosci 21:6105–6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe S, Urushido T, Konno D, Okado H, Sobue K (2001b). Rapid redistribution of the postsynaptic density protein PSD-Zip45 (Homer 1c) and its differential regulation by NMDA receptors and calcium channels. J Neurosci 21:9561–9571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y (2004). Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci 7:1104–1112. [DOI] [PubMed] [Google Scholar]
- Palay SL (1958). The morphology of synapses in the central nervous system. Exp Cell Res 5:275–293. [PubMed] [Google Scholar]
- Petersen JD, Chen X, Vinade L, Dosemeci A, Lisman JE, Reese TS (2003). Distribution of postsynaptic density (PSD)-95 and Ca2+/calmodulin-dependent protein kinase II at the PSD. J Neurosci 23:11270–11278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qualmann B, Boeckers TM, Jeromin M, Gundelfinger ED, Kessels MM (2004). Linkage of the actin cytoskeleton to the postsynaptic density via direct interactions of Abp1 with the ProSAP/Shank family. J Neurosci 24:2481–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala C, Piech V, Wilson NR, Passafaro M, Liu G, Sheng M (2001). Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron 31:115–130. [DOI] [PubMed] [Google Scholar]
- Sheng M, Kim MJ (2002). Postsynaptic signaling and plasticity mechanisms. Science 298:776–780. [DOI] [PubMed] [Google Scholar]
- Sheng M, Sala C (2001). Pdz domains and the organization of supramolecular complexes. Annu Rev Neurosci 24:1–29. [DOI] [PubMed] [Google Scholar]
- Shiraishi Y, Mizutani A, Bito H, Fujisawa K, Narumiya S, Mikoshiba K, Furuichi T (1999). Cupidin, an isoform of Homer/Vesl, interacts with the actin cytoskeleton and activated rho family small GTPases and is expressed in developing mouse cerebellar granule cells. J Neurosci 19:8389–8400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Star EN, Kwiatkowski DJ, Murthy VN (2002). Rapid turnover of actin in dendritic spines and its regulation by activity. Nat Neurosci 5:239–246. [DOI] [PubMed] [Google Scholar]
- Stein V, House DR, Bredt DS, Nicoll RA (2003). Postsynaptic density-95 mimics and occludes hippocampal long-term potentiation and enhances long-term depression. J Neurosci 23:5503–5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama Y, Kawabata I, Sobue K, Okabe S (2005). Determination of absolute protein numbers in single synapses by a GFP-based calibration technique. Nat Methods 2:677–684. [DOI] [PubMed] [Google Scholar]
- Tadokoro S, Tachibana T, Imanaka T, Nishida W, Sobue K (1999). Involvement of unique leucine-zipper motif of PSD-Zip45 (Homer1c/vesl-1L) in group 1 metabotropic glutamate receptor clustering. Proc Natl Acad Sci USA 93:1540–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topinka JR, Bredt DS (1998). N-terminal palmitoylation of PSD-95 regulates association with cell membranes and interaction with K+ channel Kv1.4. Neuron 20:125–134. [DOI] [PubMed] [Google Scholar]
- Truskey GA, Yuan F, Katz DF (2004). In: Transport phenomena in biological systems Upper Saddle River, NJ: Person Prentice Hall.
- Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P, Doan A, Aakalu VK, Lanahan AA, Sheng M, Worley PF (1999). Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron 23:583–592. [DOI] [PubMed] [Google Scholar]
- Usui S, Konno D, Hori K, Maruoka H, Okabe S, Fujikado T, Tano Y, Sobue K (2003). Synaptic targeting of PSD-Zip45 (Homer 1c) and its involvement in the synaptic accumulation of F-actin. J Biol Chem 278:10619–10628. [DOI] [PubMed] [Google Scholar]
- Valtschanoff JG, Weinberg RJ (2001). Laminar organization of the NMDA receptor complex within the postsynaptic density. J Neurosci 21:1211–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Fukaya M, Sakimura K, Manabe T, Mishina M, Inoue Y (1998). Selective scarcity of NMDA receptor channel subunits in the stratum lucidum (mossy fibre-recipient layer) of the mouse hippocampal CA3 subfield. Eur J Neurosci 10:478–487. [DOI] [PubMed] [Google Scholar]
- Weed SA, Karginov AV, Schafer DA, Weaver AM, Kinley AW, Cooper JA, Parsons JT (2000). Cortactin localization to sites of actin assembly in lamellipodia requires interactions with F-actin and the Arp2/3 complex. J Cell Biol 151:29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyszynski M, Lin J, Rao A, Nigh E, Beggs AH, Craig AM, Sheng M (1997). Competitive binding of alpha-actinin and calmodulin to the NMDA receptor. Nature 385:439–442. [DOI] [PubMed] [Google Scholar]
- Yao I, Iida J, Nishimura W, Hata Y (2003). Synaptic localization of SAPAP1, a synaptic membrane-associated protein. Genes Cells 8:121–129. [DOI] [PubMed] [Google Scholar]