

Abstract
The rate of Ca2+ clearance from the neuronal cytoplasm affects the amplitude, duration, and localization of Ca2+ signals and influences a variety of Ca2+-dependent functions. We reported previously that activation of protein kinase C (PKC) accelerates Ca2+ efflux in rat sensory neurons mediated by the plasma membrane Ca2+-ATPase isoform 4 (PMCA4). Here we show that sarco-endoplasmic reticulum Ca2+-ATPase (SERCA)-mediated Ca2+ uptake into intracellular stores is also accelerated by PKC activation. The rate of intracellular Ca2+ concentration ([Ca2+]i) clearance was studied after small (<350 nm) action potential-induced Ca2+ loads in rat dorsal root ganglion neurons. Under these conditions, mitochondrial Ca2+ uptake and Na+/Ca2+ exchange do not significantly influence [Ca2+]i recovery. Phorbol dibutyrate (PDBu) increased the rate of [Ca2+]i clearance by 71% in a manner sensitive to the selective PKC inhibitors GF109203x (2-[1-(3-dimethylaminopropyl)-1H-indol-3-yl]-3-(1H-indol-3-yl)maleimide) and calphostin. PKC-dependent acceleration was still observed (∼39%) when the PKC-sensitive PMCA isoform was knocked down by expression of an antisense PMCA4 cDNA (AS4). Direct measurement of Ca2+ in the endoplasmic reticulum (ER) lumen revealed that PKC increased the rate of store refilling more than twofold after depletion by treatment with cyclopiazonic acid. ER refilling was less complete in PDBu-treated cells, although, in AS4-expressing cells, PDBu accelerated the rate without reducing the ER capacity, suggesting that PMCA and SERCA compete for Ca2+. Thus, activation of PKC accelerates the clearance of Ca2+ from the cytoplasm by the concerted stimulation of Ca2+ sequestration and Ca2+ efflux.
Keywords: PKC, SERCA, PMCA, ER, Ca2+ uptake, Mag-indo-1, sensory neuron
Introduction
Phosphorylation of voltage- and ligand-gated Ca2+ channels is a critical means to regulate Ca2+ signaling and neuronal plasticity (Catterall, 2000; Salter and Kalia, 2004). In contrast, little is known about the modulation of the Ca2+ clearance mechanisms counterbalancing Ca2+ influx pathways. The high-affinity Ca2+ pumps in the endoplasmic reticulum (ER) and plasma membrane are especially important for recovery of intracellular Ca2+ concentration ([Ca2+]i) after brief electrical stimuli (Benham et al., 1992; Werth et al., 1996). Recently, we found that Ca2+ extrusion was stimulated by activation of protein kinase C (PKC) in sensory neurons, and that this effect was mediated by the plasma membrane Ca2+ ATPase (PMCA) isoform 4b (Usachev et al., 2002). In this report, we examine the possibility that Ca2+ sequestration might also be stimulated by PKC.
Ca2+ uptake mediated by the sarco-endoplasmic reticulum Ca2+ ATPase (SERCA) significantly contributes to Ca2+ clearance in neurons. Accordingly, the rate of Ca2+ sequestration into the ER modulates the amplitude and duration of cytosolic Ca2+ signals (Neering and McBurney, 1984; Friel and Tsien, 1992; Shmigol et al., 1994; Fierro et al., 1998; Suzuki et al., 2002). SERCA also functions to replenish ER stores with releasable Ca2+ (Garaschuk et al., 1997; Usachev and Thayer, 1999), which determines the ability of the stores to amplify cytosolic Ca2+ signals. Ca2+ release via ryanodine or inositol 1,4,5-trisphosphate (IP3) receptors regulates excitability, neuronal differentiation, and some forms of synaptic plasticity (for review, see Berridge, 1998; Verkhratsky, 2005). Furthermore, Ca2+ sequestered within the ER is essential for maintaining protein synthesis (Brostrom and Brostrom, 2003), whereas chronic Ca2+ overload of the stores is implicated in some neurological disorders (Paschen, 2003; Verkhratsky, 2005). Thus, SERCA-mediated Ca2+ sequestration is a critical component of neuronal signaling.
In neurons, modulation of SERCA-type Ca2+ pumps by kinases has not been described. However, in muscle, protein kinase A (PKA) and calmodulin-dependent kinase (CaMK) phosphorylate phospholamban, a SERCA accessory protein, to accelerate SERCA-mediated Ca2+ uptake into the sarcoplasmic reticulum (Tada and Toyofuku, 1998; MacLennan and Kranias, 2003). Phospholamban is not expressed in neurons (Plessers et al., 1991), although analogous proteins may be present (Dou and Joseph, 1996). Modulation of Ca2+ sequestration in the ER by phosphorylation has also been reported in blood platelets and in Xenopus oocytes (Lacabaratz-Porret et al., 1998; Roderick et al., 2000). Thus, although little is known about the modulation of SERCAs in neurons, in other cell types, SERCA-mediated Ca2+ uptake is modulated by protein kinases.
Here, we found that activation of PKC stimulated Ca2+ clearance from the cytoplasm of sensory neurons. PKC-dependent facilitation of Ca2+ extrusion (Usachev et al., 2002) could not alone account for this marked acceleration of [Ca2+]i recovery. In addition, SERCA-mediated Ca2+ uptake significantly contributed to the PKC effect. Direct monitoring of Ca2+ concentration in the ER revealed a more than twofold increase in the rate of Ca2+ sequestration into the lumen of the ER. Thus, the activation of PKC results in a coordinated acceleration of multiple Ca2+ clearance mechanisms in sensory neurons.
Materials and Methods
Materials. Indo-1 AM, Mag-indo-1 AM, and Pluronic F-127 were obtained from Invitrogen (Carlsbad, CA). GF109203x (2-[1-(3-dimethylaminopropyl)-1 H-indol-3-yl]-3-(1 H-indol-3-yl)maleimide), Ro31-8220 (3-[1-(3-(amidinothio)propyl)-1H-indol-3-yl]-3-(1-methylindol-3-yl)maleimide), calphostin, and phorbol dibutyrate (PDBu) were obtained from Calbiochem (San Diego, CA). All other reagents were purchased from Sigma (St. Louis, MO).
Cell culture. Rat DRG neurons were grown in culture as described previously (Werth et al., 1996). In brief, 1- to 3-d-old Sprague Dawley rats were killed under a protocol approved by the University of Minnesota Institutional Animal Care and Use Committee. DRGs were dissected from the thoracic and lumbar segments and incubated at 37°C in collagenase–dispase (0.8 and 6.4 U/ml, respectively) for 45 min. Ganglia were dissociated by trituration through a flame-constricted pipette and then plated onto laminin-coated (50 μg/ml) glass coverslips (25 mm diameter). Cells were grown in Ham's F-12 media supplemented with 5% heat-inactivated horse serum and 5% fetal bovine serum, 50 ng/ml NGF, 4.4 mm glucose, 2 mm l-glutamine, modified Eagle's medium vitamins, and penicillin–streptomycin (100 U/ml and 100 μg/ml, respectively). Cultures were maintained at 37°C in a humidified atmosphere of 5% CO2. Neurons with cell body diameters of 18–27 μm were used within 2–3 d of plating.
[Ca2+]i measurements. Instrumentation for [Ca2+]i recording from single DRG neurons using indo-1- or Mag-indo-1-based microfluorimetry was similar to that described previously (Werth et al., 1996). Cells were placed in a flow-through chamber that was mounted on the stage of an inverted epifluorescence microscope equipped with a 70× objective (numerical aperture, 1.15; Leitz, Wetzlar, Germany). To introduce the indicators into cells, the culture was incubated with the AM form of indo-1 at a concentration of 10 μm at room temperature (22°C) for 30 min or the AM form of Mag-indo-1 at a concentration of 7.5 μm in 0.02% Pluronic F-127 at 37°C for 30 min. The cells were washed for 30 min in dye-free HEPES-buffered Hank's salt solution (HHSS) at 37°C before initiating recording. HHSS had the following composition (in mm): 10 HEPES, 140 NaCl, 5 KCl, 1.3 CaCl2, 0.4 MgSO4, 0.5 MgCl2, 0.4 KH2PO4, 0.6 Na2HPO4, 3 NaHCO3, and 10 glucose, pH 7.35 with NaOH (310 mOsm/kg with sucrose). Ca2+-free solution was obtained by substituting 0.1 mm EGTA for Ca2+. Indo-1 and Mag-indo-1 were excited at 350 nm (10 nm bandpass), and emission was detected at 405 (20) and 490 (20) nm. Fluorescence was monitored by a pair of photomultiplier tubes (Thorn EMI, Fairfield, NJ) operating in photon-counting mode. Changes in the fluorescence of indo-1 were converted to [Ca2+]i by using the formula [Ca2+]i = Kdβ(R – Rmin)/(Rmax –R), where R is 405/490 nm fluorescent intensity ratio (Grynkiewicz et al., 1985). The dissociation constant used for indo-1 was 250 nm, and β was the ratio of fluorescence emitted at 490 nm and measured in the absence and presence of Ca2+. Rmin, Rmax, and β were determined in intact cells by applying 10 μm ionomycin in Ca2+-free buffer (1 mm EGTA) and saturating Ca2+ (5 mm Ca2+). Values for Rmin, Rmax, and β were 1.16, 10.4, and 4.3, respectively. Potential cytoplasmic contamination with Mag-indo-1 prevented us from calibrating the indicator; thus, we reported [Ca2+]ER as the ratio (R) of fluorescence intensity of the Ca2+-bound (405 nm) relative to the Ca2+-free form (490 nm) of the dye. To evoke action potentials in intact neurons, extracellular field stimulation was used (Piser et al., 1994). Exponential functions were fitted to the data using a nonlinear, least-squares curve fitting algorithm (Origin 4.1 software; OriginLab, Northampton, MA). Data are presented as mean ± SEM.
Antisense experiments. Gene transfer into DRG neurons was performed using a biolistic particle delivery system as described previously (Usachev et al., 2000). A mammalian expression plasmid (pCI-neo) harboring cDNAs encoding nucleotides 71–443 of PMCA4 in the antisense orientation (AS4) was generated as described previously (Garcia et al., 2001). The pCI-neo-based construct was mixed in a 4:1 ratio with a plasmid encoding enhanced green fluorescent protein (pEGFP-C1; Clontech, Mountain View, CA) and precipitated on 1.6 μm gold particles. After 48 h, transfected cells were identified by green fluorescence [excitation, 480(10) nm; emission, 540(25) nm]. Effective knockdown of PMCA4 was confirmed by immunohistochemistry with PMCA4-specific antibody JA9, as described previously (Usachev et al., 2002).
Results
In sensory neurons, a small increase in [Ca2+]i recovers to basal levels by the concerted action of Ca2+ efflux and sequestration processes (Benham et al., 1992; Werth et al., 1996; Usachev and Thayer, 1999). Rat DRG neurons in culture were challenged with small Ca2+ loads by firing brief trains of action potentials (3–5 s, 6–10 Hz) using electric field stimulation (Piser et al., 1994), and changes in [Ca2+]i were monitored with indo-1-based photometry (Werth and Thayer, 1994). This stimulus is within the physiological range for DRG neurons (Matthews, 1931; Lawson, 2002), evokes a rapid increase in [Ca2+]i that minimizes the effect of Ca2+ buffering on amplitude, and results in peak [Ca2+]i between 200 and 350 nm, which is the range in which ATPases dominate Ca2+ clearance. The rate constant for recovery (k), a value reciprocal to the time constant, was calculated for each field stimulation-evoked [Ca2+]i signal by fitting the [Ca2+]i recovery phase to a monoexponential decay function. As shown in Figure 1, a brief train of action potentials evoked increases in [Ca2+]i that were reproducible in amplitude and recovery rate. We have shown previously that mitochondrial Ca2+ uptake and Na+/Ca2+ exchange do not significantly influence the rate of recovery from these small (<350 nm) increases in [Ca2+]I (Werth and Thayer, 1994; Usachev et al., 2002). However, when PMCA- and SERCA-type Ca2+ pumps were poisoned with vanadate, [Ca2+]i recovery kinetics slowed by more than ninefold (Usachev et al., 2002).
Figure 1.

Activation of PKC accelerates recovery from action potential-induced increases in [Ca2+]i. [Ca2+]i was recorded from single rat DRG neurons using indo-1-based photometry as described in Materials and Methods. A, Brief trains of action potentials (3–5 s, 6–10 Hz) were delivered every 3 min as indicated by the filled triangles, and 0.5 μm PDBu was applied to the bath during the time indicated by the horizontal bar. To maintain comparable Ca2+ loads throughout the experiment, stimulation frequency in the presence of PDBu was increased by 20%. B, Representative [Ca2+]i responses in the absence and presence of PDBu, indicated by arrows in A, were superimposed on an expanded timescale. C, Rate constants describing the recovery kinetics for individual responses from experiments such as the one shown in A were normalized to the first response and plotted versus time. PDBu was applied after the fourth stimulus. Experiments were performed in the absence (filled circles; n = 11) and presence (open circles; n = 5) of 5μm GF109203x (GF) applied at time 0. Data points are means ± SEM.
Activation of PKC accelerates Ca2+ clearance from DRG neurons
Treatment of DRG neurons with 500 nm PDBu, an activator of PKC, significantly accelerated the clearance of Ca2+ from the cytoplasm (Fig. 1A,B). PDBu inhibited whole-cell Ca2+ currents by 32 ± 3% (n = 4), consistent with previous reports (Rane and Dunlap, 1986; Gross and MacDonald, 1989; Boland et al., 1991; Diversepierluissi and Dunlap, 1993) (but see Hall et al., 1995). When peak [Ca2+]i was maintained constant by increasing the stimulus intensity in the presence of PDBu, the rate of Ca2+ recovery (k) increased from 4.3 ± 0.4 to 7.3 ± 0.7 min–1 over ∼20 min (n = 11; p < 0.001 for t ≥ 15 min; repeated measures ANOVA, Bonferroni's post hoc test). This corresponds to an increase in rate constant to 171 ± 10% of control (Fig. 1C). When stimulus strength was held constant throughout the recording, the amplitude of the [Ca2+]i response decreased from 272 ± 39 nm (t = 0) to 221 ± 35 nm (t = 24 min; n = 7), and the rate constant for Ca2+ clearance increased to 175 ± 14% (n = 7) during treatment with PDBu. The stimulation of Ca2+ clearance was completely blocked by the PKC antagonists GF109203x (5 μm; n = 5) (Fig. 1C) and calphostin (300 nm; n = 4; data not shown). Another phorbol ester, phorbol-12-myristate-13-acetate (PMA) (1 μm; 18 min) produced a similar increase in the [Ca2+]i recovery rate (158 ± 15%; n = 7). In contrast, the inactive analog 4-α-PMA (1 μm) was without effect (105 ± 3%; n = 4).
We have shown previously that activation of PKC accelerated Ca2+ efflux by ∼45% via PMCA4 in DRG neurons with SERCAs blocked by treatment with cyclopiazonic acid (CPA) (Usachev et al., 2002). For small Ca2+ loads, CPA reduced the rate constant from 4.9 ± 0.5 to 1.9 ± 0.3 min–1 (n = 8), suggesting that, under these conditions, SERCA accounted for ∼60% of Ca2+ clearance in DRG neurons. The remaining Ca2+ was primarily removed from the cytoplasm by PMCAs (Werth and Thayer, 1994; Usachev et al., 2002). If PMCA was the only Ca2+ transporter targeted by PKC, then the effect of PDBu should be diluted when SERCA was not blocked by CPA. In contrast, we found that, in the absence of CPA, PKC-dependent acceleration of [Ca2+]i recovery was more pronounced (171 ± 10% of control; n = 11) (Fig. 1C), suggesting that PKC might also stimulate another Ca2+ clearance process.
Activation of PKC stimulates PMCA4 and SERCA-dependent Ca2+ clearance
To determine the contribution of SERCA-mediated Ca2+ uptake into the ER to PKC-stimulated acceleration of Ca2+ clearance from the cytoplasm, we examined [Ca2+]i recovery kinetics in cells in which PMCA4, the PKC-sensitive plasma membrane Ca2+ pump isoform, was knocked down by expression of a PMCA4 antisense cDNA (AS4). We have shown previously that, in DRG neurons cotransfected with EGFP reporter and AS4 expression plasmids, PMCA4 immunoreactivity was undetectable (Usachev et al., 2002). In Figure 2, A and B, we show that activation of PKC increased the rate of [Ca2+]i recovery to 139 ± 4% of control (n = 5) in cells with reduced PMCA4 protein. Basal [Ca2+]i in control (63 ± 7 nm; n = 11) and AS4-expressing (66 ± 5 nm; n = 5) cells were similar. In AS4-expressing cells, [Ca2+]i recovery kinetics were not affected by the mitochondrial uncoupling agent FCCP (carbonyl cyanide p-trifluoromethoxyphenylhydrazone) (1 μm) in either the absence (5.2 ± 0.3 and 5.2 ± 0.2 min–1, without or with FCCP, respectively; n = 4) or presence (7.6 ± 0.5 and 7.4 ± 0.7 min–1, without or with FCCP, respectively; n = 4) of PDBu. In PMCA4-deficient cells, CPA essentially blocked stimulation by PDBu (104 ± 9%; n = 11) (Fig. 2C). In contrast, in cells with normal levels of PMCA4, CPA reduced the maximal response in PDBu from 171 ± 10% (n = 11) to 143 ± 5% (n = 14). Thus, activation of PKC stimulates PMCA4-mediated Ca2+ efflux and SERCA-mediated Ca2+ uptake (Fig. 2D).
Figure 2.

PDBu stimulates SERCA-mediated recovery from action potential-induced increases in [Ca2+]i. [Ca2+]i was recorded from single rat DRG neurons using indo-1-based photometry. A–D, DRG neurons were cotransfected with plasmids containing an EGFP reporter construct and an antisense PMCA4 cDNA (AS4). Recordings are from EGFP-positive cells. A, Brief trains of action potentials (3–5s, 6–10 Hz) were delivered every 4 min to an AS4-expressing cell as indicated by the filled triangles. PDBu at 0.5 μm was applied to the bath during the time indicated by the horizontal bar. B, Representative [Ca2+]i responses in the absence and presence of PDBu, indicated by arrows in A, were superimposed on an expanded timescale. C, Rate constants describing the recovery kinetics for individual responses from experiments such as the one shown in A were normalized to the first response and plotted versus time. PDBu was applied after the fourth stimulus. Experiments were performed in the absence (open circles; n = 5) and presence (filled circles; n = 11) of 5 μm CPA applied 30 min before beginning the recording. Data points are means ± SEM. *p < 0.05 relative to the same time point when CPA was not added, one-way ANOVA with Bonferroni's post hoc test. Results in the presence of CPA are replotted from Usachev et al. (2002). D, Bar graph summarizes results of various combinations of SERCA and PMCA4 block on the PDBu effect. *p < 0.05, relative to untreated control (no AS4 and no CPA); #p < 0.05 relative to cells expressing AS4 (no CPA); Student's t test.
Activation of PKC increases the rate of Ca2+ sequestration
We tested further the hypothesis that activation of PKC stimulates Ca2+ uptake into the ER by measuring ER Ca2+ levels ([Ca2+]ER) directly. DRG neurons were loaded with the low-affinity Ca2+ indicator Mag-indo-1 (Kd ≈ 35 μm) under conditions that preferentially loaded the ER, similar to the protocols using another low-affinity Ca2+ dye Mag-fura-2 to monitor [Ca2+]ER in neurons (Fujiwara et al., 2001; Solovyova et al., 2002; Solovyova and Verkhratsky, 2003). Because of the low affinity of this indicator, any dye localized to the cytoplasm did not appreciably contribute to Ca2+-dependent changes in fluorescence. Thus, when the ER was depleted and refilled with Ca2+, indo-1-based [Ca2+]i and Mag-indo-1-based [Ca2+]ER recordings displayed complimentary waveforms (Fig. 3A). Application of CPA (5 μm) in Ca2+-free media produced a slow leak of Ca2+ from the ER into the cytoplasm (Camello et al., 2002). [Ca2+]i returned to a lower baseline as Ca2+ was pumped out of the cell across the plasma membrane. After washing CPA from the cell, return of Ca2+ to the media produced a small increase in [Ca2+]i and a marked refilling of the ER lumen. The [Ca2+]i and [Ca2+]ER traces in Figure 3A were each collected from a separate cell and aligned temporally. We also performed simultaneous [Ca2+]i and [Ca2+]ER measurements using fura-2 and Magfluo-4. The combined recordings were in excellent agreement with Figure 3A and are displayed in supplemental Figure 1 (available at www.jneurosci.org as supplemental material). These data show that the two indicators were clearly detecting Ca2+ in different pools. Changes in [Ca2+]i correlated with the rate of Ca2+ mobilization from intracellular stores (d[Ca2+]ER/dt) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). This observation suggests that [Ca2+]i is the net result of simultaneous Ca2+ mobilization from ER stores and Ca2+ extrusion across the plasma membrane. The rate constant of [Ca2+]ER recovery was derived from an exponential equation fit to the time course of refilling and used as a quantitative index of the rate of Ca2+ uptake into the ER. The mean rate constant (k) for refilling was 0.33 ± 0.05 min–1 (n = 15) and is in good agreement with the rate of store refilling assessed by discharge with caffeine (Usachev and Thayer, 1999).
Figure 3.

Direct recording of [Ca2+]ER to measure SERCA function. [Ca2+] in the lumen of the ER was recorded using Mag-indo-1, and [Ca2+]i was recorded using indo-1. A, Representative [Ca2+]i (top trace) and [Ca2+]ER (bottom trace) recordings obtained from two different cells are plotted on the same timescale. Depleting the Ca2+ stores by blocking SERCA with 5μm CPA in Ca2+-free buffer (indicated by horizontal bars) produced a leak of Ca2+ from the ER, resulting in a decrease in [Ca2+]ER and a corresponding increase in [Ca2+]i. Return of Ca2+ to the media allowed the ER to refill with Ca2+. B, Representative [Ca2+]ER recording shows that the refilling process could be evoked repeatedly. C, If CPA was not removed before the second application of Ca2+ to the bath, refilling was completely blocked. D, The two recovery phases from B were superimposed. The [Ca2+]ER recovery was well described by a single-exponential equation (heavy black line).
We used a paired protocol to assess the modulation of refilling (Fig. 3B). The ER could be repeatedly depleted, and the rate of refilling was remarkably reproducible for a given cell (k2/k1 = 1.07 ± 0.08; n = 15) (Fig. 3D). The initial [Ca2+]ER varied widely from cell to cell, although the amplitude of [Ca2+]ER changes (Δ[Ca2+]ER), defined as the difference between the stable asymptote reached after refilling and the [Ca2+] in depleted stores, reached consistent values under the controlled conditions of the store replenishment protocol. After the second refilling period, Δ[Ca2+]ER reached 118 ± 8% (n = 15) of the value after the first refilling. This Ca2+ reuptake by the stores was mediated by SERCA-type Ca2+ pumps because this process was blocked by 5 μm CPA (Fig. 3C) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). We next used this method to determine the effects of PKC activation on SERCA-mediated refilling of the ER with Ca2+.
Application of PDBu (0.5 μm) before the second refilling in the paired protocol significantly accelerated the rate of Ca2+ uptake (Fig. 4A,B). In the presence of PDBu, the rate of refilling increased to 275 ± 32% (n = 10) of the initial control rate. Interestingly, PDBu also reduced the level of the Ca2+ store replenishment. In Figure 4B, refilling traces in the absence and presence of PDBu are shown superimposed. [Ca2+]ER returned to only 77 ± 4% (n = 10) of control levels in the presence of PDBu (p < 0.01; n = 10) (Fig. 4E). These effects of PDBu resulted from the activation of PKC as indicated by a complete block of the acceleration in the presence of the PKC antagonist Ro31-8220 (Fig. 4C,D).
Figure 4.

Activation of PKC accelerates Ca2+ uptake into the ER. [Ca2+] in the lumen of the ER was recorded using Mag-indo-1-based photometry. A, Representative recording shows the effects of PDBu on the rate of [Ca2+]ER recovery using the refilling protocol described in Figure 3B. CPA at 5μm, Ca2+-free buffer, and 0.5μm PDBu were applied to the bath by superfusion at the times indicated by the horizontal bars. B, [Ca2+]ER recordings from A were superimposed on an expanded timescale. Heavy lines show fitted single-exponential curves. C, [Ca2+]ER recordings from an experiment similar to that in A, except that the PKC antagonist Ro31–8220 (2μm) was applied 10 min before the application of PDBu. The initial control response and the response in the presence of Ro31-8220 plus PDBu were superimposed on an expanded time-scale. Heavy lines show fitted single-exponential curves. D, Bar graph summarizes the effects of PKC activation on refilling kinetics. ***p < 0.001 relative to untreated control, Student's t test. E, Bar graph illustrates difference in completeness of refilling in the absence (control) and presence (0.5 μm) of PDBu. Δ[Ca2+]ER was defined as the difference between the stable asymptote reached after refilling and the [Ca2+] in depleted stores. **p < 0.01 relative to untreated control in same cell, Student's paired t test. F, Representative recording shows the rapid depletion of Ca2+ stores by the application of 30μm CPA in Ca2+-free media. The rate of Ca2+ leaking from the store was quantified by measuring the slope of a linear regression fit to the first 5 min of the [Ca2+]ER trace in CPA (fits are indicated by heavy lines).
To determine whether PKC increased SERCA-mediated Ca2+ transport into the ER or whether it reduced the leak of Ca2+ out of the ER (Camello et al., 2002), we examined the effects of PDBu on the mobilization of Ca2+ from loaded Ca2+ stores. [Ca2+]ER was recorded from DRG neurons after application of a high concentration (30 μm) of CPA to rapidly and completely block Ca2+ uptake (Fig. 4F). The loss of ER Ca2+ appeared to initially follow a linear process. PDBu had no effect on the slope of the linear regression fit to the initial phase of ER Ca2+ mobilization. In control recordings, the slope of the second release phase was 83 ± 2% (n = 4) of the first. When PDBu was added before and during the second release phase, the slope was 80 ± 4% (n = 3) of the first phase, which was not significantly different from control. Thus, PDBu does not appear to influence the Ca2+ leak from the ER, suggesting that PKC activation stimulates SERCA.
PKC-dependent competition between Ca2+ extrusion and Ca2+ sequestration
The traces in Figure 4B indicate that the activation of PKC increased the rate of Ca2+ uptake into the ER but also reduced the total amount of Ca2+ sequestered. We tested the hypothesis that PDBu was stimulating the PMCA, resulting in reduced Ca2+ available for uptake into the ER. We first confirmed that less Ca2+ was taken up into the ER when PKC was activated by using an alternative approach complimentary to the direct [Ca2+]ER measurements in Figure 4. Ca2+ release from the ER was evoked with CPA, and the increase in [Ca2+]i was used as an index of the amount of stored Ca2+. As shown in Figure 5A, 5 μm CPA elicited reproducible elevations in [Ca2+]i when the cells were treated with the same refilling protocol as shown in Figure 3B. Treatment with PDBu during the second refilling phase (the same protocol as in Fig. 4A) decreased the amplitude of the CPA-evoked response (Fig. 5A,B). Thus, activation of PKC reduces the amount of Ca2+ taken up into the ER. Resting [Ca2+]i in naive cells was 53 ± 6 nm (n = 16) and declined significantly to 40 ± 5 nm during treatment with PDBu (p < 0.001, paired Student's t test). PMCA isoform 4 is also stimulated by PKC (Usachev et al., 2002). To determine whether PMCA4-mediated Ca2+ efflux contributed to the reduced refilling of ER Ca2+ stores in the presence of PDBu, PMCA4 was knocked down by expression of a PMCA4 antisense cDNA (AS4). In AS4-treated cells, the CPA-evoked [Ca2+]i increase was comparable with nontransfected cells and cells transfected with EGFP expression vector alone (Fig. 5C). However, when PMCA4 expression was downregulated, PDBu no longer reduced the amplitude of the CPA-evoked response (Fig. 5C). Resting [Ca2+]i in AS4 cells was 57 ± 9 nm and did not change significantly (50 ± 6 nm; p = 0.25, paired Student's t test) during treatment with PDBu (n = 8). Thus, PMCA4 appears to compete with the SERCA, depriving the ER of Ca2+ when stimulated by PKC (Fig. 5D).
Figure 5.
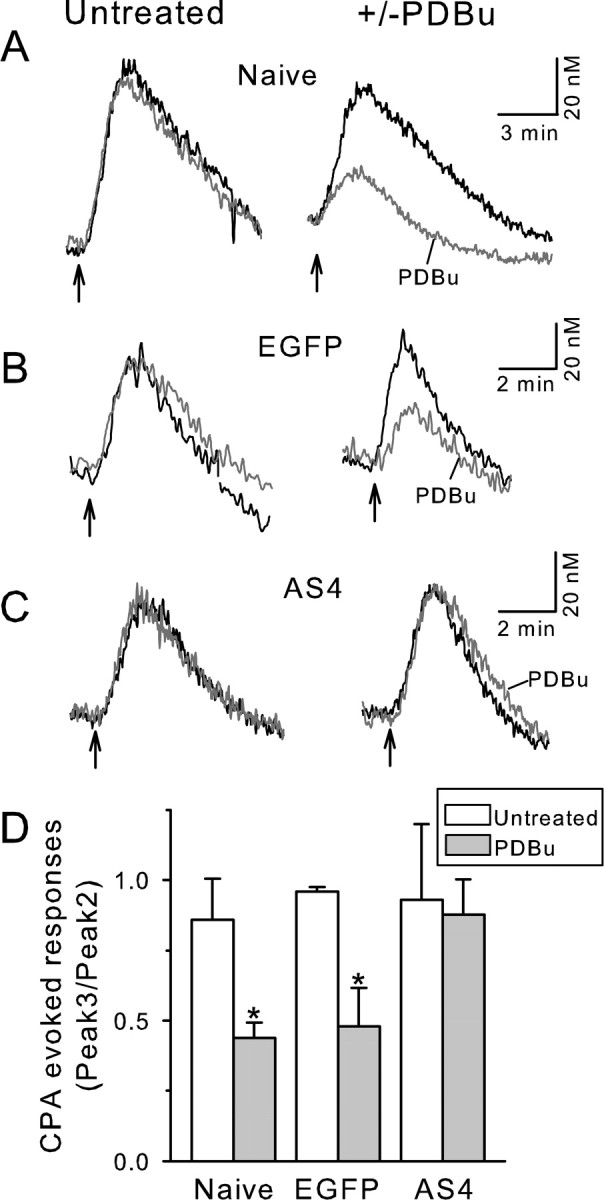
PKC activation reduces ER refilling by stimulating PMCA4. A–C, [Ca2+]i was recorded using indo-1-based photometry. CPA (5 μm) was applied in Ca2+-free media at the times indicated by the arrows. Because the refilling status of the Ca2+ stores varied at the start of the recording, the [Ca2+]i transients shown in A–C are the second and third CPA-evoked responses and correspond to the refilling status of the store after the first and second refilling under controlled conditions. The duration of the treatments are identical to those in Figures 3 (left column) and 4 (right column). Representative traces from nontransfected (Naive) cells (A), cells transfected with EGFP (B), and cells transfected with EGFP plus AS4 (C) are shown. The second and third CPA-evoked responses were superimposed for untreated (left column) and PDBu-treated (right column) cells. D, Bar graph summarizes the changes in the amplitude of the CPA-evoked response in the presence and absence of PDBu. *p < 0.05, PDBu compared with untreated, Student's t test.
If both PMCA4- and SERCA-mediated Ca2+ clearance were accelerated by activation of PKC, then in AS4-treated cells PDBu should accelerate the rate of ER Ca2+ refilling without reducing the capacity of the ER. In AS4-treated cells the rate and completeness of refilling were reproducible (Fig. 6A). Furthermore, as our hypothesis predicted, activation of PKC in these cells accelerated the rate of Ca2+ uptake into the ER (k2/k1 = 1.9 ± 0.3; n = 5), without diminishing the steady level of refilling (Fig. 6B). In AS4-treated cells, the steady-state [Ca2+]ER in the presence of PDBu was 126 ± 11% (n = 5) of control. Thus, both SERCA-mediated Ca2+ sequestration and PMCA-mediated Ca2+ efflux were accelerated by PKC, and these processes compete for Ca2+.
Figure 6.

Activation of PKC accelerates the rate of ER refilling with Ca2+ without reducing capacity in cells lacking the PKC-sensitive PMCA isoform. [Ca2+]ER was recorded using Magindo-1-based photometry. DRG neurons were cotransfected with plasmids containing an EGFP reporter construct and an antisense PMCA4 cDNA. Recordings are from EGFP-positive cells. A, Representative traces show [Ca2+]ER in AS4-treated DRG neuron during repeated application of the refilling protocol described in Figure 3. The rate and degree of refilling were reproducible. B, [Ca2+]ER traces recorded from AS4-treated DRG neurons before and after PDBu application were superimposed. PDBu accelerated the rate of refilling and increased the steady-state [Ca2+]ER.
Discussion
We have shown that activation of PKC markedly accelerates the clearance of Ca2+ from the cytoplasm of rat DRG neurons after physiological Ca2+ loads. Our previous study identified Ca2+ extrusion as one of the PKC targets (Usachev et al., 2002). Here we demonstrate PKC-dependent acceleration of Ca2+ accumulation into the ER. This novel mechanism for regulating the [Ca2+]i in neurons identifies ER Ca2+ uptake as a point of crosstalk between Ca2+ signaling and other second-messenger pathways. Furthermore, because PKC also modulates other elements of the Ca2+ signaling system, these results indicate that Ca2+ regulatory processes are controlled in a coordinated manner.
We used two complimentary approaches to study the acceleration of [Ca2+]i clearance after activation of PKC. We first showed that the rate of [Ca2+]i recovery from action potential-induced increases in [Ca2+]i was increased by PDBu (Fig. 1) in a manner reversed by the PKC antagonists GF109203x and calphostin. The brief train of action potentials (3–5 s, 6–10 Hz) used as a stimulus is well within the physiological range of firing for sensory neurons (Matthews, 1931; Lawson, 2002), suggesting that PKC-dependent acceleration of Ca2+ uptake is relevant to normal signal processing in these cells. The PKC-dependent acceleration of Ca2+ clearance could be entirely accounted for by two processes: stimulation of PMCA4, a PKC-sensitive plasma membrane Ca2+ pump isoform that was inhibited by antisense knockdown, and stimulation of a SERCA-mediated component that was inhibited by CPA (Fig. 2). Indeed, PDBu failed to accelerate Ca2+ clearance in antisense 4-expressing cells treated with CPA. Note that recovery from the small Ca2+ loads introduced by the stimulus used here are not significantly influenced by low-affinity Ca2+ clearance mechanisms such as mitochondria and Na+/Ca2+ exchange (Werth and Thayer, 1994; Usachev et al., 2002). Recovery kinetics were not influenced by response amplitude within the range of peak [Ca2+]i used in this study (Usachev et al., 2002).
The acceleration of [Ca2+]i recovery kinetics via a CPA-sensitive process predicted that the rate of Ca2+ uptake into the ER would also be increased. We tested this hypothesis by directly measuring [Ca2+]ER with the low-affinity dye Mag-indo-1. The low affinity of this dye for Ca2+ (Kd ≈ 35 μm) renders it virtually insensitive to the small changes in [Ca2+]i observed in the protocols used here. Thus, even if some dye contaminated the cytoplasm, it could not account for the described effects of PDBu. Furthermore, our principal observation that PDBu increased the rate of rise of [Ca2+]ER (Fig. 4) was opposite to the increased rate of recovery of [Ca2+]i (Figs. 1, 2). The refilling protocol was initiated by the return of extracellular Ca2+ to the bath. Thus, Ca2+ influx across the plasma membrane could potentially influence the refilling rate. However, refilling kinetics were slower than the rise in [Ca2+]i (Fig. 3A), and PDBu does not increase capacitative Ca2+ influx in these cells (Y. M. Usachev and S. A. Thayer, unpublished observations). Indeed, activation of PKC generally inhibits capacitative Ca2+ influx (Venkatachalam et al., 2004). The effect of PKC on Ca2+ sequestration in the ER was more dramatic in the protocols that measured [Ca2+]ER directly (Fig. 4B,D) relative to protocols that examined [Ca2+]i recovery kinetics in PMCA4-depleted cells (Fig. 2). This quantitative difference can be explained in part by expression in DRG neurons of another PMCA isoform, PMCA2 (Usachev et al., 2002). This isoform is not stimulated by PKC (Enyedi et al., 1997). Thus, the effect of accelerated SERCA function on [Ca2+]i recovery was diluted by the presence of PMCA2 (Fig. 2). Additionally, the stores were filled with Ca2+ by repeated electrical stimulation in the indo-1 recordings (Fig. 2), whereas the rate of Ca2+ uptake into the empty stores was assayed in the Mag-indo-1 experiments (Fig. 4). SERCAs are known to be affected by luminal Ca2+ in various cell types (John et al., 1998; Mogami et al., 1998; Gyorke et al., 2002; Li and Camacho, 2004), including DRG neurons (Usachev and Thayer, 1999). In summary, the PKC-mediated increase in Ca2+ clearance rate corresponded to an increased uptake rate as indicated by complimentary measurements of [Ca2+]i and [Ca2+]ER.
The mechanism by which PKC activation leads to accelerated Ca2+ uptake in DRG neurons is unclear. The results presented in Figure 4F suggest that PKC is stimulating the Ca2+ pump rather than inhibiting the leak of Ca2+ from the ER. Data on the regulation of SERCA-type Ca2+ pumps by direct phosphorylation are limited. Phosphorylation of SERCA2a and SERCA2b by CaMK, but not by PKC or PKA, was reported for cardiac and smooth muscle (Hawkins et al., 1994; Allen and Katz, 1996; Grover et al., 1996). However, a more common mechanism is phosphorylation of accessory proteins such as phospholamban in cardiac and slow-twitch skeletal muscle (Tada and Toyofuku, 1998; MacLennan and Kranias, 2003). PKA- or CaMK-mediated phosphorylation of phospholamban reduces the inhibitory interaction of the protein with SERCA2a, resulting in stimulated pump and increased contractility. Phospholamban is also phosphorylated by PKC, although the physiological outcome of direct phosphorylation by this kinase is unclear (Allen and Katz, 1996). Activation of PKC-α removes inhibition of protein phosphatase-1, leading to dephosphorylation of phospholamban and inhibition of SERCA2 in heart muscle (Braz et al., 2004). Although phospholamban has not been detected in neurons (Plessers et al., 1991), the presence of an analogous protein in brain has been suggested (Dou and Joseph, 1996). Alternatively, phosphorylation of a cytoplasmic domain on calnexin, an ER transmembrane protein that acts to chaperone protein folding within the ER, alters SERCA2b activity when overexpressed in Xenopus oocytes (Roderick et al., 2000). Calnexin and SERCA isoform 2b are ubiquitously expressed in brain (Krijnse-Locker et al., 1995; Baba-Aissa et al., 1998), making it likely that calnexin–SERCA2b interaction is involved in shaping Ca2+ signals in neurons. Another ER chaperone protein, calreticulin, can also modulate SERCA2b activity, although it is not clear whether calreticulin is regulated by phosphorylation (John et al., 1998; Li and Camacho, 2004). Future studies will determine the target of PKC phosphorylation that is responsible for the increased Ca2+ sequestration described here.
In sensory neurons, activation of PKC orchestrates a coordinated reduction in cytoplasmic Ca2+ levels by inhibiting influx (Rane and Dunlap, 1986; Gross and MacDonald, 1989; Boland et al., 1991; Diversepierluissi and Dunlap, 1993), facilitating Ca2+ efflux (Usachev et al., 2002), and, as shown here, accelerating Ca2+ sequestration. Such coordinated modulation of the Ca2+ signal resembles phosphorylation-dependent processes in cardiac muscle, in which changes in the levels of cAMP triggered by activation of β adrenergic receptors regulate Ca2+ influx, uptake, and release to produce coordinated chronotropic and inotropic responses (Petrashevskaya et al., 2002). We suggest that this type of coordinated modulation of Ca2+ signaling processes may prove common in neurons.
Accelerated Ca2+ clearance may affect many functions in sensory neurons, including excitability, secretion, gene expression, and even survival. An increased rate of [Ca2+]i recovery reduces prominent Ca2+-activated K+ currents in DRG neurons (Sah, 1996). The resulting attenuation of the slow afterhyperpolarization in these cells is predicted to reduce spike frequency adaptation and to increase excitability (Abdulla and Smith, 1997; Cordoba-Rodriguez et al., 1999; Bahia et al., 2005). The shape of the presynaptic [Ca2+]i transient has profound effects on neurotransmitter release as does the level of residual Ca2+ (Kamiya and Zucker, 1994; Chen and Regehr, 1999; Muschol and Salzberg, 2000; Korogod et al., 2005). Thus, accelerated [Ca2+]i clearance would be expected to reduce the duration of the secretory response and improve the fidelity of high-frequency synaptic transmission (Talbot et al., 2003). A more rapid refilling of the ER with Ca2+ will also affect Ca2+ release from ryanodine- and IP3-sensitive Ca2+ stores (Berridge, 1998), as well as the store-operated Ca2+ influx present in DRG neurons (Usachev and Thayer, 1999). An increase in SERCA-mediated Ca2+ uptake could influence neurotoxic processes. Accelerating [Ca2+]i clearance might protect from Ca2+ overload (Nicholls and Budd, 2000; Orrenius et al., 2003; Verkhratsky and Toescu, 2003). Alternatively, full ER Ca2+ stores are a prerequisite for some apoptotic pathways, so accelerated refilling of the ER could exacerbate toxic processes (Szabadkai and Rizzuto, 2004). Finally, activation of Ca2+-dependent transcription factors is sensitive to the amplitude and duration of transient increases in [Ca2+]i (Berridge, 1997; Bito et al., 1997; Fields et al., 2001), and, thus, enhanced sequestration could affect gene expression.
In summary, we found that activation of PKC accelerated SERCA-mediated Ca2+ uptake into the ER. Kinase-dependent modulation of SERCA function, which has not been described previously for neurons, may influence a number of ER functions, including protein processing, apoptosis, and Ca2+ signaling. The coordinated stimulation of Ca2+ sequestration and extrusion mechanisms enables PKC to tune [Ca2+]i responses to the changing signaling needs of the cell.
Footnotes
This work was supported by National Science Foundation Grant IBN0110409 and National Institutes of Health Grants DA07304 and DA11806 (S.A.T.). Y.M.U. was supported by American Heart Association National Scientist Development Grant 0535240N. We thank Dr. Emanuel Strehler for providing us with the PMCA4 antisense plasmid.
Correspondence should be addressed to Stanley A. Thayer, Department of Pharmacology, University of Minnesota, 6-120 Jackson Hall, 321 Church Street SE, Minneapolis, MN 55455-0217. E-mail: sathayer@umn.edu.
DOI:10.1523/JNEUROSCI.2920-05.2006
Copyright © 2006 Society for Neuroscience 0270-6474/06/260311-08$15.00/0
References
- Abdulla FA, Smith PA (1997) Ectopic α2-adrenoceptors couple to N-type Ca2+ channels in axotomized rat sensory neurons. J Neurosci 17: 1633–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen BG, Katz S (1996) Phosphorylation of cardiac junctional and free sarcoplasmic reticulum by PKC alpha, PKC beta, PKA and the Ca2+/calmodulin-dependent protein kinase. Mol Cell Biochem 155: 91–103. [DOI] [PubMed] [Google Scholar]
- Baba-Aissa F, Raeymaekers L, Wuytack F, Dode L, Casteels R (1998) Distribution and isoform diversity of the organellar Ca2+ pumps in the brain. Mol Chem Neuropathol 33: 199–208. [DOI] [PubMed] [Google Scholar]
- Bahia PK, Suzuki R, Benton DCH, Jowett AJ, Chen MX, Trezise DJ, Dickenson AH, Moss GWJ (2005) A functional role for small-conductance calcium-activated potassium channels in sensory pathways including nociceptive processes. J Neurosci 25: 3489–3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benham CD, Evans ML, McBain CJ (1992) Ca2+ efflux mechanisms following depolarization evoked calcium transients in cultured rat sensory neurones. J Physiol (Lond) 455: 567–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ (1997) The AM and FM of calcium signalling. Nature 386: 759–760. [DOI] [PubMed] [Google Scholar]
- Berridge MJ (1998) Neuronal calcium signaling. Neuron 21: 13–26. [DOI] [PubMed] [Google Scholar]
- Bito H, Deisseroth K, Tsien RW (1997) Ca2+-dependent regulation of gene expression. Curr Opin Neurobiol 7: 419–429. [DOI] [PubMed] [Google Scholar]
- Boland LM, Allen AC, Dingledine R (1991) Inhibition by bradykinin of voltage-activated barium current in a rat dorsal root ganglion cell line: role of protein kinase C. J Neurosci 11: 1140–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky R, Kimball TF, Lorenz JN, Nairn AC, Liggett SB, Bodi I, Wang S, Schwartz A, Lakatta EG, DePaoli-Roach AA, Robbins J, Hewett TE, Bibb JA, Westfall MV, Kranias EG, Molkentin JD (2004) PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nat Med 10: 248–254. [DOI] [PubMed] [Google Scholar]
- Brostrom MA, Brostrom CO (2003) Calcium dynamics and endoplasmic reticular function in the regulation of protein synthesis: implications for cell growth and adaptability. Cell Calcium 34: 345–363. [DOI] [PubMed] [Google Scholar]
- Camello C, Lomax R, Petersen OH, Tepikin AV (2002) Calcium leak from intracellular stores—the enigma of calcium signalling. Cell Calcium 32: 355–361. [DOI] [PubMed] [Google Scholar]
- Catterall WA (2000) Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol 16: 521–555. [DOI] [PubMed] [Google Scholar]
- Chen C, Regehr WG (1999) Contributions of residual calcium to fast synaptic transmission. J Neurosci 19: 6257–6266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordoba-Rodriguez R, Moore KA, Kao JPY, Weinreich D (1999) Calcium regulation of a slow post-spike hyperpolarization in vagal afferent neurons. Proc Natl Acad Sci USA 96: 7650–7657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diversepierluissi M, Dunlap K (1993) Distinct, convergent 2nd messenger pathways modulate neuronal calcium currents. Neuron 10: 753–760. [DOI] [PubMed] [Google Scholar]
- Dou D, Joseph R (1996) Cloning of human neuronatin gene and its localization to chromosome-20q 11.2–12: the deduced protein is a novel “proteolipid.” Brain Res 723: 8–22. [DOI] [PubMed] [Google Scholar]
- Enyedi A, Elwess NL, Filoteo AG, Verma AK, Paszty K, Penniston JT (1997) Protein kinase C phosphorylates the a forms of plasma membrane Ca2+ pump isoforms 2 and 3 and prevents binding of calmodulin. J Biol Chem 272: 27525–27528. [DOI] [PubMed] [Google Scholar]
- Fields RD, Eshete F, Dudek S, Ozsarac N, Stevens B (2001) Regulation of gene expression by action potentials: dependence on complexity in cellular information processing. Novartis Found Symp 239: 160–172; discussion 172–176, 234–240. [DOI] [PubMed] [Google Scholar]
- Fierro L, DiPolo R, Llano II (1998) Intracellular calcium clearance in Purkinje cell somata from rat cerebellar slices. J Physiol (Lond) 510: 499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friel DD, Tsien RW (1992) A caffeine- and ryanodine-sensitive Ca2+ store in bullfrog sympathetic neurones modulates effects of Ca2+ entry on [Ca2+]. J Physiol (Lond) 450: 217–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara A, Hirose K, Yamazawa T, Iino M (2001) Reduced IP3 sensitivity of IP3 receptor in Purkinje neurons. NeuroReport 12: 2647–2651. [DOI] [PubMed] [Google Scholar]
- Garaschuk O, Yaari Y, Konnerth A (1997) Release and sequestration of calcium by ryanodine-sensitive stores in rat hippocampal neurones. J Physiol (Lond) 502: 13–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia ML, Usachev YM, Thayer SA, Strehler EE, Windebank AJ (2001) Plasma membrane calcium ATPase plays a role in reducing Ca2+-mediated cytotoxicity in PC12 cells. J Neurosci Res 64: 661–669. [DOI] [PubMed] [Google Scholar]
- Gross RA, MacDonald RL (1989) Activators of protein kinase C selectively enhance inactivation of a calcium current component of cultured sensory neurons in a pertussis toxin-sensitive manner. J Neurophysiol 61: 1259–1269. [DOI] [PubMed] [Google Scholar]
- Grover AK, Xu A, Samson SE, Narayanan N (1996) Sarcoplasmic reticulum Ca2+ pump in pig coronary artery smooth muscle is regulated by a novel pathway. Am J Physiol 271: C181–C187. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260: 3440–3450. [PubMed] [Google Scholar]
- Gyorke S, Gyorke I, Lukyanenko V, Terentyev D, Viatchenko-Karpinski S, Wiesner TF (2002) Regulation of sarcoplasmic reticulum calcium release by luminal calcium in cardiac muscle. Front Biosci 7: d1454–d1463. [DOI] [PubMed] [Google Scholar]
- Hall KE, Browning MD, Dudek EM, Macdonald RL (1995) Enhancement of high threshold calcium currents in rat primary afferent neurons by constitutively active protein kinase C. J Neurosci 15: 6069–6076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins C, Xu A, Narayanan N (1994) Sarcoplasmic reticulum calcium pump in cardiac and slow twitch skeletal muscle but not fast twitch skeletal muscle undergoes phosphorylation by endogenous and exogenous Ca2+/calmodulin-dependent protein kinase. Characterization of optimal conditions for calcium pump phosphorylation. J Biol Chem 269: 31198–31206. [PubMed] [Google Scholar]
- John LM, Lechleiter JD, Camacho P (1998) Differential modulation of SERCA2 isoforms by calreticulin. J Cell Biol 142: 963–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya H, Zucker RS (1994) Residual Ca2+ and short-term synaptic plasticity. Nature 371: 603–606. [DOI] [PubMed] [Google Scholar]
- Korogod N, Lou X, Schneggenburger R (2005) Presynaptic Ca2+ requirements and developmental regulation of posttetanic potentiation at the calyx of Held. J Neurosci 25: 5127–5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijnse-Locker J, Parton RG, Fuller SD, Griffiths G, Dotti CG (1995) The organization of the endoplasmic reticulum and the intermediate compartment in cultured rat hippocampal neurons. Mol Biol Cell 6: 1315–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacabaratz-Porret C, Corvazier E, Kovacs T, Bobe R, Bredoux R, Launay S, Papp B, Enouf J (1998) Platelet sarco/endoplasmic reticulum Ca2+ ATPase isoform 3b and Rap 1b: interrelation and regulation in physiopathology. Biochem J 332: 173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson SN (2002) Phenotype and function of somatic primary afferent nociceptive neurones with C-, Adelta- or Aalpha/beta-fibres. Exp Physiol 87: 239–244. [PubMed] [Google Scholar]
- Li Y, Camacho P (2004) Ca2+-dependent redox modulation of SERCA 2b by ERp57. J Cell Biol 164: 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLennan DH, Kranias EG (2003) Phospholamban: a crucial regulator of cardiac contractility. Nat Rev Mol Cell Biol 4: 566–577. [DOI] [PubMed] [Google Scholar]
- Matthews BHC (1931) The response of a single end organ. J Physiol (Lond) 71: 64–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogami H, Tepikin AV, Petersen OH (1998) Termination of cytosolic Ca2+ signals: Ca2+ reuptake into intracellular stores is regulated by the free Ca2+ concentration in the store lumen. EMBO J 17: 435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muschol M, Salzberg BM (2000) Dependence of transient and residual calcium dynamics on action-potential patterning during neuropeptide secretion. J Neurosci 20: 6773–6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neering IR, McBurney RN (1984) Role for microsomal Ca storage in mammalian neurones? Nature 309: 158–160. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL (2000) Mitochondria and neuronal survival. Physiol Rev 80: 315–360. [DOI] [PubMed] [Google Scholar]
- Orrenius S, Zhivotovsky B, Nicotera P (2003) Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol 4: 552–565. [DOI] [PubMed] [Google Scholar]
- Paschen W (2003) Endoplasmic reticulum: a primary target in various acute disorders and degenerative diseases of the brain. Cell Calcium 34: 365–383. [DOI] [PubMed] [Google Scholar]
- Petrashevskaya NN, Koch SE, Bodi I, Schwartz A (2002) Calcium cycling, historic overview and perspectives. Role for autonomic nervous system regulation. J Mol Cell Cardiol 34: 885–896. [DOI] [PubMed] [Google Scholar]
- Piser TM, Lampe RA, Keith RA, Thayer SA (1994) ω-Grammotoxin blocks action-potential-induced Ca2+ influx and whole-cell Ca2+ current in rat dorsal root ganglion neurons. Pflügers Arch 426: 214–220. [DOI] [PubMed] [Google Scholar]
- Plessers L, Eggermont JA, Wuytack F, Casteels R (1991) A study of the organellar Ca2+-transport ATPase isozymes in pig cerebella Purkinje neurons. J Neurosci 11: 650–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rane SG, Dunlap K (1986) Kinase C activator 1,2-oleoylacetylglycerol attenuates voltage-dependent calcium current in sensory neurons. Proc Natl Acad Sci USA 83: 184–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roderick HL, Lechleiter JD, Camacho P (2000) Cytosolic phosphorylation of calnexin controls intracellular Ca2+ oscillations via an interaction with SERCA2b. J Cell Biol 149: 1235–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P (1996) Ca2+-activated K+ currents in neurones: types, physiological roles and modulation. Trends Neurosci 19: 150–154. [DOI] [PubMed] [Google Scholar]
- Salter MW, Kalia LV (2004) Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci 5: 317–328. [DOI] [PubMed] [Google Scholar]
- Shmigol A, Kostyuk P, Verkhratsky A (1994) Role of caffeine-sensitive Ca2+ stores in Ca2+ signal termination in adult mouse DRG neurones. NeuroReport 5: 2073–2076. [DOI] [PubMed] [Google Scholar]
- Solovyova N, Verkhratsky A (2003) Neuronal endoplasmic reticulum acts as a single functional Ca2+ store shared by ryanodine and inositol-1,4,5-trisphosphate receptors as revealed by intra-ER [Ca2+] recordings in single rat sensory neurones. Pflügers Arch 446: 447–454. [DOI] [PubMed] [Google Scholar]
- Solovyova N, Veselovsky N, Toescu EC, Verkhratsky A (2002) Ca2+ dynamics in the lumen of the endoplasmic reticulum in sensory neurons: direct visualization of Ca2+-induced Ca2+ release triggered by physiological Ca2+ entry. EMBO J 21: 622–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Osanai M, Mitsumoto N, Akita T, Narita K, Kijima H, Kuba K (2002) Ca2+-dependent Ca2+ clearance via mitochondrial uptake and plasmalemmal extrusion in frog motor nerve terminals. J Neurophysiol 87: 1816–1823. [DOI] [PubMed] [Google Scholar]
- Szabadkai G, Rizzuto R (2004) Participation of endoplasmic reticulum and mitochondrial calcium handling in apoptosis: more than just neighborhood? FEBS Lett 567: 111–115. [DOI] [PubMed] [Google Scholar]
- Tada M, Toyofuku T (1998) Molecular regulation of phospholamban function and expression. Trends Cardiovasc Med 8: 330–340. [DOI] [PubMed] [Google Scholar]
- Talbot JD, David G, Barrett EF (2003) Inhibition of mitochondrial Ca2+ uptake affects phasic release from motor terminals differently depending on external [Ca2+]. J Neurophysiol 90: 491–502. [DOI] [PubMed] [Google Scholar]
- Usachev YM, Thayer SA (1999) Ca2+ influx in resting rat sensory neurones that regulates and is regulated by ryanodine-sensitive Ca2+ stores. J Physiol (Lond) 519: 115–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usachev YM, Khammanivong A, Campbell C, Thayer SA (2000) Particle-mediated gene transfer to rat neurons in primary culture. Pflügers Arch 439: 730–738. [DOI] [PubMed] [Google Scholar]
- Usachev YM, DeMarco SJ, Campbell C, Strehler EE, Thayer SA (2002) Bradykinin and ATP accelerate Ca2+ efflux from rat sensory neurons via protein kinase C and the plasma membrane Ca2+ pump isoform 4. Neuron 33: 113–122. [DOI] [PubMed] [Google Scholar]
- Venkatachalam K, Zheng F, Gill DL (2004) Control of TRPC and store-operated channels by protein kinase C. Novartis Found Symp 258: 172–185; discussion 185–278, 263–276. [PubMed] [Google Scholar]
- Verkhratsky A (2005) Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol Rev 85: 201–279. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Toescu EC (2003) Endoplasmic reticulum Ca2+ homeostasis and neuronal death. J Cell Mol Med 7: 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werth JL, Thayer SA (1994) Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. J Neurosci 14: 348–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werth JL, Usachev YM, Thayer SA (1996) Modulation of calcium efflux from cultured rat dorsal root ganglion neurons. J Neurosci 16: 1008–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]