

Abstract
The insulin-like growth factor-II/mannose-6-phosphate (IGF-II/M6P) receptor is a single-pass transmembrane glycoprotein that plays an important role in the intracellular trafficking of lysosomal enzymes and endocytosis-mediated degradation of IGF-II. However, its role in signal transduction after IGF-II binding remains unclear. In the present study, we report that IGF-II/M6P receptor in the rat brain is coupled to a G-protein and that its activation by Leu27IGF-II, an analog that binds rather selectively to the IGF-II/M6P receptor, potentiates endogenous acetylcholine release from the rat hippocampal formation. This effect is mediated by a pertussis toxin (PTX)-sensitive GTP-binding protein and is dependent on protein kinase Cα (PKCα)-induced phosphorylation of downstream substrates, myristoylated alanine-rich C kinase substrate, and growth associated protein-43. Additionally, treatment with Leu27IGF-II causes a reduction in whole-cell currents and depolarization of cholinergic basal forebrain neurons. This effect, which is blocked by an antibody against the IGF-II/M6P receptor, is also sensitive to PTX and is mediated via activation of a PKC-dependent pathway. These results together revealed for the first time that the single transmembrane domain IGF-II/M6P receptor expressed in the brain is G-protein coupled and is involved in the regulation of central cholinergic function via the activation of specific intracellular signaling cascades.
Keywords: cholinergic neurons, G-protein-coupled receptor, intracellular signaling, insulin-like growth factors, neurotransmitter release, protein kinase C
Introduction
The insulin-like growth factor-II (IGF-II) receptor, which is identical to the cation-independent mannose-6-phosphate (M6P) receptor, is a single-pass transmembrane glycoprotein containing a large extracellular domain and a small cytoplasmic tail (Morgan et al., 1987; MacDonald et al., 1988; Jones and Clemmons, 1995; Ghosh et al., 2003). The receptor binds IGF-II with higher affinity than IGF-I and does not bind insulin. The receptor also interacts, via distinct sites, with M6P-bearing lysosomal enzymes and a variety of other ligands, such as transforming growth factor-β (TGFβ), leukemia inhibitory factor (LIF), proliferin, and retinoic acid (Dore et al., 1997; Kang et al., 1997; Blanchard et al., 1999). A majority of IGF-II/M6P receptors are expressed within trans-Golgi network/endosomal compartments, in which they segregate newly synthesized lysosomal enzymes for subsequent sorting to endosomes and lysosomes (Kornfeld, 1992; Braulke, 1999). A subset of receptors located on the plasma membrane regulates endocytosis of secreted lysosomal enzymes, mediates internalization and subsequent degradation of IGF-II, LIF, and proliferin, and potentiates activation of latent TGFβ (Hille-Rehfeld, 1995; Braulke, 1999).
Unlike its participation in intracellular trafficking, the role of the IGF-II/M6P receptor in the transmembrane signaling of IGF-II remains unclear. The biological effects of IGF-II are mostly mediated via the IGF-I receptor or insulin receptor isoform A receptor, whereas the IGF-II/M6P receptor acts as a “clearance receptor” to stabilize local IGF-II concentration (Frasca et al., 1999; Hawkes and Kar, 2004). In some nonneuronal tissues, however, the IGF-II/M6P receptor mediates specific biological responses after IGF-II binding, including amino acid uptake (Shimizu et al., 1986), motility of human rhabdomyosarcoma cells (Minniti et al., 1992), and stimulation of Na+/H+ exchange and inositol triphosphate (IP3) production (Rogers et al., 1990). The underlying mechanisms whereby the IGF-II/M6P receptor mediates these effects remain unclear. Nevertheless, some studies, including the demonstration of a putative G-protein binding site within the cytoplasmic domain of the receptor, suggest a possible role for G-proteins in the ligand-induced responses of the receptor (Murayama et al., 1990; Ikezu et al., 1995). However, failure of the receptor to couple with a G-protein under certain conditions (Sakano et al., 1991; Körner et al., 1995) has raised doubt about the role of the receptor in mediating biological responses after IGF-II binding (Kiess et al., 1994; Dahms and Hancock, 2002).
In the adult rat brain, IGF-II/M6P receptor is known to be distributed in various regions, including cortex, striatum, and hippocampus (Couce et al., 1992; Kar et al., 1993a; Nagano et al., 1995; Hawkes and Kar, 2003). Evidence for the distinct distribution of the IGF-II/M6P receptor from that of IGF-I or insulin receptors and specific alterations in IGF-II/M6P receptor levels in response to various pharmacological/surgical manipulations (Kar et al., 1993b, 1997a; Breese et al., 1996; Walter et al., 1999) raises the possibility that this receptor may have a role in the regulation of certain brain functions. The present study shows that the single transmembrane domain IGF-II/M6P receptor is coupled to a G-protein in the brain and is involved in the potentiation of acetylcholine (ACh) release by activating a G-protein-sensitive, protein kinase Cα (PKCα)-dependent pathway.
Materials and Methods
Materials. Adult (3 months) and postnatal (21-25 d) male Sprague Dawley rats (Charles River Canada, Montreal, Quebec, Canada) were kept in accordance with Institutional and Canadian Council on Animal Care Guidelines. Recombinant IGF-I was purchased from ICN Biomedicals (Montreal, Quebec, Canada), whereas Leu27IGF-II was obtained from GroPep (Adelaide, Australia) and also as a gift from Dr. K. Sakano (Daiichi Pharmaceutical, Tokyo, Japan). Nonhydrolyzable GTP analogs, guanosine-5′-[γ-thio]triphosphate (GTPγS) and guanosine 5′-[β,γ-imido]triphosphate [Gpp(NH)p], were purchased from Roche Diagnostics (Laval, Quebec, Canada), whereas IGF-II, insulin, pertussis toxin (PTX), cholera toxin (CTX), and polymyxin B (PMB) were from Calbiochem (San Diego, CA). Labeled [125I]IGF-I, [125I]IGF-II, [125I]insulin (2000 Ci/mmol), and [3H]cAMP kit were purchased from Amersham Biosciences (Toronto, Ontario, Canada). [γ-32P]ATP, [3H]choline (75 Ci/mmol), and [14C]acetyl-CoA were obtained from PerkinElmer (Mississauga, Ontario, Canada). Tetrodotoxin (TTX), cGMP, adenosine 5′-[β,γ-imido]triphosphate [App(NH)p], isoproterenol, vesicular ACh transporter (VAChT) antibody, and iberiotoxin (IBTX) were obtained from Sigma (St. Louis, MO), IGF-I receptor antagonist JB-1 was from Bachem (Torrance, CA), and anti-IGF-I receptor antibody was purchased from Oncogene (San Diego, CA). Polyclonal rat IGF-II/M6P receptor antibodies and the purified receptor were from our laboratory, as well as from Dr. S. P. Nissley (National Institutes of Health, Bethesda, MD). Anti-phosphotyrosine, anti-PKCα, anti-phospho-myristoylated alanine-rich C kinase substrate (MARCKS), anti-Giα2, anti-Gqα, anti-Gsα, horseradish peroxidase-conjugated antibodies, purified Giα2, and protein A/G-PLUS agarose were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and oligo-dT, Superscript II RT, and Taq polymerase were from Invitrogen (Burlington, Ontario, Canada). Anti-phospho-PKCα and anti-phospho-PKCϵ antibodies were purchased from Upstate Biotechnology (Lake Placid, NY), anti-growth associated protein-43 (GAP-43) antibody was from Chemicon (Temecula, CA), and fluorescence-conjugated secondary antibodies were from Jackson ImmunoResearch (West Grove, PA). All other chemicals of analytical grade were purchased from Sigma, Invitrogen, or Fisher Scientific (Pitts-burgh, PA).
Receptor binding assays. Animals were killed by decapitation, and their brains were processed for either membrane binding assay or receptor autoradiography as described previously (Jafferali et al., 2000). For [125I]IGF-I or [125I]IGF-II membrane binding, hippocampal regions were dissected out, homogenized in 50 mm Tris-HCl, pH 7.4, centrifuged, and then incubated either with 25 pm [125I]IGF-I at 22°C for 2 h in 50 mm Tris-HCl buffer containing 10 mm MgCl2, 0.1% bovine serum albumin (BSA), and 0.1% Bacitracin or with 25 pm [125I]IGF-II at 4°C for 18 h in 50 mm Tris-HCl buffer containing 0.025% BSA. For the insulin binding, hippocampal P2 synaptosomal fractions were incubated with 50 pm [125I]insulin at 4°C for 6 h in 10 mm HEPES containing 0.5% BSA, 0.025% Bacitracin, 0.0125% N-ethylmaleimide (NEM), and 100 kIU/ml aprotinin. For each radioligand, competition binding was performed in the presence of 10-6 to 10-12 m IGF-I, IGF-II, Leu27IGF-II, and insulin. Incubations were terminated either by filtration or centrifugation and then radioactivity was measured. All experiments were performed three to five times, each in triplicate, and data were analyzed using the GraphPad Prism software (GraphPad Software, San Diego, CA).
For in vitro receptor autoradiography, frozen brain sections (20 μm) through the hippocampus were processed as described previously (Kar et al., 1993a, 1997a). In brief, for [125I]IGF-I binding, sections were incubated with 25 pm [125I]IGF-I for 2 h at 22°C in Tris-HCl (50 mm), pH 7.4, buffer containing 10 mm MgCl2, 0.1% BSA, and 0.1% Bacitracin with or without 10-12 to 10-6 m IGF-I, IGF-II, Leu27IGF-II, or insulin. The slides were then washed and exposed against Hyperfilm for 4 d. For [125I]IGF-II binding, sections were incubated with 25 pm [125I]IGF-II in 50 mm Tris-HCl buffer containing 0.025% BSA with or without 10-12 to 10-6 m IGF-I, IGF-II, Leu27IGF-II, or insulin. After 18 h at 4°C, sections were washed and exposed for 3 d against Hyperfilm. The insulin binding was performed by incubating sections with 50 pm [125I]insulin for 18 h at 4°C in 10 mm HEPES, pH 7.4, containing 0.5% BSA, 0.025% Bacitracin, 0.0125% NEM, and 100 kIU/ml aprotinin with or without 10-12 to 10-6 m IGF-I, IGF-II, Leu27IGF-II, or insulin. Sections were washed and then exposed to Hyperfilm for 5 d. All autoradiograms were analyzed using a microcomputer imaging device (MCID) image analysis system (Kar et al., 1997a).
Affinity cross-linking. For cross-linking experiments, hippocampal membranes were incubated with either 25 pm [125I]IGF-II in 50 mm Tris-HCl for 18 h at 4°C or 25 pm [125I]IGF-I in 50 mm Tris-HCl for 2 h at 22°C in the presence or absence of 10-7 m IGF-I, IGF-II, or Leu27IGF-II as mentioned in receptor binding assays. The bound ligands were then cross-linked to their respective receptors by incubating for an additional 50 min at 4°C with 2.5-7.5 × 10-4 m disuccinimidyl suberate (MacDonald, 1991). The reactions were quenched with 500 μl of 0.1 m Tris-HCl, pH 7.4, centrifuged, and then separated by 4-20% SDS-PAGE. Gels were dried and exposed to Hyperfilm for 7-14 d as described previously (MacDonald, 1991).
IGF-II/M6P receptor and G-protein. To establish whether the IGF-II/M6P receptor is linked to a G-protein, hippocampal membranes were prepared as described previously and then incubated with 25 pm [125I]IGF-II in 50 mm Tris-HCl, pH 7.4, buffer containing 0.025% BSA with or without 10-7 to 10-3 m GTPγS, Gpp(NH)p, cGMP, App(NH)p, 5-20 μg/ml preactivated (1 h incubation in 50 mm DTT) PTX or 10-8 to 10-6 m CTX. Nonspecific binding was determined in the presence of 10-7 m unlabeled IGF-II or Leu27IGF-II. In parallel, [125I]IGF-I binding was also performed in the presence of 10-7 to 10-3 m GTPγS and Gpp(NH)p. The binding reaction in both cases was terminated by rapid filtration, and radioactivity was measured with a gamma counter. In certain instances, cross-linking experiments were performed after [125I]IGF-II binding assay with or without GTPγS, Gpp(NH)p, IGF-II, or Leu27IGF-II as described above.
IGF-II/M6P receptor Western blotting and immunocytochemistry. Adult male rats were decapitated, and their brain areas of interest [i.e., medial septum/diagonal band of Broca (DBB), lateral septum, nucleus basalis of Meynert (NBM), and hippocampus] were homogenized in Tris-lysis buffer [50 mm Tris-HCl, pH 7.4, 10 mm MgCl2, 0.1% BSA, 5 mm phenyl-methylsulfonyl fluoride (PMSF), 10 μg/ml leupeptin, and 10 μg/ml aprotinin] as described previously (Hawkes and Kar, 2003). Proteins were separated by gel electrophoresis and then incubated with anti-IGF-II/M6P receptor antibody (1:8000). Membranes were exposed to anti-rabbit IgG and visualized using an ECL kit. For immunolabeling, Bouin's-fixed brain sections were incubated overnight with a cholinergic marker, VAChT antibody (1:250), and then exposed to FITC-conjugated anti-goat IgG for 2 h. The sections were reincubated with anti-IGF-II/ M6P receptor antibody (1:250) and then exposed to Texas Red-conjugated anti-rabbit IgG for 2 h (Hawkes and Kar, 2003). Mounted sections were examined under a Nikon (Tokyo, Japan) PCM2000 confocal microscope.
Determination of ACh release. Brain slices from adult rats were superfused as described previously (Kar et al., 1997b; Seto et al., 2002). Briefly, rats were decapitated, and hippocampus and striatum were dissected out, sliced at 400 μm, and then superfused with oxygenated Krebs' buffer (in mm: 120 NaCl, 4.6 KCl, 2.4 CaCl2, 1.2 KH2PO4, 1.2 MgSO4, 9.9 d-glucose, and 25 NaHCO3, adjusted to pH 7.4) using Brandel (Gaithersburg, MD) superfusion apparatus. The buffer also contained physostigmine (30 μm) to prevent degradation of ACh and choline chloride (10 μm) to support a constant supply of precursor for the synthesis of ACh. Effluents for the first 45 min were discarded to establish a stable basal efflux of ACh, and thereafter samples were collected every 20 min. After 1 h basal efflux, tissues were stimulated with high-K+ Krebs' buffer (25 mm KCl with equimolar reduction in NaCl to conserve isotonicity) for an additional 1 h in either the presence or absence of 10-12 to 10-8 m Leu27IGF-II or 10-9 m IGF-II. In some experiments, hippocampal slices were superfused with 25 mm K+ Krebs' buffer alone or containing 10-8 m Leu27IGF-II with or without 10 μg TTX, 25 μg/ml PTX, 25 μm PMB, or 20 μg/ml JB-1. The JB-1 concentration (i.e., 20 μg/ml) used in this study was based on previous results (Fernandez et al., 1999; Quesada and Micevych, 2004). The use of IGF-II/M6P receptor blocking antibody was not feasible because of the permeability problem across 400 μm brain slices as reported previously (Ronn et al., 1995). The superfusates collected every 20 min were processed to detect ACh levels using radioenzymatic assay as described previously (Kar et al., 1997b). Evoked release represents the net release above the basal efflux and is expressed as picomoles of ACh per minute per milligram of protein. The data, which are presented as mean ± SEM, were analyzed using two-tailed Student's t test, with significance set at p < 0.05.
Immunoprecipitation. Rat hippocampal slices were prepared as described for in vitro ACh release and exposed to 10-8 m Leu27IGF-II, 10-7 m IGF-I, 1 μm isoproterenol, 10-7 m IGF-I plus 20 μg/ml JB-1, or 10-8 m Leu27IGF-II plus 25 μg/ml PTX. Tissue slices were then lysed in cold radioimmunoprecipitation assay (RIPA) buffer [50 mm Tris-HCl, 150 mm NaCl, 1 mm EDTA, 1% Igepal CA-630, 0.1% SDS, 50 mm NaF, 1 mm NaVO3, 5 mm PMSF, 10 μg/ml leupeptin, and 10 μg/ml aprotinin] and processed for IGF-II/M6P receptor/Giα2/Gsα/Gqα immunoprecipitation by incubating supernatant as well as pellets at 4°C overnight with either IGF-II/M6P receptor, mouse Giα2, Gsα, or Gqα antibodies. The immune complexes were precipitated by protein A/G PLUS-agarose, separated by gel electrophoresis, and then blotted with anti-Giα2/Gsα/Gqα or IGF-II/M6P receptor antisera, respectively (Zheng et al., 2000). As for Gsα control, isoproterenol-treated slices were immunoprecipitated with an β-adrenergic receptor antibody (1:5000) and then blotted with anti-Gsα antibody. For IGF-I receptor immunoprecipitation, the supernatant was incubated with an anti-IGF-I receptor, and then IGF-I receptor phosphorylation was determined with an anti-phosphotyrosine antibody as described previously (Seto et al., 2002).
Intracellular signaling. Hippocampal slices were superfused for 5, 10, 20, 40, or 60 min with or without 10-8 m Leu27IGF-II and then processed for radioimmunoassay to measure cAMP levels. Additionally, hippocampal slices exposed to either 10-8 m Leu27IGF-II or 10-8 m Leu27IGF-II plus 25 μm PMB for 5, 10, 20, 40, or 60 min were simultaneously processed for Western blotting to evaluate PKC/MARCKS/GAP-43 levels. For [3H]cAMP assay, hippocampal slices were homogenized in 0.5 m Tris-EDTA buffer, pH 7.4, and then assayed as per the instructions of the manufacturer. For Western blotting, tissues were homogenized in RIPA lysis buffer, and subcellular membrane and cytosolic fractions were prepared (Zheng et al., 2000). Proteins were subsequently separated by 4-20% SDS-PAGE and incubated overnight with anti-PKCα (1:5000), anti-phospho-PKCα (1:5000), anti-PKCγ (1:500), anti-phospho-PKCϵ (1:5000), anti-phospho-MARCKS (1:1000), or anti-phospho-GAP-43 (1:5000) antibodies. Membranes were then exposed to the secondary antibody, visualized using an ECL detection kit, and quantified using an MCID system (Hawkes and Kar, 2003).
Dissociation of neurons for electrophysiology. Brains were removed from decapitated male 21-25 d postnatal rats, and the area containing the DBB was dissected out as described previously (Jassar et al., 1999). Acutely dissociated neurons were then prepared by treating the 350 μm slices with trypsin (0.65 mg/ml) at 30°C, followed by mechanical trituration. Cells were then plated on poly-l-lysine-coated coverslips and viewed under a Zeiss (Oberkochen, Germany) inverted microscope.
Voltage- and current-clamp recordings. All whole-cell patch-clamp recordings were performed under visual guidance using either Axopatch-1D or Axopatch 200B amplifiers (Molecular Devices, Union City, CA). Patch electrodes were flame polished to yield resistances of 3-6 MΩ. Whole-cell patch-clamp recordings were obtained using pipette solution that contained the following (in mm): 140 K-methylsulfate, 10 EGTA, 5 MgCl2, 1 CaCl2, 10 HEPES, 2.2 Na2-ATP, and 0.3 Na-GTP, pH 7.2. After whole-cell configuration was established with voltage-clamp mode (holding potential of -80 mV), we waited at least 5 min for the cell to stabilize then either started voltage-clamp studies or switched to current-clamp recording mode. All treatments (i.e., Leu27IGF-II, PTX, PMB, JB-1, IBTX, preimmune Ig, and anti-IGF-II/M6P receptor antibody) were applied either via bath perfusion or through a focal applicator from a micromanifold tip. The current and membrane voltages were recorded using a low-pass filter at 5 kHz and digitized at 10 kHz. Cells were held in voltage clamp at -80 mV, which was close to the resting membrane potential of DBB neurons (Jhamandas et al., 2003). Based on the previous observations (Jassar et al., 1999), to record whole-cell currents, we used a voltage-ramp protocol in which the cells were held at -80 mV and received a 1 s hyperpolarizing pulse (to remove K+ channel inactivation) before being subjected to a slow voltage ramp from -110 to +30 mV at the rate of 20 mV/s. All data, which were analyzed using pClamp6 and 9 software, are presented as mean ± SEM. Student's two-tailed t test was used for determining significance of effect.
Single-cell reverse transcription-PCR. Neurons were harvested after electrophysiological recordings for reverse transcription (RT)-PCR as described previously (Jhamandas et al., 2003). Primer sequences for choline acetyltransferase (ChAT) and glutamate decarboxylase (GAD) have been described previously (Jhamandas et al., 2003), and that for β-actin was obtained from GenBank (the lower primer, 5′-GAT AGA GCC ACC AAT CCA C; the upper primer, 5′-CCA TGT ACG TAG CCA TCC A). All primers were synthesized at the Department of Biochemistry (University of Alberta). The contents were mixed together and placed in the thermal cycler (Jhamandas et al., 2003). A portion of the product was run on a 2% Tris-acetate-EDTA agarose gel, and then the gel was placed in 2 μg/ml ethidium bromide. After 10 min, DNA bands were visualized with ultraviolet transilluminator and photographed.
Results
Characterization of Leu27IGF-II in the rat brain
To determine the receptor specificity of Leu27IGF-II, we first performed competition binding studies in the rat hippocampus, which is enriched with cholinergic terminals and receptors for IGF-I, IGF-II/M6P, and insulin (Hawkes and Kar, 2004). Specific [125I]IGF-II binding sites were competed potently by IGF-II > Leu27IGF-II > IGF-I ≫ insulin (Fig. 1A). The most potent competitors were IGF-II (IC50 of 0.2 nm) and Leu27IGF-II (IC50 of 1 nm), whereas IGF-I was much less potent (IC50 of 15 nm), followed by insulin (IC50 of 3 μm). In contrast, specific [125I]IGF-I binding was inhibited in a dose-dependent manner by IGF-I > IGF-II > insulin > Leu27IGF-II (Fig. 1B). The IC50 values for IGF-I, IGF-II, insulin, and Leu27IGF-II were 0.3 nm, 4.8 nm, 0.2 μm, and 0.8 μm, respectively. Competition studies with [125I]insulin binding sites revealed that insulin (IC50 of 1.4 nm) competed potently than IGF-II (IC50 of 15 nm) > IGF-I (IC50 of 60 nm) ≫ Leu27IGF-II (IC50 of 8 μm) (Fig. 1C).
Figure 1.

Comparative competition binding profiles of IGF-I, IGF-II, insulin, and Leu27IGF-II (L27IGF-II) against [125I]IGF-II (A), [125I]IGF-I (B), and [125I]insulin (C) in adult rat hippocampal membrane preparations. The binding profiles indicate that each ligand bound its respective receptor with higher affinity than related peptides. Leu27IGF-II competed with high affinity for [125I]IGF-II but rather poorly for [125I]IGF-I and [125I]insulin receptor binding sites. Each point represents the mean ± SEM of data obtained from three to five experiments, each performed in triplicate and expressed as percentage of specific binding.
At the regional level, [125I]IGF-II, [125I]IGF-I, and [125I]insulin receptor binding sites exhibited distinct distribution profiles in the hippocampus (Fig. 2A-C). High levels of [125I]IGF-II binding sites, as reported previously (Lesniak et al., 1988; Kar et al., 1993a), were evident in the pyramidal cell layer (12.5 ± 1.7 fmol/mg tissue) of the Ammon's horn and the granular cell layer (17.5 ± 2.3 fmol/mg tissue) of the dentate gyrus (DG), whereas all other layers showed low levels (2.5 ± 0.3 to 3.8 ± 0.5 fmol/mg tissue) of specific labeling (Fig. 2A). The [125I]IGF-II binding was competed potently by IGF-II > Leu27IGF-II > IGF-I ≫ insulin (Fig. 2D,G,J,M). High amounts of [125I]IGF-I binding sites, which were localized in the CA2-CA3 subfields and in the molecular layer of the DG, were competed potently by IGF-I > IGF-II > insulin > Leu27IGF-II (Fig. 2B,E,H,K,N). As for [125I]insulin binding, a high density of binding was evident in the molecular layer of the DG (Fig. 2C), whereas moderate labeling was noted in other layers of the Ammon's horn. Specific [125I] insulin binding was competed potently by insulin > IGF-II > IGF-I ≫ Leu27IGF-II (Fig. 2F,I,L,O).
Figure 2.
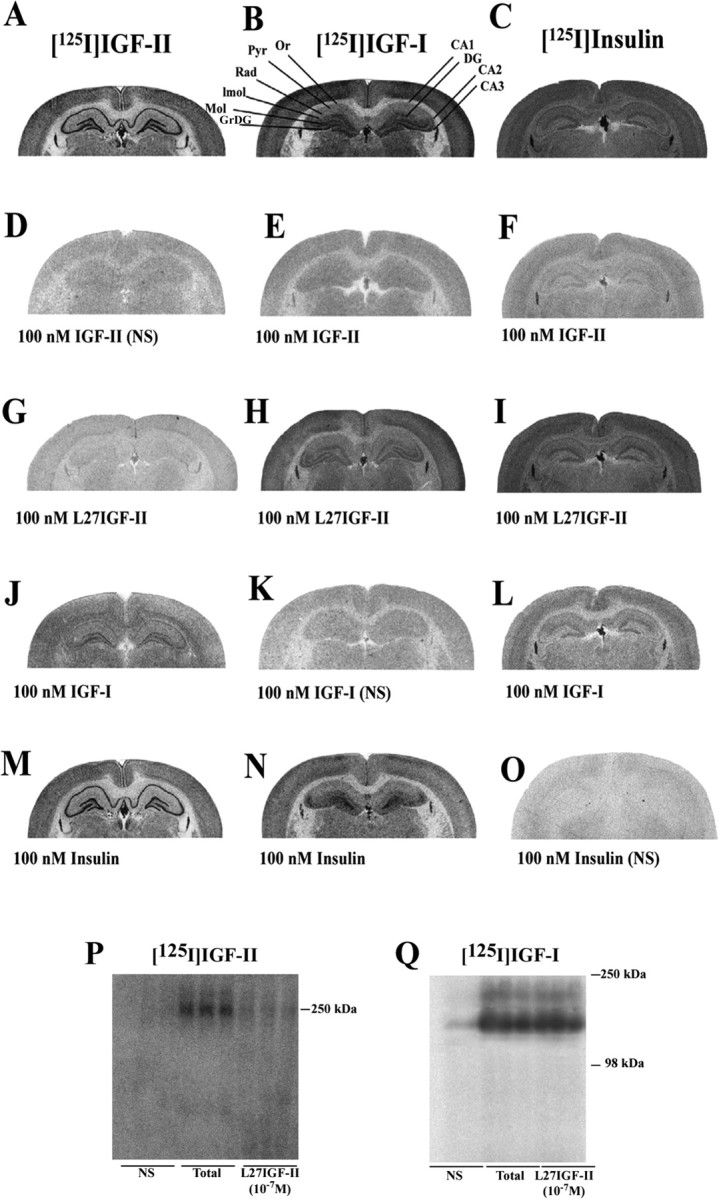
A-Q, Photomicrographs showing the autoradiographic distribution of [125I]IGF-II (A, D, G, J, M), [125I]IGF-I (B, E, H, K, N), and [125I]insulin (C, F, I, L, O) binding sites in the absence or presence of 100 nm IGF-II, Leu27IGF-II (L27IGF-II), IGF-I, and insulin in the adult rat hippocampus. [125I]IGF-II binding in the hippocampus was competed potently by IGF-II > Leu27IGF-II > IGF-I (D, G, J) but not much by insulin (M). [125I]IGF-I binding was competed potently by IGF-I > IGF-II > insulin > Leu27IGF-II (K, E, N, H). [125I] Insulin binding was competed by insulin > IGF-II > IGF-I ≫ Leu27IGF-II (O, F, L, I). P, Q, Affinity cross-linking of [125I]IGF-II (P) and [125I]IGF-I (Q) to rat hippocampal membranes showing that Leu27IGF-II displaces radiolabel from the 250 kDa band corresponding to the IGF-II/M6P receptor but not from the 240 or 135 kDa bands bound by [125I]IGF-I. NS, Nonspecific binding; Or, stratum oriens; Pyr, pyramidal cell layer; Rad, stratum radiatum; lmol, stratum lacunosum moleculare; Mol, molecular layer of the DG; GrDG, granular cell layer of the DG.
Affinity cross-linking experiments with rat hippocampal membranes revealed that [125I]IGF-II bound to a 250 kDa band corresponding to the IGF-II/M6P receptor (MacDonald, 1991), which was completely displaced by 10-7 m IGF-II or Leu27IGF-II (Fig. 2P). Affinity labeling with [125I]IGF-I showed two bands of ∼135 and 240 kDa, corresponding to the size of the IGF-I receptor α-subunit and α-subunit dimers, respectively (Zeeh et al., 1997). Radiolabel was displaced from both bands by 10-7 m IGF-I but not with 10-7 m Leu27IGF-II (Fig. 2Q).
IGF-II/M6P receptor and PTX-sensitive G-protein
To establish the possible link of the IGF-II/M6P receptor to a G-protein, [125I]IGF-II binding assays were performed in hippocampal membranes with or without 10-7 to 10-3 m cGMP, App(NH)p, GTPγS, and Gpp(NH)p. Both Gpp(NH)p and GTPγS, which promote the shift of receptors from the coupled to the uncoupled form (Stiles et al., 1984), but not cGMP or App(NH)p, inhibited [125I]IGF-II receptor binding with IC50 values of 230 and 270 μm (Fig. 3A), respectively, as reported for seven-transmembrane G-protein-coupled receptors (Inui et al., 1989). In contrast, binding of ligands by the tyrosine kinase IGF-I receptor was unaffected by either analog (Fig. 3B). The interaction of the IGF-II/M6P receptor with Gpp(NH)p and GTPγS was further supported by our cross-linking data, which showed that binding of [125I]IGF-II to its receptor is dose dependently inhibited by GTP analogs (Fig. 3C). To determine the subset of G-proteins associated with the IGF-II/M6P receptor, competition binding experiments were performed using ADP-ribosyltransferase toxins, PTX and CTX, which inhibit Gi/Go- and Gs/G12-proteins, respectively (Yamane and Fung, 1993). Incubation of hippocampal membranes with CTX did not alter [125I]IGF-II receptor binding, whereas PTX decreased binding in a dose-dependent manner (Fig. 3D). To substantiate these data, rat hippocampal proteins were immunoprecipitated with anti-IGF-II/M6P receptor antibody after exposure to 10-8 m Leu27IGF-II and then blotted with an anti-Giα2, anti-Gsα, or anti-Gqα antibody. Our results demonstrate that a Giα2 protein (∼15%), but not Gsα or Gqα protein, coimmunoprecipitated with the IGF-II/M6P receptor (Fig. 3E, G,I) (Gqα data not shown). We were also able to detect IGF-II/M6P receptor (∼18%) in hippocampal proteins after coimmunoprecipitation with an anti-Giα2 antibody (Fig. 3F, H). As for control, a Gsα protein, as expected, was coimmunoprecipitated with a β-adrenergic receptor antibody after isoproterenol stimulation (Fig. 3I) (Gurdal et al., 1997). The interaction between IGF-II/M6P receptor and the Giα2 protein is validated by the observation that PTX treatment reduced the receptor level coimmunoprecipitated by Giα2 antibody (Fig. 3J).
Figure 3.

A, B, Competition binding experiments showing that GTPγS and Gpp(NH)p, but not cGMP or App(NH)p, produced a dose-dependent decrease in [125I]IGF-II binding in adult rat hippocampal membranes (A). [125I]IGF-I binding was unaffected by either GTPγS or Gpp(NH)p (B). C, Affinity cross-linking of [125I]IGF-II to rat hippocampal membranes depicting that GTPγS and IGF-II displace radiolabel from the 250 kDa band corresponding to the IGF-II/M6P receptor. D, Binding experiments showing that PTX, but not CTX, decreased [125I]IGF-II binding in a dose-dependent manner. E-H, Western blots depicting reciprocal coimmunoprecipitation of the IGF-II/M6P receptor and Giα2-protein from hippocampal samples of four different adult rats. These panels show the results of immunoprecipitation (IP) by anti-IGF-II/M6P receptor, followed by Western blotting with anti-Giα2 antibody (E) and the reciprocal immunoprecipitation Western blotting experiment (F). Purified Giα2 (E, far right lane) or IGF-II/M6P receptor (F, far right lane) was loaded as controls. Antibody specificity was confirmed by immunoprecipitation of either IGF-II/M6P receptor followed blotting with anti-IGF-II receptor antibody (G) or Giα2 followed by blotting with anti-Giα2 (H). I, Western blot showing that Gsα-protein can be coimmunoprecipitated from hippocampal samples by a specificβ-adrenergic receptor antibody after isoproterenol stimulation (far right lane) but not by an IGF-II/M6P receptor antibody after Leu27IGF-II treatment (left 4 lanes). J, Western blot showing that PTX treatment reduced the IGF-II/M6P receptor level coimmunoprecipitated by the Giα2 antibody. All competition binding data, which are presented as the mean ± SEM of percentage of specific binding, were obtained from three to five experiments, each performed in triplicate. Nonspecific (NS) and Leu27IGF-II (L27IGF-II) binding detected in the presence of 10-7 m IGF-II and 10-7 m Leu27IGF-II, respectively.
IGF-II/M6P receptor in cholinergic neurons
As a prelude to examining the effects of Leu27IGF-II on cholinergic function, we determined the presence of IGF-II/M6P receptor in the basal forebrain nuclei, which provide cholinergic projection to the hippocampus (Fig. 4A). Our immunoblotting results reveal that the IGF-II/M6P receptor is expressed in the NBM, lateral septum, medial septum/DBB, and hippocampus (Fig. 4B). At the cellular level, IGF-II/M6P receptor immunoreactivity is evident in neurons and their processes in the septum, striatum, NBM, DBB, and hippocampus (Fig. 4C,E). Additionally, all VAChT-positive cholinergic neurons located in the basal forebrain are found to express the receptor (Fig. 4C-E).
Figure 4.
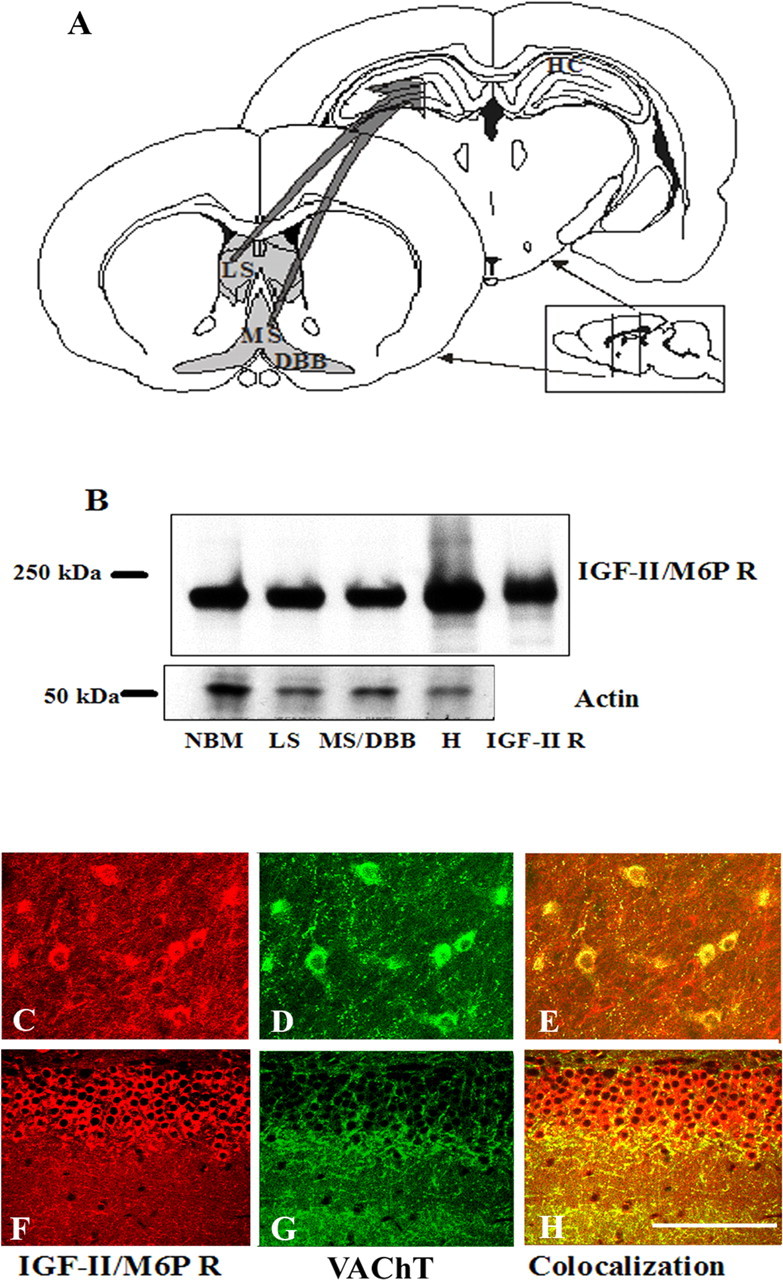
A, Schematic diagram of septohippocampal cholinergic projections in the adult rat brain. B, Western blot showing the presence of IGF-II/M6P receptor in the NBM, lateral septum (LS), medial septum/diagonal band complex (MS/DBB), and hippocampus (H) of the adult rat brain, with purified IGF-II/M6P receptor loaded as control. C-H, Immunofluorescence photomicrographs showing the IGF-II/M6P receptor (C, F) and VAChT immunoreactivities (D, G) and their colocalization (E, H) in the basal forebrain (C-E) and hippocampal granule cell layers (F-H) of the adult rat brain. Scale bar, 10 μm.
Effect of Leu27IGF-II on ACh release
Based on the abundance of IGF-II/M6P receptors in cholinergic neurons, we sought to examine the effect of Leu27IGF-II and IGF-II on ACh release by stimulating hippocampal slices with 25 mm K+ buffer. Leu27IGF-II significantly (p < 0.01) increased ACh release in a dose-dependent manner (Fig. 5A). The potentiation of ACh release was apparent during the final phase of stimulation with 10-8 and 10-9 m Leu27IGF-II (Fig. 5B), whereas no alterations in evoked release were evident at lower concentrations (i.e., 10-10 to 10-12 m). IGF-II (10-9 m), as observed for Leu27IGF-II, was also found to significantly potentiate evoked ACh release from rat hippocampal slices (Fig. 5C). Apart from hippocampus, Leu27IGF-II at 10-8 m augmented evoked ACh release in rat striatal slices (Fig. 5D). The stimulatory response of Leu27IGF-II was insensitive to TTX but sensitive to PTX (Fig. 5E). To determine whether Leu27IGF-II potentiates release by regulating ACh synthesis, the effects of Leu27IGF-II on ChAT activity and high-affinity choline uptake (HACU) were examined (Collier, 1988). However, neither ChAT activity nor HACU was altered by Leu27IGF-II (supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material). Finally, to confirm that the potentiation of ACh release by Leu27IGF-II is not mediated by the IGF-I receptor, hippocampal slices were exposed to 10-8 m Leu27IGF-II or 10-7 m IGF-I and then processed for IGF-I receptor immunoprecipitation, followed by blotting with phosphotyrosine antibodies. Our data reveal that, although IGF-I increased tyrosine phosphorylation of the IGF-I receptor, Leu27IGF-II did not (Fig. 5F). Furthermore, the well characterized IGF-I receptor antagonist JB-1 did not alter the potentiating effect of 10-8 m Leu27IGF-II on hippocampal ACh release (Fig. 5C).
Figure 5.

A, Dose-dependent potentiating effects of Leu27IGF-II (L27IGF-II) on 25 mm K+-evoked ACh release from adult rat hippocampal slices. B, Time course effects of Leu27IGF-II on the evoked ACh release from the hippocampus. Tissue slices were stimulated with 25 mm K+ buffer in the presence (dotted lines) or absence (solid lines) of 10-8 m Leu27IGF-II, and evoked release was potentiated during late phase of stimulation. The inset in B represents total release as percentage of control over the 60 min stimulation period. C, Histograms showing that Leu27IGF-II and IGF-II can potentiate ACh release from adult rat hippocampal slices and the effect of Leu27IGF-II was unaltered in presence of IGF-I receptor antagonist JB-1 (20 μg/ml). D, Time course effects of Leu27IGF-II on the evoked ACh release from the striatum. Tissue slices were stimulated with 25 mm K+ buffer in the presence (dotted lines) or absence (solid lines) of 10-8 m Leu27IGF-II and evoked release was potentiated during late phase of stimulation. The inset in D represents total release as percentage of control over the 60 min stimulation period. E, Histograms showing that 10-8 m Leu27IGF-II-mediated potentiation of evoked ACh release was unaffected by 10 μm TTX but attenuated in the presence of 25 μg/ml PTX in rat hippocampal slices. F, Effect of 10-7 m IGF-I and 10-8 m Leu27IGF-II on tyrosine phosphorylation of the hippocampal IGF-I receptor as determined by immunoprecipitation followed by Western blotting. IGF-I, but not Leu27IGF-II, increased the tyrosine phosphorylation of the IGF-I receptor. The blot shown in the bottom panel represents IGF-I receptor level after reprobing. The blots are representative of experiments that were replicated three times. All ACh release results are expressed as the mean ± SEM (n = 15-18). *p < 0.05; **p < 0.01.
Intracellular pathway involved in Leu27IGF-II-mediated ACh release
IGF-II binding to the IGF-II receptor has been shown to alter the activity of adenylate cyclase and PKC, two intracellular substrates involved in the regulation of ACh release (Zhang et al., 1997; McKinnon et al., 2001). In our study, Leu27IGF-II did not alter [3H]cAMP levels during the course of the experiment (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material). Interestingly, Leu27IGF-II also did not alter either membrane or cytosolic PKCα levels at any time over the 1 h course of the experiment (data not shown), but membrane levels of phospho-PKCα (Fig. 6A) and its downstream substrates MARCKS and GAP-43 (Newton, 2001; Rhee, 2001) (Fig. 6B, C) were significantly increased during the initial 10 min of stimulation and then subsequently declined with their translocation into the cytosol. However, unlike phospho-PKCα or MARCKS, translocation of phospho-GAP-43 into the cytosol did not occur until 20 min exposure to Leu27IGF-II. In contrast to phospho-PKCα, no alterations in phospho-PKCϵ or PKCγ was evident after Leu27IGF-II exposure (Fig. 6D). To confirm the involvement of PKC, hippocampal slices were superfused with 10-8 m Leu27IGF-II in the presence or absence of 25 μm PMB, a PKC inhibitor that has been shown to inhibit ACh release (Iannazzo et al., 2000). Although PMB itself did not alter evoked release, it did block the potentiation of ACh release induced by Leu27IGF-II (Fig. 6E). Additionally, altered levels of phospho-PKCα (Fig. 6F), MARCKS, and GAP-43 (data not shown), observed after treatment with Leu27IGF-II, were also found to be inhibited by PMB.
Figure 6.

A-D, Leu27IGF-II (L) significantly increased membrane levels of phospho-PKCα (A), phospho-MARCKS (B), and phospho-GAP-43 (C) within the first 10 min of stimulation compared with K+-treated (K) controls (top blots). This activation induced their translocation into the cytosolic fraction (bottom blots). Leu27IGF-II had no effect on phospho-PKCϵ levels (D). Quantification of blots was performed as described previously, and histograms were compiled by dividing the optical density value of Leu27IGF-II-treated samples by that of the corresponding K+-treated control samples, for every time point. E, Histogram showing that the PKC inhibitor PMB attenuated Leu27IGF-II-mediated potentiation of hippocampal ACh release. Results are expressed as the mean ± SEM (n = 12-15). **p < 0.01. F, Western blots of phospho-PKCα levels in membrane (top blots) and cytosolic (bottom blots) fractions of hippocampal tissues treated with either high-K+ buffer alone (K) or in combination with Leu27IGF-II (L), PMB (P), or PMB plus Leu27IGF-II (P+L). Leu27IGF-II-induced increase in membrane levels of phospho-PKCα was attenuated in the presence of PMB.
Effects of Leu27IGF-II on DBB neurons
The effects of Leu27IGF-II on ACh release were further examined using whole-cell patch-clamp recordings in dissociated DBB neurons (Figs. 7, 8). In 17 DBB neurons, application of 50 nm Leu27IGF-II resulted in a reversible, significant decrease (p < 0.01) in the outward whole-cell currents in the voltage range from -30 to +30 mV (Fig. 7A), and the response did not desensitize. In five DBB neurons, Leu27IGF-II did not evoke a significant change in peak current at +30 mV (control, 5.4 ± 0.6 nA; Leu27IGF-II, 5.5 ± 0.5 nA; p = 0.58). Under current-clamp conditions, Leu27IGF-II evoked a reversible depolarization of the DBB neurons (11.2 ± 3.4 mV; n = 6) (Fig. 7F). Single-cell RT-PCR analysis revealed that Leu27IGF-II-responsive cells expressed ChAT mRNA-derived product, whereas non-responsive cells expressed GAD mRNA (Fig. 7B).
Figure 7.

A, I-V relationship from 17 DBB neurons showing whole-cell currents evoked under control conditions, in the presence of 50 nm Leu27IGF-II (L27IGF-II), and after recovery. B, Examples of single-cell RT-PCR analysis from two DBB neurons tested with Leu27IGF-II. The Leu27IGF-II-responsive neuron is cholinergic (ChAT positive), whereas the nonresponsive is GABAergic (GAD positive). For each cell, the presence of β-actin acts as a positive control. Lengths of fragments arising from amplification of mRNAs of β-actin of ∼515 bp, ChAT of ∼308 bp, and GAD of ∼400 bp are indicated. C, I-V relationship of mean whole-cell currents evoked by applying voltage ramps under control conditions, in the presence of calcium-activated potassium channel blocker IBTX (25 nm) alone, and Leu27IGF-II (50 nm) with IBTX. IBTX application resulted in a reduction in the outward currents in the voltage range from -30 to +30 mV. In the presence of IBTX, the Leu27IGF-II response is significantly attenuated. In D, voltage protocol for recording IK is depicted on the left with holding potential of -80 mV and a 150 ms conditioning pulse to -40 mV. In this protocol, outward currents are mediated mostly through IK (delayed rectifier), and the effects of Leu27IGF-II (50 nm) on a DBB neuron are shown in the inset (right). Voltage-dependent changes in mean IK from four cells are shown in an I-V plot. E, In the same neuron as D, Leu27IGF-II (50 nm) did not affect the transient outward K+ current (IA) (inset, right). IA was obtained as difference currents by subtracting the currents obtained by the voltage protocol shown in D from that obtained by applying the voltage protocol shown in E (inset, left), in which cells were held at -80 mV and a 150 ms conditioning pulse to -120 mV was applied. Voltage-dependent changes in mean IK from five cells are shown in an I-V plot. F, Under whole-cell current-clamp recording conditions, focal application of Leu27IGF-II depolarized a DBB neuron and evoked action potential generation. The dashed line represents the resting membrane potential of this cell, -70 mV.
Figure 8.

A, I-V relationship from DBB neurons (n = 9) pretreated with IGF-II/M6P receptor antibody (1:100), which showed a drastically reduced response to Leu27IGF-II (L27IGF-II; 50 nm) in whole-cell currents. B, I-V relationship from DBB neurons (n = 12) pretreated with PTX (1 μg/ml), in which Leu27IGF-II (50 nm) did not evoke a significant reduction in whole-cell currents. C, Histograms show the effects of Leu27IGF-II alone (n = 17) and in the cells pretreated with PTX (n = 12) as a percentage of control whole-cell currents at +30 mV; *p < 0.01. D, I-V relationship from DBB neurons (n = 7) in which Leu27IGF-II evoked a significant and reversible reduction in whole-cell currents. In the same cells, inclusion of PMB (10 μm) in the perfusion medium blocks the Leu27IGF-II-induced reduction of whole-cell currents. E, Histograms depict the effects of Leu27IGF-II, PMB, and Leu27IGF-II in the presence of PMB (n = 7) as a percentage of control whole-cell currents at +30 mV; *p < 0.01.
Effects of blocking calcium-activated potassium currents (IC) on the Leu27IGF-II response
The contribution of IK,Ca or IC (BK channels) to the voltage-activated currents can be determined by using the selective blockers of this family of voltage-sensitive calcium-activated potassium channels. The more sensitive and specific one is IBTX (IC50 of 0.25 nm), a snail toxin from Buthus tamulus (Galvez et al., 1990). Figure 7C shows an average of voltage-ramp relationship of currents from four cells evoked under control conditions, with 25 nm IBTX alone and with Leu27IGF-II plus IBTX. IBTX applied alone caused a reduction in outward currents in the same voltage range as Leu27IGF-II. When Leu27IGF-II was applied in the presence of IBTX, there was no additional significant reduction in whole-cell currents than evoked by Leu27IGF-II alone (IBTX, 4.5 ± 0.6 nA; Leu27IGF-II plus IBTX, 4.3 ± 0.5 nA; p > 0.05). The inset in Figure 7C illustrates Leu27IGF-II-evoked reduction of whole-cell currents under control conditions and in the presence of IBTX. Thus, 81% of the Leu27IGF-II-evoked decrease in whole-cell currents is composed of an IBTX-sensitive calcium-activated potassium conductance.
Effects of Leu27IGF-II on transient outward (IA) and the delayed rectifier (IK) potassium currents
IA and IK are voltage-sensitive currents, and their activation and inactivation are strongly voltage-dependent. IA requires the holding potential to be relatively hyperpolarized (approximately -110 mV) for removal of its inactivation, whereas it is inactivated at -40 mV. Conversely, IK is not inactivated at -40 mV. These biophysical properties of IA and IK can, thus, be used to isolate these currents. Therefore, a conditioning pulse to -40 mV will activate IK without any significant contamination by IA (Connor and Stevens, 1971; Jhamandas et al., 2001). A conditioning pulse to -120 mV will activate both IA and IK. The difference currents obtained by subtracting the currents evoked by depolarizing pulses after a conditioning pulse to -40 mV from those evoked after a conditioning pulse to -120 mV provide an accurate estimate of IA. Figure 7D, inset, shows the currents recorded from a DBB neuron with a conditioning pulse to -40 mV for 150 ms, representing mainly IK, under control conditions and in the presence of Leu27IGF-II. Figure 7E shows the difference currents recorded from the same neuron representing mainly IA, under control conditions and in the presence of Leu27IGF-II. Figure 7, D and E, shows the current-voltage (I-V) relationships of averaged peak IK (n = 4) and IA (n = 5), respectively, using the voltage step protocols shown in the insets of this figure. Leu27IGF-II reduced IK by 11% but did not affect IA (peak currents measured at +30 mV).
Specificity of the Leu27IGF-II response in DBB neurons
To establish that the effects of Leu27IGF-II on DBB neurons were mediated via the IGF-II/M6P receptor, dissociated neurons were pretreated with or without a specific IGF-II/M6P receptor blocking antibody (1:100) (for antibody characterization, see supplemental Fig. 1D, available at www.jneurosci.org as supplemental material), and then their response to Leu27IGF-II was assessed. Neurons treated with the IGF-II/M6P receptor antibody, but not preimmune Ig (1:100), exhibited a drastically reduced response to Leu27IGF-II compared with the untreated neurons (untreated neurons, 20.0 ± 1.5 vs antibody-treated neurons, 6.8 ± 0.8; p < 0.01) (Fig. 8A) (supplemental Fig. 1E, available at www.jneurosci.org as supplemental material). In addition, the response of 50 nm Leu27IGF-II was found to be completely maintained in the presence of the IGF-I receptor antagonist JB-1 (20 μg/ml) (supplemental Fig. 1F, available at www.jneurosci.org as supplemental material).
Effects of Leu27IGF-II on DBB neurons in the presence of PTX and PKC inhibitor
To determine whether the Leu27IGF-II-evoked effects on DBB neurons were mediated by a G-protein-coupled mechanism, neurons were pretreated with and without PTX (1 μg/ml) for 5-8 h and assessed for their response to Leu27IGF-II. Leu27IGF-II did not evoke a significant reduction in whole-cell currents in PTX-treated neurons (control, 5.1 ± 0.4 nA; Leu27IGF-II plus PTX, 5.0 ± 0.4 nA; n = 12; p = 0.42), whereas the analog caused a significant reduction in whole-cell currents in neurons not exposed to PTX (Fig. 8B, C). Additionally, Leu27IGF-II had no effect on whole-cell currents in the presence of the PKC inhibitor PMB (10 μm) when compared with control (control, 4.4 ± 0.7 nA; Leu27IGF-II alone, 2.4 ± 0.2 nA; PMB alone, 4.0 ± 0.3 nA; Leu27IGF-II plus PMB, 3.9 ± 0.2 nA; p = 0.51 for control vs Leu27IGF-II plus PMB) (Fig. 8D, E).
Discussion
Using a combination of experimental approaches, the present study provides the very first evidence that the single-pass transmembrane IGF-II/M6P receptor expressed in the brain is G-protein coupled and is involved in the regulation of central cholinergic function by activating PKC-dependent signaling cascades. Traditional G-protein-coupled receptors exhibit a seven-transmembrane spanning conformation that mediate receptor-G-protein interactions (Neves et al., 2002; Pierce et al., 2002). Previous studies done in a cell-free non-neuronal system have suggested a possible interaction between the IGF-II/M6P receptor and G-proteins in response to IGF-II binding (Okamoto et al., 1990; Takahashi et al., 1993; Ikezu et al., 1995). However, failure of the IGF-II/M6P receptor to interact with a G-protein in mouse L-cell membrane and phospholipid vesicles (Sakano et al., 1991; Körner et al., 1995) has challenged the relevance of these results (Dahms and Hancock, 2002). The present study clearly demonstrates an interaction between rat hippocampal IGF-II/M6P receptors and a G-protein. This is supported by four different lines of evidence. First, only GTPγS and Gpp(NH)p, which promote affinity reduction of ligand/receptor binding (Stiles et al., 1984), inhibited [125I]IGF-II interaction with its receptor. Second, PTX, which causes ADP-ribosylation of a cysteine residue in the Gi/o-proteins (Yamane and Fung, 1993), inhibited [125I]IGF-II receptor binding. Third, Giα proteins can be coimmunoprecipitated with the IGF-II/M6P receptors and are sensitive to PTX treatment. Fourth, pretreatment with PTX abolished Leu27IGF-II-mediated ACh release and the response of dissociated cholinergic neurons to this peptide. These results, however, did not provide any evidence of direct interaction between the IGF-II/M6P receptor and the G-protein. Thus, it is possible that the IGF-II/M6P receptor and PTX-sensitive G-proteins may constitute part of a larger protein complex, and the interaction observed in the present study is mediated indirectly via other protein(s). Although other receptors that lack seven transmembrane domains have also been shown to interact with G-proteins (Cunha et al., 1999; Dalle et al., 2001), our results provide compelling evidence that the single transmembrane domain IGF-II/M6P receptor in the adult rat brain is linked to and can mediate cell signaling by activating a G-protein.
The presence of IGF-I, IGF-II, and insulin receptors on most cell types and their ability to bind insulin, IGF-I, and IGF-II has made it difficult to delineate a specific role for the IGF-II/M6P receptor in the mediation of a given biological response. The development of the Leu27IGF-II analog, which interacts selectively with the IGF-II/M6P receptor in a variety of non-neuronal studies, has provided an opportunity to reexamine the role of IGF-II/M6P receptor in cell signaling (Burgisser et al., 1991; Minniti et al., 1992; McKinnon et al., 2001). Our receptor binding assays show that specific [125I]IGF-I, [125I]IGF-II, and [125I]insulin binding sites are concentrated in distinct hippocampal regions. The IGF-I and insulin receptors bind preferentially their own ligands and interact with related ligands at lower affinity. Conversely, the IGF-II/M6P receptor recognizes IGF-II with higher affinity than IGF-I and does not interact with insulin. Leu27IGF-II binds the IGF-II/M6P receptor with high affinity and does not interact substantially with either the IGF-I or insulin receptors. This is supported by our cross-linking studies, which revealed that Leu27IGF-II competes for the IGF-II/M6P receptor but not the IGF-I receptor. These results show that Leu27IGF-II in the adult rat brain, as reported for other tissues (Beukers et al., 1991; Roth et al., 1991; Rosenthal et al., 1994), acts as a rather selective analog for the IGF-II/M6P receptor. To substantiate further that the effects of Leu27IGF-II are mediated by the IGF-II/M6P receptor, we have used a well characterized IGF-II/M6P receptor blocking antibody (MacDonald et al., 1989) that has been shown to distinguish a biological role for the receptor in non-neuronal tissues (Minniti et al., 1992; McKinnon et al., 2001).
Our electrophysiological data reveal that Leu27IGF-II evokes a reduction of whole-cell currents in DBB cholinergic neurons within a voltage range in which a suite of potassium conductances is activated. Among the potassium conductances affected, Leu27IGF-II mostly decreases the calcium-activated potassium conductance, IC, with lesser effects on the delayed rectifier potassium conductance, IK, and no discernable effects on the transient outward potassium conductance, IA. Because IC provides a drive for the repolarization phase of the action potential and plays an important role in the process of spike frequency adaptation (Vergara et al., 1998), the Leu27IGF-II-induced blockade of IC could explain the increase in excitability and depolarization observed under current-clamp conditions. In fact, we have shown previously that, in DBB neurons, inhibition of IC by IBTX, a specific blocker of IC or BK channels, results in membrane depolarization (Jhamandas et al., 2002).
The increase in the cellular activity of cholinergic neurons is consistent with the observed potentiation of ACh release evoked by Leu27IGF-II in brain slices. The latter effect was TTX insensitive, indicating that, within the hippocampus, Leu27IGF-II may possibly act either in close proximity or directly on the cholinergic terminals. The localization of the IGF-II/M6P receptors on cholinergic terminals may provide an anatomical substrate for a direct action of Leu27IGF-II, but it is possible that potentiation of ACh release can be mediated indirectly via other neurotransmitters, because Leu27IGF-II requires prolonged depolarization to potentiate ACh release and IGF-II/M6P receptors in the hippocampus are located primarily on the cell bodies rather than in the cholinergic terminals. Because neither ChAT activity nor HACU is affected, it appears that IGF-II/M6P receptor activation modulates ACh release without altering ACh synthesis. Furthermore, given the evidence that Leu27IGF-II binds rather selectively to the IGF-II/M6P receptor and the analog-induced depolarization of DBB neurons can be blocked by an IGF-II/M6P receptor antibody but not by preimmune serum or an IGF-I receptor antagonist, it is apparent that the potentiation of ACh release by Leu27IGF-II is mediated via this receptor rather than IGF-I receptor. This is further supported by three distinct lines of evidence: (1) Leu27IGF-II, unlike IGF-I, does not induce tyrosine phosphorylation of the IGF-I receptor, (2) the IGF-I receptor antagonist JB-1 did not affect Leu27IGF-II-mediated potentiation of ACh release, and (3) IGF-I has been shown to inhibit hippocampal ACh release indirectly via GABAergic neurons (Seto et al., 2002). The PTX-sensitive effects of Leu27IGF-II on the reduction of whole-cell currents and the potentiation of ACh release not only reinforce our receptor binding data on G-protein-sensitive toxin but also implicate a role for the Gi/o-protein in the regulation of IGF-II/M6P receptor function.
Synaptic release of ACh is modulated by a variety of intracellular messengers, including cAMP and PKC, two molecules known to be regulated by IGF-II (Zhang et al., 1997; McKinnon et al., 2001). Our results indicate that cAMP level is not altered, but phospho-PKCα level and its downstream signaling molecules MARCKS and GAP-43 are time dependently increased in membrane and then in cytosolic fractions after exposure to Leu27IGF-II. These results are in agreement with the established role of PKC in the regulation of ACh release (Vaughan et al., 1999; Iannazzo et al., 2000). Cellular subfractionation studies have demonstrated the recruitment of inactive cytosolic PKCα to the plasma membrane after the generation of second messengers [i.e., IP3, diacyl glycerol (DAG), and Ca2+] for subsequent activation (Newton, 2001). This translocation occurs within 10 min of stimulation and is then gradually down-regulated (Stable and Parker, 1991). IGF-II/M6P receptor activation has been reported to increase the levels of IP3 via phospholipase C (PLC), the enzyme that catalyzes IP3 and DAG production (Rogers et al., 1990; Poiraudeau et al., 1997). Given the evidence that PLC can be directly activated by the βγ subunits of G-proteins (Singer et al., 1997; Rhee, 2001), it is likely that, in our paradigm, Leu27IGF-II stimulates PKCα phosphorylation after IGF-II/M6P receptor-mediated coupling to a G-protein. Activated PKCα subsequently phosphorylates membrane-associated MARCKS, which then translocates into the cytoplasm, allowing for a partial breakdown of the actin cytoskeleton barrier (Vaughan et al., 1999). At the same time, phospho-PKCα also activates membrane-associated GAP-43 proteins, facilitating calcium/calmodulin-dependent kinase activation and increased release of vesicular ACh. The observation that the PKC inhibitor PMB blocked Leu27IGF-II-induced potentiation of ACh release as well as activation of phospho-PKCα, MARCKS, and GAP-43 reinforces a role for the PKC-dependent pathway in mediating response of the IGF-II/M6P receptor in cholinergic neurons. This is further substantiated by our electrophysiological data, which showed that PMB can inhibit Leu27IGF-II-mediated response of whole-cell currents in dissociated cholinergic neurons.
The present study suggests that the IGF-II/M6P receptor can regulate central cholinergic function by activating a specific intracellular signaling pathway. These results are underscored by in vitro data that have shown that IGF-II upregulates the expression of ChAT enzyme in septal cultured neurons by activating the IGF-II/M6P receptor (Konishi et al., 1994) and promotes proliferation/survival of ChAT/calbindin-positive neurons in rat septal cultures (Silva et al., 2000). We also recently reported that IGF-II/M6P receptor level is decreased, as a function of apolipoprotein E ϵ4 allele, in the hippocampus of Alzheimer's disease brain (Kar et al., 2006), in which loss of cholinergic markers have been associated with impairment of cognitive functions (Francis et al., 1999). These results together suggest that the IGF-II/M6P receptor may have a role not only in the normal brain but also in degenerative disorders characterized by preferential loss of cholinergic neurons.
Footnotes
This work was supported by grants from Natural Sciences and Engineering Research Council of Canada (S.K.), Canadian Institutes of Health Research (S.K., J.H.J.), and by National Institutes of Health Grant CA91885 (R.G.M.). We thank Drs. R. Quirion, S. Ahmad, D. Seto, and P. R. Albert, as well as M. Hildebrand for useful discussions during the course of this work. C.H. is a recipient of an Alzheimer Society studentship award. J.H.J. is a recipient of Canada Research Chair in Medicine (Neurology), and S.K. is a recipient of Canada Research Chair in Medicine and Psychiatry and a Senior Scholar award from the Alberta Heritage Foundation for Medical Research.
Correspondence should be addressed to Dr. Satyabrata Kar, Departments of Medicine (Neurology) and Psychiatry, 1E7.44 Walter Mackenzie Center, University of Alberta, Edmonton, Alberta, Canada T6G 2B7. E-mail: skar@ualberta.ca.
Copyright © 2006 Society for Neuroscience 0270-6474/06/260585-12$15.00/0
References
- Beukers MW, Oh Y, Zhang H, Ling N, Rosenfeld RG (1991) [Leu27] insulin-like growth factor II is highly selective for the type-II IGF receptor in binding, cross-linking and thymidine incorporation experiments. Endocrinology 128: 1201-1203. [DOI] [PubMed] [Google Scholar]
- Blanchard F, Duplomb L, Raher S, Vusio P, Hoflack B, Jacques Y, Godard A (1999) Mannose 6-phosphate/insulin-like growth factor II receptor mediates internalization and degradation of leukemia inhibitory factor but not signal transduction. J Biol Chem 274: 24685-24693. [DOI] [PubMed] [Google Scholar]
- Braulke T (1999) Type-2 IGF receptor: a multiple-ligand binding protein. Horm Metab Res 31: 242-246. [DOI] [PubMed] [Google Scholar]
- Breese CR, D'Costa A, Rollins YD, Adams C, Booze RM, Sonntag WE, Leonard S (1996) Expression of insulin-like growth factor-1 (IGF-1) and IGF-binding protein 2 (IGF-BP2) in the hippocampus following cytotoxic lesion of the dentate gyrus. J Comp Neurol 369: 388-404. [DOI] [PubMed] [Google Scholar]
- Burgisser DM, Roth BV, Giger R, Luthi C, Weigl S, Zarn J, Humbel RE (1991) Mutants of human insulin-like growth factor II with altered affinities for the type 1 and type 2 insulin-like growth factor receptor. J Biol Chem 266: 1029-1033. [PubMed] [Google Scholar]
- Collier B (1988) The synthesis and storage of acetylcholine in mammalian cholinergic nerve terminals. In: Neurotransmitters and cortical function: from molecules to mind (Avoli M, Reader TA, Dykes RW, Gloor P, eds), pp 261-276. New York: Plenum.
- Connor JA, Stevens CF (1971) Voltage clamp studies of a transient outward membrane current in gastropod neural somata. J Physiol (Lond) 299: 289-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couce M, Weatherington A, McGinty JF (1992) Expression of insulin-like growth factor-II (IGF-II) and IGF-II/Mannose-6-phosphate receptor in the rat hippocampus: an in situ hybridization and immunocytochemical study. Endocrinology 131: 1636-1642. [DOI] [PubMed] [Google Scholar]
- Cunha RA, Malva JO, Ribeiro JA (1999) Kainate receptors coupled to Gi/Go proteins in the rat hippocampus. Mol Pharmacol 56: 429-433. [DOI] [PubMed] [Google Scholar]
- Dahms NM, Hancock MK (2002) P-type lectins. Biochim Biophys Acta 1572: 317-340. [DOI] [PubMed] [Google Scholar]
- Dalle S, Ricketts W, Imamura T, Vollenweider P, Olefsky JM (2001) Insulin and insulin-like growth factor I receptors utilize different G protein signaling components. J Biol Chem 276: 15688-15695. [DOI] [PubMed] [Google Scholar]
- Dore S, Kar S, Quirion R (1997) Rediscovering an old friend, IGF-I: potential use in the treatment of neurodegenerative diseases. Trends Neurosci 20: 326-331. [DOI] [PubMed] [Google Scholar]
- Fernandez AM, Gonzalez de la Vega A, Planas B, Torres-Aleman I (1999) Neuroprotective actions of peripherally administered insulin-like growth factor-I in the injured olivo-cerebellar pathway. Eur J Neurosci 11: 2019-2030. [DOI] [PubMed] [Google Scholar]
- Francis PT, Palmer AM, Snape M, Wilcock GK (1999) The cholinergic hypothesis of Alzheimer's disease: a review of progress. J Neurol Neurosurg Psychiatry 66: 137-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frasca F, Pandini G, Scalia P, Sciacca L, Mineo R, Costantino A, Goldfine ID, Belfiore A, Vigneri R (1999) Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol Cell Biol 19: 3278-3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvez A, Gimenez-Gallego G, Reuben JP, Roy-Contancin L, Feigenbaum P, Kaczorowski GJ, Garcia ML (1990) Purification and characterization of a unique, potent, peptide probe for the high conductance calcium-activated potassium channel from venom of the scorpion Buthus tamulus J Biol Chem 265: 11083-11090. [PubMed] [Google Scholar]
- Ghosh P, Dahms NM, Kornfeld S (2003) Mannose 6-phosphate receptors: new twists in the tale. Nat Rev Mol Cell Biol 4: 202-212. [DOI] [PubMed] [Google Scholar]
- Gurdal H, Bond RA, Johnson MD, Friedman E, Onaran HO (1997) An efficacy-dependent effect of cardiac overexpression of beta2-adrenoceptor on ligand affinity in transgenic mice. Mol Pharmacol 52: 187-194. [DOI] [PubMed] [Google Scholar]
- Hawkes C, Kar S (2003) Insulin-like growth factor-II/mannose-6-phosphate receptor: widespread distribution in neurons of the central nervous system including those expressing cholinergic phenotype. J Comp Neurol 458: 113-127. [DOI] [PubMed] [Google Scholar]
- Hawkes C, Kar S (2004) The insulin-like growth factor-II/mannose-6-phosphate receptor: structure, distribution and function in the central nervous system. Brain Res Rev 44: 117-140. [DOI] [PubMed] [Google Scholar]
- Hille-Rehfeld A (1995) Mannose 6-phosphate receptors in sorting and transport of lysosomal enzymes. Biochim Biophys Acta 1241: 177-194. [DOI] [PubMed] [Google Scholar]
- Iannazzo L, Kotsonis P, Majewski H (2000) Modulation of acetylcholine release from mouse cortex by protein kinase C, dependence on stimulation intensity. Life Sci 67: 31-38. [DOI] [PubMed] [Google Scholar]
- Ikezu T, Okamoto T, Giambarella U, Yokota T, Nishimoto I (1995) In vivo coupling of insulin-like growth factor II/mannose 6-phosphate receptor to heteromeric G proteins. Distinct roles of cytoplasmic domains and signal sequestration by the receptor. J Biol Chem 270: 29224-29228. [DOI] [PubMed] [Google Scholar]
- Inui A, Okita M, Inoue T, Sakatani N, Oya M, Morioka H, Shii K, Yokono K, Mizuno N, Baba S (1989) Characterization of peptide YY receptors in the brain. Endocrinology 124: 402-409. [DOI] [PubMed] [Google Scholar]
- Jafferali S, Dumont Y, Sotty F, Robitaille Y, Quirion R, Kar S (2000) Insulin-like growth factor-I and its receptor in the frontal cortex, hippocampus and cerebellum of normal human and Alzheimer's disease brains. Synapse 38: 450-459. [DOI] [PubMed] [Google Scholar]
- Jassar BS, Harris KH, Ostashewski PM, Jhamandas JH (1999) Ionic mechanisms of action of neurotensin in acutely dissociated neurons from the diagonal band of Broca of the rat. J Neurophysiol 81: 234-246. [DOI] [PubMed] [Google Scholar]
- Jhamandas JH, Cho C, Jassar B, Harris K, MacTavish D, Easaw J (2001) Cellular mechanisms for amyloid beta-protein activation of rat cholinergic basal forebrain neurons. J Neurophysiol 86: 1312-1320. [DOI] [PubMed] [Google Scholar]
- Jhamandas JH, Harris KH, MacTavish D, Jassar BS (2002) Novel excitatory actions of galanin on rat cholinergic basal forebrain neurons: implications for its role in Alzheimer's disease. J Neurophysiol 87: 696-704. [DOI] [PubMed] [Google Scholar]
- Jhamandas JH, Harris KH, Cho C, Fu W, MacTavish D (2003) Human amylin actions on rat cholinergic basal forebrain neurons: antagonism of beta-amyloid effects. J Neurophysiol 89: 2923-2930. [DOI] [PubMed] [Google Scholar]
- Jones JI, Clemmons DR (1995) Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev 16: 3-34. [DOI] [PubMed] [Google Scholar]
- Kang JX, Li Y, Leaf A (1997) Mannose-6-phosphate/insulin-like growth factor-II receptor is a receptor for retinoic acid. Proc Natl Acad Sci USA 94: 13671-13676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar S, Chabot JG, Quirion R (1993a) Quantitative autoradiographic localization of [125I]insulin-like growth factor I, [125I]insulin-like growth factor II and [125I]insulin receptor binding sites in developing and adult rat brain. J Comp Neurol 333: 375-397. [DOI] [PubMed] [Google Scholar]
- Kar S, Baccichet A, Quirion R, Poirier J (1993b) Entorhinal cortex lesion induces differential responses in [125I]insulin-like growth factor I, [125I]insulin-like growth factor II and [125I]insulin receptor binding sites in the rat hippocampal formation. Neuroscience 55: 69-80. [DOI] [PubMed] [Google Scholar]
- Kar S, Seto D, Dore S, Chabot J-G, Quirion R (1997a) Systemic administration of kainic acid induces selective time dependent decrease in [125I]insulin-like growth factor I, [125I]insulin-like growth factor II and [125I]insulin receptor binding sites in adult rat hippocampal formation. Neuroscience 80: 1041-1055. [DOI] [PubMed] [Google Scholar]
- Kar S, Seto D, Doré S, Hanisch UK, Quirion R (1997b) Insulin-like growth factors-I and -II differentially regulate endogenous acetylcholine release from the hippocampal formation. Proc Natl Acad Sci USA 94: 14054-14059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar S, Poirier J, Guevara J, Dea D, Hawkes C, Robitaille Y, Quirion R (2006) Cellular distribution of insulin-like growth factor-II/mannose-6-phosphate receptor in normal human brain and its alteration in Alzheimer's disease pathology. Neurobiol Aging, in press. [DOI] [PubMed]
- Kiess W, Yang Y, Kessler U, Hoeflich A (1994) Insulin-like growth factor II (IGF-II) and the IGF-II/mannose-6-phosphate receptor: the myth continues. Horm Res 41: 66-73. [DOI] [PubMed] [Google Scholar]
- Konishi T, Takahashi K, Chui DH, Rosenfeld R, Himeno M, Tabira T (1994) Insulin-like growth factor II promotes in vitro cholinergic development of mouse septal neurons: comparison with the effects of insulin-like growth factor I. Brain Res 649: 53-61. [DOI] [PubMed] [Google Scholar]
- Körner C, Nurnberg B, Uhde M, Braulke T (1995) Mannose 6-phosphate/insulin-like growth factor II receptor fails to interact with G-proteins, analysis of mutant cytoplasmic receptor domains. J Biol Chem 270: 287-295. [DOI] [PubMed] [Google Scholar]
- Kornfeld S (1992) Structure and function of the mannose 6-phosphate/insulin-like growth factor II receptors. Annu Rev Biochem 61: 307-330. [DOI] [PubMed] [Google Scholar]
- Lesniak M, Hill J, Kiess W, Rojeski M, Pert C, Roth J (1988) Receptors for insulin-like growth factors I and II: autoradiographic localization in rat brain and comparison to receptors for insulin. Endocrinology 123: 2089-2099. [DOI] [PubMed] [Google Scholar]
- MacDonald RG (1991) Mannose-6-phosphate enhances cross-linking efficiency between insulin-like growth factor-II (IGF-II) and IGF-II/mannose-6-phosphate receptors in membranes. Endocrinology 128: 413-421. [DOI] [PubMed] [Google Scholar]
- MacDonald RG, Pfeffer SR, Coussens L, Tepper MA, Brocklebank CM, Mole JE, Anderson JK, Chen E, Czech MP, Ullrich A (1988) A single receptor binds both insulin-like growth factor II and mannose-6-phosphate. Science 239: 1134-1137. [DOI] [PubMed] [Google Scholar]
- MacDonald RG, Tepper MA, Clairmont KB, Perregaux SB, Czech MP (1989) Serum form of the rat insulin-like growth factor II/mannose 6-phosphate receptor is truncated in the carboxyl-terminal domain. J Biol Chem 264: 3256-3261. [PubMed] [Google Scholar]
- McKinnon T, Chakraborty C, Gleeson LM, Chidiac P, Lala PK (2001) Stimulation of human extravillous trophoblast migration by IGF-II is mediated by IGF type 2 receptor involving inhibitory G protein(s) and phosphorylation of MAPK. J Clin Endocrinol Metab 86: 3665-3674. [DOI] [PubMed] [Google Scholar]
- Minniti CP, Kohn EC, Grubb JH, Sly WS, Oh Y, Muller HL, Rosenfeld RG, Helman LJ (1992) The insulin-like growth factor II (IGF-II)/mannose 6-phosphate receptor mediates IGF-II-induced motility in human rhabdomyosarcoma cells. J Biol Chem 267: 9000-9004. [PubMed] [Google Scholar]
- Morgan DO, Edman JC, Standring DN, Fried VA, Smith MC, Roth RA, Rutter WJ (1987) Insulin-like growth factor II receptor as a multifunctional binding protein. Nature 329: 301-307. [DOI] [PubMed] [Google Scholar]
- Murayama Y, Okamoto T, Ogata E, Asano T, Iiri T, Katada T, Ui M, Grubb JH, Sly WS, Nishimoto I (1990) Distinctive regulation of the functional linkage between the human cation-independent mannose 6-phosphate receptor and GTP-binding proteins by insulin-like growth factor II and mannose 6-phosphate. J Biol Chem 265: 17456-17462. [PubMed] [Google Scholar]
- Nagano T, Sato M, Mori Y, Du Y, Takagi H, Tohyama M (1995) Regional distribution of messenger RNA encoding in the insulin-like growth factor type 2 receptor in the rat lower brainstem. Mol Brain Res 32: 14-24. [DOI] [PubMed] [Google Scholar]
- Neves SR, Ram PT, Iyengar R (2002) G protein pathways. Science 296: 1636-1639. [DOI] [PubMed] [Google Scholar]
- Newton A (2001) Protein kinase C: structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem Rev 101: 2353-2364. [DOI] [PubMed] [Google Scholar]
- Okamoto T, Katada T, Murayama Y, Ui M, Ogata E, Nishimoto I (1990) A simple structure encodes G protein-activating function of the IGF-II/mannose 6-phosphate receptor. Cell 62: 709-717. [DOI] [PubMed] [Google Scholar]
- Pierce KL, Premont RT, Lefkowitz RJ (2002) Seven-transmembrane receptors. Nat Rev Mol Cell Biol 3: 639-650. [DOI] [PubMed] [Google Scholar]
- Poiraudeau S, Lieberherr M, Kergosie N, Corvol MT (1997) Different mechanisms are involved in intracellular calcium increase by insulin-like growth factors 1 and 2 in articular chondrocytes: voltage-gated calcium channels, and/or phospholipase C coupled to a pertussis-sensitive G-protein. J Cell Biochem 64: 414-422. [PubMed] [Google Scholar]
- Quesada A, Micevych PE (2004) Estrogen interacts with the IGF-1 system to protect nigrostriatal dopamine and maintain motoric behavior after 6-hydroxdopamine lesions. J Neurosci Res 75: 107-116. [DOI] [PubMed] [Google Scholar]
- Rhee S-G (2001) Regulation of phosphoinositide-specific phospholipase C. Annu Rev Biochem 70: 281-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers SA, Purchio AF, Hammerman MR (1990) Mannose 6-phosphate-containing peptides activate phospholipase C in proximal tubular baso-lateral membranes from canine kidney. J Biol Chem 265: 9722-9727. [PubMed] [Google Scholar]
- Ronn LC, Bock E, Linnemann D, Jahnsen H (1995) NCAM-antibodies modulate induction of long-term potentiation in rat hippocampal CA1. Brain Res 677: 145-151. [DOI] [PubMed] [Google Scholar]
- Rosenthal SM, Hsiao D, Silverman LA (1994) An insulin-like growth factor-II (IGF-II) analog with highly selective affinity for IGF-II receptors stimulates differentiation, but not IGF-I receptor down-regulation in muscle cells. Endocrinology 135: 38-44. [DOI] [PubMed] [Google Scholar]
- Roth BV, Burgisser DM, Luthi C, Humbel RE (1991) Mutants of human insulin-like growth factor II: expression and characterization of analogs with a substitution of TYR27 and/or a deletion of residues 62-67. Biochem Biophys Res Commun 181: 907-914. [DOI] [PubMed] [Google Scholar]
- Sakano K, Enjoh T, Numata F, Fujiwara H, Marumoto Y, Higashihashi N, Sato Y, Perdue JF, Fujita-Yamaguchi Y (1991) The design, expression, and characterization of human insulin-like growth factor II (IGF-II) mutants specific for either the IGF-II/cation-independent mannose 6-phosphate receptor or IGF-I receptor. J Biol Chem 266: 20626-20635. [PubMed] [Google Scholar]
- Seto D, Zheng W-H, McNicoll A, Collier B, Quirion R, Kar S (2002) Involvement of GABA in insulin-like growth factor-I mediated inhibition of acetylcholine release from rat hippocampal formation. Neuroscience 115: 603-612. [DOI] [PubMed] [Google Scholar]
- Shimizu M, Webster C, Morgan D, Blau H, Roth R (1986) Insulin and insulin-like growth factor receptors and responses in cultured human muscle cells. Am J Physiol 215: E611-E615. [DOI] [PubMed] [Google Scholar]
- Silva A, Montague J, Lopez T, Mudd L (2000) Growth factor effects on survival and development of calbindin immunopositive cultured septal neurons. Brain Res Bull 51: 35-42. [DOI] [PubMed] [Google Scholar]
- Singer WD, Brown HA, Sternweis PC (1997) Regulation of eurkaryotic phosphatidylionositol-specific phospholipase C and phospholipase D. Annu Rev Biochem 66: 475-509. [DOI] [PubMed] [Google Scholar]
- Stable S, Parker PJ (1991) Protein kinase C. Pharmacol Ther 51: 71-95. [DOI] [PubMed] [Google Scholar]
- Stiles GL, Caron MG, Lefkowitz RJ (1984) Beta-adrenergic receptors: Biochemical mechanisms of physiological regulation. Physiol Rev 64: 661-743. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Murayama Y, Okamoto T, Tokata T, Ikezu T, Takahashi S, Giambarella U, Ogata E, Nishimoto I (1993) Conversion of G-protein specificity of insulin-like growth factor II/mannose 6-phosphate receptor by exchanging of a short region with β-adreneergic receptor. Proc Natl Acad Sci USA 90: 11772-11776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan PFT, Walker JH, Peers C (1999) The regulation of neurotransmitter secretion by protein kinase C. Mol Neurobiol 18: 125-155. [DOI] [PubMed] [Google Scholar]
- Vergara C, Latorre R, Marrion NV, Adelman JP (1998) Calcium-activated potassium channels. Curr Opin Neurobiol 8: 321-329. [DOI] [PubMed] [Google Scholar]
- Walter HJ, Berry M, Hill DJ, Cwyfan-Hughes S, Holly JM, Logan A (1999) Distinct sites of insulin-like growth factor (IGF)-II expression and localization in lesioned rat brain: possible roles of IGF binding proteins (IG-FBPs) in the mediation of IGF-II activity. Endocrinology 140: 520-532. [DOI] [PubMed] [Google Scholar]
- Yamane HK, Fung BK (1993) Covalent modifications of G-proteins. Annu Rev Pharmacol Toxicol 33: 201-241. [DOI] [PubMed] [Google Scholar]
- Zeeh JM, Ennes HS, Hoffmann P, Procaccino F, Eysselein VE, Snape WJ, McRoberts JA (1997) Expression of insulin-like growth factor I receptors and binding proteins by colonic smooth muscle cells. Am J Physiol 272: G481-G487. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Tally M, Larsson O, Kennedy R, Huang L, Hall K, Berggren P-O (1997) Insulin-like growth factor-II signaling through the insulin-like growth factor-II/mannose 6-phosphate receptor promotes exocytosis of insulin-secreting cells. Proc Natl Acad Sci USA 94: 6232-6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng WH, Kar S, Quirion R (2000) Stimulation of protein kinase C modulates insulin-like growth factor-1-induced akt activation in PC12 cells. J Biol Chem 275: 13377-13385. [DOI] [PubMed] [Google Scholar]