

Abstract
Postsynaptic alterations have been suggested to account for NMDA receptor (NMDAR)-dependent long-term depression (LTD) and long-term potentiation of synaptic strength, although there is substantial evidence supporting changes in presynaptic release. Direct chemical activation of either NMDA or group I metabotropic glutamate receptor (mGluR1) elicits LTD of similar magnitudes, but it is unknown whether they share common expression mechanisms. Using dual-photon laser-scanning microscopy of FM1-43 [N-(3-triethylammoniumpropyl)-4-(4-(dibutylamino)styryl)pyridinium dibromide] to directly visualize presynaptic vesicular release from the rapidly recycling vesicle pool (RRP) at Schaffer collateral terminals in field CA1 of rat hippocampal slices, we found that a persistent reduction in vesicular release from the RRP is induced by NMDA-LTD but not by mGluR1-LTD. Variance-mean analyses of Schaffer collateral release probability (Pr) at varying extracellular calcium concentrations confirmed that NMDA-LTD was associated with reduced Pr, whereas mGluR1-LTD was not. Pharmacological isolation of NMDAR-dependent and mGluR-dependent forms of stimulus-evoked LTD revealed that both are composed of a combination of presynaptic and postsynaptic alterations. However, when group I mGluR-dependent LTD was isolated by combining an NMDAR blocker with a group II mGluR antagonist, this form of LTD was purely postsynaptic. The nitric oxide synthase inhibitor Nω-nitro-l-arginine blocked the induction of NMDA-LTD but did not alter mGluR-LTD, consistent with a selective role for nitric oxide as a retrograde messenger mediating NMDA-LTD. These data demonstrate that single synapses can express multiple forms of LTD with different sites of expression, that NMDA-LTD is a combination of presynaptic and postsynaptic alterations, but that group I mGluR-LTD appears to be expressed entirely postsynaptically.
Keywords: CA1, hippocampus, long-term depression, metabotropic glutamate receptors, NMDA, presynaptic, rapidly recycling vesicle pool, Schaffer collateral, transmitter release
Introduction
Activity-dependent, long-term changes in synaptic strength, such as long-term potentiation (LTP) and long-term depression (LTD), are believed to be important for information storage, neural network development, fine-tuning of synaptic connections, learning, and memory (Bienenstock et al., 1982; Bailey et al., 1996; Katz and Shatz, 1996; Martin et al., 2000; Braunewell and Manahan-Vaughan, 2001). Studies of mechanisms underlying both LTP and LTD have supplied evidence of presynaptic and postsynaptic long-term alterations (Malenka and Nicoll, 1999; Malinow et al., 2000; Pavlidis et al., 2000). Postsynaptic alterations associated with LTP and LTD include changes in AMPA receptor-gated channel conductances (Benke et al., 1998; Lee et al., 1998), insertion/removal of AMPA receptors (Carroll et al., 1999; Shi et al., 1999), and changes in dendritic spine shape (Engert and Bonhoeffer, 1999; Toni et al., 1999). Evidence of presynaptic alterations has been derived primarily from quantal analysis studies of pairs of monosynaptically connected neurons (Bekkers and Stevens, 1990; Malinow and Tsien, 1990; Bolshakov and Siegelbaum, 1994), vesicular antibody uptake (Malgaroli et al., 1995), and postsynaptic drug infusion (Reyes and Stanton, 1996; Yeckel et al., 1999; Mellor and Nicoll, 2001). More recently, direct two-photon excitation of the vesicle-specific styryl dye FM1-43 [N-(3-triethylammoniumpropyl)-4-(4-(dibutylamino)styryl)pyridinium dibromide] has demonstrated directly that LTP and LTD can both be associated with long-term changes in transmitter release (Stanton et al., 2001, 2003, 2005; Zakharenko et al., 2001), but that partial LTP can also be induced without any change in presynaptic release probability (Stanton et al., 2005), supporting the notion that postsynaptic and presynaptic long-term changes associated with synaptic plasticity can occur independently and be mediated by different cellular biochemical cascades.
There are multiple experimental protocols, both stimulus and chemical, that can elicit LTD at Schaffer collateral–CA1 (Bolshakov and Siegelbaum, 1994; Gage et al., 1997; Oliet et al. 1997; Palmer et al., 1997; Lee et al., 1998; Reyes-Harde et al., 1999; Santschi et al., 1999, 2006) and other synapses (O'Mara et al., 1995; Domenici et al., 1998; Pöschel et al., 2005). Previous studies have shown that stimulus protocols pairing different stimulus frequencies and levels of postsynaptic depolarization can evoke Schaffer collateral LTDs that selectively require either NMDA or metabotropic glutamate receptor (mGluR) activation, but not both (Oliet et al., 1997). Brief bath application of NMDA (Lee et al., 1998) or the mGluR1-selective agonist (S)-3,5–3,5-dihydroxyphenylglycine [(S)-DHPG] (Palmer et al., 1997; Huber et al., 2000, 2001) can each, on their own, elicit long-lasting depressions of Schaffer collateral–CA1 transmission. However, although contradictory data suggest that mGluR1-LTD may be expressed postsynaptically (Otani and Connor, 1998; Nosyreva and Huber, 2005), or presynaptically (Bolshakov and Siegelbaum, 1994; Watabe et al., 2002), it has yet to be directly demonstrated whether long-term presynaptic changes underlie a component of activity-dependent LTD that is selectively the provenance of NMDA- or mGluR-LTD. Furthermore, although recent work has suggested that the rapidly recycling vesicle pool (RRP) is the selective target of presynaptic changes underlying stimulus-evoked LTD (Stanton et al., 2003) and LTP (Stanton et al., 2005), it is unknown whether pharmacological glutamate receptor activation is sufficient to persistently alter RRP release, and which receptors are involved.
Materials and Methods
Slice preparation and electrophysiology.
Fourteen- to 19-d-old Sprague Dawley rats (Taconic, Germantown, NY) were decapitated under deep isoflurane anesthesia, the brains were quickly removed and hemisected, and a tissue block containing the hippocampus was prepared. The block was glued to a stage immersed in ice-cold oxygenated artificial CSF (ACSF; 2–4°C), and 300-μm-thick transverse hippocampal slices were cut with a vibratome (DSK DTK-1000). Slices were placed in an interface holding chamber at 32 ± 1°C for at least 1 h and then kept at room temperature until transfer to an interface chamber for recording at 32°C. Slices were perfused with ACSF (4 ml/min; in mm: 126 NaCl, 3 KCl, 1.25 NaH2PO4, 1.3 MgCl, 2.5 CaCl2, 26 NaHCO3, and 10 glucose) saturated with 95%O2/5%CO2, and all drugs were bath applied. All experiments, except those using FM1-43, were performed in 25 μm bicuculline, and a cut was made in stratum radiatum between CA3 and CA1 to prevent spontaneous synaptic excitation. Schaffer collateral/commissural axons in stratum radiatum were stimulated using a bipolar platinum stimulating electrode (FHC, Bowdoinham, ME) at 0.033 Hz, with baseline intensities chosen to evoke half-maximal field EPSPs in field CA1.
Whole-cell recordings.
Whole-cell patch-clamp recordings were performed in CA1 pyramidal neurons using standard techniques. Patch pipettes (r = 3–4 MΩ) were filled with recording solution containing the following (in mm): 135 CsMeSO2, 8 NaCl, 10 HEPES, 2 Mg-ATP, 0.3 Na-GTP, 0.5 EGTA, and 1 QX-314 [N-(2,6-dimethylphenylcarbamoylmethyl)-triethylammonium bromide], 275 mOsm, pH 7.25 adjusted with Cs(OH)2. Access resistance was carefully monitored, and only cells with stable access resistance (<5% change) were included in analyses. CA1 pyramidal cells were recorded under voltage clamp using a MultiClamp 700B (Molecular Devices, Union City, CA) with Clampex (version 9), filtered at 3 kHz, and digitized at 10 kHz. Sampled data were analyzed off-line with Clampfit (version 9) and OriginPro (version 6.1). Neurons were clamped at −65 mV, and Schaffer collateral-evoked EPSCs were triggered by MultiClamp and delivered to the bipolar stimulating electrode via a stimulus isolator (ISO-Flex; A.M.P.I., Jerusalem, Israel). During induction of NMDA receptor-dependent LTD by either NMDA application or electrical stimulation during mGluR blockade, the membrane potential was held at −35 mV to relieve the Mg2+ block of NMDA channels.
Induction of stimulus-evoked NMDA-LTD and mGluR-LTD.
Different trains of Schaffer collateral stimuli were applied to elicit LTD, depending on which receptor antagonists were present. NMDA receptor-dependent LTD was induced in the presence of the nonspecific mGluR antagonist (S)-α-methyl-4-carboxyphenylglycine [(S)-MCPG], with a simple 2 Hz, 10 min stimulus train (1200 total stimuli). mGluR-dependent LTD was induced in the presence of the NMDA antagonist 2-amino-5-phosphonopentanoic acid (AP-5), by a 2 Hz, 10 min train of paired-pulse stimuli, with an interstimulus interval of 50 ms for each pair (2400 total stimuli). Group I mGluR-dependent LTD was induced in the presence of AP-5 plus the group II mGluR blocker (2S)-α-ethylglutamic acid (EGLU), by a 2 Hz, 10 min train of three pulse bursts of stimuli, with an interstimulus interval of 30 ms between each pulse in a burst (3600 total stimuli). All stimulus pulse durations were 150 μs.
Loading of the rapidly recycling vesicle pools.
After inducing either NMDA-LTD (10 μm, 3 min bath application of NMDA) or mGluR-LTD [25 μm, 5 min bath application of (S)-DHPG], 10 μm 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) was bath applied for the rest of the experiments to prevent synaptically driven action potentials from accelerating dye release. Presynaptic boutons were loaded by bath-applying 5 μm FM1-43 (Invitrogen, Eugene, OR) in hypertonic ACSF supplemented with sucrose to 800 mOsm for 25 s, which selectively loads the rapidly recycling vesicle pool (RRP) (Stanton et al., 2003), and rapidly returning to normal ACSF. In separate control experiments, the loading protocol did not produce any long-term changes in amplitude or shape of Schaffer collateral-evoked synaptic EPSPs, or in pyramidal neuron membrane properties. Stimulus-induced destaining was measured after 30 min in dye-free ACSF, evoked by 1 s bursts of 10 Hz stimulation, applied once each 10 s. These short, discontinuous bursts produced a slower time course of release than does a continuous stimulus train in either brain slices (Stanton et al., 2001, 2003; Zakharenko et al., 2001) or cultured neurons (Rosenmund and Stevens, 1996; Pyle et al., 2000). At the end of each experiment, complete depolarization-induced destaining was evoked by 85 mm [K+] ACSF.
Two-photon imaging.
FM1-43 fluorescence was visualized using a Leica (Nussloch, Germany) DM LFS E upright microscope, two-photon excitation, a water-immersion ultraviolet APO L 40×/0.80 W objective, and a Leica multispectral confocal laser scan unit. The light source was a Millenia 5 W diode laser source pumping a Tsunami Ti:sapphire laser (Spectra-Physics, Fremont, CA) that provided ≈130 fs pulses at 82 MHz, 840 nm center wavelength. Bandpass-filtered epifluorescence was detected with non-descanned photomultiplier tubes behind the objective and a 1.3 numerical aperture oil condensor, optimized for signal over background (540–600 nm) based on spectral analysis with the confocal laser scan head with pinhole maximally open. Laser intensity was controlled with a variable beam splitter exploiting polarization of the laser light and neutral density filters. Although there were no signs of photodamage, we always used the lowest intensity necessary for an adequate signal-to-noise ratio. Pixel images (512 × 512) were acquired, 0.15 μm/pixel in the x–y axes. In off-line analyses, rectangular regions of interest (ROIs; ∼2–4 μm2) were selected around centers of bright, punctate fluorescence spots, and 12–16 boutons and three to four background ROIs were measured per slice. If lateral displacement of a bouton beyond the ROI occurred, the data set was discarded. Moreover, in separate experiments to control for z-axis drift, fluorescent beads (0.5 μm) were injected into hippocampal slices, and their movement was monitored in the same slice chamber. The z-axis drift for a typical data set was estimated at not >0.15 μm/3 min, which was the time period of calculation of initial destaining rates from destaining curves. Only puncta that showed stimulus-dependent unloading were analyzed (∼90% fulfilled this criteria; the nonreleasing sites showed ∼4% destaining over the entire 20 min time course). All fields imaged were 25–60 μm deep in the slice and 40–60 μm away from the bipolar stimulating electrode. Destaining time courses were generated by normalizing each ROI time course to starting intensity, averaging the background fields to produce a dye bleaching time course (2 h bleaching, −12.1 ± 1.0%), and dividing each ROI by bleaching at corresponding time points. The half-time of decay of intensity during unloading (t1/2) was calculated for each punctum from single exponential fits to the first 5 points in destaining curves. Vertical bars are SEMs for the average of all normalized and corrected boutons across experiments.
Statistical analyses.
All data were analyzed by ANOVA or independent Student's t test using SPSS software (SPSS, Chicago, IL). The significance level was preset to p < 0.05. Data are presented as mean ± SEM across experiments.
Chemicals.
AP-5, (−)-bicuculline methobromide, CNQX, (S)-DHPG, EGLU, and (S)-MCPG were obtained from Tocris Bioscience (Ellisville, MO); NMDA and salts were from Sigma-Aldrich (St. Louis, MO), and FM1-43 was from Invitrogen.
Results
LTD in the CA1 region can be induced by short-term activation of either NMDA receptors or group I mGluRs
There is substantial evidence for the expression of at least two forms of hippocampal LTD expressed at Schaffer collateral–CA1 synapses: an NMDA receptor-dependent form (Dudek and Bear, 1992; Mulkey and Malenka, 1992; Oliet et al., 1997) and a group I mGluR-dependent form (Oliet et al., 1997; Huber et al., 2000, 2001; Rush et al., 2002; Tan et al., 2003). Indeed, a few minutes bath application of either NMDA (Lee et al., 1998) or the group I mGluR agonist DHPG (Camodeca et al., 1999; Huber et al., 2000, 2001) have each been shown to elicit LTD at these and other synapses. However, the loci of expression of these two forms of LTD are still debatable. To test whether presynaptic alterations are involved in either form of LTD, we used a number of different methods of sensing presynaptic alterations during LTD induced by bath application of either NMDA or DHPG.
Constant stimulation of Schaffer collaterals at a low frequency of 0.1 Hz was delivered to evoke EPSCs in CA1 pyramidal neurons. After a stable recording period of at least 15 min, the slice was perfused with ASCF containing 10 μm NMDA for 3 min, while holding the membrane potential at −35 mV, and then returned to a holding potential of −65 mV and washed with normal ACSF. As Figure 1A illustrates, peak amplitudes of EPSCs were dramatically decreased after the short-term exposure to NMDA. Although EPSCs partially recovered in amplitude after washout of NMDA, they only reached an EPSC peak amplitude of 47 ± 9% of their original predrug control levels. Similarly, 5 min exposure of slices to 25 μm DHPG produced similar long-term reductions in EPSC peak amplitudes, to 48 ± 10% of predrug baseline amplitudes (Fig. 1B). These results indicate that short-term activation of either NMDA receptors or group I mGluRs can each reliably induce LTD of Schaffer collateral–CA1 synaptic transmission and that the magnitude of these LTDs is quite similar.
Figure 1.

NMDA and the group I mGluR agonist DHPG both elicit LTD of synaptic transmission at Schaffer collateral–CA1 synapses. A, Time course of LTD induced by bath-applied NMDA (10 μm, 3 min; filled bar), plotting normalized Schaffer collateral-evoked EPSC peak amplitudes recorded with whole-cell patch clamp from CA1 pyramidal cells (n = 6). B, Time course of LTD induced by bath-applied (S)-DHPG (25 μm, 5 min; filled bar), plotting normalized Schaffer collateral-evoked EPSC peak amplitudes recorded with whole-cell patch clamp from CA1 pyramidal cells (n = 6). Each point is mean ± SEM. EPSC peak amplitude is from n cells. Insets, Sample averaged evoked EPSCs (n = x) recorded at the times indicated by bars 1 and 2.
NMDA receptor-induced, but not DHPG-induced, LTD is associated with increased paired-pulse facilitation, suggesting different expression loci
Neuronal short-term synaptic plasticity is classically assessed with “paired-pulse stimulation,” two stimuli in close succession (Andersen and Lømo, 1967; Zucker, 1989). One form of paired-pulse modulation, paired-pulse facilitation (PPF), is typically attributed to an increase of release probability (Pr) during the second stimulus, arising from previous accumulation of residual Ca2+ near active zones or a lingering effect of Ca2+ on a Ca2+ sensor (Zucker, 1989; Neher, 1998). Therefore, it is believed that a decrease in initial Pr should be associated with an increase in PPF. As illustrated in Figure 2, A and C, PPF in CA1 pyramidal neurons was readily elicited by two Schaffer collateral stimuli applied with a 30 ms interpulse interval, before drug exposure (solid lines). Thirty minutes after NMDA-LTD was induced by a 3 min application of 10 μm NMDA, PPF was significantly enhanced from a ratio of 2.2 ± 0.3 to 3.2 ± 0.5 (Fig. 2B) (p < 0.05, paired t test; n = 6). In contrast, DHPG-LTD measured 30 min after a 5 min application of 25 μm DHPG did not show any significant change in the magnitude of PPF (Fig. 2D) (2.2 ± 0.2 vs 2.3 ± 0.2; p > 0.20, paired t test; n = 7). These data suggest that NMDA-LTD is associated with presynaptic alterations in Pr, whereas DHPG-LTD is not.
Figure 2.

NMDA-LTD is associated with a long-term increase in PPF, whereas mGluR-LTD is not. A, Average EPSCs (mean of 8–10 traces) in a single CA1 pyramidal neuron evoked by paired-pulse stimuli (30 ms interstimulus interval) before (solid trace) and 30 min after (dotted trace) induction of NMDA-LTD by bath application of 10 μm NMDA for 3 min. Calibration: 50 pA, 25 ms. B, Plot of PPF in each individual experiment (open circles) and mean ± SEM. PPF (filled circles) before versus 30 min after induction of NMDA-LTD (10 μm, 3 min). NMDA-LTD was associated with a significant increase in PPF (*p < 0.05, paired t test). C, Average EPSCs (mean of 8–10 traces) in a single CA1 pyramidal neuron evoked by paired-pulse stimuli (30 ms interstimulus interval) before (solid trace) and 30 min after (dotted trace) induction of mGluR-LTD by bath application of 25 μm (S)-DHPG for 5 min. Calibration: 100 pA, 25 ms. D, Plot of PPF in each individual experiment (open circles) and mean ± SEM. PPF (filled circles) before (Control) and 30 min after induction of mGluR-LTD [25 μm (S)-DHPG, 5 min]. mGLuR-LTD was not associated with any change in PPF (p > 0.20, paired t test).
Estimation of release probability using the paired-pulse refractory period
Based on the observation that individual synapses exhibit a short period (∼5–6 ms) after transmitter release during which the presynaptic terminal is refractory to further release (Stevens and Wang, 1995), Hjelmstad et al. (1997) developed a simple method to estimate Pr by comparing EPSCs evoked with paired pulses separated by 5 and 30 ms intervals. With this method, Pr is given by the following: Pr(estimate) = 1 − EPSC(5)/EPSC(30), where EPSC(5) and EPSC(30) are the amplitudes of the second paired-pulse response at 5 and 30 ms, respectively, after subtracting the EPSCs evoked by the first stimulus. The logic of this method is that, with the 5 ms paired-pulse stimulus, any terminals that release on the first stimulus will be refractory on the second, whereas this will not be the case with the 30 ms interpulse interval. Thus, the amplitude ratio of the second EPSCs at the two intervals cancels the effect of PPF, leaving the reduction in EPSC(5) amplitude compared with EPSC(30) a direct function of Pr. Using this method, we analyzed EPSCs from whole-cell recordings, as shown in schematic diagrams of the method of assaying Pr using this PPF refractory period paradigm (Fig. 3, A,B). Figure 3, C and D, plots the estimated Pr before and during LTD in seven cells exposed to 25 μm DHPG and six cells from 10 μm NMDA, respectively. Neither mGluR-LTD nor NMDA-LTD was associated with any significant change in mean Pr as estimated with this refractory period method.
Figure 3.

Synaptic refractory period provides a measure of release probability (Pr) but does not detect any change in Pr associated with either NMDA-LTD or mGluR-LTD. A, Typical Schaffer collateral-evoked EPSCs (means of 15 traces each) recorded from a CA1 pyramidal neuron using three paired-pulse interstimulus intervals of 0, 5, and 30 ms. B, The top trace shows the subtraction of the first EPSC from the second one evoked at a 5 ms interval, whereas the bottom trace is the subtraction of the first response from that at the 30 ms interstimulus interval. Because presynaptic sites that release on the first stimulus are absolutely refractory after 5 ms but not 30 ms, the ratio of second-response amplitudes at 5 ms/30 ms is a direct function of Pr [see Results and Hjelmstad et al. (1997)]. Calibration: 25 pA, 25 ms. C, Plot of individual (open circles) and mean ± SEM (filled circles; n = 7) estimated Pr before (Control) and 20 min after induction of mGluR-LTD [25 μm (S)-DHPG, 5 min]. D, Plot of individual (open circles) and mean ± SEM (filled circles; n = 6) estimated Pr before (Control) and 20 min after induction of NMDA-LTD. ISI, Interstimulus interval.
Estimation of presynaptic and postsynaptic contributions to expression of LTD by variance-mean analysis
Variance-mean (VM) analysis according to a binomial model of synaptic transmission is a method that has been used to study a growing variety of synapses (Clements and Silver, 2000; Silver, 2003). It is mainly applied to steady-state sequences of evoked EPSCs recorded under a variety of conditions by varying extracellular [Ca2+] or delivering long repetitive trains of stimulation of different frequencies, each resulting in a range of mean response size (Silver et al., 1998; Reid and Clements, 1999; Oleskevich et al., 2000; Foster and Regehr, 2004).
Assuming that the mean amplitude of the synaptic response (Imean) is given by the following:
where N is the total number of release sites, P is the release probability, and Q is the quantal size of the postsynaptic response. The binomial model yields a variance in the size of the synaptic current (σB2) as follows:
This results in a parabolic relationship between the variance and Imean, defined as follows:
Here, Q and N can be obtained from the initial slope and the width of a parabolic fit to a VM plot. Alternatively, this parabola can be written in a linear form as follows:
where Q and N can be determined from the y-axis intercept and the slope of a linear fit to a plot of variance/mean versus mean, respectively.
In this study, we used three ratios of [Ca2+]/[Mg2+] in ACSF (4:1, 2:2, and 1:4 mm) to alter release probability at Schaffer collateral synapses. A typical experiment began with establishing stable whole-cell recording from a CA1 pyramidal neuron and perfusing the slice with 4:1 [Ca2+]/[Mg2+] ACSF. Cells were voltage clamped at −65 mV, and 100 μs constant-current stimulus pulses were delivered to Schaffer collateral/commissural fiber axons every 10 s to evoke an EPSC. Stable recordings for 8–10 min were made in 4:1 [Ca2+]/[Mg2+], before replacing the perfusate with 1:4 mm [Ca2+]/[Mg2+] ACSF. After EPSCs decreased in amplitude and restabilized, which usually took 5–8 min, EPSCs were recorded for an additional 8 min. Slices were then perfused with 2:2 mm [Ca2+]/[Mg2+] ACSF. After EPSC amplitudes had again stabilized, another 8 min of recordings were made. To induce LTD, Slices were exposed to either 10 μm NMDA or 25 μm DHPG in 2:2 mm [Ca2+]/[Mg2+] ACSF for 3 or 5 min, respectively, durations that reliably induced LTD lasting hours. After drug exposure, slices were continuously perfused with 2:2 mm [Ca2+]/[Mg2+] ACSF for at least 30 min, to verify continued expression of LTD, and the same sequence of [Ca2+]/[Mg2+] ACSF applications was repeated. To ensure that postsynaptic AMPA receptors were responding to a nonsaturating concentration of glutamate, a requirement for VM analysis, all experiments were conducted in a low concentration of the selective AMPA receptor antagonist 6,7-dinitroquinoxaline-2,3-dione (100 nm).
Figure 4 illustrates a typical experiment analyzing the long-term effects of NMDA exposure on CA1 synaptic transmission. Figure 4A shows individual CA1 pyramidal neuron EPSC amplitudes recorded in a representative slice during the stable periods after each change of extracellular [Ca2+]o, before and 30 min after induction of NMDA-LTD. The stability of the data recorded was assessed by fitting a straight line to the amplitudes in each recording condition plotted versus repetition number. For analysis, only data that displayed <20% change in the regression line over at least 30 data points were selected for additional VM analysis (Oleskevich et al., 2000). As shown in Figure 4, C and D, the VM relationship obtained by varying [Ca2+]o is parabolic. After exposure to 10 μm NMDA, the mean amplitudes at different Ca2+ concentrations were reduced along the same parabolic fit (Fig. 4B,C), consistent with a reduction in presynaptic release probability. Figure 4D explicitly shows this relationship, converted to Pr, whereas Figure 4E shows the associated variance/mean versus mean linear plot, from which it can be seen that the linear fits before and during NMDA-LTD differ in slope, consistent with a presynaptic site of expression.
Figure 4.

Typical VM analysis of peak EPSCs before and during NMDA receptor-dependent LTD. A, EPSC peak amplitudes (in picoamperes) recorded at 1, 2, and 4 mm [Ca2+]o as denoted, before (left) and 30 min after (right) exposure to 10 μm NMDA for 3 min. B, EPSC trace envelopes before (Control) and 30 min after (30 min After 10 μm NMDA) induction of NMDA-LTD (arrows indicate stimulus onset) C, A typical plot of variance versus mean peak EPSC amplitudes at [Ca2+]o of 1, 2, and 4 mm as indicated, before (filled circles) and 30 min after (open circles) exposure to 10 μm NMDA for 3 min. The line is the best parabolic fit forced to pass through 0,0. All data points before and during NMDA-LTD were well fit by the same parabola, consistent with a presynaptic reduction in Pr. D, An example of estimating release probability by parabolic fitting. Mean amplitudes on the x-axis are normalized with the estimated cross between the parabolic curve and x-axis. The normalized scale on the x-axis thus represents estimated Pr. The fact that data points before (filled circles) and during (open circles) NMDA-LTD fall on the same parabola indicates that this form of LTD is associated with a reduction in presynaptic Pr. E, Plot of variance/mean ratio versus mean EPSC amplitude (in picoamperes), which converts the parabolic relationship between mean and variance to a linear one. The number of release sites can be easily derived by estimating the slope of the linear fit, whereas the y-intercept denotes the quantal size of the EPSC. The increase in slope indicates that NMDA-LTD is associated with a reduction in presynaptic Pr.
Figure 5 illustrates results of VM analyses for a typical experiment analyzing the long-term effects of (S)-DHPG exposure on CA1 synaptic transmission. The VM relationship obtained by varying extracellular [Ca2+] is parabolic, and after exposure to 25 μm (S)-DHPG, mean amplitudes were reduced (Fig. 5A) and fit by a different parabola (Fig. 5B). Figure 5C shows this relationship, converted to Pr, in which Pr was not significantly different after induction of LTD by activation of group I mGluRs. Figure 5D shows the associated variance/mean versus mean linear plot in which the linear fits before and during group I mGluR-LTD have the same slope but different intercepts, consistent with a postsynaptic site of expression.
Figure 5.

Typical VM relationship of peak EPSCs before and during group I mGluR receptor-dependent LTD. The numbers in B and C denote [Ca2+]o when recordings were made. A, EPSC trace envelopes before (Control) and 30 min after (30 min after 25 μm DHPG) induction of mGluR-LTD (arrows indicate stimulus onset). B, Filled circles represent the variance versus mean of EPSCs recorded before application of 25 μm (S)-DHPG for 5 min, whereas open circles depict data from the same cells 30 min after (S)-DHPG. Note that the data before and during mGluR-LTD fall on two distinct parabolas. C, Pr estimated by normalizing means with expected x-intercepts of both parabolas, indicating that release probabilities remain the same at corresponding [Ca2+]o before and during mGluR-LTD. D, Plot of variance/mean ratios versus mean EPSC amplitudes at 1, 2, and 4 mm [Ca2+]o shows the linear relationships between the mean and variance/mean estimated before and after application of (S)-DHPG. The fact that these lines have the same slope and different y-intercepts indicates that mGluR-LTD was associated with a purely postsynaptic reduction in quantal size.
VM analysis shows that NMDA-LTD is associated with reduced transmitter release probability, whereas group I mGluR-LTD induced by (S)-DHPG is not
VM analysis using whole-cell recordings from CA1 pyramidal neurons was applied in hippocampal slices before and 30 min after the induction of long-lasting depression by bath application of either NMDA (10 μm for 3 min) or the group I mGluR agonist (S)-DHPG (25 μm for 5 min). Figure 6 summarizes the results of these analyses for both NMDA-LTD and mGluR-LTD. Plotting variance versus mean of predrug baseline (Fig. 6A, filled circles) and 30 min after induction of NMDA-LTD (open circles), at varying [Ca2+]o, shows that both sets of points are well fit by the same parabola, consistent with a long-term reduction in Pr associated with NMDA-LTD. Figure 6B shows the same data after normalization of mean/variance to plot Pr shifts directly. In contrast, a variance/mean plot of predrug baselines (Fig. 6C, filled circles), compared with 30 min after induction of DHPG-LTD (open circles), shows that these data need to be fit by two distinct parabolas, consistent with purely postsynaptic sites of expression for DHPG-LTD. Figure 6D shows the same data after normalization to yield Pr. Summary data of estimated long-term changes in Pr at different [Ca2+]o is shown in Figure 6E, where NMDA-LTD was associated with a significant long-term decrease in Pr at 1, 2, and 4 mm [Ca2+]o (open squares; *p < 0.05, one-way ANOVA with multiple measures), whereas DHPG-LTD (open circles) was not.
Figure 6.

NMDA-induced LTD at Schaffer collateral–CA1 pyramidal cell synapses is associated with a reduction in release probability assessed by VM analysis, whereas (S)-DHPG-induced mGluR-LTD is not. A, VM relationships determined by altering [Ca2+]o to 1, 2, and 4 mm before (filled circles) and 20 min after (open circles) exposure to 10 μm NMDA for 3 min to induce NMDA-dependent LTD (n = 5 cells). Both means and variances are normalized to the maximal peak amplitude recorded at 4 mm [Ca2+]o before NMDA application. Data before and after NMDA application were well fit by a single parabola, consistent with a presynaptic reduction in Pr. B, Calculation of Pr from the data in A confirms the presynaptic NMDA-induced reduction in Pr. C, VM relationships determined as in A before (filled circles) and 20 min after (open circles) exposure to 25 μm (S)-DHPG for 5 min to induce mGluR-LTD (n = 6 different cells). D, Calculation of Pr from the data in C. In both C and D, the data collected before and after exposure to (S)-DHPG fell on two different parabolas, demonstrating that mGluR-LTD is not associated with a change in Pr. E, Estimated change in Pr during NMDA-LTD (open squares) and mGluR-LTD (open circles) at each [Ca2+]o. *P < 0.05, paired t test compared with pre-LTD control values.
VM analysis shows that Schaffer collateral stimulus-evoked NMDA receptor- and mGluR-dependent LTDs are each associated with reductions in both release probability and quantal content
Although the previous experiments indicate clear differences in sites of expression for pharmacologically induced NMDA-LTD versus mGluR1-LTD, they do not speak directly to the question of expression sites of these components in stimulus-evoked activity-dependent LTD. To address this question, we used selective glutamate receptor blockers to isolate NMDA receptor- and mGluR-dependent LTDs in response to Schaffer collateral synaptic stimulation. When a low-frequency (2 Hz, 10 min) train of paired-pulse Schaffer collateral stimuli (50 ms interstimulus interval, 2400 stimuli) was applied in the presence of the NMDA receptor blocker AP-5 (50 μm), it elicited a stable mGLuR-LTD [65.7 ± 4.9% of pre-paired-pulse low-frequency stimulation (LFS) baseline; n = 13], and mean-variance analysis shows that this LTD was accompanied by both reductions in postsynaptic quantal size (Fig. 7A) and in presynaptic Pr (Fig. 7B). Similarly, when a low-frequency stimulus train (2 Hz, 10 min, 1200 stimuli) was applied to slices bathed in the broad-spectrum mGluR antagonist (S)-MCPG (200 μm), LTD was also elicited (55.3 ± 5.3% of pre-LFS baseline; n = 12), and this NMDA-LTD was also expressed as a mixture of postsynaptic reduction of quantal size (Fig. 7C) and of presynaptic Pr (Fig. 7D).
Figure 7.

Pharmacologically isolated stimulus-evoked mGluR-dependent LTD and NMDA receptor-dependent LTD are associated with reductions in both presynaptic release probability and postsynaptic reduction in quantal size, and the presynaptic component of changes associated with stimulus-evoked mGluR-dependent LTD require group II mGluR activation. A, VM relationships determined by altering [Ca2+]o to 1, 2, and 4 mm before (filled circles) and 30 min after (open circles) application of low-frequency Schaffer collateral stimulation (2 Hz, 10 min) in the presence of the NMDA receptor blocker AP-5 (50 μm; n = 7 cells). Both means and variances are normalized to the maximal peak amplitude recorded at 4 mm [Ca2+]o before LFS. After LFS, variances at all [Ca2+]o were shifted to the left, whereas data were fit by a lower-amplitude parabola, consistent with both a presynaptic reduction in Pr and postsynaptic reduction in quantal size. B, Calculation of Pr from the data in A confirms that mGluR-dependent LTD is composed of both presynaptic and postsynaptic alterations. C, VM relationships determined as in A before (filled circles) and 30 min after (open circles) application of a low-frequency train of paired-pulse stimuli (50 ms paired-pulse interval × 2 Hz, 10 min) in the presence of the mGluR receptor blocker MCPG (200 μm; n = 8 cells). D, Calculation of Pr from the data in C. In both C and D, the data collected before and after induction of mGluR-LTD fell on two different parabolas and showed reductions in Pr, indicating that mGluR-LTD is also a combination of presynaptic and postsynaptic alterations. E, VM relationship determined as in A before (filled circles) and 30 min after (open circles) application of a low-frequency train of paired-pulse stimuli (50 ms paired-pulse interval × 2 Hz, 10 min) in the presence of the NMDA receptor blocker AP-5 (50 μm) plus the group II mGluR blocker EGLU (250 μm; n = 7 cells). F, Calculation of Pr from the data in E. In both E and F, the data collected before and after induction of mGluR-LTD fell on two different parabolas with no differences in Pr, indicating that once group II mGluRs are blocked, group I-dependent mGluR-LTD is a purely postsynaptic phenomenon.
The presynaptic component of mGluR-dependent LTD requires group II mGluR activation
The above experiments indicate that the pharmacological isolation of both NMDA receptor- and mGluR-dependent stimulus-evoked LTD results in phenomenon that are a combination of presynaptic and postsynaptic alterations. In a recent study, we showed that stimulus-evoked LTD at Schaffer collateral synapses can be attenuated by blockade of group II mGluRs, which are localized primarily presynaptically, where they inhibit adenylate cyclase in the presynaptic terminal (Santschi et al., 2006). Therefore, we tested the hypothesis that the presynaptic component of pharmacologically isolated mGluR-LTD requires activation of group II mGluRs, whereas the postsynaptic form might be purely group I mGluR dependent. We bath applied the NMDA receptor antagonist AP-5 along with the group II-selective mGluR antagonist EGLU before inducing LTD with a train of low-frequency bursts that consisted of three pulses at 30 ms interstimulus intervals, applied twice each second for 10 min (3600 stimuli). This variant of LFS elicited stable LTD (63.7 ± 3.7% of pre-LFS baseline; n = 12) with both NMDA and group II mGluRs blocked, and mean-variance analysis shows that this form of group I mGluR-LTD was associated with only postsynaptic reduction in quantal size (Fig. 7E), without any change in presynaptic Pr (Fig. 7F). Table 1 summarizes the changes in quantal size and Pr associated with the different forms of LTD examined, indicating that, although both NMDA and mGluR-dependent LTD induced by synaptic stimulation are expressed as a mixture of presynaptic and postsynaptic alterations, the group I mGluR component of LTD, like DHPG-LTD, is expressed purely postsynaptically.
Table 1.
Summary of Pr and quantal content measured by VM analysis before and during different forms of LTD
| LTD form | Release probabilities |
Quantal sizes |
Receptor numbers |
|||
|---|---|---|---|---|---|---|
| Before LTD | After LTD | Before LTD | After LTD | Before LTD | After LTD | |
| mGluR (n = 7) | 1 [Ca2+]o: 0.13 ± 0.01 | 0.11 ± 0.01 | 1.83 ± 0.09 | 1.54 ± 0.15* | 291 ± 80 | 280 ± 89 |
| 2 [Ca2+]o: 0.49 ± 0.04 | 0.35 ± 0.04* | |||||
| 4 [Ca2+]o: 0.69 ± 0.04 | 0.52 ± 0.05* | |||||
| NMDA (n = 8) | 1 [Ca2+]o: 0.23 ± 0.03 | 0.12 ± 0.03* | 2.13 ± 0.26 | 1.30 ± 0.17* | 208 ± 58 | 289 ± 81 |
| 2 [Ca2+]o: 0.46 ± 0.05 | 0.22 ± 0.04* | |||||
| 4 [Ca2+]o: 0.63 ± 0.05 | 0.36 ± 0.07* | |||||
| Group I mGluR (n = 7) | 1 [Ca2+]o: 0.21 ± 0.06 | 0.22 ± 0.03 | 2.82 ± 0.46 | 1.62 ± 0.28* | 201 ± 109 | 121 ± 23 |
| 2 [Ca2+]o: 0.45 ± 0.06 | 0.44 ± 0.04 | |||||
| 4 [Ca2+]o: 0.69 ± 0.02 | 0.67 ± 0.07 | |||||
Data are presented as means ± SEM. *p < 0.05, significant paired t test.
Nitric oxide synthase activity is required for induction of NMDA-LTD but not DHPG-LTD
There is substantial evidence that particular forms of both stimulus-evoked LTP (Schuman and Madison, 1991; Bon and Garthwaite, 2001) (but see Bannerman et al., 1994; Cummings et al., 1994) and LTD (Izumi and Zorumski, 1993; Gage et al., 1997) may depend on the postsynaptic production of the intercellular gaseous messenger nitric oxide (NO), which diffuses to the presynaptic terminal and activates soluble guanylyl cyclase, triggering cGMP–protein kinase G-dependent presynaptic events leading to long-term plasticity. NO synthase (NOS), the enzymes that generate NO, are a Ca2+/calmodulin-dependent enzyme that can, in principle, be activated by any glutamate receptors that produce elevations in intracellular [Ca2+]. However, it is less clear whether different glutamate receptors and sources of elevations in [Ca2+]I are more or less effective in activating forms of NOS necessary for plasticity.
Therefore, we tested the ability of the selective NOS inhibitor Nω-nitro-l-arginine (L-NA) to block the induction of NMDA-LTD or DHPG-LTD. L-NA (100 μm) was bath applied to slices 15 min before and during the application of either NMDA (10 μm for 3 min) or DHPG (25 μm for 5 min) to induce LTD, before a 30 min washout of all drugs in drug-free ACSF. Field recordings in CA1 stratum radiatum show that blockade of NOS activity had no effect on the magnitude or duration of DHPG-LTD (Fig. 8A, filled circles) compared with untreated control DHPG-LTD (open circles). In contrast, blockade of NOS activity converted NMDA-LTD (Fig. 8B, open circles) to a transient depression that completely reversed by 30 min washout after NMDA application (filled circles). These data demonstrate that NMDA receptor activation is a much more effective method of eliciting NOS-dependent LTD at Schaffer collateral–CA1 synapses. In contrast, group I mGluR-dependent LTD does not appear to require NOS, suggesting that Ca2+ released from intracellular stores is much less effective at activating NOS, does not generate NO in the proper locus, and/or is more effective at activating other enzymes that lead to NO-independent, postsynaptic LTD. Differing locations (synaptic vs extrasynaptic) of NMDA receptors and mGluRs, magnitude, and location of [Ca2+] increases could all be important reasons for these differences.
Figure 8.

The NOS inhibitor Nω-nitro-l-arginine (L-NA) blocks induction of NMDA-LTD, but not mGluR-LTD, of synaptic transmission at Schaffer collateral–CA1 synapses. A, Time course of LTD of Schaffer collateral-evoked field EPSP slopes induced by 25 μm (S)-DHPG (filled bar) in slices pretreated for 30 min before addition of (S)-DHPG with bath-applied L-NA (filled circles; 100 μm; n = 8) versus untreated control slices (open circles; n = 8). B, Time course of LTD of Schaffer collateral-evoked field EPSP slopes induced by 10 μm NMDA (filled bar) in slices pretreated for 30 min before the addition of NMDA with bath-applied L-NA (filled circles; 100 μm; n = 8) versus untreated control slices (open circles; n = 8). All points are means ± SEM.
NMDA-LTD, but not DHPG-LTD, elicits long-term reduction of transmitter release from the RRP in Schaffer collateral terminals
We have used two-photon excitation to visualize release of the styryl dye FM1-43 from the RRPs of Schaffer collateral–CA1 terminals in hippocampal slices and demonstrated that stimulus-evoked LTP and LTD of synaptic transmission at Schaffer collateral–CA1 synapses can be associated with long-term alterations in the rates of vesicular uptake and release that are selective to the RRP (Stanton et al., 2003, 2005). In one of these studies (Stanton et al., 2005), we found that, although the generation of near-saturating LTP produced a persistent increase in presynaptic RRP release rates, weaker partial LTP could be evoked without any presynaptic alterations, consistent with the existence of distinct presynaptic and postsynaptic forms of LTP. In the current study, we used the same two-photon methodology to test whether NMDA-LTD or DHPG-LTD is associated with presynaptic alterations in RRP release.
Figure 9 contrasts the long-term effects of NMDA-LTD and DHPG-LTD on vesicular release kinetics. Figure 9A shows representative fields in CA1 stratum radiatum immediately before (0′) and after 5 min application of Schaffer collateral stimulus bursts (5′), in a control slice (control) versus slices in which DHPG-LTD or NMDA-LTD had been induced (30 min) before loading the RRP with FM1-43 by brief hypertonic shock (see Materials and Methods). Although the rate of destaining evoked by the stimulus bursts (10 Hz, 1 s each 10 s) was the same during DHPG-LTD as in the control slice, destaining was markedly slower in the slice in which NMDA-LTD had been induced. Figure 9B shows the time course of destaining in all slices in which NMDA-LTD (filled diamonds; n = 5) or DHPG-LTD (filled circles; n = 5) had been induced compared with control slices (open circles; n = 5). NMDA-LTD was associated with a marked slowing in the rate of FM1-43 stimulus-evoked destaining, indicating a strong presynaptic LTD of release from the RRP. In contrast, DHPG-LTD release rates were unchanged from controls, consistent with group I mGluR-dependent LTD being expressed at entirely postsynaptic sites.
Figure 9.
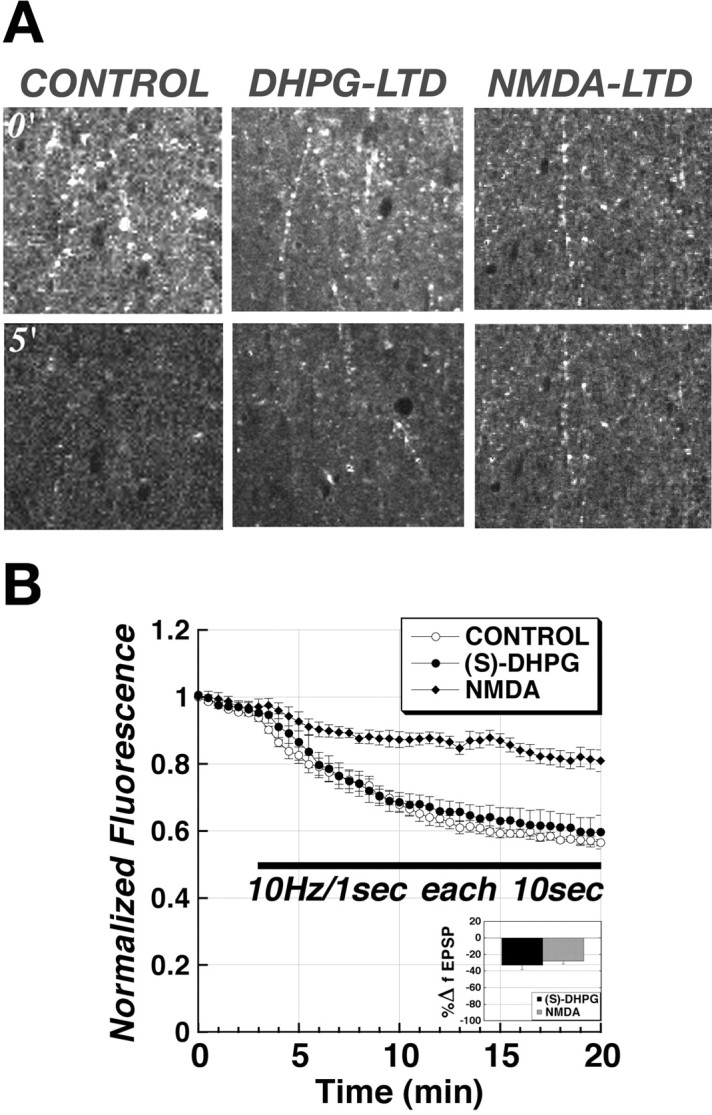
Direct two-photon imaging of FM1-43 release from the RRP of Schaffer collateral terminals confirms that NMDA-LTD but not mGluR-LTD is associated with a reduction in rate of release from the RRP. A, Two-photon excitation fluorescent images of RRP puncta in the same field of CA1 stratum radiatum immediately before (0′; top row) and 5 min after (5′; bottom row) the start of unloading Schaffer collateral stimulation in a control slice versus slices pretreated with either (S)-DHPG (25 μm, 5 min; mGluR-LTD) or NMDA (10 μm, 3 min; NMDA-LTD). B, Time courses of Schaffer collateral stimulus-evoked (filled bar; 10 Hz, 1 s bursts each 10 s) FM1-43 destaining from the RRP in control slices (open circles; n = 5) versus slices in which LTD had been induced either by bath application of (S)-DHPG (filled circles; 25 μm; n = 6) or NMDA (filled diamonds; 10 μm; n = 5) 30 min before start of destaining stimulation. Inset, Mean ± SEM. LTD of a field EPSP recorded in the same slices 15 min after bath application of either (S)-DHPG (black bar) or NMDA (gray bar) is shown.
Although postsynaptic NMDA receptors must be activated for the induction of both NMDA receptor-dependent LTP and LTD (Kamiya et al., 1993; Cummings et al., 1996), bath application of NMDA can also activate presynaptic NMDA receptors that may play a role in LTD (Sjostrom et al., 2003). To determine whether presynaptic NMDA receptor activation plays any role in the induction of LTD of release, we exploited the method of Alici et al. (1996), who showed that prolonged (90–120 min) exposure of hippocampal slices to glucose-free ACSF severely damages and silences CA1 pyramidal neurons, eliminating postsynaptic responses, while leaving presynaptic terminal ionic fluxes and transmitter release relatively unaffected. When we pretreated slices for 90 min with glucose-free ACSF and bath applied NMDA (10 μm, 3 min), NMDA no longer induced any LTD of FM1-43 release compared with control slices after glucose-free ACSF (control 1/t1/2, 0.054 ± 0.005; NMDA 1/t1/2, 0.044 ± 0.003; p > 0.10, Student's t test; data not shown).
Discussion
Although there are those who have contended that long-term plasticity is expressed as either presynaptic or postsynaptic alterations, it is increasingly evident that there are multiple forms of both LTP and LTD evoked by different mechanisms, consist of different alterations within a single synapse, which can be expressed either simultaneously or separately. Studies of activity-dependent LTP and LTD suggest there are both presynaptic and postsynaptic expression sites, and both NMDA receptor and mGluR-dependent forms of LTP and LTD. In the present study, we used two-photon laser-scanning microscopy and paired-pulse and VM analyses to test whether either of these forms of LTD, elicited by direct receptor agonist application, are associated with long-term alterations in presynaptic glutamate release probability. All of these methods indicate that NMDA-LTD elicited by NMDA application is associated with clear reductions in presynaptic release probability, whereas group I mGluR-LTD elicited by DHPG is not. At Schaffer collateral–CA1 synapses, NMDA receptor activation acts via a mechanism requiring NO generation to produce LTD of presynaptic release that is revealed by both paired-pulse and VM analyses and that appears to target the RRP loaded selectively by hypertonic shock. In contrast, group I mGluR-LTD was expressed without any alterations in presynaptic release that could be detected with any of these methods. Finally, coactivation of group I and II mGluRs produces a mixed LTD consisting of both postsynaptic and presynaptic long-term alterations.
Previous studies have indicated that, like LTP, the induction of LTD requires activation of postsynaptic dendritic NMDA receptors and Ca2+ influx. The fact that there is substantial data indicating that both LTP and LTD can be associated with long-term alterations in presynaptic transmitter release implies a requirement for a retrograde, perhaps membrane-permeable, messenger to diffuse from activated postsynaptic dendritic spines to the presynaptic terminals responsible for the Ca2+ influx. Our current evidence is that a form of LTD that uses such a retrograde messenger is NMDA-LTD and that one of the diffusible intercellular messengers involved is NO. Because NO is a rapidly, spherically diffusible molecule that can cover distances up to 50–100 μm despite its short half-life (Wood and Garthwaite, 1994), it remains to be determined whether active terminals, perhaps by virtue of residual higher [Ca2+] after action potential invasion, are more prone to NO-dependent presynaptic LTD, or whether local synapse specificity is not strictly maintained.
There is evidence that very immature synapses (postnatal days 1–2) exhibit a form of mGluR-dependent LTD that is induced postsynaptically and expressed presynaptically (Bolshakov and Siegelbaum 1994). In this study, mGluR activation had to be paired with L-type voltage-dependent calcium channel activation. It is unclear whether this form of presynaptic LTD, at a time when NMDA receptors are not yet expressed at significant levels, also requires NO, or whether it shares any mechanism with the long-term presynaptic changes associated with NMDA-LTD in older animals.
However, other studies have indicated that NO, acting by raising [cGMP], is a necessary component of stimulus-evoked LTD at Schaffer collateral–CA1 synapses in older, adolescent animals (Izumi and Zorumski, 1993; Gage et al., 1997; Reyes-Harde et al., 1999). We found that pairing elevated [cGMP] with inhibition of adenylate cyclase, however this is achieved, is sufficient to induce LTD (Santschi et al., 1999, 2006; Stanton et al., 2001) that is associated with presynaptic reductions in release probability (Stanton et al., 2001, 2003), and this stimulus-evoked LTD of presynaptic release is reduced by extracellular scavenging of NO (Stanton et al., 2003). We now conclude that NMDA receptors are an effective means of activating postsynaptic NO synthase that generates NO, which diffuses to presynaptic terminals and activates soluble guanylyl cyclase. Considered together with studies showing postsynaptic NMDA receptor-dependent alterations during LTP (Andrasfalvy and Magee, 2004; Leutgeb et al., 2005) and LTD (Lee et al., 1998; Carroll et al., 1999), it seems clear that NMDA receptors contribute to both presynaptic and postsynaptic forms of LTD.
Similarly, there are studies indicating that mGluR-dependent LTD is associated with both presynaptic (Fitzjohn et al., 2001; Nosyreva and Huber, 2005; Moult et al., 2006) and postsynaptic (Huber et al., 2000, 2001; Nosyreva and Huber, 2005) alterations. Our data suggest that the presynaptic form of mGluR-LTD uses activation of group II mGluRs as a way to inhibit presynaptic adenylate cyclase and group I mGluR activation to release Ca2+ from intracellular stores and activate NO synthase (as well as many other postsynaptic calcium-dependent enzymes). When we block group II mGluR activation, all that seems to be left is a group I mGluR-selective LTD that is induced and expressed purely postsynaptically.
Our use of PPF and refractory period methods to assess alterations in presynaptic function produced data somewhat at odds with previous reports. Lee at al. (1998), in a study that emphasized postsynaptic alterations associated with LTD elicited by the same NMDA bath application protocol we used here, also stated that PPF was not altered 60 min after NMDA application. A number of variables were different between their study and ours, such as slice preparation (removing CA3 vs not, which could affect presynaptic function), interstimulus interval (unknown vs 30 ms), age (21–35 vs 14–19 d old), and rat strain (Long–Evans vs Sprague Dawley). The robust presence of presynaptic forms of LTD in younger animals suggests that developmental age could be one important factor in the relative amount of presynaptic versus postsynaptic alterations elicited by bath-applied NMDA. Although PPF has often been assumed to be a function of release probability, many investigators have also reported that an animal's age and starting Pr are additional determinants of the magnitude and direction of shifts in PPF (Schulz et al., 1995; Santschi and Stanton, 2003), making the absence of a PPF shift weak evidence of the absence of a presynaptic alteration.
When Hjelmstad et al. (1997) first exploited the presynaptic refractory period method of measuring changes in release probability, they also reported no change in Pr during either stimulus-evoked LTP or LTD. Although they did show that this measurement was sensitive to other ways of changing Pr, such as altering extracellular [Ca2+], their and our data suggest that the method may be less sensitive to presynaptic changes associated with LTP and LTD. For example, the method assumes that the number of release sites does not change with induction of plasticity and will not detect silent presynaptic release sites that turn on or sites that turn completely off, if the Pr of remaining terminals does not change. Although debate continues over presynaptic sites of expression in both LTP and LTD, it seems clear that this particular measurement method is relatively insensitive to presynaptic activity-dependent plasticity.
There is continuing debate concerning whether or not the induction of low-frequency stimulus-evoked LTD, which may contain both NMDA receptor and mGluR-dependent components, requires ongoing protein synthesis. Whereas in vitro studies at Schaffer collateral–CA1 synapses have variously reported that LTD is not (Huber et al., 2000) and is (Sajikumar and Frey, 2003) protein synthesis dependent, an in vivo study reported blockade of LTD by the translational inhibitor anisomycin, but not the transcriptional blocker actinomycin D (Manahan-Vaughan et al., 2000). In field CA1, group I mGluR-dependent LTD induced by DHPG appears to be dependent on protein, but not mRNA, synthesis (and is absent in mGluR5 knock-out mice) (Huber et al., 2000, 2001). At perforant path–dentate granule cell synapses, group I mGluR-LTD has also been reported to be protein synthesis dependent (Naie and Manahan-Vaughan, 2005), but LTD induced by group II or III mGluR agonists was not (Naie and Manahan-Vaughan, 2005; Pöschel and Manahan-Vaughan, 2005), although NMDA receptor-dependent LTD has yet to be examined. Interestingly, a recent study (Nosyreva and Huber, 2005) found that, although DHPG-induced LTD was protein synthesis dependent in slices from 21-d-old rats, it was not in slices from 8- to 15-d-old animals. Moreover, this and other studies (Bolshakov and Siegelbaum, 1994; Zakharenko et al., 2002) supply evidence that DHPG-induced LTD in very young (2–9 d old) neonatal rats is associated with alterations in presynaptic glutamate release, but that there is a developmental switch that occurs, after which group I mGluR-LTD appears to be purely postsynaptic in expression, consistent with our findings from 14- to 19-d-old rats.
Considering our data in light of previous work, we conclude that presynaptic LTD, at least at Schaffer collateral–CA1 synapses, depends on activation of group I mGluRs early in development (before expression of NMDA receptors) and switches to being elicited preferentially by NMDA receptor activation later. Furthermore, it appears that mGluR-dependent LTD contains both presynaptic and postsynaptic expression components, with the presynaptic changes requiring paired activation of group I mGluRs with group II mGluRs that inhibit adenylate cyclase, whereas postsynaptic LTD probably consists of both NMDA receptor and group I mGluR-dependent components that, at least in part, require intact protein synthesis for their long-term maintenance.
Footnotes
This work was supported by the Alexander von Humboldt Foundation, National Institutes of Health Grant NS44421 (P.K.S.), and Deutsche Forschungsgemeinschaft Grants GRK 238 (J.W.) and Mu 809/6-3 (W.M.). We thank Hong Peng for expert technical assistance.
This work is dedicated to the memory of Lewis N. Stanton Sr, Gary L. Stanton, and John M. Sarvey.
References
- Alici K, Gloveli T, Weber-Luxenburger G, Motine V, Heinemann U. Comparison of effects induced by toxic applications of kainate and glutamate by glucose deprivation on area CA1 of rat hippocampal slices. Brain Res. 1996;738:109–120. doi: 10.1016/0006-8993(96)00766-4. [DOI] [PubMed] [Google Scholar]
- Andersen P, Lømo T. Control of hippocampal output by afferent volley frequency. Structure and function of the limbic system. Prog Brain Res. 1967;27:400–412. doi: 10.1016/S0079-6123(08)63112-X. [DOI] [PubMed] [Google Scholar]
- Andrasfalvy BK, Magee JC. Changes in AMPA receptor currents following LTP induction on rat CA1 pyramidal neurons. J Physiol (Lond) 2004;559:543–554. doi: 10.1113/jphysiol.2004.065219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CH, Bartsch D, Kandel ER. Toward a molecular definition of long-term memory storage. Proc Natl Acad Sci USA. 1996;93:13445–13452. doi: 10.1073/pnas.93.24.13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannerman DM, Chapman PF, Kelly PA, Butcher SP, Morris RG. Inhibition of nitric oxide synthase does not prevent the induction of long-term potentiation in vivo. J Neurosci. 1994;14:7415–7425. doi: 10.1523/JNEUROSCI.14-12-07415.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. Presynaptic mechanism for long-term potentiation in the hippocampus. Nature. 1990;346:724–729. doi: 10.1038/346724a0. [DOI] [PubMed] [Google Scholar]
- Benke TA, Luthi A, Isaac JT, Collingridge GL. Modulation of AMPA receptor unitary conductance by synaptic activity. Nature. 1998;393:793–797. doi: 10.1038/31709. [DOI] [PubMed] [Google Scholar]
- Bienenstock EL, Cooper LN, Munro PW. Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. J Neurosci. 1982;2:32–48. doi: 10.1523/JNEUROSCI.02-01-00032.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolshakov VY, Siegelbaum SA. Postsynaptic induction and presynaptic expression of hippocampal long-term depression. Science. 1994;264:1148–1152. doi: 10.1126/science.7909958. [DOI] [PubMed] [Google Scholar]
- Bon CL, Garthwaite J. Exogenous nitric oxide causes potentiation of hippocampal synaptic transmission during low-frequency stimulation via the endogenous nitric oxide-cGMP pathway. Eur J Neurosci. 2001;14:585–594. doi: 10.1046/j.0953-816x.2001.01680.x. [DOI] [PubMed] [Google Scholar]
- Braunewell KH, Manahan-Vaughan D. Long-term depression: a cellular basis for learning? Rev Neurosci. 2001;12:121–140. doi: 10.1515/revneuro.2001.12.2.121. [DOI] [PubMed] [Google Scholar]
- Camodeca N, Breakwell NA, Rowan MJ, Anwyl R. Induction of LTD by activation of group I mGluR in the dentate gyrus in vitro. Neuropharmacology. 1999;18:1597–1606. doi: 10.1016/s0028-3908(99)00093-3. [DOI] [PubMed] [Google Scholar]
- Carroll RC, Lissin DV, von Zastrow M, Nicoll RA, Malenka RC. Rapid redistribution of glutamate receptors contributes to long-term depression in hippocampal cultures. Nat Neurosci. 1999;2:454–460. doi: 10.1038/8123. [DOI] [PubMed] [Google Scholar]
- Clements JD, Silver RA. Unveiling synaptic plasticity: a new graphical and analytical approach. Trends Neurosci. 2000;23:105–113. doi: 10.1016/s0166-2236(99)01520-9. [DOI] [PubMed] [Google Scholar]
- Cummings JA, Nicola SM, Malenka RC. Induction in the rat hippocampus of long-term potentiation (LTP) and long-term depression (LTD) in the presence of a nitric oxide synthase inhibitor. Neurosci Lett. 1994;176:110–114. doi: 10.1016/0304-3940(94)90883-4. [DOI] [PubMed] [Google Scholar]
- Cummings JA, Mulkey RM, Nicoll RA, Malenka RC. Ca2+-signaling requirements for long-term depression in the hippocampus. Neuron. 1996;16:825–833. doi: 10.1016/s0896-6273(00)80102-6. [DOI] [PubMed] [Google Scholar]
- Domenici MR, Berretta N, Cherubini E. Two distinct forms of long-term depression coexist at the mossy fiber-CA3 synapse in the hippocampus during development. Proc Natl Acad Sci USA. 1998;95:8310–8315. doi: 10.1073/pnas.95.14.8310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek S, Bear MF. Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-d-aspartate receptor blockade. Proc Natl Acad Sci USA. 1992;89:4363–4367. doi: 10.1073/pnas.89.10.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- Fitzjohn SM, Palmer MJ, May JE, Neeson A, Morris SA, Collingridge GL. A characterization of long-term depression induced by metabotropic glutamate receptor activation in the rat hippocampus in vitro. J Physiol (Lond) 2001;537:421–430. doi: 10.1111/j.1469-7793.2001.00421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster KA, Regehr WG. Variance-mean analysis in the presence of a rapid antagonist indicates vesicle depletion underlies depression at the climbing fiber synapse. Neuron. 2004;43:119–131. doi: 10.1016/j.neuron.2004.06.022. [DOI] [PubMed] [Google Scholar]
- Gage AT, Reyes M, Stanton PK. Nitric oxide-guanylyl cyclase-dependent and -independent components of multiple forms of long-term synaptic depression. Hippocampus. 1997;7:286–295. doi: 10.1002/(SICI)1098-1063(1997)7:3<286::AID-HIPO4>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Hjelmstad GO, Nicoll RA, Malenka RC. Synaptic refractory period provides a measure of probability of release in the hippocampus. Neuron. 1997;19:1309–1318. doi: 10.1016/s0896-6273(00)80421-3. [DOI] [PubMed] [Google Scholar]
- Huber KM, Kayser MS, Bear MF. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288:1254–1257. doi: 10.1126/science.288.5469.1254. [DOI] [PubMed] [Google Scholar]
- Huber KM, Roder JC, Bear MF. Chemical induction of mGLuR5- and protein synthesis-dependent long-term depression in hippocampal area CA1. J Neurophysiol. 2001;86:321–325. doi: 10.1152/jn.2001.86.1.321. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Zorumski CF. Nitric oxide and long-term synaptic depression in the rat hippocampus. NeuroReport. 1993;4:1131–1134. [PubMed] [Google Scholar]
- Kamiya H, Sawada S, Yamomoto C. Long-lasting potentiation of synaptic transmission in the Schaffer collateral-commissural pathway of the guinea pig hippocampus by activation of postsynaptic N-methyl-D-aspartate receptor. Synapse. 1993;13:186–194. doi: 10.1002/syn.890130210. [DOI] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- Lee HK, Kameyama K, Huganir RL, Bear MF. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron. 1998;21:1151–1162. doi: 10.1016/s0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- Leutgeb JK, Frey JU, Behnisch T. Single cell analysis of activity-dependent cyclic AMP-responsive element-binding protein phosphorylation during long-lasting long-term potentiation in area CA1 of mature rat hippocampal-organotypic cultures. Neuroscience. 2005;131:601–610. doi: 10.1016/j.neuroscience.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. Long-term potentiation—a decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- Malgaroli A, Ting AE, Wendland B, Bergamaschi A, Villa A, Tsien RW, Scheller RH. Presynaptic component of long-term potentiation visualized at individual hippocampal synapses. Science. 1995;268:1624–1628. doi: 10.1126/science.7777862. [DOI] [PubMed] [Google Scholar]
- Malinow R, Tsien RW. Presynaptic enhancement shown by whole-cell recordings of long-term potentiation in hippocampal slices. Nature. 1990;346:177–180. doi: 10.1038/346177a0. [DOI] [PubMed] [Google Scholar]
- Malinow R, Mainen ZF, Hayashi Y. LTP mechanisms: from silence to four-lane traffic. Curr Opin Neurobiol. 2000;10:352–357. doi: 10.1016/s0959-4388(00)00099-4. [DOI] [PubMed] [Google Scholar]
- Manahan-Vaughan D, Kulla A, Frey JU. Requirement of translation but not transcription for the maintenance of long-term depression in the CA1 region of freely moving rats. J Neurosci. 2000;20:8572–8576. doi: 10.1523/JNEUROSCI.20-22-08572.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin KC, Barad M, Kandel ER. Local protein synthesis and its role in synapse-specific plasticity. Curr Opin Neurobiol. 2000;10:587–592. doi: 10.1016/s0959-4388(00)00128-8. [DOI] [PubMed] [Google Scholar]
- Mellor J, Nicoll RA. Hippocampal mossy fiber LTP is independent of postsynaptic calcium. Nat Neurosci. 2001;4:125–126. doi: 10.1038/83941. [DOI] [PubMed] [Google Scholar]
- Moult PR, Gladding CM, Sanderson TM, Fitzjohn SM, Bashir ZI, Molnar E, Collingridge GL. Tyrosine phosphatases regulate AMPA receptor trafficking during metabotropic glutamate receptor-mediated long-term depression. J Neurosci. 2006;26:2544–2554. doi: 10.1523/JNEUROSCI.4322-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey RM, Malenka RC. Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron. 1992;9:967–975. doi: 10.1016/0896-6273(92)90248-c. [DOI] [PubMed] [Google Scholar]
- Naie K, Manahan-Vaughan D. Investigations of the protein synthesis dependency of mGluR-induced long-term depression in the dentate gyrus of freely moving rats. Neuropharmacology. 2005;49:35–44. doi: 10.1016/j.neuropharm.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Neher E. Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron. 1998;20:389–399. doi: 10.1016/s0896-6273(00)80983-6. [DOI] [PubMed] [Google Scholar]
- Nosyreva ED, Huber KM. Developmental switch in synaptic mechanisms of hippocampal metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2005;25:2992–3001. doi: 10.1523/JNEUROSCI.3652-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleskevich S, Clements J, Walmsley B. Release probability modulates short-term plasticity at a rat giant terminal. J Physiol (Lond) 2000;524:513–523. doi: 10.1111/j.1469-7793.2000.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SH, Malenka RC, Nicoll RA. Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron. 1997;18:969–982. doi: 10.1016/s0896-6273(00)80336-0. [DOI] [PubMed] [Google Scholar]
- O'Mara SM, Rowan MJ, Anwyl R. Metabotropic glutamate receptor-induced homosynaptic long-term depression and depotentiation in the dentate gyrus of the rat hippocampus in vitro. Neuropharmacology. 1995;34:983–989. doi: 10.1016/0028-3908(95)00062-b. [DOI] [PubMed] [Google Scholar]
- Otani S, Connor JA. Requirement of rapid Ca2+ entry and synaptic activation of metabotropic glutamate receptors for the induction of long-term depression in adult rat hippocampus. J Physiol (Lond) 1998;511:761–770. doi: 10.1111/j.1469-7793.1998.761bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer MJ, Irving AJ, Seabrook GR, Jane DE, Collingridge GJ. The group I mGluR receptor agonist DHPG induces a novel form of LTD in the CA1 region of the hippocampus. Neuropharmacology. 1997;36:1517–1532. doi: 10.1016/s0028-3908(97)00181-0. [DOI] [PubMed] [Google Scholar]
- Pavlidis P, Montgomery J, Madison DV. Presynaptic protein kinase activity supports long-term potentiation at synapses between individual hippocampal neurons. J Neurosci. 2000;20:4497–4505. doi: 10.1523/JNEUROSCI.20-12-04497.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pöschel B, Wroblewska B, Heinemann U, Manahan-Vaughan D. The metabotropic glutamate receptor mGluR3 is critically required for hippocampal long-term depression and modulates long-term potentiation in the dentate gyrus of freely moving rats. Cereb Cortex. 2005;15:1414–1423. doi: 10.1093/cercor/bhi022. [DOI] [PubMed] [Google Scholar]
- Pyle JL, Kavalali ET, Piedras-Renteria ES, Tsien RW. Rapid resuse of readily releasable pool vesicles at hippocampal synapses. Neuron. 2000;28:221–231. doi: 10.1016/s0896-6273(00)00098-2. [DOI] [PubMed] [Google Scholar]
- Reid CA, Clements JD. Postsynaptic expression of long-term potentiation in the rat dentate gyrus demonstrated by variance-mean analysis. J Physiol (Lond) 1999;518:121–130. doi: 10.1111/j.1469-7793.1999.0121r.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes M, Stanton PK. Induction of hippocampal long-term depression requires release of Ca2+ from separate presynaptic and postsynaptic intracellular stores. J Neurosci. 1996;16:5951–5960. doi: 10.1523/JNEUROSCI.16-19-05951.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Harde M, Potter BVL, Galione A, Stanton PK. Induction of hippocampal LTD requires nitric-oxide-stimulated PKG activity and Ca2+ release from cyclic ADP-ribose-sensitive stores. J Neurophysiol. 1999;82:1569–1576. doi: 10.1152/jn.1999.82.3.1569. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily-releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Rush AM, Wu J, Rowan MJ, Anwyl R. Group I metabotropic glutamate receptor (mGluR)-dependent long-term depression mediated via p38 mitogen-activated protein kinase is inhibited by previous high-frequency stimulation and activation of mGluRs and protein kinase C in the rat dentate gyrus in vitro. J Neurosci. 2002;22:6121–6128. doi: 10.1523/JNEUROSCI.22-14-06121.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajikumar S, Frey JU. Anisomycin inhibits the late maintenance of long-term depression in rat hippocampal slices in vitro. Neurosci Lett. 2003;338:147–150. doi: 10.1016/s0304-3940(02)01400-3. [DOI] [PubMed] [Google Scholar]
- Santschi LA, Stanton PK. A paired-pulse facilitation analysis of long-term synaptic depression at excitatory synapses in rat hippocampal CA1 and CA3 regions. Brain Res. 2003;962:78–91. doi: 10.1016/s0006-8993(02)03846-5. [DOI] [PubMed] [Google Scholar]
- Santschi LA, Reyes-Harde M, Stanton PK. Chemically-induced, activity-independent LTD elicited by simultaneous activation of PKG and inhibition of PKA. J Neurophysiol. 1999;82:1577–1589. doi: 10.1152/jn.1999.82.3.1577. [DOI] [PubMed] [Google Scholar]
- Santschi LA, Zhang XL, Stanton PK. Activation of receptors negatively coupled to adenylate cyclase is required for induction of long-term synaptic depression at Schaffer collateral-CA1 synapses. J Neurobiol. 2006;66:205–219. doi: 10.1002/neu.20213. [DOI] [PubMed] [Google Scholar]
- Schulz PE, Cook EP, Johnston D. Using paired-pulse facilitation to probe the mechanisms for long-term potentiation (LTP) J Physiol (Paris) 1995;89:3–9. doi: 10.1016/0928-4257(96)80546-8. [DOI] [PubMed] [Google Scholar]
- Schuman EM, Madison DV. A requirement for the intercellular messenger nitric oxide in long-term potentiation. Science. 1991;254:1503–1506. doi: 10.1126/science.1720572. [DOI] [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Petralia R, Zaman S, Wenthold R, Svoboda K, Malinow R. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284:1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- Silver RA. Estimation of nonuniform quantal parameters with multiple-probability fluctuation analysis: theory, application and limitations. J Neurosci Methods. 2003;130:127–141. doi: 10.1016/j.jneumeth.2003.09.030. [DOI] [PubMed] [Google Scholar]
- Silver RA, Momiyama A, Cull-Candy SG. Locus of frequency-dependent depression identified with multiple-probability fluctuation analysis at rat climbing fibre-Purkinje cell synapses. J Physiol (Lond) 1998;510:881–902. doi: 10.1111/j.1469-7793.1998.881bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjostrom P, Turrigiano GG, Nelson SB. Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron. 2003;39:641–654. doi: 10.1016/s0896-6273(03)00476-8. [DOI] [PubMed] [Google Scholar]
- Stanton PK, Heinemann U, Müller W. FM1-43 imaging reveals cGMP-dependent long-term depression of presynaptic transmitter release. J Neurosci. 2001;21:RC167. doi: 10.1523/JNEUROSCI.21-19-j0002.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton PK, Winterer J, Bailey CP, Kyrozis A, Raginov I, Laube G, Veh RW, Nguyen CQ, Müller W. Long-term depression of presynaptic release from the readily releasable vesicle pool induced by NMDA receptor-dependent retrograde nitric oxide. J Neurosci. 2003;23:5936–5944. doi: 10.1523/JNEUROSCI.23-13-05936.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton PK, Winterer J, Müller W. Imaging LTP of presynaptic release of FM1-43 from the rapidly-recycling vesicle pool at Schaffer collateral-CA1 synapses in rat hippocampal slices. Eur J Neurosci. 2005;22:2451–2461. doi: 10.1111/j.1460-9568.2005.04437.x. [DOI] [PubMed] [Google Scholar]
- Stevens CF, Wang Y. Facilitation and depression at single central synapses. Neuron. 1995;14:795–802. doi: 10.1016/0896-6273(95)90223-6. [DOI] [PubMed] [Google Scholar]
- Tan Y, Hori N, Carpenter DO. The mechanism of presynaptic long-term depression mediated by group I metabotropic glutamate receptors. Cell Mol Neurobiol. 2003;23:187–203. doi: 10.1023/A:1022949922364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toni N, Buchs PA, Nikonenko I, Bron CR, Mullre D. LTP promotes formation of multiple spine synapses between a single axon terminal and a dendrite. Nature. 1999;402:421–425. doi: 10.1038/46574. [DOI] [PubMed] [Google Scholar]
- Watabe AM, Carlisle HJ, O'Dell TJ. Postsynaptic induction and presynaptic expression of group I mGluR-dependent LTD in the hippocampal CA1 region. J Neurophysiol. 2002;87:1395–1403. doi: 10.1152/jn.00723.2001. [DOI] [PubMed] [Google Scholar]
- Wood J, Garthwaite J. Models of the diffusional spread of nitric oxide: implications for neural nitric oxide signaling and its pharmacological properties. Neuropharmacology. 1994;33:1235–1244. doi: 10.1016/0028-3908(94)90022-1. [DOI] [PubMed] [Google Scholar]
- Yeckel MF, Kapur A, Johnston D. Multiple forms of LTP in hippocampal CA3 neurons use a common postsynaptic mechanism. Nat Neurosci. 1999;2:625–633. doi: 10.1038/10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakharenko SS, Zablow L, Siegelbaum SA. Visualization of changes in presynaptic function during long-term synaptic plasticity. Nat Neurosci. 2001;4:711–717. doi: 10.1038/89498. [DOI] [PubMed] [Google Scholar]
- Zakharenko SS, Zablow L, Siegelbaum SA. Altered presynaptic vesicle release and cycling during mGluR-dependent LTD. Neuron. 2002;35:1099–1110. doi: 10.1016/s0896-6273(02)00898-x. [DOI] [PubMed] [Google Scholar]
- Zucker RS. Short-term plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]