

Abstract
Mammalian corticogenesis substantially depends on migration and axon projection of newborn neurons that are coordinated by a yet unidentified molecular mechanism. Dual leucine zipper kinase (DLK) induces activation of c-Jun N-terminal kinase (JNK), a molecule that regulates morphogenesis in various organisms. We show here, using gene targeting in mice, that DLK is indispensable for establishing axon tracts, especially those originating from neocortical pyramidal neurons of the cerebrum. Direct and quantitative analysis of radial migration of pyramidal neurons using slice culture and a time-lapse imaging system revealed that acceleration around the subplate was affected by DLK gene disruption and by administration of a JNK inhibitor. Phosphorylation of JNK substrates, including c-Jun and doublecortin, and of JNK itself at the activation loop were partially affected in brains of DLK-deficient mouse embryos. These data suggest that DLK plays a significant role in the coordinated regulation of radial migration and axon projection by modulating JNK activity.
Keywords: cerebral cortex, axon, migration, pyramidal cell, c-Jun, microtubules
Introduction
Radial migration of postmitotic neural cells to their appropriate positions is indispensable for the development of the laminated cerebral cortex (Rakic, 1990; Hatten, 1999). An increasing number of studies on molecular mechanism regulating radial migration have been reported (Trommsdorff et al., 1999; Caspi et al., 2000; Niethammer et al., 2000; Magdaleno and Curran, 2001; Ohshima et al., 2001; Assadi et al., 2003; Schaar et al., 2004; Tanaka et al., 2004). However, it remains difficult to draw a unified signaling network regulating radial migration for two main reasons. First, radial migration involves diverse modes. At least three different modes of radial migration have been identified and are named somal translocation, locomotion (Nadarajah et al., 2001), and multipolar migration (Tabata and Nakajima, 2003). Second, radial migration often takes place in parallel with axon growth (Stensaas and Stensaas, 1968; Hatanaka and Murakami, 2002; Noctor et al., 2004). A common requirement for cytoskeletal modulators, such as microtubule-associated proteins (MAPs) and Lissencephaly 1 (Lis1), in these cellular events has been reported (Teng et al., 2001; Tsai et al., 2005). However, the signaling pathway involved in coordinated regulation of radial migration and axon growth in the developing neocortex remains to be identified.
c-Jun N-terminal kinase (JNK) is one of the key factors involved in morphogenesis in various multicellular organisms. It regulates planar cell polarity, cell migration, and apoptosis (Martín-Blanco, 1997; Xia and Karin, 2004). In the telencephalon of mouse embryos later than 14 d of gestation, JNK is significantly activated in the intermediate zone (IZ), which is mainly composed of migrating neurons (Hirai et al., 2002; Kawauchi et al., 2003; Gdalyahu et al., 2004). This layer-specific activation of JNK indicates its role in differentiation or migration of cortical neurons. Actually, in utero gene transfer experiments using an activator or suppressor of JNK showed the possible involvement of JNK in the regulation of radial migration of cortical neurons (Hirai et al., 2002; Kawauchi et al., 2003). More recently, doublecortin (DCX), a product of the gene responsible for human X-linked lissencephaly, was identified as a JNK substrate in developing cortical neurons, which might act as a signal linkage between JNK and radial migration (Gdalyahu et al., 2004). However, the molecular basis of the regulation of JNK activity in developing cortical neurons is essentially unknown.
Dual leucine zipper kinase (DLK), also called MUK (MAPK-upstream protein kinase) or ZPK (leucine zipper protein kinase), is a member of the mixed-lineage kinases that can function as an MAP triple kinase (MAPKKK) in the JNK pathway (Gallo and Johnson, 2002). Expression of DLK protein is highly specific to neural tissues in mouse embryos (Hirai et al., 2005). In the developing cerebrum, DLK expression is restricted to cell layers in which JNK activity is potently induced (Hirai et al., 2002). Here, we took a genetic approach to test the significance of DLK in the development of mouse neural tissues. DLK gene disruption resulted in decreases in JNK activity and in phosphorylation of known JNK substrates in the embryonic brain. Under these conditions, axon growth and radial migration of neocortical pyramidal neurons were significantly impaired.
Materials and Methods
Gene targeting.
The mice used in these experiments were housed in accordance with protocols approved by the Institutional Animal Care and Use Committee at Yokohama City University, School of Medicine. The DLK gene of TT2 embryonic stem (ES) cells derived from F1 generation of C57BL/6N female and CBA male mice was disrupted by homologous recombination using targeting vectors. The result was replacement of the NcoI–EcoRI segment of the DLK gene encoding 225 N-terminal amino acids with an artificial gene cassette composed of a tandemly oriented internal ribosome entry site (IRES), the bacterial LacZ gene, a first loxP sequence, the MC1 promoter, a neomycin resistance gene (neo), and a second loxP sequence. A polyadenylation signal was not included in this gene cassette. The length of homologous regions in targeting vectors were 6.5 and 2.6 kb at the 5′ and 3′ ends of the gene cassette, respectively (Fig. 1a). Targeted ES clones were first identified with PCR and confirmed with Southern blotting (Fig. 1b). Chimeric male mice were generated by injecting three ES clones into the eight-cell embryos of ICR donor mice and were crossed with C57BL/6N female mice. Two lines of F1 heterozygous offspring, 20-1 and 23B-14, originating from different ES cell lines were obtained. The male offspring were crossed with CAG–Cre recombinase (Cre) transgenic female mice (Sakai and Miyazaki, 1997) to remove the MC1–neo expression cassette from the DLK mutated locus in ovo. F1 heterozygous mice, lacking the CAG–Cre transgene and the MC1–neo expression cassette, were bred with C57BL/6N mice for six to eight generations. Because typical phenotypes of homozygous DLK mutants (viability and histological features of embryos as shown in Figs. 2, 3) obtained with the two different mutant lines were indistinguishable from each other, only one mutant line (20-1) was used for additional analysis, including assays for protein expression and cell migration. The genotype of the DLK locus was routinely determined by PCR using three primers: FC2 (CTACTGCATCCTTATGGAATTCTGC), L3 (GTGGCGACGACTCCTGGAGCCCGTC), and BB1R (ACATCTTGGTGCTCTTGTCACTCAG) (Fig. 1a). FC2 and BB1R gave a 481 bp fragment derived from the wild-type allele. L3 and BB1R gave a 746 bp fragment derived from the DLK mutated allele.
Figure 1.

Generation of DLK mutant mice. a, Schematic representation of the DLK gene targeting strategy. The exon encoding 224 N-terminal amino acids including the ATP binding motif of the kinase domain (amino acid sequence is indicated at the top) was first replaced with tandemly arrayed IRES–LacZ and a floxed MC1–neo cassette (Targeted allele). The MC1–neo cassette was then removed (Targeted allele Δneo) by crossing with CAG–Cre gene transgenic mice (see Materials and Methods). DLK-CT indicates the region of DLK protein used as antigenic sequence for polyclonal and monoclonal antibodies. The homologous region included in the targeting vector is also shown with a gray line. FC2, BB1R, and L3 indicate annealing sites of primers used for genotype analysis. B, BamHI; E, EcoRI; Ev, EcoRV. b, Southern blot analysis of genomic DNA obtained from liver of wild-type or mutant embryos. Regions of genomic DNA corresponding to the cDNA probes used for Southern blot analysis (BS and EE) are shown in a, at the bottom. Arrows indicate sizes of DNA fragments hybridized with each probe. EE probe detected 9.1 kb BamHI fragment of targeted allele including the neo cassette and 7.7 kb BamHI fragment of the targeted allele without the neo cassette.
Figure 2.

Loss of DLK expression in homozygous mutant mice. a, Western blot analysis of DLK protein immunoprecipitated from E16 brain lysates of wild-type (Dlk+/+), heterozygous mutant (Dlk+/−), and homozygous mutant (Dlk−/−) mice. A mouse monoclonal antibody raised against the C-terminal part of the DLK protein (Fig. 1a) was used for immunoprecipitation, and a rabbit polyclonal antibody raised against the C-terminal part of DLK protein was used for Western blot analysis. No residual protein reacting with these antibodies was detected in lysates prepared from brains of homozygous mutants. b, Sagittal sections of wild-type (Dlk+/+) or homozygous mutant (Dlk−/−) E14 embryos were stained with hematoxylin and eosin (HE), DLK polyclonal antibody, or βIII-tubulin, as indicated. Positive DLK antibody staining of neural tissues observed in sections of wild-type embryos was not visible in mutants, whereas generations of neural tissues including CNS and peripheral nervous system and non-neural tissues were not affected. Tc, Telencephalon; Mc, mesencephalon; Cb, cerebellar anlage; Sp, spinal cord; Ht, heart; Lu, lung; Lv, liver; In, intestine; St, stomach; Kd, kidney; CG, cervical ganglion; DRG, dorsal root ganglion. Scale bar, 1 mm.
Figure 3.

Loss of DLK affects fiber tract development. a, DLK protein in coronal sections of the cerebrum prepared from heterozygous (left hemisphere) and homozygous mutant (right hemisphere) embryos were stained with the anti-DLK polyclonal antibody. b, Coronal sections of wild-type (Dlk+/+) or homozygous mutant (Dlk−/−) head portions at E19 were stained with cresyl violet. Two sections (top and bottom) separated by ∼700 μm from each other revealed commissure neuron dysgenesis. The corpus callosum was particularly reduced in size at the front part (top row) in mutants. As for the anterior commissure, the anterior part was hardly seen, and the posterior part was reduced in mutants. c, The structure of olfactory bulb shown by Nissl staining (top row) and DiI labeling of lateral olfactory tract (bottom rows). Although the layer structure of olfactory bulb was preserved in homozygous mutants (Dlk−/−), thickness of glomerular layer (arrows) and lateral olfactory tract (arrowheads) was reduced. d, Rostrocaudal sections of heterozygous mutant (Dlk+/−) or homozygous mutant (Dlk−/−) brains at E18 were stained with a neurofilament antibody. In homozygous mutant brains, a reduction of neural fibers in the cingulum (arrowheads) and internal capsule (arrows) was obvious. Neural fibers along the hippocampus were apparently not affected. e, DiI labeling of ventrally extended axons from cortical neurons (arrows), which are reduced in mutant (Dlk−/−). f, Structures of the hippocampus and its associated neural fibers are shown as horizontal sections stained with cresyl violet. Structures of the psalterium, fimbria, and the hippocampus itself were indistinguishable between wild-types and mutants. CC, Corpus callosum; aAC, anterior part of the anterior commissure; pAC, posterior part of the anterior commissure; OB, olfactory bulb; HI, hippocampus; LOT, lateral olfactory tract; FIM, fimbria; PS, psalterium. Asterisks indicate DiI injection sites. Scale bars, 1 mm.
Immunohistochemistry.
Whole mouse embryos or isolated brains at embryonic day 14 (E14) to E19 were fixed overnight in 4% paraformaldehyde (PFA) at 4°C and then embedded in paraffin wax. The morning on which a vaginal plug was detected was designated as E0.5. Paraffin sections (6 μm thick) were dewaxed and hydrated before being heated at 120°C for 20 min in 10 mm sodium citrate, pH 6.0, and then immunohistochemically stained using a standard protocol. Briefly, sections were pretreated with 10% goat serum in Tris-buffered saline and Tween 20, pH 8.0, at room temperature for 30 min and were first incubated with affinity-purified anti-DLK rabbit primary antibody raised against the C-terminal part of DLK (Fig. 1a), rabbit anti-neurofilament M (dilution, 1:5000; Chemicon, Temecula, CA), mouse anti-vimentin (dilution, 1:1000; Sigma, St. Louis, MO), rabbit anti-reelin antibody (dilution, 1:300; Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-calretinin (dilution, 1:3000; Chemicon), or mouse anti-class III β-tubulin (dilution, 1:3000; Babco, Richmond, CA) at 4°C overnight. Subsequently, the sections were incubated with alkaline phosphatase-conjugated anti-rabbit or mouse IgG (TAGO) secondary antibodies for 2 h at room temperature. The enzymatic activity of alkaline phosphatase was detected with BM purple (Roche, Indianapolis, IN). Cyanine-3-conjugated (Jackson ImmunoResearch, West Grove, PA) and Alexa488-conjugated (Invitrogen, Carlsbad, CA) secondary antibodies were used in some experiments for antibody detection using fluorescence microscopy.
DiI labeling.
Brains of E19 embryo were isolated and fixed overnight in 4% PFA at 4°C. After washing the fixed brain with PBS for 3 h, 0.2% DiI solution in dimethylformamide was injected into the cortex or olfactory bulb. Brains were stored in the dark at room temperature in PBS for 4 weeks, embedded in 2% agarose in PBS, and sectioned by vibratome (200-μm-thick sections). The labeled sections were examined under a fluorescence microscope with a rhodamine filter set.
Birthdating experiments.
Pregnant mice at 15 d postconception (dpc) were injected intraperitoneally with bromodeoxyuridine (BrdU) (30 mg/kg body weight). Embryos were dissected and fixed at 19 dpc (E19). The sections were stained to detect BrdU-incorporated nuclei using an anti-BrdU monoclonal antibody (MBL, Woburn, MA) as described above with some modifications, i.e., sections were incubated with 1N HCl for 30 min and were neutralized by repeated PBS washes before application of the primary antibody. For staining, a biotinylated secondary antibody, streptavidin-conjugated horseradish peroxidase, and 3,3′-4,4′-diaminobenzidine were used.
Western blotting.
Whole brains from E16 embryos were dissected in ice-cold PBS. The meninges were removed, and brains were homogenized in 1 ml of SDS-PAGE sample buffer. These samples were appropriately diluted to give equal protein amounts and were used for SDS-PAGE. For immunoprecipitation (IP) of DLK protein, brains were homogenized in 1 ml of IP buffer containing 150 mm NaCl, 20 mm HEPES, pH 7.5, 1 mm EGTA, 25 mm β-glycerophosphate, 50 mm NaF, 1 mm Na3VO4, and a protease inhibitor cocktail (Sigma). A mouse monoclonal antibody (5F4) raised against the C-terminal part of DLK (Fig. 1a) was used for immunoprecipitation. Western blot analysis was performed according to standard protocols using the following antibodies: rabbit anti-DLK (dilution, 1:300), rabbit anti-active JNK (dilution, 1:1000; Promega, Madison, WI), rabbit c-Jun (dilution, 1:200; Cell Signaling Technology, Beverly, MA), rabbit phospho-c-Jun (Ser73; dilution, 1:300; Cell Signaling Technology), goat doublecortin (dilution, 1:1000; Santa Cruz Biotechnology), phospho-doublecortin (Thr331, Ser334; dilution, 1:5000) (Gdalyahu et al., 2004), mouse anti-MAP2 (dilution, 1:200; Sigma), mouse anti-glycogen synthase kinase 3β (GSK3β) (dilution, 1:2000; BD), rabbit anti-phospho-GSK3β (Ser9; dilution, 1:2000; Cell Signaling Technology), mouse anti-cyclin-dependent kinase 5 (Cdk5) (dilution, 1:200 Santa Cruz Biotechnology), and rabbit anti-phospho-cell division cycle 2 (Cdc2) (Tyr15; dilution, 1:1000; Cell Signaling Technology). To detect JNKs, a mixture of mouse anti-JNK1 (dilution, 1:1000; PharMingen, San Diego, CA) and mouse anti-JNK2 (dilution, 1:2000; Santa Cruz Biotechnology) was used. For secondary antibodies, horseradish peroxidase-conjugated anti-rabbit or mouse Igs (dilution, 1:2000 to 1:5000; Amersham Biosciences, Arlington Heights, IL) were used. Enzyme activity was detected with the ECL plus system (Amersham Biosciences), and luminescence was quantified with a Fuji (Tokyo, Japan) Las1000 plus luminescence image analyzer.
Monitoring of radial migration in slice cultures.
Green fluorescent protein (GFP) expression vectors driven by the α1-tubulin promoter were introduced into E13.5 or E14.5 mouse embryonic telencephalons by electroporation (Tabata and Nakajima, 2003), and slice cultures were made at E16.5, as described previously (Miyata et al., 2001). Briefly, dissected telencephalons were manually sliced in the coronal plane (200–300 μm thick) in silicon rubber-coated Petri dishes containing DMEM. Slices were mounted in collagen gel containing culture medium supplemented with epidermal growth factor (10 ng/ml), basic fibroblast growth factor (10 ng/ml), horse serum (5%), and fetal calf serum (5%) and were placed in an incubator (37°C, 5% CO2, 40% O2) for 30 min to solidify the gel. For experiments using the JNK inhibitor, slices were soaked in culture medium containing 20 μm SP600125 (anthra[1,9-cd]pyrazol-6(2H)-one) or vehicle alone [2 μl/ml dimethylsulfoxide (DMSO)] for 30 min and were placed on 35 mm dishes separated into two areas, for inhibitor treatment or vehicle treatment, using silicon rubber plates. Slices were then mounted in collagen gel containing the inhibitor or the vehicle alone and were incubated for an additional 30 min for solidification. Time-lapse recording to monitor radial migration of GFP-labeled cells was performed for 8 h at 15 min intervals, using an inverted epifluorescence microscope (IX70; Olympus Optical, Tokyo, Japan) equipped with a 10× objective lens (LCPlanF1; Olympus Optical) and a CCD camera (CoolSNAP; QImaging, Burnaby, British Columbia, Canada). During that period, the culture dish was placed on a heating stage (37°C) and was supplied with water-saturated gas containing 5% CO2 and 40% O2.
Statistical analysis.
To quantify migration rates of GFP-labeled cells, positions of all detectable cell somata on the slice 2 h after starting image acquisition were marked on tracing paper fixed to the video monitor, and migration during the next 6 h was traced for all cells that could be continuously followed on the movie. Positions of cells in each slice were determined by merging the fluorescence image with the phase-contrast view of the slice culture (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). To assess the effects of DLK gene knock-out on cell migration, 11 wild-type slices and 14 mutant slices were used in total. To assess the effects of the JNK inhibitor, 12 pairs of slices were used for vehicle (DMSO) or JNK inhibitor (20 μm SP600125) treatment. For the inhibitor experiment, pairs of cortical slices adjacent or close to each other (estimated by shapes of slices and distribution of GFP-labeled cells in slices) were selected and were used for vehicle or inhibitor treatment. In both cases, essentially the same results were obtained by two independent sets of experiments performed on different days. Other details for statistical analysis of video images are described in the text.
For quantitative Western blot analysis, brain extracts were prepared from five sets of wild-type (Dlk+/+), heterozygous (Dlk+/−), and homozygous (Dlk−/−) E16 embryos. To minimize interference with data by stage aberrations in embryos, sample preparation and statistical analysis were performed as follows: (1) three embryos of each set (Dlk+/+, Dlk+/−, Dlk−/−) were selected from littermates, (2) signal intensity obtained from a single sample was expressed as a percentage of the sum of signal intensities obtained from three embryos of the same set, and (3) this value was used to calculate the average and SD for each genotype (n = 5). To avoid type I errors, significant differences between heterozygous and homozygous embryos and between wild-type and heterozygous embryos were not taken into account, when no significant difference was observed between wild-type and homozygous embryos. (No such case was observed in the present work.)
Statistical significance was evaluated by a two-tailed Student's t test, and a p value <0.05 was regarded as significant.
Results
Disruption of DLK in mice
We disrupted the DLK gene by LacZ replacement of the coding exons 1, 2, and a part of 3, which encode 224 N-terminal amino acids including an ATP binding motif of the kinase domain (Fig. 1a,b). The remaining part of the DLK gene encoded 664 C-terminal amino acids of the DLK protein, which may not have any kinase activity. In the brain of homozygous DLK mutants (Dlk−/−), we detected no full-length or truncated forms of DLK proteins by immunoprecipitation and Western blotting, using antibodies raised against the C-terminal region of DLK (Fig. 2a). Therefore, we concluded that this mutant allele of DLK induced a strong loss of function.
Heterozygous mutant animals (Dlk+/−) were apparently normal and fertile. Homozygous mutant animals (Dlk−/−) survived during embryonic stages without showing any gross tissue abnormality (Fig. 2b) but they died perinatally, and no homozygous mutant survived until weaning.
DLK gene disruption affects specific axon tracts in the cerebrum
In wild-type E14 embryos, DLK is widely expressed in neural tissues, including the CNS and peripheral nervous system (Hirai et al., 2005). Disruption of the DLK gene resulted in the total loss of DLK expression in nervous tissues, including the brain, spinal cord, and ganglia of sensory, sympathetic, and vagus nerves (Fig. 2b). However, nervous tissue sections from E14 DLK homozygous mutants appeared to be normal (Fig. 2b; data not shown). At later stages, E18 or E19, DLK expression was evident in commissural neurons developing in the cerebrum of wild-type embryos (Fig. 3a). In mutant embryos, the anterior part of the anterior commissure was barely detectable, and the corpus callosum and the posterior part of the anterior commissure were reduced in size (Fig. 3b) (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). The gross structure of the olfactory bulb in the mutant was normal, whereas the thickness of the glomerular layer was reduced and the formation of the lateral olfactory tract was partly impaired (Fig. 3c). Neurofilament staining revealed that neural fibers of the cingulum and internal capsule were also reduced (Fig. 3d). The reduction of cortical axons was also shown by the injection of DiI into dorsolateral regions of cortex. In control E19 brain, DiI labeled a bundle of ventrally extended axons that may have been directed toward the ventrolaterally positioned internal capsule (Fig. 3e, left) (Richards et al., 1997). In the mutant, however, such extension of axons was greatly diminished (Fig. 3e, right). Conversely, structures of the hippocampus and the associated axonal tract of fimbria and psalterium in mutant embryos were indistinguishable from those of wild-type embryos (Fig. 3f). Together, these observations indicated that DLK gene disruption particularly affected axons that mainly originated from pyramidal neurons of the neocortex.
Neural cell migration is retarded by DLK gene disruption
Development of the cerebral cortex normally proceeded in mutant embryos at an early stage (E14), as the cortical plate was formed in the preplate, splitting it into the marginal zone and the subplate (data not shown). At a later stage, there were some abnormalities in the structure of the cortical plate. The laminated cortical structure was partly formed at E19, and layer-specific differences in Nissl-staining and cell density were prominent in the cortical plate of wild-type embryos (Fig. 4a). However, in mutant embryos, such differential patterns of different types of cells were not clearly seen (Fig. 4c). Partial disruption of the layered structure was also seen with calretinin staining of cortical neurons at E19. Although calretinin-positive neurons were localized to the upper layer of the cortical plate in control embryos (Fig. 4f,h) (Schmid et al., 2004), they had a more diffusible distribution in the mutant (Fig. 4g,h). This might reflect defects in the migration of postmitotic neurons. To evaluate effects of DLK gene disruption on neural migration, we first performed birthdating experiments in which nuclei of cortical neurons produced at approximately E15 were labeled with BrdU; then, at E19, positions of the labeled nuclei in the cortical plate were examined (Fig. 4b,d). In the dorsal cortex of wild-type embryos, a large proportion of labeled nuclei (57% of labeled nuclei found in the cortical plate) were located in the outermost layer of the cortical plate adjacent to the marginal zone (bin 1) at E19. In mutant embryos, BrdU-labeled nuclei were more dispersed in the cortical plate, and a smaller proportion (39%) of labeled nuclei was found in the outermost layer. A decrease in the proportion of labeled nuclei in the outermost layer was also significant in other cortical regions, including the dorsolateral and lateral cortices (Fig. 4e). These observations indicated that radial migration was partly impaired in mutant embryos. Notably, the overall architecture of radial glial fibers and the expression patterns of reelin were not affected by DLK gene disruption (Fig. 4i–l). Therefore, cell positioning in the cortical plate of mutant embryos is most likely affected by DLK gene disruption in migrating neurons. This notion was also supported by the predominant expression of DLK in migrating neurons (Hirai et al., 2002).
Figure 4.
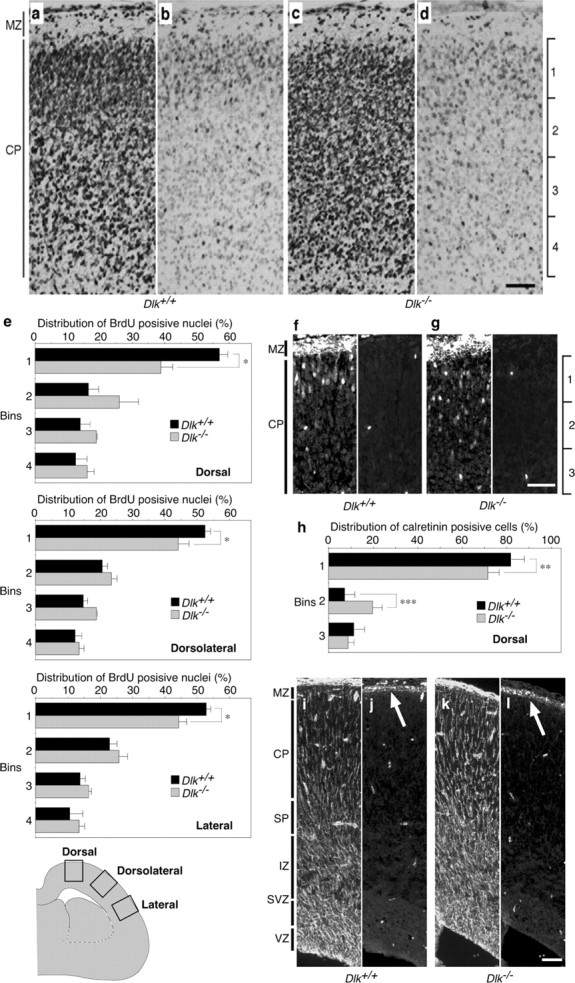
Radial migration in the neocortical region is affected by disruption of the DLK gene. a-d, BrdU was administered at E15, and the brain was fixed at E19. Coronal sections of wild type (a, b) or mutant (c, d) dorsal neocortex beneath bregma, stained with cresyl violet (a, c) or an anti-BrdU antibody (b, d). e, To quantify the distribution of BrdU-positive nuclei, the cortical plate was divided into four equal bins from the pial side to the ventricular side. The number of nuclei in each bin was counted and are shown as percentages of the total nuclei number in the cortical plate. Coronal sections of dorsal, dorsolateral, and lateral neocortices beneath bregma were used for analysis. *p < 0.05; n = 3. f, g, Immunofluorescent staining (cyanine 3) of coronal sections of E19 dorsal neocortex with anti-calretinin. Panels accompanied by f and g show nonspecific interaction of secondary antibody (Alexa488) by blood cells. Calretinin-positive neurons in the cortical plate were more dispersed in mutants (g). h, Numbers of calretinin-positive cells in each bin were counted and are shown as percentages of the total count. The deep layer of cortical plate enriched with calretinin-positive neural fibers was excluded from this analysis. **p < 0.01, ***p < 0.001; n = 8 sections. i–l, Immunofluorescent staining of coronal sections of the dorsal neocortex with anti-vimentin (i, k) and anti-reelin (j, l) antibodies. Radial glial architecture and reelin expression (arrow) in wild types (i, j) were preserved in mutants (k, l). The bright scattered signals correspond to nonspecific staining of blood cells by the secondary antibodies. Arrows indicate white belts located at the marginal zone. MZ, Marginal zone; VZ, ventricular zone. Scale bars, 50 μm.
To directly compare radial migration in the cerebral cortex of mutant and wild-type embryos, we used a slice culture system combined with in vivo labeling of cortical neurons by introducing GFP expression vectors. A GFP expression vector driven by the neuron-specific α1-tubulin promoter was introduced into neural progenitors in the ventricular zone at E13.5 by electroporation. Cortical slices were made 3 d after transfection, i.e., at E16.5, when a proportion of GFP-labeled cells migrated into the cortical plate. Migration of labeled cells in cultured slices was monitored with fluorescence microscopy, combined with a time-lapse imaging system. After making a movie file from the still photographs, the migration routes of soma of each labeled cell were monitored for 6 h (supplemental movies 1, 2, available at www.jneurosci.org as supplemental material). The start and finish positions were connected by a straight line, indicating the direction and distance of net displacement of cell soma. The array of lines drawn for cells in the wild-type embryo slice cultures correctly revealed changes in modes of radial migration (Fig. 5a, left). Short and randomly oriented lines might correspond to “multipolar migration,” which was characterized by slow or nondirectional migration and was often found in the intermediate zone (Tabata and Nakajima, 2003). Despite a slow rate of net replacement, cells were actively moving within a small area by frequently changing their shape. Long radially arrayed lines might correspond to “locomotion” or “somal translocation,” which was characterized by relatively fast and directed migration (Nadarajah et al., 2001). The array of lines drawn for cells of mutant embryos showed a similar profile, but it was more difficult to classify the lines into multipolar-type slow migration and locomotion- or soma translocation-type fast migrations (Fig. 5a, right). To quantify differences in cell migration profiles, we first classified the labeled cells into two groups depending on their position in the slice culture at time 0. Because the slow migrating cells were mainly located in the IZ and the fast migrating cells were in the subplate and cortical plate (SP–CP), we drew the border at the ventricular side edge of the subplate to separate the two groups of cells (Fig. 5a) (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Then, the length of each line (distance of net replacement) was measured, and an average for IZ cells or SP–CP cells in individual slices was used to calculate an average for all slices (n = 11 for controls; n = 14 for mutants). As shown in Figure 5b, the mean value of net replacement in SP–CP cells (52.5 μm/6 h) was double that in IZ cells (25.0 μm/6 h) in control slices. The value for SP–CP cells of mutant slices (38.5 μm/6 h) was significantly lower than that of control slices, whereas differences in values for IZ cells (25.9 μm/6 h for mutant slices) were not significant (Fig. 5b).
Figure 5.

Acceleration of radial migration at the subplate/cortical plate region is affected by disruption of the DLK gene. a, For quantitative analysis of radial migration, migration of individual GFP-labeled cells in slice cultures of E16.5 cortices were traced on a movie screen (for details, see Results). The starting points for each cell are shown by dots, and net displacement (distance and direction) during 6 h is shown by linear lines. b, The average values of net displacement of GFP-labeled cells located in different areas including the IZ and the CP were first calculated for each cultured slice. Then, averages and SDs of net displacement for wild-type slices (n = 11) and mutant slices (n = 14) were calculated and are shown graphically. Error bars indicate SDs. ***p < 0.001. c, The frequency distributions of net displacement for individual cells are shown as histograms. Median values for cells in different areas are shown with dotted lines. d, The percentage of migrating cells in SP–CP region. Cells whose soma changed position within 30 min were counted as migrating cells. Values were calculated for each slice, and average and SD for wild-type or mutant slices are shown graphically. *p < 0.01.
When the frequency distribution of net displacement for individual cells in all slices was illustrated using histograms, IZ cells of control slices had a peak of 10–20 μm/6 h, and >70% of these cells were within the range of 0–30 μm/6 h (Fig. 5c, left). In contrast, only 20% of SP–CP cells of control slices were in this range, and the remaining cells showed a broad distribution with a peak at ∼40–70 μm/6 h. This difference was reflected in median values for net displacements of IZ cells and SP–CP cells, which were 20.0 and 53.0 μm/6 h, respectively. These results indicated that most of migrating cells in control slices increased speed when they crossed the subplate. In mutant slices, net displacement of a relatively large population (36%) of SP–CP cells was in the range of 0–30 μm/6 h, giving a relatively low median value (38.0 μm/6 h) (Fig. 5c, right). Nevertheless, effects of DLK gene disruption on frequency distribution of net displacement for IZ cells was subtle, i.e., >70% of IZ cells in mutant slices were found in the range of 0–30 μm/6 h, and the median value was 21.7 μm/6 h (Fig. 5c).
In the locomotion-type cell migration, cells do not move continuously, and they are almost stopped for 40% of the total time period (Nadarajah et al., 2001). When we calculated the percentage of migrating SC–CP cells, it was significantly reduced in DLK mutant slices (Fig. 5d). Therefore, this reduction of moving time period or increase of pausing time period explains at least in part the sluggishness of radial migration in mutants. Regardless of this sluggishness, mutant cells migrated along the radial axis in SP–CP region (Fig. 5a) (supplemental movies 1, 2, available at www.jneurosci.org as supplemental material) with radially oriented leading process, as observed with control cells (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). Together, these quantitative analyses of live-imaging data revealed that radial migration of the majority of neurons in control slices accelerated in the subplate region and that disruption of the DLK gene affected the acceleration of radial migration.
JNK activity is reduced in DLK homozygous mutant brains
Because DLK is a MAPKKK for the JNK pathway, complete loss of DLK protein may result in a decrease in JNK activity. To evaluate changes in JNK activity in mutant mouse brain, we analyzed phosphorylation levels of JNK at the Thr–Pro–Tyr motif of the activation loop by Western blotting (Hirai et al., 2002), using whole-brain extracts of mutant or wild-type embryos. As shown in Figure 6b, JNK activity in the brain of E16 homozygous mutants decreased to 75.5% (p46 JNK) or 67.9% (p55 JNK) of that in wild-type controls. Protein level of JNK was slightly increased in mutant brains. Although DLK protein level in heterozygous mutants decreased to 41.8% compared with wild-type controls (Fig. 6a), the decrease in JNK activity in heterozygous mutants was not significant.
Figure 6.

Loss of DLK expression results in a decrease in JNK activity. a, DLK protein level in whole brain (E16) extracts was quantified by Western blotting using a rabbit polyclonal antibody raised against the C-terminal part of DLK. DLK protein level was significantly reduced in heterozygous mutants (***p < 0.001) and was absent in homozygous mutants. The faint bands detected in the lysate from homozygous mutants corresponded to nonspecific interaction of the secondary antibody, which were removed by immunoprecipitation using the anti-DLK monoclonal antibody (Fig. 2a). b, JNK activity was monitored by Western blotting using an anti-active (pThr–Pro–pTyr) JNK antibody. Both the p46 fraction and the p55 fraction of active JNK were significantly decreased in homozygous mutants. The total amount of p46 JNK was also quantified and was found to be slightly increased in mutants. **p < 0.01, ***p < 0.001; n = 5.
The transcription factor c-Jun and some microtubule-associated proteins have been identified as JNK substrates (Chang et al., 2003; Gdalyahu et al., 2004; Xia and Karin, 2004; Yoshida et al., 2004; Björkblom et al., 2005). We evaluated JNK activity by examining phosphorylation levels of these JNK substrates. The level of the phosphorylated form of c-Jun at Ser73 in homozygous mutant brains decreased to less than half that of the wild-type control (Fig. 7a). The effects of DLK gene disruption on the phosphorylation of DCX at Thr331 and Ser334 (Gdalyahu et al., 2004) were relatively moderate but statistically significant (p < 0.05) (Fig. 7b). Phosphorylation levels of MAP2c, estimated by mobility delay on SDS-PAGE (supplemental Fig. S4, available at www.jneurosci.org as supplemental material), were also significantly affected by DLK gene disruption (Fig. 7c). However, we could not detect any significant effect on the phosphorylation level of tau protein (Fig. 7d). We detected a slight decrease in phosphorylation of the Tyr15 residue conserved in some Cdk-related proteins, including Cdc2, Cdk2, and Cdk5 and a slight increase in the phosphorylation of GSK3β at Ser9; the latter observation was statistically significant (supplemental Fig. S5, available at www.jneurosci.org as supplemental material). This indicated a weak, indirect interaction of the DLK–JNK pathway with other signaling pathways regulating cell migration or axon growth (Jiang et al., 2005; Yoshimura et al., 2005). Together, these results indicate that DLK plays a significant role in activation of JNK in the embryonic brain, although there are extensive redundant pathways. Moreover, the DLK–JNK pathway is connected to multiple downstream targets, which might contribute to the regulation of radial migration or axon growth.
Figure 7.

Phosphorylation levels of JNK substrates decreased in E16 brains of DLK homozygous mutants. a, Protein levels of c-Jun phosphorylated at Ser73 and c-Jun in total were quantified by Western blot analysis. The amount of phospho-c-Jun was decreased in homozygous mutants to less than half that of wild-type embryos. b, Protein levels of DCX phosphorylated at Thr331 and Ser334 and DCX in total were quantified by Western blotting. The amount of the phospho-DCX was significantly decreased in homozygous mutants, although the difference with wild types was not significant. c, The phosphorylation level of MAP2c was estimated from the ratio of mobility-delayed bands (pMAP2c) in total MAP2c bands. These mobility delayed bands were detected with an anti-phospho-Thr–Pro antibody and disappeared after phosphatase treatment in vitro (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). The relative amount of pMAP2c was also slightly decreased in homozygous mutants. d, No significant change was observed in tau protein level or its phosphorylation level when calculated by Western blotting using an anti-tau antibody, an anti-dephosphorylated tau antibody (tau-1), and an anti-tau protein phosphorylated at Ser202 (AT-8). *p < 0.05, **p < 0.01, ***p < 0.001, respectively; n = 5.
Inhibition of JNK affects acceleration of radial migration at the subplate
If JNK is a major downstream target for DLK in the regulation of radial migration, inhibition of JNK activity might also affect acceleration of migration around the subplate as observed with disruption of the DLK gene. To test this possibility, we examined effects of SP600125 (Bennett et al., 2001), a JNK inhibitor, on migration of cortical neurons in slice culture. The suppressive effects of a JNK inhibitor on radial cell migration in cortical slice cultures have been shown previously by comparing positions of GFP-labeled neurons before and after 28 h of cultivation (Kawauchi et al., 2003). However, quantitative analysis of position-dependent speed change is required for an accurate comparison with the phenotype of DLK mutants. For such analysis, α1-tubulin-driven GFP expression vectors were transfected into ventricular cells of E14.5 wild-type embryos, and cortical slices were made at E16.5. Slices were then cultured in the presence or absence of 20 μm SP600125, and the migration of GFP-labeled cells was monitored using a time-lapse imaging system (supplemental movies 3, 4, available at www.jneurosci.org as supplemental material). As already shown in Figure 5, fast migrating cells were mainly found in SP–CP, and slow migrating cells were in IZ. To separate ventricular side slow-migrating cells and pial side fast-migrating cells, we drew a border line, as described in supplemental Figure S6 (available at www.jneurosci.org as supplemental material), which was mostly located in the subplate area. The mean value of the net displacement for cells located at the pial side of the border line (56.3 μm/6 h) was more than three times bigger than that of ventricular side cells (16.8 μm/6 h) in control slice cultures (Fig. 8a). The frequency distribution of net displacement for individual cells in control cultures showed a clear segregation between slow-migrating cells in the ventricular side and fast-migrating cells in the pial side (Fig. 8b, left), thus demonstrating the appropriate location of the border line.
Figure 8.

Inhibition of JNK activity significantly impairs radial migration in cortical slice cultures. a, The average values of net displacement of GFP-labeled cells located in different areas, i.e., in the ventricular side and the pial side (for area definition, see supplemental Fig. S6, available at www.jneurosci.org as supplemental material), were first calculated for each cultured slice. Then, averages and SDs of net displacement for slices treated with vehicle (DMSO; n = 12) or JNK inhibitor (SP600125; n = 12) were calculated and are shown graphically. Error bars indicate SDs. ***p < 0.001. b, The frequency distributions of net displacement of individual cells are shown using histograms. Median values for cells in different areas are shown with dotted lines. c, Percentage of migrating cells in pial side. Cells whose soma changed position within 30 min were counted as migrating cells. Values were calculated for each slice, and the average and SD for slices treated with vehicle or JNK inhibitor are shown graphically. ***p < 0.001.
Administration of JNK inhibitors to cultures resulted in a remarkable reduction in migration rate on the pial side of the border, whereas it showed no significant effect on cells of the ventricular side (Fig. 8a). As a result, improvement of mean migration rate in the pial side compared with the ventricular side was only 1.5 times. A change in frequency distribution of net displacement was also remarkable on the pial side. Addition of JNK inhibitors caused a shift of frequency distribution of pial cells to a lower range, which heavily overlapped with the distribution of ventricular side cells (Fig. 8b, right). This was also seen as a decrease in the median value of net displacement of cells in the pial side (27.0 μm/6 h for inhibitor-treated slices vs 59.0 μm/6 h for control slices). In contrast, inhibitor treatment did not affect the median value for cells in the ventricular side (18.0 μm/6 h). The percentage of migrating cells in the pial side was also significantly reduced (Fig. 8c). These quantitative data indicate that JNK plays an important role in acceleration of radial migration at the “speed-changing” line located close to the subplate.
Discussion
JNK activity is maintained at an extremely high level in the embryonic brain compared with other MAP kinase-related enzymes (Hirai et al., 2002). However, the biological significance of such high activity of JNK and the molecular basis for the maintenance of high JNK activity are obscure. We show here that DLK gene disruption affects the maintenance of high JNK activity and phosphorylation levels of JNK target proteins in the embryonic mouse brain. Concurrently, axon tracts that originated from pyramidal neurons of the neocortex and acceleration of radial migration at the subplate region in the cerebral cortex were affected by DLK gene disruption. Given that most of the radially migrating cells generated in the cortical ventricular zone are pyramidal neurons (Parnavelas, 2000), the DLK–JNK pathway may contribute to both axon growth regulation and changes in modes of radial migration of pyramidal neurons of the neocortex.
Acceleration of radial migration at the subplate
Quantitative analysis of movies that monitored migration of GFP-labeled cells in slice cultures showed the presence of a speed-change zone around the subplate, clearly segregating slow migrating cells to the ventricular side and fast migrating cells to the pial side. Mean values of migration rate calculated from net displacement of soma during 6 h (Fig. 8) were 2.8 μm/h for ventricular cells and 9.4 μm/h for pial cells. Such differences in migration rates might be attributable to changes in modes of radial migration from multipolar migration to locomotion or somal translocation (Nadarajah et al., 2001; Tabata and Nakajima, 2003). Accordingly, those migration rates were comparable with mean values for multipolar migration (2.2 μm/h) and locomotion (9–47 μm/h), respectively.
DLK protein and active JNK are relatively abundant in the IZ region. Nevertheless, DLK gene disruption and JNK inhibition specifically affected fast migration in the SP–CP region. Slow migration in the IZ region was not significantly affected. Moreover, constitutive high level expression of DLK is suppressive for radial migration (Hirai et al., 2002). These observations imply that DLK protein and JNK activity are required for neurons to prepare for changes in modes of cell migration rather than for fast migration itself. However, we cannot rule out the possibility that relatively low levels of DLK proteins and JNK activity in neurons of the SP–CP region would support fast migration.
What can trigger changes in migration rates or modes? Because expressions of DLK and active JNK are relatively high in most cells on the ventricular side of the speed change zone (Hirai et al., 2002; Kawauchi et al., 2003; Gdalyahu et al., 2004), they might not be a primary trigger for acceleration. Instead, calcium signaling triggered by the NMDA receptor and interaction with extracellular factors produced by subplate neurons might play such a role (Godfraind et al., 1988; Kerjan et al., 2005; Kholmanskikh et al., 2006). Moreover, because migrating neurons in the intermediate zone often accompanied by a primary axon extend toward the ventricular zone, it is possible that such cellular processes transmit a signal that regulates migration mode (Noctor et al., 2004).
DLK is a JNK activator in vivo
Gene disruption of DLK caused a decrease in JNK activity in the embryonic brain, indicating that endogenous DLK acted as a MAPKKK for the JNK pathway. JNK activity remaining in the brain of DLK mutant embryos may depend on spontaneous activity of the MAPKK class of JNK activators such as MKK7 and MKK4 and other MAPKKKs for the JNK pathway present in the brain (Widmann et al., 1999; Gallo and Johnson, 2002). Disruption of one such MAPKKKs, MEKK4, has been shown to cause a defect in neural tube closure as well as periventricular heterotopia as a result of cell migration defects in the ventricular zone (Chi et al., 2005). Inactivation of two of three JNK genes, JNK1 and JNK2, also results in defects in neural tube closure and dysregulation of apoptosis in the neuroepithelium of the brain region (Kuan et al., 1999). These observations indicate possible roles of MEKK4 and JNK in the neuroepithelium or ventricular zone of the developing brain composed of undifferentiated neural progenitors. However, extensive investigation of their function in neurons that moved out from the ventricular zone was limited by severe malformation of the brain or the death of embryos before the start of pronounced neurogenesis in the cerebrum. Because DLK is almost exclusively expressed in neurons (Hirai et al., 2005), disruption of this gene in mice uncovered the JNK function in these cells.
What regulates DLK activity? Distribution of DLK proteins showed layer specificity in the developing neocortex, a clear contrast with JNK protein, which shows a rather uniform distribution (Hirai et al., 2002). However, distribution of active JNK shows clear layer specificity considerably overlapping with that of DLK (Hirai et al., 2002; Gdalyahu et al., 2004). Therefore, DLK activity to induce JNK activation might simply depend on its protein level, which could be regulated by the ubiquitin-proteasome system, as suggested for DLK regulation in the synapse of Caenorhabditis elegans (Nakata et al., 2005). In addition, JNK interacting protein (JIP), a scaffold for the DLK–JNK pathway, might be involved in the regulation of DLK-dependent JNK activation. Notably, disruption of JIP-3 gene caused commissure defects in the cerebrum (Kelkar et al., 2003). The Wnt (wingless-type MMTV integration site family) pathway is also a candidate as a DLK regulator because it has been shown to regulate process formation in neural cells via JNK (Rosso et al., 2004; Tissir et al., 2005). It is clear that additional studies are required to identify the molecular basis of regulation of DLK-dependent JNK activation in the context of axonal growth and radial cell migration.
Downstream targets of DLK for the regulation of axon growth and radial migration
Although molecular mechanisms underlying regulation of radial migration and axon growth by the DLK–JNK pathway remain to be elucidated, some downstream targets of JNK that are thought to be directly involved in these cellular events have been reported. For example, DCX is the product of the gene responsible for type 1 lissencephaly in humans (Gleeson and Walsh, 2000), and it can be phosphorylated by JNK mainly at Thr321, Thr331, and Ser334. Mutations in these JNK phosphorylation sites on DCX affect migration of cerebellar neurons, indicating that DCX is a good candidate downstream target of JNK to regulate radial migration (Gdalyahu et al., 2004). In fact, in the present study, we showed a slight decrease in phosphorylation level of DCX at Thr331 and Ser334 in DLK mutant mice brains. MAP2 might also be a JNK target regulating modes of neural cell migration and maintenance of axons by modulating microtubule organization (Teng et al., 2001; Chang et al., 2003; Björkblom et al., 2005). A slight decrease in MAP2c phosphorylation level in brains of DLK mutant embryos supports this possibility. Tau protein is another microtubule-associated protein regulating microtubule architecture. It can also be phosphorylated by JNK (Yoshida et al., 2004). However, we could not detect any significant difference in phosphorylation level of tau protein at one of the JNK target sites (Ser202) between wild-type and DLK mutant embryo brains. This might be attributable to multiple JNK phosphorylation sites on tau protein, i.e., even if phosphorylation was significantly diminished in total, the effect on individual phosphorylation sites might be limited. We cannot conclude that these rather small changes in phosphorylation levels of microtubule modulators are sufficient to explain the phenotype of DLK mutant mice described in the present study. However, cooperation between these changes may result in some kind of dysregulation in microtubule organization, which might affect modes of cell migration and axon growth.
In addition to these microtubule modulators, the transcription factor c-Jun is a good substrate for JNK, whose activity is enhanced by phosphorylation at its N-terminal part, including the two JNK phosphorylation sites Ser63 and Ser73 (Hibi et al., 1993). Although the c-Jun protein might not directly be involved in regulation of cell migration or axon growth, a relatively large effect of DLK mutation on the phosphorylation level of c-Jun Ser73 suggests a certain contribution of c-Jun to expression of genes functioning in these cellular events (Martín-Blanco, 1997; Xia and Karin, 2004).
Coordination of cell migration and axon growth
An increasing number of reports have shown that radially migrating neurons often have a long axon-like trailing process (Stensaas and Stensaas, 1968; Miyata et al., 2001; Noctor et al., 2004). Moreover, it has been shown that the axon-like process grows during radial migration of neocortical pyramidal neurons (Hatanaka and Murakami, 2002). This coordination of cell migration and axon growth may depend on a particular molecular mechanism that is able to modulate both activities. This notion is supported by observations showing that some cell surface molecules, such as Thy1 (Rege and Hagood, 2006), semaphorin 4D/plexin-B1 (Tamagnone and Comoglio, 2004), and neuregulin-1 (López-Bendito et al., 2006), function in both axon growth and cell migration. Moreover, a common requirement for microtubules modulators, including MAPs (Teng et al., 2001), Lis1 (Tsai et al., 2005), DCX/DCX-like kinase (Deuel et al., 2006; Koizumi et al., 2006), and superior cervical ganglion 10 (Tararuk et al., 2006), for these cellular events has been reported. Although a molecular mechanism coordinating the migration and axon growth of neocortical pyramidal neurons remains obscure, one can speculate that it is driven by protein kinases known to modify the function of such microtubules modulators, e.g., Cdk5 (Niethammer et al., 2000; Tanaka et al., 2004), GSK3β (Jiang et al., 2005; Yoshimura et al., 2005), MARK (MAP/microtubule affinity-regulating kinase 1), PKA (Drewes et al., 1997; Schaar et al., 2004), and JNK (Gdalyahu et al., 2004; Tararuk et al., 2006). Our finding that the disruption of the DLK gene, a JNK activator, affects both radial migration and axon growth of pyramidal neurons supports the presence of such a coordinating mechanism and provides a new clue for investigating its molecular base.
Footnotes
This work was supported by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan. We thank T. Ohshima for his helpful comments on this manuscript and K. Saito for her technical assistance on time-lapse recording of cell migration. A special thanks to O. Reiner for providing the phospho-doublecortin (Thr331, Ser334) antibody.
References
- Assadi AH, Zhang G, Beffert U, McNeil RS, Renfro AL, Niu S, Quattrocchi CC, Antalffy BA, Sheldon M, Armstrong DD, Wynshaw-Boris A, Herz J, D'Arcangelo G, Clark GD. Interaction of reelin signaling and Lis1 in brain development. Nat Genet. 2003;35:270–276. doi: 10.1038/ng1257. [DOI] [PubMed] [Google Scholar]
- Bennett BL, Sasaki DT, Murray BW, O'Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci USA. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björkblom B, Östman N, Hongisto V, Komarovski V, Filén J-J, Nyman TA, Kallunki T, Courtney MJ, Coffey ET. Constitutively active cytoplasmic c-Jun N-terminal kinase 1 is a dominant regulator of dendritic architecture: role of microtubule-associated protein 2 as an effector. J Neurosci. 2005;25:6350–6351. doi: 10.1523/JNEUROSCI.1517-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi M, Atlas R, Kantor A, Sapir T, Reiner O. Interaction between LIS1 and doublecortin, two lissencephaly gene products. Hum Mol Genet. 2000;9:2205–2213. doi: 10.1093/oxfordjournals.hmg.a018911. [DOI] [PubMed] [Google Scholar]
- Chang L, Jones Y, Ellisman MH, Goldstein LSB, Karin M. JNK1 is required for maintenance of neuronal microtubules and controls phosphorylation of microtubule-associated proteins. Dev Cell. 2003;4:521–533. doi: 10.1016/s1534-5807(03)00094-7. [DOI] [PubMed] [Google Scholar]
- Chi H, Sarkisian MR, Rakic P, Flavell RA. Loss of mitogen-activated protein kinase kinase kinase 4 (MEKK4) results in enhanced apoptosis and defective neural tube development. Proc Natl Acad Sci USA. 2005;102:3846–3851. doi: 10.1073/pnas.0500026102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuel TAS, Liu JS, Corbo JC, Yoo S-Y, Rorke-Adams LB, Walsh CA. Genetic interactions between Doublecortin and Doublecortin-like kinase in neuronal migration and axon outgrowth. Neuron. 2006;49:41–53. doi: 10.1016/j.neuron.2005.10.038. [DOI] [PubMed] [Google Scholar]
- Drewes G, Ebneth A, Preuss U, Mandelkow E-M, Mandelkow E. MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell. 1997;89:297–308. doi: 10.1016/s0092-8674(00)80208-1. [DOI] [PubMed] [Google Scholar]
- Gallo KA, Johnson GL. Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat Rev Mol Cell Biol. 2002;3:663–672. doi: 10.1038/nrm906. [DOI] [PubMed] [Google Scholar]
- Gdalyahu A, Ghosh I, Levy T, Sapir T, Sapoznik S, Fishler Y, Azoulai D, Reiner O. DCX, a new mediator of the JNK pathway. EMBO J. 2004;23:823–832. doi: 10.1038/sj.emboj.7600079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleeson JG, Walsh CA. Neuronal migration disorders: from genetic diseases to developmental mechanisms. Trends Neurosci. 2000;23:352–359. doi: 10.1016/s0166-2236(00)01607-6. [DOI] [PubMed] [Google Scholar]
- Godfraind C, Schachner M, Goffinet AM. Immunohistological localization of cell adhesion molecules L1, J1, N-CAM and their common carbohydrate L2 in the embryonic cortex of normal and reeler mice. Brain Res. 1988;470:99–111. doi: 10.1016/0165-3806(88)90205-2. [DOI] [PubMed] [Google Scholar]
- Hatanaka Y, Murakami F. In vitro analysis of the origin, migratory behavior, and maturation of cortical pyramidal cells. J Comp Neurol. 2002;454:1–14. doi: 10.1002/cne.10421. [DOI] [PubMed] [Google Scholar]
- Hatten ME. Central nervous system neuronal migration. Annu Rev Neurosci. 1999;22:511–539. doi: 10.1146/annurev.neuro.22.1.511. [DOI] [PubMed] [Google Scholar]
- Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinae that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- Hirai S, Kawaguchi A, Hirasawa R, Baba M, Ohnishi T, Ohno S. MAPK-upstream protein kinase (MUK) regulates the radial migration of immature neurons in telencephalon of mouse embryo. Development. 2002;129:4483–4495. doi: 10.1242/dev.129.19.4483. [DOI] [PubMed] [Google Scholar]
- Hirai S, Kawaguchi A, Suenaga J, Ono M, Cui DF, Ohno S. Expression of MUK/DLK/ZPK, an activator of the JNK pathway, in the nervous system of the developing mouse embryo. Gene Expr Patterns. 2005;5:517–523. doi: 10.1016/j.modgep.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Jiang H, Guo W, Liang X, Rao Y. Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3β and its upstream regulators. Cell. 2005;120:123–135. doi: 10.1016/j.cell.2004.12.033. [DOI] [PubMed] [Google Scholar]
- Kawauchi T, Chihama K, Nabeshima Y, Hoshino M. The in vivo roles of STEF/Tiam1, Rac1 and JNK in cortical neuronal migration. EMBO J. 2003;22:4190–4201. doi: 10.1093/emboj/cdg413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelkar N, Delmotte M-H, Weston CR, Barrett T, Sheppard BJ, Flabell RA, Davis RJ. Morphogenesis of the telencephalic commissure requires scaffold protein JNK-interacting protein 3 (JIP3) Proc Natl Acad Sci USA. 2003;100:9843–9848. doi: 10.1073/pnas.1733944100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerjan G, Dolan J, Haumaitre C, Schneider-Maunoury S, Fujisawa H, Mitchell K, Chédotal A. The transmembrane semaphoring Sema6A controls cerebellar granule cell migration. Nat Neurosci. 2005;8:1516–1524. doi: 10.1038/nn1555. [DOI] [PubMed] [Google Scholar]
- Kholmanskikh SS, Koeller HB, Wynshaw-Boris A, Gomez T, Letourneau PC, Ross ME. Calcium-dependent interaction of Lis1 with IQGAP1 and Cdc42 promotes neuronal motility. Nat Neurosci. 2006;9:50–57. doi: 10.1038/nn1619. [DOI] [PubMed] [Google Scholar]
- Koizumi H, Tanaka T, Gleeson J. Doublecortin-like kinase functions with doublecortin to mediate fiber tract decussation and neuronal migration. Neuron. 2006;49:55–66. doi: 10.1016/j.neuron.2005.10.040. [DOI] [PubMed] [Google Scholar]
- Kuan CY, Yang DD, Samanta Roy DR, Davis RJ, Rakic P, Flavell RA. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron. 1999;22:667–676. doi: 10.1016/s0896-6273(00)80727-8. [DOI] [PubMed] [Google Scholar]
- López-Bendito G, Cautinat A, Sánchez JA, Bielle F, Flames N, Garratt AN, Talmage DA, Role LW, Charnay P, Marín O, Garel S. Tangential neuronal migration controls axon guidance: a role for neuregulin-1 in thalamocortical axon navigation. Cell. 2006;125:127–142. doi: 10.1016/j.cell.2006.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magdaleno SM, Curran T. Brain development: integrins and the Reelin pathway. Curr Biol. 2001;11:R1032–R1035. doi: 10.1016/s0960-9822(01)00618-2. [DOI] [PubMed] [Google Scholar]
- Martín-Blanco E. Regulation of cell differentiation by the Drosophila Jun kinase cascade. Curr Opin Genet Dev. 1997;7:666–671. doi: 10.1016/s0959-437x(97)80015-9. [DOI] [PubMed] [Google Scholar]
- Miyata T, Kawaguchi A, Okano H, Ogawa M. Asymmetric inheritance of radial glial fibers by cortical neurons. Neuron. 2001;31:727–741. doi: 10.1016/s0896-6273(01)00420-2. [DOI] [PubMed] [Google Scholar]
- Nadarajah B, Brunstrom JE, Grutzendler J, Wong ROL, Pearlman AL. Two modes of radial migration in early development of the cerebral cortex. Nat Neurosci. 2001;4:143–150. doi: 10.1038/83967. [DOI] [PubMed] [Google Scholar]
- Nakata K, Abrams B, Grill B, Goncharov A, Huang X, Chisholm AD, Jin Y. Regulation of a DLK-1 and p38 MAP kinase pathway by the ubiquitin ligase RPM-1 is required for presynaptic development. Cell. 2005;120:407–420. doi: 10.1016/j.cell.2004.12.017. [DOI] [PubMed] [Google Scholar]
- Niethammer M, Smith DS, Ayala R, Peng J, Ko J, Lee M-S, Morabito M, Tsai L-H. NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron. 2000;28:697–711. doi: 10.1016/s0896-6273(00)00147-1. [DOI] [PubMed] [Google Scholar]
- Noctor S, Martínes-Cerdeño V, Ivic L, Kriegstein AR. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci. 2004;7:136–144. doi: 10.1038/nn1172. [DOI] [PubMed] [Google Scholar]
- Ohshima T, Ogawa M, Veeranna, Hirasawa M, Longenecker G, Ishiguro K, Pant HC, Brady RO, Kulkarni AB, Mikoshiba K. Synergistic contributions of cyclin-dependent kinase 5/p35 and Reelin/Dab1 to the positioning of cortical neurons in the developing mouse brain. Proc Natl Acad Sci USA. 2001;98:2764–2769. doi: 10.1073/pnas.051628498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnavelas JG. The origin and migration of cortical neurons: new vistas. Trends Neurosci. 2000;23:126–131. doi: 10.1016/s0166-2236(00)01553-8. [DOI] [PubMed] [Google Scholar]
- Rakic P. Principles of neural cell migration. Experientia. 1990;46:882–891. doi: 10.1007/BF01939380. [DOI] [PubMed] [Google Scholar]
- Rege TA, Hagood JS. Thy-1 as a regulator of cell-cell and cell-matrix interactions in axon regeneration, apoptosis, adhesion, migration, cancer, and fibrosis. FASEB J. 2006;20:1045–1054. doi: 10.1096/fj.05-5460rev. [DOI] [PubMed] [Google Scholar]
- Richards LJ, Koester SE, Tuttle R, O'Leary DM. Directed growth of early cortical axons is influenced by a chemoattractant released from an intermediate target. J Neurosci. 1997;17:2445–2458. doi: 10.1523/JNEUROSCI.17-07-02445.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosso SB, Sussman D, Wynshaw-Boris A, Salinas PC. Wnt signaling through Dishevelled, Rac and JNK regulates dendritic development. Nat Neurosci. 2004;8:34–42. doi: 10.1038/nn1374. [DOI] [PubMed] [Google Scholar]
- Sakai K, Miyazaki J. A transgenic mouse line that retains Cre recombinase activity in mature oocytes irrespective of the cre transgene transmission. Biochem Biophys Res Commun. 1997;237:318–324. doi: 10.1006/bbrc.1997.7111. [DOI] [PubMed] [Google Scholar]
- Schaar BT, Kinoshita K, McConnell SK. Doublecortin microtubule affinity is a regulated by a balance of kinase and phosphatase activity at the leading edge of migrating neurons. Neuron. 2004;41:203–213. doi: 10.1016/s0896-6273(03)00843-2. [DOI] [PubMed] [Google Scholar]
- Schmid RS, Shelton S, Stanco A, Yokota Y, Kreidberg JA, Anton ES. α3β1 integrin modulates neuronal migration and placement during early stages of cerebral cortical development. Development. 2004;131:6023–6031. doi: 10.1242/dev.01532. [DOI] [PubMed] [Google Scholar]
- Stensaas LJ, Stensaas SS. An electron microscope study of cells in the matrix and intermediate laminae of the cerebral hemisphere of the 45 mm rabbit embryo. Z Zellforsch Mikrosk Anat. 1968;91:341–365. doi: 10.1007/BF00440763. [DOI] [PubMed] [Google Scholar]
- Tabata H, Nakajima K. Multipolar migration: the third mode of radial neuronal migration in the developing cerebral cortex. J Neurosci. 2003;23:9996–10001. doi: 10.1523/JNEUROSCI.23-31-09996.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamagnone L, Comoglio PM. To move or not to move? EMBO Rep. 2004;5:356–361. doi: 10.1038/sj.embor.7400114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Serneo FF, Tseng H-C, Kulkarni AB, Tsai L-H, Gleeson JG. Cdk5 phosphorylation of Doublecortin Ser297 regulates its effect on neuronal migration. Neuron. 2004;41:215–227. doi: 10.1016/s0896-6273(03)00852-3. [DOI] [PubMed] [Google Scholar]
- Tararuk T, Östman N, Li W, Björkblom B, Padzik A, Zdrojewska J, Hongisto V, Herdegen T, Konopka W, Courtney MJ, Coffey ET. JNK1 phosphorylation of SCG10 determines microtubule dynamics and axodendritic length. J Cell Biol. 2006;173:265–277. doi: 10.1083/jcb.200511055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng J, Takei Y, Harada A, Nakata T, Chen J, Hirokawa N. Synergistic effects of MAP2 and MAP1B knockout in neuronal migration, dendritic outgrowth, and microtubule organization. J Cell Biol. 2001;155:65–76. doi: 10.1083/jcb.200106025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissir F, Bar I, Jossin Y, DeBacker O, Goffinet AM. Protocadherin Celsr3 is crucial in axonal tract development. Nat Neurosci. 2005;9:451–457. doi: 10.1038/nn1428. [DOI] [PubMed] [Google Scholar]
- Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, Hammer RE, Richardson JA, Herz J. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell. 1999;97:689–701. doi: 10.1016/s0092-8674(00)80782-5. [DOI] [PubMed] [Google Scholar]
- Tsai J-W, Chen Y, Kriegstein AR, Vallee RB. Lis1 RNA interference blocks neural stem cell division, morphogenesis, and motility at multiple stages. J Cell Biol. 2005;170:935–945. doi: 10.1083/jcb.200505166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev. 1999;79:143–180. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- Xia Y, Karin M. The control of cell motility and epithelial morphogenesis by Jun kinas. Trends Cell Biol. 2004;14:94–101. doi: 10.1016/j.tcb.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Hastie CJ, McLauchlan H, Cohen P, Goedert M. Phosphorylation of microtubule-associated protein tau by isoforms of c-Jun N-terminal kinase (JNK) J Neurochem. 2004;90:352–358. doi: 10.1111/j.1471-4159.2004.02479.x. [DOI] [PubMed] [Google Scholar]
- Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K. GSK-3β regulates phosphorylation of CRMP-2 and neuronal polarity. Cell. 2005;120:137–149. doi: 10.1016/j.cell.2004.11.012. [DOI] [PubMed] [Google Scholar]