

Abstract
To study the functional role of activated astrocytes in glutamate homeostasis in vivo, we used a model of sustained astrocytic activation in the rat striatum through lentiviral-mediated gene delivery of ciliary neurotrophic factor (CNTF). CNTF-activated astrocytes were hypertrophic, expressed immature intermediate filament proteins and highly glycosylated forms of their glutamate transporters GLAST and GLT-1. CNTF overexpression produced a redistribution of GLAST and GLT-1 into raft functional membrane microdomains, which are important for glutamate uptake. In contrast, CNTF had no detectable effect on the expression of a number of neuronal proteins and on the spontaneous glutamatergic transmission recorded from striatal medium spiny neurons. These results were replicated in vitro by application of recombinant CNTF on a mixed neuron/astrocyte striatal culture. Using microdialysis in the rat striatum, we found that the accumulation of extracellular glutamate induced by quinolinate (QA) was reduced threefold with CNTF. In line with this result, CNTF significantly increased QA-induced [18F]-fluoro-2-deoxyglucose uptake, an indirect index of glutamate uptake by astrocytes. Together, these data demonstrate that CNTF activation of astrocytes in vivo is associated with marked phenotypic and molecular changes leading to a better handling of increased levels of extracellular glutamate. Activated astrocytes may therefore be important prosurvival agents in pathological conditions involving defects in glutamate homeostasis.
Keywords: activated astrocytes, CNTF, glutamate transporters, rafts, excitotoxicity, glucose uptake
Introduction
Astrocytes clear the excitatory neurotransmitter glutamate from the synapse through their transporters GLAST and GLT-1 (Danbolt, 2001). This function is crucial to optimize glutamatergic transmission and to prevent a rise in extracellular glutamate concentration that can trigger excitotoxic cascades (Maragakis and Rothstein, 2004). Numerous regulatory mechanisms of glutamate transporter efficiency have been reported so far, including transcriptional regulation or trafficking to the membrane (Danbolt, 2001; Gegelashvili et al., 2001). A recent report also indicates that glutamate transporters are associated with cholesterol-rich lipid raft microdomains of the plasma membrane and that such association is of great importance for their function (Butchbach et al., 2004).
Astrocytes are activated in response to acute brain insults and in the course of chronic neurodegenerative diseases; they become hypertrophic and overexpress glial fibrillary acidic protein (GFAP) (Kalman, 2004). In vitro activation of astrocytes by growth factors or serum either increases the level of expression of glutamate transporters (Figiel et al., 2003) or changes their activity by modulating their phosphorylation level (Vermeiren et al., 2005). However, little is known about how activated astrocytes ensure glutamate homeostasis during pathological conditions in vivo.
The molecular events leading to astrocytic activation after an acute or chronic insult are not fully understood, and several members of the cytokine family may be involved, such as ciliary neurotrophic factor (CNTF). An increase in CNTF concentration leads to astrocyte hypertrophy and GFAP overexpression in vitro (Levison et al., 1998) and in vivo (Kahn et al., 1995; Winter et al., 1995; Lisovoski et al., 1997). In addition, the levels of endogenous CNTF are increased in conditions associated with astrocytic activation such as mechanical injury (Ip et al., 1993) or excitotoxic lesion (Haas et al., 2004). The induction of CNTF receptors and the activation of the CNTF intracellular signaling pathway [the Janus kinase (JAK)–signal transducer and activator of transcription (STAT) pathway] are also observed in activated astrocytes after axotomy (Xia et al., 2002) or neurochemical insult (Sriram et al., 2004). Finally, astrocyte response to injury is altered in CNTF-knock out mice, despite compensation by other cytokines (Martin et al., 2003). Therefore, in addition to its well known neuroprotective function (Sendtner et al., 1994; Sleeman et al., 2000), CNTF appears as a potent endogenous activator of astrocytes.
To study the role of activated astrocytes in glutamate homeostasis, we have developed an in vivo model of astrocytic activation using lentivirus-mediated CNTF overexpression in the rat striatum. CNTF-activated astrocytes were hypertrophic, had an immature phenotype and expressed highly glycosylated forms of both GLAST and GLT-1 that were significantly redistributed into raft microdomains. All these changes were also observed on mixed astrocyte/neuron striatal cultures treated with recombinant CNTF. During an intrastriatal injection of quinolinic acid in vivo, overexpression of CNTF was associated with an improved handling of extracellular glutamate and an increased energetic supply. These changes may underlie, at least in part, the strong neuroprotective effects of CNTF.
Materials and Methods
Animals and products.
Three-month-old male Lewis rats (weight, ∼300 g; IFFA Credo, Arbresle, France) were used in this study. All experimental procedures were performed in strict accordance with the recommendations of the European Economic Community (86/609/EEC) for care and use of laboratory animals. All products were purchased from Sigma (St. Louis, MO), unless otherwise specified.
Injection of lentiviruses.
We used self-inactivated (SIN) lentiviruses that encode either the human CNTF gene (“lenti-CNTF”) with the export sequence of Ig or the β-galactosidase gene (“lenti-LacZ”) under the control of the mouse phosphoglycerate kinase 1 promoter. The construction, purification, and titration of these lentiviruses and their in vivo bioactivity have been described previously (Hottinger et al., 2000; de Almeida et al., 2001). Lentiviruses were diluted in vehicle (Veh) (0.1 m PBS with 1% bovine serum albumin) at a final concentration of 105 ng p24/ml. Rats were anesthetized with a mixture of ketamine (15 mg/kg) and xylazine (1.5 mg/kg). Suspensions of lenti-CNTF, lenti-LacZ, or Veh were injected stereotaxically into the striatum using a 10 μl Hamilton syringe via a 28 gauge blunt needle [stereotaxic coordinates: anteroposterior (AP), +0.5 mm; lateral (L), ±3.0 mm from bregma; ventral (V), −4.5 mm from the dura, with tooth bar set at −3.3 mm]. Rats received a total volume of 2 μl per striatum at a rate of 0.2 μl/min. Injections were made bilaterally with the same virus (Lenti-CNTF, Lenti-LacZ, or Veh rats) or with lenti-CNTF and lenti-LacZ in the left and right striatum, respectively (lenti-CNTF/lenti-LacZ rats). Control rats received Veh and lenti-LacZ in the left and right striatum, respectively (Veh/lenti-LacZ rats). At the end of injection, the needle was left in place for 1 min before being slowly removed. The skin was sutured and rats were allowed to recover.
Time course.
Preliminary studies have demonstrated that, using this viral titer of lenti-CNTF, the effects were stable for several months without any sign of downregulation. All experiments were performed 2–6 months after infection. In addition, immunoblot and immunohistochemical experiments were also done at 2 weeks after infection.
Primary striatal neuron/astrocyte cultures.
Primary striatal neuron/astrocyte cocultures were made from embryonic day 14 (E14)–E15 rat embryos and prepared as described previously (Zala et al., 2005). Timed-pregnant Sprague Dawley rats were killed with a lethal dose of pentobarbital, and embryos were quickly removed and dissected on cooled Hank's balanced sodium salts without Ca2+ and Mg2+. Ganglionic eminences were isolated and incubated for 15 min at 37°C in 0.3 mg/ml DNase I. Tissues were mechanically dissociated with a fire-polished Pasteur pipette and debris removed after decantation of the suspension. Cells were finally concentrated by centrifugation (20°C, 10 min, 1500 × g) and resuspended in serum-free Neurobasal medium supplemented with 2% B27 supplement (Invitrogen, Carlsbad, CA), 1% antibiotic-antifungal mixture (Invitrogen), and 0.5 mm l-glutamine. Cells were plated at a density of 400,000 cells/well in 24-well plates coated with 50 μg/ml 30–70 kDa poly-d-lysine. The cultures were kept in a humid incubator (5% CO2, 37°C) and half of the medium was changed once per week. Human recombinant CNTF (R & D Systems, Minneapolis, MN) was diluted in PBS and 0.1% BSA at a stock concentration of 25 μg/ml. CNTF (50 ng/ml, dilution 1:500) was added at 21 d in vitro (DIV) and at 23 DIV. Control wells were treated with vehicle (final concentration, 2.5 μg/ml BSA in PBS). Cell cultures were studied at 25 DIV for all experiments and also at 23 DIV for Western blot characterization (see below).
Biochemical analysis.
The striatum of lenti-CNTF, lenti-LacZ, or Veh rats was rapidly dissected out on ice from fresh slices (2 slices, 2 mm thick) made in the coronal plane using a steel rat-brain matrix. Tissue samples were homogenized using a glass potter (400 rpm, 20 strokes) in 300 μl of buffer [50 mm Tris-HCl, pH 7.4, 100 mm NaCl, 1% SDS, 10 mm β-glycerophosphate, and 1 mm orthovanadate with phosphatase inhibitor cocktail 1 (1:100) and protease inhibitor cocktail (Roche, Indianapolis, IN)]. Proteins from cell cultures were homogenized in 250 μl of the same buffer after scraping of the well.
Western blots were performed using ECL detection as described previously (Herard et al., 2005). The antibodies used were raised against actin (1:10,000), β-III-tubulin (1:1000), calbindin D (1:1000), connexin 43 (1:250; BD Biosciences, San Jose, CA), dopamine and cAMP-regulated phosphoprotein (DARPP-32; 1:1000; Chemicon, Temecula, CA), excitatory amino acid carrier 1 (EAAC1; 1:1000; Alpha Diagnostics International, San Antonio, TX), flotilin-1 (1:500; BD Biosciences), GFAP (1:2000; Dako, High Wycombe, UK), glutamic acid decarboxylase (GAD; 1:3000; Chemicon), glutamate receptor subunit 2 (GluR2; 1:500; Chemicon), nestin (1:1000; BD Biosciences), NMDA receptor subunit 2A (NR2A; 1:500; Santa Cruz Biotechnology, Santa Cruz, CA), NR2B (1:500; Santa Cruz Biotechnology), proliferating cell nuclear antigen (PCNA) (1:1000), STAT3 (1:1000; Cell Signaling Technology, Beverly, MA), phospho-STAT3 (Tyr705; 1:1000; Cell Signaling Technology), synaptophysin (1:250), vesicular glutamate transporter 1 (VGLUT1; 1:1000; a kind gift from Prof. S. El Mestikawy, Institut Mondor de Médecine Moléculaire, Créteil, France) and vimentin (1:200; Dako). Antibodies directed to the C terminus of GLAST and GLT-1 (dilution, 1:10,000) were a kind gift from Prof. K. Tanaka (Tokyo Medical and Dental University, Tokyo, Japan).
In vitro deglycosylation of proteins by peptide N-glycosidase F (PNGase F) was made following manufacturer's instructions (New England Biolabs, Ipswich, MA). All reactions were made on ice to limit aggregation of glutamate transporters.
Raft preparation.
Raft purification is based on properties of resistance to nonionic detergent at low temperature and their floatability in low-density fractions during gradient centrifugation. Rafts were purified in basal conditions and after stimulation with the excitotoxin quinolinate (QA). Rats infected bilaterally with lenti-CNTF or lenti-LacZ (n = 3/group) were injected stereotaxically with either 40 nmol (40 mm in 1 μl) of QA or PBS in the right and left striatum, respectively. Fifteen minutes later, the striata were dissected out, and rafts were purified by 1 h incubation in 1% Brij-58 at 4°C and fractionation through a discontinuous sucrose gradient following the protocol of Butchbach et al. (2004). From the top of the gradient, 10 fractions (400 μl) and the pellet were collected and protein abundance was quantified in each fraction. An equal volume of each fraction (30 μl) was diluted in a loading buffer and used for immunoblotting as described before. Fractions corresponding to raft microdomains (fraction numbers 4 and 5) and fractions corresponding to detergent-soluble material (fraction numbers 8, 9 and 10) were pooled before loading on the same acrylamide gel.
A similar protocol was applied for cell cultures except that homogenates were incubated for 30 min in 1% Brij-58. Six wells were pooled for each condition, and only the fractions corresponding to raft and soluble proteins were studied.
Western blot quantification.
Films were scanned, and optical density (OD) was measured using the Total Lab image analysis software (version 3.1; Amersham Biosciences, Piscataway, NJ). Specific ODs were normalized to actin abundance for experiments on total homogenates. For raft experiments, specific OD was normalized according to the amount of protein (in micrograms) loaded on the corresponding lane. A partition ratio was calculated as the ratio of normalized abundance of the protein in raft fractions to the normalized abundance in raft and soluble fractions (i.e., total abundance) and was expressed as a percentage.
Immunochemistry.
Brains from lenti-CNTF, lenti-LacZ, and Veh rats (n = 5–6/group) were postfixed in paraformaldehyde (4% in 0.1 m phosphate buffer) for 1 week and cryoprotected in sucrose solutions. Coronal brain sections (40 μm thick) were cut on a freezing microtome, collected serially (interspace, 400 μm), and stored at −20°C until analysis.
Brain sections were processed for immunohistochemistry with tyramine amplification, as described previously (Mittoux et al., 2002). The primary antibodies used were directed against calbindin D (1:10,000), CD-11b (1:5000; Serotec, Oxford, UK), GFAP (1:100,000), nestin (1:1000), PCNA (1:5000) and vimentin (1:5000; Calbiochem, La Jolla, CA).
Some brain sections were double labeled and studied with confocal microscopy (LSM 510; Zeiss, Thornwood, NY). After blocking, primary antibodies were incubated overnight at 4°C. Fluorescent secondary antibodies were then incubated for 1 h at room temperature at a dilution of 1:200 (anti-rabbit AlexaFluor 488, anti-mouse AlexaFluor 594; Invitrogen, Eugene, OR). The same protocol was applied for cell cultures plated on glass coverslips and fixed for 30 min in paraformaldehyde 4% except that primary antibodies were incubated for 3 h, and secondary antibodies were incubated at a dilution of 1:500. Some wells were treated with 5 mm methyl-β-cyclodextrin for 30 min at 37°C to disrupt raft domains (Ottico et al., 2003) before fixation. The primary antibodies used were directed against: GFAP (1:1000), GLAST (1:500), MAP2 (1:200), nestin (1:500), neuronal nuclei protein (NeuN; 1:1000; Chemicon), S100β (1:1000), STAT3 (1:200), and vimentin (1:1000).
Electron microscopy.
Lenti-CNTF and lenti-LacZ rats (n = 1/group) were deeply anesthetized with an intraperitoneal injection of pentobarbital and perfused immediately with 0.2% glutaraldehyde and 2% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4, followed by 2.5% glutaraldehyde and 2% paraformaldehyde in the same buffer. One hour after perfusion, the brains were removed and stored overnight in PBS at 4°C. Sixty-micrometer vibratome (VT100; Leica, Nussloch, Germany) sections were cut through the striatal region spanning the lesion site by ∼1 mm. After washing in cacodylate buffer (0.1 m, pH 7.4), the sections were postfixed for 40 min in 1.5% potassium ferrocyanide and 1% osmium tetroxide and then 1 h in 1% osmium tetroxide alone, followed by 40 min in 1% uranyl acetate in 70% ethanol. They were then dehydrated through increasing concentrations of ethanol and finally embedded in Durcapan resin. In both the experimental and control brains, serial thin sections, of 50 nm thickness, were taken at a position ∼600 μm lateral to the ventralmost extent of the needle track. Sections were contrasted with lead citrate, and images were captured digitally on a CCD camera inside a Philips (Aachen, Germany) CM12 electron microscope at a filament voltage of 80 kV.
Electrophysiology.
Experiments were performed as described previously (Gubellini et al., 2002). Briefly, corticostriatal coronal slices (200–250 μm thick) were prepared from hemibrains of Veh (n = 4), lenti-LacZ (n = 6), and lenti-CNTF (n = 10) rats by means of a vibratome (Leica VT1000S). The slices were then kept at 35°C in artificial CSF (aCSF) whose composition was the following (in mm): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 1.2 NaH2PO4, 2.4 CaCl2, 11 glucose, and 25 NaHCO3, pH 7.4, gassed with O2/CO2 (95/5%). Whole-cell patch-clamp microelectrodes (4–5 MΩ) were filled with a solution containing the following (in mm): 125 K+-gluconate, 10 NaCl, 1.0 CaCl2, 2.0 MgCl2, 0.5 1,2-bis (2-aminophenoxy)-ethane-N,N,N,N-tetraacetic acid, 19 HEPES, 0.3 GTP, 1.0 Mg-ATP, adjusted to pH 7.3 with KOH. Striatal medium spiny neurons were visually selected by means of a BX51WI infrared videomicroscope (Olympus, Tokyo, Japan) and their identity confirmed by analyzing their electrophysiological properties (resting membrane potential, firing, voltage/current properties). Neurons were clamped at −85 mV and glutamatergic spontaneous EPSCs (sEPSCs) were recorded in the presence of 3 μm bicuculline by means of an Axopatch 200B amplifier and pClamp 8.2 software (Molecular Devices, Union City, CA). Afterward, sEPSCs were analyzed off-line in terms of frequency and amplitude by MiniAnalysis 6.0 software (Synaptosoft, Decatur, GA). Recorded neurons showing instable membrane potential, unpaired depolarization-induced firing, or an unstable seal were discarded from the analysis. The sEPSC detection threshold (3–4 pA) was set to twice the baseline noise.
Microdialysis and HPLC.
The effects of QA on the extracellular levels of glutamate and other amino acids were assessed in vivo with a microdialysis study according to the protocol of Popoli et al. (2004) with some modifications. Lenti-CNTF/lenti-LacZ rats (n = 5) or Veh/lenti-LacZ rats (n = 4) were anesthetized with isoflurane and placed in a stereotaxic frame. The body temperature was stabilized at 37°C throughout the experiment with a thermostatic blanket. A microdialysis probe (CMA/12, 4 mm, cutoff 20 kDa; CMA Microdialysis, North Chelmsford, MA) was implanted in each striatum (coordinates: AP, +0.8 mm; L, ±3.5 mm from bregma; V, −6.5 mm from dura). Probes were perfused with aCSF (in mm: 147 NaCl, 2.7 KCl, 1.2 CaCl2, and 0.85 MgCl2) at a rate of 2 μl/min. Rats recovered for 2 h and then microdialysates were collected every 10 min into a refrigerated fraction collector and frozen at −80°C until analysis. After 30 min of baseline measurement, the probe was perfused with QA (5 mm in aCSF) for 30 min and with aCSF for an additional 60 min to return to baseline. For each rat, the probe location inside the striatum was controlled at the end of the experiment. The amino acid content of samples was determined by reverse-phase HPLC coupled to a fluorometric detector. Each standard solution or microdialysis sample was precolumn derivatized with o-phthalaldehyde before being injected into a 20 μl sample loop (Rheodyne, Rohnert Park, CA). The HPLC system included a solvent delivery pump (Spectra-Physics, Mountain View, CA), a C18, 5 μm, 125 × 4 mm column (Macherey-Nagel, Easton, PA), and a fluorescent detector (Jasco, Easton, MD) set at λex = 340 nm and λem = 460 nm. For the kinetic study of extracellular glutamate, we performed a 10 min binary gradient according to the method of Kehr (1998). Other amino acids were separated using a 35 min binary gradient according to the following scheme (time in min − %A/%B): 0 − 100/0; 8 − 90/10; 12 − 75/25; 15 − 60/40; 20 − 40/60; 24 − 0/100; 26 − 100/0. Solvent A was composed of 0.1 m sodium acetate, pH 6.95, methanol, tetrahydrofuran (92.5:5:2.5) and solvent B of methanol, tetrahydrofuran (97.5:2.5). Only the samples corresponding to baseline (T0–T10–T20 min) and to the maximum QA-induced increase (T 60 min) were analyzed.
[18F]-fluoro-2-deoxyglucose uptake during quinolinate injection.
Lenti-CNTF, lenti-LacZ, or Veh rats (n = 4–6/group) were used to evaluate [18F]-fluoro-2-deoxyglucose (FDG) uptake (Sokoloff et al., 1977) just after QA injection. Rats were anesthetized, and a catheter was implanted in both the femoral artery and vein. Then, after being placed in a stereotaxic frame, they were injected with 40 nmol of QA in the right striatum and PBS in the left. Fifteen minutes later, they received an intravenous injection of FDG (1–1.5 mCi in 700 μl of normal saline) and after an additional 45 min, a lethal dose of pentobarbital. The brains were then rapidly removed, frozen in isopentane at −40°C, cut coronally into 20 μm sections with a cryostat (interspace, 100 μm), and dried on a 60°C hot plate. Slides were then processed for autoradiography on BioMax MR films (Kodak, Rochester, NY) for 6 h at room temperature. Autoradiograms were digitized, and striatal OD was measured in both striata for each animal using an image analysis system (Total Lab). The QA-induced increase in FDG uptake was expressed as a percentage of the contralateral, PBS-injected striatum for each section. For each animal, the mean percentage from 20 successive sections that correspond to the maximum QA-induced increase in FDG uptake was calculated.
Body temperature was maintained at 37°C throughout the experiment using a thermostatic blanket. Arterial blood samples were taken before and 60 min after QA injection to measure blood glucose levels using a Onetouch glucose meter (Lifescan, Milpitas, CA) and arterial pH, PO2, and PCO2 (ABL5; Radiometer, Copenhagen, Denmark). All of these parameters were in the physiological range during the experiment and were not significantly different between groups (data not shown).
Statistical analysis.
Results are expressed as mean values ± SEM. Statistical analysis included one-way ANOVA followed by a post hoc Scheffé's test. Student's paired t test was performed for left–right comparisons. The partition ratio obtained for raft experiments (i.e., proportion), was normalized using the arcsine transformation before performing ANOVA and Scheffé's test (Zar, 1974; Wong and Schlichter, 2004). The significance level was set at p < 0.05.
Results
CNTF overexpression induces a sustained activation of astrocytes
The lentiviral constructs used in this study (lenti-CNTF or lenti-LacZ) have been characterized previously (de Almeida et al., 2001) and were shown to target striatal neurons almost exclusively. CNTF is fused to a signal peptide sequence that leads to its constitutive release by infected cells and to striatal CNTF levels reaching up to 1 ng/mg of protein 1 week after injection and at a titer of 2.105 ng p24/ml (de Almeida et al., 2001).
After striatal injection, we did not detect any lesion or microglial activation using gallocyanin staining and CD-11b immunolabeling, except for the needle track attributable to the local mechanical disruption (data not shown). Release of CNTF by transduced neurons induced a noticeable change in astrocyte phenotype in a large part of the striatum, up to 2–3 mm from the injection site. This change was visible 15 d after infection and lasted for >6 months without any sign of diminishing. GFAP immunolabeling was more intense in the lenti-CNTF-infected striatum compared with control rats injected with lenti-LacZ (Fig. 1A). CNTF overexpression also led to the re-expression of vimentin and nestin, two intermediate filament proteins that are characteristic of immature astrocytes (Fig. 1B,C). Nestin- or vimentin-positive astrocytes were absent in the striatum of lenti-LacZ rats except in the vicinity of the needle track and the border of the lateral ventricle, whereas in the lenti-CNTF group they were present at very high density (Fig. 1B,C). CNTF-activated astrocytes appeared hypertrophic with enlarged soma and numerous processes compared with lenti-LacZ rats (Fig. 1D). Immunoblot experiments confirmed that GFAP was overexpressed (+68 ± 2%; p < 0.0001, Scheffé's test; n = 4–5/group) and that the expression of vimentin and nestin was induced in the lenti-CNTF-infected striatum (Fig. 1E). Activated astrocytes also overexpressed the gap junction forming connexin 43 as compared with control groups (+127 ± 17%; p < 0.01, Scheffé's test; n = 4–5/group) (Fig. 1E). Importantly, we did not observe any difference between rats injected with lenti-LacZ or its vehicle (Veh), demonstrating that lentiviral infection and transgenic protein overexpression had no intrinsic effect on the phenotype of astrocytes.
Figure 1.
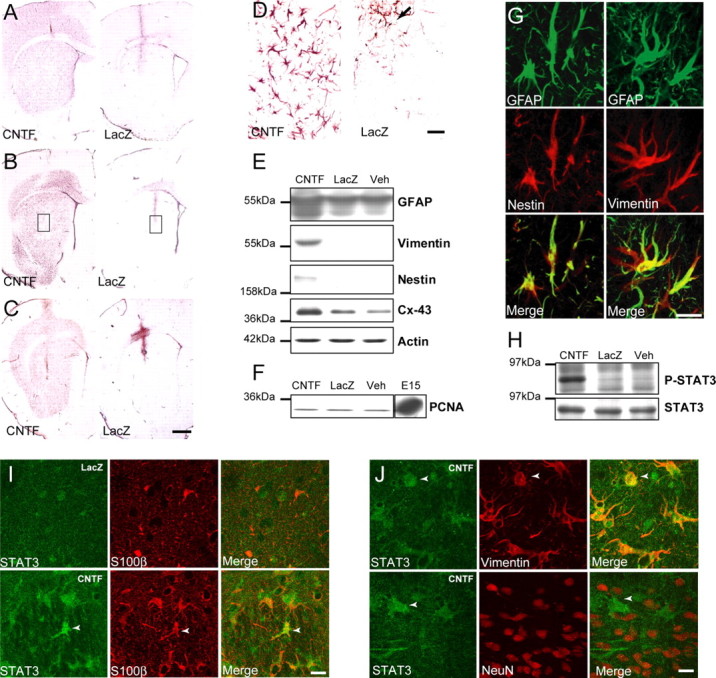
Anatomical and biochemical characterization of CNTF-activated astrocytes. A–C, GFAP (A), vimentin (B), and nestin (C) immunohistochemical staining of rat brain sections after intrastriatal injection of lentiviruses encoding either CNTF or LacZ. CNTF increases the level of expression of GFAP (A) and induces the expression of both vimentin (B) and nestin (C) compared with the control striatum, injected with lenti-LacZ. D, Enlargement of boxes in B shows that CNTF-activated vimentin-positive astrocytes are hypertrophic and have numerous large processes compared with labeled astrocytes in the lenti-LacZ striatum that are only visible along the needle track (arrow). E, Immunoblot analysis confirms that the level of expression of GFAP is significantly increased in the lenti-CNTF striatum as compared with striata of rats injected with lenti-LacZ or vehicle (Veh). Vimentin and nestin are only detected in the lenti-CNTF group. CNTF-activated astrocytes also significantly overexpress connexin 43 (Cx-43). F, The level of expression of the cyclin PCNA is low and unchanged between groups. By comparison, the embryonic E15 brain (positive control) shows a high level of expression of PCNA resulting from active proliferation. G, Left, Confocal analysis of immunofluorescent double labeling of nestin (red) and GFAP (green) in the lenti-CNTF striatum shows that both filaments are coexpressed in the same cell. G, Right, Vimentin (red) and GFAP (green) are also colocalized in the same astrocytes. These results suggest that CNTF alters the phenotype of the vast majority of striatal astrocytes without inducing major proliferation or formation of new astroglial cells. H, The phosphorylated form of STAT3 is only detected in the lenti-CNTF group, whereas total STAT3 levels are similar in all groups. I, In control conditions, STAT3 has a neuropil-like localization. In the lenti-CNTF group, several cells expressing S100β present an intense perinuclear staining with STAT3 (arrowhead), which may reflect STAT3 translocation toward the nucleus in astrocytes. J, The cells showing a perinuclear location of STAT3 coexpress vimentin but not NeuN, suggesting that CNTF signaling pathways are specifically activated in astrocytes. Images are representative of four to five animals per group at 2 weeks and 2–6 months after infection. Scale bars: (in C) A–C, 1 mm; D, 50 μm; G, I, J, 20 μm.
To evaluate whether activated astrocytes were new astrocytes originating from progenitor cell proliferation, we studied the cyclin PCNA, whose level of expression is increased during mitosis. We could not detect any rise in PCNA levels in the lenti-CNTF group, in contrast to the positive control consisting of E15 brain (Fig. 1F), and we did not observe any PCNA-positive cells in the lenti-CNTF striatum by immunohistochemistry (data not shown). Dual-label confocal microscopy also demonstrated that vimentin or nestin proteins were coexpressed with GFAP in astrocytes at all time points (from 2 weeks to 6 months after infection) (Fig. 1G). These results suggested that activated astrocytes in the lenti-CNTF-infected striatum did not result from the proliferation of progenitor cells that would progressively differentiate into new astrocytes but rather corresponded to a sustained transformation of resident striatal astrocytes toward a more immature and activated phenotype.
Although the total level of expression of STAT3 was the same in all groups, we detected the active, phosphorylated form of STAT3 only in the lenti-CNTF group (Fig. 1H). To determine the cell type that was responsive to CNTF, we performed dual-labeling experiments. Because phospho-specific STAT3 antibodies did not work in our conditions [in accordance with another study performed in the striatum (Sriram et al., 2004)], we labeled total STAT3, which translocates to the nucleus when activated. Brain sections from lenti-LacZ rats had a diffuse STAT3 immunolabeling, whereas numerous cells with an intense, perinuclear staining were visible in the lenti-CNTF group (Fig. 1I). Cells that displayed STAT3 relocalization coexpressed S100β and vimentin, but not NeuN, suggesting that CNTF overproduction specifically activated the JAK–STAT pathway in astrocytes (Fig. 1I,J).
We then used electron microscopy to study CNTF-induced changes in astrocyte morphology at the ultrastructural level. CNTF-activated astrocytes were hypertrophic, filled with bundles of intermediate filaments that occupied a large part of cytoplasm (Fig. 2). All capillaries (∼15 observed) were found to be wrapped with prominent and enlarged astrocytic endfeet in the lenti-CNTF group. Other structures within the striatum appeared normal at this level of resolution with no obvious changes in the ultrastructure (Fig. 2).
Figure 2.
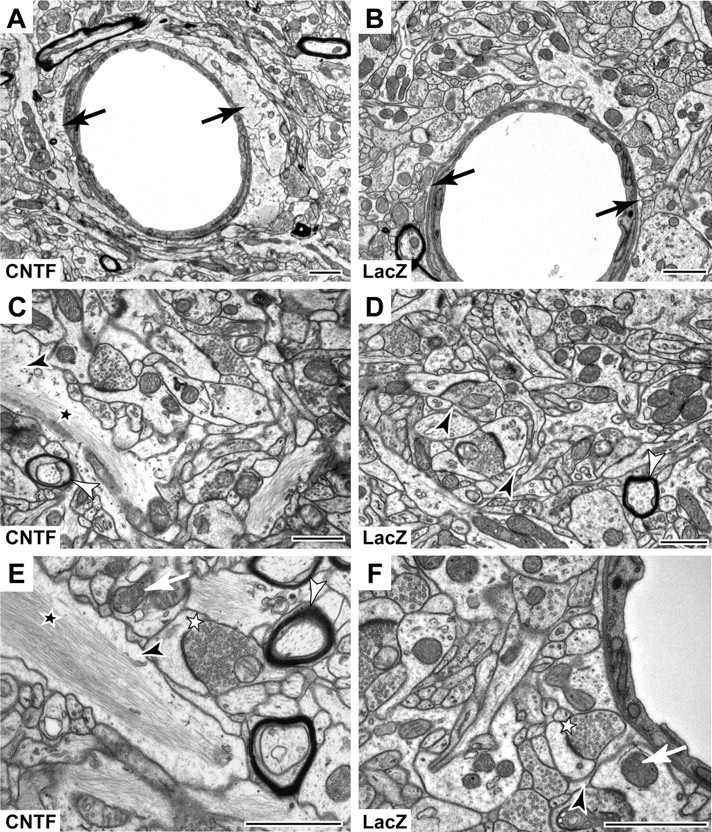
Ultrastructural characterization of CNTF-activated astrocytes. Electron micrographs show enlarged astrocytic elements containing an abundance of filaments in the lenti-CNTF striatum (A, C, E), compared with the lenti-LacZ striatum (B, D, F). A, The blood capillary is partially surrounded by a large astrocytic endfoot (black arrow) compared with that observed in lenti-LacZ striatum (B). At higher magnification, large astrocytic processes (black arrowhead) filled with bundles of filaments (black star) are visible only in the lenti-CNTF striatum (C, E), whereas astrocytic processes appear as small elements surrounding synapses in the control striatum (D, F). Qualitative assessment of other ultrastructural elements such as synapses (white star), mitochondria (white arrow), or myelin sheets (white arrowhead) does not evidence any change between groups. Images are taken from lenti-CNTF or lenti-LacZ rats (n = 1 per group) at 6 months after infection. Scale bars, 1 μm.
The expression of all neuronal proteins studied appeared to be unchanged by CNTF overexpression at all time points (p > 0.05 for all proteins, ANOVA; n = 4–5/group). In particular, CNTF had no detectable effect on the level of expression of several neuronal proteins such as calbindin, a calcium-binding protein, synaptophysin, and β-III-tubulin, as well as other striatal specific markers such as DARPP-32, a protein involved in striatal dopamine signaling and GAD, the GABA-producing enzyme. Similarly, we detected no difference between groups regarding the expression of neuronal proteins involved in the glutamatergic system such as ionotropic glutamate receptor subunits (NR2A, NR2B, GluR2), the vesicular glutamate transporter VGLUT1 and the neuronal glutamate transporter EAAC1 (Fig. 3A). Immunodetection of calbindin and NeuN on brain sections from lenti-CNTF and lenti-LacZ-injected rats did not show any difference (Fig. 3B). Finally, in vitro patch-clamp experiments were performed to assess the effects of CNTF overexpression on the spontaneous activity of striatal medium spiny neurons. In our recording conditions, i.e., −85 mV membrane potential and 1.2 mm extracellular Mg2+, sEPSCs in the striatum are mainly mediated by AMPA glutamate receptor, in response to glutamate released by corticostriatal afferences. The average amplitude and frequency of sEPSCs recorded from neurons of Veh, lenti-lacZ, and lenti-CNTF rats were not significantly different (p = 0.89 and 0.30 for amplitude and frequency, respectively; n = 8–10 neurons per group; ANOVA) (Fig. 3C), suggesting that CNTF did not induce any major change in basal spontaneous glutamatergic transmission in the striatum.
Figure 3.
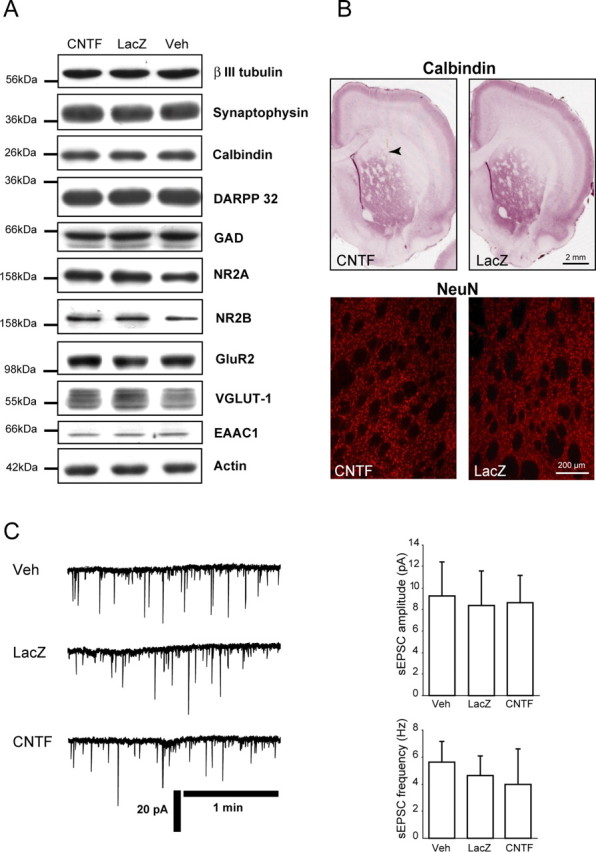
CNTF does not alter the level of expression of several neuronal markers and does not modify the spontaneous glutamatergic activity of striatal medium spiny neurons. A, Immunoblots were performed to compare the level of expression of several neuronal proteins in the lenti-CNTF, lenti-LacZ, and Veh groups. β-III-Tubulin, calbindin, synaptophysin, DARPP-32, and GAD are all expressed at the same level in all groups. Similarly, glutamate receptor subunits NR2A, NR2B, and GluR2, the vesicular glutamate transporter VGLUT-1, and the neuronal glutamate transporter EAAC1, which are all involved in glutamatergic neurotransmission, are unaltered by CNTF overexpression. Blots are representative of four to five animals per group at 2 weeks and 2–6 months after infection. B, Immunohistological detection of calbindin- and NeuN-expressing neurons does not show any qualitative or quantitative difference of labeling between lenti-CNTF and lenti-LacZ brain sections. Images are representative of four to five animals per group at 2 weeks and 2–6 months after infection. C, Patch-clamp electrophysiological recordings (left traces) show that glutamatergic spontaneous activity is similar in striatal spiny neurons of Veh, lenti-LacZ, and lenti-CNTF rats. Accordingly, average sEPSC amplitude and frequency (right graphs) are not significantly different between the three groups. Data are mean ± SEM. Experiments were done at 2–6 months after infection on n = 8, 8, and 10 neurons for Veh, lenti-LacZ, and lenti-CNTF groups, respectively.
Activated astrocytes express highly glycosylated forms of glutamate transporters
Astroglial glutamate transporters GLAST and GLT-1 are responsible for the majority of glutamate removal from the extracellular space. Immunoblots using specific antibodies directed against these transporters showed a different immunoblot pattern inj> lenti-CNTF striata compared with control groups. Both proteins had a higher apparent molecular weight of ∼5 kDa without any significant change in their total level of expression (p = 0.11 and 0.18 for GLT-1 and GLAST, respectively; n = 4–5/group; ANOVA) (Fig. 4). Because glutamate transporters are glycosylated, we tested whether the higher apparent molecular weight of GLT-1 and GLAST was attributable to a change in their level of glycosylation. After in vitro PNGaseF treatment, deglycosylated glutamate transporters from the three experimental groups showed a similar immunoblot pattern, indicating that CNTF overexpression increased the level of glycosylation of GLAST and GLT-1 (Fig. 4).
Figure 4.

CNTF-activated astrocytes express highly glycosylated glutamate transporters. Immunoblots show that the two main glial glutamate transporters, GLAST and GLT-1, present a higher apparent molecular weight of ∼5 kDa in the lenti-CNTF group compared with the two control groups. This shift is observed with the monomeric forms (m) and the dimeric form (d) of the transporters. This higher molecular weight is attributable to a higher level of glycosylation, as demonstrated after treatment with PNGase F, which cleaves N-glycosylation. Deglycosylated forms of GLAST and GLT-1 present a reduced molecular weight (resulting from the loss of glycosylated chains) and have a similar pattern in all groups. Blots are representative of four to five animals per group at 2 weeks and 2–6 months after infection.
Because the pattern of protein glycosylation is a targeting signal (van Vliet et al., 2003), we wanted to assess whether the localization of the highly glycosylated transporters was altered. However, studying the subcellular localization of glutamate transporters using immunofluorescence is difficult to perform in vivo. We therefore developed an in vitro model of CNTF-mediated astrocytes activation. We treated rat primary striatal neuron/astrocyte cultures with 50 ng/ml of human recombinant CNTF for 48 or 96 h at 21 DIV. GFAP, vimentin, and nestin were overexpressed in cell cultures exposed to CNTF (Fig. 5A). Using immunofluorescence, we also found that astrocytes displayed a different phenotype in the presence of CNTF. They had enlarged processes and cell bodies, whereas the expression of the neuronal marker MAP2 was not changed (Fig. 5B). Application of CNTF in vitro also resulted in a similar shift of the apparent molecular weight for GLAST (Fig. 5C). No data are available for GLT-1 in vitro because it is expressed at a very low level. Using immunofluorescence detection of GLAST and nestin in these cell cultures, we observed a change in the labeling pattern of GLAST in the presence of CNTF. GLAST labeling was less diffuse in CNTF-treated cultures, and the shape of astrocytes became more recognizable (Fig. 5D). At a higher magnification, the labeling at the membrane was more pronounced with CNTF, and we observed larger clusters (Fig. 5E).
Figure 5.

CNTF activates astrocytes and increases the level of glycosylation of GLAST in vitro. A, Treatment of striatal primary cultures of neurons and astrocytes with 50 ng/ml CNTF (C) for 96 h increases the level of expression of GFAP, vimentin, and nestin compared with vehicle (V). Vimentin is already overexpressed after a 48 h treatment with CNTF. B, CNTF-activated astrocytes labeled with GFAP and S100β antibodies are hypertrophic and have enlarged processes in culture. The pattern of expression of MAP2, a component of neuronal dendrites, is not altered by CNTF treatment (B). C, CNTF increases the apparent molecular weight of GLAST in culture. This effect is more prominent after a 96 h treatment. D, CNTF also changes the pattern of labeling of GLAST, which appears less diffuse. E, At higher magnification, larger clusters are visible along nestin-positive processes in CNTF-activated astrocytes (arrowhead) compared with vehicle treated astrocytes. After treatment with methyl-β-cyclodextrin (MBCD), these clusters are lost and GLAST becomes evenly distributed at the astrocytic membrane (arrow). Scale bars: B, 50 μm; D, 20 μm; E, 5 μm.
It has recently been suggested that glutamate transporters are found predominantly in lipid raft compartments (Butchbach et al., 2004). The direct association of glutamate transporters with cholesterol-enriched domains is important for their activity (Shouffani and Kanner, 1990; Butchbach et al., 2004). After 30 min treatment with 5 mm methyl-β-cyclodextrin, a cholesterol-depleting agent, clusters were no longer visible and GLAST distribution became homogenous (Fig. 5E). These results suggest that, in CNTF-activated astrocytes, GLAST has a different subcellular localization, in close association with raft domains.
Glutamate transporters are redistributed into raft microdomains of activated astrocytes
We further evaluated whether the distribution of glutamate transporters between raft and nonraft membrane domains was different in CNTF-activated astrocytes. Because glutamate exposure induces a rapid increase in cell-surface expression of GLAST (Duan et al., 1999), we also studied transporter dynamic regulation in conditions of acute rise in glutamate levels. We used the excitotoxin QA, which induces a rapid increase in endogenous extracellular glutamate (Popoli et al., 2004).
Rafts were prepared from the striatum of lenti-CNTF and lenti-LacZ rats 15 min after an intrastriatal injection of QA or PBS (n = 3/group). Fractionation profiles were obtained, and the majority of proteins were recovered in fractions corresponding to detergent-soluble material as seen with Ponceau red staining (Fig. 6A). Both GLAST and GLT-1 were recovered in low-density fractions corresponding to the raft domains (R), as confirmed by the presence of the protein flotilin-1 (Fig. 6A). Immunoblots using pooled fractions corresponding to rafts and detergent-soluble material clearly showed that a large majority of both GLAST and GLT-1 were found in raft fractions (Fig. 6B,C). The partition ratio calculated for each transporter (see Materials and Methods) indicated that 80% of GLAST and 76% of GLT-1 were in raft fractions in control conditions (Fig. 6B,C). GLAST enrichment into raft fractions was significantly increased in the lenti-CNTF group compared with lenti-LacZ in basal conditions (p < 0.01, Scheffé's test) (Fig. 6B). In addition, QA tended to induce a redistribution of GLAST into raft domains in both lenti-LacZ and lenti-CNTF groups, although it did not reach significance. GLAST partition ratio was still significantly higher after QA injection in the lenti-CNTF group than in the lenti-LacZ group, with a nearly complete recruitment into rafts (p < 0.05, Scheffé's test). GLT-1 was also significantly enriched into raft domains from astrocytes in the lenti-CNTF group compared with lenti-LacZ (p < 0.05, Scheffé's test) (Fig. 6C). However, in contrast to GLAST, QA did not produce any additional effect on GLT-1 recruitment into rafts in both groups. To control for the specificity of GLAST and GLT-1 redistribution into raft domains, we performed the same quantification for flotilin-1 and for total proteins. The partition ratio of flotilin-1 was not different between groups (p = 0.43, ANOVA) (Fig. 6D), as was the partition ratio of total proteins (p = 0.94, ANOVA) (Fig. 6E).
Figure 6.
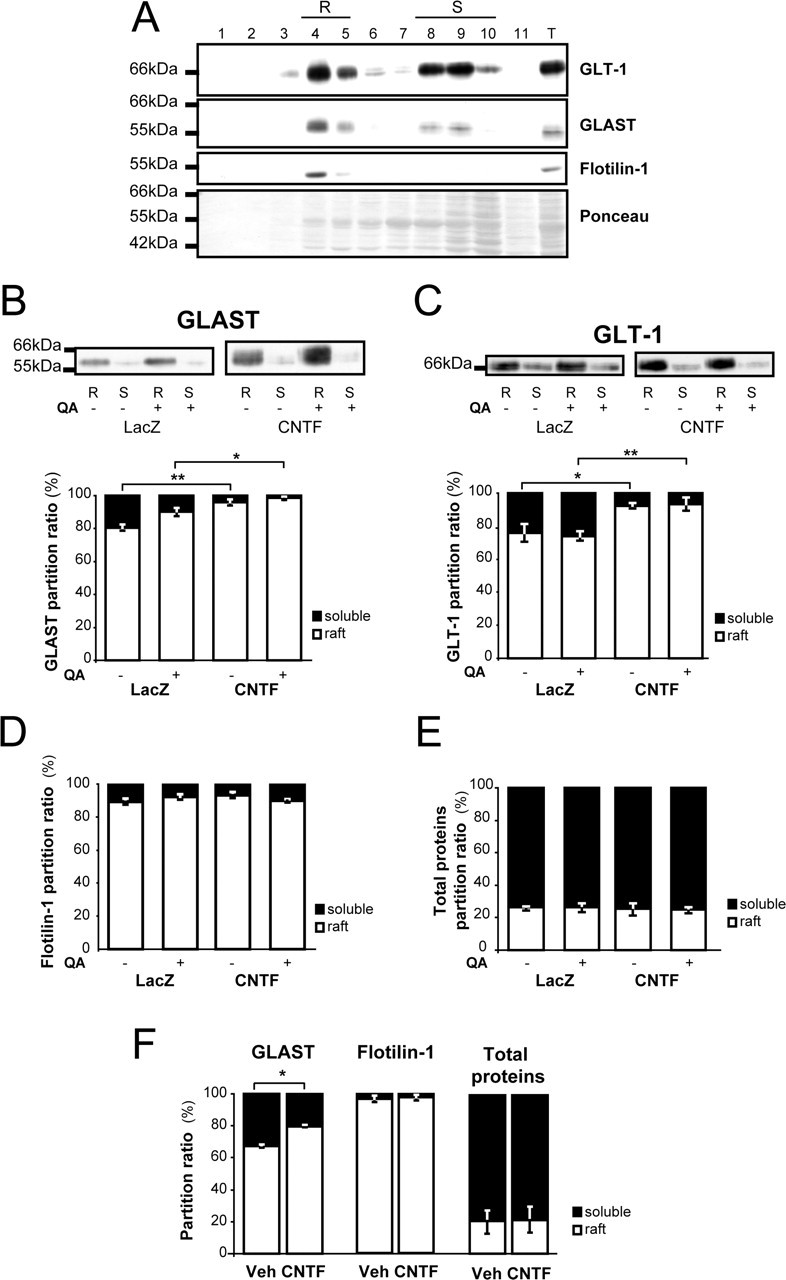
Glutamate transporters are preferentially redistributed in raft domains of CNTF-activated astrocytes. A, Fractionation profiles for GLT-1 and GLAST representative of a control rat. Raft domains (R) are recovered in low-density fractions 4 and 5, which are enriched in flotilin-1, a component of lipid rafts. Fractions 8, 9, and 10 correspond to “detergent-soluble” material (S), and fraction 11 is made of detergent-insoluble, nonraft, pellet material. An aliquot of total homogenate (T) used to load the discontinuous gradient is also shown on the same gel. The majority of proteins are found in detergent-soluble fractions even if a detectable amount of proteins is observed in raft fractions (see Ponceau red staining). Both GLAST and GLT-1 are enriched in raft domains. B, C, Pooled fractions corresponding to raft domain membranes (R) and detergent-soluble material (S) were loaded on the same gel. B, The partition ratio of GLAST is significantly increased in the lenti-CNTF group. In addition, QA injection tends to induce a relocalization of GLAST into rafts in both lenti-LacZ and lenti-CNTF groups. C, GLT-1 is also significantly enriched in rafts in the lenti-CNTF group, but its distribution is not modified by QA injection. D, E, The partition ratios of flotillin-1 (D) and of total proteins (E) are similar between groups. F, CNTF also significantly increases GLAST partition ratio in vitro but has no effect on flotillin-1 and total proteins distribution. Data are mean ± SEM ∗p < 0.05 and ∗∗p < 0.01, ANOVA and Scheffé's test. Statistical tests were performed on normalized data after arcsine transformation. In vivo experiments were performed at 2–6 months after infection on three rats per group. In vitro experiments were made in duplicate from 96 h treated cultures by pooling six wells per condition.
When rafts were prepared from primary striatal neuron/astrocyte mixed cultures, application of recombinant CNTF (50 ng/ml for 4 d) also induced a significant redistribution of GLAST into raft microdomains (p < 0.05, n = 2/group, Scheffé's test) (Fig. 6F). Again, this effect was specific for GLAST because flotilin-1 and total proteins were not redistributed into raft domains.
These results demonstrate that CNTF redistributes glutamate transporters into raft microdomains of activated astrocytes, in which they have a greater ability to take up glutamate (Butchbach et al., 2004). We thus evaluated the capacity of glutamate buffering in vivo during an intrastriatal injection of QA.
CNTF improves glutamate handling in vivo
Microdialysis experiments were performed to measure extracellular glutamate concentrations in vivo. Lenti-CNTF/lenti-LacZ rats (n = 5) were implanted with a microdialysis probe in each striatum. Basal glutamate levels corrected for probe efficiency were not different between groups (5.26 ± 0.93 μm for lenti-LacZ and 7.18 ± 1.72 μm for lenti-CNTF p = 0.20, Student's paired t test). After recovery, they were perfused with QA for 30 min to evoke glutamate release. QA induced a steep rise in extracellular glutamate levels. Although the concentration of glutamate increased by 900% in the lenti-LacZ-injected striatum, it was only increased by 330% in the lenti-CNTF-injected striatum (p < 0.05, Student's paired t test) (Fig. 7A). After the end of QA infusion, glutamate levels returned to baseline more rapidly in the lenti-CNTF striatum (20 min compared with 40 min in the lenti-LacZ-injected striatum). In control animals (Veh/lenti-LacZ rats, n = 4), both striata showed a similar response to QA infusion, in terms of kinetics and maximum glutamate increase (+800 and +940%, respectively; not significant; data not shown).
Figure 7.

QA-induced glutamate increase is reduced in the lenti-CNTF group. Microdialysis experiments were performed on lenti-CNTF/lenti-LacZ rats (n = 5) to evaluate the handling of extracellular glutamate and other amino acids during perfusion with QA in vivo. A, QA perfusion through the microdialysis probe (between 30 and 60 min after the start of the experiment; black bar) induces a strong increase in microdialysate glutamate concentrations in the lenti-LacZ striatum (black square). The QA-induced increase is significantly reduced in the lenti-CNTF striatum (open circle). B, The microdialysate concentrations of asparagine (ASN), serine (SER), threonine (THR), and arginine (ARG) were measured in basal conditions and at the end of QA perfusion. QA perfusion has similar effects on the levels of these amino acids. Data are mean ± SEM, expressed as percentage versus baseline levels. ∗p < 0.05, ∗∗p < 0.01 versus lenti-CNTF, Student's paired t test. Experiments were performed at 2–6 months after infection.
To test for the specificity of CNTF effects toward glutamate levels, we measured the concentrations of other amino acids in basal and QA-stimulated conditions. We found that the extracellular baseline levels of asparagine, serine, threonine, and arginine were not significantly different between experimental groups (p = 0.19–0.69, Student's paired t test). The effects of QA on the levels of these amino acids were not significantly different between lenti-CNTF and lenti-LacZ groups (p = 0.31–0.97 vs basal concentrations, Student paired t test) (Fig. 7B).
These results indicate that glutamate homeostasis is specifically improved in the lenti-CNTF group.
Overexpression of CNTF is associated with an increased glucose uptake during QA injection
This improvement in glutamate handling could either be caused by a decrease in QA-induced glutamate release or by an increase in glutamate uptake by glial transporters. Glutamatergic activity is tightly linked to glucose uptake (Sibson et al., 1998), through a mechanism involving glutamate uptake by glial transporters (Pellerin and Magistretti, 1994). We thus evaluated striatal FDG uptake after QA injection, as an in vivo index of both glutamate activity and glutamate transporter function.
Rats injected bilaterally with lenti-CNTF, lenti-LacZ or Veh received an injection of QA and PBS in the right and left striatum, respectively. Then, they were intravenously injected with FDG. In all groups, QA induced a significant increase in striatal FDG uptake in a large part of the striatum as compared with PBS (Fig. 8A). QA-induced increase in FDG uptake was similar between lenti-LacZ and Veh groups, but was significantly higher in the lenti-CNTF group (p < 0.01 vs Veh, n = 4–6/group, Scheffé's test) (Fig. 8B). This result suggests that the improvement in glutamate homeostasis was not caused by a decrease in QA-induced glutamate release but more likely to an increased activity of glutamate transporters.
Figure 8.

The increase in FDG uptake during QA injection is enhanced in the lenti-CNTF group. Measurement of FDG uptake in vivo was performed by autoradiography after intrastriatal injection of QA. A, QA injection induces a marked increase in FDG uptake in a large part of the striatum in all groups, compared with a PBS injection. B, In the lenti-CNTF group (n = 6), QA-induced increase in FDG uptake is higher compared with control groups. Data are mean ± SEM. ∗∗p < 0.01 versus Veh group (n = 4) and p = 0.052 versus lenti-LacZ (n = 5). ANOVA and Scheffé's test. Scale bar, 2 mm. Experiments were performed at 2–6 months after infection.
Discussion
In vivo model of sustained astrocytic activation by CNTF
We have developed a model of astrocytic activation using lentivirus-mediated CNTF overexpression in the rat striatum to explore the functional role of activated astrocytes in vivo. We used the cytokine CNTF, which is an endogenous activator of astrocytes through the JAK–STAT3 pathway. We found sustained phosphorylation of STAT3 only in activated astrocytes, indicating that these cells were indeed the main target of CNTF released by infected neurons. There was no effect of CNTF overexpression on neuronal phenotypes and spontaneous striatal glutamatergic synaptic transmission.
CNTF-activated astrocytes were hypertrophic and expressed a different set of glial proteins as compared with control groups. CNTF-activated astrocytes also overexpressed connexin 43, the main constituent of immature glial gap junctions (Nagy and Rash, 2000), suggesting that CNTF overexpression may also have modified the functional characteristics of astrocytic networks.
Alteration of astrocytic phenotype was stable and lasted for several months without an obvious increase in their proliferation, demonstrating the ability of CNTF to sustain astrocytes in an immature and activated state. Part of these phenotypic changes has been reported previously at shorter time periods using injections of recombinant CNTF (see Introduction) and adenovirus-mediated gene transfer of CNTF in the rat striatum (Lisovoski et al., 1997) and retina (van Adel et al., 2005). In addition, the changes in the phenotype of astrocytes were reproduced in our in vitro model of primary striatal neuron/astrocyte cultures, demonstrating that whatever the delivery system used, extracellular CNTF is indeed the triggering signal for the activation of astrocytes. This model of sustained astrocytic activation in the whole striatum offered the possibility to study in vivo several complex brain functions that are regulated by astrocytes, such as glutamate homeostasis.
Glutamate transporters are highly glycosylated in CNTF-activated astrocytes, and they are enriched in raft domains
We found that, in CNTF-activated astrocytes, GLAST and GLT-1 were highly glycosylated without any alteration in their total level of expression. In contrast, the neuronal glutamate transporter EAAC1 was not altered by CNTF. We also demonstrated that GLAST and GLT-1 were enriched into raft microdomains of CNTF-activated astrocytes and that CNTF had the same effect on GLAST in primary cultures of neurons and astrocytes.
The increase in the glycosylation level represents a new posttranslational modification of glutamate transporters. Removal of glycosylation sites on glutamate transporters does not directly affect transport properties but can reduce the stability of GLT-1 (Raunser et al., 2005), decrease the formation of the multimeric, active forms of GLAST (Conradt et al., 1995) or decrease the cell-surface expression of GLT-1 (Trotti et al., 2001). Indeed, N-linked glycans play a pivotal role in protein sorting to subcellular compartments (van Vliet et al., 2003) and could be involved in protein targeting to raft domains (Simons and Ikonen, 1997; Fullekrug and Simons, 2004). We speculate that glutamate transporter redistribution into rafts could be mediated by a change in their pattern of glycosylation. Testing this hypothesis would require a specific inhibitor of glutamate transporter hyperglycosylation that does not affect other glycosylated proteins or the normal N-glycan chains on transporters. Unfortunately, pharmacological tools targeted at specific proteins and specific enzymes involved in the glycosylation process are still lacking (Schachter, 2005).
We observed that CNTF can induce a specific re-localization of transporters into functional raft microdomains in which glutamate uptake is more efficient (Butchbach et al., 2004). This may represent a new and efficient mechanism of regulation of glutamate transporters activity. Other mechanisms reported thus far include modulation of mRNA and protein levels of glutamate transporters by several agents such as growth factors, cAMP analogs or neuron-derived factors and modulation of uptake activity by phosphorylation or recruitment to plasma membrane (Danbolt, 2001; Gegelashvili et al., 2001). In purified raft fractions, glutamate uptake is more than 10 times higher than in nonraft membranes and when these rafts are disrupted after cholesterol removal, uptake is significantly reduced both in cortical culture and purified plasma membrane vesicles (Butchbach et al., 2004). In addition, recent data indicate that the formation of glutamate transporter clusters in glial cell processes, as we observed in vitro under CNTF treatment, plays important roles in regulating glutamate uptake in astrocytes (Zhou and Sutherland, 2004). Glutamate transporter enrichment in rafts of CNTF-activated astrocytes could therefore have strong functional consequences on the efficiency of glutamate homeostasis.
CNTF improves glutamate handling and energy supply
Using microdialysis in situ, we directly evaluated the power of glutamate buffering in response to QA injection. We found that the rise in extracellular glutamate during QA perfusion was significantly reduced when astrocytes were activated by CNTF. QA triggers excitotoxic processes by overactivating postsynaptic glutamate NMDA receptors and by inducing a massive glutamate outflow through stimulation of presynaptic terminals (Popoli et al., 2004). Two mechanisms could be involved in the reduction in extracellular glutamate rise. CNTF overexpression and/or astrocytic activation could decrease QA primary effects (i.e., NMDA receptors stimulation and glutamate release) or increase glutamate clearance by transporters. Our data argue against the first hypothesis. First, the expression of several neuronal proteins involved in the glutamatergic neurotransmission was not changed by CNTF. Second, the increase in the extracellular concentration of other amino acids during QA perfusion (Connick and Stone, 1988) was not significantly different between lenti-CNTF and lenti-LacZ groups. Combined with the third observation that QA induced a significant increase in FDG uptake in all groups, these results demonstrate that the early stimulatory effects of QA on neuronal activity and glutamate release have indeed occurred. We found that the increase in FDG uptake during QA injection was significantly higher in the lenti-CNTF group. It has been proposed that glutamate uptake by glial transporters, which accounts for 80% of total uptake in the striatum (Rothstein et al., 1996), is coupled to glucose uptake from blood stream (Pellerin and Magistretti, 1994; Voutsinos-Porche et al., 2003) by a stoichiometry close to 1:1 (Sibson et al., 1998). Therefore, the higher FDG uptake during QA injection in the lenti-CNTF group may reflect an enhanced glutamate uptake by activated astrocytes. Astrocytes have been recently shown to regulate capillary diameter (Zonta et al., 2003; Takano et al., 2006). Because CNTF-activated astrocytes have enlarged endfeet around capillaries, it may be worthwhile to examine the effects of astrocyte activation on blood flow regulation. Finally, such increase in glucose uptake should help neurons cope with the excitotoxic challenge by providing more energetic substrates.
The neuroprotective potential of CNTF-activated astrocytes
Glutamate excitotoxic neuronal death is involved in many pathological situations (Choi, 1988). CNTF overexpression attenuated the rise in glutamate levels induced by QA. This could explain its neuroprotective effects against QA in the rat striatum (Anderson et al., 1996; de Almeida et al., 2001) and the primate caudate–putamen (Emerich et al., 1997). CNTF also protects striatal neurons against metabolic impairment (Mittoux et al., 2000, 2002), a pathological situation in which glutamate excitotoxicity is thought to play a key role (Brouillet et al., 2005). Therefore, CNTF-activated astrocytes appear beneficial for neuronal function, in particular during an excitotoxic process, and could be involved in CNTF neuroprotective effects, although additional mechanisms may participate. CNTF is not the only activator of astrocytes in pathological conditions (Kalman, 2004). Whether the observed changes in astrocytic function toward a neuroprotective phenotype also apply with other molecules known to activate astrocytes remains to be determined.
In conclusion, activation of astrocytes could provide an alternative neuroprotective approach because these pleiotropic cells control several key brain functions that are crucial for neuronal survival and recovery.
Footnotes
We thank Dr. P. Gervais for providing [18F]-fluoro-2-deoxyglucose and Dr. J. M. Hermel and A. Christophe for their technical help. We thank Dr. J. Y. Thuret for providing technical advice on confocal analysis, Dr. L. Rouquier on microdialysis, and A. Abbood and Drs. M. Taverna and T. Tran on HPLC. We are grateful to Prof. M. Peschanski for providing access to the safety level 3 laboratory. We acknowledge Prof. K. Tanaka for sharing GLAST and C-terminal GLT-1 antibodies and Prof. S. El Mestikawy for VGLUT-1 antibody. We thank Dr. L. Pellerin for critical reading of a previous version of this manuscript.
References
- Anderson KD, Panayotatos N, Corcoran TL, Lindsay RM, Wiegand SJ (1996). Ciliary neurotrophic factor protects striatal output neurons in an animal model of Huntington disease. Proc Natl Acad Sci USA 93:7346–7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouillet E, Jacquard C, Bizat N, Blum D (2005). 3-Nitropropionic acid: a mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in Huntington's disease. J Neurochem 95:1521–1540. [DOI] [PubMed] [Google Scholar]
- Butchbach ME, Tian G, Guo H, Lin CL (2004). Association of excitatory amino acid transporters, especially EAAT2, with cholesterol-rich lipid raft microdomains: importance for excitatory amino acid transporter localization and function. J Biol Chem 279:34388–34396. [DOI] [PubMed] [Google Scholar]
- Choi DW (1988). Glutamate neurotoxicity and diseases of the nervous system. Neuron 1:623–634. [DOI] [PubMed] [Google Scholar]
- Connick JH, Stone TW (1988). Quinolinic acid effects on amino acid release from the rat cerebral cortex in vitro and in vivo. Br J Pharmacol 93:868–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conradt M, Storck T, Stoffel W (1995). Localization of N-glycosylation sites and functional role of the carbohydrate units of GLAST-1, a cloned rat brain L-glutamate/L-aspartate transporter. Eur J Biochem 229:682–687. [DOI] [PubMed] [Google Scholar]
- Danbolt NC (2001). Glutamate uptake. Prog Neurobiol 65:1–105. [DOI] [PubMed] [Google Scholar]
- de Almeida LP, Zala D, Aebischer P, Deglon N (2001). Neuroprotective effect of a CNTF-expressing lentiviral vector in the quinolinic acid rat model of Huntington's disease. Neurobiol Dis 8:433–446. [DOI] [PubMed] [Google Scholar]
- Duan S, Anderson CM, Stein BA, Swanson RA (1999). Glutamate induces rapid upregulation of astrocyte glutamate transport and cell-surface expression of GLAST. J Neurosci 19:10193–10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerich DF, Cain CK, Greco C, Saydoff JA, Hu ZY, Liu H, Lindner MD (1997). Cellular delivery of human CNTF prevents motor and cognitive dysfunction in a rodent model of Huntington's disease. Cell Transplant 6:249–266. [DOI] [PubMed] [Google Scholar]
- Figiel M, Maucher T, Rozyczka J, Bayatti N, Engele J (2003). Regulation of glial glutamate transporter expression by growth factors. Exp Neurol 183:124–135. [DOI] [PubMed] [Google Scholar]
- Fullekrug J, Simons K (2004). Lipid rafts and apical membrane traffic. Ann NY Acad Sci 1014:164–169. [DOI] [PubMed] [Google Scholar]
- Gegelashvili G, Robinson MB, Trotti D, Rauen T (2001). Regulation of glutamate transporters in health and disease. Prog Brain Res 132:267–286. [DOI] [PubMed] [Google Scholar]
- Gubellini P, Picconi B, Bari M, Battista N, Calabresi P, Centonze D, Bernardi G, Finazzi-Agro A, Maccarrone M (2002). Experimental parkinsonism alters endocannabinoid degradation: implications for striatal glutamatergic transmission. J Neurosci 22:6900–6907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas SJ, Ahrens A, Petrov S, Schmitt O, Wree A (2004). Quinolinic acid lesions of the caudate putamen in the rat lead to a local increase of ciliary neurotrophic factor. J Anat 204:271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herard AS, Dubois A, Escartin C, Tanaka K, Delzescaux T, Hantraye P, Bonvento G (2005). Decreased metabolic response to visual stimulation in the superior colliculus of mice lacking the glial glutamate transporter GLT-1. Eur J Neurosci 22:1807–1811. [DOI] [PubMed] [Google Scholar]
- Hottinger AF, Azzouz M, Deglon N, Aebischer P, Zurn AD (2000). Complete and long-term rescue of lesioned adult motoneurons by lentiviral-mediated expression of glial cell line-derived neurotrophic factor in the facial nucleus. J Neurosci 20:5587–5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip NY, Wiegand SJ, Morse J, Rudge JS (1993). Injury-induced regulation of ciliary neurotrophic factor mRNA in the adult rat brain. Eur J Neurosci 5:25–33. [DOI] [PubMed] [Google Scholar]
- Kahn MA, Ellison JA, Speight GJ, de Vellis J (1995). CNTF regulation of astrogliosis and the activation of microglia in the developing rat central nervous system. Brain Res 685:55–67. [DOI] [PubMed] [Google Scholar]
- Kalman M (2004). Glial reaction and reactive glia. In: Non neuronal cells of the nervous system: function and dysfunction (Hertz L, ed.) pp. 787–835. Amsterdam: Elsevier.
- Kehr J (1998). Determination of glutamate and aspartate in microdialysis samples by reversed-phase column liquid chromatography with fluorescence and electrochemical detection. J Chromatogr B Biomed Sci Appl 708:27–38. [DOI] [PubMed] [Google Scholar]
- Levison SW, Hudgins SN, Crawford JL (1998). Ciliary neurotrophic factor stimulates nuclear hypertrophy and increases the GFAP content of cultured astrocytes. Brain Res 803:189–193. [DOI] [PubMed] [Google Scholar]
- Lisovoski F, Akli S, Peltekian E, Vigne E, Haase G, Perricaudet M, Dreyfus PA, Kahn A, Peschanski M (1997). Phenotypic alteration of astrocytes induced by ciliary neurotrophic factor in the intact adult brain, as revealed by adenovirus-mediated gene transfer. J Neurosci 17:7228–7236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maragakis NJ, Rothstein JD (2004). Glutamate transporters: animal models to neurologic disease. Neurobiol Dis 15:461–473. [DOI] [PubMed] [Google Scholar]
- Martin A, Hofmann HD, Kirsch M (2003). Glial reactivity in ciliary neurotrophic factor-deficient mice after optic nerve lesion. J Neurosci 23:5416–5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittoux V, Joseph JM, Conde F, Palfi S, Dautry C, Poyot T, Bloch J, Deglon N, Ouary S, Nimchinsky EA, Brouillet E, Hof PR, Peschanski M, Aebischer P, Hantraye P (2000). Restoration of cognitive and motor functions by ciliary neurotrophic factor in a primate model of Huntington's disease. Hum Gene Ther 11:1177–1187. [DOI] [PubMed] [Google Scholar]
- Mittoux V, Ouary S, Monville C, Lisovoski F, Poyot T, Conde F, Escartin C, Robichon R, Brouillet E, Peschanski M, Hantraye P (2002). Corticostriatopallidal neuroprotection by adenovirus-mediated ciliary neurotrophic factor gene transfer in a rat model of progressive striatal degeneration. J Neurosci 22:4478–4486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy JI, Rash JE (2000). Connexins and gap junctions of astrocytes and oligodendrocytes in the CNS. Brain Res Brain Res Rev 32:29–44. [DOI] [PubMed] [Google Scholar]
- Ottico E, Prinetti A, Prioni S, Giannotta C, Basso L, Chigorno V, Sonnino S (2003). Dynamics of membrane lipid domains in neuronal cells differentiated in culture. J Lipid Res 44:2142–2151. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ (1994). Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci USA 91:10625–10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popoli P, Pintor A, Tebano MT, Frank C, Pepponi R, Nazzicone V, Grieco R, Pezzola A, Reggio R, Minghetti L, De Berardinis MA, Martire A, Potenza RL, Domenici MR, Massotti M (2004). Neuroprotective effects of the mGlu5R antagonist MPEP towards quinolinic acid-induced striatal toxicity: involvement of pre- and post-synaptic mechanisms and lack of direct NMDA blocking activity. J Neurochem 89:1479–1489. [DOI] [PubMed] [Google Scholar]
- Raunser S, Haase W, Bostina M, Parcej DN, Kuhlbrandt W (2005). High-yield expression, reconstitution and structure of the recombinant, fully functional glutamate transporter GLT-1 from Rattus norvegicus J Mol Biol 351:598–613. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF (1996). Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16:675–686. [DOI] [PubMed] [Google Scholar]
- Schachter H (2005). The search for glycan function: fucosylation of the TGF-{beta}1 receptor is required for receptor activation. Proc Natl Acad Sci USA 102:15721–15722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sendtner M, Carroll P, Holtmann B, Hughes RA, Thoenen H (1994). Ciliary neurotrophic factor. J Neurobiol 25:1436–1453. [DOI] [PubMed] [Google Scholar]
- Shouffani A, Kanner BI (1990). Cholesterol is required for the reconstruction of the sodium- and chloride-coupled, gamma-aminobutyric acid transporter from rat brain. J Biol Chem 265:6002–6008. [PubMed] [Google Scholar]
- Sibson NR, Dhankhar A, Mason GF, Rothman DL, Behar KL, Shulman RG (1998). Stoichiometric coupling of brain glucose metabolism and glutamatergic neuronal activity. Proc Natl Acad Sci USA 95:316–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K, Ikonen E (1997). Functional rafts in cell membranes. Nature 387:569–572. [DOI] [PubMed] [Google Scholar]
- Sleeman MW, Anderson KD, Lambert PD, Yancopoulos GD, Wiegand SJ (2000). The ciliary neurotrophic factor and its receptor, CNTFR alpha. Pharm Acta Helv 74:265–272. [DOI] [PubMed] [Google Scholar]
- Sokoloff L, Reivich M, Kennedy C, Des Rosiers MH, Patlak CS, Pettigrew KD, Sakurada O, Shinohara M (1977). The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. J Neurochem 28:897–916. [DOI] [PubMed] [Google Scholar]
- Sriram K, Benkovic SA, Hebert MA, Miller DB, O'Callaghan JP (2004). Induction of gp130-related cytokines and activation of JAK2/STAT3 pathway in astrocytes precedes up-regulation of glial fibrillary acidic protein in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of neurodegeneration: key signaling pathway for astrogliosis in vivo? J Biol Chem 279:19936–19947. [DOI] [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Libionka W, Han X, Nedergaard M (2006). Astrocyte-mediated control of cerebral blood flow. Nat Neurosci 9:260–267. [DOI] [PubMed] [Google Scholar]
- Trotti D, Aoki M, Pasinelli P, Berger UV, Danbolt NC, Brown Jr RH, Hediger MA (2001). Amyotrophic lateral sclerosis-linked glutamate transporter mutant has impaired glutamate clearance capacity. J Biol Chem 276:576–582. [DOI] [PubMed] [Google Scholar]
- van Adel BA, Arnold JM, Phipps J, Doering LC, Ball AK (2005). Ciliary neurotrophic factor protects retinal ganglion cells from axotomy-induced apoptosis via modulation of retinal glia in vivo. J Neurobiol 63:215–234. [DOI] [PubMed] [Google Scholar]
- van Vliet C, Thomas EC, Merino-Trigo A, Teasdale RD, Gleeson PA (2003). Intracellular sorting and transport of proteins. Prog Biophys Mol Biol 83:1–45. [DOI] [PubMed] [Google Scholar]
- Vermeiren C, Najimi M, Vanhoutte N, Tilleux S, de Hemptinne I, Maloteaux JM, Hermans E (2005). Acute up-regulation of glutamate uptake mediated by mGluR5a in reactive astrocytes. J Neurochem 94:405–416. [DOI] [PubMed] [Google Scholar]
- Voutsinos-Porche B, Bonvento G, Tanaka K, Steiner P, Welker E, Chatton JY, Magistretti PJ, Pellerin L (2003). Glial glutamate transporters mediate a functional metabolic crosstalk between neurons and astrocytes in the mouse developing cortex. Neuron 37:275–286. [DOI] [PubMed] [Google Scholar]
- Winter CG, Saotome Y, Levison SW, Hirsh D (1995). A role for ciliary neurotrophic factor as an inducer of reactive gliosis, the glial response to central nervous system injury. Proc Natl Acad Sci USA 92:5865–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong W, Schlichter LC (2004). Differential recruitment of Kv1.4 and Kv4.2 to lipid rafts by PSD-95. J Biol Chem 279:444–452. [DOI] [PubMed] [Google Scholar]
- Xia XG, Hofmann HD, Deller T, Kirsch M (2002). Induction of STAT3 signaling in activated astrocytes and sprouting septal neurons following entorhinal cortex lesion in adult rats. Mol Cell Neurosci 21:379–392. [DOI] [PubMed] [Google Scholar]
- Zala D, Benchoua A, Brouillet E, Perrin V, Gaillard MC, Zurn AD, Aebischer P, Deglon N (2005). Progressive and selective striatal degeneration in primary neuronal cultures using lentiviral vector coding for a mutant huntingtin fragment. Neurobiol Dis 20:785–798. [DOI] [PubMed] [Google Scholar]
- Zar J (1974). Biostatistical analysis. Englewood Cliffs, NJ: Prentice-Hall.
- Zhou J, Sutherland ML (2004). Glutamate transporter cluster formation in astrocytic processes regulates glutamate uptake activity. J Neurosci 24:6301–6306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, Carmignoto G (2003). Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci 6:43–50. [DOI] [PubMed] [Google Scholar]