

Abstract
Astrocytic gap junctions have been suggested to contribute to spatial buffering of potassium in the brain. Direct evidence has been difficult to gather because of the lack of astrocyte-specific gap junction blockers. We obtained mice with coupling-deficient astrocytes by crossing conditional connexin43-deficient mice with connexin30−/− mice. Similar to wild-type astrocytes, genetically uncoupled hippocampal astrocytes displayed negative resting membrane potentials, time- and voltage-independent whole-cell currents, and typical astrocyte morphologies. Astrocyte densities were also unchanged. Using potassium-selective microelectrodes, we assessed changes in potassium buffering in hippocampal slices of mice with coupling-deficient astrocytes. We demonstrate that astrocytic gap junctions accelerate potassium clearance, limit potassium accumulation during synchronized neuronal firing, and aid in radial potassium relocation in the stratum lacunosum moleculare. Furthermore, slices of mice with coupling-deficient astrocytes displayed a reduced threshold for the generation of epileptiform events. However, it was evident that radial relocation of potassium in the stratum radiatum was not dependent on gap junctional coupling. We suggest that the perpendicular array of individual astrocytes in the stratum radiatum makes these cells ideally suited for spatial buffering of potassium released by pyramidal cells, independent of gap junctions. In general, a surprisingly large capacity for K+ clearance was conserved in mice with coupling-deficient astrocytes, indicating that gap junction-dependent processes only partially account for K+ buffering in the hippocampus.
Keywords: Cx43, Cx30, epileptiform events, potassium buffering, hippocampus, astrocyte
Introduction
The role of astrocytes in the CNS has come under intense scrutiny in recent years. Intriguingly, astrocytes have been recognized to actively modulate neuronal communication (Volterra and Meldolesi, 2005), in addition to their more established functions in regulating the extracellular milieu. Despite these exciting novel findings, the molecular basis of elementary homeostatic functions of astrocytes, such as the regulation of the extracellular potassium (K+) concentration ([K+]o), have remained incompletely understood. K+ accumulates in the extracellular space during neuronal activity. If K+ regulation fails, pathological events such as spreading depression, anoxic depolarization, and epileptiform activity may be initiated (Somjen, 2001; Seifert et al., 2006). The mechanisms proposed to underlie astrocytic K+ buffering can be categorized as either spatial redistribution of K+, termed spatial buffering, or as net uptake of K+ (Walz, 2000b; Kofuji and Newman, 2004). Net uptake has been associated with the activity of the Na+-K+-ATPase, Na+-K+-2Cl− cotransporters, or separate K+ and Cl− channels. K+ spatial buffering, in contrast, requires exclusive uptake of K+, cytosolic diffusion, and subsequent release at remote sites. At sites of maximal [K+]o accumulation, K+ entry is driven by the difference between the glial membrane potential and the K+ equilibrium potential. Because of electrotonic propagation of the K+-induced depolarization, a driving force for K+ efflux results at sites in which local depolarization exceeds the K+ equilibrium potential. It has been argued that a single elongated astrocyte might redistribute K+ from sites of maximal accumulation to sites of lower [K+]o (Ransom, 1996). Indeed, the seminal work of Newman (Newman et al., 1984; Newman, 1993) has demonstrated that the expression pattern of inwardly rectifying K+ channels along the longitudinal axis of retinal Müller cells enables uptake and redistribution of extracellular K+, a process called K+ siphoning.
However, in other CNS areas, extensive linkage of astrocytes via gap junctions has been assumed to be essential for spatial buffering (Orkand et al., 1966). In addition, gap junctions might also benefit extracellular K+ homeostasis attributable to net K+ uptake by stabilizing intracellular ion concentrations (Rose and Ransom, 1997). Direct examination of these concepts has been difficult, primarily because selective inhibition of gap junctions is currently not possible (Rozental et al., 2001). Furthermore, it has been impossible to differentiate between astrocytic and neuronal gap junctions, which may play differential roles. In contrast to expression of connexin36 (Cx36) in neurons, Cx43 is widely expressed in brain astrocytes. In addition, astrocytic expression of Cx30 and Cx26 has been described (Nagy et al., 2004). To specifically target astrocytic gap junctions, mice with Cx43-deficient astrocytes have been generated. However, inactivation of Cx43 in astrocytes only partially inhibited astroglial tracer coupling (Theis et al., 2003).
Here, we show that additional deficiency of Cx30 results in complete disruption of coupling. Mice with Cx30/Cx43-deficient astrocytes were fertile and showed no gross behavioral abnormalities. Focusing on the hippocampus, we combined patch-clamp recordings with field potential analyses and [K+]o measurements to investigate properties of uncoupled astrocytes and unravel the specific role of astroglial gap junctions in K+ buffering.
Materials and Methods
Animals.
Experiments were performed on hippocampal slices of wild-type (wt) mice and transgenic mice with conditional deletion of Cx43 in astrocytes (Theis et al., 2003), as well as with additional, unrestricted deletion of Cx30 (Teubner et al., 2003). Their genotypes were single deficient Cx43fl/fl:hGFAP-Cre, heterozygous double-deficient Cx30+/−, Cx43fl/fl:hGFAP-Cre, and homozygous double-deficient Cx30−/−, Cx43fl/fl:GFAP-Cre [double knock-out (dko)] mice, all aged 30–90 d.
Patch clamp.
Mice were anesthetized with 50% CO2 and decapitated. Brains were removed, and 300-μm-thick transversal slices were cut on a vibratome (VT1000S; Leica, Nussloch, Germany). During slicing, the tissue was submerged in ice-cold solution containing the following (in mm): 87 NaCl, 2.5 KCl, 1.25 NaH2PO4, 7 MgCl2, 0.5 CaCl2, 25 NaHCO3, 25 glucose, and 25 sucrose (bubbled with 95% O2/5% CO2). Thereafter, slices were maintained in the same solution at 35°C for 20 min, cooled down to room temperature, and placed for at least 30 min into standard artificial CSF (ACSF) containing the following (in mm): 126 NaCl, 3 KCl, 2 MgSO4, 2 CaCl2, 10 glucose, 1.25 NaH2PO4, and 26 NaHCO3, equilibrated to a pH of 7.4 (with 95% O2/5% CO2). A slice was transferred to a recording chamber and continuously perfused with bubbled ACSF at room temperature. Cells were visualized at 600-fold magnification with a microscope (Axioskop FS2; Zeiss, Oberkochen, Germany). Whole-cell voltage-clamp recordings were obtained in visually identified “passive” astrocytes [also termed glutamate transporter cells (Steinhäuser et al., 1994; Matthias et al., 2003; Wallraff et al., 2004)] of the CA1 stratum radiatum. Pipettes (from borosilicate capillaries; Hilgenberg, Malsfeld, Germany) had resistances of 3–6 MΩ when filled with an internal solution containing the following (in mm): 130 K-gluconate, 1 MgCl2, 3 Na2-ATP, 20 HEPES, and 10 EGTA, pH 7.2. Currents were recorded with an EPC9 amplifier (HEKA Elektronik, Lambrecht, Germany). Data were stored on computer hard disk using TIDA software (HEKA Elektronik), sampled at 1–30 kHz, and filtered at 3–10 kHz. Only one astrocyte was recorded in any individual slice.
Evaluation of gap junctional coupling.
As a measure of gap junctional coupling, we used the extent of diffusion of a tracer, as described previously (Theis et al., 2003; Wallraff et al., 2004). To this end, N-biotinyl-l-lysine (biocytin; Sigma, Taufkirchen, Germany) was added to the internal solution (0.5%), and recordings were limited to exactly 20 min. Immediately after recordings, slices were fixed in 4% paraformaldehyde in 0.1 m PBS, pH 7.4, at 4°C. The next steps were performed at room temperature, pH 7.4, unless otherwise stated. For better resolution, after cryoprotection (30% sucrose in PBS), the 300 μm slices were cut in 60-μm-thick slices (Fig. 1E) using a microtome (Microm, Walldorf, Germany). Before biocytin visualization, endogenous peroxidase was blocked (1% H2O2 in 0.1 m Tris buffer, pH 7.4, 2 h) and tissue was permeabilized [0.25% dimethylsulfoxide (DMSO) and 2% bovine serum albumin (BSA) in 0.05 m Tris-buffered saline (TBS), pH 7.4, 1 h]. Incubation with the Elite ABC kit (Vector Laboratories, Burlingame, CA) was performed for 36 h (1:500 in 0.1 m TBS with 0.5% BSA and 0.25% DMSO) at 4°C. For the peroxidase reaction (limited to 30 min), we used 0.002% H2O2, 0.025% diaminobenzidine (DAB), and 0.005% NiNH4SO4 in 0.1 m Tris buffer, pH 7.6. Slices were mounted in Vectashield (Vector Laboratories) and inspected in a Zeiss Axiophot microscope. All DAB-positive cells located in the first three slices were counted (Fig. 1E). Images were taken with a digital SPOT camera (Diagnostic Instruments, Sterling Heights, MI), and MetaView software (Universal Imaging Corporation, West Chester, PA) was used to combine optical sections, at 1 μm intervals through the depth of a slice, to a final image.
Figure 1.
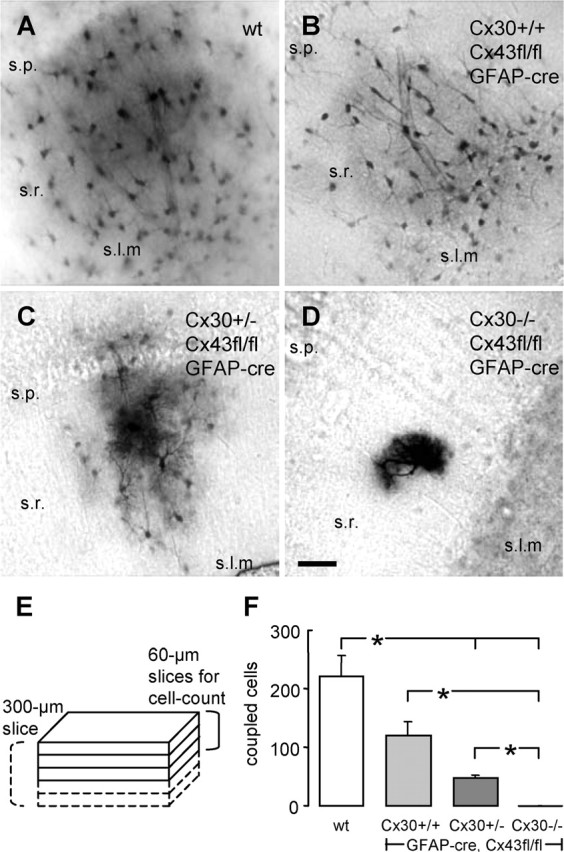
Deletion of Cx30 in addition to Cx43 (dko) in hippocampal astrocytes abolishes tracer coupling. A, Tracer-coupled astrocytes in a wt hippocampal slice, as obtained after biocytin diffusion from a single cell, held in whole-cell voltage clamp for 20 min. B and C display the reduced numbers of tracer-coupled astrocytes in hippocampal slices of a Cx43fl/fl:hGFAP-Cre mouse and a Cx30+/−, Cx43fl/fl:hGFAP-Cre mouse, respectively. D, Absence of tracer coupling in a hippocampal slice of a Cx30−/−, Cx43fl/fl:hGFAP-Cre (dko) mouse. Note that, with strongly reduced (C) or abolished (D) tracer coupling, fine astrocytic processes become visible. Scale bar, 50 μm. E, Scheme for quantification of tracer coupling. Slices, 60 μm thick, obtained from a 300 μm slice, were processed for biocytin visualization. In the top three slices, all biocytin-positive cells were counted. F, Summary of the amount of tracer coupling in the different genotypes. *p < 0.5. Error bars represent SEM. s.p., Stratum pyramidale; s.r., stratum radiatum; s.l.m., stratum lacunosum moleculare.
Determination of astrocyte density.
Transversal hippocampal slices (60 μm thick, cut on a vibratome) of six wt and three dko mice were stained free floating for glial fibrillary acidic protein (GFAP) [primary antibody, 1:1000 mouse monoclonal anti-GFAP (Chemicon, Hofheim, Germany); secondary antibody, 1:800 goat anti-mouse Alexa Fluor 594 (Invitrogen, Carlsbad, CA)]. Antibodies were diluted in 0.1 m PBS, containing 2% normal goat serum and 0.2% Triton X-100, pH 7.4. Slices were mounted in Vectashield (Vector Laboratories), and stacks of optical sections at 2 μm intervals through the depth of a slice were obtained with a confocal laser scanning microscope (Leica TCS; Leica, Pulheim, Germany). Forty and 63× lenses were used for obtaining images in the stratum radiatum and stratum lacunosum moleculare, respectively. After combining image stacks to a final image (MetaView software; Universal Imaging Corporation), the volumes for cell counts were 250 × 250 × 60 μm for stratum radiatum and 159 × 159 × 60 μm for stratum lacunosum moleculare. Total numbers of counted GFAP-positive astrocytes were 1192 and 891 for wt and dko mice, respectively.
Extracellular recordings.
Mice were anesthetized using ether and decapitated; brains were removed and placed in ice-cold ACSF containing the following (in mm): 129 NaCl, 3 KCl, 1.8 MgSO4, 2 CaCl2, 10 glucose, 1.25 NaH2PO4, and 21 NaHCO3 (bubbled with 95% O2/5% CO2). Horizontal hippocampal slices (400 μm) were prepared using a vibratome (Campden Instruments, Leicester, UK). Slices were transferred to an interface chamber, perfused at 1.8 ml/min with prewarmed (34°C) ACSF in a humidified atmosphere of 95% O2/5% CO2. Before recordings, 30 μm l-APV, 30 μm CNQX, and 5 μm bicuculline were added to block synaptic transmission, and slices were allowed to equilibrate for 1 h. Changes in [K+]o and field potential were measured at a depth of 100 μm with double-barreled K+-selective/reference microelectrodes. Electrodes were prepared and tested as described (Lux and Neher, 1973) and displayed voltage responses of 59 ± 1 mV (n = 34) per decade increase in [K+]o. The ion-sensitive barrel was tip filled with Fluka (Neu-Ulm, Germany) 60031 ionophore and backfilled with 100 mm KCl; the reference barrel was filled with 150 mm NaCl. Using a custom-made amplifier equipped with negative capacitance compensation, the signal at the reference barrel was subtracted from the signal at the K+-selective barrel to obtain a signal correlating to [K+]o. This voltage signal was converted to K+ concentration (millimolar) using a modified Nernst equation: log[Ion]1 = Em × (s × v)exp(−1) + log[Ion]o, where Em is the recorded potential, s is the electrode slope obtained at calibration, v is the valence of the specific ion, [Ion]o is the ion concentration at rest, and [Ion]1 is the ion concentration during stimulation. Recording electrodes were placed at the border of stratum pyramidale and stratum oriens in the middle of the CA1 subfield. Bipolar stimulation electrodes (platinum wires of 30 μm diameter) were positioned in the alveus in area CA1 (close to the subiculum) to elicit antidromic population spikes (see Fig. 4A). We applied paired stimuli (0.1 ms, 50 ms interval) or trains of stimuli (10 s, 20 Hz), programmed with Master-8 (A.M.P.I., Jerusalem, Israel). Intensities were selected to evoke population spike amplitudes at 25, 50, 75, and 100% of maximal response to use stimulation of similar efficacy in all experiments. For analysis of the poststimulus decline of [K+]o, the decay beyond the first 10% of decrease to the trough of the undershoot was fitted to a biexponential function using a least-squares fitting routine (Levenberg–Marquardt algorithm, Clampfit, pClamp 9.2; Molecular Devices, Munich, Germany). To obtain a laminar profile of changes in [K+]o, we shifted the recording electrodes from the stratum pyramidale through stratum radiatum to the hippocampal fissure (step size of 100 μm set with a micromanipulator; MPC-100; Sutter Instruments, Novato, CA) (see Fig. 6A). Data were recorded using Spike 2 software (Cambridge Electronic Design, Cambridge, UK) and sampled at 10 kHz (field potentials, 3 kHz filter) or 100 Hz ([K+]o measurements, 1.6 Hz filter), respectively.
Figure 4.

Elevated stimulation-induced [K+]o levels in dko mice. A, Experimental setup for [K+]o measurements. The stimulation electrode (stim.) was placed in the alveus (a.), and the recording electrode (rec.) was placed in the CA1 stratum pyramidale (s.p.). B, Typical examples of rises in [K+]o elicited by three paired pulses (0.1 ms, 50 ms interval, 30 s interspike interval) at 100% stimulation intensity. Left, wt; right, dko. Scale bar, 5 s. C, Typical examples of rises in [K+]o elicited by trains of stimuli (10 s, 20 Hz) at 25, 50, 75, and 100% stimulation intensity, respectively. Left, wt; right, dko. Scale bar, 5 s. Note that, at maximal stimulation intensity, [K+]o reaches a higher level in dko compared with wt mice. D, Summary of evoked [K+]o rises during double-pulse stimulation. Mean and SEM values of responses to three double pulses were calculated and normalized to amplitudes of concomitant population spikes. White bars, wt; gray bars, dko. Asterisks indicate significant differences (p = 0.015). E, Summary of rises in [K+]o evoked through trains of stimuli, normalized to amplitudes of respective population spikes. White bars, wt; gray bars, dko. Bars represent mean and SEM, and asterisks indicate significant differences (p = 0.001). Note that only wt mice showed a decrease in variation with increasing stimulation strength.
Figure 6.
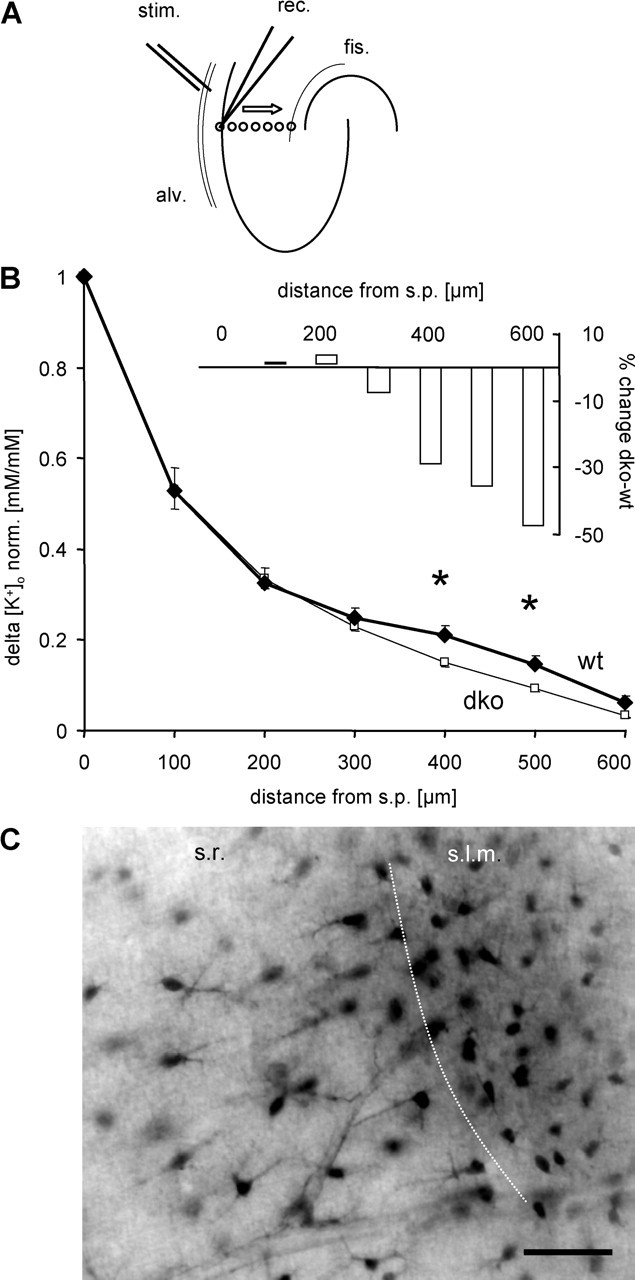
Impaired spatial buffering in the stratum lacunosum moleculare of dko mice. A, Experimental setup for the analysis of laminar profiles of changes in [K+]o. Stimulation was performed in the alveus (20 Hz, 100%) while the recording electrode was stepped from the stratum pyramidale to the hippocampal fissure (100 μm step size). B, Mean rises in [K+]o (normalized to rise at the stratum pyramidale) plotted against the distance from stratum pyramidale (thin line, dko, n = 8 animals, 15 slices; thick line, wt, n = 8 animals, 13 slices). Normalized [K+]o in dko mice reached lower levels at 400 and 500 μm from the stratum pyramidale compared with wt. For relative changes, see inset. C demonstrates the difference in astrocyte morphology and orientation in the stratum radiatum versus stratum lacunosum moleculare, as obtained after biocytin injection into a stratum radiatum astrocyte proximal to the stratum lacunosum moleculare. Note the small size and random orientation of cells in the stratum lacunosum moleculare. Scale bar, 50 μm. alv., Alveus; fis., fissure; stim., stimulation electrode; rec. recording electrode; s.r. stratum radiatum; s.l.m., stratum lacunosum moleculare.
For recording spontaneous and evoked activity, slices were placed in an interface chamber and perfused with ACSF containing the following (in mm): 124 NaCl, 3 KCl, 1.25 NaH2PO4, 1.6 CaCl2, 1.8 MgSO4, 26 NaHCO3, and 10 glucose, ph 7.4 (34°C, monitored throughout recordings). Field potentials were recorded (direct current, filtered at 2 kHz, custom-made amplifier), using glass micropipettes (1 MΩ) filled with ACSF placed in CA1 stratum pyramidale. Spontaneous epileptiform activity was assessed for 10 min. Stimulation electrodes were placed in the stratum radiatum of the CA2/CA1 border to stimulate Schaffer collaterals ∼300 μm from recording electrode (single stimuli, 50 μs, 30 s intervals). Stimulus intensity was adjusted to yield half-maximal responses. Slices were then exposed to ACSF containing no Mg2+, and the latency to the appearance of epileptiform events was established. Their frequency was determined during the last 10 min of 60 min exposure to 0 Mg2+.
Statistical evaluation.
Data are expressed as mean values ± SEM. Data obtained from different slices of a single animal were pooled to yield a single data point. Data were examined for statistical significance using Student’s two-tailed t test. ANOVA was performed for examination of data with k > 2 (post hoc Dunnett’s test) and for comparison of regression curves. Differences were considered significant at p < 0.05, tested with SPSS 12.0 software package (SPSS, Munich, Germany).
Results
Genetic deletion of astrocytic gap junctions
In the absence of selective pharmacological blockers of gap junctions, we sought to determine the role of astrocytic gap junctions in K+ buffering using transgenic mice with gap junction-deficient astrocytes. In these mice, the extent of coupling was estimated by evaluating transcellular tracer diffusion after a fixed period of whole-cell patch-clamp recordings (see Materials and Methods and Fig. 1). As shown previously, stratum radiatum astrocytes remain coupled to a considerable extent in mice that exhibit conditional deficiency of Cx43, the major astrocytic connexin, although coupling is reduced (Theis et al., 2003). In wt mice, tracer spread from single injected astrocytes to 222 ± 34 encircling cells (n = 8 injections) (Fig. 1A) as opposed to 119 ± 24 cells in conditional Cx43-deficient mice (n = 7) (Fig. 1B). Because residual coupling might have been caused by Cx30, another Cx expressed by astrocytes, the conditional Cx43-deficient mice were crossed with Cx30−/− mice (Teubner et al., 2003). Additional deletion of one allele of Cx30 (Cx30+/−, Cx43fl/fl:hGFAP-Cre) further diminished tracer coupling to 20% of control (47 ± 6 cells; n = 11) (Fig. 1C). Elimination of the second Cx30 allele (dko mice) completely suppressed tracer coupling (n = 16) (Fig. 1D). Data are summarized in Figure 1F.
Uncoupled cells show typical astrocytic morphology, spatial orientation, and current pattern
We next examined whether the morphology or electrophysiological properties of astrocytes were altered in dko mice and whether astrocyte density was changed. Protoplasmic astrocytes typically exhibit thick primary processes and a dense net of higher-order processes (Bushong et al., 2002; Wallraff et al., 2004). Figure 2A demonstrates these characteristic morphological attributes of a wt astrocyte with capillary end feet. ACSF with low bicarbonate (Fig. 3) blocked gap junctional coupling, and thereby biocytin was confined to the injected cell, allowing visualization of fine processes. Astrocytes with genetic ablation of Cx43 and Cx30 qualitatively displayed similar morphological features and retained processes giving rise to capillary end feet (Fig. 2B,C). Immunostaining against GFAP and cell counts revealed that the density of GFAP-positive astrocytes was similar in dko and wt mice, in both stratum radiatum (dko, 10,593 ± 399 cells/mm−3, n = 3 mice; wt, 10,211 ± 438 cells/mm−3, n = 6 mice) (Fig. 2E,F) and stratum lacunosum moleculare (dko, 21,388 ± 576 cells/mm−3, n = 3 mice; wt, 20,241 ± 449 cells/mm−3, n = 5 mice) (Fig. 2E,F, insets). In each genotype, the density of GFAP-positive astrocytes was higher in stratum lacunosum moleculare compared with radiatum. We have to acknowledge, however, that GFAP staining is not likely to label all astrocytes, and we cannot exclude alterations in density of putative GFAP-negative astrocytes in the dko animals. The primary processes of mature astrocytes in the stratum radiatum were often oriented perpendicularly to the pyramidal cell layer (Nixdorf-Bergweiler et al., 1994), and this orientation appeared to be preserved in mice with Cx-deficient astrocytes (Fig. 2F). Figure 2D shows an example of a biocytin-filled astrocyte in a dko mouse that had a striking bipolar morphology and a perpendicular orientation, spanning approximately one-third of the stratum radiatum. Furthermore, disruption of gap junctional coupling also preserved the typical negative resting membrane potential of astrocytes. In fact, we observed a slightly more negative resting membrane potential in dko astrocytes (dko, −92.5 ± 0.5 mV, n = 22; wt, −90.3 ± 0.5 mV, n = 19). Whole-cell currents of dko astrocytes were time and voltage independent, as described for wt astrocytes (Fig. 2, insets, B–D compared with A). This observation further supports the idea that the passive current pattern is not solely attributable to gap junction-dependent currents but, in addition, reflects an intrinsic property of the astrocytic cell membrane. The membrane resistance of dko astrocytes was enhanced (to 160%; dko, 4 ± 0.3 MΩ, n = 10; wt, 2.5 ± 0.7 MΩ, n = 8), which would be expected if the gap junction-dependent conductance is abolished. Collectively, these results show that, in dko mice, fundamental morphological and functional properties of astrocytes are unchanged.
Figure 2.
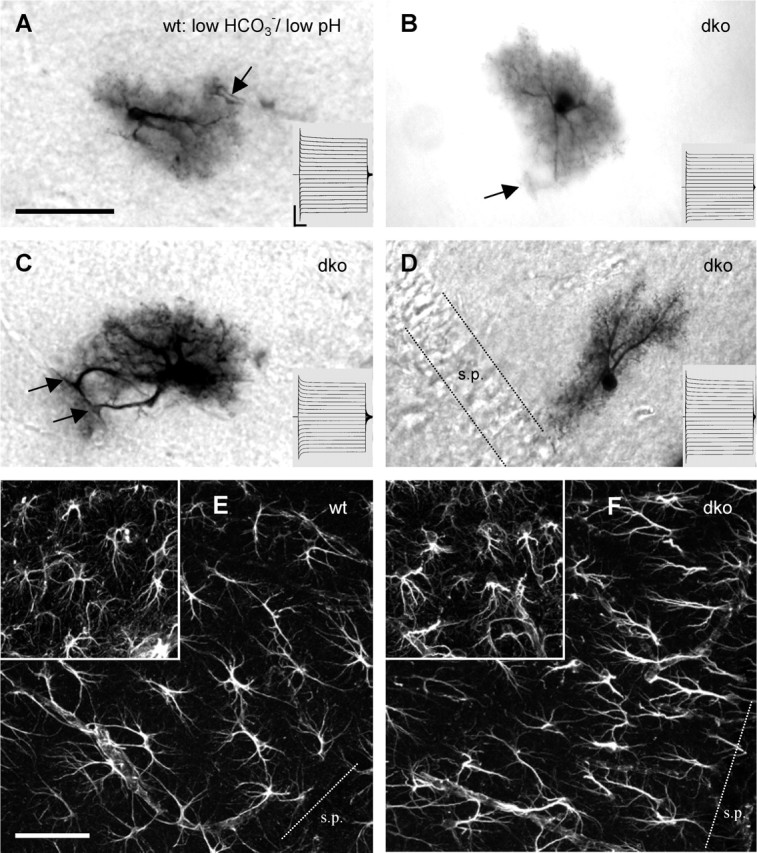
dko astrocytes and wt astrocytes share similar morphology and whole-cell currents. A, Tracer-filled astrocyte in a wt hippocampal slice. Low bicarbonate bath solution, pH 6.4, was applied 30 min before and during whole-cell recordings (20 min) to block gap junctional coupling. Under these conditions, this cell was tracer coupled only with two other astrocytes. Note the prominent primary processes and the dense net of fine processes. The arrow indicates a blood vessel encircled by an astrocytic end foot. Scale bar, 50 μm. The inset represents current responses elicited by 50 ms voltage steps from −180 to 0 mV (Vhold of −90 mV). Calibration: 10 ms, 4 nA. B–D represent examples of tracer-filled astrocytes in slices of dko mice, using standard ACSF. Arrows denote astrocytic pericapillary end feet. Insets and calibration as in A. Note that the astrocyte in D displays a bipolar morphology with an orientation perpendicular to the stratum pyramidale (s.p.). E and F depict immunofluorescent anti-GFAP stainings of the stratum radiatum of wt and dko mice, respectively. The insets show stainings of the stratum lacunosum moleculare. Densities of GFAP-positive cells were similar in wt and dko mice, each with a higher density in stratum lacunosum moleculare compared with the stratum radiatum. Note that major processes of stratum radiatum astrocytes often were oriented perpendicularly to the stratum pyramidale. Scale bar, 50 μm.
Figure 3.
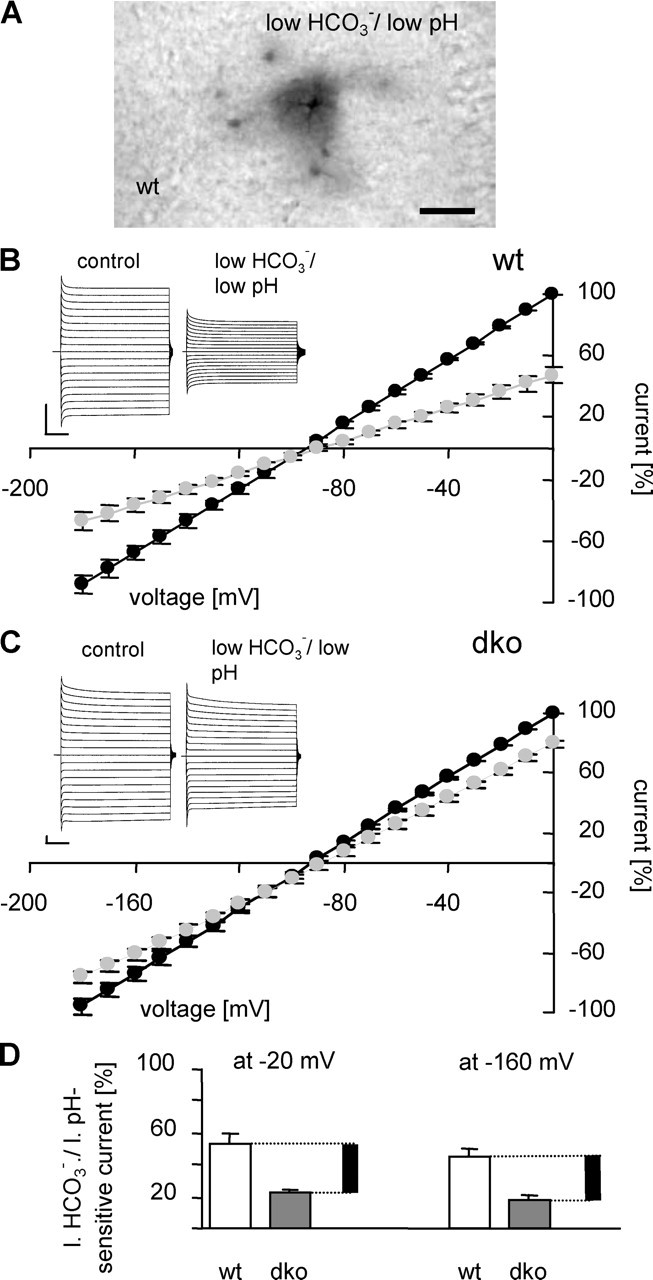
Gap junctions account for ∼30% of whole-cell currents in wt astrocytes. A illustrates the blocking action of l.b./l.pH bath solution, pH 6.4, on tracer coupling in a wt hippocampal slice (30 min preapplication, 20 min recordings). Scale bar, 50 μm. B and C demonstrate the effect of l.b./l.pH solution on whole-cell currents in wt and dko astrocytes, respectively. The insets show representative examples (voltage steps as in Fig. 2); current–voltage relationships in control ACSF (black circles) and l.b./l.pH solution (gray circles) represent means and SEM of five cells. Current amplitudes were normalized to the amplitude recorded at 0 mV. Calibration: 1 nA, 10 ms. D, The amount of bicarbonate-sensitive currents in wt (white bars; 53 ± 5% at −20 mV and 45 ± 5% at −160 mV; n = 5) and dko (dark gray bars; 22 ± 3% at −20 mV and 18 ± 3% at −160 mV; n = 5) mice. For every holding potential, current amplitude in standard ACSF was considered 100%. Black bars represent the extrapolated amount of gap junction-mediated current in wt astrocytes.
Gap junction-mediated currents represent ∼30% of whole-cell currents in wt astrocytes
Next, we sought to quantify the contribution of gap junction-mediated currents to the overall membrane conductance of wt astrocytes. This question has been a matter of debate in studies using pharmacological blockade of gap junctions (Blomstrand et al., 2004; Wallraff et al., 2004). To resolve this issue, we preapplied (25–35 min) low-bicarbonate/low-pH (l.b./l.pH) bath solution (3.6 mm bicarbonate, pH 6.4). In wt hippocampal slices, this treatment virtually eliminated gap junctional coupling (Fig. 3A) and reduced tracer spread to 6% of the control (13 ± 6 cells; n = 3 injections). Moreover, application of l.b./l.pH bath solution during recordings of wt astrocytes led to a decline of current amplitudes to ∼50% of the control condition (n = 5) (Fig. 3B, gray circles), whereas the resting potential of the cells remained almost unchanged (shift by +6.4 ± 2.2 mV). To test for potential unspecific, gap junction-independent effects of l.b./l.pH solution, we applied it to dko astrocytes and observed a 20% reduction of current amplitudes (n = 5; shift of the resting potential by +4.2 ± 1.9 mV) (Fig. 3C, gray circles). Subtracting the l.b./l.pH-sensitive current component of dko astrocytes (22 ± 3% at −20 mV; 18 ± 3% at −160 mV) (Fig. 3D, gray bars) from that of wt astrocytes (53 ± 5% at −20 mV; 45 ± 5% at −160 mV) (Fig. 3D, white bars) allowed us to estimate that gap junction-mediated currents account for ∼30% of overall currents of wt astrocytes (Fig. 3D, black bars), which is in good agreement with the increased input resistance of dko astrocytes (see previous paragraph).
Altered [K+]o amplitudes in the hippocampus of dko mice
Then we set out to examine how astrocytic gap junctions are involved in limiting local [K+]o accumulation caused by neuronal activity. [K+]o rises were elicited by alvear stimulation of CA1 pyramidal neurons (Fig. 4A). To obtain defined and temporally stable [K+]o elevations, caused exclusively by action potential firing of CA1 neurons, synaptic activity was suppressed by applying antagonists for ionotropic glutamate and GABA receptors. Stimulation intensity was set to levels eliciting 25, 50, 75, and 100% of maximal population spike amplitudes. Maximal population spike amplitudes did not differ significantly between wt and dko mice (14.5 ± 0.4 mV, n = 8 animals, 12 slices; 12.8 ± 0.9 mV, n = 9 animals, 14 slices, respectively).
Paired-pulse stimulation (0.1 ms, 50 ms interval), applied to the alveus, elicited small rises in [K+]o that were well above baseline noise for 50, 75, and 100% stimulation intensity. Consecutive responses to three paired pulses (as depicted in Fig. 4B for 100% stimulation intensity) were averaged. To evaluate rises in [K+]o independent of the degree of neuronal activation, population spike amplitudes were used to normalize concomitant rises in [K+]o. At lower stimulation intensities, normalized [K+]o rises of both genotypes did not differ. In contrast, maximal stimulation intensity produced a significantly larger normalized [K+]o rise in dko compared with wt mice (dko, 34 ± 2 μm/mV, n = 8 animals; wt, 26 ± 2 μm/mV, n = 8 animals) (Fig. 4D), suggesting a deficiency in [K+]o clearance in the absence of astrocytic gap junctions. Because higher stimulation intensities activate larger numbers of nerve fibers, both the amount of released K+ and the area containing activated neurons increase. To test whether the contribution of gap junctions to buffering is stimulated by higher K+ loads per se or rather depends on the size of the area of increased [K+]o (as indicated by the population spike amplitude), we applied trains of stimuli (10 s, 20 Hz). Even at 25% stimulus intensity, evoked [K+]o responses were larger than all responses to double pulses (Fig. 4C). However, there were no significant differences in normalized [K+]o rises for 25, 50, or 75% stimulus intensities between genotypes. Again, only maximal stimulation led to larger normalized [K+]o rises in dko mice compared with control (dko, 0.65 ± 0.04 mm/mV, n = 9 animals; wt, 0.47 ± 0.01 mm/mV, n = 8 animals) (Fig. 4E). Furthermore, at maximal stimulation intensity, [K+]o did not exceed 12 mm in wt mice (13 of 13 slices), a value described previously as the “ceiling level” in the hippocampus (Benninger et al., 1980; Krnjevic et al., 1980). Interestingly, we could record substantially higher [K+]o peaks in dko mice in 4 of 15 slices (14–17 mm). In addition, we tested 50% intensity stimulation at different frequencies (5, 10, 20, and 50 Hz) and found no difference in [K+]o accumulation between genotypes (data not shown). Together, these results indicate that astrocytic gap junctional coupling is equally important for K+ homeostasis at very high K+ levels (as obtained with stimulation trains) and at more physiological levels (as obtained with paired-pulse stimulation). However, in both conditions, larger [K+]o rises at the stratum pyramidale were observed only with antidromic stimulation that activates most of the CA1 neurons in the vicinity of the recording electrode (100% stimulation intensity).
Slower decay of elevated [K+]o in dko mice
To determine whether K+ buffering was slowed in dko mice, we analyzed the decline of [K+]o subsequent to stimulus trains (10 s, 20 Hz). [K+]o initially decayed very rapidly, followed by a prolonged phase of slower decay, which was often associated with an undershoot (Heinemann and Lux, 1975). To only consider the fast decay phase, we initially determined the time at which [K+]o had decayed to 1/e of the maximal value (t1/e), providing a first descriptive parameter for the decay process. It was immediately apparent that increasing peak [K+]o values by increasing stimulation strength caused t1/e values to become shorter (Fig. 5A). This phenomenon was seen independently of the genotype. Because of this dependence, t1/e was plotted against amplitudes of rises in [K+]o. This revealed an inverse correlation, best described by a power function: Y = aXexp(b) (wt, r = −0.55, n = 52 pairs of variates; dko, r = −0.51, n = 59 pairs of variates) (Fig. 5B). After double-logarithmic transformation for linearization, analysis of covariance disclosed equal slopes for wt and dko mice (F = 0.001), indicating that larger K+ rises accelerated buffering in wt and dko mice equally. However, comparison of y-axis intercepts yielded a significant shift of the [K+]o/t1/e relationship toward slower decay times in slices from dko mice (by 0.84 s; F = 14.22), demonstrating that removal of any K+ load was slowed in the absence of astrocytic gap junctions.
Figure 5.

Recovery of stimulation-induced increase of [K+]o in stratum pyramidale is slower in dko mice. A, Decline of representative, matched [K+]o transients after stimulus trains (20 Hz) evoked with high (wt, 100%; dko, 75%) and low (wt and dko, 50%) stimulus intensity. For the wt traces, t1/e (the time after which [K+]o amplitude has decayed to 1/e of its initial value) is indicated. Note that decay is faster for larger rises in [K+]o (dotted line, t1/e = 1.93 s; dashed line, t1/e = 2.35 s). B, t1/e values of [K+]o recovery after a stimulus train (20 Hz; 25, 50, 75, and 100% stimulation intensities) plotted against [K+]o at the end of the stimulus (wt, open diamonds; dko, filled squares). The inverse relationships between [K+]o and t1/e were best described by a power function (thin line for wt, y = 5.272x exp(−0.417); thick line for dko mice, y = 6.068x exp(−0.412). y-axis intercepts differed significantly (asterisk; see Results). C, Semilogarithmic plots of the decline of representative, matched [K+]o transients (from 90% to the trough of the undershoot; gray dots) after 100% stimulus trains (20 Hz). Top, wt; bottom, dko. Two exponential components contribute to this decline and are plotted individually (straight black lines; wt, τfast = 1.46 s, τslow = 11.3 s; dko, τfast = 1.15 s, τslow = 10.5 s). Their sum provides an excellent fit of the data (black curves). Note that the relative amplitude of τfast (τfast amplitude fraction) is larger in the wt (84%) compared with the dko [K+]o decay (70%). D, τfast amplitude fractions plotted against [K+]o at the end of the stimulus (wt, open diamonds; dko, filled squares). The linear relationship was significantly (asterisk) shifted toward smaller τfast amplitude fractions in dko mice (thin line, wt, linear equation: y = 0.0445x + 0.39; thick line, dko, y = 0.0346x + 0.38).
Because decay of [K+]o has been suggested to occur with two clearly separable rate constants in rat optic nerve (Ransom et al., 2000) and rat dentate gyrus (Santhakumar et al., 2003), the time course of [K+]o decay was analyzed in a more quantitative manner by fitting with a biexponential function using a least-squares fitting routine (fitting range from 90% maximal amplitude to the trough of the undershoot) (Fig. 5C). Neither τfast nor τslow were significantly different for dko versus wt mice throughout stimulation categories, and there was also no correlation with [K+]o amplitudes (data not shown). However, the τfast amplitude fraction was linearly correlated with amplitudes of rises in [K+]o (wt, r = 0.69; dko, r = 0.6) (Fig. 5D). Again, covariance analysis yielded equal slopes for the two genotypes but a shift in y-axis intercept (F = 15.82), indicating that, in dko mice, the τfast amplitude fraction for a given rise in [K+]o was reduced by 7.8% (Fig. 5D). Hence, in both genotypes, faster decay of larger [K+]o transients apparently resulted from an increased contribution of fast buffering mechanisms. The reduced τfast amplitude fraction in dko mice might indicate that fast buffering mechanisms contribute to buffering to a lesser extent in dko mice.
Because undershoots indicate active K+ uptake (Heinemann and Lux, 1975), it was of interest to see whether the amplitudes of undershoots were different in both animal groups: We compared undershoots at 100% stimulation intensity and found that they were equally proportional to the preceding rise in [K+]o in both genotypes (dko, 0.036 ± 0.004 mm/mm, n = 8 animals; wt, 0.030 ± 0.004 mm/mm, n = 8 animals).
To resolve in dko mice the apparent discrepancy between a slower recovery of [K+]o at all stimulation intensities (Fig. 5) and a selective increase in normalized [K+]o amplitudes at maximal stimulation (Fig. 4), we also determined 1/e times for [K+]o rises. Interestingly, we observed that the rise time of [K+]o was slowed in dko mice at 25% (dko, 2.5 ± 0.2 s; wt, 1.9 ± 0.1 s), 50% (dko, 2 ± 0.2 s; wt, 1.6 ± 0.05 s), and 75% (dko, 1.9 ± 0.2 s; wt, 1.5 ± 0.1 s) stimulation intensities, whereas there was no difference from wt mice at 100% stimulation intensity (dko, 1.6 ± 0.2 s; wt, 1.3 ± 0.1 s).
dko mice reveal impaired spatial buffering in stratum lacunosum moleculare
We sought to challenge the long standing hypothesis of glial spatial K+ buffering (Orkand et al., 1966). To this end, the laminar profile of stimulus-induced rises in [K+]o from stratum pyramidale throughout stratum radiatum and stratum lacunosum moleculare to the hippocampal fissure was investigated (Fig. 6A) as described previously (Gabriel et al., 1998a,b). To resolve small K+ changes at distant sites, maximal stimulation intensity was used throughout experiments (10 s train, 20 Hz). The population spikes, elicited by antidromic stimulation, were clearly visible at 100 μm from stratum pyramidale, declining steeply toward the stratum lacunosum moleculare. This behavior was similar in wt and dko mice, indicating that invasion of action potential into dendrites was similar in both genotypes. We hypothesized that the redistribution of [K+]o from the major efflux sites at the soma to the dendritic field might be impaired in dko mice, leading to a steeper decline of [K+]o responses toward the fissure. Therefore, we compared [K+]o changes at different distances from the stratum pyramidale, normalized to those obtained at the stratum pyramidale (Fig. 6B). Interestingly, up to 300 μm away from the stratum pyramidale, this parameter did not differ between genotypes, indicating that, in the stratum radiatum (∼350 μm wide), radial [K+]o relocation was not altered in dko compared with wt mice. In contrast, at a distance of 400–500 μm from the stratum pyramidale, anatomically corresponding to the stratum lacunosum moleculare, normalized [K+]o rises in dko mice were much smaller than those in wt mice (Fig. 6B). In view of these findings, we suspect unequal contribution of gap junctions to radial transport of [K+]o, depending on the location within hippocampal layers. This idea is supported by the unequal size and orientation of astrocytes in stratum radiatum versus stratum lacunosum moleculare (Fig. 6C) (for quantitative evaluation, see Nixdorf-Bergweiler et al., 1994): single astrocytes in the stratum radiatum span much larger areas than those in the stratum lacunosum moleculare. Specifically, cells in the stratum radiatum often show a bipolar shape, with an orientation perpendicular to stratum pyramidale, whereas cells in stratum lacunosum moleculare are round and have no particular orientation.
dko mice show increased susceptibility for the generation of epileptiform events
To analyze whether ablation of gap junctions in hippocampal astrocytes leads to neuronal hyperactivity, we recorded field potentials in the absence of synaptic blockers over 10 min periods. Indeed, only slices of dko mice developed spontaneous epileptiform events (dko, 6 of 10 slices; wt, 0 of 10 slices) (Fig. 7A). Moreover, only slices of dko mice generated epileptiform activity during low-intensity Schaffer collateral stimulation (dko, 4 of 6 slices; wt, 0 of 10 slices) (Fig. 7B).
Figure 7.
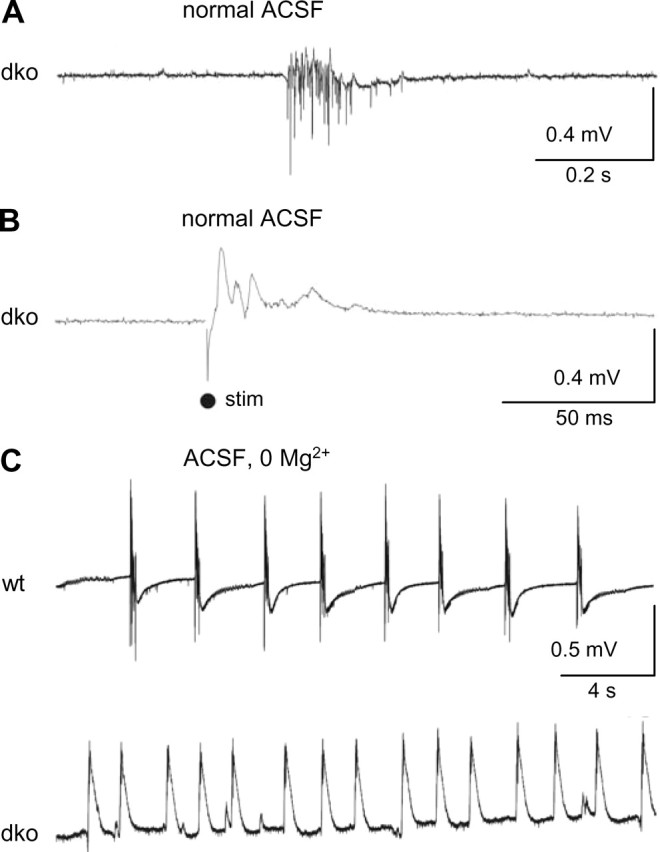
Epileptiform field potentials (EFP) in CA1 stratum pyramidale of dko mice. A, Spontaneous epileptiform field potentials (CA1, stratum pyramidale) occurred only in slices from dko mice (representative trace) but not in slices from wt mice (data not shown; p = 0.001). B, Low-intensity Schaffer-collateral stimulation gave rise to EFP only in dko slices (representative sample trace) but not in slices from wt mice (data not shown) (p = 0.007). C, EFP induced by washout of Mg2+ (0 Mg2+) were more frequent in dko slices and occurred with shorter latency. Original traces of a wt (top) and dko (bottom) slice.
K+ buffering by astrocytes may become particularly important under conditions of synchronized high-frequency neuronal firing, such as during seizure-like discharges. To test whether uncoupling of astrocytes aggravates epileptiform activity, spontaneous seizure-like discharges were recorded in the CA1 stratum pyramidale after switching slice perfusion from 1.6 to 0 mm Mg2+ [0 Mg2+ model (Mody et al., 1987)]. In hippocampal slices from dko mice, these discharges occurred with a shorter latency (dko, 21.7 ± 0.7 min, n = 3 animals, 10 slices; wt, 34.3 ± 3.2 min, n = 3 animals, 10 slices) and displayed a higher frequency (dko, 24.6 ± 4.1 discharges/min; wt, 7.9 ± 2.9 discharges/min) (Fig. 7C).
Discussion
Experimental evidence supports two major mechanisms of extracellular K+ homeostasis during periods of raised [K+]o: net accumulation of K+ within astrocytes (Coles and Tsacopoulos, 1979; Ballanyi et al., 1987) and spatial buffering (Orkand et al., 1966). Although gap junctions have been assumed necessary for extracellular K+ homeostasis by abetting spatial buffering, this has never been tested conclusively (Ransom, 1996). It should be added that gap junctions might also benefit extracellular K+ homeostasis attributable to net K+ uptake by stabilizing intracellular ion concentrations (Rose and Ransom, 1997). We generated mice lacking coupled astrocytes, allowing us to quantitatively determine the importance of junctional communication for K+ homeostasis. These mice displayed changes in spatial and temporal dynamics of stimulus-induced [K+]o transients and a lowered threshold for generating hyperactivity. However, a surprisingly large capacity for K+ redistribution was conserved in these mice, indicating that gap junction-dependent processes only partially account for K+ buffering in the hippocampus.
Composition of astrocytic gap junctions
Cx43 and Cx30 are expressed in astrocytes but not in neurons (Nagy et al., 2004). Cx26 was localized in subcortical astrocytes (Nagy et al., 2001) (but see Filippov et al., 2003). The present study indicates that residual biocytin coupling in Cx43-deficient astrocytes (Theis et al., 2003) was related to transfer through Cx30 homotypic junctions, because it was abolished in Cx43/Cx30 dko mice (Fig. 1D). In view of the low expression of Cx30 in stratum radiatum of control mice (Söhl et al., 2004), this study confirms that Cx30 is upregulated in Cx43-deficient astrocytes, as shown previously by Western blot (Theis et al., 2003), and that it can partly compensate for loss of Cx43. Cx26 does not contribute to astrocytic tracer coupling in CA1 stratum radiatum.
Properties of uncoupled astrocytes
Fundamental astrocyte-specific properties as well as astrocyte density were preserved in dko mice, indicating that they represent a valuable model to investigate the role of astrocytic coupling. We also confirmed the presence of intrinsic time- and voltage-independent astrocytic whole-cell currents (Blomstrand et al., 2004; Wallraff et al., 2004). Such passive currents had occasionally been regarded as recording artifacts attributable to junctional coupling (Walz, 2000a), although they were also observed in isolated astrocytes (Seifert and Steinhäuser, 1995). The junctional contribution to passive current patterns amounted to ∼30% of total whole-cell currents. This substantial contribution is consistent with K+ flow through gap junctions during spatial buffering.
Astrocytic K+ buffering
Many groups have produced evidence in favor of net uptake of K+ or spatial buffering, which could coexist and vary in different brain regions (Ransom, 1996; Walz, 2000b; Kofuji and Newman, 2004). Involvement of gap junctions in K+ buffering, however, has never been tested conclusively. We chose three parameters to characterize K+ homeostasis in mice with gap junction-deficient astrocytes: the maximal increase of [K+]o at stratum pyramidale, the rate of decay of [K+]o after stimulation, and the laminar profile of [K+]o increases from stratum pyramidale to the hippocampal fissure. Throughout all stimulation intensities, [K+]o decay rates in dko mice were slowed (Fig. 5). In contrast, larger [K+]o rises at stratum pyramidale were observed only with antidromic stimulation at 100% intensity (Fig. 4D,E). This applied to both [K+]o transients obtained with double pulses and train stimulation. This finding is puzzling because the magnitude of [K+]o transients to maximal double pulses is smaller than the [K+]o increases induced by low-stimulation trains (25–75%). The most plausible explanation for this discrepancy is that weak antidromic stimulation causes action potential generation in a limited population of CA1 neurons, not necessarily congruent with the site of K+ recordings. In this case, spread of K+ from the site of maximal neuronal activity to the recording site will contribute significantly to the amplitudes of the recorded [K+]o transients. Because equilibration of [K+]o is significantly slower in dko mice (Fig. 5), we would predict that, at low stimulation intensities, [K+]o at the recording site rises more slowly in dko mice. This was in fact observed (see Results). Accordingly, at lower stimulation intensities, this effect could have attenuated differences in [K+]o amplitudes between wt and dko animals.
The slower decay of [K+]o transients in dko mice (Fig. 5) was not attributable to altered time constants, suggesting that similar fast and slow buffering processes occurred as in wt mice. However, the proportion of K+ removed by fast clearance was smaller in dko mice (Fig. 5C,D). Slow buffering processes mainly represent net uptake of K+ via the neuronal Na+/K+ ATPase because they are strophantidine sensitive (Ransom et al., 2000) and because the undershoot (Heinemann and Lux, 1975) is not seen when neuronal activity is blocked and K+ is applied iontophoretically (Jauch et al., 2002). Indeed, amplitudes of undershoots in dko mice were unchanged (see Results). The proportion of fast versus slow K+ buffering was governed, in both genotypes, by the K+ load at stratum pyramidale (Fig. 5D), suggesting a significant contribution of spatial buffering to fast clearance because it would be facilitated by a steep concentration gradient. The shift in the relationship of fast clearance fraction to [K+]o in dko mice indicates that a smaller proportion of [K+]o is dissipated through spatial buffering. Gap junction-independent fast buffering processes preserved in dko mice (i.e., astrocytic K+ sequestration or spatial buffering through single astrocytes) appear to require higher K+ loads than gap junction-dependent mechanisms to contribute to K+ clearance. Interestingly, in rat optic nerve, τfast became smaller as a function of [K+]o (Ransom et al., 2000). An attractive interpretation of this difference would be that unequal buffering processes take place in white versus gray matter: maximal stimulation in optic nerve leads to uniform K+ accumulation (i.e., there is no gradient to facilitate spatial buffering). Here, fast clearance primarily represents K+ uptake by the glial Na+/K+ ATPase (Ransom et al., 2000).
Pyramidal cell activation in the CA1 region entails a steep [K+]o gradient from stratum pyramidale toward the hippocampal fissure (Benninger et al., 1980; Gabriel et al., 1998a), consistent with a spatial buffering mechanism. Spatial relocation is strongly impaired by Ba2+, which blocks K+ channels necessary for passive uptake, leading to larger [K+]o amplitudes at stratum pyramidale and a steeper decline throughout strata radiatum and lacunosum moleculare (Gabriel et al., 1998a). Surprisingly, we found equal slopes of the laminar [K+]o profiles of wt and dko mice in stratum radiatum, using maximal stimulation intensity, but a steeper decline in stratum lacunosum moleculare of dko mice (Fig. 6B). This indicates that other factors, not gap junctions, mediate K+ redistribution in stratum radiatum, at least in the direction perpendicular to the neuronal cell layer. Because astrocytes in stratum radiatum display a preferentially perpendicular orientation to stratum pyramidale (Nixdorf-Bergweiler et al., 1994; this study) (Figs. 2E,F, 6B), single cells could be well suited to move K+ radially, without the participation of gap junctions, especially if part of the K+ is released from apical dendrites, at some distance from stratum pyramidale. In addition, partial overlap of elongated astrocytic processes might facilitate release of K+ from one astrocytic pole and uptake at the next, via K+ channels, a mechanism coined as “indirect coupling” (Newman, 1995; Ransom, 1996). In stratum lacunosum moleculare, on the contrary, relocation of K+ in radial direction was impaired in the absence of astrocytic gap junctions, demonstrating that junctional coupling can, indeed, contribute to spatial buffering. Lacunosum astrocytes show no preferential orientation, and their processes are less extended compared with radiatum astrocytes (Nixdorf-Bergweiler et al., 1994; this study) (Figs. 2E,F, 6B), which might explain why, in this region, gap junctions play a role in radial transport of K+.
However, we demonstrate that buffering of [K+]o, released at stratum pyramidale, is impaired in dko mice (Figs. 4, 5). It is conceivable that astrocytic gap junctions help buffer K+ from stratum pyramidale by facilitating net uptake (proposed by Newman, 1995; Ransom, 1996) through spatial buffering in stratum lacunosum moleculare (Fig. 6B) or by promoting spatial buffering in stratum radiatum in parallel to the neuronal cell layer (as discussed above).
Pathophysiological implications and perspectives
Pharmacological evidence suggests that gap junction inhibitors may be effective anticonvulsants (Nemani and Binder, 2005). However, we show for the first time that genetic ablation of gap junctions in astrocytes lowers the threshold for generation of epileptiform discharges. The anticonvulsant action of gap junction blockers, therefore, probably stems from blockade of neuronal gap junctions and could be increased by developing blockers selectively recognizing neuronal connexins. Our findings might explain the inconsistency in the literature regarding the role of Cx43 in human or experimental epilepsy (Steinhäuser and Seifert, 2002) and indicates that local alterations in gap junction function may have variable effects on the actual level of hyperactivity depending on the local astrocytic population. Upregulation of Cx43 in epileptic tissue might reflect adaptive consequences rather than a causation of seizure activity.
Availability of mice with ablation of astrocytic connexins might also further a better understanding of the proposed roles of Cx30/Cx43 in other glia-related transcellular signaling pathways, such as propagation of astroglial Ca2+ waves (Cotrina et al., 1998), release of gliotransmitters (Ye et al., 2003), or astrocytic interactions with the vasculature (Simard et al., 2003).
Footnotes
This work was supported by German Research Association Grants Sonderforschungsbereich (SFB)/TR3, SFB400, SFB645, JA942/4, and SE747/3. We gratefully acknowledge Ingrid Schroedter for technical assistance and Dr. Siegrun Gabriel for excellent advice on [K+]o measurements and discussion of results. We thank Dr. Rolf Fimmers for help with nonlinear regression analysis and Dr. Herbert Siegmund for sharing his analysis software for Spike.
References
- Ballanyi K, Grafe P, ten Bruggencate G (1987). Ion activities and potassium uptake mechanisms of glial cells in guinea-pig olfactory cortex slices. J Physiol (Lond) 382:159–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benninger C, Kadis J, Prince DA (1980). Extracellular calcium and potassium changes in hippocampal slices. Brain Res 187:165–182. [DOI] [PubMed] [Google Scholar]
- Blomstrand F, Venance L, Siren AL, Ezan P, Hanse E, Glowinski J, Ehrenreich H, Giaume C (2004). Endothelins regulate astrocyte gap junctions in rat hippocampal slices. Eur J Neurosci 19:1005–1015. [DOI] [PubMed] [Google Scholar]
- Bushong EA, Martone ME, Jones YZ, Ellisman MH (2002). Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci 22:183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles JA, Tsacopoulos M (1979). Potassium activity in photoreceptors, glial cells and extracellular space in the drone retina: changes during photostimulation. J Physiol (Lond) 290:525–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotrina ML, Lin JHC, Alves-Rodrigues A, Liu S, Li J, Azmi-Ghadimi H, Kang J, Naus CCG, Nedergaard M (1998). Connexins regulate calcium signaling by controlling ATP release. Proc Natl Acad Sci USA 95:15735–15740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippov MA, Hormuzdi SG, Fuchs EC, Monyer H (2003). A reporter allele for investigating connexin 26 gene expression in the mouse brain. Eur J Neurosci 18:3183–3192. [DOI] [PubMed] [Google Scholar]
- Gabriel S, Eilers A, Kivi A, Kovacs R, Schulze K, Lehmann TN, Heinemann U (1998a). Effects of barium on stimulus induced changes in extracellular potassium concentration in area CA1 of hippocampal slices from normal and pilocarpine-treated epileptic rats. Neurosci Lett 242:9–12. [DOI] [PubMed] [Google Scholar]
- Gabriel S, Kivi A, Eilers A, Kovács R, Heinemann U (1998b). Effects of barium on stimulus-induced rises in [K+]o in juvenile rat hippocampal area CA1. NeuroReport 9:2583–2587. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Lux HD (1975). Undershoots following stimulus-induced rises of extracellular potassium concentration in cerebral cortex of cat. Brain Res 93:63–76. [DOI] [PubMed] [Google Scholar]
- Jauch R, Windmuller O, Lehmann TN, Heinemann U, Gabriel S (2002). Effects of barium, furosemide, ouabaine and 4,4′-diisothiocyana-tostilbene-2,2′-disulfonic acid (DIDS) on ionophoretically-induced changes in extracellular potassium concentration in hippocampal slices from rats and from patients with epilepsy. Brain Res 925:18–27. [DOI] [PubMed] [Google Scholar]
- Kofuji P, Newman EA (2004). Potassium buffering in the central nervous system. Neuroscience 129:1045–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krnjevic K, Morris ME, Reiffenstein RJ (1980). Changes in extracellular Ca2+ and K+ activity accompanying hippocampal discharges. Can J Physiol Pharmacol 58:579–582. [DOI] [PubMed] [Google Scholar]
- Lux HD, Neher E (1973). The equilibration time course of [K+]o in cat cortex. Exp Brain Res 17:190–205. [DOI] [PubMed] [Google Scholar]
- Matthias K, Kirchhoff F, Seifert G, Hüttmann K, Matyash M, Kettenmann H, Steinhäuser C (2003). Segregated expression of AMPA-type glutamate receptors and glutamate transporters defines distinct astrocyte populations in the mouse hippocampus. J Neurosci 23:1750–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mody I, Lambert JD, Heinemann U (1987). Low extracellular magnesium induces epileptiform activity and spreading depression in rat hippocampal slices. J Neurophysiol 57:869–888. [DOI] [PubMed] [Google Scholar]
- Nagy JI, Li X, Rempel J, Stelmack G, Patel D, Staines WA, Yasumura T, Rash JE (2001). Connexin26 in adult rodent central nervous system: demonstration at astrocytic gap junctions and colocalization with connexin30 and connexin43. J Comp Neurol 441:302–323. [DOI] [PubMed] [Google Scholar]
- Nagy JI, Dudek FE, Rash JE (2004). Update on connexins and gap junctions in neurons and glia in the mammalian nervous system. Brain Res Brain Res Rev 47:191–215. [DOI] [PubMed] [Google Scholar]
- Nemani VM, Binder DK (2005). Emerging role of gap junctions in epilepsy. Histol Histopathol 20:253–259. [DOI] [PubMed] [Google Scholar]
- Newman EA (1993). Inward-rectifying potassium channels in retinal glial (Müller) cells. J Neurosci 13:3333–3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA (1995). Glial cell regulation of extracellular potassium. In: Neuroglia (Kettenmann H, Ransom BR, eds) pp. 717–731. New York: Oxford UP.
- Newman EA, Frambach DA, Odette LL (1984). Control of extracellular potassium levels by retinal glial cell K+ siphoning. Science 225:1174–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixdorf-Bergweiler BE, Albrecht D, Heinemann U (1994). Developmental changes in the number, size, and orientation of GFAP-positive cells in the CA1 region of rat hippocampus. Glia 12:180–195. [DOI] [PubMed] [Google Scholar]
- Orkand RK, Nicholls JG, Kuffler SW (1966). Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J Neurophysiol 29:788–806. [DOI] [PubMed] [Google Scholar]
- Ransom BR (1996). Do glial gap junctions play a role in extracellular ion homeostasis? In: Gap junction in the nervous system (Spray DC, Dermietzel R, eds) pp. 159–173. Austin, TX: RG Landes Company.
- Ransom CB, Ransom BR, Sontheimer H (2000). Activity-dependent extracellular K+ accumulation in rat optic nerve: the role of glial and axonal Na+ pumps. J Physiol (Lond) 522:427–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose CR, Ransom BR (1997). Gap junctions equalize intracellular Na+ concentration in astrocytes. Glia 20:299–307. [DOI] [PubMed] [Google Scholar]
- Rozental R, Srinivas M, Spray DC (2001). How to close a gap junction channel. Efficacies and potencies of uncoupling agents. Methods Mol Biol 154:447–476. [DOI] [PubMed] [Google Scholar]
- Santhakumar V, Voipio J, Kaila K, Soltesz I (2003). Post-traumatic hyperexcitability is not caused by impaired buffering of extracellular potassium. J Neurosci 23:5865–5876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert G, Steinhäuser C (1995). Glial cells in the mouse hippocampus express AMPA receptors with an intermediate Ca2+ permeability. Eur J Neurosci 7:1872–1881. [DOI] [PubMed] [Google Scholar]
- Seifert G, Schilling K, Steinhäuser C (2006). Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat Rev Neurosci 7:194–206. [DOI] [PubMed] [Google Scholar]
- Simard M, Arcuino G, Takano T, Liu QS, Nedergaard M (2003). Signaling at the gliovascular interface. J Neurosci 23:9254–9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söhl G, Odermatt B, Maxeiner S, Degen J, Willecke K (2004). New insights into the expression and function of neural connexins with transgenic mouse mutants. Brain Res Rev 47:245–259. [DOI] [PubMed] [Google Scholar]
- Somjen GG (2001). Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol Rev 81:1065–1096. [DOI] [PubMed] [Google Scholar]
- Steinhäuser C, Seifert G (2002). Glial membrane channels and receptors in epilepsy: impact for generation and spread of seizure activity. Eur J Pharmacol 447:227–237. [DOI] [PubMed] [Google Scholar]
- Steinhäuser C, Jabs R, Kettenmann H (1994). Properties of GABA and glutamate responses in identified glial cells of the mouse hippocampal slice. Hippocampus 4:19–36. [DOI] [PubMed] [Google Scholar]
- Teubner B, Michel V, Pesch J, Lautermann J, Cohen-Salmon M, Söhl G, Jahnke K, Winterhager E, Herberhold C, Hardelin JP, Petit C, Willecke K (2003). Connexin30 (Gjb6)-deficiency causes severe hearing impairment and lack of endocochlear potential. Hum Mol Genet 12:13–21. [DOI] [PubMed] [Google Scholar]
- Theis M, Jauch R, Zhuo L, Speidel D, Wallraff A, Doring B, Frisch C, Söhl G, Teubner B, Euwens C, Huston J, Steinhäuser C, Messing A, Heinemann U, Willecke K (2003). Accelerated hippocampal spreading depression and enhanced locomotory activity in mice with astrocyte-directed inactivation of connexin43. J Neurosci 23:766–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volterra A, Meldolesi J (2005). Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci 6:626–640. [DOI] [PubMed] [Google Scholar]
- Wallraff A, Odermatt B, Willecke K, Steinhäuser C (2004). Distinct types of astroglial cells in the hippocampus differ in gap junction coupling. Glia 48:36–43. [DOI] [PubMed] [Google Scholar]
- Walz W (2000a). Controversy surrounding the existence of discrete functional classes of astrocytes in adult gray matter. Glia 31:95–103. [DOI] [PubMed] [Google Scholar]
- Walz W (2000b). Role of astrocytes in the clearance of excess extracellular potassium. Neurochem Int 36:291–300. [DOI] [PubMed] [Google Scholar]
- Ye ZC, Wyeth MS, Baltan-Tekkok S, Ransom BR (2003). Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J Neurosci 23:3588–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]