

Abstract
During a critical period in the developing mammalian brain, there is a major switch in the nature of GABAergic transmission from depolarizing and excitatory, the pattern of the neonatal brain, to hyperpolarizing and inhibitory, the pattern of the mature brain. This switch is believed to play a major role in determining neuronal connectivity via activity-dependent mechanisms. The GABAergic developmental switch may also be particularly vulnerable to dysfunction leading to seizure disorders. The developmental GABA switch is mediated primarily by KCC2, a neuronal K+/Cl− cotransporter that determines the intracellular concentration of Cl− and, hence, the reversal potential for GABA. Here, we report that kazachoc (kcc) mutations that reduce the level of the sole K+/Cl− cotransporter in the fruitfly Drosophila melanogaster render flies susceptible to epileptic-like seizures. Drosophila kcc protein is widely expressed in brain neuropil, and its level rises with developmental age. Young kcc mutant flies with low kcc levels display behavioral seizures and demonstrate a reduced threshold for seizures induced by electroconvulsive shock. The kcc mutation enhances a series of other Drosophila epilepsy mutations indicating functional interactions leading to seizure disorder. Both genetic and pharmacological experiments suggest that the increased seizure susceptibility of kcc flies occurs via excitatory GABAergic signaling. The kcc mutants provide an excellent model system in which to investigate how modulation of GABAergic signaling influences neuronal excitability and epileptogenesis.
Keywords: seizure, epilepsy, K+/Cl− cotransporter, GABAA receptor, genetics, Drosophila
Introduction
Synaptic inhibition in Drosophila, as in mammals, is mediated primarily by the neurotransmitter GABA (Mody et al., 1994; Hosie et al., 1997). GABAergic neurons are found throughout the CNS of Drosophila adults and at all stages of development (Buchner et al., 1988; Jackson et al., 1990; Harrison et al., 1996). Inhibitory GABAergic signaling occurs primarily via ionotropic GABAA receptors encoded by the Resistance to dieldrin (Rdl) gene (Hosie et al., 1997; Lee et al., 2003). The homomeric Rdl GABAA channel is the target of several commercially important insecticides; a single Rdl point mutation is responsible for nearly all known cases of resistance to the series of insecticides that target GABAA receptors (Buckingham et al., 2005). As with other GABAA receptors, binding of GABA to Rdl leads to the opening of an internal channel that conducts primarily Cl− ions (Buckingham et al., 2005). Consequently, synaptic inhibition is contingent on the electrochemical gradient for Cl− of the GABAergic neuron, which determines the GABAergic reversal potential of the neuron (E GABA; the voltage at which GABAergic currents change their direction).
Recent investigations of GABAergic inhibition in the mammalian CNS have focused considerable interest on the interplay between the GABAA receptor and a K+/Cl− cotransporter termed KCC2. KCC2 is the neuronal member of a family of four vertebrate KCCs, all of which concomitantly extrude K+ and Cl− from the cell (Mount et al., 1998; Hebert et al., 2004). Because the level of KCC2 affects EGABA, expression of KCC2 greatly influences signaling mediated by GABAA receptors (Miles, 1999; Lee et al., 2005; Zhu et al., 2005). In normal adult cortical neurons, KCC2 activity produces low intracellular Cl− levels (Rivera et al., 1999; Stein et al., 2004). GABA-mediated opening of GABAA Cl− channels thus produces Cl− efflux, resulting in hyperpolarization that, in turn, reduces the ability of the neuron to fire action potentials (Qian and Sejnowski, 1990; Staley and Mody, 1992; Mitchell and Silver, 2003). In immature neurons or under certain pathophysiological conditions, reduced expression of the KCC2 GABA switch results in an elevated intracellular Cl− concentration (Katchman et al., 1994; van den Pol et al., 1996; Ben-Ari, 2002; Nabekura et al., 2002; Payne et al., 2003). GABA-mediated activation of GABAA receptors can then lead to depolarizing outward Cl− currents and, in some instances, produce synaptic excitation rather than inhibition (Luhmann and Prince, 1991; Yuste and Katz, 1991; Wang et al., 1994; Obrietan and van den Pol, 1995; Chen et al., 1996; Owens et al., 1996). Both reduced KCC2 expression and excitatory GABAergic signaling have been linked to temporal lobe epilepsy (Köhling et al., 1998; Cohen et al., 2002; Shinnar and Glauser, 2002; Baulac et al., 2004; Palma et al., 2006).
This paper describes a K+/Cl− cotransporter gene from Drosophila called kazachoc (kcc) that is homologous to mammalian KCC2. Complete loss-of-function kcc mutations are lethal, indicating that kcc is an essential gene. Phenotypic characterization of partial loss-of-function mutants that reduce kcc level reveals neurological excitability defects that render the kcc flies especially susceptible to epileptic-like seizures. The seizure susceptibility of kcc flies shows a pronounced age dependence that mirrors a developmental increase in kcc level. The seizure sensitivity of kcc mutants is mediated by Rdl GABAA receptors, thereby linking kcc seizures with dysfunction of the GABAergic inhibitory system. The kcc mutation is a potent seizure enhancer, interacting with a series of other neurological mutations to exacerbate seizure disorder phenotypes. We discuss a model in which K+/Cl− cotransporters play a central role in polygenic inheritance of seizure phenotypes, the most common mode of inheritance of idiopathic generalized epilepsies in humans. The K+/Cl− cotransporter may provide an important link between idiopathic epilepsy and perturbation of GABAergic inhibition.
Materials and Methods
Fly stocks.
A list of Drosophila stocks used in this study is given in Table 1. Stocks were maintained on standard cornmeal–molasses medium in vials on an upper shelf at room temperature (∼24°C) (Ashburner, 1989). Crosses were performed at 22°C if progeny were to be tested for bang sensitivity (unless otherwise specified); all other crosses were performed at 25°C. Three bang-sensitive (BS) mutations are included: eas, bss, and sda. The eas gene is located at cytological region 14B and encodes an ethanolamine kinase (Pavlidis et al., 1994). The recessive easPC80 allele carries a frameshift mutation and probably constitutes a null allele. The bss gene is located at 1–54.6 (corresponding to approximately cytological region 12F); its gene product has not been described (Ganetzky and Wu, 1982). The bss1 allele is a semidominant mutation. The sda gene is located at 97D and encodes an aminopeptidase (Zhang et al., 2002). The recessive sdaiso7.8 mutation occurs in the 5′ noncoding region and greatly diminishes the level of sda transcript. The genotypes w easPC80 f, w bss1 f, and w sdaiso7.8 are abbreviated as eas, bss, and sda, respectively, in the text. The deficiency stocks carrying Df(2R)OV1 (59F5;60A1), Df(2R)b23 (59F8;60A3), and Df(2R)bwDRa (59E1;60A4-5) were kindly provided by Kristi Wharton (Brown University, Providence, RI). Deficiency stocks carrying Df(2R)tid (59F4;59F5), Df(2R)egl2 (59E;60A1), Df(2R)bwS46 (59D8-11;60A7), Df(2R)Chig230 (60A3-7;60B4), Df(2R)106 (60A3;60A7), and Df(2R)PX1 (60B8-10;60D1) were obtained from the Bloomington Drosophila Stock Center, as was the Dp(2;Y)bw+ (58F1-3;60E11-12) duplication stock. Lethal alleles of Nap1 [Nap1KO1 (Lankenau et al., 2003)] and CG4882 [l(2)SH2263 (Oh et al., 2003)] were provided by Dirk Lankenau (University of Heidelberg, Heidelberg, Germany) and Steven Hou (National Institutes of Health, Bethesda, MD), respectively.
Table 1.
Drosophila stocks
| Stock no. | Genotype |
|---|---|
| CS-5 | Wild type |
| D505 | w1118; kccDHS1/SM5 Cy; P{w + LacZ}CG9924J |
| D506 | w1118; kccDHS1/CyO |
| U036 | w; SM5 Cy; TM3/apXa |
| D225 | w; TM3/TM6B |
| D245 | w1118; nub b nocSco lt stw3/CyO |
| 2(A) | y ac w; P(YES:YBB = y+ CG9925+) |
| D572 | w; kccDHS1/CyO; sdaiso7.8/TM6B |
| MR047 | w easPC80 f |
| MR068 | w bss1 f |
| D547 | w; SM5 Cy; sdaiso7.8/apXa |
| D548 | w easPC80 f; +/CyO |
| D508 | w bss1 f; CyO;+/apXa |
| D250 | al dp b pr c px sp |
| D253 | w; Sp/CyO; Dr Δ2-3/TM3 |
| D254 | y; cn bw sp |
| 5206 | dpov1 b1 cn1 l(2)60A-CP20–180/SM6a Cy |
| 5207 | dpov1 b1 cn1 l(2)60A-CAd-4/SM6a Cy |
| D562 | w; dpov1 b1 cn1 l(2)60A-CAd-4/CyO |
| 5224 | Df(2R)106/SM5 Cy |
| 16887 | y1 w67c23; P{w+ =EPgy2}CG5594EY08304 |
| EP2164 | w1118; P{w+ =EP}CG10413EP2164 |
| 13216 | y1 w67c23; P{y+w+ =SUPor-P}KG02390 |
| D577 | w; kccML1/CyO |
| D569 | w; kccDHS1/SM5 Cy; Rdl1/TM6B |
| D582 | w; kccDHS1/SM5 Cy; Gad1L352F es/TM6B |
Behavioral testing.
Testing for BS paralysis was performed on flies reared at 22°C and 1–2 d after eclosion unless otherwise specified. Flies were anesthetized with CO2 and collected <1 d after eclosion, held at 18°C overnight, and tested the following day unless otherwise specified. Approximately 15 flies were transferred to a clean vial (Applied Scientific, Eugene, OR) without agitation and immediately vortexed at maximum speed for 10 s. Bang-sensitive flies displayed a period of paralysis followed by a period of hyperactivity. The χ2 test was used to determine the p values for differences in percent bang sensitivity between test flies and their sibling controls.
Identification of the initial kazachoc (kcc) mutation and construction of a kcc stock.
We initially identified kcc as a spontaneous mutation present on the second chromosome of one of our sda-enhancer stocks, D505 (Zhang et al., 2002), which also carries an unlinked P-element insertion, P(w+ LacZ)CG9925J, on its third chromosome. To determine whether the P-element insertion upstream of the CG9925 gene was responsible for the strain's recessive BS phenotype, we asked whether this phenotype could be rescued by a P-element construct carrying the complete CG9925 gene inserted into the second chromosome. Accordingly, we first crossed non-Cy virgin females from our D505 stock to U036 males. We then crossed what we presumed to be w1118; +/SM5 Cy; P(w+)CG9925/TM3 male progeny from this cross to virgin 2(A) females. The 2(A) stock, kindly provided by Gary Landis (University of Southern California, Los Angeles, CA), has a P-element carrying the CG9925 gene inserted into the second chromosome (Landis et al., 1997). Finally, we crossed what we assumed to be P(CG9925+)/SM5 Cy; P(w+)CG9925/TM3 males from this cross to virgin females from our original D505 stock and analyzed the progeny for bang sensitivity. To our surprise, we observed no bang sensitivity in any of the homozygous P(w+)CG9925 progeny: neither the P(CG9925+) ones that carried the rescue construct nor the +/SM5 Cy; P(w+)CG9925 ones that lacked it were BS. We postulated that the original D505 strain must carry an unmarked mutation on its second chromosome that was necessary for its recessive BS phenotype. The presence of a deleterious second chromosomal mutation in D505 would also be consistent with the observation that most of the flies in our D505 stock retained their SM5 balancer chromosome. We termed this new BS mutation kazachoc (kcc) and the spontaneous allele kccDHS1. The “kazachoc” is a Slavic dance that involves squatting with the alternate kicking out of legs, a process that to some extent resembles a BS fly experiencing a bang-induced seizure. The kccDHS1 mutation apparently arose as a spontaneous mutation in the w1118 stock used in a screen for enhancers of sda/+.
We obtained a balanced kcc stock, D506, as follows: non-Cy D505 males (w1118; kccDHS1; P(w+)CG9925) were crossed to U036 virgin females. Virgin female progeny of the genotype w1118; kccDHS1SM5 Cy; P(w+)CG9925/TM3 were then crossed to D245 males. Male and virgin female progeny of the genotype w1118; kccDHS1/CyO; +/TM3 were then crossed and their homozygous w1118; kccDHS1 progeny mated to create an initial stock. Several bang-sensitive males from this initial w1118; kccDHS1 stock were then crossed to D245 virgin females. Finally, male and virgin female progeny of the genotype w1118; kccDHS1/CyO were crossed to create our D506 balanced kccDHS1 stock
Recombination mapping of kcc.
In an initial recombination mapping experiment, virgin females from a multiply marked second chromosome mapping strain (D250) were crossed to D506 males. Groups of two +/w1118; al dp b pr c px sp/kccDHS1 virgin female progeny were then crossed to D250 males. Recombinant male progeny were then individually crossed to D506 and the progeny scored for bang sensitivity. These experiments revealed that kcc is very near sp [at 107 map units (m.u.)].
We then performed three-point mapping experiments to further refine the position of the kcc gene. In these experiments, D506 virgin females were crossed to D254 males. The resultant kccDHS1/cn bw sp female progeny were then crossed in groups of three to D506 males, and the male progeny were tested for bang sensitivity. The 115 bang-sensitive (kccDHS1/kccDHS1) male progeny were then individually crossed to D254 virgin females, and the progeny were scored for the bw and sp markers. Five recombinants (three bw kcc and two kcc sp) were identified, indicating that the gene order is as follows: bw kcc sp.
Creation of a new kcc allele by imprecise P-element excision.
The kccML1 allele was produced by imprecise excision of a white+ P-element (SUPOr-P) located 5 bp downstream of the kcc 3′-untranslated region (UTR) as follows: First, no. 13216 virgin females were crossed to D253 males. Next, the resulting w−; P(w+)/CyO; Δ2–3/+ male progeny were crossed to U036 virgin females, and the male progeny were screened for those with white eyes, which presumably had undergone excision of the P(w+) element. From ∼2400 male progeny examined, we obtained 41 independently derived white-eyed males. Each of these white-eyed males was then separately crossed to D562 virgin females at 23°C, and both the Cy and non-Cy progeny were screened for bang sensitivity. Six of these crosses yielded some bang-sensitive non-Cy (but not Cy) progeny, and another produced few non-Cy progeny. Balanced stocks for each of the seven lines were created as follows: First, the seven original white-eyed males of interest were individually crossed to D245 virgin females. CyO male and virgin female progeny were then crossed to create the balanced stock. A fraction of the homozygous flies from four of the resultant lines were bang sensitive (8B, 8%; 19A, 1%; 21A, 2%; 23A, 2%); another produced no homozygous flies, indicating the presence of a lethal mutation on the second chromosome.
Other genetic analysis.
In our deficiency mapping experiments, D506 virgin females were crossed to balanced Df(2R) males and at least 30 of the nonbalanced kccDHS1/Df(2R) progeny were tested for bang-sensitive paralysis. We concluded that a deficiency uncovered kcc if a significant fraction of the kccDHS1/Df(2R) progeny were bang sensitive. In our duplication mapping experiment, D506 virgin females were first crossed to y′ wa/Dp(2:Y)bw+;E(wa)/CyO males (Bloomington stock no. 1024). Individual pairs of the resulting w1118/y wa; kccDHS1/CyO virgin females and w1118/Dp(2:Y)bw+; kccDHS1/CyO (or w1118/Dp(2:Y)bw+; E(wa)/CyO) males were then crossed. For those crosses that yielded bang-sensitive progeny, the percent bang sensitivity of the duplication-bearing males was significantly higher than that of their female siblings, indicating that kcc is covered by Dp(2:Y)bw+.
We determined that kccDHS1 enhances sda, bss, and eas by comparing the percent bang sensitivity at 22°C of at least 60 of the indicated flies: (sda): kccDHS1/kccDHS1; sda/TM6B and kccDHS1/CyO; sda/TM6B D572 flies, as well as the +/CyO; sda/+ progeny of a cross of CS-5 females × D547 males; (bss): bss/+; kccDHS1/kccDHS1 and bss/+; kccDHS1/CyO female progeny of D506 females × bss; kccDHS1/CyO males as well as bss/+; +/CyO females progeny of a cross of CS-5 females × D508 males; and (eas): eas/+; kccDHS1/kccDHS1 and eas/+; kccDHS1/CyO female progeny of a cross of D506 females × D548 males as well as eas/+; +/CyO females progeny of a cross of CS-5 females × D548 males.
To show that Rdl1 suppressed kccDHS1, we crossed D506 virgin females to D569 males and compared the fraction of bang-sensitive flies among the resulting kccDHS1/kccDHS1; Rdl1/+ and kccDHS1/kccDHS1; +/TM6B progeny. We assayed suppression of eas, sda, or bss by Rdl1 by crossing D506 virgin females to MR047, MR063, or MR068 males, respectively. We then compared the fraction of BS flies among the resultant eas/+; Rdl1/+ versus eas; +/TM6B female progeny, Rdl1/sda versus sda/TM6B progeny, or bss; Rdl1/+ versus bss; +/TM6B female progeny, respectively. Similarly, to show that Gad1L352F suppressed kccDHS1, we crossed D506 virgin females to D582 males and compared the fraction of bang-sensitive flies among the resulting kccDHS1/kccDHS1; Gad1L352F/+ and kccDHS1/kccDHS1; +/TM6B progeny. We assayed suppression of eas or bss by Rdl1 by crossing D506 virgin females to MR047 or MR068 males, respectively, and compared the fraction of BS flies among the resultant eas/+; Rdl1/+ versus eas; +/TM6B or bss; Rdl1/+ versus bss; +/TM6B female progeny, respectively.
Picrotoxin feeding.
Homozygous kccDHS1/kccDHS1 flies from D506 bottles grown at 22°C were collected <24 h after eclosion. Flies were then placed in groups of 10 in vials containing three 2.4 cm Whatman GF/A filters saturated with 600 μl of 5% sucrose containing green food coloring with or without 1 mm picrotoxin (PTX) (Sigma, St. Louis, MO). The flies were allowed to ingest either the control or picrotoxin-containing sucrose for ∼24 h at 22°C and then tested for bang-sensitive paralysis. Green-colored abdomens indicated that the flies had ingested the corresponding picrotoxin-laden or control sucrose solution.
DNA molecular biology.
Genomic DNA was prepared from aliquots of ∼30 flies essentially as described in the Berkeley Drosophila Genome Project Methods (http://www.fruitfly.org/about/methods) except that the buffer used to resuspend the final DNA pellets contained 20 μg/ml RNase A. PCR amplifications were performed on approximately one fly-equivalent of DNA. The following PCR conditions were used: 94°C for 4 min, followed by 35 cycles of 94°C for 45 s, 55°C for 1 min, and 72°C for 2 min, and then one cycle of 72°C for 7 min. AmpliTaq DNA polymerase (Roche, Basel, Switzerland) was used according to the manufacturer's instructions. Twelve primer pairs, which lead to the production of 12 overlapping ∼600 bp products, were used to amplify almost the entire kcc gene from homozygous kccDHS1/kccDHS1 D506 flies. (The ∼1 kb intronic regions flanking each of the alternative exons were not examined.) Two primers, CG14-5 (5′-CAATTGTAGACCACCATTATGACGC) and Pwht1 (5′-GTAACGCTAATCACTCCGAACAGGTCACA), were used to amplify the kcc gene/P-element junction in homozygous kccML1/kccML1 D577 flies. CG14-5′ is in an alternative exon ∼1 kb from the 3′ end of kcc, whereas Pwht1 occurs in SUPor-P ∼10 kb downstream of the kcc gene. As anticipated, no CG14-5/Pwht1 PCR product was obtained using DNA from the original no. 13216 stock. However, a 1.5 kb CG14-5/Pwht1 PCR product was obtained using D577 DNA, indicating an imprecise excision that removed the 3′ portion of the P-element (the end proximal to the kcc gene). All PCR products were purified using the QIAquick PCR Purification kit (Qiagen, Valencia, CA) and sequenced at the University of California, Berkeley, DNA Sequencing Facility using the corresponding 5′ and 3′ PCR primers
RT-PCR analysis.
Homozygous kccDHS1 D506 and CS-5 control flies were reared at 22°C, tested for bang sensitivity at <2 d after eclosion, collected using CO2, and frozen at –80°C. Fly heads were detached by vigorous shaking of the frozen flies. For each strain, equivalent numbers of male and female heads were collected in the presence of liquid nitrogen, and total RNA was prepared using the Trizol Reagent (Invitrogen Life Technologies, San Diego, CA) according to the manufacturer's instructions. RT-PCR was performed using the Titan One Tube RT-PCR System (Roche) essentially according to the manufacturer's instructions. The kcc primers were kcc-15-5 (5′-CCAGAAGAGCATACCCATCG) and kcc-15-3 (5′-AATGATGGCCATTCCCATAG), and the Act79B primers were act-rtf2 (5′-ATGGAGAAGGTCTGGCACCA) and act-rtfb2 (5′-TCTGCTGGAAGGTGGACAAC). The kcc15-5 and 15-3 primers are located in the kcc exons (11 and 13, respectively) that span intron 11, where the 13 bp kccDHS1 insertion is located. The thermocycling program used was as follows: 50°C for 30 min; followed by 94°C for 2 min; and then 33 cycles of 94°C for 10 s, 50°C for 30 s, and 68°C for 45 s, with the extension time increased by 5 s/cycle starting with the ninth cycle; and finally 68°C for 7 min. To determine the linear range for both the kcc and control RT-PCRs, we varied the total RNA concentration from 10 ng to 1 μg and the number of PCR amplification cycles from 33 to 35. Ultimately, we found that RT-PCR using a total RNA concentration of 50 ng and 33 PCR cycles fell within the linear range, and these were the conditions used for the quantitation. An inverse image of the ethidium bromide-stained gel of the CS-5 and kccDHS1 RT-PCR products shown in Figure 7 was quantified using ImageJ 1.34 (http://rsb.info.nih.gov/ij). The relative kcc transcript levels in the test and control lanes were determined by normalizing the signal intensity of the kcc band to that of the corresponding Act79B band. A separate set of RT-PCRs was performed using 1 μg of each total RNA sample, the kcc (but not Act79B) primers and 35 PCR cycles. The resulting RT-PCR products were excised from a 0.9% agarose gel stained with ethidium bromide, purified using the QIAquick PCR Purification kit (Qiagen), and sequenced at the University of California, Berkeley, DNA Sequencing Facility using the kcc-15-5 and kcc-15-3 primers
Figure 7.

RT-PCR reveals a reduction in kcc transcript level in the kccDHS1 mutant. Shown are the results of a RT-PCR analysis of RNA prepared from the heads of kccDHS1 and control flies that used kcc primers located in exons that span the intron carrying the kccDHS1 insertion. Positions of both the kcc and Act79B control bands (at 0.6 and 0.82 kb, respectively) are indicated with arrows. The heads of kccDHS1 flies display a significant (2.3-fold) reduction in the level of kcc transcript. Sequence analysis of kccDHS1 and wild-type RT-PCR products revealed no alteration in the splicing of intron 11 in the kccDHS1 mutant (data not shown).
Western analysis.
For the initial Western blot (see Fig. 8), flies were reared at room temperature (∼24°C), collected using CO2, and frozen at –80°C. For the developmental Westerns (see Fig. 9), flies were reared at 22°C, collected using CO2 <4 h after eclosion, and returned to 22°C for 0–4 additional days before freezing at –80°C. Fly heads were detached by vigorous shaking of flies frozen at –80°C. For each strain, equal numbers of male and female heads were collected in the presence of liquid nitrogen and again stored at –80°C. Subsequently, extraction buffer (140 mm NaCl, 10 mm Tris-HCl, pH 7.5, 1 mm EDTA, 1% Triton X-100, and 100 μm PMSF) was added to each set of heads, and the heads were homogenized at 4°C using a Teflon pestle. The homogenates were microfuged at 4°C for 30 min, and the supernatants were collected. For each set of heads, the clarified homogenate from three fly-head equivalents was separated on a 7% SDS-PAGE gel and transferred to Hybond ECL nitrocellulose membrane (Amersham Biosciences, Piscataway, NJ) by standard methods (Sambrook et al., 1989). The kcc protein bands were visualized using the Protoblot Western Blot AP System (Promega, Madison, WI), according to the manufacturer's instructions with two modifications: the blocking step was done for 2 d at 4°C, and the incubation with the primary antibody was done overnight at 4°C. The primary antibody was a polyclonal rabbit antiserum, α-SA24 (Su et al., 1999), directed against a peptide (RGGGREVITIYS) found at the C terminus of mammalian KCC1, as well as Drosophila kcc (Payne et al., 1996; Filippov et al., 2003), and was diluted 1:5000. The secondary antibody, alkaline phosphatase-conjugated goat α-rabbit IgG (Promega), was diluted 1:7500. This same Western blot was subsequently reblocked and incubated with a mouse anti-α-tubulin monoclonal antibody (Sigma), diluted 1:5000, and then HRP-conjugated goat α-mouse IgG. The signal was visualized with 0.5 mg/ml DAB and 0.03% hydrogen peroxide. For the developmental Western shown in Figure 9 A, the initial Western blot was subsequently incubated with peroxidase-conjugated horse α-goat IgG and peroxidase-conjugated horse α-mouse IgG (both 1:1000 and obtained from Vector Laboratories, Burlingame, CA), and proteins were detected using the ECL system (Amersham Biosciences) according to the manufacturer's directions. Band signal intensities were quantified using ImageJ 1.34 (http://rsb.info.nih.gov/ij). We then obtained an approximation of the relative kcc protein levels in each lane by normalizing the signal intensity of the kcc band to that of the corresponding α-tubulin band.
Figure 8.

The kcc protein level is reduced in heads of kcc mutants. Shown is a Western blot of protein homogenates from the heads of wild-type (+/+) flies and various kcc mutants. The positions of both kcc and α-tubulin (α-tub) control bands are indicated by arrows. All of the strains produce an ∼125 kDa kcc protein. However, the heads of flies homozygous for the seizure-sensitive kccDHS1 mutation show a significant (∼4-fold) reduction in kcc protein. The heads of flies homozygous for the weaker kccML1 mutation also display a more modest (∼1.9-fold) reduction in kcc level. Flies heterozygous for the strong, lethal kccAd4 mutation show a threefold reduction in the level of kcc protein; those heterozygous for kccP20–180 show a more modest (1.6-fold) reduction in kcc protein. Flies carrying one copy of Df(2R)106, which removes the kcc gene, and one wild-type kcc allele (lane 6) display the expected twofold reduction in kcc level, suggesting that the protein levels used for quantitation are within the linear range.
Figure 9.

Both wild-type and kccDHS1 mutant flies display an age-dependent increase in kcc protein. A, Western analysis of protein extracts from kccDHS1 heads (lanes 2–6) reveals a progressive increase in kcc level in aging flies. After 4 d (lane 6), the level of kcc in kccDHS1 heads is ∼13-fold higher than that observed in the heads of newly eclosed flies (lane 2). Nonetheless, it is still significantly lower than that seen in 4-d-old wild-type flies (+; lane 1). B, Wild-type flies also show an increase (1.9-fold) in the level of kcc protein after the first 4 d of life. The loading control is again α-tubulin (α-tub).
Immunohistochemistry.
Heads from 4- to 5-d-old CS-5 flies reared at 22°C were manually dissected and frozen in OCT embedding medium (TissueTek). Microtome sections (8 μm) were collected on lysine-coated slides and fixed for 15 min in Histochoice (Electron Microscopy Sciences, Fort Washington, PA). Immunohistochemical staining was performed using the Vectastain ABC kit (Vector Laboratories) according to the manufacturer's directions except that both the blocking and the primary antibody incubation steps were done overnight at 4°C. The α-SA24 primary antibody (Su et al., 1999) was diluted 1:150.
Electrophysiology.
Homozygous kccDHS1/kccDHS1 and heterozygous kccDHS1/CyO flies from D506 bottles grown at 22°C were collected <24 h after eclosion for electrophysiological testing. Brain stimulation and recording of both giant fiber (GF)-driven muscle potentials and seizures was performed essentially as described previously (Kuebler and Tanouye, 2000) except that the flies were mounted using soft wax (Godenschwege et al., 2002). Both single-pulse stimuli and high frequency (HF) wave trains were delivered to the fly's brain using bipolar tungsten stimulating electrodes. Single-pulse stimuli (0.5 ms duration; 0.8 Hz) were used to drive the GF, and the GF-driven muscle potentials were recorded from the dorsal longitudinal muscles using tungsten recording electrodes. The GF threshold was considered to be the lowest voltage at which the GF pathway responded to a single pulse stimulus. The GF was stimulated continuously to assess circuit function during the course of each experiment, and flies were discarded if GF function appeared compromised. Seizures consist of HF activity in at least seven different muscle groups and over 30 muscle fibers in the thorax, and reflect the HF firing of the innervating motoneurons (Kuebler and Tanouye, 2000). We attempted to elicit seizures by delivering short wave trains of HF electrical stimuli (0.5 ms pulses delivered at 200 Hz for 300 ms) to the fly's brain. The lowest intensity HF stimulus required to elicit a seizure was designated the “seizure threshold.” The two-tailed t test was used to determine the p values for differences in seizure threshold between the kccDHS1/kccDHS1 flies and their kccDHS1/CyO sibling controls.
Results
A novel neurological mutant with seizure phenotypes called kazachoc (kcc)
A novel Drosophila neurological mutant was identified serendipitously in a screen for seizure-enhancer mutations (Zhang et al. 2002). Flies from a putative seizure-enhancer line called D505 carry a P-element insertion upstream of the CG9925 gene in cytological region 88A on the third chromosome (Zhang et al. 2002). Genetic analysis revealed that the seizure-enhancer phenotype of D505 flies resulted primarily from an unmarked mutation on the second chromosome rather than the P-element insertion. We named the novel second chromosomal mutation kazachoc (kccDHS1). The kccDHS1 mutation confers an incompletely penetrant BS paralytic phenotype, a behavioral indication of seizure sensitivity. At room temperature (23°C), 27% of homozygous kccDHS1/kccDHS1 flies display BS paralysis (Fig. 1). The incomplete penetrance of the kccDHS1/kccDHS1 BS phenotype appears to be a stochastic phenomenon: only 27% of flies that were BS after 1–2 d were still BS when retested the following day, and 9% of flies that were not BS when initially tested were subsequently found to be BS when retested the next day (data not shown). The recovery time for homozygous kccDHS1 flies is relatively short: 19.8 ± 5.9 s (14 s of paralysis followed by 5.8 s of recovery seizure). In contrast, the BS mutants sda, eas, and bss have significantly longer recovery times of 37, 52, and 198 s, respectively; they show 100% penetrance of the BS phenotype (Tan et al., 2004). Heterozygous kccDHS1/+ flies are not BS, indicating that the kccDHS1 mutation is recessive (Fig. 1). Heterozygotes carrying both kccDHS1 and a deletion (Δ) that uncovers it (described later) display 40% bang sensitivity (Fig. 1). Because the phenotype of the kccDHS1/Δ flies is more severe than that of the homozygous kccDHS1/kccDHS1 flies, the kccDHS1 mutation likely represents a partial loss-of-function (hypomorph).
Figure 1.

The kazachoc (kcc) mutation produces bang-sensitive behavioral phenotype. The kccDHS1 mutation produces an incompletely penetrant, recessive bang-sensitive paralytic phenotype. The phenotype is more penetrant if the flies are grown at a lower temperature, and if the temperature is dropped to 18°C, homozygous kccDHS1/kccDHS1 flies are inviable. Flies heterozygous for kccDHS1 and a deficiency, Df(2R)106, which completely removes the kcc gene, show a more severe phenotype than do the homozygous kccDHS1/kccDHS1 flies, indicating that the kccDHS1 mutation produces a partial loss of function. The “kcc/kcc” and “kcc/+” flies were kccDHS1/kccDHS1 and kccDHS1/CyO siblings, respectively, from our D506 stock (n > 200). The “kcc/Δ” flies were unbalanced kccDHS1/Df(2R)106 progeny produced by a cross of D506 virgin females to 5224 males (n > 80). At temperatures at which no unbalanced progeny were observed (“Dead”), >300 balanced progeny were counted.
The variable penetrance of the kccDHS1 seizure phenotype is especially sensitive to both rearing temperature and age. The kccDHS1 mutation is more severe at lowered temperatures (Fig. 1). Homozygous kccDHS1/kccDHS1 flies show a significant increase in the BS paralytic phenotype at 22°C compared with 23°C (44 vs 27% BS paralysis, respectively). At 18°C, homozygous kccDHS1/kccDHS1 flies are inviable. The kccDHS1/Δ flies display a more extreme temperature sensitivity; they are inviable at temperatures of 22°C and lower. This cold-sensitive lethality implies that kcc is an essential gene. At any given temperature, the BS paralysis produced by kccDHS1 is more pronounced in younger flies (Fig. 2). The fraction of kccDHS1 flies that display the BS phenotype decreases significantly after 2–3 d after eclosion and thereafter, and few flies exhibit BS paralysis.
Figure 2.

The kazachoc (kcc) mutation displays an age-dependent decline in bang sensitivity. The fraction of kccDHS1 flies that display the BS phenotype decreases after 2–3 d (from 38 to 22% BS) and then falls precipitously (to 3% BS) 1 d later. Homozygous kccDHS1 flies were collected <1 d after eclosion from a D506 population reared at 22°C. The kccDHS1 flies (n = 65) were then placed in food vials at 22°C and tested for bang-sensitive paralysis at ∼24 h intervals.
The kcc mutant displays a reduced seizure threshold
The bang-sensitive behavioral phenotype is a useful measure of seizure sensitivity in Drosophila: flies that show a strong BS phenotype also display significantly reduced seizure thresholds at the electrophysiological level, whereas mutations that decrease behavioral bang sensitivity raise the fly's seizure threshold (Pavlidis and Tanouye, 1995; Kuebler and Tanouye, 2000; Kuebler et al., 2001; Glasscock and Tanouye, 2005; Glasscock et al., 2005; Hekmat-Scafe et al., 2005; Song and Tanouye, 2006). Examination of seizure susceptibility showed that the seizure threshold of kccDHS1 flies is almost one-third that of wild-type and control genotypes. Thus, kccDHS1 shows a seizure threshold of 13.1 ± 4.67 V high-frequency stimulus (HFS), whereas their kccDHS1/+ sibling controls show a seizure threshold of 32.7 ± 0.23 V HFS, comparable with wild-type CS flies [34.3 ± 1.9 V HFS after J. S. Tan et al. (2004)]. In contrast, flies carrying the fully penetrant BS mutations bss, eas, or sda have seizure thresholds of (3.2 ± 0.6, 3.4 ± 0.5, and 6.2 ± 0.8 V HFS, respectively), 5- to 10-fold lower than wild-type (Kuebler et al., 2001). Figure 3 A shows a seizure recorded from the dorsal longitudinal muscle of a homozygous kccDHS1 fly in response to a 17 V HF stimulus. Abnormal HF muscle potentials (>100 Hz) are observed and reflect seizure activity of the single motoneuron innervating this muscle fiber (Kuebler and Tanouye, 2000). This seizure activity is similar in appearance and time course to those previously observed in other BS mutants (Pavlidis and Tanouye, 1995; Kuebler and Tanouye, 2000; Zhang et al., 2002). Figure 3 B shows a dorsal longitudinal muscle recording from a heterozygous kccDHS1/+ fly in response to a 30 V HF stimulus. In this instance, no seizure activity can be seen, indicating that this higher intensity HF stimulus was not of sufficient strength to elicit a seizure in the heterozygote.
Figure 3.

kcc reduces seizure threshold. A, A seizure is elicited in a homozygous kccDHS1/kccDHS1 fly by a high-frequency stimulus at its seizure threshold (17.0 ± 1.4 V). This seizure activity is similar in appearance and time course to those previously observed in other BS mutants (Pavlidis and Tanouye, 1995; Kuebler and Tanouye, 2000; Zhang et al., 2002). The HF stimulus is a short wave train (0.5 ms pulses at 200 Hz for 300 ms) of electrical stimuli delivered to the brain. The vertical calibration bar is 20 mV, and the horizontal bar is 200 ms. B, In contrast, a higher-intensity HF stimulus (30 V) fails to elicit a seizure in a kccDHS1/+ sibling control fly, because it is below the fly's seizure threshold (32.7 ± 0.23 V). The seizure threshold of the kccDHS1/+ control flies is comparable with the 34.3 V threshold of wild-type CS-5 flies (J. S. Tan et al., 2004), whereas the homozygous kccDHS1/kccDHS1 mutation produces almost a threefold reduction in seizure threshold.
The seizure susceptibility of a variety of seizure-prone flies is increased by kccDHS1
The kccDHS1 mutation was uncovered in a screen for enhancers of sda/+. Here, we examined more explicitly seizure-enhancer functions of kccDHS1 (Fig. 4). We showed that approximately one-half (51%) of doubly heterozygous kccDHS1/+; sda/+ flies are BS. Because the singly heterozygous sda/+ flies fail to show a BS phenotype and heterozygous kccDHS1/+ are also not BS, the indication is that there is a genetic interaction in the double heterozygous mutations that enhances the BS phenotype. We suggest that kccDHS1 acts as an enhancer of sda. A more extreme phenotype attributable to interaction is observed in kccDHS1/kccDHS1; sda/+ flies with 82% of flies showing BS phenotype. The mutations also display synthetic lethality: the double mutant combinations sda/sda; kccDHS1/+ and sda/sda; kccDHS1/kccDHS1 are inviable at temperatures ranging from 18 to 25°C (data not shown).
Figure 4.

The kcc mutation enhances a variety of other bang-sensitive mutations. Flies of the indicated different genotypes were examined for the bang-sensitive paralytic behavioral phenotype in response to a 10 s mechanical bang. The data reveal that the kccDHS1 mutation produces significant enhancement of the BS mutations sda, bss, and eas. The kccDHS1 mutation is recessive, and kcc/+ flies are normally not BS (Fig. 1). At 22°C, our sda, bss, and eas mutations are also recessive, and consequently sda/+, bss/+, and eas/+ flies are also not BS. However, the double heterozygotes of kcc with either sda or bss (kcc/+; sda/+ or bss/+; kcc/+) display significant bang sensitivity (51 and 48%, respectively). If the sda/+ or bss/+ heterozygotes are instead homozygous for the kccDHS1 mutation (kcc/kcc; sda/+ or bss/+; kcc/kcc), their bang sensitivity is even more pronounced (82 or 77%, respectively). Flies of the genotype eas/+; kcc/kcc show a level of bang sensitivity (64%) that is greater than that of flies carrying either the eas/+ or kcc/kcc mutations alone (0 or 44%, respectively), indicating moderate enhancement.
The kccDHS1 mutation also enhances other BS mutations including bang-senseless (bss) and easily shocked (eas) (Fig. 4). Both heterozygous and homozygous kccDHS1 mutations produce significant enhancement of bss. Whereas 0% of either heterozygotes kccDHS1/+ or bss/+ flies are BS, almost one-half (48%) of the double heterozygotes (bss/+; kccDHS1/+) are BS. An even greater fraction (77%) of bss/+; kccDHS1/kccDHS1 flies are BS. The kccDHS1 enhancement of the BS phenotype for eas is modest. None of the double heterozygotes (eas/+; kccDHS1/+) are BS. However, there is genetic interaction because 64% of eas/+; kccDHS1/kccDHS1 flies are BS (Fig. 4). This proportion is higher than that of kccDHS1/kccDHS1 flies at this temperature (40%) (Fig. 1). The percent bang sensitivity of homozygous kccDHS1/kccDHS1 flies is further increased by either the P-element insertion in CG9925 (identified in line J) (Zhang et al., 2002) and the jitterbug (jbug) mutation identified in the same screen (data not shown). The jbug gene encodes Drosophila filamin; mutations in human filamin are associated with periventricular heterotopia, which presents with epilepsy (X. Ren and M. A. Tanouye, personal communication) (Fox et al., 1998).
Mapping and identifying the kcc gene
The kcc gene resides in a 57 kb segment at region 60A near the distal tip of chromosome 2R as revealed by genetic mapping. Recombination mapping of the BS phenotype shows that kcc is located between brown (bw) at 104.5 m.u. and speck (sp) at 107 m.u.; the apparent position of kcc is 106.5 m.u. Duplication and deficiency mapping with a number of aneuploid chromosomes showed that kcc lies between the distal break point of Df(2R)b23, which fails to uncover kcc, and Df(2R)bwDra, which uncovers kcc. Superposition of our deficiency mapping with a molecular map of the region revealed that the kcc gene must map to a 57 kb region of chromosomal region 60A (Fig. 5 B).
Figure 5.
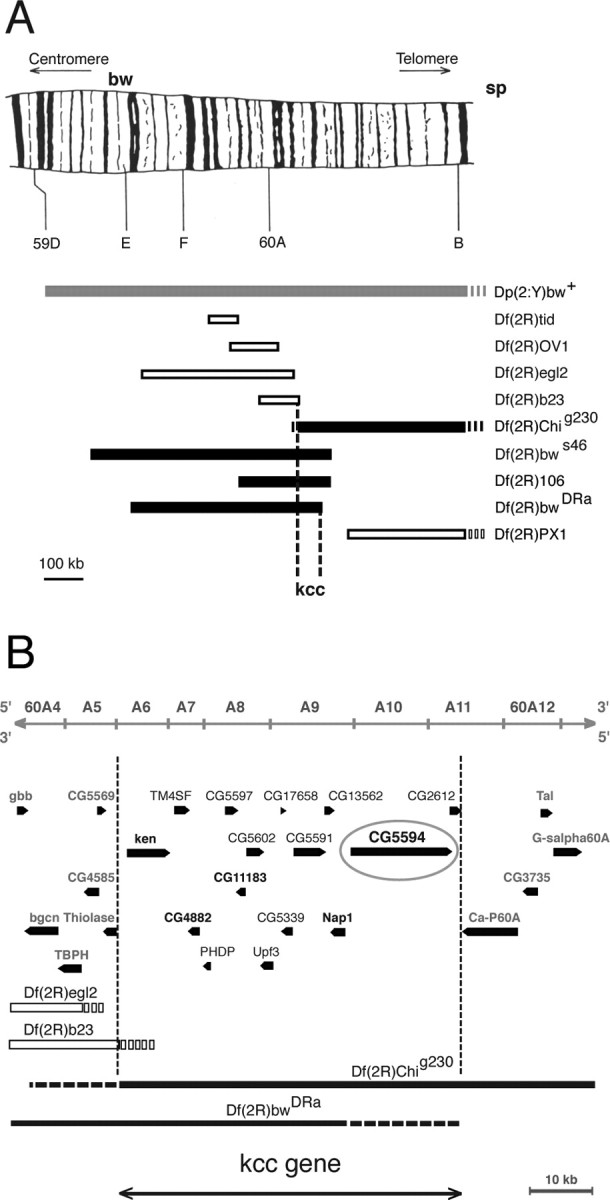
Genetic mapping of the kazachoc (kcc) gene. A, Initial recombination mapping experiments revealed that the kazachoc gene is located between the markers bw and sp on the distal tip of chromosome 2R. Subsequent duplication and deficiency mapping confirmed this result and further narrowed down the location of the kcc gene to a small cytological region (60A). A duplication of genomic DNA containing this region onto the Y chromosome (Dp(2;Y)bw+; 58F1-3; 60E11-12) produced a significant reduction in the bang sensitivity of homozygous kccDHS1 flies, indicating the presence of the kcc gene on the duplicated segment (gray box). Four of the deficiencies examined failed to complement kccDHS1 and therefore included the kcc gene (black boxes) (Df(2R)bwS46, Df(2R)Chig230, Df(2R)106, and Df(2R)Dra), whereas five others fully complemented kccDHS1 and therefore lacked the kcc gene (open boxes) (Df(2R)tid, Df(2R)OV1, Df(2R)egl2, Df(2R)b23, and Df(2R)PX1). The schematic drawing of chromosomal region 59D–60A has been reproduced with permission from Kurzik-Dumke et al. (1992). B, Superposition of the results of our deficiency mapping experiments onto a molecular map of the 60A cytological region (from Flybase) is shown. Deficiencies that uncover kcc are depicted as black boxes, and those that do not uncover kcc are depicted as white boxes; regions of uncertainty are shown as broken lines. The location of the kcc gene is bounded by the distal break points of Df(2R)b23, which does not uncover kcc, and those of Df(2R)bwDRa, which uncovers kcc. Hence, the kcc gene maps to a 57 kb region of 60A, which carries 15 genes. The distal break points shown for Df(2R)egl2 and Df(2R)b23, as well as the proximal break point for Df(2R)Chig230, are from Wharton et al. (1999). The position of the distal Df(2R)bwDRa break point takes into account the observations that this deficiency fails to uncover Ca-P60A (Periz and Fortini, 1999) but does uncover lethal mutations in CG4882, Nap1, or CG5594 (data not shown).
The DNA segment where kcc maps contains 15 genes (Fig. 5 B). Our focus for kcc candidates was on the five essential genes because of kcc lethal phenotypes. Three candidates (ken, CG4882, and Nap1) were eliminated by complementation analysis (data not shown). Of the remaining two candidates (CG5594 and CG11183), kcc was shown to correspond to CG5594. This identification was based on a series of mutations that all failed to complement in different heterozygote combinations, indicating that they are allelic (Table 2). These include the mutations kccDHS1, kccML1, CG5594EY08304, l(2)60A-CAd4, and l(2)60A-CP20–180. Molecular lesions for several of the mutations have been localized to the same transcription unit (Fig. 6). Subsequently, we will use a consolidated terminology that refers to kcc lethal alleles as kccEY08304, kccAd4, and kccP20–180.
Table 2.
Complementation tests reveal kcc is allelic to CG5594 and l(2)60A-C
| kccDHS1 | kccML1 | CG5594EY08304 | l(2)60A-CAd4 | l(2)60A-CP20–180 | + | |
|---|---|---|---|---|---|---|
| kccDHS1 | 44% | 13% | 42% | 34% | 12% | 0% |
| kccML1 | 8% | 41% | 15% | 0% | 0% | |
| CG5594EY08304 | Lethal | Lethal | Lethal | 0% | ||
| l(2)60A-Cad4 | Lethal | Lethal | 0% | |||
| l(2)60A-CP20–180 | Lethal | 0% | ||||
| + | 0% |
Heterozygous flies (n > 100) carrying the corresponding pair of mutations listed on the horizontal and vertical axes were examined for a bang-sensitive paralytic phenotype. The data reveal that mutations in kcc, CG5594, and l(2)60A-C fail to complement each other, indicating that they are alleles of the same gene. We propose calling this gene kazachoc (kcc) and renaming the CG5594EY08304, l(2)60A-CAd4, and l(2)60A-CP20–180 alleles kccEY08304, kccAd4, and kccP20–180, respectively.
Figure 6.

The kcc K+/Cl− cotransporter gene. Shown is a diagram of the kcc (CG5594) transcription unit. The gene is alternatively spliced at two points generating four possible transcripts. A database search revealed a large number of cDNAs corresponding to two of the alternative splice forms, A and B, in libraries prepared from Drosophila embryos or adult heads, respectively (data not shown). Positions of the kccEY08304, kccML1, and kccDHS1 mutations are indicated. The lethal kccEY08304 mutation results from a P-element insertion into the last exon of the CG5594 gene. The kccML1 mutation was produced by the imprecise excision of a nearby P(w+) element, which resulted in the deletion of 88 bp of genomic DNA. This deletion truncated the kcc 3′-UTR by 83 bp. The original kccDHS1 mutation carries a 13 bp insertion (ACTATGCTACTGT) after the seventh base pair in the 11th intron of the kcc gene. Note that our diagram of the kcc gene lacks the large 412 transposable element within the 13th intron included in the Flybase annotation of CG5594, because sequencing of the genomic PCR products revealed no 412 element in any of the strains we examined.
The kcc gene encodes a K+/Cl− cotransporter
The Drosophila kcc gene has been previously identified as a cation cotransporter closely related to the mammalian KCC2 cotransporter (Filippov et al., 2003). The kcc gene has four splice variants (A–D) (Drysdale et al., 2005) shown in Figure 6. The product of each of these splice variants has 12 predicted transmembrane domains, which is a canonical feature of vertebrate KCCs (Delpire and Mount, 2002), with which they share 53–59% amino acid identity. The major kcc splice variants appear to be B and D, which constitute the preponderance of cDNAs identified in a BLAST search (http://flybase.net/blast) of Drosophila expressed sequence tags (dbESTs). Our dbEST BLAST search indicated that the B variant is enriched in libraries prepared from adult heads, whereas the D variant is enriched in embryonic libraries. Proteins predicted for the A and C transcripts have a different N terminus than those predicted for the B and D transcripts. The N termini of KCCs are located intracellularly and apparently regulate cotransport (Delpire and Mount, 2002). The C and D variants have an additional exon not present in either the A or B forms. Consequently, the C and D products would carry an additional 31 aa in their C-terminal tails. The mammalian KCC2 differs from the other KCCs by carrying an additional ∼100 aa domain at approximately the same C-terminal region (Payne et al., 1996; Hebert et al., 2004).
Sequence analysis identified molecular lesions within the kcc gene associated with kccEY08304, kccML1, and kccDHS1 mutants (Fig. 6). The lethal kccEY08304 allele results from a P-element insertion within the last exon of the kcc gene causing a truncation of 47 C-terminal amino acid residues. In the mammalian ortholog KCC1, C-terminal truncation is known to completely abolish function (Casula et al., 2001). The kccEY08304 product also lacks a conserved tyrosine known to be essential in KCC2, KCC1, and KCC4 (Strange et al., 2000). The kccML1 allele is a deletion that removes 83 bp of 3′-UTR from the last kcc exon. The kccDHS1 mutation is a 13 bp insertion (ACTATGCTACTGT) after the seventh base pair in intron 11 of the kcc gene. Analysis of kcc RT-PCR products revealed that splicing of intron 11 is unaltered, but there is a 2.3-fold reduction in kcc transcript levels in the heads of kccDHS1 mutants relative to those of wild-type controls (Fig. 7).
kcc protein is reduced in kcc mutants
Western blot analysis showed that kcc protein is reduced in the heads of both kccDHS1 and kccML1 flies (Fig. 8). Wild-type flies show a prominent band of apparently 125 kDa, slightly larger than the expected size of 114–119 kDa. This size difference may reflect posttranslational modification of the kcc protein: mammalian KCCs are extensively glycosylated and phosphorylated (Payne et al., 1996; Mount et al., 1998, 1999; Su et al., 1999). An estimate of the relative kcc levels shows that compared with wild-type flies, kcc protein is reduced ∼4-fold and 1.9-fold in the kccDHS1 and kccML1 mutants, respectively. Heterozygotes carrying one copy of the lethal kccAd4 allele and one wild-type allele also show a threefold reduction in kcc protein relative to wild-type flies.
The level of kcc protein in the heads of kccDHS1 flies progressively increases with developmental age (Fig. 9 A). By 4 d after eclosion, the level of kcc in the heads of kccDHS1 flies has risen ∼13-fold. This rise in kcc level likely explains the progressive decrease in bang sensitivity of the kccDHS1 mutant (Fig. 2). Wild-type flies also display a developmental increase in kcc level, which nearly doubles in the fly's first 4 d (Fig. 9 B).
Immunohistochemical staining of Drosophila heads revealed that kcc is widely expressed in brain neuropil (Fig. 10). This pattern is consistent with kcc expression in neuronal dendrites and/or axons, although we cannot exclude the possibility of additional glial expression. Intense and specific immunostaining was observed in the protocerebrum, deutocerebrum, central brain (protocerebral bridge, ellipsoid body, and fan-shaped body), antennal lobe, and optic lobe (lamina, medulla, lobula, and lobula plate). In contrast to the strong immunostaining observed in most other neuropil regions, there was little or no kcc immunostaining in the mushroom body (Fig. 10 A,B), a structure involved in learning and memory (Heisenberg, 2003). The kcc expression pattern in brain neuropil is strikingly similar to that observed previously for the Rdl GABAA receptor except that Rdl also displays strong expression in mushroom body (Aronstein and ffrench-Constant, 1995; Harrison et al., 1996).
Figure 10.

Immunohistochemical staining reveals kcc expression in brain neuropil. A, In this frontal section, kcc immunostaining is observed in much of the brain neuropil regions including the protocerebrum (pr) and deutocerebrum (deu), as well as the ellipsoid body (eb) and fan-shaped body (fb) of the central brain. In contrast, little staining is observed in the mushroom body peduncle (pd). B, This frontal section shows strong kcc immunostaining in the protocerebral bridge (pb) of the central brain. A low, but discernible, level of staining is observed in the mushroom body calyx (ca), particularly in the upper, dendritic region. C, This horizontal section reveals that kcc is expressed in all four optic neuropils of the optic lobe: lamina (l), medulla (m), lobula (lob), and lobula plate (lp), as well as the optic chiasma connecting the lamina and medulla. Distinct neuronal layers of immunostaining are particularly evident in the medulla. Magnification, 125× in all sections.
kcc-mediated seizure susceptibility requires GABAergic signaling
The kcc seizure phenotype depends on signaling via the GABAA receptor suggesting that dysfunction is attributable to a disruption of the normal Cl− gradients that underlie GABAergic inhibition in the CNS. GABAA receptor involvement was initially tested using PTX. PTX is a GABAA receptor blocker and also a convulsant drug in mammals (Usunoff et al., 1969). PTX is also found here to act as a convulsant in wild-type Drosophila. Feeding wild-type flies PTX (1 mm) produces a low level of seizure susceptibility (1% BS) (Fig. 11 A). This dose of PTX is ultimately lethal for wild-type flies with death occurring in 1.5 d (t 1/2). Feeding PTX to kcc flies produces a surprising result: PTX acts as an anticonvulsant rather than a convulsant. PTX (1 mm) reduces the level of BS paralysis of kcc flies: 16% of PTX-fed kcc flies are BS, whereas 38% of control-fed kcc siblings are BS. PTX feeding is less toxic for kcc flies than for their wild-type counterparts: kcc flies die with a longer t 1/2 of 3.5 d (data not shown). Thus, a PTX block of GABAA receptors has differential effects on wild-type and kcc flies. In wild-type animals, partial loss of GABAA receptor function acts to enhance the seizure phenotype, whereas in kcc, partial loss of GABAA receptor function acts to suppress seizures.
Figure 11.

Reduced levels of GABAA receptor suppress the kcc mutation. A, Feeding wild-type (WT) flies (n = 120) the convulsant PTX, which is a competitive inhibitor of the GABAA receptor, produces a low-level bang sensitivity (1% BS). In contrast, PTX-fed kccDHS1 flies (n = 74) displayed a significant reduction in bang sensitivity relative to their sibling controls (n = 65). Whereas 38% of control kcc flies were bang sensitive, only 16% of their PTX-fed siblings were bang sensitive (p < 0.05). B, The bang sensitivity of kccDHS1 flies is suppressed in Rdl11/+ flies, which carry one null allele of the Rdl GABAA receptor gene. Whereas 26% of control kccDHS1 flies (n = 231) were bang sensitive, only 1% of their kccDHS1; Rdl1/+ siblings (n = 301) were BS (p < 0.05).
GABAA receptor function was also reduced genetically using a Resistance to dieldrin (Rdl1) mutation. Rdl encodes a Drosophila GABAA receptor that is sensitive to block by organophosphate toxins such as dieldrin and to picrotoxin (Zhang et al., 1995; Hosie et al., 1997). Seizure sensitivity of kcc flies is significantly suppressed when the dosage of the Rdl GABAA receptor gene is reduced twofold by the null Rdl1 mutation (Fig. 11 B). Here, 26% of homozygous kccDHS1/kccDHS1 flies are BS. However, only ∼1% of their kccDHS1/kccDHS1; Rdl1/+ siblings show the BS phenotype (p < 0.001). Reducing the dosage of the GABA biosynthetic enzyme glutamic acid decarboxylase via the Gad1L352F null mutation similarly reduces the seizure sensitivity of kcc flies (Table 3). Whereas 28% of homozygous kccDHS1/kccDHS1 flies are BS, only 3% of their kccDHS1/kccDHS1; Gad1L352F/+ siblings display the BS phenotype (p < 0.001). We observed that a decreased dosage of the Rdl GABAA receptor also produces modest reductions in the seizure susceptibility of heterozygous bss/+ and sda/+ flies and that bss/+ is partially suppressed by the Gad1 mutation (Table 3).
Table 3.
Reduced dosage of GABAA receptor gene Rdl or GABA biosynthetic gene Gad1 suppresses kccDHS1 and some other BS mutations
| Genotype | % Bang sensitivity |
p value | |||
|---|---|---|---|---|---|
| Test | (n) | Control | (n) | ||
| kccDHS1; Rdl1/+ | 1 | (301) | 26 | (231) | 3.7 × 10−19 |
| bss1/+; Rdl1/+ | 53 | (146) | 78 | (137) | 1.0 × 10−5 |
| sdaiso7.8/+; Rdl1/+ | 1 | (144) | 2 | (80) | 1.9 × 10−9 |
| easPC20/+; Rdl1/+ | 0 | (104) | 0 | (94) | |
| kccDHS1; Gad1L352F/+ | 3 | (72) | 28 | (47) | 1.0 × 10−4 |
| bss1 f/+; Gad1L352F/+ | 19 | (145) | 53 | (110) | 1.9 × 10−7 |
| easPC20/+; Gad1L352F/+ | 0 | (85) | 0 | (49) | |
Genotypes for Rdl1/+ and Gad1L352F/+ test flies (labeled “Test”) are indicated; control siblings (labeled “Control”) carried the TM6B balancer chromosome rather than the Rdl1 null allele of Rdl. Results are highly significant (p < 0.001) for all BS genotypes listed, except eas/+. Rdl1/+ produced slight (4%) suppression of hemizygous bss (p = 0.06).
Discussion
We find that Drosophila kcc null mutations cause recessive lethal phenotypes, indicating that kcc is an essential gene. Partial loss-of-function kcc mutations cause seizure-sensitive phenotypes and act as potent seizure-enhancer mutations. Seizure sensitivity of kcc flies appears to depend on transmission by the ionotropic GABAA receptor, the PTX-sensitive product of the Rdl gene. These observations confirm and extend recent investigations of mammalian KCC2 in mouse and humans. Reduced KCC2 function has also been found to cause lethality (null mutation) and epilepsy (partial loss-of-function mutations) (Hübner et al., 2001; Woo et al., 2002; Tornberg et al., 2005). Thus, the results in the present paper provide a link between human epilepsy and Drosophila and mouse models of seizure disorder. Furthermore, the present investigation suggests that seizure disorders in kcc and KCC2 mutants result from a dysfunction in GABAergic inhibition. We suggest that epilepsy phenotypes, in many instances, might be traced back to a perturbation of the biology underlying the ontogenetic switch from excitatory to inhibitory GABAergic signaling.
Drosophila kcc mutations resemble those described for mouse KCC2. KCC2 is an essential gene that causes lethal phenotypes in knock-out mutants (Hübner et al., 2001). Seizure phenotypes are observed in mouse KCC2 knock-down mutants that reduce the normal level of neuronal K+/Cl− cotransporter (Woo et al., 2002; Tornberg et al., 2005). In the more severe knock-down mutant (5% normal KCC2 level), generalized seizures are frequently induced by the mild mechanical stimulation that occurs during handling (Woo et al., 2002). Hippocampal slices from heterozygous KCC2 disruption mice display a twofold increase in seizure susceptibility (Woo et al., 2002). The level of KCC2 is known to decrease after brain trauma in vertebrate models of epilepsy, which may explain why head trauma frequently produces seizures in humans (Payne et al., 2003). For example, reduced KCC2 protein level is seen after focal cerebral ischemia–exotoxicity (Thomas-Crusells et al., 2000). The resulting increase in intracellular Cl− is associated with depolarizing GABAA receptor-mediated responses after brain trauma (Katchman et al., 1994; van den Pol et al., 1996; Fukuda et al., 1998; Nabekura et al., 2002). Excitatory GABAergic transmission is also found in the mature hippocampus, a plastic structure which is also a frequent location for epileptic foci (Leinekugel et al., 1995; Obrietan and van den Pol, 1995; Khazipov et al., 1997; Fujiwara-Tsukamoto et al., 2003).
The seizure susceptibility of the Drosophila kcc mutants shows a marked age and temperature dependence (Figs. 1, 2). Mutant kccDHS1 flies become progressively less bang sensitive with increasing age (Fig. 2). A plausible explanation for this observation is the concomitant increase in the level of kcc protein that occurs as the flies age (Fig. 9 A). This may recapitulate the ontogenetic switch from excitatory to inhibitory GABAergic signaling that occurs during mammalian development (Miles, 1999; Ben-Ari, 2002). It is currently unclear why the kcc phenotype becomes more pronounced as the developmental temperature is reduced (Fig. 1). This may reflect the fact that Drosophila resting potentials become progressively more depolarized as the temperature decreases (Hosler et al., 2000). However, virtually all neural processes are temperature sensitive (Montgomery and MacDonald, 1990). It would be of interest to examine whether the mouse KCC2 mutants (Woo et al., 2002; Tornberg et al., 2005) display similar age- and temperature-dependent alterations in seizure susceptibility.
Pharmacological agents that act as convulsants increase overall CNS excitability by blocking synaptic inhibition or by promoting excess excitability (LaRoche and Helmers, 2004). Conversely, anticonvulsants work by decreasing excitation or by promoting inhibition (Woodbury, 1980). An unexpected finding in this study is that PTX, a potent GABAA receptor blocker, can act as either a convulsant or an anticonvulsant in Drosophila depending on the genetic background (Fig. 11). As expected, in wild-type Drosophila, PTX acts as a convulsant. We expect that PTX interferes with GABAergic synaptic inhibition by blocking GABAA receptors, thereby promoting overall CNS excitability (Usunoff et al., 1969; Zhang et al., 1995). The surprising finding is that PTX acts as an anticonvulsant to suppress seizures in a kcc genetic background. At present, the anticonvulsant properties of PTX are difficult to explain. We assume that PTX continues to act by blocking GABAA receptors. We assume further that anticonvulsant properties reflect an overall decrease in CNS excitability. An attractive hypothesis is that, in kcc mutants, GABAergic transmission is mostly excitatory. This excitatory GABAergic transmission could act to promote seizure sensitivity and enhance the seizure susceptibility of other seizure-sensitive mutations. If so, then PTX could act as an anticonvulsant by blocking GABAA receptors and thereby reducing overall CNS excitability. Taken alone, our PTX results alone must be interpreted with some caution, because electrophysiological recordings of wild-type flies after short-term PTX feeding suggest that PTX may induce an atypical seizure-like state characterized by periodic bursts rather than stereotypical electrophysiological seizures (Lee and Wu, 2002). However, consistent with our hypothesis that PTX acts by reducing GABAergic excitation in our kcc mutants, we observed that a genetic reduction of GABAA receptor also suppressed kcc seizures (Fig. 11). Additional experimental evidence will depend on the development of a good electrophysiological preparation in Drosophila for examining synaptic inhibition; such a preparation is currently lacking. However, such paradoxical seizure suppression after application of a GABAA receptor antagonist has been observed previously in brain slices from patients with temporal lobe epilepsy as well as in rodent models of epilepsy in which excitatory GABAergic signaling is believed to underlie seizure activity (Cohen et al., 2002; Shinnar and Glauser, 2002; Baulac et al., 2004).
An especially challenging problem in neurobiology is to determine how genetic and environmental factors interact to cause the expression of seizure phenotypes in epilepsy. The major difficulties are a combination of polygenic inheritance and incomplete penetrance: inheritance of particular combinations of mutations may predispose, but not cause epilepsy in a given individual (Noebels, 2003). For example, recent interest has focused on the genetics of juvenile myoclonic epilepsy (JME), an idiopathic generalized epilepsy characterized by myoclonic jerks and generalized tonic-clonic seizures (Zifkin et al., 2005). JME, like most idiopathic generalized epilepsies, displays multifactorial inheritance that reflects the additive effects of multiple susceptibility genes interacting with environmental factors to produce the final phenotype (Berkovic et al., 1998). Polymorphisms in a number of genes are suspected to predispose individuals to JME, including GABRA1, the α1 subunit of the GABAA receptor (Wallace et al., 2001; Cossette et al., 2002), CICN2, a voltage-gated Cl− channel primarily expressed in cerebral neurons inhibited by GABA (D'Agostino et al., 2004), and the KCNQ3 K+ channel (Vijai et al., 2003). However, even in these cases of apparent monogenic inheritance, the phenotypic variation between families and even family members suggests that modifying genes and environmental factors interact with the predisposing mutations to produce JME, as well as other forms of idiopathic generalized epilepsy (Zifkin et al., 2005). Although genetic interactions appear to play a central role in several idiopathic generalized epilepsies, there are surprisingly few experimental observations providing confirmation (N. C. K. Tan et al., 2004). The Drosophila kcc mutation appears to provide an excellent model in which to investigate the ways interacting genes contribute to seizure disorder. The kcc seizure-enhancer mutation may be used to facilitate the identification of genes that contribute to seizure phenotypes via double mutant analysis. Contributing genes may then be separated from kcc and basal phenotypes examined to determine the extent to which phenotypes are dependent on interactions. In addition, the kcc mutant provides a model for how nongenetic factors such as age and temperature contribute to overall phenotypic expression of the seizure phenotype.
Our results suggest that seizure susceptibility in Drosophila is determined, in large part, by disruption of GABAergic signaling and, in particular, the kcc GABA switch. Genetic alterations that perturb the GABAergic pathway ameliorate the seizure susceptibility conferred by a variety of BS mutations (Table 3). The partial loss-of-function kccDHS1 and kccML1 mutations, which reduce the level of the kcc GABA switch, increase seizure sensitivity. Indeed, the degree of seizure sensitivity increases as the kcc level falls (Figs. 2, 8, 9 A; Table 2). Both Rdl and kcc are expressed in similar regions of brain neuropil with the exception of mushroom body, where only low levels of kcc are found (Fig. 10). Consequently, the mushroom body could be particularly sensitive to a reduction in kcc level, resulting in GABAergic excitation. Mutations that reduce kcc function might increase seizure susceptibility, particularly if the level of kcc is already low. This may explain why kccDHS1 acts as a seizure enhancer for a large variety of other BS mutations (Fig. 4). The BS mutations that interact genetically with both kcc and Rdl could reduce kcc function by any of number of molecular or cell biological processes including the following: inefficient transport activity because of abnormalities in phospholipid environment (i.e., loss of eas function), mislocalization because of deficiencies in cytoskeletal scaffolding (i.e., loss of jbug filamin function), destabilization of the extracellular environment of the synapse (i.e., loss of the sda aminopeptidase), transcriptional, translational, or posttranslational effects on molecules affecting kcc expression levels or subcellular localization. Likewise, seizure resistance and seizure suppression could presumably be attributable to factors that increase kcc function, thereby decreasing intracellular Cl−. This could have the net effect of acting to sharpen or refine inhibitory GABAA function in the nervous system. For these reasons, we suggest that modulation of GABAergic signaling by kcc could be central to both seizure sensitivity and seizure resistance.
Footnotes
This work was supported by grants from the Epilepsy Foundation and the National Institute of Neurological Disorders and Stroke (NS-31231) and by an undergraduate research fellowship to M.Y.L. from the University of California, Berkeley, Biology Fellows Program, which is funded by the Howard Hughes Medical Institute. We are most grateful to Kristi Wharton (Brown University, Providence, RI), Goran Periz, and Mark Fortini (National Cancer Institute, Frederick, MD), Dirk Lankenau (University of Heidelberg, Heidelberg, Germany), Steven Hou (National Institutes of Health, Bethesda, MD), Gary Landis (University of Southern California, Los Angeles, CA), Todd Laverty (University of California, Berkeley, CA), and the Bloomington Drosophila Stock Center for Drosophila stocks, and Seth Alper (Harvard Medical School, Boston, MA) for his generous gift of anti-KCC1 antiserum. We gratefully acknowledge Pooja Kadakia, who performed a preliminary kcc recombination mapping experiment, and Ann Way-Na Lee, who helped collect Drosophila heads for the developmental Western blots. Steve de Belle (University of Nevada, Las Vegas, NV) and Pavel Masek (University of California, Berkeley, CA) generously shared their neuroanatomical expertise, and William Leiserson (Yale University, New Haven, CT) provided helpful comments on this manuscript.
References
- Aronstein, ffrench-Constant R. Immunocytochemistry of a novel GABA receptor subunit Rdl in Drosophila melanogaster. Invert Neurosci. 1995;1:25–31. doi: 10.1007/BF02331829. [DOI] [PubMed] [Google Scholar]
- Ashburner M. Drosophila: a laboratory handbook. Cold Spring Harbor, NY: Cold Spring Harbor; 1989. [Google Scholar]
- Baulac S, Gourfinkel-An I, Nabbout R, Huberfeld G, Serratosa J, Leguern E. Fever, genes, and epilepsy. Lancet. 2004;3:421–430. doi: 10.1016/S1474-4422(04)00808-7. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y. Excitatory actions of GABA during development: the nature of the nurture. Nature. 2002;3:728–739. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
- Berkovic SF, Howell RA, Day DA, Hopper JL. Epilepsies in twins: genetics of the major epilepsy syndromes. Ann Neurol. 1998;43:435–445. doi: 10.1002/ana.410430405. [DOI] [PubMed] [Google Scholar]
- Buchner E, Bader R, Buchner S, Cox J, Emson PC, Flory E, Heizmann CW, Hemm S, Hofbauer A, Oertel WH. Cell-specific immuno-probes for the brain of normal and mutant Drosophila melanogaster. Cell Tissue Res. 1988;253:357–370. doi: 10.1007/BF00222292. [DOI] [PubMed] [Google Scholar]
- Buckingham SD, Biggin PC, Sattelle BM, Brown LA, Sattelle DB. Insect GABA receptors: splicing, editing, and targeting by antiparasitics and insecticides. Mol Pharmacol. 2005;68:942–951. doi: 10.1124/mol.105.015313. [DOI] [PubMed] [Google Scholar]
- Casula S, Shmukler BE, Wilhelm S, Stuart-Tilley AK, Su W, Chernova MN, Brugnara C, Alper SL. A dominant negative mutant of the KCC1 K-Cl cotransporter: both N- and C-terminal cytoplasmic domains are required for K-Cl cotransport activity. J Biol Chem. 2001;276:41870–41878. doi: 10.1074/jbc.M107155200. [DOI] [PubMed] [Google Scholar]
- Chen G, Trombley PQ, van den Pol AN. Excitatory actions of GABA in developing rat hypothalmic neurones. J Physiol (Lond) 1996;494:451–464. doi: 10.1113/jphysiol.1996.sp021505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen I, Navarro V, Clemenceau S, Baulac M, Miles R. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science. 2002;298:1418–1421. doi: 10.1126/science.1076510. [DOI] [PubMed] [Google Scholar]
- Cossette P, Liu L, Brisebois K, Dong H, Lortie A, Vanasse M, Saint-Hilaire JM, Carmant L, Verner A, Lu WY, Wang YT, Rouleau GA. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat Genet. 2002;31:184–189. doi: 10.1038/ng885. [DOI] [PubMed] [Google Scholar]
- D'Agostino D, Bertelli M, Gallo S, Cecchin S, Albiero E, Garofalo PG, Gambardella A, St Hilaire JM, Kwiecinski H, Andermann E, Pandolfo M. Mutations and polymorphisms of the CICN2 gene in idiopathic epilepsy. Neurology. 2004;63:1500–1502. doi: 10.1212/01.wnl.0000142093.94998.1a. [DOI] [PubMed] [Google Scholar]
- Delpire E, Mount DB. Human and murine phenotypes associated with defects in cation-chloride cotransport. Annu Rev Physiol. 2002;64:803–843. doi: 10.1146/annurev.physiol.64.081501.155847. [DOI] [PubMed] [Google Scholar]
- Drysdale RA, Crosby MA, Consortium TF. FlyBase: genes and gene models. Nucleic Acids Res. 2005;33:D390–D395. doi: 10.1093/nar/gki046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippov V, Aimanova K, Gill SS. Expression of an Aedes aegypti cation-chloride cotransporter and its Drosophila homologoues. Insect Mol Biol. 2003;12:319–331. doi: 10.1046/j.1365-2583.2003.00415.x. [DOI] [PubMed] [Google Scholar]
- Fox JW, Lamperti ED, Eksioglu YZ, Hong SE, Feng Y, Graham DA, Scheffer IE, Dobyns WB, Hirsch BA, Radtke RA, Berkovic SF, Huttenlocher PR, Walsh CA. Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron. 1998;21:1315–1325. doi: 10.1016/s0896-6273(00)80651-0. [DOI] [PubMed] [Google Scholar]
- Fujiwara-Tsukamoto Y, Isomura Y, Nambu A, Takada M. Excitatory GABA input directly drives seizure-like rhythmic synchronization in mature hippocampal CA1 pyramidal cells. Neuroscience. 2003;119:265–275. doi: 10.1016/s0306-4522(03)00102-7. [DOI] [PubMed] [Google Scholar]
- Fukuda A, Muramatsu K, Okabe A, Shimano Y, Hida H, Fujimoto I, Nishino H. Changes in intracellular Ca2+ induced by GABAA receptor activation and reduction in Cl− gradient in neonatal rat neocortex. J Neurophysiol. 1998;79:439–446. doi: 10.1152/jn.1998.79.1.439. [DOI] [PubMed] [Google Scholar]
- Ganetzky B, Wu C-F. Indirect suppression involving behavioral mutants with altered nerve excitability in Drosophila melanogaster. Genetics. 1982;100:597–614. doi: 10.1093/genetics/100.4.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasscock E, Tanouye MA. Drosophila couch potato mutants exhibit complex neurological abnormalities including epilepsy phenotypes. Genetics. 2005;169:2137–2149. doi: 10.1534/genetics.104.028357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasscock E, Singhania A, Tanouye MA. The mei-P26 gene encodes a RING finger B-box coiled-coil-NHL protein that regulates seizure susceptibility in Drosophila. Genetics. 2005;170:1677–1689. doi: 10.1534/genetics.105.043174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godenschwege TA, Simpson JH, Shan X, Bashaw GJ, Goodman CS, Murphey RK. Ectopic expression in the giant fiber system of Drosophila reveals distinct roles for roundabout (Robo), Robo2, and Robo3 in dendritic guidance and synaptic connectivity. J Neurosci. 2002;22:3117–3129. doi: 10.1523/JNEUROSCI.22-08-03117.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison JB, Chen HH, Sattelle E, Barker PJ, Huskisson NS, Rauh JJ, Bai D, Sattelle DB. Immunocytochemical mapping of a C-terminus anti-peptide antibody to the GABA receptor subunit, RDL in the nervous system of Drosophila melanogaster. Cell Tissue Res. 1996;284:269–278. doi: 10.1007/s004410050587. [DOI] [PubMed] [Google Scholar]
- Hebert SC, Mount DB, Gamba G. Molecular physiology of cation-coupled Cl− cotransport: the SLC12 family. Pflügers Arch. 2004;447:580–593. doi: 10.1007/s00424-003-1066-3. [DOI] [PubMed] [Google Scholar]
- Hekmat-Scafe DS, Dang KN, Tanouye MA. Seizure suppression by gain-of-function escargot mutations. Genetics. 2005;169:1477–1493. doi: 10.1534/genetics.104.036558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heisenberg M. Mushroom body memoir: from maps to models. Nat Rev Neurosci. 2003;4:266–275. doi: 10.1038/nrn1074. [DOI] [PubMed] [Google Scholar]
- Hosie AM, Aronstein K, Sattelle DB, ffrench-Constant RH. Molecular biology of insect neuronal GABA receptors. Trends Neurosci. 1997;20:578–583. doi: 10.1016/s0166-2236(97)01127-2. [DOI] [PubMed] [Google Scholar]
- Hosler JS, Burns JE, Esch HE. Flight muscle resting potential and species-specific differences in chill-coma. J Insect Physiol. 2000;46:621–627. doi: 10.1016/s0022-1910(99)00148-1. [DOI] [PubMed] [Google Scholar]
- Hübner CA, Stein V, Hermans-Borgmeyer I, Meyer T, Ballanyi K, Jentsch TJ. Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron. 2001;30:515–524. doi: 10.1016/s0896-6273(01)00297-5. [DOI] [PubMed] [Google Scholar]
- Jackson FR, Newby LM, Kulkarni SJ. Drosophila GABAergic systems: sequence and expression of glutamic acid decarboxlyase. J Neurochem. 1990;54:1068–1078. doi: 10.1111/j.1471-4159.1990.tb02359.x. [DOI] [PubMed] [Google Scholar]
- Katchman AN, Vicini S, Hershkowitz N. Mechanism of early anoxia-induced suppression of the GABAA-mediated inhibitory postsynaptic current. J Neurophysiol. 1994;71:1128–1138. doi: 10.1152/jn.1994.71.3.1128. [DOI] [PubMed] [Google Scholar]
- Khazipov R, Leinekugel X, Khalilov I, Gaiarsa JL, Ben-Ari Y. Synchronization of GABAergic interneuronal network in CA3 subfield of neonatal rat hippocampal slices. J Physiol (Lond) 1997;498:763–772. doi: 10.1113/jphysiol.1997.sp021900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhling R, Lücke A, Straub H, Speckmann E-J, Tuxhorn I, Wolf P, Pannek H, Oppel F. Spontaneous sharp waves in human neocortical slices excised from epileptic patients. Brain. 1998;121:1073–1087. doi: 10.1093/brain/121.6.1073. [DOI] [PubMed] [Google Scholar]
- Kuebler D, Tanouye MA. Modifications of seizure susceptibility in Drosophila. J Neurophysiol. 2000;83:998–1009. doi: 10.1152/jn.2000.83.2.998. [DOI] [PubMed] [Google Scholar]
- Kuebler D, Zhang H, Ren X, Tanouye MA. Genetic suppression of seizure susceptibility in Drosophila. J Neurophysiol. 2001;86:1211–1225. doi: 10.1152/jn.2001.86.3.1211. [DOI] [PubMed] [Google Scholar]
- Kurzik-Dumke U, Phannavong B, Gundacker D, Gateff E. Genetic, cytogenetic and developmental analysis of the Drosophila melanogaster tumor suppressor gene lethal(2)tumorous imaginal discs (l(2)tid) Differentiation. 1992;51:91–104. doi: 10.1111/j.1432-0436.1992.tb00685.x. [DOI] [PubMed] [Google Scholar]
- Landis G, Kelley R, Spradling AC, Tower J. The k43 gene, required for chorion gene amplification and diploid cell chromosome replication, encodes the Drosophila homolog of yeast origin recognition complex subunit 2. Proc Natl Acad Sci USA. 1997;94:3888–3892. doi: 10.1073/pnas.94.8.3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lankenau S, Barnickel T, Marhold J, Lyko F, Mechler BM, Lankenau DH. Knockout targeting of the Drosophila nap1 gene and examination of DNA repair tracts in the recombination products. Genetics. 2003;163:611–623. doi: 10.1093/genetics/163.2.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRoche SM, Helmers SL. The new antiepileptic drugs. JAMA. 2004;291:605–614. doi: 10.1001/jama.291.5.605. [DOI] [PubMed] [Google Scholar]
- Lee D, Su H, O'Dowd DK. GABA receptors containing Rdl subunits mediate fast inhibitory synaptic transmission in Drosophila neurons. J Neurosci. 2003;23:4625–4634. doi: 10.1523/JNEUROSCI.23-11-04625.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Chen CX-Q, Liu Y-J, Aizenman E, Kandler K. KCC2 expression in immature rat cortical neurons is sufficient to switch the polarity of GABA responses. Eur J Neurosci. 2005;21:2593–2599. doi: 10.1111/j.1460-9568.2005.04084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Wu C-F. Electroconvulsive seizure behavior in Drosophila: analysis of the physiological repertoire underlying a stereotyped action pattern in bang-sensitive mutants. J Neurosci. 2002;22:11065–11079. doi: 10.1523/JNEUROSCI.22-24-11065.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinekugel S, Tseeb V, Ben-Ari Y, Bregestovski P. Synaptic GABAA activation induces Ca2+ rise in pyramidal cells and interneurons from rat neonatal hippocampal slices. J Physiol (Lond) 1995;487:319–329. doi: 10.1113/jphysiol.1995.sp020882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhmann HJ, Prince DA. Postnatal maturation of the GABAergic system in rat neocortex. J Neurophysiol. 1991;65:247–263. doi: 10.1152/jn.1991.65.2.247. [DOI] [PubMed] [Google Scholar]
- Miles R. A homeotic switch. Nature. 1999;397:215–216. doi: 10.1038/16604. [DOI] [PubMed] [Google Scholar]
- Mitchell SJ, Silver RA. Shunting inhibition modulates neuronal gain during synaptic excitation. Neuron. 2003;38:433–445. doi: 10.1016/s0896-6273(03)00200-9. [DOI] [PubMed] [Google Scholar]
- Mody I, De Koninck YD, Otis TS, Soltesz I. Bridging the cleft at GABA synapses in the brain. Trends Neurosci. 1994;17:517–525. doi: 10.1016/0166-2236(94)90155-4. [DOI] [PubMed] [Google Scholar]
- Montgomery JC, MacDonald JA. Effects of temperature on nervous system: implications for behavioral performance. Am J Physiol. 1990;259:R191–R196. doi: 10.1152/ajpregu.1990.259.2.R191. [DOI] [PubMed] [Google Scholar]
- Mount DB, Delpire E, Gamba G, Hall AE, Poch E, Hoover RS, Herbert SC. The electroneutral cation-chloride cotransporters. J Exp Biol. 1998;201:2091–2102. doi: 10.1242/jeb.201.14.2091. [DOI] [PubMed] [Google Scholar]
- Mount DB, Mercado A, Song L, Xu J, George AL, Delpire E, Gamba G. Cloning and characterization of KCC3 and KCC4, new members of the cation-chloride cotransporter gene family. J Biol Chem. 1999;274:16355–16362. doi: 10.1074/jbc.274.23.16355. [DOI] [PubMed] [Google Scholar]
- Nabekura J, Ueno T, Okabe A, Furuta A, Iwaki T, Shimizu-Okabe C, Fukuda A, Akaike N. Reduction of KCC2 expression and GABAA receptor-mediated excitation after in vivo axonal injury. J Neurosci. 2002;22:4412–4417. doi: 10.1523/JNEUROSCI.22-11-04412.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noebels JL. The biology of epilepsy genes. Annu Rev Neurosci. 2003;26:599–625. doi: 10.1146/annurev.neuro.26.010302.081210. [DOI] [PubMed] [Google Scholar]
- Obrietan K, van den Pol AN. GABA neurotransmission in the hypothalamus: developmental reversal from Ca2+ elevating to depressing. J Neurosci. 1995;15:5065–5077. doi: 10.1523/JNEUROSCI.15-07-05065.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SW, Kingsley T, Shin HH, Zheng Z, Chen HW, Chen X, Wang H, Ruan P, Moody M, Hou SX. A P-element insertion screen identified mutations in 455 novel essential genes in Drosophila. Genetics. 2003;163:195–201. doi: 10.1093/genetics/163.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens DF, Boyce LH, Davis MB, Kriegstein AR. Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. J Neurosci. 1996;16:6414–6423. doi: 10.1523/JNEUROSCI.16-20-06414.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palma E, Amici M, Sobrero F, Spinelli G, Di Angelantonio S, Ragozzino D, Mascia A, Scoppetta C, Esposito V, Miledi R, Eusebi F. Anomalous levels of Cl− transporters in the hippocampal subiculum from temporal lobe epilepsy patients make GABA excitatory. Proc Natl Acad Sci USA. 2006;103:8465–8468. doi: 10.1073/pnas.0602979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlidis P, Tanouye MA. Seizures and failures in the giant fiber pathway of Drosophila bang-sensitive paralytic mutants. J Neurosci. 1995;15:5810–5819. doi: 10.1523/JNEUROSCI.15-08-05810.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlidis P, Ramaswami M, Tanouye MA. The Drosophila easily shocked gene: a mutation in a phospholipid synthetic pathway causes seizure, neuronal failure, and paralysis. Cell. 1994;79:23–33. doi: 10.1016/0092-8674(94)90397-2. [DOI] [PubMed] [Google Scholar]
- Payne JA, Stevenson TJ, Donaldson LF. Molecular characterization of a putative K-Cl cotransporter in rat brain. J Biol Chem. 1996;271:16245–16252. doi: 10.1074/jbc.271.27.16245. [DOI] [PubMed] [Google Scholar]
- Payne JA, Rivera C, Voipio J, Kaila K. Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 2003;26:199–206. doi: 10.1016/S0166-2236(03)00068-7. [DOI] [PubMed] [Google Scholar]
- Periz G, Fortini ME. Ca2+-ATPase function is required for intracellular trafficking of the Notch receptor in Drosophila. EMBO J. 1999;18:5983–5993. doi: 10.1093/emboj/18.21.5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian N, Sejnowski TJ. When is an inhibitory synapse effective? Proc Natl Acad Sci USA. 1990;87:8145–8149. doi: 10.1073/pnas.87.20.8145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor; 1989. [Google Scholar]
- Shinnar S, Glauser TA. Febrile seizures. J Child Neurol. 2002;17([Suppl 1]):S44–S52. doi: 10.1177/08830738020170010601. [DOI] [PubMed] [Google Scholar]
- Song J, Tanouye MA. Seizure suppression by shakB2, a gap junction connexin mutation in Drosophila. J Neurophysiol. 2006;95:627–635. doi: 10.1152/jn.01059.2004. [DOI] [PubMed] [Google Scholar]
- Staley KJ, Mody I. Shunting of excitatory input to dentate gyrus granule cells by a depolarizing GABAA receptor-mediated postsynaptic conductance. J Neurophysiol. 1992;68:197–212. doi: 10.1152/jn.1992.68.1.197. [DOI] [PubMed] [Google Scholar]
- Stein V, Hermans-Borgmeyer I, Jentsch TJ, Hübner CA. Expression of the KCl cotransporter KCC2 parallels neuronal maturation and the emergence of low intracellular chloride. J Comp Neurol. 2004;468:57–64. doi: 10.1002/cne.10983. [DOI] [PubMed] [Google Scholar]
- Strange K, Singer TD, Morrison R, Delpire E. Dependence of KCC2 K-Cl cotransporter activity on a conserved carboxy terminus tyrosine residue. Am J Physiol. 2000;279:C860–C867. doi: 10.1152/ajpcell.2000.279.3.C860. [DOI] [PubMed] [Google Scholar]
- Su W, Shmukler BE, Chernova MN, Stuart-Tilley AK, De Franceschi L, Brugnara C, Alper SL. Mouse K-Cl cotransporter KCC1: cloning, mapping, pathological expression, and functional regulation. Am J Physiol. 1999;277:C899–C912. doi: 10.1152/ajpcell.1999.277.5.C899. [DOI] [PubMed] [Google Scholar]
- Tan JS, Lin F, Tanouye MA. Potassium bromide, an anticonvulsant, is effective at alleviating seizures in the Drosophila bang-sensitive mutant bang senseless. Brain Res. 2004;1020:45–52. doi: 10.1016/j.brainres.2004.05.111. [DOI] [PubMed] [Google Scholar]
- Tan NCK, Mulley JC, Berkovic SF. Genetic association studies in epilepsy: the truth is out there. Epilepsia. 2004;45:1429–1442. doi: 10.1111/j.0013-9580.2004.22904.x. [DOI] [PubMed] [Google Scholar]
- Thomas-Crusells J, Vieira A, Airaksinen MS, Saarma M, Kaila K, Rivera C. Downregulation of the neuronal K–Cl cotransporter KCC2 in basal CA1 dendrites following epileptic activity in vitro is a consequence of a fast turn-over rate and slowing down of KCC2 protein transport. Soc Neurosci Abstr. 2000;26:233.236. [Google Scholar]
- Tornberg J, Voikar V, Savilahti H, Rauvala H, Airaksinen MS. Behavioural phenotypes of hypomorphic KCC2-deficient mice. Eur J Neurosci. 2005;21:1327–1337. doi: 10.1111/j.1460-9568.2005.03959.x. [DOI] [PubMed] [Google Scholar]
- Usunoff G, Atsev E, Tchavdarov D. On the mechanisms of picrotoxin epileptic seizures (macro- and micro-electrode investigations) Electroencephalogr Clin Neurophysiol. 1969;27:444. [PubMed] [Google Scholar]
- van den Pol AN, Obrietan K, Chen G. Excitatory actions of GABA after neuronal trauma. J Neurosci. 1996;16:4283–4292. doi: 10.1523/JNEUROSCI.16-13-04283.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijai J, Kapoor A, Ravishankar HM, Cherian PJ, Girija AS, Rajendran B, Rangan G, Jayalakshmi S, Mohandas S, Radhakrishnan K, Anand A. Genetic association analysis of KCNQ3 and juvenile myoclonic epilepsy in a South Indian population. Hum Genet. 2003;113:461–463. doi: 10.1007/s00439-003-1003-8. [DOI] [PubMed] [Google Scholar]
- Wallace RH, Marini C, Petrou S, Harkin LA, Bowser DN, Panachal RG, Williams DA, Sutherland GR, Mulley JC, Scheffer IE, Berkovic SF. Mutant GABAA receptor γ2-subunit in childhood absence epilepsy and febrile seizures. Nat Genet. 2001;28:48–52. doi: 10.1038/ng0501-49. [DOI] [PubMed] [Google Scholar]
- Wang J, Reichling DB, Kyrozis A, MacDermott AB. Developmental loss of GABA- and glycine-induced depolarization and Ca2+ transients in embryonic rat dorsal horn neurons in culture. Eur J Neurosci. 1994;6:1275–1280. doi: 10.1111/j.1460-9568.1994.tb00317.x. [DOI] [PubMed] [Google Scholar]
- Wharton KA, Cook JM, Torres-Schumann S, de Castro K, Borod E, Phillips DA. Genetic analysis of the bone morphogenetic protein-related gene, gbb, identifies multiple requirements during Drosophila development. Genetics. 1999;152:629–640. doi: 10.1093/genetics/152.2.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo N-S, Lu J, England R, McClellan R, Dufour S, Mount DB, Deutch AY, Lovinger DM, Delpire E. Hyperexcitability and epilepsy associated with disruption of the mouse neuronal-specific K-Cl cotransporter gene. Hippocampus. 2002;12:258–268. doi: 10.1002/hipo.10014. [DOI] [PubMed] [Google Scholar]
- Woodbury DM. Convulsant drugs: mechanisms of action. Adv Neurol. 1980;27:249–303. [PubMed] [Google Scholar]
- Yuste R, Katz LC. Control of postsynaptic Ca2+ influx in developing neocortex by excitatory and inhibitory neurotransmitters. Neuron. 1991;6:334–344. doi: 10.1016/0896-6273(91)90243-s. [DOI] [PubMed] [Google Scholar]
- Zhang H, Tan J, Reynolds E, Kuebler D, Faulhaber S, Tanouye M. The Drosophila slamdance gene: a mutation in an aminopeptidase can cause seizure, paralysis and neuronal failure. Genetics. 2002;162:1283–1299. doi: 10.1093/genetics/162.3.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H-G, Lee H-J, Rocheleau T, ffrench-Constant RH, Jackson MY. Subunit composition determines picrotoxin and bicuculline sensitivity of Drosophila γ-aminobutyric acid receptors. Mol Pharmacol. 1995;48:835–840. [PubMed] [Google Scholar]
- Zhu L, Lovinger D, Delpire E. Cortical neurons lacking KCC2 expression show impaired regulation of intracellular chloride. J Neurophysiol. 2005;93:1557–1568. doi: 10.1152/jn.00616.2004. [DOI] [PubMed] [Google Scholar]
- Zifkin B, Andermann E, Andermann F. Mechanisms, genetics, and pathogenesis of juvenile myoclonic epilepsy. Curr Opin Neurol. 2005;18:147–153. doi: 10.1097/01.wco.0000162856.75391.b1. [DOI] [PubMed] [Google Scholar]