

Abstract
Fibroblast growth factor receptors (Fgfr) comprise a widely expressed family of developmental regulators implicated in oligodendrocyte (OL) maturation of the CNS. Fgfr2 is expressed by OLs in myelinated fiber tracks. In vitro, Fgfr2 is highly upregulated during OL terminal differentiation, and its activation leads to enhanced growth of OL processes and the formation of myelin-like membranes. To investigate the in vivo function of Fgfr2 signaling by myelinating glial cells, we inactivated the floxed Fgfr2 gene in mice that coexpress Cre recombinase (cre) as a knock-in gene into the OL-specific 2′,3′-cyclic nucleotide phosphodiesterase (Cnp1) locus. Surprisingly, no obvious defects were detected in brain development of these conditional mutants, including the number of OLs, the onset and extent of myelination, the ultrastructure of myelin, and the expression level of myelin proteins. However, unexpectedly, a subset of these conditional Fgfr2 knock-out mice that are homozygous for cre and therefore are also Cnp1 null, displayed a dramatic hyperactive behavior starting at ∼2 weeks of age. This hyperactivity was abolished by treatment with dopamine receptor antagonists or catecholamine biosynthesis inhibitors, suggesting that the symptoms involve a dysregulation of the dopaminergic system. Although the molecular mechanisms are presently unknown, this novel mouse model of hyperactivity demonstrates the potential involvement of OLs in neuropsychiatric disorders, as well as the nonpredictable role of genetic interactions in the behavioral phenotype of mice.
Keywords: oligodendrocyte; myelin; FGF receptor; CNP; fibroblast growth factor; 2′,3′-cyclic nucleotide phosphodiesterase
Introduction
Fibroblast growth factors are important regulators of nervous system development and physiology. This signaling is mediated by four FGF receptors (Fgfr1–Fgfr4) and a subset of ligand FGFs expressed in the brain from a family of 22 members (Orr-Urtreger et al., 1993; Miyake et al., 1996; Ford-Perriss et al., 2001; Bansal et al., 2003). These FGF ligand–receptor complexes play diverse roles in the regulation of proliferation, migration, differentiation, and survival of both neurons and glia (Vaccarino et al., 1999; Ford-Perriss et al., 2001), including oligodendrocytes (OLs), the myelin-forming cells of the CNS (for review, see Bansal, 2002; Fortin et al., 2005).
A key aspect of this regulation is the differential expression of three FGF receptors during OL development (Bansal et al., 1996; Fortin et al., 2005); Fgfr1 is expressed at all stages of the OL lineage, Fgfr2 appears only in differentiated OLs, and Fgfr3 expression increases from the early to late progenitor stage and then is downregulated as OLs begin terminal differentiation. The expression pattern of Fgfr2 and Fgfr3 observed in vivo suggest that they are developmentally regulated in a manner consistent with the pattern of their expression in vitro (Bansal et al., 2003). To investigate mechanisms of OL developmental regulation by FGF, we have undertaken an extensive study of transgenic mice lacking FGF receptors in OLs. Previously, we have shown that Fgfr3 null mice display delayed OL differentiation and myelination (Oh et al., 2003). In the present study, we investigated the in vivo role of Fgfr2.
Fgfr2 is of particular interest because it is abundantly expressed in vitro by mature OLs (but not progenitors) coincidentally with major myelin proteins (Bansal et al., 1996; Cohen and Chandross, 2000; Yim et al., 2001; Fortin et al., 2005). In vivo Fgfr2 is expressed by OLs in myelinated fiber tracts of adult rodent brain, spinal cord, and optic nerve and is present in purified myelin, whereas expression of Fgfr2 by neurons and astrocytes is low or absent (Yazaki et al., 1994; Miyake et al., 1996; Cohen and Chandross, 2000; Messersmith et al., 2000; Bansal et al., 2003; Fortin et al., 2005). In contrast to Fgfr2, in mature OLs, Fgfr3 and Fgfr4 proteins are absent, and Fgfr1 is found at lower levels (Yazaki et al., 1994; Bansal et al., 1996; Miyake et al., 1996; Oh et al., 2003). We also showed that selective activation of Fgfr2 leads to stimulation of OL process outgrowth and myelin-like membrane formation in cultures, whereas inhibition of Fgfr2 function by blocking antibodies leads to attenuation of these responses (Fortin et al., 2005). Together, these data strongly suggested that FGF signaling via Fgfr2 plays a role in CNS myelination.
The in vivo analysis of OL development and myelination in Fgfr2 null mice is complicated by their embryonic lethality (Arman et al., 1998). We therefore used the Cre recombinase (Cre)/loxP system to conditionally inactivate Fgfr2 specifically in myelinating OLs and Schwann cells. We report here that, surprisingly, conditional Fgfr2 knock-out mice have no obvious defects of OL development or myelination. However, a significant subset of these mice display a dramatic hyperactive behavior.
Materials and Methods
Generation of Fgfr2 conditional knock-out mice
The generation of floxed Fgfr2flox/+ mice (129SV strain) and CNPcre/+ mice (C57BL/6 strain) has been described previously (Lappe-Siefke et al., 2003; Yu et al., 2003).
Fgfr2flox/+ transgenic mice. The loxP sites flank Fgfr2 exons 8–10 that encode a portion of ligand binding IgIII domain and the transmembrane domain of the receptor. Deletion of this region renders Fgfr2 inactive (Yu et al., 2003). For genotyping the Fgfr2flox/+ mice, PCR was performed on genomic tail DNA using primers FR2.5 (5′-ATAGGAGCAACAGGCGG) and FR2.3 (5′- TGCAAGAGGCGACCAGTCAG), yielding 200 and 140 bp fragments as described previously (Yu et al., 2003).
CNPcre/+ transgenic mice. 2′,3′-Cyclic nucleotide phosphodiesterase (CNP) is a myelin protein expressed by OLs, OL progenitors, and Schwann cells, with low expression in non-neural tissues such as thymus and heart (for review, see Gravel et al., 1998; Braun et al., 2004). CNPcre/+ mice, generated by replacing the entire reading frame of CNP gene by the coding sequence of Cre recombinase, are normal as heterozygote, whereas homozygote develops axonal degeneration at ∼6–7 months of age (Lappe-Siefke et al., 2003). Cre-mediated recombination capability has been established by crosses with floxed LacZ indicator A-LacZ-R mice (Lappe-Siefke et al., 2003); it was reported that Cre and LacZ expression after Cre-mediated recombination occurred in OLs (Lappe-Siefke et al., 2003). Mice that lack the ability to synthesize cholesterol have also been successfully generated using these CNPcre/+ mice (Saher et al., 2005). One targeted allele of cre was enough for loxP recombination in the indicator A-LacZ-R mice and cholesterol-deficient mice. For genotyping the CNPcre/+ mice, PCR was performed on genomic tail DNA using primers, antisense (5′-AAATCAGGTGGAAGGCA), sense (5′-GCCTTCAAACTGTCCATCTC), 5′EcoIN2 (5′-GATGGGGCTTACTCTTGC), and puro3 (5′-CATAGCCTGAAGAACGAGA), yielding 496 and 894 bp fragments as described previously (Lappe-Siefke et al., 2003).
Generation of Fgfr2flox/flox,cre/+ and Fgfr2flox/flox,cre,cre transgenic mice. Fgfr2flox/+ and CNPcre/+ transgenic mice were suitably crossed to produce progeny in which CNP promoter-driven cre activity recombines loxP sites in the gene for Fgfr2 to render an Fgfr2 null mutation in OLs and Schwann cells. This final mating produced two Fgfr2 null genotypes, with either one cre allele (and one Cnp1 allele) termed FFC+ (Fgfr2flox/flox,cre/+) or two cre alleles (Cnp1 null) termed FFCC (Fgfr2flox/flox,cre/cre). Littermates with the genotype Fgfr2flox/flox,+/+ (FF++) were used as controls.
The efficiency of Fgfr2 recombination was assessed by various methods. Double immunostaining with anti-Cre and anti-myelin basic protein (MBP) (Fig. 1A) showed overlap in cerebellar white matter and corpus callosum of mutant mice, confirming that Cre was expressed by OLs. PCR analysis of genomic DNA purified from different regions of CNS and PNS identified the expected 500 bp fragment indicative of Cre-mediated Fgfr2 recombination in FFC+ but not in wild-type or control mice (Fig. 1B). Even after deletion of the floxed portion of the Fgfr2 gene, presumably nonfunctional Fgfr2 transcripts (and nonfunctional proteins) may be expressed and detected by riboprobes (and antibodies). Thus, we performed reverse transcription (RT)-PCR with primers that would show Fgfr2 recombination. We found a corresponding band only in mice with either one or two alleles of cre (FFCC and FFC+) but not in control mice (Fig. 1C). Immunoblot analysis of homogenates of FFC+ and FFCC mice showed that the characteristic Fgfr2-specific protein doublet was absent in myelin and hindbrain and markedly reduced in forebrain and cerebellum from both genotypes (Fig. 1D); the low residual levels of Fgfr2 protein were presumably attributable to expression of Fgfr2 by astrocytes and cerebellar Purkinje neurons (Bansal et al., 2003) (see Fig. 4). Importantly, no new protein fragment was detected in mutant extracts, suggesting that the truncated Fgfr2 gene was either not transcribed or the encoded protein was rapidly degraded (data not shown). Unfortunately, direct labeling of histological sections with anti-Fgfr2 was impossible, because the available antibody also recognized other proteins on immunoblots. Elevation of the levels of Fgfr1 and Fgfr3 was not observed in the mutant mice (data not shown). Body size of FFC+ mice (29 ± 1.3 g) was similar to controls (31 ± 1.7 g), whereas FFCC (22 ± 1 g) mice were 30% smaller (p < 0.002) (n = 6 in each group) (Fig. 1E).
Figure 1.
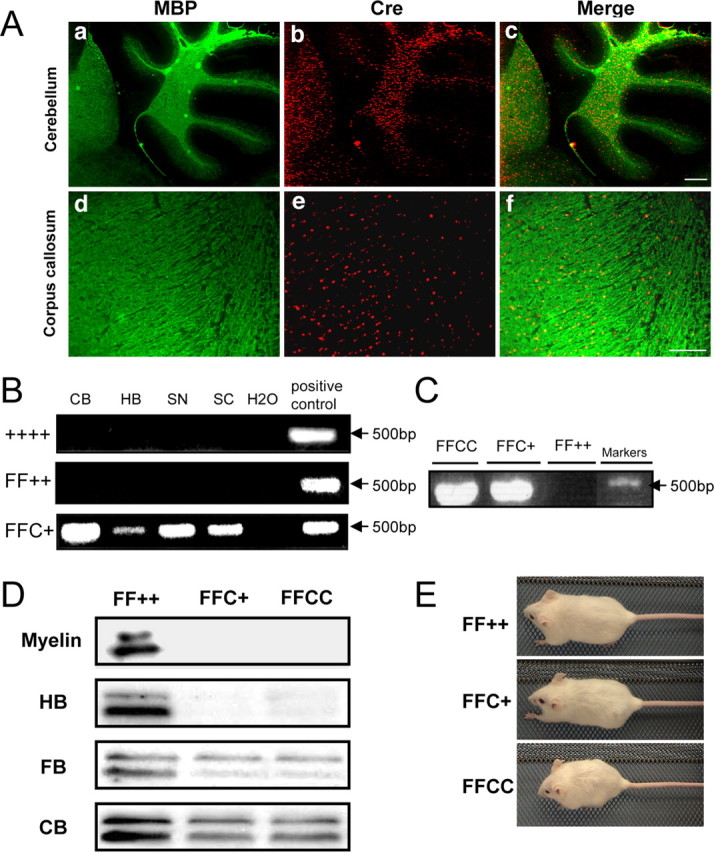
Generation of Fgfr2 conditional knock-out mice. A, Immunolabeling of control cerebellum and corpus callosum with anti-MBP and anti-Cre. Nuclear CNP–Cre expression overlapped with MBP in the cerebellar white matter and corpus callosum, showing that the target cells are oligodendrocytes. Scale bars: a–c, 200 μm; d–f, 100 μm. B, PCR of genomic DNA from wild type (++++), control (FF++; Fgfr2flox/flox,+/+), and FFC+ (Fgfr2flox/flox,cre/+). Primers amplify a 500 bp DNA fragment indicative of the expected deletion in tissue taken from different regions of the CNS and PNS, showing that the Fgfr2 gene sequence has been successfully disrupted in FFC+ mutant mice. CB, Cerebellum; HB, hindbrain; SN, sciatic nerve; SC, spinal cord. C, RT-PCR with primers that identify transcripts with the expected deletion shows that the deleted Fgfr2 mRNA is present in mutant mice with either one (FFC+) or two (FFCC) alleles of cre but not in control mice (FF++). D, Immunoblot analyses of different regions of the brain from mutant mice (FFC+ and FFCC) and control mice (FF++) mice show that the Fgfr2-specific doublet is missing (myelin and myelin-rich hindbrain) or present at only low levels (forebrain and cerebellum, tissues in which other cell types presumably contribute Fgfr2) in the mutant mice. E, At 7 months of age, the body sizes of FFC+ and FF++ mice are comparable, whereas those of FFCC mice are significantly smaller.
Figure 4.
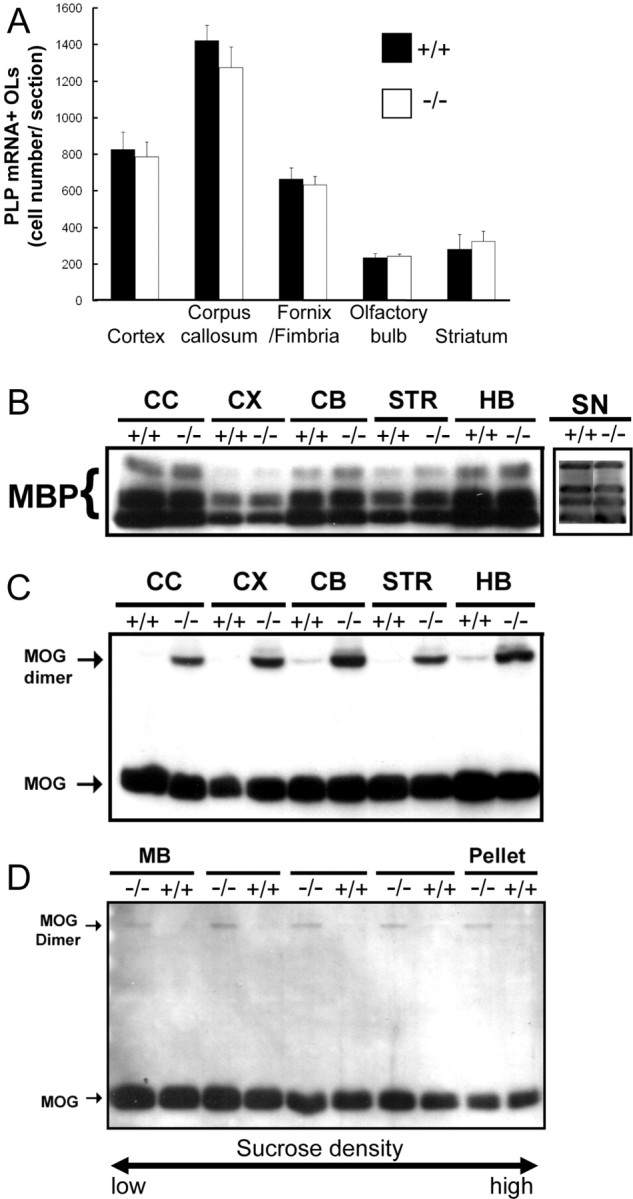
Oligodendrocyte differentiation and levels of myelin proteins in different regions of Fgfr2 conditional knock-out mouse brain. A, PLP mRNA+ cells in the entire regions specified were counted for each section. Four to 12 sections each from two to three mice in each group were analyzed at P13. No statistically significant differences between the number of PLP mRNA+ cells from control (+/+) and FFC+ mice were observed. B, C, Homogenates from different brain regions of control (+/+) and FFCC (−/−) mice at 2 months of age were analyzed by immunoblot analysis for the expression of MBP (B) or MOG (C). No differences were observed in MBP expression in any of the regions of the brain analyzed or in P18 sciatic nerve. However, a dimer of MOG is observed in mutant but not in control mice. CC, Corpus callosum; CX, cerebral cortex; CB, cerebellum; STR, striatum; HB, hindbrain; SN, sciatic nerve. D, Immunoblot analysis of MOG from myelin fractions separated on sucrose density gradients from control (+/+) and FFCC (−/−) mice. Similar to homogenates, a dimer of MOG was also observed in all of the fractions of purified myelin. MB, Main band of myelin.
These data together show that conditional Fgfr2 ablation in oligodendrocytes and Schwann cells was achieved in both FFC+ and FFCC mice.
Preparation of tissue sections
Mice were anesthetized and perfused with 4% paraformaldehyde (PFA). The brains and spinal cords were removed, postfixed overnight in 4% PFA at 4°C, and cryoprotected in 10% sucrose, followed by 30% sucrose, each performed overnight at 4°C. Brains were cut sagittally at the midline, embedded in Tissue-Tek OCT mounting media, and frozen at −80°C. Parasagittal cryostat sections of brains and transversal sections of lumbar spinal cords (15 μm thick) were used for in situ hybridization and immunohistochemistry.
Optic nerves and sciatic nerves from 2-month-old control and mutant mice were fixed with 4% PFA in 0.1 m phosphate buffer (PB), equilibrated in 20% sucrose in 0.1 m PB, and frozen at −30°C in OCT. Sections (optic nerves, 10 μm; sciatic nerves, 25 μm) were placed in 0.1 m PB and spread on gelatin-coated coverslips for immunohistochemistry.
In situ hybridization
In situ hybridization for proteolipid protein (PLP) mRNA was performed as described previously (Oh et al., 2003). A riboprobe specific for PLP mRNA covering the entire cording region was used (a gift from B. Fuss and W. B. Macklin, Cleveland, OH). Briefly, sections were fixed with 4% PFA for 15 min, washed in PBS, and incubated in 1 μg/ml proteinase K at 37°C for 30 min. After the sections were fixed with 4% PFA and washed with PBS again, hybridization for PLP mRNA was performed overnight at 65°C by using digoxigenin-labeled antisense cRNA probes, containing a solution of 50% formamide, 5× SSC, and 1% SDS. After hybridization, the sections were washed in 50% formamide, 2× SSC, and 1% SDS at 65°C for 2–3 h, followed by rinses in 2× SSC and 0.2× SSC at room temperature and 0.1× SSC at 60°C. After blocking for nonspecific binding in Tris-buffered saline, pH 7.4, with 1% Tween 20 and 1% normal goat serum (1 h), alkaline phosphatase-conjugated anti-digoxigenin antibody (Roche Diagnostics, Penzberg, Germany) was used at 1:5000 in the same blocking buffer for 2 h. Color development in the presence of 4-nitroblue tetrazolium chloride/5-bromo-4-chloro-3-indolylphosphate was performed in the dark at room temperature. The sections were washed in TE buffer containing 10 mm Tris, pH7.5, and 10 mm EDTA, pH 8, incubated in Hoechst blue dye 3342 (1 μg/ml; Sigma, St. Louis, MO) to counterstain the nuclei, fixed in 3.7% formaldehyde, and mounted with 90% glycerol.
Comparative analyses of PLP mRNA-positive cells of control and mutant mice. Analyses and cell counting were performed as described previously (Bansal et al., 1999; Oh et al., 2003). Comparison between mutant and controls were made within the same litters. Whole brains were cut parasagittally in the same plane (15-μm-thick sections), located ∼300 μm from the midline. Transverse 15-μm-thick spinal cord sections were cut in the lumbar enlargement. Control and mutant mice sections were matched by viewing Hoechst dye-stained sections so that they were equidistant from the midline. The total numbers of PLP mRNA-positive (RNA+) cells present in the whole corpus callosum, cerebral cortical area, olfactory bulb, thalamus, striatum, and fornix/fimbria of the brain sections were counted systematically using a grid and 20× objective. In the spinal cord, we counted the number of the cells in white matter and gray matter, measured each area with NIH Image analysis program and calculated cell densities in each area. The comparison of cell numbers between control and mutant mice were analyzed by unpaired Student's t test.
Immunohistochemistry
Tissue preparation and sectioning was performed as described above. For MBP and myelin oligodendrocyte glycoprotein (MOG) immunolabeling, sections were immersed in 100% ethanol, washed in PBS, and blocked for 1 h in a buffer consisting of 10% normal goat serum (NGS), 5% bovine serum albumin (BSA), 0.05% NaN3, and 0.1% gelatin in PBS. For glial fibrillary acidic protein (GFAP) immunolabeling, sections were blocked in 3% NGS and 0.1% Triton X-100 in PBS. Sections were incubated overnight at 4°C in polyclonal anti-rabbit MBP (1:3000; S. E. Pfeiffer), monoclonal anti-mouse IgG MOG (1:1000; Dr. C. Linington, Aberdeen, UK), polyclonal anti-rabbit GFAP (1:500; DakoCytomation, Carpinteria, CA), polyclonal anti-tyrosine hydroxylase (1:500; ImmunoStar, Hudson, WI), or monoclonal anti-mouse IgG Cre (1:100; Babco, Berkeley, CA). After being washed in PBS, the sections were incubated for 1 h with goat anti-rabbit IgG conjugated to Alexa 488 (1:200; Invitrogen, Carlsbad, CA) or goat anti-mouse IgG conjugated to cyanine 3 (1:600; Jackson ImmunoResearch, West Grove, PA) and Hoechst Blue 33342 (1:1000), washed in PBS, mounted in 1,4-diazobicyclo-(2,2,2)-octane in glycerol, and analyzed with an epifluorescent microscope (Axiovert microscope; Zeiss, Thornwood, NY).
For optic nerves and sciatic nerves, sections were permeabilized in 0.3% Triton X-100 and 10% NGS in 0.1 m PB, incubated overnight at room temperature with polyclonal anti-rabbit contactin-associated protein (Caspr) (1:300; M. Rasband) and monoclonal pan anti-mouse IgG sodium channel (Nav; 1:500) (Rasband and Trimmer, 2001), washed, incubated for 1 h with second antibodies, washed, mounted on slides with anti-fade mounting medium, and analyzed.
Immunoblotting
Corpus callosum, cerebral cortex, cerebellum, striatum, and hindbrain were dissected and stored at −80°C before analysis. Tissue samples were homogenized in lysis buffer (10 mm Tris-HCl, 150 mm NaCl, 0.1% SDS, 1% deoxycholate, and 1% NP-40, pH 7.4) with protease inhibitors (2 mm PMSF, 2 μg/ml leupeptin, and 2 μg/ml aprotinin). The homogenates were then incubated (30 min, on ice) and centrifuged (15,000 × g, 10 min, 4°C). The protein concentration was assayed with the DC Protein Assay kit (Bio-Rad, Hercules, CA). Aliquots of equal amounts of total protein from different experimental conditions were subjected to electrophoresis on 12% SDS polyacrylamide gels and transferred onto polyvinylidene difluoride membranes. The membranes were blocked for 1 h (Tris-buffered saline, 5% nonfat powdered milk, and 0.2% Tween 20 or 10% BSA) and incubated for 1 h in primary antibodies: anti-FGFR2 (1:500; Santa Cruz Biotechnology, Santa Cruz, CA), anti-MOG (1:3000), anti-MBP (1:10,000), anti-myelin-associated glycoprotein (MAG) (1:5000; J. Rodor, University of Toronto, Toronto, Ontario, Canada), anti-PLP/DM20 (1:1000; M. Lees, Shriver Center, Waltham, MA), anti-Nav (1:500), anti-Caspr (1:2), anti-Kv (1:500), or anti-neurofascin 155 (NF155) (1:1000; M. Rasband). The membranes were incubated for 30 min in either anti-rabbit IgG (1:10,000; Santa Cruz Biotechnology) or anti-mouse IgG (1:10,000; Transduction Laboratories, Lexington, KY), both conjugated to horseradish peroxidase. The membranes were developed with ECL Plus kit (GE Healthcare, Little Chalfont, UK).
Electron microscopy
For electron microscopic analysis, mice were anesthetized with pentobarbital and fixed by transcardiac perfusion with 3% glutaraldehyde/2% formaldehyde (freshly made) in 0.1 m cacodylate buffer, pH 7.3. Optic nerves were dissected, rinsed, postfixed with osmic acid, dehydrated, and embedded in Araldite. Sections (1 μm) were stained with toluidine blue for light microscopy, and selected areas were further sectioned at 0.1 μm for transmission electron microscopy. Thin sections were double stained with permanganate and uranyl acetate and examined in a Philips (Aachen, Germany) 300 electron microscope at 60 kV or a Jeol (Peabody, MA) 1200EX instrument at 80 kV.
Behavioral analysis
Mutants and their littermate control mice were analyzed for motor function and strength of forelimbs. We recorded the time that mice remained balanced on a round bar of 15 mm diameter and the time the mice held to the wire turned upside down (25 cm square wire with 12 mm grids) (Crawley, 2000).
Locomotor activity was measured using the automated Columbus Instruments (Columbus, OH) Videomex-V system. The animals were placed in a 9 × 19 inch open-field chamber and monitored in multiple 5 min sessions. The Videomex System converts the image to a pixel array that allows tracking of a moving image and counts the distance moved. In addition, the video-tracking system provided an image of the tracking movements of the mice proportional to the size of the open field. Drugs obtained from Sigma were dissolved in sterile saline or water and injected intraperitoneally as described for each experiment. Effective doses were selected according to previous studies (Ralph et al., 2001a): d-Amphetamine sulfate (1.25 mg/kg), α-methyl-p-tyrosine (250 mg/kg), apomorphine (2 mg/kg), eticlopride and Sch 23390 [R(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride] (0.01–1 mg/kg body weight). Measures of the locomotor activity were made both before (baseline) and after drug injection.
Results
Generation of Fgfr2 conditional knock-out mice
Fgfr2flox/+ and CNPcre/+ transgenic mice were crossed to produce progeny with an Fgfr2 null mutation in myelinating cells (see Materials and Methods). The final mating produced two Fgfr2 null genotypes with either one cre allele (and thus one Cnp1 allele; Fgfr2flox/flox, cre/+) termed FFC+, or two cre alleles (therefore, Cnp1 null; Fgfr2flox/flox,cre/cre) termed FFCC. Conditional Fgfr2 ablation was achieved in both genotypes (Fig. 1) (see Materials and Methods), which yielded similar results with regards to myelination. Therefore, except when otherwise indicated, data for FFC+ are shown. Littermates with the genotype Fgfr2flox/flox,+/+ (FF++) were used as controls.
Oligodendrocyte differentiation in Fgfr2 conditional knock-out mouse brain
Fgfr2 mRNA and protein are highly upregulated as OLs differentiate (Bansal et al., 1996; Fortin et al., 2005). We therefore examined the influence of Fgfr2 inactivation on OL differentiation.
Parallel parasagittal sections from control and FFC+ littermates were used for in situ hybridization of PLP mRNA to identify and count the number of OLs as a function of age. PLP mRNA+ OLs first appear in the corpus callosum at approximately postnatal day 7 (P7) and then spread progressively into the cerebral cortex (Oh et al., 2003). As shown previously, in control brains at P8, P13, and P60, the expression of PLP mRNA+ cells in these regions increases with age (Fig. 2Aa,Ac,Ae). FFC+ mice showed a similar pattern (Fig. 2Ab,Ad,Af). No statistically significant differences were observed between control and FFC+ mice in the total numbers of PLP mRNA+ OLs in matched sections at any of the ages examined, except for a slight reduction at P17 (Fig. 2B). Furthermore, no differences were observed between FFC+ and FFCC mice in the numbers of PLP mRNA+ cells (supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material).
Figure 2.
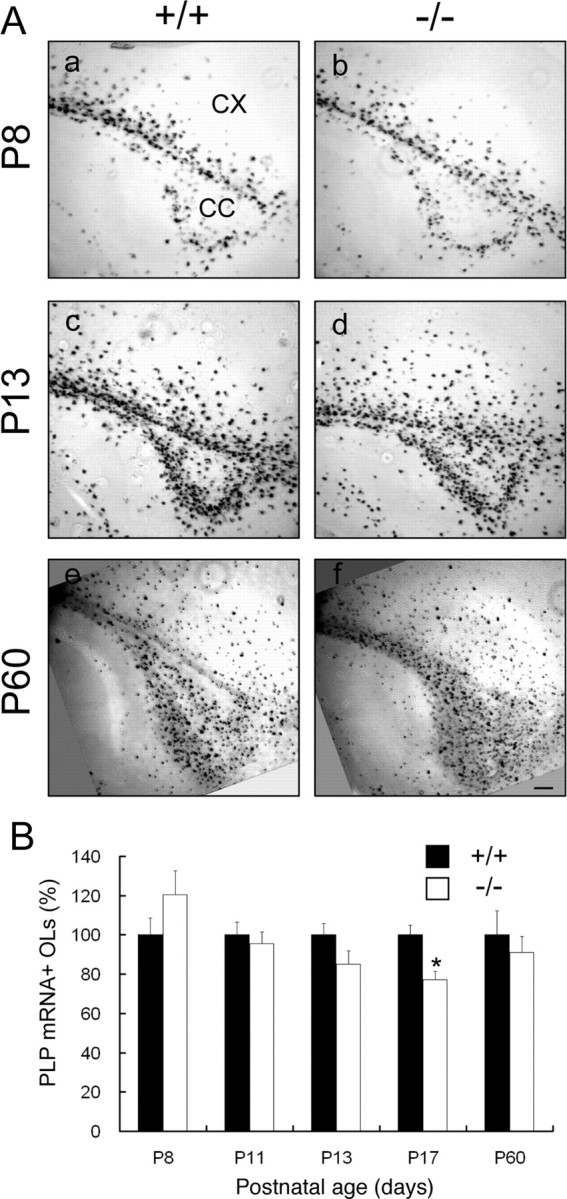
Oligodendrocyte differentiation in Fgfr2 conditional knock-out mouse brain. A, Parasagittal sections from matched regions of forebrains of control (a, c, e) and FFC+ (b, d, f) mice at P8 (a, b), P13 (c, d), and P60 (e, f) were analyzed by in situ hybridization for the OL marker PLP mRNA. PLP mRNA+ cells showed similar patterns of expression in FFC+ (−/−) compared with control (+/+) mice. CX, Cerebral cortex; CC, corpus callosum. Scale bar, 100 μm. B, Quantification of the number of PLP mRNA+ cells that differentiated as a function of time in the corpus callosum of control and FFC+ mice. All PLP mRNA+ cells in the entire corpus callosum region were counted for each section. Four to 10 sections each from four mice from each group and age were analyzed. No significant differences were found between control and FFC+ mice at all ages, except for a slight decrease at P17 (p < 0.05, Student's t test). Black bar, Control (+/+); white bar, FFC+ (−/−). Error bars indicate SEM (n = 4).
Myelination in Fgfr2 conditional knock-out mouse brain
Because Fgfr2 is expressed by mature OLs and is present in purified myelin, we analyzed FFC+ mice to evaluate the effect of eliminating Fgfr2 function on myelin formation. We examined MBP and MOG expression as myelin markers by immunohistochemistry on parasagittal sections of whole brain from control and mutant mice as a function of development (Fig. 3A, P13 is shown). At P13, when myelination progresses rapidly, myelinated fibers in the white matter immunolabeled with anti-MBP or anti-MOG showed similar patterns in normal and FFC+ mice (Fig. 3A). Similar results were obtained at P7, P17, P60, and 7 months (data not shown). Furthermore, no differences were observed between FFC+ and FFCC mice in the expression of MOG or MBP (supplemental Fig. 1Aa–Ad, available at www.jneurosci.org as supplemental material). These results were confirmed by immunoblotting for the myelin markers MOG, PLP/DM20, MBP, and MAG in homogenates of forebrain obtained at postnatal day 18 and 3 months of age and in whole brain homogenates of 12-month-old mice. Consistent with the immunohistochemical results, the expression of these myelin proteins showed no differences between control and FFC+ mice (Fig. 3B). Note that, unlike the numerical decrease of PLP mRNA+ OLs at P17, PLP protein expression did not show a corresponding reduction at a similar age.
Figure 3.
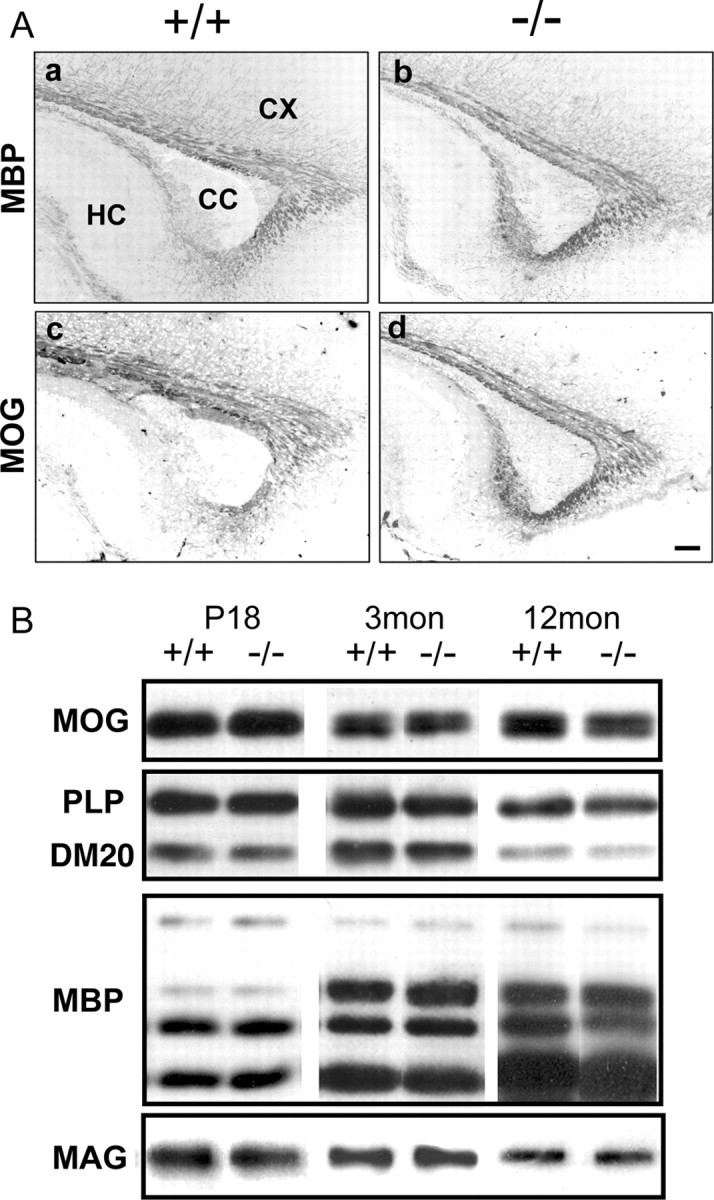
Myelination in Fgfr2 conditional knock-out mouse brain. A, Parallel parasagittal sections from P13 forebrain of control and FFC+ mice were analyzed by immunohistochemistry for MBP and MOG as markers of myelinated fibers. In the forebrain of FFC+ mouse (−/−), both MBP (a, b) and MOG (c, d) expressions appeared similar to control mice (+/+). Three to six sections each from two to four mice were analyzed from each group. CC, Corpus callosum; CX, cortex; HC, hippocampus. Scale bar, 100 μm. B, Immunoblot analysis of brain homogenates from control (+/+) and FFC+ (−/−) mice for the myelin proteins MOG, PLP/DM20, MBP, and MAG. No significant differences were observed between +/+ and −/− at 18 d, 3 months, or 12 months of age. A representative blot is shown.
Oligodendrocyte differentiation and myelination in different brain regions of Fgfr2 conditional knock-out mice
Because various mouse mutants exhibit deficits in the number of OLs and/or myelination only in specific regions of the CNS [e.g., fyn (Sperber et al., 2001); pdgf-A (Fruttiger et al., 1999); Golli MBP (Jacobs et al., 2005); chemokine receptor Cxcr2 (Padovani-Claudio et al., 2006)], we examined multiple regions (cortex, corpus callosum, fornix/fimbria, olfactory bulb, striatum, thalamus, brainstem, and cerebellum) of Fgfr2 conditional knock-out mice for defects in OL differentiation and myelination (Fig. 4). P13 brains showed no statistically significant differences in the numbers of PLP mRNA+ OLs between control and FFC+ mice for any of the regions examined by in situ hybridization (Fig. 4A). By immunoblot analysis, no significant difference appeared between control and mutant mice (FFCC genotype is shown) either in the levels of MBP or of the major band of MOG from brain homogenates (Fig. 4B,C) or MOG from purified myelin (Fig. 4D). Curiously, the amount of MOG dimer was dramatically elevated in brain homogenate and in all fractions of purified myelin from mutants compared with control mice (Fig. 4C,D).
CNP–cre is also expressed in Schwann cells, the myelin-forming cells of the peripheral nervous system, rendering sciatic nerves of these mice also deficient in Fgfr2 (Fig. 1B). As for the CNS, no difference was observed in the levels of MBP in the homogenates from sciatic nerves compared with control (Fig. 4B).
Oligodendrocyte differentiation and myelination of Fgfr2 conditional knock-out mouse spinal cord
It has been reported that Fgfr2 is expressed by spinal cord OL and that OL differentiation is effected in the spinal cords of mice lacking FGF-2 (Messersmith et al., 2000; Murtie et al., 2005). However, whether FGF-2 directly regulates OL-lineage cells or indirectly through other cell types that also respond to FGF-2 is not clear. We therefore examined OL differentiation and myelin formation in spinal cords of Fgfr2 knock-out mice (Fig. 5). In transverse sections of spinal cord at P16, the density of PLP mRNA+ cells (average ± SE cell numbers per square millimeter) in the white matter was 71 ± 5 in control compared with 82 ± 5 (FFC+) and 74 ± 6 (FFCC) in mutant mice. In spinal gray matter, it was 20 ± 3 in controls and 26 ± 2 (FFC+) and 31 ± 5 (FFCC) in mutant mice (Fig. 5B). Thus, differences in cell density were not statistically significant between control and mutant mice of either genotype (FFC+ or FFCC). Consistent with this, MBP and MOG immunostainings were also similar in both control and mutant mice (Fig. 5Ac–Af).
Figure 5.
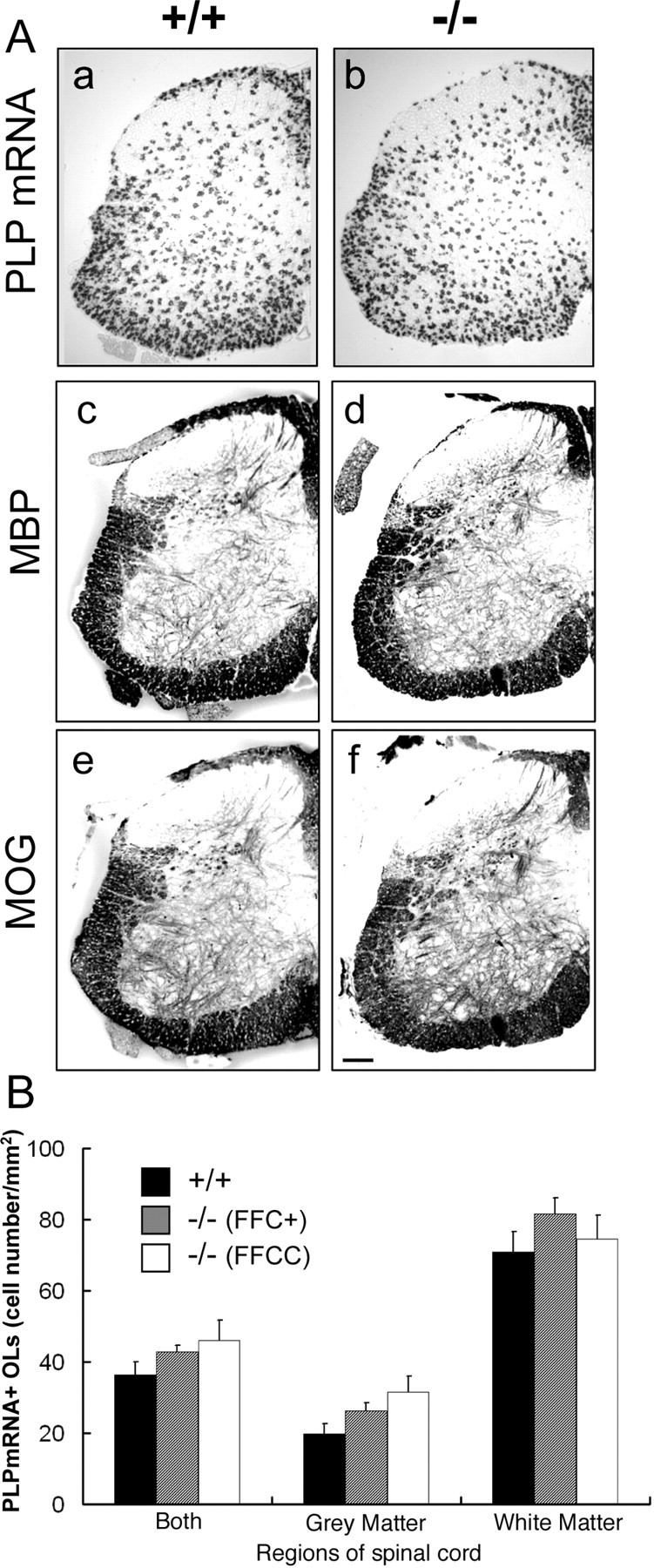
Oligodendrocyte differentiation and myelination in Fgfr2 conditional knock-out mouse spinal cord. A, Transverse sections of P16 lumbar spinal cord were analyzed by in situ hybridization for PLP mRNA (a, b) and immunohistochemistry for MBP (c, d) and MOG (e, f). No differences were observed in the expression pattern of either PLP mRNA+ cells or MBP/MOG+ myelinated fibers between control (+/+) and FFC+ (−/−) mice. Scale bar, 100 μm. B, Quantitative analysis of the density of PLP mRNA+ cells in the spinal cord at P16. The total numbers of PLP mRNA-positive cells in the gray or white matter regions were divided by the area of these regions. Three sections each from three mice of each group were analyzed. No statistically significant differences were observed in the cell density between control and mutant mice (FFC+, FFCC) from either white matter or gray matter. Error bars indicate SEM.
Myelin ultrastructure and structure of node of Ranvier and paranodes of the Fgfr2 conditional knock-out mice
The ultrastructure of myelin was examined by electron microscopy in cross and transverse sections of optic nerves from control and mutant mice (Fig. 6A). No differences in myelin structure were found (Fig. 6Aa–Ac) in either young adults (∼3–4.5 months) or 12-month-old FFC+ mice. Although there was a small increase in the number of redundant myelin profiles (Fig. 6Ab, arrow and asterisks), such profiles are well known to occur in the normal CNS as well, although somewhat less frequently (Rosenbluth, 1966). Paranodal loops that indent the axon and axoglial junctions with regularly spaced transverse bands were normal in all cases (Fig. 6Ac,Ad). There was no apparent increase in everted loops or detached loops.
Figure 6.
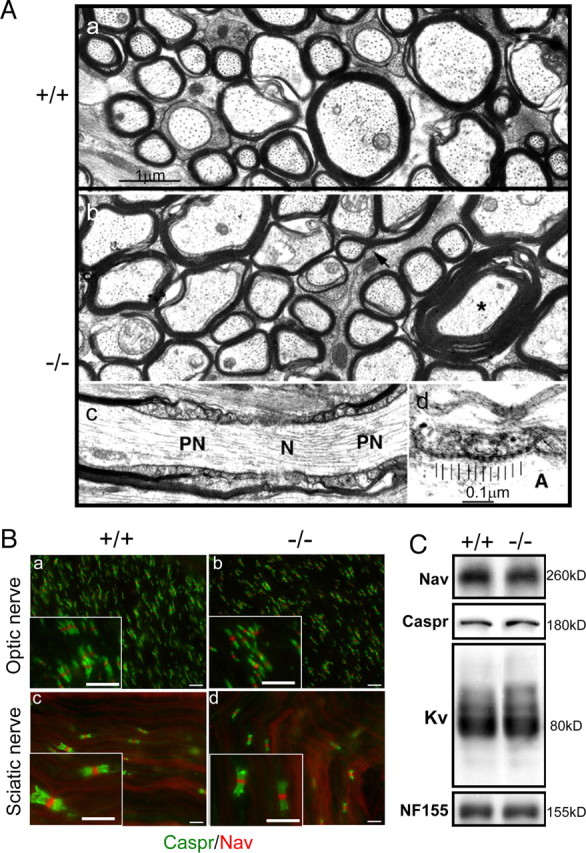
Myelin ultrastructure and structure of node of Ranvier and paranodes of the Fgfr2 conditional knock-out mice. A, Electron micrographs are shown of sections of optic nerve from a 4-month-old control (+/+) mouse (a) and a 1-year-old FFC+ (−/−) mouse (b–d). Transverse sections show myelinated fibers of various sizes. Myelin sheaths appear to be of normal structure. The arrow and asterisk indicate redundant myelin profiles that are comparable with those present in normal CNS. c, A longitudinal section shows a node of Ranvier (N) flanked by paranodes (PN). The overlapping paranodal terminal loops are of normal conformation with no evidence of eversion or detachment. d, Detail of a paranodal junction shows regularly repeating “transverse bands” (indicated by vertical bars) in the space between the axolemma and the membrane of the terminal loops. Scale bars: a–c, 1 μm; d, 0.1 μm. A, Axon. B, Immunohistochemical analysis of the nodes of Ranvier and paranodes in optic nerves (a, b) and sciatic nerves (c, d) of FFC+ mice. Longitudinal sections of optic nerve and sciatic nerve of 2-month-old mice were immunolabeled with markers for nodes, Na channels (Nav), and paranodes (Caspr). There were no differences in the clustering of either Nav or Caspr between control (+/+) (a, c) and FFC+ (−/−) (b, d) mice. Scale bars, 10 μm. C, Whole-brain homogenates from 12-month-old control and FFC+ mice analyzed by immunoblotting for Nav, Caspr, Kv, and NF155 showed comparable patterns of staining between the two groups.
Myelin nodes, paranodes, and juxtaparanodes are specialized regions of myelinated axons that constitute a critical component of axon–glial communication. We investigated the formation of nodes of Ranvier, paranodes, and juxtaparanodes, using immunohistochemistry and immunoblot analysis (Fig. 6B,C). No differences were detected in the localization of Nav channels (markers of nodes) or Caspr (paranodal marker) in either optic nerve (Fig. 6Ba,Bb) or sciatic nerve (Fig. 6Bc,Bd). Immunoblot analysis of Nav, Caspr, NF155 (a marker of paranodes), and Kv (a marker of juxtaparanodes) from whole-brain homogenate of FFC+ also showed similar levels of proteins compared with control animals even at 12 months of age (Fig. 6C).
Behavioral studies
Despite the apparently normal OL and myelin content, we considered the possibility that subtle myelin defects (below the levels of our histochemical and molecular detection) could lead to behavioral changes. Therefore, we evaluated mutant mice for myelin-related behavioral defects. Specifically, control and mutant littermates were judged for hindlimb strength by determining the length of time they could stay on a rod or wire mesh. Three- to 5-month-old control and mutant littermates (n = 8) from two different litters were analyzed. Each mouse was tested five times. No differences were observed in the length of time that the mice from different groups stayed on the rod or wire (data not shown). Overall, these mice did not display any obvious behavioral impairments characteristic of myelin mutants. In FFCC homozygous cre mice that were also CNP null (++CC), hindlimb weakness appeared at later ages (6 months), as has been shown previously for ++CC mice (Lappe-Siefke et al., 2003).
However, at ∼2 weeks of age, a subgroup of the mutant mice, the majority of which were FFCC (95%), became dramatically hyperactive during minor environmental stimuli, such as moving the cage. This hyperactivity was displayed by 20 of 52 FFCC mice (38%), born over a period of ∼2 years. This hyperactivity was very rare in FFFC+ mice (4 of 148 mice analyzed) and not observed in ++CC mice. Both male and female hyperactive FFCC and FFC+ mice were found.
To quantify this hyperactivity, we analyzed locomotor and exploratory behavior in an open-field test (Fig. 7Aa–Ac) (video recording of locomotion is presented as supplemental movie 1, available at www.jneurosci.org as supplemental material). Activity was recorded as distance (centimeters) traveled per 5 min (Fig. 7Ad) and total numbers of rotations (both clockwise and counterclockwise) per 50 min (Fig. 7Ae). Sixfold and 10-fold increases were observed, respectively, in hyperactive FFCC mice compared with mice of either FF++ or ++CC genotypes. The nonhyperactive FFCC and FFC+ mice were identical to control FF++ (data not shown) with respect to their locomotion. In addition, the locomotor patterns of the hyperactive mice were also different. Control mice usually explored the perimeter of the open field in a circular pattern (Fig. 7Aa). In contrast, locomotion of these hyperactive mice included both circular and cross-field motion (Fig. 7Ac).
Figure 7.

Hyperactive locomotor behavior in FFCC mice. Baseline locomotor activity was recorded in an open-field test. A, Tracking images of the locomotion of mice proportional to the size of the open field are shown for a single 5 min session for control (FF++), CNP-null (++CC), and the hyperactive Fgfr2/CNP double knock-out mice (FFCC) (a–c). Activity of each animal was recorded and summed every 5 min for 10 consecutive sessions (50 min) and expressed either as distance traveled in centimeters per 5 min (d) or rotations per 50 min (e). Error bars indicate SEM; n = 6. B, Amphetamine (1.25 mg/kg) was injected and locomotor activity was recorded as centimeters traveled per 5 min; n = 6 for each group. Fgfr2/CNP double knock-out mice show hyperactive behavior, and this activity is not attenuated by amphetamine.
Hyperactive behavior is observed in children with attention deficient hyperactivity disorder in children. Administration of psychostimulants such as amphetamines leads to a normalization of distractibility and thus a decrease of hyperactivity. We assessed the response of the mutant mice to amphetamine (Fig. 7B). No decrease in hyperactivity was observed by this treatment, and, in fact, there was an increase over baseline in both controls and mutant mice, a normal response to amphetamine attributable to catecholamine release.
Because dopamine neurotransmission plays a major role in motor activity, we asked whether hyperlocomotion of these mice involved dysregulation of the dopamine system. We evaluated the effect of the administration of dopamine receptor antagonists on locomotor activity of control and hyperactive mice in an open-field test (Fig. 8A). Injection of dopamine D1 receptor antagonist Sch 23390 reduced the locomotor activity of hyperactive mice as a function of dose, with 1 mg/kg completely abolishing the hyperactivity. Similarly, injection of the D2 receptor antagonist eticlopride antagonized the hyperactive behavior, albeit at a 10-fold higher dose. This suggests that both D1 and D2 receptors are involved in the behavioral abnormality such that the inhibition of either one can be used to correct the abnormal behavior.
Figure 8.
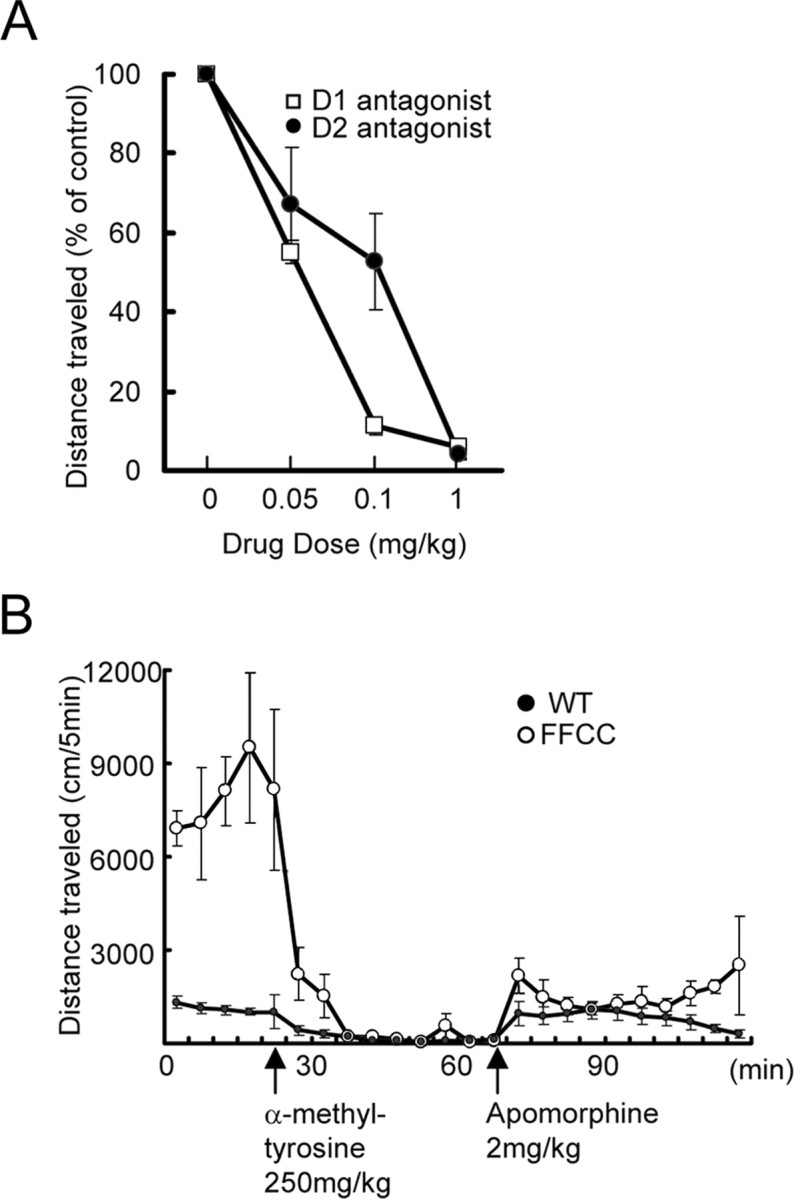
Hyperactivity of FFCC mice in relation to the dopaminergic system. A, Activity was recorded in an open-field test in response to dopamine receptor antagonists. Hyperactive mice were injected with different doses of either Sch 23390, a specific D1 dopamine receptor antagonist, or eticlopride, a specific D2 dopamine antagonist. For each dose (0.01–1 mg/kg), the mice were given the antagonist at baseline level of hyperactivity (set at 100%) and tested for an additional 50 min. The percentage change during the drug session is plotted against the dose used. The different doses were separately tested for each animal after a gap of 7–14 d. Error bars are SEM; n = 3. B, Effect of dopamine synthesis inhibition on the activity of hyperactive mice. The mice were allowed access to the open field for a baseline measure of hyperactivity (first 20 min). They were then injected intraperitoneally with 250 mg/kg α-methyl-tyrosine, an inhibitor of tyrosine hydroxylase. Activity was monitored for next 10 5-min sessions, during which negligible activity was recorded. Each animal was then given a dose of apomorphine (2.0 mg/kg, i.p.), a dopamine receptor agonist. Activity monitored for an additional 10 5-min sessions showed a recovery of activity. Each point is data collected over 5 min periods for a total of 120 min. Error bars are SEM for control group (n = 6) and for mutants (n = 4). Note that inhibition of tyrosine hydroxylase results in abolishment of hyperactivity, followed by recovery of the activity to control animal levels by administration of dopamine agonist. WT, Wild type.
To further confirm the involvement of the dopamine system, we examined the effect of inhibition of dopamine synthesis (Fig. 8B) as described previously for dopamine transporter knock-out mice (Gainetdinov et al., 1999). When α-methyl-tyrosine (an inhibitor of tyrosine hydroxylase, the rate-limiting enzyme in catecholamine biosynthesis) was injected intraperitoneally, the hyperactivity of the FFCC mice was greatly attenuated within 30 min and remained so for the 50 min duration of the test. These mice were not asleep or sedated; if disturbed, they moved to a new location.
We next asked whether the apparently high levels of dopamine released had resulted in the downregulation of dopamine receptors in the mutant mice. Thus, after a 50 min test session after α-methyl-tyrosine administration (while tyrosine hydroxylase was still inhibited and catecholamine synthesis still blocked), mutant and control mice were injected with apomorphine, the nonselective agonist of all dopamine receptors (Fig. 8B). The mice regained activity, suggesting that the dopamine receptors were not downregulated. Furthermore, the level and pattern of apomorphine-induced locomotor activity in mutants was more similar to controls than to their original characteristic hyperactive pattern. This suggests that high-level release of dopamine is still inhibited and that the resulting normal activity observed is simply attributable to activation of the receptors by the normal level of agonist supplied exogenously.
We then asked whether there was a change in the numbers or survival of midbrain dopaminergic neurons (Fig. 9A). Immunolabeling of parallel sections with anti-tyrosine hydroxylase to identify dopaminergic neurons reveled no obvious differences in the staining pattern of control and mutant mice, nor were differences observed in the numbers of apoptotic cells (data not shown). Interestingly, an FGFR1 conditional knock-out mouse also displays hyperactive locomotion (Shin et al., 2004). This was attributed to the loss of glutamatergic pyramidal neurons in the frontal and temporal cortical areas. No such abnormality was observed in the cortical structure or the pattern of pyramidal neurons in the FFCC hyperactive mouse (data not shown). Thus, the lack of structural abnormalities in either dopamine neurons or the cerebral cortical areas linked to motivational and motor activity suggests that functional, rather than anatomical, abnormalities in the dopaminergic system are the likely cause for the hyperactive behavior.
Figure 9.
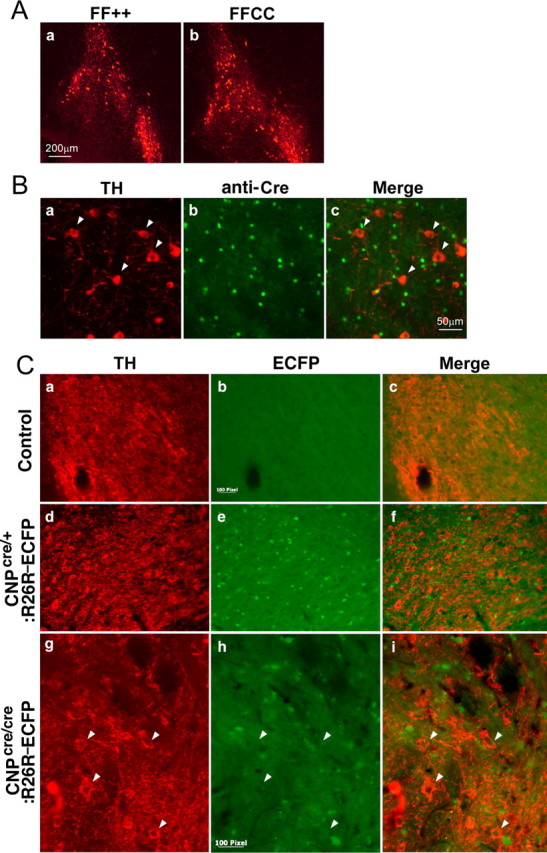
Immunohistochemistry with anti-tyrosine hydroxylase and CNP–Cre expression. A, Anti-tyrosine hydroxylase immunolabeling showing no change in the expression pattern of midbrain tyrosine hydroxylase-positive dopaminergic neurons of FFCC mice compared with controls. B, Double labeling of midbrain dopaminergic neurons with anti-tyrosine hydroxylase (TH) (a) and anti-Cre (b) in FFCC hyperactive mice shows that Cre is not coexpressed with tyrosine hydroxylase-positive dopaminergic neurons. C, Tyrosine hydroxylase (TH) immunolabeling of control (a–c) or CNP–Cre;ROSA26–ECFP (R26R–ECFP) reporter mice heterozygous (d–f) or homozygous (g–i) for the Cre allele. Note the mutually exclusive expression of ECFP and TH in mice with either one or two alleles of Cre. Scale bars: 100 pixel = 50 μm.
We next considered whether the functional abnormalities were in dopaminergic neurons or in OLs. In CNP–Cre conditional knock-out mice, Fgfr2 functions should in principle be eliminated only from OLs. However, although Cnp1 is selectively expressed by OL-lineage cells, it is possible that a low level of Cnp1 promoter-driven Cre recombinase expression could also occur in midbrain dopaminergic neurons. We used three different approaches to examine this possibility, immunostaining midbrain dopaminergic neurons with anti-tyrosine hydroxylase from (1) FFCC mutant mice double labeled with anti-Cre (Fig. 9B), (2) CNP–enhanced green fluorescent protein (EGFP) transgenic mice (Aguirre and Gallo, 2004) (unpublished data), or (3) mice derived from intercrosses of CNP–Cre mice with the ROSA26 strain of reporter mice carrying a floxed STOP allele of enhanced cyan fluorescent protein (ECFP) (Srinivas et al., 2001) in which ECFP is synthesized only in cells in which Cre activity is present (Fig. 9C). In all three conditions, CNP/Cre-expressing cells were distinct from neurons expressing tyrosine hydrolase; that is, no evidence was found for the expression of CNP/Cre in midbrain dopaminergic neurons.
Discussion
Multiple lines of evidence predict an important role for Fgfr2 in OL differentiation and myelin membrane formation (Bansal, 2002; Fortin et al., 2005). Thus, the absence of defects of OL development and myelination in Fgfr2 conditional knock-out mice was unexpected. Nevertheless, a significant subset of these mutants, almost exclusively mice lacking both CNP and Fgfr2 expression (FFCC), displayed dramatic hyperactivity, with even minor environmental stimuli causing rapid and prolonged hyperlocomotion. This behavior is apparently related to the dopaminergic system because administration of dopamine receptor antagonists or dopamine synthesis inhibitors attenuate this hyperactivity.
Although OLs have dopamine receptors (Bongarzone et al., 1998; Howard et al., 1998; Rosin et al., 2005), how modulation of these by Fgfr2 and CNP elimination would lead to hyperactivity is not clear. It is likely that the hyperactive behavior of FFCC mice is related to functional abnormalities in dopaminergic neurons rather than dopamine receptor-mediated alterations in OLs. In principle, Fgfr2 function in these conditional knock-out mice should be eliminated from only CNP-expressing OLs. Nevertheless, Cre-mediated recombination might also occur in neurons if low, but effective, levels of CNP promoter activity were present in them. Consistent with this idea, CNP promoter activity has been reported in olfactory bulb interneurons in transgenic (CNP–EGFP) mice (Aguirre and Gallo, 2004). However, we did not observe EGFP expression in midbrain dopaminergic neurons of these CNP–EGFP mice. Furthermore, in our mutant mice generated by a knock-in procedure, we also did not detect Cre immunoreactivity in dopaminergic neurons. Finally, in CNP–Cre;ROSA26–ECFP reporter mice, no CNP–Cre activity was identified in midbrain dopaminergic neurons, regardless of the presence of one or two alleles of Cre. Thus, a twofold Cre gene dosage in FFCC mice is also unlikely to cause recombination in neurons. However, CNP–Cre-mediated neuronal recombination, should it be present, would still fail to explain why, almost exclusively, homozygous Fgfr2/CNP–Cre mice are affected. Furthermore, the organization of neuronal layers of other regions linked to motivational and motor activity, such as frontal and temporal cerebral cortex, also did not show any abnormalities in these mice.
We therefore favor the notion that the hyperactivity of FFCC mice is caused by the combined elimination of Fgfr2 and CNP function in OLs, which then indirectly affects neuronal function leading to hyperactive behavior. Exactly how CNP and Fgfr2 signaling interact in cis and how loss of these proteins in OLs affects neurons remains to be defined. Bidirectional signaling between neurons and myelinating glia is a current forefront of myelin biology (for review, see Menon et al., 2003; Sherman and Brophy, 2005). For example, genetic defects of OLs, including Cnp1 function, can cause progressive axonal loss without disrupting myelination (Griffiths et al., 1998; Lappe-Siefke et al., 2003). Furthermore, the elimination of the OL/myelin lipids GalC and sulfatide leads to disorganized neuronal proteins at the nodes of Ranvier and axonal loss but grossly normal compact myelin (Coetzee et al., 1996; Garcia-Fresco et al., 2006). Thus, we envision that OL function could be perturbed in FFCC mice and lead to abnormal OL–neuron interactions without significant structural changes of myelin itself. However, because the axons of dopaminergic neurons are unmyelinated, neuronal functions would, in this model, be influenced by OLs independently of myelin. Indeed, the cell bodies of dopaminergic neurons are in close association with OLs (Fig. 9). Interestingly, a polymorphism of the human CNP1 gene, defined by downregulation of CNP (and Fgfr2) mRNA expression, has been associated recently with schizophrenia, a condition also intimately related to the brain dopamine system (Peirce et al., 2006) [Dr. V. Haroutunian (Bronx Veterans Administration, Bronx, NY), personal communications].
Although hyperactivity occasionally has been observed in certain knock-out mice, the high levels and intensity of the hyperactivity in our affected animals is matched only by reports of the dopamine transporter knock-out (Giros et al., 1996; Jones et al., 1998; Spielewoy et al., 2000; Ralph et al., 2001a), knock-outs related to dopamine signaling such as G-protein Go (Jiang et al., 1998), or phosphodiesterase 1B (Reed et al., 2002). Specifically, the motor hyperactivity with disrupted locomotor habituation and exploratory behavior in response to external stimuli of dopamine transporter-null mice is attributable to abnormally high extracellular levels of dopamine.
Mammalian dopaminergic neurons fire action potentials in two modes, a slow steady rate when in a nonstressful state (“tonic” mode) or rapid bursting spikes (“phasic” mode) in a stressful or rewarding stimulation (Schultz et al., 1992; Rebec et al., 1997). In the absence of arousal stimuli, the FFCC mice engaged in normal locomotion similar to that of control animals. It was only after environmental stimuli that they displayed hyperactive behavior. Thus, the hyperactivity appears to involve phasic levels of dopamine release correlated in previous studies with physiological bursting effects at the level of the cell body (Floresco et al., 2003). How FGF signaling, hyperactivity, and the dopamine system are related is not clear. However, the ligands FGF1 and FGF2 are present in regions pertinent to the dopamine system during normal development and are expressed in adult brain (Bean et al., 1992) at levels that can be experimentally modified (Chadi et al., 1994; Flores et al., 1998, 2000; Moroz et al., 2003; Bustos et al., 2004; Fumagalli et al., 2006). Furthermore, hyperactivity in mice has also been associated with attenuation of Fgfr1 signaling and loss of glutamatergic pyramidal neurons in frontal and temporal cortex (Shin et al., 2004), and transgenic mice expressing dominant-negative Fgfr1 in dopaminergic neurons display schizophrenia-like syndromes associated with high dopamine levels and hyperactivity (Klejbor et al., 2006).
Our finding that only a subset of FFCC mice displayed hyperactivity could in principle be attributable to an unlinked autosomal-recessive gene defect generated inadvertently by embryonic stem cell selection. However, the hyperactive phenotype was not lost over several generations of breeding over a period of more than 2 years. Moreover, this phenotype was never observed in the parental homozygous CNP-null or Fgfr2 floxed mice. Thus, any other mutation could not be on a separate chromosome but, if present at all, would have to be closely linked to either the Cnp1 or the Fgfr2 locus to be retained over several generations. However, the observed concordance of only ∼38% would contradict such a close linkage. Thus, it is far more likely that the Cnp1 and Fgfr2 genes interact functionally in OLs but that the penetrance of the double mutation is partial at the level of behavior. Possible explanations include the effect of behaviorally relevant modifier genes that may differ among strains of mice (Ralph et al., 2001a; Linder, 2006).
Why does the absence of Fgfr2 result in no major alterations of myelination? Functional compensation by Fgfr1 or Fgfr3 is certainly possible. In support of this, FGFR1, FGFR-2, and FGFR-3 have overlapping binding specificities and significant homology in their signaling domains, and signaling redundancy has been observed for Fgfr1 and Fgfr2 (Wang et al., 1994; Hebert, 2005). The generation of mice lacking both Fgfr1 and Fgfr2 is expected to help resolve this issue. Furthermore, Fgfr2 signaling may cooperate with other growth factors (Bansal et al., 2003) and play an “enhancing” rather than “essential” function in myelination. Nevertheless, there is evidence of significant specificity in FGF receptor signaling as well. The expression of the three FGF receptors is differentially regulated, playing different roles during OL-lineage progression from progenitors to myelin-producing cells (Bansal et al., 1996; Fortin et al., 2005). For example, activation of Fgfr1 in mature OLs leads to downregulation of myelin proteins and reentry into the cell cycle, whereas activation of Fgfr2 resulted in process elongation (Fortin et al., 2005). Complete knock-outs of specific FGF receptors have indicated that signals mediated by one receptor are not rescued by those mediated by another (Deng et al., 1994; Arman et al., 1998). The fact that OL differentiation remains unaffected in mice lacking Fgfr2 (present study), but was delayed in mice lacking Fgfr3 (Oh et al., 2003), further shows the specificity of FGF receptor signaling in the OL-lineage cells.
One interesting difference between control and Fgfr2 knock-out mice stands out. Despite normal levels of myelin proteins, the level of MOG dimer was significantly elevated in these mice. Based on native PAGE and crystalographic studies, it has been proposed that MOG can form a homodimer, possibly acting as an adhesion molecule linking neighboring myelinated fibers (Clements et al., 2003). Cross-linking MOG leads to cascades of signal transduction (Marta et al., 2005); a role for Fgfr2 signaling in this process, although currently unspecified, seems possible.
In summary, conditional Fgfr2 knock-out mice exhibit an unexpected absence of major defects related to OL development or myelination, suggesting that compensatory signaling mechanisms exist to facilitate myelination. However, a significant subset of Fgfr2 mutant mice that also lack CNP display a dramatic hyperactive behavior related to dysregulation of the dopaminergic system, suggesting that the hyperactivity may be attributable to a combinatorial lack of both Fgfr2 and CNP. Nevertheless, dopamine appears to be an important component of the hyperactivity and may provide insights into human conditions that feature manic behavior.
Footnotes
This work was supported by National Institutes of Health Grant NS 38878 and the Deutsche Forschungsgemeinschaft Center for Molecular Physiology of the Brain. We thank F. Kim for helping with the immunoblots, Drs. P. Maye and D. Rowe (University of Connecticut Medical School, Farmington, CT) for providing Rosa26–ECFP mouse and helping with the photography, Dr. V. Gallo (Children's National Medical Center, Washington, DC) for supplying us with adult brain from CNP–EGFP transgenic mouse, and Drs. K. Morest and S. Clark (University of Connecticut Medical School, Farmington, CT) for their insightful comments.
References
- Aguirre A, Gallo V. Postnatal neurogenesis and gliogenesis in the olfactory bulb from NG2-expressing progenitors of the subventricular zone. J Neurosci. 2004;24:10530–10541. doi: 10.1523/JNEUROSCI.3572-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arman E, Haffner-Krausz R, Chen Y, Heath JK, Lonai P. Targeted disruption of fibroblast growth factor (FGF) receptor 2 suggests a role for FGF signaling in pregastrulation mammalian development. Proc Natl Acad Sci USA. 1998;95:5082–5087. doi: 10.1073/pnas.95.9.5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal R. Fibroblast growth factors and their receptors in oligodendrocyte development: implications for demyelination and remyelination. Dev Neurosci. 2002;46:24–35. doi: 10.1159/000064944. [DOI] [PubMed] [Google Scholar]
- Bansal R, Kumar M, Murray K, Morrison RS, Pfeiffer SE. Regulation of FGF receptors in the oligodendrocyte lineage. Mol Cell Neurosci. 1996;7:263–275. doi: 10.1006/mcne.1996.0020. [DOI] [PubMed] [Google Scholar]
- Bansal R, Winkler S, Bheddah S. Negative regulation of oligodendrocyte differentiation by galactosphingolipids. J Neurosci. 1999;19:7913–7924. doi: 10.1523/JNEUROSCI.19-18-07913.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal R, Lakhina V, Remedios R, Tole S. Expression of FGF receptors 1, 2, 3 in the embryonic and postnatal mouse brain compared with Pdgfralpha, Olig2 and Plp/dm20: implications for oligodendrocyte development. Dev Neurosci. 2003;25:83–95. doi: 10.1159/000072258. [DOI] [PubMed] [Google Scholar]
- Bean AJ, Oellig C, Pettersson RF, Hokfelt T. Differential expression of acidic and basic FGF in the rat substantia nigra during development. NeuroReport. 1992;3:993–996. doi: 10.1097/00001756-199211000-00012. [DOI] [PubMed] [Google Scholar]
- Bongarzone ER, Howard SG, Schonmann V, Campagnoni AT. Identification of the dopamine D3 receptor in oligodendrocyte precursors: potential role in regulating differentiation and myelin formation. J Neurosci. 1998;18:5344–5353. doi: 10.1523/JNEUROSCI.18-14-05344.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun PE, Lee J, Gravel M. 2′, 3′-Cyclic nucleotide 3′-phosphodiesterase: structure, biology, and function In: Myelin biology and disorders. In: Lazzarini RA, editor. London: Elsevier Science; 2004. pp. 499–522. [Google Scholar]
- Bustos G, Abarca J, Campusano J, Bustos V, Noriega V, Aliaga E. Functional interactions between somatodendritic dopamine release, glutamate receptors and brain-derived neurotrophic factor expression in mesencephalic structures of the brain. Brain Res Brain Res Rev. 2004;47:126–144. doi: 10.1016/j.brainresrev.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Chadi G, Cao Y, Pettersson RF, Fuxe K. Temporal and spatial increase of astroglial basic fibroblast growth factor synthesis after 6-hydroxydopamine-induced degeneration of the nigrostriatal dopamine neurons. Neuroscience. 1994;61:891–910. doi: 10.1016/0306-4522(94)90411-1. [DOI] [PubMed] [Google Scholar]
- Clements CS, Reid HH, Beddoe T, Tynan FE, Perugini MA, Johns TG, Bernard CC, Rossjohn J. The crystal structure of myelin oligodendrocyte glycoprotein, a key autoantigen in multiple sclerosis. Proc Natl Acad Sci USA. 2003;100:11059–11064. doi: 10.1073/pnas.1833158100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee T, Fujita N, Dupree J, Shi R, Blight A, Suzuki K, Popko B. Myelination in the absence of galactocerebroside and sulfatide: normal structure with abnormal function and regional instability. Cell. 1996;86:209–219. doi: 10.1016/s0092-8674(00)80093-8. [DOI] [PubMed] [Google Scholar]
- Cohen RI, Chandross KJ. Fibroblast growth factor-9 modulates the expression of myelin related proteins and multiple fibroblast growth factor receptors in developing oligodendrocytes. J Neurosci Res. 2000;61:273–287. doi: 10.1002/1097-4547(20000801)61:3<273::AID-JNR5>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Crawley JN. Motor functions. In: What's wrong with my mouse? Behavioral phenotype of transgenic and knockout mice. In: Crawley JN, editor. New York: Wiley; 2000. pp. 47–65. [Google Scholar]
- Deng CX, Wynshaw-Boris A, Shen MM, Daugherty C, Ornitz DM, Leder P. Murine FGFR-1 is required for early postimplantation growth and axial organization. Genes Dev. 1994;8:3045–3057. doi: 10.1101/gad.8.24.3045. [DOI] [PubMed] [Google Scholar]
- Flores C, Rodaros D, Stewart J. Long-lasting induction of astrocytic basic fibroblast growth factor by repeated injections of amphetamine: blockade by concurrent treatment with a glutamate antagonist. J Neurosci. 1998;18:9547–9555. doi: 10.1523/JNEUROSCI.18-22-09547.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores C, Samaha AN, Stewart J. Requirement of endogenous basic fibroblast growth factor for sensitization to amphetamine. J Neurosci. 2000;20(RC55):1–5. doi: 10.1523/JNEUROSCI.20-02-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floresco SB, West AR, Ash B, Moore H, Grace AA. Afferent modulation of dopamine neuron firing differentially regulates tonic and phasic dopamine transmission. Nat Neurosci. 2003;6:968–973. doi: 10.1038/nn1103. [DOI] [PubMed] [Google Scholar]
- Ford-Perriss M, Abud H, Murphy M. Fibroblast growth factors in the developing central nervous system. Clin Exp Pharmacol Physiol. 2001;28:493–503. doi: 10.1046/j.1440-1681.2001.03477.x. [DOI] [PubMed] [Google Scholar]
- Fortin D, Rom E, Sun H, Yayon A, Bansal R. Distinct fibroblast growth factor (FGF)/FGF receptor signaling pairs initiate diverse cellular responses in the oligodendrocyte lineage. J Neurosci. 2005;25:7470–7479. doi: 10.1523/JNEUROSCI.2120-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruttiger M, Karlsson L, Hall AC, Abramsson A, Calver AR, Bostrom H, Willetts K, Bertold CH, Heath JK, Betsholtz C, Richardson WD. Defective oligodendrocyte development and severe hypomyelination in PDGF-A knockout mice. Development. 1999;126:457–467. doi: 10.1242/dev.126.3.457. [DOI] [PubMed] [Google Scholar]
- Fumagalli F, Pasquale L, Racagni G, Riva MA. Dynamic regulation of fibroblast growth factor 2 (FGF-2) gene expression in the rat brain following single and repeated cocaine administration. J Neurochem. 2006;96:996–1004. doi: 10.1111/j.1471-4159.2005.03627.x. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Jones SR, Caron MG. Functional hyperdopaminergia in dopamine transporter knock-out mice. Biol Psychiatry. 1999;46:303–311. doi: 10.1016/s0006-3223(99)00122-5. [DOI] [PubMed] [Google Scholar]
- Garcia-Fresco GP, Sousa AD, Pillai AM, Moy SS, Crawley JN, Tessarollo L, Dupree JL, Bhat MA. Disruption of axo–glial junctions causes cytoskeletal disorganization and degeneration of Purkinje neuron axons. Proc Natl Acad Sci USA. 2006;103:5137–5142. doi: 10.1073/pnas.0601082103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- Gravel M, Di Polo A, Valera PB, Braun PE. Four-kilobase sequence of the mouse CNP gene directs spatial and temporal expression of lacZ in transgenic mice. J Neurosci Res. 1998;53:393–404. doi: 10.1002/(SICI)1097-4547(19980815)53:4<393::AID-JNR1>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Griffiths I, Klugmann M, Anderson T, Yool D, Thomson C, Schwab MH, Schneider A, Zimmermann F, McCulloch M, Nadon N, Nave KA. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science. 280:1610–1613. doi: 10.1126/science.280.5369.1610. [DOI] [PubMed] [Google Scholar]
- Hebert JM. Unraveling the molecular pathways that regulate early telencephalon development. Curr Top Dev Biol. 2005;69:17–37. doi: 10.1016/S0070-2153(05)69002-3. [DOI] [PubMed] [Google Scholar]
- Howard S, Landry C, Fisher R, Bezouglaia O, Handley V, Campagnoni A. Postnatal localization and morphogenesis of cells expressing the dopaminergic D2 receptor gene in rat brain: expression in non-neuronal cells. J Comp Neurol. 1998;391:87–98. doi: 10.1002/(sici)1096-9861(19980202)391:1<87::aid-cne8>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Jacobs EC, Pribyl TM, Kampf K, Campagnoni C, Colwell CS, Reyes SD, Martin M, Handley V, Hiltner TD, Readhead C, Jacobs RE, Messing A, Fisher RS, Campagnoni AT. Region-specific myelin pathology in mice lacking the golli products of the myelin basic protein gene. J Neurosci. 2005;25:7004–7013. doi: 10.1523/JNEUROSCI.0288-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Gold M, Boulay G, Spicher K, Peyton M, Brabet P, Srinivasan Y, Rudolph U, Ellison G, Birnbaumer L. Multiple neurological abnormalities in mice deficient in the G protein Go. Proc Natl Acad Sci USA. 1998;95:3269–3274. doi: 10.1073/pnas.95.6.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Jaber M, Giros B, Wightman RM, Caron MG. Profound neuronal plasticity in response to inactivation of the dopamine transporter. Proc Natl Acad Sci USA. 1998;95:4029–4034. doi: 10.1073/pnas.95.7.4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klejbor I, Myers JM, Hausknecht K, Corso TD, Gambino AS, Morys J, Maher PA, Hard R, Richards J, Stachowiak EK, Stachowiak MK. Fibroblast growth factor receptor signaling affects development and function of dopamine neurons—inhibition results in a schizophrenia-like syndrome in transgenic mice. J Neurochem. 2006;97:1243–1258. doi: 10.1111/j.1471-4159.2006.03754.x. [DOI] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet. 2003;33:366–374. doi: 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- Linder CC. Genetic variables that influence phenotype. ILAR J. 2006;47:132–140. doi: 10.1093/ilar.47.2.132. [DOI] [PubMed] [Google Scholar]
- Marta CB, Montano MB, Taylor CM, Taylor AL, Bansal R, Pfeiffer SE. Signaling cascades activated upon antibody cross-linking of myelin oligodendrocyte glycoprotein: potential implications for multiple sclerosis. J Biol Chem. 2005;280:8985–8993. doi: 10.1074/jbc.M413174200. [DOI] [PubMed] [Google Scholar]
- Menon K, Rasband MN, Taylor CM, Brophy P, Bansal R, Pfeiffer SE. The myelin-axolemmal complex: biochemical dissection and the role of galactosphingolipids. J Neurochem. 2003;87:995–1009. doi: 10.1046/j.1471-4159.2003.02075.x. [DOI] [PubMed] [Google Scholar]
- Messersmith DJ, Murtie JC, Le TQ, Frost EE, Armstrong RC. Fibroblast growth factor 2 (FGF2) and FGF receptor expression in an experimental demyelinating disease with extensive remyelination. J Neurosci Res. 2000;62:241–256. doi: 10.1002/1097-4547(20001015)62:2<241::AID-JNR9>3.0.CO;2-D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake A, Hattori Y, Ohta M, Itoh N. Rat oligodendrocytes and astrocytes preferentially express fibroblast growth factor receptor-2 and -3 mRNAs. J Neurosci Res. 1996;45:534–541. doi: 10.1002/(SICI)1097-4547(19960901)45:5<534::AID-JNR3>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Moroz IA, Rajabi H, Rodaros D, Stewart J. Effects of sex and hormonal status on astrocytic basic fibroblast growth factor-2 and tyrosine hydroxylase immunoreactivity after medial forebrain bundle 6-hydroxydopamine lesions of the midbrain dopamine neurons. Neuroscience. 2003;118:463–476. doi: 10.1016/s0306-4522(02)00974-0. [DOI] [PubMed] [Google Scholar]
- Murtie JC, Zhou Y-X, Le TQ, Armstrong RC. In vivo analysis of oligodendrocyte lineage development in postnatal FGF-2 null mice. Glia. 2005;49:542–554. doi: 10.1002/glia.20142. [DOI] [PubMed] [Google Scholar]
- Oh LY, Denninger A, Colvin JS, Vyas A, Tole S, Ornitz DM, Bansal R. Fibroblast growth factor receptor 3 signaling regulates the onset of oligodendrocyte terminal differentiation. J Neurosci. 2003;23:883–894. doi: 10.1523/JNEUROSCI.23-03-00883.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr-Urtreger A, Bedford MT, Burakova T, Arman E, Zimmer Y, Yayon A, Givol D, Lonai P. Developmental localization of the splicing alternatives of fibroblast growth factor receptor-2 (FGFR2) Dev Biol. 1993;158:475–486. doi: 10.1006/dbio.1993.1205. [DOI] [PubMed] [Google Scholar]
- Padovani-Claudio DA, Liu L, Ransohoff RM, Miller RH. Alterations in the oligodendrocyte lineage, myelin, and white matter in adult mice lacking the chemokine receptor CXCR2. Glia. 2006;54:471–483. doi: 10.1002/glia.20383. [DOI] [PubMed] [Google Scholar]
- Peirce TR, Bray NJ, Williams NM, Norton N, Moskvina V, Preece A, Haroutunian V, Buxbaum JD, Owen MJ, O'Donovan MC. Convergent evidence for 2′,3′-cyclic nucleotide 3′-phosphodiesterase as a possible susceptibility gene for schizophrenia. Arch Gen Psychiatry. 2006;63:18–24. doi: 10.1001/archpsyc.63.1.18. [DOI] [PubMed] [Google Scholar]
- Ralph RJ, Paulus MP, Fumagalli F, Caron MG, Geyer MA. Prepulse inhibition deficits and perseverative motor patterns in dopamine transporter knock-out mice: differential effects of D1 and D2 receptor antagonists. J Neurosci. 2001a;21:305–313. doi: 10.1523/JNEUROSCI.21-01-00305.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralph RJ, Paulus MP, Geyer MA. Strain-specific effects of amphetamine on prepulse inhibition and patterns of locomotor behavior in mice. J Pharmacol Exp Ther. 2001b;298:148–155. [PubMed] [Google Scholar]
- Rasband MN, Trimmer JS. Developmental clustering of ion channels at and near the node of Ranvier. Dev Biol. 2001;236:5–16. doi: 10.1006/dbio.2001.0326. [DOI] [PubMed] [Google Scholar]
- Rebec GV, Grabner CP, Johnson M, Pierce RC, Bardo MT. Transient increases in catecholaminergic activity in medial prefrontal cortex and nucleus accumbens shell during novelty. Neuroscience. 1997;76:707–714. doi: 10.1016/s0306-4522(96)00382-x. [DOI] [PubMed] [Google Scholar]
- Reed T, Repaske D, Snyder G, Greengard P, Vorhees C. Phosphodiesterase 1B knock-out mice exhibit exaggerated locomotor hyperactivity and DARPP-31 phosphorylation in response to dopamine agonists and display impaired spatial leaning. J Neurosci. 2002;22:5188–5197. doi: 10.1523/JNEUROSCI.22-12-05188.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbluth J. Redundant myelin sheaths and other ultrastructural features of the toad cerebellum. J Cell Biol. 1966;28:73–93. doi: 10.1083/jcb.28.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosin C, Colombo S, Calver AA, Bates TE, Skaper SD. Dopamine D2 and D3 receptor agonists limit oligodendrocyte injury caused by glutamate oxidative stress and oxygen/glucose deprivation. Glia. 2005;52:336–343. doi: 10.1002/glia.20250. [DOI] [PubMed] [Google Scholar]
- Saher G, Brugger B, Lappe-Siefke C, Mobius W, Tozawa R, Wehr MC, Wieland F, Ishibashi S, Nave KA. High cholesterol level is essential for myelin membrane growth. Nat Neurosci. 2005;8:468–475. doi: 10.1038/nn1426. [DOI] [PubMed] [Google Scholar]
- Schultz W, Apicella P, Scarnati E, Ljungberg T. Neuronal activity in monkey ventral striatum related to the expectation of reward. J Neurosci. 1992;12:4595–4610. doi: 10.1523/JNEUROSCI.12-12-04595.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman DL, Brophy PJ. Mechanisms of axon ensheathment and myelin growth. Nat Rev Neurosci. 2005;6:683–690. doi: 10.1038/nrn1743. [DOI] [PubMed] [Google Scholar]
- Shin DM, Korada S, Raballo R, Shashikant CS, Simeone A, Taylor JR, Vaccarino F. Loss of glutamatergic pyramidal neurons in frontal and temporal cortex resulting from attenuation of FGFR1 signaling is associated with spontaneous hyperactivity in mice. J Neurosci. 2004;24:2247–2258. doi: 10.1523/JNEUROSCI.5285-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperber BR, Boyle-Walsh EA, Engleka MJ, Gadue P, Peterson AC, Stein PL, Scherer SS, McMorris FA. A unique role for Fyn in CNS myelination. J Neurosci. 2001;21:2039–2047. doi: 10.1523/JNEUROSCI.21-06-02039.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielewoy C, Roubert C, Hamon M, Nosten-Bertrand M, Betancur C, Giros B. Behavioural disturbances associated with hyperdopaminergia in dopamine-transporter knockout mice. Behav Pharmacol. 2000;11:279–290. doi: 10.1097/00008877-200006000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin C-S, William CM, Tanabe Y, Jessell TM, Constantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaccarino FM, Schwartz ML, Raballo R, Rhee J, Lyn-Cook R. Fibroblast growth factor signaling regulates growth and morphogenesis at multiple steps during brain development. Curr Top Dev Biol. 1999;46:179–200. doi: 10.1016/s0070-2153(08)60329-4. [DOI] [PubMed] [Google Scholar]
- Wang JK, Gao G, Goldfarb M. Fibroblast growth factor receptors have different signaling and mitogenic potentials. Mol Cell Biol. 1994;14:181–188. doi: 10.1128/mcb.14.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazaki N, Hosoi Y, Kawabata K, Miyake A, Minami M, Satoh M, Ohta M, Kawasaki T, Itoh N. Differential expression patterns of mRNAs for members of the fibroblast growth factor receptor family, FGFR-1-FGFR-4, in rat brain. J Neurosci Res. 1994;37:445–452. doi: 10.1002/jnr.490370403. [DOI] [PubMed] [Google Scholar]
- Yim SH, Hammer JA, Quarles RH. Differences in signal transduction pathways by which platelet-derived and fibroblast growth factors activate extracellular signal-regulated kinase in differentiating oligodendrocytes. J Neurochem. 2001;76:1925–1934. doi: 10.1046/j.1471-4159.2001.00199.x. [DOI] [PubMed] [Google Scholar]
- Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, Towler DA, Ornitz DM. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development. 2003;130:3063–3074. doi: 10.1242/dev.00491. [DOI] [PubMed] [Google Scholar]