

Abstract
BACKGROUND:
Osteoarthritis (OA) is a highly prevalent degenerative joint disease involving joint cartilage and its surrounding tissues. OA is the leading cause of pain and disability worldwide. At present, there are no disease-modifying OA drugs, and the primary therapies include exercise and nonsteroidal anti-inflammatory drugs until total joint replacement at the end-stage of the disease.
METHODS:
In this review, we summarized the current state of knowledge in genetic and epigenetic associations and risk factors for OA and their potential diagnostic and therapeutic applications.
RESULTS:
Genome-wide association studies and analysis of epigenetic modifications (such as miRNA expression, DNA methylation and histone modifications) conducted across various populations support the notion that there is a genetic basis for certain subsets of OA pathogenesis.
CONCLUSION:
With recent advances in the development of genome editing technologies such as the CRISPR-Cas9 system, these genetic and epigenetic alternations in OA can be used as platforms from which potential biomarkers for the diagnosis, prognosis, drug response, and development of potential personalized therapeutic targets for OA can be approached. Furthermore, genome editing has allowed the development of “designer” cells, whereby the receptors, gene regulatory networks, or transgenes can be modified as a basis for new cell-based therapies.
Keywords: Genetics, Gene editing, Personalized medicine, Osteoarthritis
Introduction
Osteoarthritis (OA) is the most prevalent rheumatic disease worldwide, affecting an estimated 10 percent of the world’s population over the age of 60 [61]. OA is characterized by pain, stiffness, decreased function, instability, deformity and swelling due to irreversible pathological changes in the joint organ system, including articular cartilage, subchondral bone, synovium, and the infrapatellar fat pad [39, 30]. The pathogenesis of OA involves the development of cartilage degeneration, synovial inflammation, subchondral bone remodeling and sclerosis, degeneration of ligaments and meniscus, and hypertrophy of the joint capsule [9]. At present, there are no disease-modifying OA drugs (DMOADs), and the primary therapies include exercise and nonsteroidal anti-inflammatory drugs until total joint replacement at the end-stage of the disease. Clinical risk factors for the development of OA include increasing age, female sex, obesity, repetitive joint overloading, previous joint injury, lower limb deformity, smoking history, and family history of OA [5–18]. This wide array of contributing factors for the initiation and progression of OA supports the notion that OA is not simply one disease, but rather a family of conditions that have similar endpoints that involve a multitude of pathways that lead to joint failure. This diversity in the mechanisms of the etiopathogenesis of OA may contribute to the lack of viable treatment strategies.
While growing evidence suggests a genetic basis for a large proportion of OA incidence [51], it is often difficult to separate causative factors from the combined effects of these environmental contributors, as they frequently happen in combination with each other [7]. However, it is clear that both genetic and environmental risk factors contribute to OA pathogenesis. Epidemiological studies evaluating twin-pair analyses and family-based segregation studies, have demonstrated that genetic susceptibility is also one of key contributors to the development of OA [81, 75, 15–20]. However, while the genetic basis of OA etiology remains an open and active area of investigation, the characterization and analysis of genetic basis of OA pathogenesis provides an exciting platform from which potential biomarkers for the diagnosis, prognosis, drug response and development of potential therapeutic targets for future personalized biological novel treatment strategies for OA can be approached. As such, the purpose of this review is to concisely summarize the state of knowledge in the areas of genetics and genome editing to postulate opportunities for genome engineering for OA applications.
Genetic variants associated with OA
A number of studies have leveraged the opportunity for investigating the genetic underpinnings of OA, collectively reporting over 80 genetic variants subjected to candidate gene association analysis with the risk of OA [68] (Table 1). Among them, a single nucleotide polymorphism (SNP) rs143383 showed consistent and robust associations with OA after its initial report in 2007 [20–26]. The rs143383 is located in the 3′ untranslated region (3′UTR) of the growth and differentiation factor 5 gene, GDF5, which is also known as cartilage-derived morphogenetic protein 1, CDMP1, is a member of the transforming growth factor-β (TGF-β) superfamily and an extracellular signaling molecule that participates in the development, maintenance, and repair of bone, cartilage, and other tissues of the synovial joint [65, 44]. The SNP rs143383 is a common C to T transition which mediates reduced GDF5 transcription relative to the C allele [56, 74, 25]. GDF5 is an OA-associated locus from which this SNP is found. Decreased mRNA and protein levels for Gdf5 in mice can contribute to OA-like phenotype [19, 59]. More recently, OA susceptibility has been coded by a comparison of the frequency of DNA polymorphisms in individuals with osteoarthritis when compared to those without osteoarthritis (disease-free controls) in the form of candidate gene-based analyses or genome-wide association studies (GWAS). The Arthritis Research UK Osteoarthritis Genetics (arcOGEN) Consortium Study was the first GWAS for knee OA with total joint replacement to report multiple, independent association signals that replicated at a level considered significant after accounting for the multiple tests that are performed in a GWAS (p value < 5 × 10−8) [85]. This analysis identified five novel loci at genome-wide significance (GLT8D1/GNL3, ASTN2, FILIP1/SENP6, KLHDC5/PTHLH, and CHST11) and three novel loci at near genome-wide significance (TP63, FTO, and SUPT3H/CDC5L). Subsequently, several other large-scale GWAS of hand, hip, and knee OA have been published in European Caucasians, providing a dozen genome-wide significant loci that include ALDH1A2 for hand OA [78], DOT1L, NCOA3, BOP1, LRCH1, STT3B, GADL1, TGFA, PIK3R1, FGFR3, TREH, COMP, and CHADL for hip OA [31–14], and LSP1P3, GDF5, FTO, mitochondrial DNA variants for knee OA [84, 33].
Table 1.
Summary of genetic variants in OA-related GWAS studies
Epigenetics and the pathogenesis of OA
Epigenetics refers to heritable changes in gene expression or phenotype without changes in the DNA sequence [24]. Three primary mechanisms of epigenetic changes have been documented in OA pathogenesis: DNA methylation, histone modification and noncoding RNAs such as microRNAs [71]. Epigenetic changes regulate gene expression either by affecting gene transcription or by acting post-transcriptionally, leading to changes in the levels of the encoded protein. Normally functioning somatic cells, including articular chondrocytes, are subjected to epigenetic mechanisms that aid in stabilizing their phenotype [63]. These epigenetic changes can occur in chondrocytes in response to environmental factors, including diet or aging, which lead to the loss of normal epigenetic control [60]. Epigenetically modified chondrocytes can change their phenotype and function to overexpress cartilage-degenerating proteases and pro-inflammatory mediators. This phenotypic shift is thought to disrupt the homeostatic balance and contribute to the progression of OA through the changes in gene expression of transcription factors (RUNX2, NFAT1, and SOX9), pro- or anti-inflammatory cytokines (tumor necrosis factor-alpha [TNF-α], interleukin-1 beta [IL-1β], IL-6, inducible nitric oxide synthase [NOS2] and IL-8, matrix-degrading proteinases (matrix metalloproteinase-3 [MMP-3], MMP-9, MMP-13, ADAMTS-4, and ADAMTS-5) and extracellular matrix proteins (COL2A1, COL9A1, and ACAN) in articular cartilage [9, 65, 66, 6].
DNA methylation, the best characterized epigenetic mechanism, is defined by the addition of a methyl group (CH3) to a cystosine of CpG sites to form methylated cystosine by DNA methyltransferases. Studies assessing DNA methylation arrays have shown that OA and non-OA cartilage have differentially methylated genes which typically harbor CpG sites suggesting that the epigenetic regulation of gene expression via DNA methylation contributes on the pathogenesis of OA [67, 34].
Furthermore, a direct, functional relationship has been demonstrated between epigenetics and genetics at alleles implicated in OA-risk. GDF5 and DIO2 (iodothyronine deiodinase 2) has been reported to be subject to epigenetic regulation related to genotype and gene transcription. As described earlier, the GDF5 functional SNP, rs143383, is a particular risk factor for knee osteoarthritis, and also is thought to effect OA when differentially methylated [26]. Functional analyses using human normal and OA cartilage have showed that differential DNA methylation of rs143383 modulates the binding of SP1, SP3, and DEAF1 repressor proteins and therefore alters the expression differences between the C and T alleles [64]. Furthermore, a CpG site located 4 base pairs upstream of rs143383 showed highly significant demethylation in osteoarthritis knee cartilage compared with osteoarthritis hip cartilage, which correlates with reduced expression of the gene and may be responsible for the specific effect of rs143383 on knee OA [64]. DIO2 transcribes iodothyronine deiodinase 2 that catalyzes the conversion of intracellular inactive thyroxine (T4) to the bioactive thyroid hormone (T3). A common DIO2 haplotype composed of the C-allele of SNP rs225014, and the C-allele of SNP rs12885300 has been known to be associated with OA [54]. A recent functional analysis of DIO2, including DNA methylation studies, reported that differential methylation of CpGs located upstream of the gene correlated with DIO2 expression changes, and that these effects were particularly striking for individuals harboring the risk-conferring allele of rs225014 [10].
Genome and epigenome engineering tools
Genome engineering has at least two roles in elucidating the natural history of disease states: (1) identify the responsible genes for a particular disease and (2) to facilitate the functional validation of the identified genes and the development and study of disease models. The ability to precision-edit the genome of mammalian cells allows for a deeper mechanistic understanding while minimizing off-target effects [80, 35]. The era of genome editing began in the late 1970s with the successful exchange of pieces of yeast DNA via the homologous recombination system [38, 70]. With this technique, single and multiple knockouts were generated for applications in functional characterization [38, 70]. In the late 1980s, gene-targeting technologies using embryonic stem cells were created that were proficient in homologous recombination (HR) [53]. However, gene targeting was only applicable to homologous recombination-proficient cells and therefore the application to other cell types and eukaryotic systems was limited. The efficacy of homologous recombination is improved by the discovery of engineered nucleases, such as zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs), that can generate site-specific double-strand breaks (Fig. 1) [16].
Fig. 1.
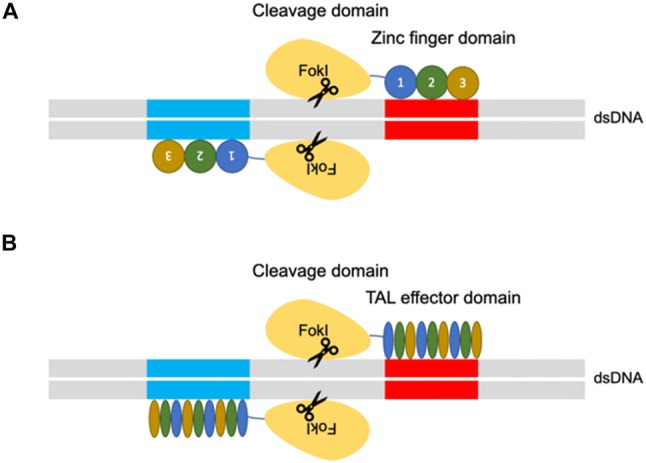
A Zinc finger nucleases (ZFNs) and B transcription activator-like effector nucleases (TALENs) genome engineering platform. The ZFNs and TALENs are engineered to bind a desired sequence, and then fused to the FokI endonuclease to create targeted double strand breaks
There are two major repair pathways for double strand breaks; non-homologous end-joining and homologous recombination (Fig. 2) [15]. The non-homologous end-joining pathways are active throughout the cell cycle and are therefore the most predominant repair mechanism in mammalian cells, despite being error-prone and resulting in insertions or deletions of nucleotides (indels), which may cause a frameshift and, effectively, a functional gene knockout when occurring in the coding region [17]. The homologous recombination pathways are highly precise because they utilize a DNA template with ends that are homologous to the break site. These techniques have been used mainly in genome editing to rewrite the DNA sequence and generate gene or protein variants. Researchers generated hybrid proteins by fusion of a DNA-binding module by altering the DNA-binding domains of transcription factors (TFs), zinc finger proteins (ZFPs) or TAL effectors with the DNA-cleaving module from the restriction endonuclease, Fok1 [45, 55]. In those nuclease platforms, a pair of ZFNs or TALENs must target adjacent sites in the genome to bring the two Fok1 domains together because the endonucleolytic domain of Fok1 acts like a dimer [45, 55]. However, protein engineering is required to obtain site specificity. The process of engineering and optimizing specific combinations of ZFNs or TALENs modules for new sequences is not trivial and is an extensive process [57].
Fig. 2.

CRISPR-Cas9 Genome Engineering Platform. Cas9 is guided by a guide RNA (sgRNA) to induce double-strand DNA breaks at a desired genomic locus, the damaged DNA can be repaired by non-homologous end-joining or homologous recombination, which results in controlled editing of the genome
CRISPR-Cas9 genome editing platform
One of the most recently discovered nuclease platforms is the clustered regularly interspaced short palindromic repeat (CRISPR)-CRISPR-associated (Cas) endonuclease system. The CRISPR-Cas9 genome editing platform has allowed for rapid and efficient precision edits in mammalian genomes [23, 46]. The CRISPR-Cas systems are essential in the prokaryote adaptive immune system. CRISPR is a family of DNA sequences in prokaryotes which contain short segments of DNA from viruses which are used to detect and destroy DNA from similar viruses at subsequent attacks as a defense mechanism. The CRISPR-Cas system has six different major types (type I–VI) based on their genetic content and structural differences of their effector proteins. Among them, type II is the most studied and consists of two main components of the system: an operon (codes for Cas9 protein) and CRISPR, which provide a template for synthesizing CRISPR RNA (crRNA) to direct nuclease activity [21]. To achieve site-specific DNA recognition and cleavage, crRNA has to bind directly to Cas9 through an accessary trans-activating CRISPR RNA (tracrRNA) [41]. To use this mechanism in genome editing, a single synthetic guide RNA was developed by combining crRNA and tracrRNA to direct the Cas9 endonuclease to its specific site in the genome in a sequence-specific manner (Fig. 1) [63–52]. Once the sgRNA hybridize to the target DNA, Cas9 is activated and cleave the targeted site [52]. A protospacer adjacent motif (PAM) which is a 5′-NGG-3′ trinucleotide sequence at the target sequence is essential for Cas9 binding to DNA [76]. While off-target effects are one key concern of Cas9 binding [5], there have been efforts to reduce off-target activity of Cas9 using gene-engineering [3]. There are two groups of gene-engineered Cas9, mutant proteins and split Cas9 proteins. Catalytically inactive, nuclease-deficient Cas9 (dCas9) is a form of mutant Cas9 and various fusions with transcriptional activator, repressor and recruitment domains have been used to modulate gene expression at targeted loci without introducing irreversible mutations to the genome [29, 62].
Applications of genome-engineering technologies to osteoarthritis treatments
One potential therapeutic application of genome editing technology is the engineering of cell-based anti-cytokine therapies under endogenous promoter sequences by editing in key transcripts for anti-inflammatory molecules. Using the successful framework of biologics in rheumatoid arthritis (RA), where a variety of protein therapies have been developed and applied, key receptors for inflammatory mediators can be inserted into cells under endogenous promoters [72]. While advancements have led to the development of several effective RA treatment modalities for RA pathogenesis, which is understood to be a systemic inflammatory disease, treatments fail or cease to benefit patients up to 50% of the time [72]. With growing appreciation that both systemic and local inflammatory mediators may also play a role in the pathogenesis of OA, there has been increasing investigation into therapeutics that may be beneficial in this context [49, 37], such as a cell-based anti-cytokine therapy that senses and responds to endogenous levels of inflammatory mediators in OA, i.e., TNF-α or IL-1 [47, 48]. However, while it is important to note that a better understanding of the individual roles for IL-1, TNF-α, and other cytokines is required in OA [42], evaluating new approaches to delivering these therapies could fill a meaningful gap in treatment for the prevention of other inflammatory conditions. A comprehensive review of genome editing for personalized arthritis therapies can be found here [1].
Genome-edited cell-based anti-cytokine therapy also may overcome several important limitations of existing biologic drugs. Continuous use of conventional disease-modifying anti-rheumatic drugs (DMARDs) and the newer biologics can lead to significant adverse effects for RA patients due to continued high-level delivery of DMARDS [79]. As the severity of symptomatic arthritic diseases and pain can fluctuate over time [39], development of specific therapeutic strategies that can sense and respond to varying degrees of endogenous inflammatory mediators in OA may mitigate unwanted adverse effects. In recent studies, CRISPR-Cas9 gene editing was used to develop an autoregulated anti-cytokine stem cell system in mouse induced pluripotent stem cells (iPSCs), which provides the opportunity to theoretically differentiate them into a variety of cell types [11, 12]. These cells were designed to transcribe the soluble receptor for TNF-α (sTNFR1), interleukin-1 receptor antagonist (IL1Ra), or luciferase (control) in a feedback-controlled manner driven by the endogenous macrophage chemoattractant protein-1 (Ccl2) promoter (Fig. 3). Given that Ccl2 and NF-kB signalling are implicated in the onset and progression of pain and structural damage OA [40], this genome-edited artificial gene circuit is an attractive therapeutic approach for evaluation in both in vitro and in vivo OA model systems. In vitro characterization of Ccl2-based genome-edited cells reveals that these cells can reduce RA-relevant inflammatory mediators when challenged with an inflammatory stimulus. Ongoing work is characterizing these cells in vivo in reponse to supra-physiological levels of inflammation, and to disease-relevent stimuli in murine models of both RA and OA.
Fig. 3.

CRISPR-Cas9 Ccl2-edited Gene Circuit Transcribing Soluble Tumor Necrosis Factor-alpha Receptor (sTNFR1) Under the Macrophage Chemoattractant-1 Locus. The transcription and synthesis of sTNFR1 in response to TNF-α-induced expression of Ccl2 results in a negative feedback system that reduces TNF-α driven inflammation. Similar systems have been developed to produce either interleukin-1 antagonist (IL1Ra) to inhibit IL-1 signaling, or produce luciferase as a control reporter system under the Ccl2 endogenous promoter
Disease modeling and drug discovery
Despite the discovery of multiple risk alleles for OA through GWAS and twin studies (as described earlier), it is difficult to separate the influence of environmental factors from genetics on OA predisposition in vivo. The use of genome-editing in iPSCs now opens up the possibility for functional interrogation of causative genetic elements, such as coding/noncoding SNPs, as the basis for more targeted—even “personalized”—drug discovery for OA. For example, recent studies have developed in vitro tissue-engineered models of human and mouse iPSC chondrogenesis that show responsiveness to OA-associated cytokines such as IL1 and TNF-α similar to native cartilage [82–83]. These in vitro modeling platforms demonstrate the potential application of iPSCs and gene editing systems such as CRISPR-Cas9 to high-throughput screening of novel DMOADs that account for various genetic backgrounds and risk alleles [50, 73].
Conclusions and applications
Due to fundamental developments in the field of genome engineering, an array of novel therapeutic options for OA are now accessible and available to explore. Specifically, the development of targeted and safe cell-based therapies that can sense and respond to endogenous levels of inflammation may provide a tremendous opportunity for the development of new therapies for OA. These cells can be differentiated into chondrocytes, tested in in vitro, and delivered in vivo for characterization and testing. Evaluating, refining and translating this potentially highly responsive therapeutic strategy for OA treatment could overcome the limitations of current anti-cytokine therapies or biologic drugs more broadly, ultimately posing less risk of adverse events to patients when compared to conventional pharmacological or biologic therapy. This approach may increase the effectiveness for OA compared to previous efforts to use biologics in this context. In addition to OA, many chronic diseases (rheumatoid arthritis, psoriatic arthritis, psoriasis, metabolic disease, diabetes) involve increased fluctuating levels of TNF-α and IL-1-mediated signaling. Long term, the scale up and applicability of this approach to other musculoskeletal disease models that could benefit from anti-cytokine therapy can be tested. The ability to deliver an auto-regulated anti-cytokine system with tunable sensitivity will allow for a range of applications in a wide variety of inflammatory conditions.
Acknowledgements
We acknowledge that this review is not exhaustive and the authors apologize to those whose work was not included due to space limitations. This study was supported in part by NIH Grants AR50245, AR48852, AG15768, AR48182, AG46927, OD10707, DK108742, EB018266, AR057235, AR073752, the Arthritis Foundation, and the Nancy Taylor Foundation for Chronic Diseases.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical statement
There are no animal experiments carried out for this article.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Pereira D, Peleteiro B, Araújo J, Branco J, Santos RA, Ramos E. The effect of osteoarthritis definition on prevalence and incidence estimates: a systematic review. Osteoarthritis Cartilage. 2011;19:1270–1285. doi: 10.1016/j.joca.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 2.Hunter DJ, McDougall JJ, Keefe FJ. The symptoms of osteoarthritis and the genesis of pain. Rheum Dis Clin North Am. 2008;34:623–643. doi: 10.1016/j.rdc.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Felson DT. An update on the pathogenesis and epidemiology of osteoarthritis. Radiol Clin North Am. 2004;42:1–9. doi: 10.1016/S0033-8389(03)00161-1. [DOI] [PubMed] [Google Scholar]
- 4.Blanco FJ, Rego-Pérez I. Editorial: Is it time for epigenetics in osteoarthritis? Arthritis Rheumatol. 2014;66:2324–2327. doi: 10.1002/art.38710. [DOI] [PubMed] [Google Scholar]
- 5.van Saase JL, van Romunde LK, Cats A, Vandenbroucke JP, Valkenburg HA. Epidemiology of osteoarthritis: Zoetermeer survey. Comparison of radiological osteoarthritis in a Dutch population with that in 10 other populations. Ann Rheum Dis. 1989;48:271–280. doi: 10.1136/ard.48.4.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson JJ, Felson DT. Factors associated with osteoarthritis of the knee in the first national Health and Nutrition Examination Survey (HANES I). Evidence for an association with overweight, race, and physical demands of work. Am J Epidemiol. 1988;128:179–189. doi: 10.1093/oxfordjournals.aje.a114939. [DOI] [PubMed] [Google Scholar]
- 7.Valdes AM, Spector TD. Genetic epidemiology of hip and knee osteoarthritis. Nat Rev Rheumatol. 2011;7:23–32. doi: 10.1038/nrrheum.2010.191. [DOI] [PubMed] [Google Scholar]
- 8.Spector TD, Cicuttini F, Baker J, Loughlin J, Hart D. Genetic influences on osteoarthritis in women: a twin study. BMJ. 1996;312:940–943. doi: 10.1136/bmj.312.7036.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Felson DT, Anderson JJ, Naimark A, Walker AM, Meenan RF. Obesity and knee osteoarthritis. The Framingham study. Ann Intern Med. 1988;109:18–24. doi: 10.7326/0003-4819-109-1-18. [DOI] [PubMed] [Google Scholar]
- 10.Felson DT, Zhang Y, Hannan MT, Naimark A, Weissman B, Aliabadi P, et al. Risk factors for incident radiographic knee osteoarthritis in the elderly: the Framingham Study. Arthritis Rheum. 1997;40:728–733. doi: 10.1002/art.1780400420. [DOI] [PubMed] [Google Scholar]
- 11.Blagojevic M, Jinks C, Jeffery A, Jordan KP. Risk factors for onset of osteoarthritis of the knee in older adults: a systematic review and meta-analysis. Osteoarthritis Cartilage. 2010;18:24–33. doi: 10.1016/j.joca.2009.08.010. [DOI] [PubMed] [Google Scholar]
- 12.Collins KH, Sharif B, Reimer RA, Sanmartin C, Herzog W, Chin R, et al. Association of Metabolic Markers with self-reported osteoarthritis among middle-aged BMI-defined non-obese individuals: a cross-sectional study. BMC Obes. 2018;5:23. doi: 10.1186/s40608-018-0201-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loughlin J. Genetic contribution to osteoarthritis development: current state of evidence. Curr Opin Rheumatol. 2015;27:284–288. doi: 10.1097/BOR.0000000000000171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berenbaum F, Griffin TM, Liu-Bryan R. Review: metabolic regulation of inflammation in osteoarthritis. Arthritis Rheumatol. 2017;69:9–21. doi: 10.1002/art.39842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kerkhof HJ, Lories RJ, Meulenbelt I, Jonsdottir I, Valdes AM, Arp P, et al. A genome-wide association study identifies an osteoarthritis susceptibility locus on chromosome 7q22. Arthritis Rheum. 2010;62:499–510. doi: 10.1002/art.27184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Panoutsopoulou K, Southam L, Elliott KS, Wrayner N, Zhai G, Beazley C, et al. Insights into the genetic architecture of osteoarthritis from stage 1 of the arcOGEN study. Ann Rheum Dis. 2011;70:864–867. doi: 10.1136/ard.2010.141473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Evangelou E, Valdes AM, Kerkhof HJ, Styrkarsdottir U, Zhu Y, Meulenbelt I, et al. Meta-analysis of genome-wide association studies confirms a susceptibility locus for knee osteoarthritis on chromosome 7q22. Ann Rheum Dis. 2011;70:349–355. doi: 10.1136/ard.2010.132787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Day-Williams AG, Southam L, Panoutsopoulou K, Rayner NW, Esko T, Estrada K, et al. A variant in MCF2L is associated with osteoarthritis. Am J Hum Genet. 2011;89:446–450. doi: 10.1016/j.ajhg.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ryder JJ, Garrison K, Song F, Hooper L, Skinner J, Loke Y, et al. Genetic associations in peripheral joint osteoarthritis and spinal degenerative disease: a systematic review. Ann Rheum Dis. 2008;67:584–591. doi: 10.1136/ard.2007.073874. [DOI] [PubMed] [Google Scholar]
- 20.Miyamoto Y, Mabuchi A, Shi D, Kubo T, Takatori Y, Saito S, et al. A functional polymorphism in the 5′ UTR of GDF5 is associated with susceptibility to osteoarthritis. Nat Genet. 2007;39:529–533. doi: 10.1038/2005. [DOI] [PubMed] [Google Scholar]
- 21.Valdes AM, Spector TD, Doherty S, Wheeler M, Hart DJ, Doherty M. Association of the DVWA and GDF5 polymorphisms with osteoarthritis in UK populations. Ann Rheum Dis. 2009;68:1916–1920. doi: 10.1136/ard.2008.102236. [DOI] [PubMed] [Google Scholar]
- 22.Evangelou E, Chapman K, Meulenbelt I, Karassa FB, Loughlin J, Carr A, et al. Large-scale analysis of association between GDF5 and FRZB variants and osteoarthritis of the hip, knee, and hand. Arthritis Rheum. 2009;60:1710–1721. doi: 10.1002/art.24524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reynard LN, Loughlin J. Genetics and epigenetics of osteoarthritis. Maturitas. 2012;71:200–204. doi: 10.1016/j.maturitas.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 24.Khan IM, Redman SN, Williams R, Dowthwaite GP, Oldfield SF, Archer CW. The development of synovial joints. Curr Top Dev Biol. 2007;79:1–36. doi: 10.1016/S0070-2153(06)79001-9. [DOI] [PubMed] [Google Scholar]
- 25.Southam L, Rodriguez-Lopez J, Wilkins JM, Pombo-Suarez M, Snelling S, Gomez-Reino JJ, et al. An SNP in the 5′-UTR of GDF5 is associated with osteoarthritis susceptibility in Europeans and with in vivo differences in allelic expression in articular cartilage. Hum Mol Genet. 2007;16:2226–2232. doi: 10.1093/hmg/ddm174. [DOI] [PubMed] [Google Scholar]
- 26.Egli RJ, Southam L, Wilkins JM, Lorenzen I, Pombo-Suarez M, Gonzalez A, et al. Functional analysis of the osteoarthritis susceptibility-associated GDF5 regulatory polymorphism. Arthritis Rheum. 2009;60:2055–2064. doi: 10.1002/art.24616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Daans M, Luyten FP, Lories RJ. GDF5 deficiency in mice is associated with instability-driven joint damage, gait and subchondral bone changes. Ann Rheum Dis. 2011;70:208–213. doi: 10.1136/ard.2010.134619. [DOI] [PubMed] [Google Scholar]
- 28.Parrish WR, Byers BA, Su D, Geesin J, Herzberg U, Wadsworth S, et al. Intra-articular therapy with recombinant human GDF5 arrests disease progression and stimulates cartilage repair in the rat medial meniscus transection (MMT) model of osteoarthritis. Osteoarthritis Cartilage. 2017;25:554–560. doi: 10.1016/j.joca.2016.11.002. [DOI] [PubMed] [Google Scholar]
- 29.arcOGEN Consortium. arcOGEN Collaborators. Zeggini E, Panoutsopoulou K, Southam L, Rayner NW, et al. Identification of new susceptibility loci for osteoarthritis (arcOGEN): a genome-wide association study. Lancet. 2012;380:815–823. doi: 10.1016/S0140-6736(12)60681-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Styrkarsdottir U, Thorleifsson G, Helgadottir HT, Bomer N, Metrustry S, Bierma-Zeinstra S, et al. Severe osteoarthritis of the hand associates with common variants within the ALDH1A2 gene and with rare variants at 1p31. Nat Genet. 2014;46:498–502. doi: 10.1038/ng.2957. [DOI] [PubMed] [Google Scholar]
- 31.Castaño Betancourt MC, Cailotto F, Kerkhof HJ, Cornelis FM, Doherty SA, Hart DJ, et al. Genome-wide association and functional studies identify the DOT1L gene to be involved in cartilage thickness and hip osteoarthritis. Proc Natl Acad Sci U S A. 2012;109:8218–8223. doi: 10.1073/pnas.1119899109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Evangelou E, Kerkhof HJ, Styrkarsdottir U, Ntzani EE, Bos SD, Esko T, et al. A meta-analysis of genome-wide association studies identifies novel variants associated with osteoarthritis of the hip. Ann Rheum Dis. 2014;73:2130–2136. doi: 10.1136/annrheumdis-2012-203114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zengini E, Hatzikotoulas K, Tachmazidou I, Steinberg J, Hartwig FP, Southam L, et al. Genome-wide analyses using UK Biobank data provide insights into the genetic architecture of osteoarthritis. Nat Genet. 2018;50:549–558. doi: 10.1038/s41588-018-0079-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Styrkarsdottir U, Helgason H, Sigurdsson A, Norddahl GL, Agustsdottir AB, Reynard LN, et al. Whole-genome sequencing identifies rare genotypes in COMP and CHADL associated with high risk of hip osteoarthritis. Nat Genet. 2017;49:801–805. doi: 10.1038/ng.3816. [DOI] [PubMed] [Google Scholar]
- 35.Castaño-Betancourt MC, Evans DS, Ramos YF, Boer CG, Metrustry S, Liu Y, et al. Novel genetic variants for cartilage thickness and hip osteoarthritis. PLoS Genet. 2016;12:e1006260. doi: 10.1371/journal.pgen.1006260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yau MS, Yerges-Armstrong LM, Liu Y, Lewis CE, Duggan DJ, Renner JB, et al. Genome-wide association study of radiographic knee osteoarthritis in North American Caucasians. Arthritis Rheumatol. 2017;69:343–351. doi: 10.1002/art.39932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fernández-Moreno M, Soto-Hermida A, Vázquez-Mosquera ME, Cortés-Pereira E, Pértega S, Relaño S, et al. A replication study and meta-analysis of mitochondrial DNA variants in the radiographic progression of knee osteoarthritis. Rheumatology (Oxford) 2017;56:263–270. doi: 10.1093/rheumatology/kew394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dupont C, Armant DR, Brenner CA. Epigenetics: definition, mechanisms and clinical perspective. Semin Reprod Med. 2009;27:351–357. doi: 10.1055/s-0029-1237423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simon TC, Jeffries MA. The epigenomic landscape in osteoarthritis. Curr Rheumatol Rep. 2017;19:30. doi: 10.1007/s11926-017-0661-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raman S, FitzGerald U, Murphy JM. Interplay of inflammatory mediators with epigenetics and cartilage modifications in osteoarthritis. Front Bioeng Biotechnol. 2018;6:22. doi: 10.3389/fbioe.2018.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peffers MJ, Balaskas P, Smagul A. Osteoarthritis year in review 2017: genetics and epigenetics. Osteoarthritis Cartilage. 2018;26:304–311. doi: 10.1016/j.joca.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roach HI, Yamada N, Cheung KS, Tilley S, Clarke NM, Oreffo RO, et al. Association between the abnormal expression of matrix-degrading enzymes by human osteoarthritic chondrocytes and demethylation of specific CpG sites in the promoter regions. Arthritis Rheum. 2005;52:3110–3124. doi: 10.1002/art.21300. [DOI] [PubMed] [Google Scholar]
- 43.de Andrés MC, Imagawa K, Hashimoto K, Gonzalez A, Roach HI, Goldring MB, et al. Loss of methylation in CpG sites in the NF-kappaB enhancer elements of inducible nitric oxide synthase is responsible for gene induction in human articular chondrocytes. Arthritis Rheum. 2013;65:732–742. doi: 10.1002/art.37806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rushton MD, Reynard LN, Barter MJ, Refaie R, Rankin KS, Young DA, et al. Characterization of the cartilage DNA methylome in knee and hip osteoarthritis. Arthritis Rheumatol. 2014;66:2450–2460. doi: 10.1002/art.38713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fernández-Tajes J, Soto-Hermida A, Vázquez-Mosquera ME, Cortés-Pereira E, Mosquera A, Fernández-Moreno M, et al. Genome-wide DNA methylation analysis of articular chondrocytes reveals a cluster of osteoarthritic patients. Ann Rheum Dis. 2014;73:668–677. doi: 10.1136/annrheumdis-2012-202783. [DOI] [PubMed] [Google Scholar]
- 46.Reynard LN, Bui C, Syddall CM, Loughlin J. CpG methylation regulates allelic expression of GDF5 by modulating binding of SP1 and SP3 repressor proteins to the osteoarthritis susceptibility SNP rs143383. Hum Genet. 2014;133:1059–1073. doi: 10.1007/s00439-014-1447-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meulenbelt I, Min JL, Bos S, Riyazi N, Houwing-Duistermaat JJ, van der Wijk HJ, et al. Identification of DIO2 as a new susceptibility locus for symptomatic osteoarthritis. Hum Mol Genet. 2008;17:1867–1875. doi: 10.1093/hmg/ddn082. [DOI] [PubMed] [Google Scholar]
- 48.Bomer N, den Hollander W, Ramos YF, Bos SD, van der Breggen R, Lakenberg N, et al. Underlying molecular mechanisms of DIO2 susceptibility in symptomatic osteoarthritis. Ann Rheum Dis. 2015;74:1571–1579. doi: 10.1136/annrheumdis-2013-204739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV, Thapar V, et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gaj T, Gersbach CA, Barbas CF., 3rd ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hinnen A, Hicks JB, Fink GR. Transformation of yeast. Proc Natl Acad Sci U S A. 1978;75:1929–1933. doi: 10.1073/pnas.75.4.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scherer S, Davis RW. Replacement of chromosome segments with altered DNA sequences constructed in vitro. Proc Natl Acad Sci U S A. 1979;76:4951–4955. doi: 10.1073/pnas.76.10.4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mansour SL, Thomas KR, Capecchi MR. Disruption of the proto-oncogene int-2 in mouse embryo-derived stem cells: a general strategy for targeting mutations to non-selectable genes. Nature. 1988;336:348–352. doi: 10.1038/336348a0. [DOI] [PubMed] [Google Scholar]
- 54.Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47:497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 55.Ceccaldi R, Rondinelli B, D’Andrea AD. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016;26:52–64. doi: 10.1016/j.tcb.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chiruvella KK, Liang Z, Wilson TE. Repair of double-strand breaks by end joining. Cold Spring Harb Perspect Biol. 2013;5:a012757. doi: 10.1101/cshperspect.a012757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci U S A. 1996;93:1156–1160. doi: 10.1073/pnas.93.3.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, et al. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol. 2011;29:143–148. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- 59.Mussolino C, Morbitzer R, Lutge F, Dannemann N, Lahaye T, Cathomen T. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 2011;39:9283–9293. doi: 10.1093/nar/gkr597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 61.Kim SJ, Kim CH, An B, Ha KS, Hong SH, Kim KS. Effects of clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated (Cas) protein 9 system-based deletion of miR-451 in mouse mmbryonic stem cells on their self-renewal and hematopoietic differentiation. Tissue Eng Regen Med. 2017;14:179–185. doi: 10.1007/s13770-017-0031-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471:602–607. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A. 2012;109:E2579–E2586. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507:62–67. doi: 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Anderson KR, Haeussler M, Watanabe C, Janakiraman V, Lund J, Modrusan Z, et al. CRISPR off-target analysis in genetically engineered rats and mice. Nat Methods. 2018;15:512–514. doi: 10.1038/s41592-018-0011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Akcakaya P, Bobbin ML, Guo JA, Malagon-Lopez J, Clement K, Garcia SP, et al. In vivo CRISPR editing with no detectable genome-wide off-target mutations. Nature. 2018;561:416–419. doi: 10.1038/s41586-018-0500-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Farhang N, Brunger JM, Stover JD, Thakore PI, Lawrence B, Guilak F, et al. CRISPR-based epigenome editing of cytokine receptors for the promotion of cell survival and tissue deposition in inflammatory environments. Tissue Eng Part A. 2017;23:738–749. doi: 10.1089/ten.tea.2016.0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Perez-Pinera P, Kocak DD, Vockley CM, Adler AF, Kabadi AM, Polstein LR, et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods. 2013;10:973–976. doi: 10.1038/nmeth.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Smolen JS, Aletaha D. Rheumatoid arthritis therapy reappraisal: strategies, opportunities and challenges. Nat Rev Rheumatol. 2015;11:276–289. doi: 10.1038/nrrheum.2015.8. [DOI] [PubMed] [Google Scholar]
- 72.Larsson S, Englund M, Struglics A, Lohmander LS. Interleukin-6 and tumor necrosis factor alpha in synovial fluid are associated with progression of radiographic knee osteoarthritis in subjects with previous meniscectomy. Osteoarthritis Cartilage. 2015;23:1906–1914. doi: 10.1016/j.joca.2015.05.035. [DOI] [PubMed] [Google Scholar]
- 73.Grunke M, Schulze-Koops H. Successful treatment of inflammatory knee osteoarthritis with tumour necrosis factor blockade. Ann Rheum Dis. 2006;65:555–556. doi: 10.1136/ard.2006.053272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ko JY, Lee J, Lee J, Im GI. Intra-articular xenotransplantation of adipose-derived stromal cells to treat osteoarthritis in a goat model. Tissue Eng Regen Med. 2017;14:65–71. doi: 10.1007/s13770-016-0010-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kobayashi T, Notoya K, Naito T, Unno S, Nakamura A, Martel-Pelletier J, et al. Pioglitazone, a peroxisome proliferator-activated receptor gamma agonist, reduces the progression of experimental osteoarthritis in guinea pigs. Arthritis Rheum. 2005;52:479–487. doi: 10.1002/art.20792. [DOI] [PubMed] [Google Scholar]
- 76.Kapoor M, Martel-Pelletier J, Lajeunesse D, Pelletier JP, Fahmi H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol. 2011;7:33–42. doi: 10.1038/nrrheum.2010.196. [DOI] [PubMed] [Google Scholar]
- 77.Adkar SS, Brunger JM, Willard VP, Wu CL, Gersbach CA, Guilak F. Genome engineering for personalized arthritis therapeutics. Trends Mol Med. 2017;23:917–931. doi: 10.1016/j.molmed.2017.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tarp S, Eric Furst D, Boers M, Luta G, Bliddal H, Tarp U, et al. Risk of serious adverse effects of biological and targeted drugs in patients with rheumatoid arthritis: a systematic review meta-analysis. Rheumatology (Oxford) 2017;56:417–425. doi: 10.1093/rheumatology/kew442. [DOI] [PubMed] [Google Scholar]
- 79.Brunger JM, Zutshi A, Willard VP, Gersbach CA, Guilak F. Genome engineering of stem cells for autonomously regulated, closed-loop delivery of biologic drugs. Stem Cell Reports. 2017;8:1202–1213. doi: 10.1016/j.stemcr.2017.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brunger JM, Zutshi A, Willard VP, Gersbach CA, Guilak F. CRISPR/Cas9 editing of murine induced pluripotent stem cells for engineering inflammation-resistant tissues. Arthritis Rheumatol. 2017;69:1111–1121. doi: 10.1002/art.39982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Im HJ, Liu Z, Li X, Bethea J. Inhibition of glial NF-KB abolishes pain in knee osteoarthritis model. Osteoarthritis Cartilage. 2016;24:S35.
- 82.Diekman BO, Christoforou N, Willard VP, Sun H, Sanchez-Adams J, Leong KW, et al. Cartilage tissue engineering using differentiated and purified induced pluripotent stem cells. Proc Natl Acad Sci USA. 2012;109:19172–19177. doi: 10.1073/pnas.1210422109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Adkar SS, Wu CL, Willard BP, Dicks A, Ettyreddy A, Steward N, et al. Step-wise chondrogenesis of human iPSCs and purification via a reporter allele generated by CRISPR-Cas9 genome editing. Stem Cells. 2018 doi: 10.1002/stem.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Willard VP, Diekman BO, Sanchez-Adams J, Christoforou N, Leong KW, Guilak F. Use of cartilage derived from murine induced pluripotent stem cells for osteoarthritis drug screening. Arthritis Rheumatol. 2014;66:3062–3072. doi: 10.1002/art.38780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lim JE, Son Y. Endogenous stem cells in homeostasis and aging. Tissue Eng Regen Med. 2017;14:679–698. doi: 10.1007/s13770-017-0097-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Son Y. Recent advances in stem cell researches and their future perspectives in regenerative medicine. Tissue Eng Regen Med. 2017;14:641–642. doi: 10.1007/s13770-017-0099-1. [DOI] [PMC free article] [PubMed] [Google Scholar]